
Submitted:
09 February 2024
Posted:
12 February 2024
You are already at the latest version
Abstract
French Guiana, located in the Guiana Shield, is a natural reservoir for many zoonotic pathogens that are of considerable medical or veterinary importance. Until now, there are few data available on the description of parasites circulating in this area, especially on protozoan belonging to the phy-lum Apicomplexa; conversely, the neighbouring countries describe a high parasitic prevalence in animals and humans. Epidemiological surveillance is necessary, as new potentially virulent strains may emerge from these forest ecosystems, such as Amazonian toxoplasmosis. However, there is no standard tool for detecting protozoa in wildlife. In this study, we developed Meat-Borne-Parasite, a high-throughput meta-barcoding workflow for detecting Apicomplexa based on Oxford Nanopore Technologies sequencing platform using the 18S gene of fourteen Apicomplexa-positive samples collected in French Guiana. Sequencing reads were then analysed with MetONTIIME pipeline. Thanks to a scoring rule, we were able to classify as Apicom-plexa-positive 10 samples out of 14 and reveal the presence of co-carriages. The same samples were also sequenced with Illumina platform, for validation purposes. On samples identified as Apicomplexa-positive by both platforms, a strong positive correlation at up to genus level was reported. Overall, the presented workflow represents a reliable method for Apicomplexa detec-tion, which may pave the way for more comprehensive biomonitoring of zoonotic pathogens.
Keywords:
Apicomplexa
; Amazon
; NGS sequencing
; Meta-barcoding
; Wild mammals
1. Introduction
The Amazon biome stretches to the extensive Amazon basin, a territory delimited by the Andean regions in the West and the Cerrado in the South. It covers part of Brazil (49%), Bolivia (11%), Peru (16%), Ecuador, Colombia, Venezuela, Guyana, Suriname and French Guiana and hosts a wide range of wild fauna inside different ecosystems. Mammals are a potential reservoir for many zoonotic pathogens, including Apicomplexa taxa such as Sarcocystis spp., Cryptosporidium spp., or Toxoplasma gondii [1,2]. This phylum, also called Sporozoa, is a diverse group of protozoan parasites, that are unicellular eukaryotes and obligate intracellular without flagella, and were detected in different mammalian organs [3,4,5]. Using their apical complex and their secretory organelle structure, they invade the host cell [6], potentially causing chronic asymptomatic diseases or severe acute diseases in endemic areas (Beck et al., 2009).
In South America, many countries describe a high prevalence of these parasites in animal meat, as Sarocystis spp. in the tongue muscles of armadillos [9], in wild birds [10] or Toxoplasma gondii and Sarcocystis spp. detected in the Alouatta guariba clamitans [5]. Except for T. gondii [3,11], there are no data on the description of such parasites circulating in French Guiana, strongly hampering the assessment of the risk of human transmission. Indeed, humans can be contaminated through activities such as hunting, fishing or consumption of soiled water, raw or uncooked game meat [12,13,14], greatly enjoyed by the population. Environmental changes induced by deforestation and urbanization enable close exchanges between the different ecosystems, causing a disruption in the sylvatic cycle. The anthropization of natural environments exposes human population to the risks of the emergence of new virulent strains from wildlife. There is therefore a risk of introduction, circulation and emergence of new parasitic species, which are usually not adapted to humans, potentially causing severe pathologies, as described for Amazonian toxoplasmosis [15].
Currently, there is no standard tool for the detection of protozoa in wild animals. Indeed, in the few studies about protozoa in meat, the authors perform conventional PCR using 18S rRNA gene general primers, or also target Apicomplexa with specific primers to amplify one parasitic species of the phylum [5,16,17]. Others also used Indirect Fluorescent Antibody Test (IFAT), with high risk of cross reactions [18]. All these conventional serological or molecular methods are not suitable for the detection of co-infections. The development of high-throughput sequencing technologies is an approach now widely used in the environmental field to assess the diversity of microorganisms [19,20]. High-throughput sequencing platforms allow to comprehensively study and better reveal the diversity of species and co-infections present in a sample [21].
In this study, we developed a low-cost, portable and fast workflow based on Nanopore sequencing, to target the 18S rRNA gene for Apicomplexa detection. Nanopore reads was analysed using MetONTIIME pipeline, which fosters reproducibility and standardization, through the underlying QIIME2 [22] and Nextflow [23] frameworks. Moreover, thanks to the sequencing of matched samples on Illumina platform, we validated the workflow and showed a high reliability of taxa abundances at genus level.
2. Materials and Methods
2.1. Sampling and samples
The animals studied were from the TBIP collection, which has been set up during PARALIM project, whose aim was to assess the dietary risks associated with the consumption of wild animals, commonly practiced in French Guiana. Therefore, it was possible to identify the potential food risks but also the ecological risks threatening the survival of animal species. It is in this interest that a massive collection of animal organs from French Guiana was built. This collection of TBIP laboratory was constituted on a voluntary basis and without financial compensation from hunters, slaughterhouses and veterinarians between 2015 and 2019. Animal species were identified using the Ivanova et al. protocol [24].
