
Submitted:
10 February 2024
Posted:
14 February 2024
You are already at the latest version
Abstract
This review provides an overview of advancements in plant genomics, emphasizing key stages in genomics projects and addressing associated challenges. Long read sequencing enables the cost-effective sequencing of plant DNA and assembly of highly continuous genome sequences - often even separating haplophases. Incorporating external hints, such as cDNA sequences from RNA-seq or full length cDNA sequencing, enhances the identification of gene models. While these steps enable high-throughput exploration of numerous plant genomes, a significant bottleneck lies in elucidating gene functions. The classical approach based on wet lab methods is impractical when dealing with thousands of genes in a new genome sequence. To overcome this challenge, computational tools harnessing existing information for cross species knowledge transfer are essential for expediting the functional annotation process. In support of researchers entering the field of plant genomics, a collection of recommended tools has been curated and is accessible at https://github.com/bpucker/ToolOverview.
Keywords:
plant genomics
; functional genomics
; gene function
; sequence comparison
; orthology
; synteny
How to Obtain a Gene Sequence?
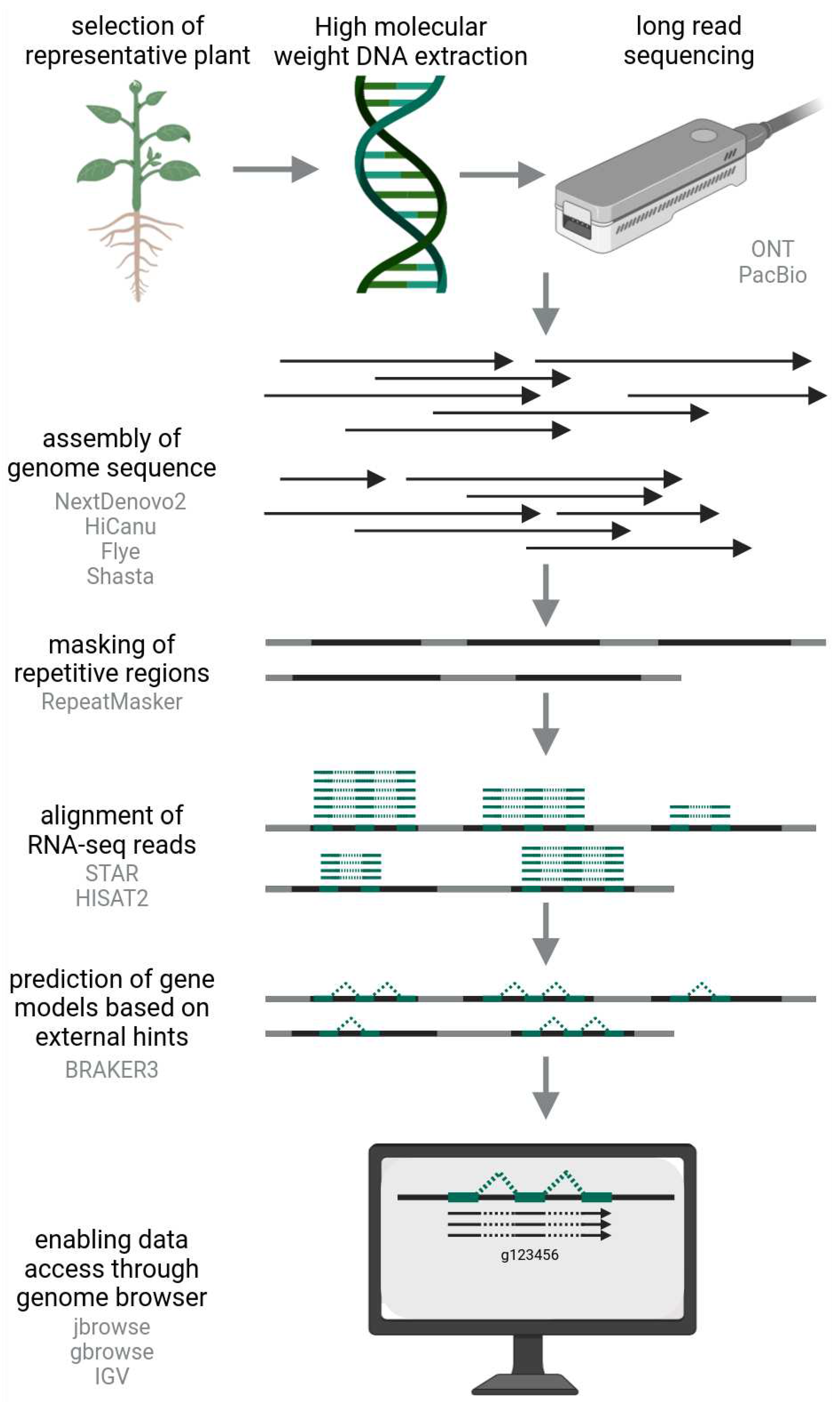
Access to the gene repertoire of a plant species is usually gained through a transcriptome assembly or a genome sequence. Given the large size of plant genomes and the large proportion of non-genic elements, transcriptome assemblies have been a cost-effective way to obtain protein encoding sequences of a species of interest (Haak et al., 2018). The rapid technological development of long read sequencing made the generation of high quality genome sequences affordable and feasible for many plant species (Marks et al., 2021; Pucker et al., 2022). Oxford Nanopore Technologies and Pacific Biosciences offer sequencing instruments that enable the continuous sequencing of long DNA molecules with high raw read accuracy of over 99%. The portability and affordable prices of ONT sequencers enable an ever increasing number of scientists to actively participate in plant genomics (Pucker et al., 2022). Sequencing data are often stored in FASTQ files that combine sequence and quality information (Cock et al., 2010). Long reads are processed by assemblers like HiCanu (Nurk et al., 2020), Flye (Kolmogorov et al., 2019), Shasta (Shafin et al., 2020), or NextDenovo2 (GrandOmics, 2023) to produce highly continuous genome sequences. These assembled sequences are called contigs and stored in a FASTA file (Lipman & Pearson, 1985). Continuous sequences (contigs) produced in the assembly process often represent chromosome arms or even entire chromosomes in some cases. The quality and completeness of the assembled sequences can be assessed with Merqury (Rhie et al., 2020) which checks the assembly for all k-mers that have been observed in the reads. Most prominent in plant genomes are repeats and transposable elements (TEs) that can be identified by tools like RepeatMasker (Smit et al., 2015), Extensive de-novo TE Annotator (EDTA) (Ou et al., 2019), or TransposonUltimate (Riehl et al., 2022). When multiple genome sequences of a species are investigated at the same time, panEDTA can be applied to benefit from the pangenome context (Ou et al., 2022). As repeats, especially in the centromeres, become more accessible with long reads, many studies are now investigating these parts of plant genomes (Naish et al., 2021; Wlodzimierz et al., 2023). Excluding positions of repeats and transposable elements (TEs) in the subsequent step of identifying protein-encoding plant genes is a common practice. This is done to ensure that transposon genes are not included in the annotation, maintaining the accuracy of the results. The gene prediction process can be performed by BRAKER3 (Gabriel et al., 2023), GeMoMa (Keilwagen et al., 2016, 2019), CAT (Fiddes et al., 2018), or Funannotate (Palmer, 2019) and results in gene models also known as structural annotation. Details about the positions and structures of all genes are typically stored in a GFF3 file. While the aforementioned tools predict only monocistronic gene models, OpenProt2021 supports polycistronic gene models in the annotation of eukaryotic genome sequences (Brunet et al., 2021). While polycistronic genes have been described in Chlamydomonas reinhardtii (Gallaher et al., 2021), there is currently limited information about the relevance of polycistronic genes in land plants (García-Ríos et al., 1997; Wang et al., 2019). RNA-seq reads are generated by sequencing fragments of cDNAs, which essentially consist of concatenated exons without the intervening presence of introns. When aligned to a genome sequence, gaps in the alignment of RNA-seq reads span the positions of introns. Therefore, RNA-seq reads can reveal the exon/intron structure of a gene and indicate which exons belong to the same gene. This process requires dedicated tools like STAR (Dobin et al., 2013; Dobin & Gingeras, 2015) or HISAT2 (Kim et al., 2019), which can accurately split alignments around introns. Reads derived from direct RNA sequencing or full length cDNA sequencing enable the annotation of distinct transcript isoforms that can be the result of alternative splicing (Amarasinghe et al., 2020; Guizard et al., 2023). Similarly, polypeptide sequences from databases can be aligned to a genome sequence to inform the gene prediction process. However, the inclusion of polypeptide sequence derived hints might lead to high rates of false-positive predictions (Vuruputoor et al., 2023). These types of external information already indicate