For this study, the samples were selected according to i) their geographical origin: from the Cayenne Island region (from Cayenne, Dégrad-des-Cannes, Matoury, Larivot and Stoupan), the Eastern region (Cacao, Saint-Georges, National Road 2 and Bélizon), the Western region (from Sinnamary to Grand-Santi), the savannahs (from Montsinery to Kourou) ii) the type of organ selected according to knowledge of parasite cycles including those consumed by the local population and iii) the species of the animals collected, the lifestyle and diet of the species that may influence the risk of exposure to parasites.
Then, we selected 20 positive samples to Apicomplexa according to PCR test for the development of Meat-Borne-Parasite, a meta-barcoding workflow based on Oxford Nanopore Technologies sequencing platform.
2.2. Molecular analysis
2.2.1. DNA extraction
DNA purification was carried out using the QIAmp DNA mini kit (Qiagen®, France) according to supplier's recommendations. Lysis was performed overnight by adding 180 µL of ATL lysis buffer and 20 µL of proteinase K to the tissue sample (< 25 mg). Then, DNA was extracted from the lysate and the eluate was collected into 200 µL of elution buffer. A negative extraction control was included in the set by replacing the biopsy with water.
2.2.2. DNA amplification
Detection of apicomplexan parasites was performed using a conventional PCR with the 800 bp DNA fragment encoding the universal eukaryote 18S ribosomal RNA (18S rRNA) gene [25]. A set of primers ApiF18Sv1v5 (5’-GCC ATG CAT GTC TAA GTA TAA GCT TT-3’) and ApiR18Sv1v5 (5’-CTT TAA CAA ATC TAA GAA TTT CACC TCT G-3’) targeting V1 to V5 regions of 18S rRNA gene were designed by TBIP laboratory.
The mixture reaction was run in a final volume of 50 µL containing: 10 µL of Hot Fire Polymerase (HFP) enzyme (Solis Biodyne, Estonia), 2 µL of primer sense and antisense (5 µM), 33 µL of water and 5 µL of the DNA sample. Using the Arktik thermocycler (ThermoFisher Scientific, USA), the following PCR conditions were used : an initial denaturation at 95 °C for 15 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 54 °C for 45 s, and an extension at 72 °C for 90 s. The last stage was a final elongation at 72 °C for 5 min. A positive and a negative control containing known genomic DNA and water, respectively, were included. The amplification products were visualized in a transilluminator after the migration of the samples by electrophoresis in 1.2% agarose TBE gel. These 20 positives PCR amplicons were purified in solution according to the FastGene kit (NIPPON Genetics EUROPE®, Germany), and were then quantified using the Qubit (Qiagen®, USA).
2.2.3. Nanopore library preparation and sequencing
Nanopore sequencing library was built following the ligation sequencing kit and native barcoding kit (SQK-LSK109 with EXP-NBD196) protocol (Oxford Nanopore Technologies®, UK) according to manufacturer’s instructions. The library was then loaded on a R9.4.1 flow-cell (FLO-MIN106D, Oxford Nanopore technologies®, UK) and sequencing was carried out on a GridION® nanopore sequencer (Oxford Nanopore technologies®, UK) for 8 hours using MinKNOW v22.10.7.
2.2.4. Illumina library preparation and sequencing
DNA extraction was performed from genomic DNA of matched samples. Then, the amplification by PCR was carried out using the primer pair P1-TAReuk454FWd1-18S (5’-CCA GCA SCY GCG GTA ATT CC-3’) and P1-TAReuk454REV3-18S (5’-ACT TTC GTT CTT GAT YRA-3’), targeting the V4 region of 18S rRNA gene [26]. A Diatomea strain was included as a positive control together with two negative controls, which were respectively the tissue extraction control and the PCR background of the total library preparation process.
- Sequencing of the amplicon libraries was performed in a single Illumina MiSeq paired-end run with 2x250 bp read chemistry according to the Metabiote® protocol for 18S gene sequencing (Genoscreen®, Lille).
2.3. Bioinformatics processing
2.3.1. Meta-barcoding pipeline for Nanopore data analysis
Nanopore reads were base-called using Guppy v6.3.9 integrated into MinKNOW v22.10.7 with “hac” model and demultiplexing was performed requiring the presence of barcodes at both ends. Reads were then analysed with a novel bioinformatic pipeline, called MetONTIIME, based on Nextflow workflow manager and QIIME2 environment [22,23]. In particular, reads with quality > 7 were filtered with NanoFilt [27] and compressed to fastq.gz format. Reads were then imported in qiime2 v.2022.8.0 using “qiime tools import” and dereplicated using “qiime vsearch dereplicate-sequences” and “qiime vsearch cluster-features-de-novo”, to obtain a set of representative sequences and the corresponding table with read counts. Representative sequences were then aligned to Silva_132_99_18S database using “qiime feature-classifier classify-consensus-vsearch” requiring a minimum alignment identity of 90%, a minimum query coverage of 80% and performing consensus taxonomy assignment among the top 3 hits. Taxonomy tables were then filtered retaining only taxa belonging to Apicomplexa phylum. A scoring rule was developed for classifying a sample as Apicomplexa positive in case the number of reads of the sample assigned to Apicomplexa represents at least a 5-fold increase compared to the average number of reads from negative controls assigned to Apicomplexa in the same sequencing run. Accordingly, samples with less than 10 reads assigned to Apicomplexa were classified as Apicomplexa negative and were dropped from the analysis. Taxonomy tables and barplots describing the taxonomic classification at each taxonomic level were generated with “qiime taxa collapse” and “qiime taxa barplot”. All scripts for running the MetONTIIME pipeline are reported in https://github.com/MaestSi/MetONTIIME repository.
2.3.2. Meta-barcoding pipeline for Illumina data analysis
Reads were analysed with QIIME2_Illumina pipeline. In particular, fastq.gz files were imported in qiime2 v2022.8.0 using “qiime tools import” after generation of manifest.txt file, and PCR primers were trimmed with “qiime cutadapt trim-paired”. Overlapping mates were then merged with “qiime dada2 denoise-paired”. A set of amplicon sequence variants (ASVs) was obtained, together with a feature table, describing the occurrence of ASVs in each sample. The database Silva_132_99_18S was then imported with “qiime tools import”, and then “qiime feature- classifier extract-reads” and “qiime feature-classifier fit-classifier-naive-bayes” were used to train a Naïve-Bayes classifier on the region of the 18S gene amplified by TAReuk454FWD1-TAReukREV3 primers. ASVs were then classified with “qiime feature-classifier classify-sklearn”. Taxonomy tables were then filtered retaining only taxa belonging to Apicomplexa phylum, and samples with no reads assigned to Apicomplexa were dropped from the analysis. Taxonomy tables and barplots were generated with “qiime taxa collapse” and “qiime taxa barplot”. All scripts for running the pipeline are reported in https://github.com/MaestSi/QIIME2_Illumina repository.
2.3.3. Comparison of Nanopore and Illumina meta-barcoding results
Bioinformatic analysis was carried out to obtain taxonomic assignments for both platforms using Silva [28] as a reference database. Feature tables reporting the relative frequencies of taxa for Apicomplexa positive samples, obtained for both Nanopore and Illumina platforms, were merged into a single feature table. Relative frequencies for all the taxa collapsed at different taxonomic levels were then compared between the two platforms, and scatterplots were produced with ggplot2 [29]. Pearson correlation was then computed between relative frequencies obtained with the two platforms, and a t-test was performed to estimate the probability of the association being null.
3. Results
3.1. Sampling and Apicomplexa-positive samples identification
In order to set-up and validate Meat-Borne-Parasite workflow, a total of 20 samples from 15 individuals were first included in the study. These samples were from 6 different animal species (Table 1) and were obtained from lung [5], heart [7], tongue [7] and brain [1] tissues. They were collected in four main regions of the country: one in the Cayenne Island region, seven in the Eastern region, one in the Western region and nine in the savannahs (Figure 1). Only 14 of them showed a clear amplicon in the gel; the remaining samples were therefore dropped and excluded from the following analyses.
3.2. Matched samples sequencing with Nanopore and Illumina platforms
The Apicomplexa-positive samples were then processed and sequenced in parallel with Nanopore and Illumina platforms. In particular, samples for Nanopore sequencing were PCR amplified with ApiF18Sv1v5 and ApiR18Sv1v5 primers and sequencing was carried out for 8 hours on a GridION device, producing 233,676 reads in total (Table 2); while, samples for Illumina sequencing were PCR amplified with P1-TAReuk454FWd1-18S and P1-TAReuk454REV3-18S primers and sequencing was carried out on a MiSeq instrument with a 2x250 paired-end mode, producing 1,569,910 reads in total (Table 2).
3.3. Taxonomy assignment and platforms comparison
Starting from a total of 20 samples, 14 samples provided enough material for sequencing after 18S PCR amplification, while the remaining 6 samples were dropped. The PCR-amplified samples were then sequenced with both Nanopore and Illumina platforms, and bioinformatic analysis was carried out with MetONTIIME and QIIME2_Illumina pipelines, respectively, to obtain taxonomic assignment. The negative controls showed no reads assigned to Apicomplexa for Illumina platform, while up to 2 reads from negative controls were assigned to Apicomplexa for Nanopore platform, possibly due to demultiplexing errors. Reads from positive controls were assigned to the expected taxa.
Only reads assigned to Apicomplexa were retained for further analyses (Figure 2). Nanopore platform classified as Apicomplexa positive 10 out of 14 samples, according to a scoring rule we developed, which classifies a sample as Apicomplexa positive in case the number of reads assigned to Apicomplexa is at least 5-fold the average number of reads assigned to Apicomplexa for negative controls. While, Illumina platform classified as Apicomplexa positive all 14 samples. In particular, the 4 samples classified as Apicomplexa negative by Nanopore platform had a very low percentage of reads assigned to Apicomplexa also in Illumina analysis, namely 0.22%, 0.09%, 0.04% and 0.02% for G0159P, G0173CR, G0173L and G0225CR1 samples, respectively.
Interestingly, we reported some cases of co-carriage. For example, in sample G0130CR2, we detected only Theileria spp. with Illumina, while Nanopore detected Theileria spp., Babesia spp. and Isospora spp. Conversely, in sample G0233CR2, Nanopore detected only Theileiria spp., while Illumina detected both Theileria spp. and Sarcocystis spp.
Co-carriers were found in 28.5% (4/14) of samples studied with Illumina sequencing and 90% (9/10) of samples studied with Nanopore technology. Among the organs studied, we found certain parasites in a single organ, such as T. gondii in the heart. On the other hand, parasites such as Babesia spp. and Theileria spp. were found in all the organs analyzed: heart, lung and tongue.