a major limitation of today’s gene prediction approaches: the structural annotation is restricted to the transcribed region of a gene and does not cover the regulatory elements in the promoter which would also be part of a plant gene. The completeness of a genome sequence and the corresponding structural annotation can be assessed with BUSCO (Simão et al., 2015; Manni et al., 2021) that checks for the presence of highly conserved single copy genes. The completeness reported by BUSCO for a genome sequence usually exceeds the reported completeness reported for the corresponding annotation. This might be due to the inclusion of pseudogenes in the completeness analysis of genome sequences, while gene prediction tools would filter out such sequences. However, both values are often >95% suggesting that high quality genomic resources are routinely generated and that missing genes are an exception. In the gene prediction process, each gene receives a unique ID that enables access to all information collected about this gene. As this ID is specific for a structural annotation, different annotation versions and sources might use different IDs for the same biological entity. Matching the IDs between different annotation versions is a frequent task that requires specific rules for complicated cases where the annotation versions deviate substantially from each other. The result of this process is a mapping table that connects each gene ID from one annotation version to zero, one, or many IDs of another annotation version. Such mapping tables are particularly important if many different annotation versions exist for the same species and are used as reference by different research groups or consortia. An example would be Vitis vinifera, for which a range of different reference genome sequences and annotation versions were developed, sometimes in parallel by different groups, and favored by different parts of the community (Velasco et al., 2007; Muñoz et al., 2014; Grimplet et al., 2014; Velt et al., 2023; Shi et al., 2023). It is important to enable the connection of biological insights reported in scientific publications based on different annotation versions to integrate all knowledge in the body of literature and to avoid redundant research endeavors. Another purpose of a gene ID is to enable users to retrieve the underlying sequence together with any attached information from a database. A practical solution to make genomic data accessible is a genome browser as implemented in jbrowse (Buels et al., 2016; Diesh et al., 2023) or gbrowse (Stein, 2013). A graphical user interface and access via the internet enable users to retrieve the desired sequences of a gene of interest. Famous examples are The Arabidopsis Information Resource (TAIR) (Lamesch et al., 2012; Berardini et al., 2015), Banana Genome Hub (Droc et al., 2022), Sol Genomics Network (Fernandez-Pozo et al., 2015), and Coffee Genome Hub (Dereeper et al., 2015) that also provide additional information besides gene and genome sequences.
Figure 1.
Workflow of a plant genomics project leading to a genome sequence and corresponding annotation that can be accessed through a genome browser.
Figure 1.
Workflow of a plant genomics project leading to a genome sequence and corresponding annotation that can be accessed through a genome browser.
How to Understand the Function of a Gene?
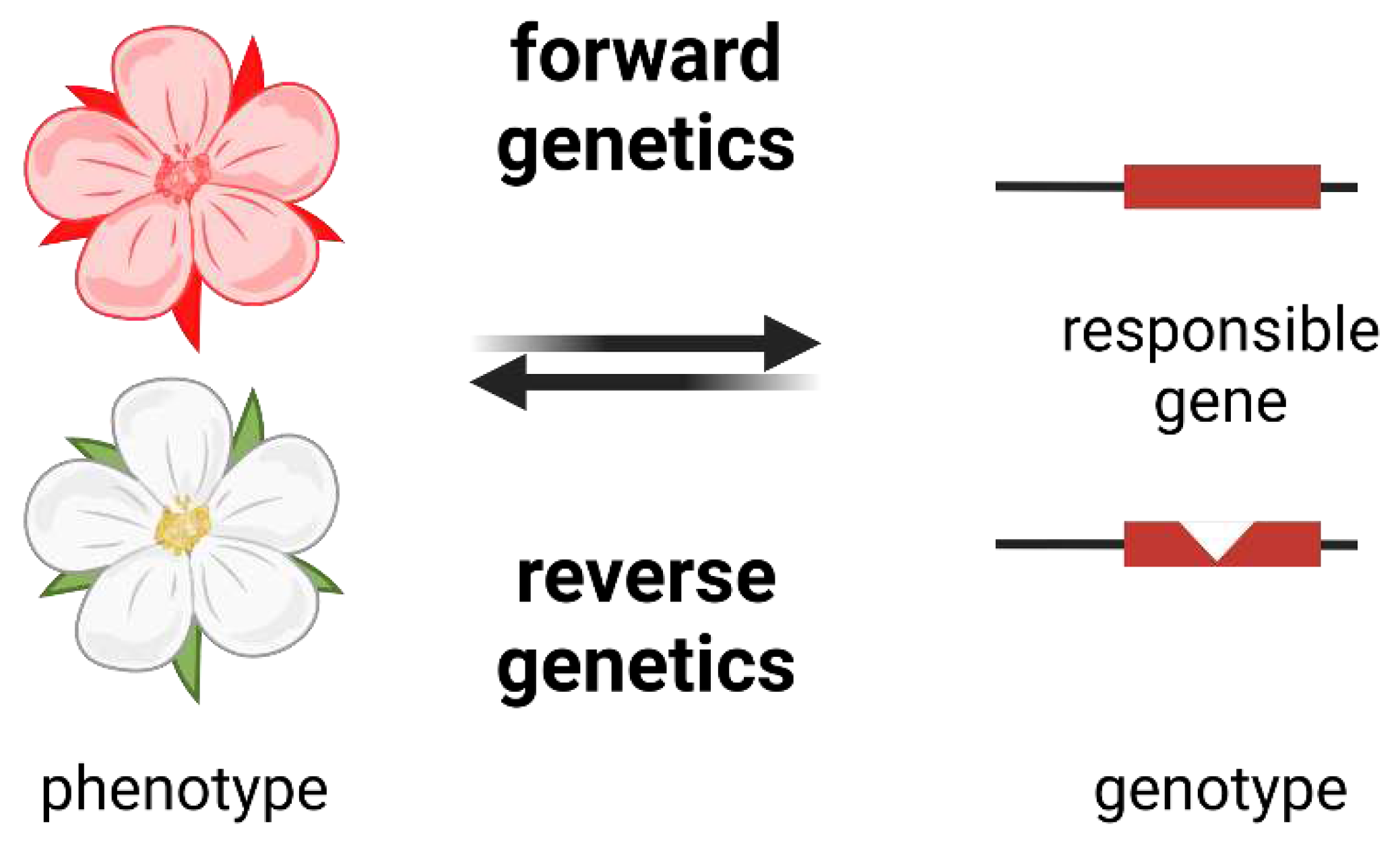
The classical reverse genetics approaches towards gene function elucidation are studying a knock-out or overexpression line. As a targeted integration of DNA into the plant genome through homologous recombination is not feasible, researchers had to rely on randomly introduced mutations during the last decades. Large collections of knock-out lines were established for model organisms like Arabidopsis thaliana (SALK, GABI-Kat) (Alonso et al., 2003; Rosso et al., 2003; O’Malley et al., 2015). These lines are based on a random integration of T-DNAs into the plant genome and a localization of the integration site in the genome (Rosso et al., 2003; Kleinboelting et al., 2012). Scientists can order a knock-out line that harbors a T-DNA inside their gene of interest through the NASC and ABRC. The floral dip method, which involves a Agrobacterium tumefaciens-mediated transfer of DNA into generative A. thaliana cells (Clough & Bent, 1998), has been a frequently applied method to generate stable transgenic lines. A careful characterization of T-DNA insertion lines is necessary as multiple T-DNA copies might affect different genes and can also trigger large-scale genomic rearrangements (Pucker et al., 2021). T-DNA based integration of genes into mutant lines can also serve as a method to characterize genes of non-model organisms in A. thaliana through complementation experiments (Lee et al., 2013; Schilbert et al., 2021; Aslam et al., 2022). The development of different CRISPR-Cas9-derived systems enables plant biologists to generate mutants in a targeted way (Grützner et al., 2021). However, this still requires the transformation of plants with the necessary constructs and a following validation of the introduced genomic changes. A gene knock-out is not possible for essential genes as their loss would be lethal. This makes knock-down a powerful alternative strategy. Initially triggered by chance in Petunia engineering experiments (Napoli et al., 1990), knock-down, i.e., reduction of transcript abundance, developed into a strategy in functional plant genomics (Samuilov et al., 2018; Debladis et al., 2020; Demirer & Landry, 2021). Virus-induced gene silencing (VIGS) emerged as a convenient tool to also knock-down genes in non-model organisms (Ruiz et al., 1998; Lu et al., 2003; Dommes et al., 2019).