Four protozoan families were identified through sequencing analysis: Piroplasmorida, Eimeriorina, Coccidia, Adeleorina, and Eugregarinorida. Among these families, 4 main genera (Sarcocystis spp., Theileria spp., Babesia spp., Toxoplasma spp.) were identified by both platforms and 5 other genera (Hepatozoon spp., Frenkelia spp., Isospora spp., Besnoitia spp. Eimeria spp.) were identified with Nanopore sequencing only.
We then focused on the 10 samples classified as Apicomplexa positive by both platforms and evaluated their relative taxa abundance at level 6 (genus level). This resulted in a strong positive correlation (r Pearson = 0.86; t-test p-value = 2.6e-11), confirming the soundness of the proposed approach (Figure 3).
4. Discussion
In this study we describe Meat-Borne-Parasite, a Nanopore sequencing-based workflow for Apicomplexa detection in wildlife samples. According to the literature, this is the first meta-barcoding study on meat targeting protozoa. An innovative matrix was used to highlight the biodiversity of microorganisms, especially from an Amazonian Forest environment. Numerous research teams are increasingly using meta-barcoding for biodiversity analysis of microbial ecology in various matrices (fish, faeces, soil, water). These innovative and powerful technologies are used in different fields for the detection of food frauds, identification of fish species or inspection of the dietary diversity of animals. However, bacteria are more frequently the target of the study, compared to parasites (20,30–33). For example, Ludwig, A. et al., and Howells et al. described the presence of microorganisms such as parasites in the meat, but it was not possible for them to establish potential co-carriage, due to the technologies used, hence the interest of our study [5,9].
Illumina MiSeq is the gold-standard and most frequently used platform in microbial ecology studies to describe biodiversity, but in recent years Oxford Nanopore Technologies has raised a lot of interest, thanks to the low price, portability, real-time features and capability to sequence long reads on the MinION device. Despite many studies showing the reliability of Nanopore sequencing for a variety of applications [34,35], the higher error rate compared to Illumina (modal accuracy with R9.4.1 chemistry is at about 96%, Figure S1) requires ad hoc bioinformatic pipelines and thorough validation studies. Moreover, advancements both in the base-calling algorithms and in the sequencing chemistry have greatly improved Nanopore sequencing accuracy. The latest “Q20+” chemistry, which was recently released on the market, allows the production of sequencing reads with sequencing accuracy higher than 99% [36,37].
Multiple studies have already focused on comparing taxa abundances provided by Nanopore and Illumina-based workflows, showing a positive correlation, although some others showed poor correlation [38,39,40,41]. In general, multiple factors concur in determining taxa abundances, as reads sequencing accuracy, bioinformatic pipeline, reference database and PCR primers used for amplification. In fact, due to Illumina fragment length limited to about 600-800 bp, different PCR primer pairs are frequently used with the two platforms, greatly reducing reciprocal overlap [42,43].
In this work, we validated our novel Meat-Borne-Parasite workflow, using Illumina platform as a gold-standard reference. In our workflow, the bioinformatic analysis is carried out by MetONTIIME, a novel QIIME2 pipeline based on Nextflow workflow manager, that exploits containerized technology for running all the steps consequentially in a resource-optimized way, enabling the streamlined analysis of multiple samples.
At genus level, we obtained a Pearson correlation value of 0.86 between the two platforms. Overall, we find a good agreement between the two platforms at up to genus level, further reinforcing the accuracy of the proposed approach. Conversely, a Pearson correlation value of 0.31 was obtained at species level, suggesting caution should be used for species-level assignments with meta-barcoding workflows.
While Illumina platform allowed to detect the presence of Apicomplexa in 14/14 samples, our Meat-Borne-Parasite workflow allowed to classify as Apicomplexa-positive 10/14 samples, based on a conservative scoring rule which requires strong evidence of reads of Apicomplexa origin, to classify a sample as positive. This rule accounts for residual demultiplexing errors, which may occur due to sequencing errors in the barcode regions. The four discordant samples were characterized by low relative abundance in Nanopore analysis, namely 0.22%, 0.09%, 0.04% and 0.02% respectively in samples G0159P, G0173CR, G0173L and G0225CR1. Therefore, the reasons why they were missed with Meat-Borne-Parasite workflow may be ascribed to lower sequencing throughput (about 15% of Illumina reads were sequenced with Nanopore), or to differential PCR primers efficiency. The lower sensitivity issue should be largely mitigated both increasing sequencing run time and using the newest R10.4.1 chemistry, which will enable higher sequencing accuracy and, in turn, further reduce demultiplexing errors.
Both platforms detected multiple co-occurring Apicomplexa genera, with a single species, Dasyprocta Leoprina, carried by a single parasite, Theileria spp.; however, this can be explained by the fact that we only studied a single individual for this animal species, and more samples from this species should be analysed to be representative. Finally, the Nanopore platform appears to be the most efficient one for global biodiversity parasite in wildlife species and can be used as a relevant tool for epidemiological or ecological surveillance on potentially human and animal pathogenic parasites, as Sarcocystis spp., Babesia spp. and Toxoplama gondi (3,13,44). Further or complementary analyses using specific qPCR should be carried out to specifically determine the prevalence of species potentially pathogenic to animals and humans.