Forward genetics is the opposite approach that can reveal the gene responsible for a certain phenotype. Frequently deployed methods to identify causal genes are genome-wide association studies (GWAS) (Lee & Lee, 2021; Gloss et al., 2022) and mapping-by-sequencing (MBS) (Schneeberger & Weigel, 2011; James et al., 2013) followed by an in-depth investigation of an identified quantitative trait locus (QTL). The concept of these approaches is to find systematic genetic differences between two groups of individuals that have been pooled based on their phenotype. These systematic genetic differences can be small sequence variants or presence/absence variants affecting entire genes. Tools like SnpEff (Cingolani et al., 2012) and NAVIP (Baasner et al., 2024) enable a prediction of the functional consequences of a sequence variant. GWAS and MBS have been deployed to study A. thaliana gene functions and to provide insights into crop genes determining important traits (Mascher et al., 2014; Sasaki et al., 2021; Schilbert et al., 2022; Naake et al., 2023; Sielemann et al., 2023).
Figure 2.
Schematic illustration of forward and reverse genetics. Forward genetics starts from the phenotype and identifies the underlying genotype. Reverse genetics starts from the genotype and aims to understand the resulting phenotype.
Figure 2.
Schematic illustration of forward and reverse genetics. Forward genetics starts from the phenotype and identifies the underlying genotype. Reverse genetics starts from the genotype and aims to understand the resulting phenotype.
How to Understand the Function of All Genes in a Genome?
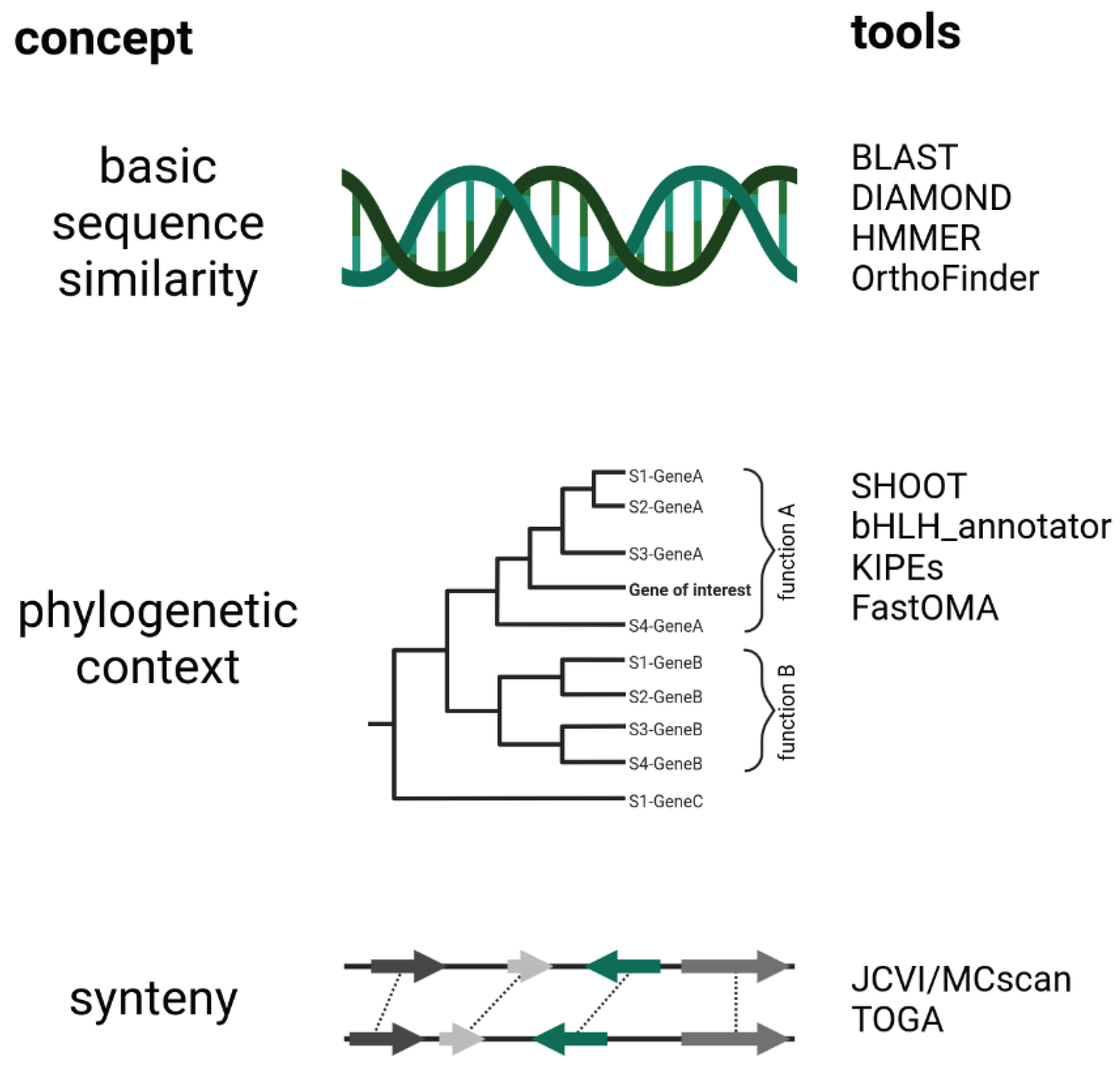
The gene function elucidation approaches described above suffer from poor scalability and are therefore not suitable for investigation of all gene functions in a newly sequenced plant genome. These have two major disadvantages: they are time intensive and costly. This restricts the investigation of gene functions through knock-out, overexpression, or knock-down lines to small numbers of genes and a small number of genetically accessible species. Given that a substantial amount of information about genes and their functions is already available in databases, individual users can access this through sequence comparison. This cross-species annotation transfer is based on the assumption that similar sequences have similar functions. While this holds true in most cases, some very similar sequences, paralogs, can differ in their function. It is generally assumed that only orthologs, i.e., the same gene in different species, are highly likely to harbor the same function (Eisen, 1998). The major challenge in functional annotation is therefore the reliable and efficient identification of orthologs i.e., to clearly distinguish between orthologs and paralogs. Three conceptually different types of analyses can be distinguished that allow the connection of genes between species. The first type utilizes only the sequence of a gene or the derived polypeptide sequence for basic similarity analyses. The second type performs a phylogenetic analysis to place the sequence of interest in an evolutionary context with similar sequences. The third type screens for genomic regions where a number of flanking genes, not just the gene of interest, show similarity between two species. This last approach utilizes information beyond the borders of the gene itself to establish a more reliable pair of orthologs.
Gene Sequence Similarity Analyses
Basic Local Alignment Search Tool (BLAST) is the most frequently used tool in life sciences and enables a quick comparison of a sequence against a comprehensive database (Altschul et al., 1990, 1997). The availability of BLAST through websites hosted by the NCBI, Phytozome (Goodstein et al., 2012), or TAIR (Lamesch et al., 2012; Berardini et al., 2015) enables life scientists to explore a gene function without computational biology skills. Annotation terms associated with the best BLAST hits can give a first impression about the function of the gene of interest. There is a large number of databases that contain valuable information about sequence functions. Examples are Plant Reactome (Naithani et al., 2020), MetaCyc (Caspi et al., 2020), Gene Ontology (GO) (Ashburner et al., 2000; Gene Ontology Consortium, 2021), Protein family (Pfam) (Mistry et al., 2021), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa & Goto, 2000; Kanehisa et al., 2023), and the large collection of sequences hosted by the International Nucleotide Sequence Database Collaboration, a consortium formed by GenBank, ENA, and DDBJ (Clark et al., 2016; Sayers et al., 2020; Arita et al., 2021). There are specific tools that can assign information of these databases to polypeptide sequences derived from a novel genome sequence. Examples are BLAST2GO (Conesa et al., 2005) that attaches GO terms and KAAS (Moriya et al., 2007) that assigns KEGG identifiers to sequences. InterProScan5 (Jones et al., 2014) assigns a range of different identifiers to novel polypeptide sequences including GO terms, KEGG identifier, Pfams, and PANTHER annotation terms. Mercator4 (Lohse et al., 2014) is available as a web service and can efficiently annotate provided polypeptide sequences by assigning them to clusters of similar sequences. BLAST is one of the most frequently used tools for the identification of similar sequences, but it is not a reliable method to identify orthologs, because only small stretches of highly similar sequences are considered for the alignment (Altschul et al., 1990). The chances of identifying bona fide orthologs can be increased by performing a reciprocal best BLAST hit analysis, which adds an additional filter layer (Pucker et al., 2016). Comparing all polypeptide sequences of a species against another species or even an entire database has huge computational costs and can result in long run times. Such tasks usually require a parallel analysis of sequences in batches on a high performance compute cluster. A faster BLAST alternative is DIAMOND (Buchfink et al., 2015, 2021), but this speed increase comes at the expense of higher memory consumption. If a large number of sequences needs to be compared against a database, users might want to use DIAMOND instead of BLAST. Eukaryotic Non-Model Transcriptome Annotation Pipeline (ENTAP) is a dedicated tool for the functional annotation of transcriptome assembly sequences through comparison against several databases (Hart et al., 2020). There are also dedicated tools for the identification of orthologs. JustOrthologs identifies putative orthologs through characteristics like gene structure, CDS length, and dinucleotide percentages in a computationally efficient way (Miller et al., 2019). SwiftOrtho is a graph-based tool for the identification of orthologs that was also developed with a focus on minimizing the computational costs (Hu & Friedberg, 2019). Since there are often not 1:1-relationships between species, a number of orthologous sequences must be collected in an orthogroup. OrthoFinder2 (Emms & Kelly, 2019) enables an automatic identification of orthogroups based on a number of polypeptide sequence sets belonging to different species. An advantage of this analysis is the ability to integrate polypeptide sequences derived from de novo transcriptome assemblies or short read-based genome sequence assemblies. Unfortunately, orthogroups might be too large and consequently not helpful in many scenarios where large gene families are of interest. Examples are the large transcription factor gene families MYB and bHLH that can have >100 members per plant species (Stracke et al., 2001; Zimmermann et al., 2004; Dubos et al., 2010; Thoben & Pucker, 2023) and are often clustered into a small number of orthogroups. As members of these gene families belong to dozens of subgroups that have individual functions (Dubos et al., 2010; Pucker et al., 2020a), a fine separation is required for accurate functional annotation transfer.
Phylogenetic Analyses
The construction of phylogenetic trees has been reported to be more accurate when predicting protein functions than the application of basic sequence similarity analyses, because they enable a more reliable identification of orthologs (Eisen, 1998; Sjölander, 2004; Brown & Sjölander, 2006; Pucker et al., 2020b). While other studies suggest that the inclusion of paralogs improves the accuracy in some pairwise comparisons (Nehrt et al., 2011; Stamboulian et al., 2020), an accurate assignment of homologous sequences across species forms the basis of the annotation transfer.
Databases like Phytozome (Goodstein et al., 2012), PANTHER (Thomas et al., 2022), OMA (Altenhoff et al., 2024), PhylomeDB (Fuentes et al., 2022), GreenPhylDB (Guignon et al., 2021), or OrthoDB (Kuznetsov et al., 2023) provide the phylogenetic relationships of all sequences belonging to the included organisms. Specific advantages and details about the underlying tools for the generation of many of these resources have been recently reviewed (de Boissier & Habermann, 2020). These phylogenies can be utilized to transfer annotation information between the included species, but would require additional processing to obtain a functional annotation file for a species of interest. A comprehensive and up-to-date API documentation facilitates the efficient utilization of these databases. Unfortunately, the representation of plant species in these databases is generally sparse.
Many computational tools can be run locally to compare sequence data sets with the objective of reliable ortholog identification for the following annotation transfer. A phylogeny-based analysis of all candidates enables an accurate annotation of members belonging to large gene families like MYBs and bHLHs (Pucker, 2022; Thoben & Pucker, 2023). The eggNOG-mapper v2 can functionally annotate all predicted polypeptide sequences of a new species through comparison against a large collection of previously computed orthogroups, the eggNOG database (Huerta-Cepas et al., 2019; Cantalapiedra et al., 2021). Unfortunately, this database does only contain a very limited number of plant datasets yet (Huerta-Cepas et al., 2019). Recently, SHOOT (Emms & Kelly, 2022) was released as a phylogenetics-informed alternative to BLAST. While the assignment of orthologs would be more reliable through SHOOT, it does not cover the entire dataset accessible through the NCBI-hosted BLAST web service. PharaohFUN assigns functional annotation information to supplied sequences based on gene trees and a data set covering a wide taxonomic diversity of plants (Ramos-González et al., 2023). FastOMA is another tool that was developed to efficiently identify orthologs in huge datasets that will be produced by the rapid generation of complete genome sequences (Majidian et al., 2024). Users might want to inspect the intermediate results that lead to the annotation of selected genes. If phylogenetic trees are generated as intermediate files by the above mentioned tools, these can be visualized in iTOL (Letunic & Bork, 2021). PhyloProfile (Tran et al., 2018) also enables users to visualize phylogenetic information associated with a gene and could provide another starting point for the manual inspection of selected cases. While online tools like PhyloFacts (Krishnamurthy et al., 2006) or OrthoVenn2 (Xu et al., 2019) could make the functional annotation process convenient, the throughput is often limited and the risk of becoming inaccessible due to broken links in the original publication is huge.
Synteny Analyses
An annotation transfer solution with very high resolution is the identification of syntelogs, i.e., genes that are located at the same genomic position (Lyons et al., 2008). This approach relies on synteny i.e., the order of genes being roughly the same in the compared plant genomes. As the order and orientation of genes is changing during evolution, this approach is limited to the comparison of species within a certain phylogenetic distance. The simultaneous analysis of multiple neighboring genes enables a more specific assignment of orthologs across species borders by including information outside the gene of interest. Tools for a synteny analysis are MCscan/JCVI (Tang et al., 2008), TBtools-II (Chen et al., 2023), and TOGA (Kirilenko et al., 2023). Synteny analyses are an excellent way to identify orthologs with high reliability and resolution, but the computational costs exceed those of a simple analysis via BLAST (Figure 3).
How Can Artificial Intelligence Improve the Functional Annotation Process?
One major challenge in genomics is the efficient exploitation of available data sets for the annotation of novel sequences. Various sequence databases show an exponential growth, which provides an excellent resource for data reuse (Sielemann et al., 2020; Marks et al., 2021). Simultaneously, it is also feasible and rewarding to automate processes that have been performed by human experts during the last years, e.g., the annotation of enzyme sequences. This increases reproducibility and allows scale-up of annotation processes. One example is Knowledge-based Identification of Pathway Enzymes (KIPEs) that automates all steps a researcher would conduct when exploring biosynthesis genes of a generally well-known pathway in a novel species (Pucker et al., 2020b; Rempel et al., 2023). This implementation of analysis steps needs to be combined with expert knowledge about the pathway of interest. When investigating enzymes, details about the functionally important amino acid residues (in the active center) could be a valuable source of information to predict whether an enzyme is active (Pucker et al., 2020b). So far, this requires an extensive body of literature about the pathway of interest, which is only available for widespread pathways like the flavonoid biosynthesis (Pucker et al., 2020b) or the carotenoid biosynthesis (Rempel et al., 2023). With increasing data availability, more pathways will be accessible through automatic processes. Also, literature research had to be done manually during the last years, but current artificial intelligence (AI) developments might enable automatic screening of publications in the near future (de la Torre-López et al., 2023). Open access publishing and the release of scientific publication in machine readable formats will pave the way to a more comprehensive cross-species transfer of knowledge. Whenever expert behavior can be described by clear rules and is not relying on ‘gut feeling’, an automation is feasible. Harnessing the full power of the scientific literature for upcoming annotation projects holds great promise.