5. Conclusions
In this study we showed the set-up of a novel workflow for Nanopore-based meta-barcoding aimed at detecting Apicomplexa infections in animal tissues. This protocol is simple and does not require extensive knowledge, such as morphological taxonomic identification skills. The strong correlation between the two sequencing platforms guarantees highly accurate genus-level results.
Moreover, this study shows a parasitic abundance in the wild fauna of French Guiana and allows the evaluation of several potential risks for humans and animals: the ecological risk, with predominant new or emerging parasitic strain that could decimate animal species and the food risk for the population, since wild meat consumption is widespread. Thanks to low-cost, portability and real-time sequencing, Nanopore-based meta-barcoding could enable local authorities to set up a surveillance system, thus contributing effectively to environmental biomonitoring tasks.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: Nanopore and Illumina relative frequency at level 6.
Author Contributions
Conceptualization M.P.D.; collection L.L., M.S.; methodology A.M., S.M., M.A.H, M.S., S.S., H.B., M.P.D.; analysis A.M., S.M., M.P.D.; writing- original draft A.M., S.M.; writing—review and editing M.A.H., M.P.D. and funding acquisition M.P.D., L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research benefited from European funding (ERDF/FEDER) within the framework of the PARALIM project (Synergie n° GY0012553) and from Investissement d’Avenir grants of the the Agence Nationale de la Recherche (CEBA: ANR-10-LABEX-25-01). This study was supported by the University of French Guiana.
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Regional Scientific Council for Natural Heritage in French Guiana (protocol approval the January 3th 2024).” for studies involving animals.
Data Availability Statement
Raw sequence reads generated in this study have been submitted to the SRA database (BioProject PRJNA1009124). Reviewers may access the data at the following link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA1009124?reviewer=e015diora6jrnc8eos2ampcjra
Acknowledgments
We acknowledge the contribution of the company Genoscreen especially Maïté LELIEVRE for performing the experimental with the Illumina technology and the Institut Pasteur de Guyane for the advice provided. We acknowledge the company Oxford Nanopore Technologies for the travel fellowship awarded to A. MATOUTE to attend the “Congres délocalisé des sociétés françaises de parasitologie et mycologie médicale” once the study was completed to promote research work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pathmanathan R, Kan SP. Three cases of human Sarcocystis infection with a review of human muscular sarcocystosis in Malaysia. Tropical and geographical medicine. 1992;44(1-2):102-8.
- Winter M, Abate SD, Pasqualetti MI, Fariña FA, Ercole ME, Pardini L, et al. Toxoplasma gondii and Trichinella infections in wild boars (Sus scrofa) from Northeastern Patagonia, Argentina. Preventive veterinary medicine. 2019;168:75-80. [CrossRef]
- Demar M, Ajzenberg D, Serrurier B, Dardé ML, Carme B. Atypical Toxoplasma gondii strain from a free-living jaguar (Panthera onca) in French Guiana. The American journal of tropical medicine and hygiene. 2008;78(2):195-7. [CrossRef]
- Dubey JP, Van Wilpe E, Calero-Bernal R, Verma SK, Fayer R. Sarcocystis heydorni, n. sp.(Apicomplexa: Sarcocystidae) with cattle (Bos taurus) and human (Homo sapiens) cycle. Parasitology Research. 2015;114:4143-7. [CrossRef]
- Ludwig A, Murer L, Santos HF dos, Ludwig A, Sangioni LA, Vogel FS. Molecular detection of Apicomplexa protozoa in tissues from Alouatta guariba clamitans. Pesquisa Veterinária Brasileira. 2021;41. [CrossRef]
- Fayer R. Sarcocystis spp. in Human Infections. Clin Microbiol Rev. oct 2004;17(4):894-902. [CrossRef]
- Beck HP, Blake D, Dardé ML, Felger I, Pedraza-Díaz S, Regidor-Cerrillo J, et al. Molecular approaches to diversity of populations of apicomplexan parasites. International journal for parasitology. 2009;39(2):175-89. [CrossRef]
- Santos A, van Aerle R, Barrientos L, Martinez-Urtaza J. Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Computational and Structural Biotechnology Journal. 2020;18:296-305. [CrossRef]
- Howells R, Carvalho A, Mello M, Rangel N. Morphological and histochemical observations on Sarcocystis from the nine-banded armadillo, Dasypus novemcinctus. Annals of Tropical Medicine & Parasitology. 1975;69(4):463-74. [CrossRef]
- Rêgo WMF do, Costa JGL, Baraviera RC de A, Pinto LV, Bessa G de L, Lopes REN, et al. Sarcocystidae em aves silvestres do sudeste do Brasil. Revista Brasileira de Parasitologia Veterinária. 2021;30. [CrossRef]
- Laure Laghoe, personal data.