Transcriptomic data sets can be a powerful resource to connect a candidate gene to members of a co-expression network if no information about any orthologs is available. JGI Plant Gene Atlas is a prime example of utilizing gene expression data to assign functional information to uncharacterized sequences (Sreedasyam et al., 2023). Coexpression networks conserved across species borders and characteristic responses of gene expression to stress treatments can be informative (Sreedasyam et al., 2023). As the resource can be updated continuously in the future, relevance will gain as more data becomes available. Well established tools to perform a co-expression analysis locally are WGCNA (Langfelder & Horvath, 2008) and GENIE3 (Huynh-Thu et al., 2010)/dynGENIE3 (Huynh-Thu & Geurts, 2018). These tools would require count tables that contain information about the activity of all genes. Downloading all RNA-seq datasets of a species and processing them is computationally intensive. Precomputed datasets might be available through Gene Expression Omnibus (GEO) (Barrett et al., 2013), which does collect some preprocessed datasets in addition to the raw RNA-seq reads. Once a co-expression network is constructed by any of the above mentioned tools, Cytoscape (Shannon et al., 2003) could be utilized to visualize the network for manual in-depth inspection. Connecting gene expression data to other omics or phenotypic data can also support the functional annotation process (Singh et al., 2022). Genes associated with a specific metabolite or a particular trait can be identified in this way. Examples are the discovery of the podophyllotoxin biosynthesis pathway in Podophyllum hexandrum (Lau & Sattely, 2015) or the montbretin A biosynthesis pathway in Crocosmia × crocosmiiflora (Irmisch et al., 2018, 2019). It is important to follow-up on these connections to distinguish between correlation and causation in these cases.
It might be feasible to exploit convergent evolution events for the transfer of annotation information. If the structure of two proteins is similar without orthology, the function might be similar too. FASSO (Andorf et al., 2022) identifies reciprocal best protein structure alignments in the identification of orthologs between two species. It might be feasible to extend such approaches to screen for 3D structural similarity rather than protein sequence similarity. Additionally, protein-protein interaction information can also help to understand the function of a protein encoding gene. STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) (Szklarczyk et al., 2015) harbors a comprehensive collection of this information, but does not cover the full taxonomic diversity of plants yet.
Large language models (LLMs) are useful in constructing annotation text that is easily accessible by humans. Given the amount of annotation terms that can be retrieved from various databases, generating a concise and accurate string of human-readable information is a major challenge. While tools like InterProScan5 (Jones et al., 2014) already compile a set of annotation details based on different databases, the tabular output requires re-structuring for readability. Previously, many annotation terms were stored as English words, but LLMs could easily enable multi language support. Generating more detailed annotation information by processing all of the available literature is another approach to advance gene function understanding. Databases connecting important pieces of information from scientific articles to genes are crucial functional genomics. The most striking example is TAIR that was built by experts curating the data (Huala et al., 2001; Lamesch et al., 2012; Berardini et al., 2015). Recently, PlantConnectome was constructed by screening over 100,000 abstracts of plant biology publications for information about the functions of genes (Fo et al., 2023). While the innovation potential of LLMs is enormous, there are also potential risks and challenges associated with their use, e.g., the requirement for appropriate filtering when collecting the database or ensuring the accuracy of generated output. A particular challenge is the avoidance of circular conclusions when research data generated with LLMs serves as a basis for the development of future LLMs.
Summary
Rapid development of long read sequencing technologies over the last years have enabled an almost routine generation of high quality genome sequences and structural annotations. The major bottleneck is currently the elucidation of gene functions. A range of different approaches transfer knowledge between orthologs across species borders. Large transcriptomic resources enable a reference-independent assignment of novel genes to biosynthesis pathways or processes based on comprehensive co-expression analyses. In the future, additional connections with other omics datasets and prediction of protein structures could establish novel annotation approaches.
References
- Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, et al.2003. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science (New York, N.Y.) 301: 653–657. [CrossRef]
- Altenhoff AM, Warwick Vesztrocy A, Bernard C, Train C-M, Nicheperovich A, Prieto Baños S, Julca I, Moi D, Nevers Y, Majidian S, et al.2024. OMA orthology in 2024: improved prokaryote coverage, ancestral and extant GO enrichment, a revamped synteny viewer and more in the OMA Ecosystem. Nucleic Acids Research 52: D513–D521. [CrossRef]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology 215: 403–410. [CrossRef]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids Research 25: 3389–3402. [CrossRef]
- Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. 2020. Opportunities and challenges in long-read sequencing data analysis. Genome Biology 21: 30. [CrossRef]
- Andorf CM, Sen S, Hayford RK, Portwood JL, Cannon EK, Harper LC, Gardiner JM, Sen TZ, Woodhouse MR. 2022. FASSO: An AlphaFold based method to assign functional annotations by combining sequence and structure orthology. : 2022.11.10.516002. [CrossRef]
- Arita M, Karsch-Mizrachi I, Cochrane G, on behalf of the International Nucleotide Sequence Database Collaboration. 2021. The international nucleotide sequence database collaboration. Nucleic Acids Research 49: D121–D124. [CrossRef]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al.2000. Gene Ontology: tool for the unification of biology. Nature Genetics 25: 25–29. [CrossRef]
- Aslam M, She Z, Jakada BH, Fakher B, Greaves JG, Yan M, Chen Y, Zheng P, Cheng Y, Qin Y. 2022. Interspecific complementation-restoration of phenotype in Arabidopsis cuc2cuc3 mutant by sugarcane CUC2 gene. BMC Plant Biology 22: 47. [CrossRef]
- Baasner J-S, Rempel A, Howard D, Pucker B. 2024. NAVIP: Unraveling the Influence of Neighboring Small Sequence Variants on Functional Impact Prediction. : 596718. [CrossRef]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al.2013. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Research 41: D991–D995. [CrossRef]
- Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E, Huala E. 2015. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. genesis 53: 474–485. [CrossRef]
- de Boissier P, Habermann BH. 2020. A Practical Guide to Orthology Resources. In: Pontarotti P, ed. Evolutionary Biology—A Transdisciplinary Approach. Cham: Springer International Publishing, 41–77. [CrossRef]
- Brown D, Sjölander K. 2006. Functional Classification Using Phylogenomic Inference. PLOS Computational Biology 2: e77. [CrossRef]
- Brunet MA, Lucier J-F, Levesque M, Leblanc S, Jacques J-F, Al-Saedi HRH, Guilloy N, Grenier F, Avino M, Fournier I, et al.2021. OpenProt 2021: deeper functional annotation of the coding potential of eukaryotic genomes. Nucleic Acids Research 49: D380–D388. [CrossRef]
- Buchfink B, Reuter K, Drost H-G. 2021. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods 18: 366–368. [CrossRef]
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nature Methods12: 59–60. [CrossRef]
- Buels R, Yao E, Diesh CM, Hayes RD, Munoz-Torres M, Helt G, Goodstein DM, Elsik CG, Lewis SE, Stein L, et al.2016. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biology 17: 66. [CrossRef]
- Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. 2021. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 38: 5825–5829. [CrossRef]
- Caspi R, Billington R, Keseler IM, Kothari A, Krummenacker M, Midford PE, Ong WK, Paley S, Subhraveti P, Karp PD. 2020. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Research 48: D445–D453. [CrossRef]
- Chen C, Wu Y, Li J, Wang X, Zeng Z, Xu J, Liu Y, Feng J, Chen H, He Y, et al.2023. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Molecular Plant 16: 1733–1742. [CrossRef]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6: 80–92. [CrossRef]
- Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2016. GenBank. Nucleic Acids Research 44: D67–D72. [CrossRef]
- Clough SJ, Bent AF. 1998. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. The Plant Journal: For Cell and Molecular Biology 16: 735–743. [CrossRef]
- Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. 2010. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research 38: 1767–1771. [CrossRef]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [CrossRef]
- Debladis E, Lee T-F, Huang Y-J, Lu J-H, Mathioni SM, Carpentier M-C, Llauro C, Pierron D, Mieulet D, Guiderdoni E, et al.2020. Construction and characterization of a knock-down RNA interference line of OsNRPD1 in rice (Oryza sativa ssp japonica cv Nipponbare). Philosophical Transactions of the Royal Society B: Biological Sciences 375: 20190338. [CrossRef]
- Demirer GS, Landry MP. 2021. Efficient Transient Gene Knock-down in Tobacco Plants Using Carbon Nanocarriers. Bio-protocol 11: e3897. [CrossRef]
- Dereeper A, Bocs S, Rouard M, Guignon V, Ravel S, Tranchant-Dubreuil C, Poncet V, Garsmeur O, Lashermes P, Droc G. 2015. The coffee genome hub: a resource for coffee genomes. Nucleic Acids Research 43 (D1). [CrossRef]
- Diesh C, Stevens GJ, Xie P, De Jesus Martinez T, Hershberg EA, Leung A, Guo E, Dider S, Zhang J, Bridge C, et al.2023. JBrowse 2: a modular genome browser with views of synteny and structural variation. Genome Biology 24: 74. [CrossRef]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [CrossRef]
- Dobin A, Gingeras TR. 2015. Mapping RNA-seq Reads with STAR. Current Protocols in Bioinformatics 51: 11.14.1-11.14.19. [CrossRef]
- Dommes AB, Gross T, Herbert DB, Kivivirta KI, Becker A. 2019. Virus-induced gene silencing: empowering genetics in non-model organisms. Journal of Experimental Botany 70: 757–770. [CrossRef]
- Droc G, Martin G, Guignon V, Summo M, Sempéré G, Durant E, Soriano A, Baurens F-C, Cenci A, Breton C, et al.2022. The banana genome hub: a community database for genomics in the Musaceae. Horticulture Research 9: uhac221. [CrossRef]
- Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. 2010. MYB transcription factors in Arabidopsis. Trends in Plant Science 15: 573–581. [CrossRef]
- Eisen JA. 1998. Phylogenomics: Improving Functional Predictions for Uncharacterized Genes by Evolutionary Analysis. Genome Research 8: 163–167. [CrossRef]
- Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20: 238. [CrossRef]
- Emms DM, Kelly S. 2022. SHOOT: phylogenetic gene search and ortholog inference. Genome Biology 23: 85. [CrossRef]
- Fernandez-Pozo N, Menda N, Edwards JD, Saha S, Tecle IY, Strickler SR, Bombarely A, Fisher-York T, Pujar A, Foerster H, et al.2015. The Sol Genomics Network (SGN)--from genotype to phenotype to breeding. Nucleic Acids Research 43: D1036-1041. [CrossRef]
- Fiddes IT, Armstrong J, Diekhans M, Nachtweide S, Kronenberg ZN, Underwood JG, Gordon D, Earl D, Keane T, Eichler EE, et al.2018. Comparative Annotation Toolkit (CAT)—simultaneous clade and personal genome annotation. Genome Research 28: 1029–1038. [CrossRef]
- Fo K, Chuah YS, Foo H, Davey EE, Fullwood M, Thibault G, Mutwil M. 2023. PlantConnectome: knowledge networks encompassing >100,000 plant article abstracts. : 2023.07.11.548541. [CrossRef]
- Fuentes D, Molina M, Chorostecki U, Capella-Gutiérrez S, Marcet-Houben M, Gabaldón T. 2022. PhylomeDB V5: an expanding repository for genome-wide catalogues of annotated gene phylogenies. Nucleic Acids Research 50: D1062–D1068. [CrossRef]
- Gabriel L, Brůna T, Hoff KJ, Ebel M, Lomsadze A, Borodovsky M, Stanke M. 2023. BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. : 2023.06.10.544449. [CrossRef]
- Gallaher SD, Craig RJ, Ganesan I, Purvine SO, McCorkle SR, Grimwood J, Strenkert D, Davidi L, Roth MS, Jeffers TL, et al.2021. Widespread polycistronic gene expression in green algae. Proceedings of the National Academy of Sciences 118: e2017714118. [CrossRef]
- García-Ríos M, Fujita T, LaRosa PC, Locy RD, Clithero JM, Bressan RA, Csonka LN. 1997. Cloning of a polycistronic cDNA from tomato encoding gamma-glutamyl kinase and gamma-glutamyl phosphate reductase. Proceedings of the National Academy of Sciences of the United States of America 94: 8249–8254. [CrossRef]
- Gene Ontology Consortium. 2021. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Research 49: D325–D334. [CrossRef]
- Gloss AD, Vergnol A, Morton TC, Laurin PJ, Roux F, Bergelson J. 2022. Genome-wide association mapping within a local Arabidopsis thaliana population more fully reveals the genetic architecture for defensive metabolite diversity. Philosophical Transactions of the Royal Society B: Biological Sciences 377: 20200512. [CrossRef]
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al.2012. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Research 40: D1178–D1186. [CrossRef]
- GrandOmics. 2023. NextDenovo.
- Grimplet J, Adam-Blondon A-F, Bert P-F, Bitz O, Cantu D, Davies C, Delrot S, Pezzotti M, Rombauts S, Cramer GR. 2014. The grapevine gene nomenclature system. BMC Genomics 15: 1077. [CrossRef]
- Grützner R, Martin P, Horn C, Mortensen S, Cram EJ, Lee-Parsons CWT, Stuttmann J, Marillonnet S. 2021. High-efficiency genome editing in plants mediated by a Cas9 gene containing multiple introns. Plant Communications 2: 100135. [CrossRef]
- Guignon V, Toure A, Droc G, Dufayard J-F, Conte M, Rouard M. 2021. GreenPhylDB v5: a comparative pangenomic database for plant genomes. Nucleic Acids Research 49: D1464–D1471. [CrossRef]
- Guizard S, Miedzinska K, Smith J, Smith J, Kuo RI, Davey M, Archibald A, Watson M. 2023. nf-core/isoseq: simple gene and isoform annotation with PacBio Iso-Seq long-read sequencing. Bioinformatics 39: btad150. [CrossRef]
- Haak M, Vinke S, Keller W, Droste J, Rückert C, Kalinowski J, Pucker B. 2018. High Quality de Novo Transcriptome Assembly of Croton tiglium. Frontiers in Molecular Biosciences 5. [CrossRef]
- Hart AJ, Ginzburg S, Xu M (Sam), Fisher CR, Rahmatpour N, Mitton JB, Paul R, Wegrzyn JL. 2020. EnTAP: Bringing faster and smarter functional annotation to non-model eukaryotic transcriptomes. Molecular Ecology Resources 20: 591–604. [CrossRef]
- Hu X, Friedberg I. 2019. SwiftOrtho: A fast, memory-efficient, multiple genome orthology classifier. GigaScience 8: giz118. [CrossRef]
- Huala E, Dickerman AW, Garcia-Hernandez M, Weems D, Reiser L, LaFond F, Hanley D, Kiphart D, Zhuang M, Huang W, et al.2001. The Arabidopsis Information Resource (TAIR): a comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Research 29: 102–105. [CrossRef]
- Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al.2019. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47: D309–D314. [CrossRef]
- Huynh-Thu VA, Geurts P. 2018. dynGENIE3: dynamical GENIE3 for the inference of gene networks from time series expression data. Scientific Reports 8: 3384. [CrossRef]
- Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. 2010. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLOS ONE 5: e12776. [CrossRef]
- Irmisch S, Jo S, Roach CR, Jancsik S, Man Saint Yuen M, Madilao LL, O’Neil-Johnson M, Williams R, Withers SG, Bohlmann J. 2018. Discovery of UDP-Glycosyltransferases and BAHD-Acyltransferases Involved in the Biosynthesis of the Antidiabetic Plant Metabolite Montbretin A. The Plant Cell 30: 1864–1886. [CrossRef]
- Irmisch S, Ruebsam H, Jancsik S, Man Saint Yuen M, Madilao LL, Bohlmann J. 2019. Flavonol Biosynthesis Genes and Their Use in Engineering the Plant Antidiabetic Metabolite Montbretin A. Plant Physiology 180: 1277–1290. [CrossRef]
- James GV, Patel V, Nordström KJV, Klasen JR, Salomé PA, Weigel D, Schneeberger K. 2013. User guide for mapping-by-sequencing in Arabidopsis. Genome Biology 14: R61. [CrossRef]
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al.2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30: 1236–1240. [CrossRef]
- Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. 2023. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Research 51: D587–D592. [CrossRef]
- Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28: 27–30. [CrossRef]