- Brasseur P, Agnamey P, Emmanuel E, Pape JW, Vaillant M, Raccurt CP. Cryptosporidium contamination of surface and water supplies in Haiti. Archives of environmental & occupational health. 2011;66(1):12-7. [CrossRef]
- Demar M, Hommel D, Djossou F, Peneau C, Boukhari R, Louvel D, et al. Acute toxoplasmoses in immunocompetent patients hospitalized in an intensive care unit in French Guiana. Clinical Microbiology and Infection. 2012;18(7):E221-31. [CrossRef]
- Demar M, Ajzenberg D, Maubon D, Djossou F, Panchoe D, Punwasi W, et al. Fatal outbreak of human toxoplasmosis along the Maroni River: epidemiological, clinical, and parasitological aspects. Clinical Infectious Diseases. 2007;45(7):e88-95. [CrossRef]
- Carme B, Bissuel F, Ajzenberg D, Bouyne R, Aznar C, Demar M, et al. Severe acquired toxoplasmosis in immunocompetent adult patients in French Guiana. Journal of clinical microbiology. 2002;40(11):4037-44. [CrossRef]
- Dahlgren SS, Gjerde B. Genetic characterisation of six Sarcocystis species from reindeer (Rangifer tarandus tarandus) in Norway based on the small subunit rRNA gene. Veterinary Parasitology. 2007;146(3-4):204-13. [CrossRef]
- Hůrková L, Modrý D. PCR detection of Neospora caninum, Toxoplasma gondii and Encephalitozoon cuniculi in brains of wild carnivores. Veterinary parasitology. 2006;137(1-2):150-4. [CrossRef]
- Moré G, Basso W, Bacigalupe D, Venturini MC, Venturini L. Diagnosis of Sarcocystis cruzi, Neospora caninum, and Toxoplasma gondii infections in cattle. Parasitology Research. 2008;102:671-5. [CrossRef]
- Liu L, Li Y, Li S, Hu N, He Y, Pong R, et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and biotechnology. 2012;2012. [CrossRef]
- Santos A, van Aerle R, Barrientos L, Martinez-Urtaza J. Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Computational and Structural Biotechnology Journal. 2020;18:296-305. [CrossRef]
- DeMone C, McClure JT, Greenwood SJ, Fung R, Hwang MH, Feng Z, et al. A metabarcoding approach for detecting protozoan pathogens in wild oysters from Prince Edward Island, Canada. International Journal of Food Microbiology. 2021;360:109315. [CrossRef]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature biotechnology. 2019;37(8):852-7. [CrossRef]
- Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nature biotechnology. 2017;35(4):316-9. [CrossRef]
- Ivanova NV, Zemlak TS, Hanner RH, Hebert PD. Universal primer cocktails for fish DNA barcoding. Molecular Ecology Notes. 2007;7(4):544-8. [CrossRef]
- Hadziavdic K, Lekang K, Lanzen A, Jonassen I, Thompson EM, Troedsson C. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PloS one. 2014;9(2):e87624. [CrossRef]
- Simon M, Jardillier L, Deschamps P, Moreira D, Restoux G, Bertolino P, et al. Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems. Environmental Microbiology. 2015;17(10):3610-27. [CrossRef]
- De Coster W, D’hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34(15):2666-9. [CrossRef]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research. 2012;41(D1):D590-6. [CrossRef]
- Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York. ISBN 978-3-319-24277-4, https://ggplot2.tidyverse.org. 2016;
- Cuscó A, Viñes J, D’Andreano S, Riva F, Casellas J, Sánchez A, et al. Using MinIONTM to characterize dog skin microbiota through full-length 16S rRNA gene sequencing approach. BioRxiv. 2017;167015. [CrossRef]