- Keilwagen J, Hartung F, Grau J. 2019. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods in Molecular Biology (Clifton, N.J.) 1962: 161–177. [CrossRef]
- Keilwagen J, Wenk M, Erickson JL, Schattat MH, Grau J, Hartung F. 2016. Using intron position conservation for homology-based gene prediction. Nucleic Acids Research 44: e89. [CrossRef]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37: 907–915. [CrossRef]
- Kirilenko BM, Munegowda C, Osipova E, Jebb D, Sharma V, Blumer M, Morales AE, Ahmed A-W, Kontopoulos D-G, Hilgers L, et al.2023. Integrating gene annotation with orthology inference at scale. Science (New York, N.Y.) 380: eabn3107. [CrossRef]
- Kleinboelting N, Huep G, Kloetgen A, Viehoever P, Weisshaar B. 2012. GABI-Kat SimpleSearch: new features of the Arabidopsis thaliana T-DNA mutant database. Nucleic Acids Research 40: D1211–D1215. [CrossRef]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology 37: 540–546. [CrossRef]
- Krishnamurthy N, Brown DP, Kirshner D, Sjölander K. 2006. PhyloFacts: an online structural phylogenomic encyclopedia for protein functional and structural classification. Genome Biology 7: R83. [CrossRef]
- Kuznetsov D, Tegenfeldt F, Manni M, Seppey M, Berkeley M, Kriventseva EV, Zdobnov EM. 2023. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Research 51: D445–D451. [CrossRef]
- Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, et al.2012. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Research 40: D1202–D1210. [CrossRef]
- Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [CrossRef]
- Lau W, Sattely ES. 2015. Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science (New York, N.Y.) 349: 1224–1228. [CrossRef]
- Lee T, Lee I. 2021. Genome-Wide Association Studies in Arabidopsis thaliana: Statistical Analysis and Network-Based Augmentation of Signals. Methods in Molecular Biology (Clifton, N.J.) 2200: 187–210. [CrossRef]
- Lee HY, Seo J-S, Cho JH, Jung H, Kim J-K, Lee JS, Rhee S, Choi YD. 2013. Oryza sativa COI Homologues Restore Jasmonate Signal Transduction in Arabidopsis coi1-1 Mutants. PLOS ONE 8: e52802. [CrossRef]
- Letunic I, Bork P. 2021. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research 49: W293–W296. [CrossRef]
- Lipman DJ, Pearson WR. 1985. Rapid and Sensitive Protein Similarity Searches. Science 227: 1435–1441. [CrossRef]
- Lohse M, Nagel A, Herter T, May P, Schroda M, Zrenner R, Tohge T, Fernie AR, Stitt M, Usadel B. 2014. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant, Cell & Environment 37: 1250–1258. [CrossRef]
- Lu R, Martin-Hernandez AM, Peart JR, Malcuit I, Baulcombe DC. 2003. Virus-induced gene silencing in plants. Methods (San Diego, Calif.) 30: 296–303. [CrossRef]
- Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Wang X, Bowers J, Paterson A, Lisch D, et al.2008. Finding and Comparing Syntenic Regions among Arabidopsis and the Outgroups Papaya, Poplar, and Grape: CoGe with Rosids. Plant Physiology 148: 1772–1781. [CrossRef]
- Majidian S, Nevers Y, Kharrazi AY, Vesztrocy AW, Pascarelli S, Moi D, Glover N, Altenhoff AM, Dessimoz C. 2024. Orthology inference at scale with FastOMA. : 2024.01.29.577392. [CrossRef]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. 2021. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38: 4647–4654. [CrossRef]
- Marks RA, Hotaling S, Frandsen PB, VanBuren R. 2021. Representation and participation across 20 years of plant genome sequencing. Nature Plants 7: 1571–1578. [CrossRef]
- Mascher M, Jost M, Kuon J-E, Himmelbach A, Aßfalg A, Beier S, Scholz U, Graner A, Stein N. 2014. Mapping-by-sequencing accelerates forward genetics in barley. Genome Biology 15: R78. [CrossRef]
- Miller JB, Pickett BD, Ridge PG. 2019. JustOrthologs: a fast, accurate and user-friendly ortholog identification algorithm. Bioinformatics 35: 546–552. [CrossRef]
- Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, et al.2021. Pfam: The protein families database in 2021. Nucleic Acids Research 49: D412–D419. [CrossRef]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Research 35: W182–W185. [CrossRef]
- Muñoz C, Di Genova A, Maass A, Orellana A, Hinrichsen P, Aravena A. 2014. VITIS VINIFERA GENOME ANNOTATION IMPROVEMENT USING NEXT-GENERATION SEQUENCING TECHNOLOGIES AND NCBI PUBLIC DATA. Acta Horticulturae: 349–356. [CrossRef]
- Naake T, Zhu F, Alseekh S, Scossa F, Perez de Souza L, Borghi M, Brotman Y, Mori T, Nakabayashi R, Tohge T, et al.2023. Genome-wide association studies identify loci controlling specialized seed metabolites in Arabidopsis. Plant Physiology: kiad511. [CrossRef]
- Naish M, Alonge M, Wlodzimierz P, Tock AJ, Abramson BW, Schmücker A, Mandáková T, Jamge B, Lambing C, Kuo P, et al.2021. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374: eabi7489. [CrossRef]
- Naithani S, Gupta P, Preece J, D’Eustachio P, Elser JL, Garg P, Dikeman DA, Kiff J, Cook J, Olson A, et al.2020. Plant Reactome: a knowledgebase and resource for comparative pathway analysis. Nucleic Acids Research 48: D1093–D1103. [CrossRef]
- Napoli C, Lemieux C, Jorgensen R. 1990. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. The Plant Cell 2: 279–289. [CrossRef]
- Nehrt NL, Clark WT, Radivojac P, Hahn MW. 2011. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS computational biology 7: e1002073. [CrossRef]
- Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM, Koren S. 2020. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Research: gr.263566.120. [CrossRef]
- O’Malley RC, Barragan CC, Ecker JR. 2015. A User’s Guide to the Arabidopsis T-DNA Insertional Mutant Collections. Methods in molecular biology (Clifton, N.J.) 1284: 323–342. [CrossRef]
- Ou S, Collins T, Qiu Y, Seetharam AS, Menard CC, Manchanda N, Gent JI, Schatz MC, Anderson SN, Hufford MB, et al.2022. Differences in activity and stability drive transposable element variation in tropical and temperate maize. : 2022.10.09.511471. [CrossRef]
- Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, Lugo CSB, Elliott TA, Ware D, Peterson T, et al.2019. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology 20: 275. [CrossRef]
- Palmer J. 2019. funannotate v1.5.3. [CrossRef]
- Pucker B. 2022. Automatic identification and annotation of MYB gene family members in plants. BMC Genomics 23: 220. [CrossRef]
- Pucker B, Holtgräwe D, Sörensen TR, Stracke R, Viehöver P, Weisshaar B. 2016. A De Novo Genome Sequence Assembly of the Arabidopsis thaliana Accession Niederzenz-1 Displays Presence/Absence Variation and Strong Synteny. PLOS ONE 11: e0164321. [CrossRef]
- Pucker B, Irisarri I, Vries J de, Xu B. 2022. Plant genome sequence assembly in the era of long reads: Progress, challenges and future directions. Quantitative Plant Biology 3: e5. [CrossRef]
- Pucker B, Kleinbölting N, Weisshaar B. 2021. Large scale genomic rearrangements in selected Arabidopsis thaliana T-DNA lines are caused by T-DNA insertion mutagenesis. BMC Genomics 22: 599. [CrossRef]
- Pucker B, Pandey A, Weisshaar B, Stracke R. 2020a. The R2R3-MYB gene family in banana (Musa acuminata): Genome-wide identification, classification and expression patterns. PLOS ONE 15: e0239275. [CrossRef]
- Pucker B, Reiher F, Schilbert HM. 2020b. Automatic Identification of Players in the Flavonoid Biosynthesis with Application on the Biomedicinal Plant Croton tiglium. Plants 9: 1103. [CrossRef]
- Ramos-González M, Ramos-González V, Arvanitidou C, Hernández-García J, García-González M, Romero-Campero FJ. 2023. PharaohFUN: PHylogenomic Analysis foR plAnt prOtein History and FUNction elucidation. : 2023.08.01.551440. [CrossRef]
- Rempel A, Choudhary N, Pucker B. 2023. KIPEs3: Automatic annotation of biosynthesis pathways. PLOS ONE 18: e0294342. [CrossRef]
- Rhie A, Walenz BP, Koren S, Phillippy AM. 2020. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21: 245. [CrossRef]
- Riehl K, Riccio C, Miska EA, Hemberg M. 2022. TransposonUltimate: software for transposon classification, annotation and detection. Nucleic Acids Research 50: e64. [CrossRef]
- Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K, Weisshaar B. 2003. An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Molecular Biology 53: 247–259. [CrossRef]
- Ruiz MT, Voinnet O, Baulcombe DC. 1998. Initiation and Maintenance of Virus-Induced Gene Silencing. The Plant Cell 10: 937–946. [CrossRef]
- Samuilov S, Rademacher N, Brilhaus D, Flachbart S, Arab L, Kopriva S, Weber APM, Mettler-Altmann T, Rennenberg H. 2018. Knock-Down of the Phosphoserine Phosphatase Gene Effects Rather N- Than S-Metabolism in Arabidopsis thaliana. Frontiers in Plant Science 9.
- Sasaki E, Köcher T, Filiault DL, Nordborg M. 2021. Revisiting a GWAS peak in Arabidopsis thaliana reveals possible confounding by genetic heterogeneity. Heredity 127: 245–252. [CrossRef]
- Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. 2020. GenBank. Nucleic Acids Research 48: D84–D86. [CrossRef]
- Schilbert HM, Pucker B, Ries D, Viehöver P, Micic Z, Dreyer F, Beckmann K, Wittkop B, Weisshaar B, Holtgräwe D. 2022. Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus. Genes 13: 1131. [CrossRef]
- Schilbert HM, Schöne M, Baier T, Busche M, Viehöver P, Weisshaar B, Holtgräwe D. 2021. Characterization of the Brassica napus Flavonol Synthase Gene Family Reveals Bifunctional Flavonol Synthases. Frontiers in Plant Science 12.
- Schneeberger K, Weigel D. 2011. Fast-forward genetics enabled by new sequencing technologies. Trends in Plant Science 16: 282–288. [CrossRef]
- Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, Armstrong J, Tigyi K, Maurer N, Koren S, et al.2020. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nature Biotechnology 38: 1044–1053. [CrossRef]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research 13: 2498–2504. [CrossRef]
- Shi X, Cao S, Wang X, Huang S, Wang Y, Liu Z, Liu W, Leng X, Peng Y, Wang N, et al.2023. The complete reference genome for grapevine (Vitis vinifera L.) genetics and breeding. Horticulture Research 10: uhad061. [CrossRef]
- Sielemann K, Hafner A, Pucker B. 2020. The Reuse of Public Datasets in the Life Sciences: Potential Risks and Rewards.
- Sielemann K, Pucker B, Orsini E, Elashry A, Schulte L, Viehöver P, Müller AE, Schechert A, Weisshaar B, Holtgräwe D. 2023. Genomic characterization of a nematode tolerance locus in sugar beet. BMC Genomics 24: 748. [CrossRef]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. [CrossRef]
- Singh KS, van der Hooft JJJ, van Wees SCM, Medema MH. 2022. Integrative omics approaches for biosynthetic pathway discovery in plants. Natural Product Reports 39: 1876–1896. [CrossRef]
- Sjölander K. 2004. Phylogenomic inference of protein molecular function: advances and challenges. Bioinformatics (Oxford, England) 20: 170–179. [CrossRef]
- Smit A, Hubley R, Green P. 2015. RepeatMasker.
- Sreedasyam A, Plott C, Hossain MS, Lovell JT, Grimwood J, Jenkins JW, Daum C, Barry K, Carlson J, Shu S, et al.2023. JGI Plant Gene Atlas: an updateable transcriptome resource to improve functional gene descriptions across the plant kingdom. Nucleic Acids Research 51: 8383–8401. [CrossRef]
- Stamboulian M, Guerrero RF, Hahn MW, Radivojac P. 2020. The ortholog conjecture revisited: the value of orthologs and paralogs in function prediction. Bioinformatics 36: i219–i226. [CrossRef]
- Stein LD. 2013. Using GBrowse 2.0 to visualize and share next-generation sequence data. Briefings in Bioinformatics 14: 162–171. [CrossRef]
- Stracke R, Werber M, Weisshaar B. 2001. The R2R3-MYB gene family in Arabidopsis thaliana. Current Opinion in Plant Biology 4: 447–456. [CrossRef]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al.2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research 43: D447-452. [CrossRef]
- Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. 2008. Synteny and Collinearity in Plant Genomes. Science 320: 486–488. [CrossRef]
- Thoben C, Pucker B. 2023. Automatic annotation of the bHLH gene family in plants. : 2023.05.02.539087. [CrossRef]
- Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou L-P, Mi H. 2022. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Science 31: 8–22. [CrossRef]
- de la Torre-López J, Ramírez A, Romero JR. 2023. Artificial intelligence to automate the systematic review of scientific literature. Computing 105: 2171–2194. [CrossRef]
- Tran N-V, Greshake Tzovaras B, Ebersberger I. 2018. PhyloProfile: dynamic visualization and exploration of multi-layered phylogenetic profiles. Bioinformatics 34: 3041–3043. [CrossRef]
- Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, FitzGerald LM, Vezzulli S, Reid J, et al.2007. A High Quality Draft Consensus Sequence of the Genome of a Heterozygous Grapevine Variety. PLoS ONE 2: e1326. [CrossRef]
- Velt A, Frommer B, Blanc S, Holtgräwe D, Duchêne É, Dumas V, Grimplet J, Hugueney P, Kim C, Lahaye M, et al.2023. An improved reference of the grapevine genome reasserts the origin of the PN40024 highly homozygous genotype. G3 GenesGenomesGenetics 13: jkad067. [CrossRef]
- Vuruputoor VS, Monyak D, Fetter KC, Webster C, Bhattarai A, Shrestha B, Zaman S, Bennett J, McEvoy SL, Caballero M, et al.2023. Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes. Applications in Plant Sciences 11: e11533. [CrossRef]
- Wang K, Wang D, Zheng X, Qin A, Zhou J, Guo B, Chen Y, Wen X, Ye W, Zhou Y, et al.2019. Multi-strategic RNA-seq analysis reveals a high-resolution transcriptional landscape in cotton. Nature Communications 10: 4714. [CrossRef]
- Wlodzimierz P, Rabanal FA, Burns R, Naish M, Primetis E, Scott A, Mandáková T, Gorringe N, Tock AJ, Holland D, et al.2023. Cycles of satellite and transposon evolution in Arabidopsis centromeres. Nature 618: 557–565. [CrossRef]
- Xu L, Dong Z, Fang L, Luo Y, Wei Z, Guo H, Zhang G, Gu YQ, Coleman-Derr D, Xia Q, et al.2019. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Research 47: W52–W58. [CrossRef]
- Zimmermann IM, Heim MA, Weisshaar B, Uhrig JF. 2004. Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. The Plant Journal: For Cell and Molecular Biology 40: 22–34. [CrossRef]
Figure 3.
Methods for the high-throughput assignment of functional annotation to novel genes. Three different concepts can be distinguished: (A) analysis of basic sequence similarity, (B) analysis in phylogenetic context, and (C) analysis of synteny. The listed tools are only examples. See GitHub repository https://github.com/bpucker/ToolOverview for an extended list of tools for the functional annotation.
Figure 3.
Methods for the high-throughput assignment of functional annotation to novel genes. Three different concepts can be distinguished: (A) analysis of basic sequence similarity, (B) analysis in phylogenetic context, and (C) analysis of synteny. The listed tools are only examples. See GitHub repository https://github.com/bpucker/ToolOverview for an extended list of tools for the functional annotation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