- Daugaliyeva A, Daugaliyeva S, Ashanin A, Kanatbayev S, Beltramo C, Peletto S. Study of cattle microbiota in different regions of Kazakhstan using 16S metabarcoding analysis. Scientific Reports. 2022;12(1):16410. [CrossRef]
- Ho JK, Puniamoorthy J, Srivathsan A, Meier R. MinION sequencing of seafood in Singapore reveals creatively labelled flatfishes, confused roe, pig DNA in squid balls, and phantom crustaceans. Food Control. 2020;112:107144. [CrossRef]
- Quéméré E, Aucourd M, Troispoux V, Brosse S, Murienne J, Covain R, et al. Unraveling the dietary diversity of Neotropical top predators using scat DNA metabarcoding: A case study on the elusive Giant Otter. Environmental DNA. 2021;3(5):889-900. [CrossRef]
- Maestri S, Cosentino E, Paterno M, Freitag H, Garces JM, Marcolungo L, et al. A rapid and accurate MinION-based workflow for tracking species biodiversity in the field. Genes. 2019;10(6):468. [CrossRef]
- Marcolungo L, Passera A, Maestri S, Segala E, Alfano M, Gaffuri F, et al. Real-time on-site diagnosis of quarantine pathogens in plant tissues by nanopore-based sequencing. Pathogens. 2022;11(2):199. [CrossRef]
- Luo J, Meng Z, Xu X, Wang L, Zhao K, Zhu X, et al. Systematic benchmarking of nanopore Q20+ kit in SARS-CoV-2 whole genome sequencing. Frontiers in microbiology. 2022;4059. [CrossRef]
- Wagner GE, Dabernig-Heinz J, Lipp M, Cabal A, Simantzik J, Kohl M, et al. Real-time nanopore Q20+ sequencing enables extremely fast and accurate core genome MLST typing and democratizes access to high-resolution bacterial pathogen surveillance. Journal of Clinical Microbiology. 2023;61(4):e01631-22. [CrossRef]
- Acharya K, Khanal S, Pantha K, Amatya N, Davenport RJ, Werner D. A comparative assessment of conventional and molecular methods, including MinION nanopore sequencing, for surveying water quality. Scientific reports. 2019;9(1):15726. [CrossRef]
- Egeter B, Veríssimo J, Lopes-Lima M, Chaves C, Pinto J, Riccardi N, et al. Speeding up the detection of invasive bivalve species using environmental DNA: A Nanopore and Illumina sequencing comparison. Molecular Ecology Resources. 2022;22(6):2232-47. [CrossRef]
- Heikema AP, Horst-Kreft D, Boers SA, Jansen R, Hiltemann SD, de Koning W, et al. Comparison of illumina versus nanopore 16S rRNA gene sequencing of the human nasal microbiota. Genes. 2020;11(9):1105. [CrossRef]
- Srivathsan A, Loh RK, Ong EJ, Lee L, Ang Y, Kutty SN, et al. Network analysis with either Illumina or MinION reveals that detecting vertebrate species requires metabarcoding of iDNA from a diverse fly community. Molecular Ecology. 2022. [CrossRef]
- Iadarola B, Xumerle L, Lavezzari D, Paterno M, Marcolungo L, Beltrami C, et al. Shedding light on dark genes: enhanced targeted resequencing by optimizing the combination of enrichment technology and DNA fragment length. Scientific Reports. 2020;10(1):9424. [CrossRef]
- Maestri S, Maturo MG, Cosentino E, Marcolungo L, Iadarola B, Fortunati E, et al. A long-read sequencing approach for direct haplotype phasing in clinical settings. International Journal of Molecular Sciences. 2020;21(23):9177. [CrossRef]
- Bilhassi TB, Oliveira HN, Ibelli AM, Giglioti R, Regitano LC, Oliveira-Sequeira TC, et al. Quantitative study of Babesia bovis infection in beef cattle from São Paulo state, Brazil. Ticks and tick-borne diseases. 2014;5(3):234-8. [CrossRef]
Figure 1.
Geographical origin distribution of the individuals analyzed.
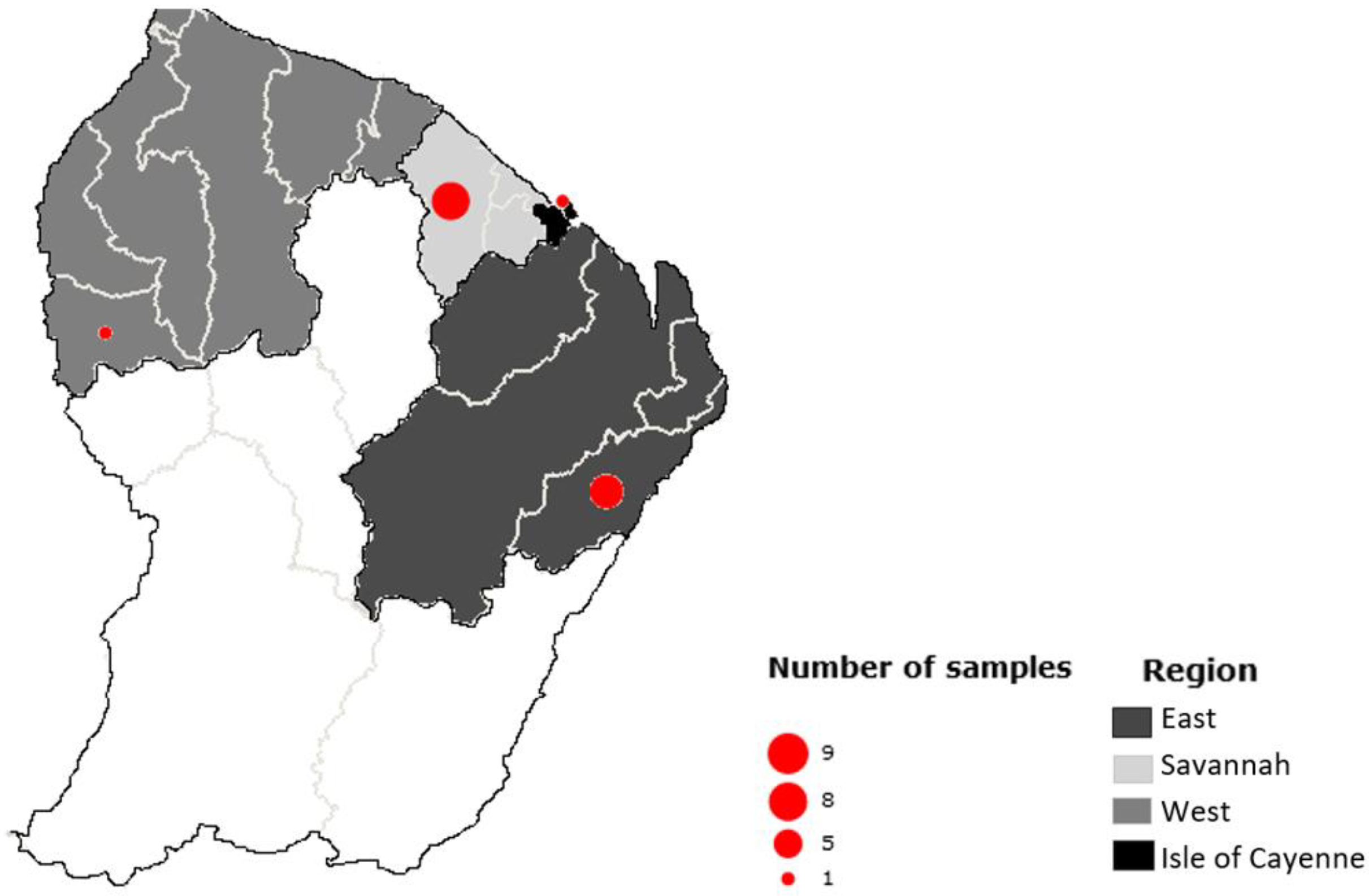
Figure 2.
Apicomplexa relative frequency in wildlife samples. For each sample, the relative frequency of reads assigned to Apicomplexa for Illumina and Nanopore matched samples at level 6 is reported. G0***L1 indicates the animal's ID, the first four elements indicate its code, followed by the letter representing the organ studied (P: lung; L or LD : tongue; CR : heart; R : spleen) and then the number representing the organ number.
Figure 2.
Apicomplexa relative frequency in wildlife samples. For each sample, the relative frequency of reads assigned to Apicomplexa for Illumina and Nanopore matched samples at level 6 is reported. G0***L1 indicates the animal's ID, the first four elements indicate its code, followed by the letter representing the organ studied (P: lung; L or LD : tongue; CR : heart; R : spleen) and then the number representing the organ number.
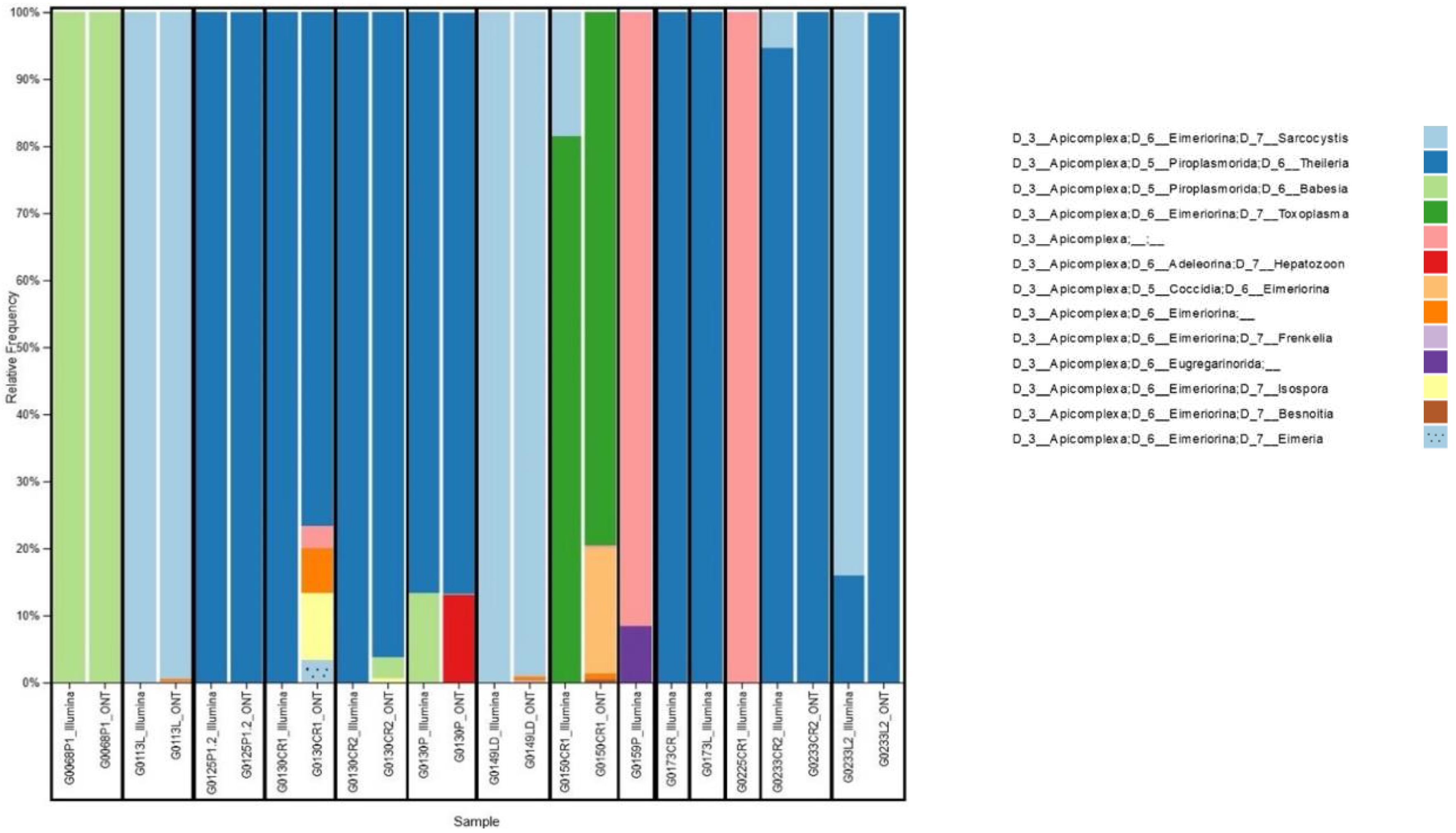
Figure 3.
Nanopore and Illumina relative frequency at level 6. For each sample, the relative frequency of genera identified by Nanopore and Illumina platforms was reported and labelled with the genus name. Dots are coloured by sample.
Figure 3.
Nanopore and Illumina relative frequency at level 6. For each sample, the relative frequency of genera identified by Nanopore and Illumina platforms was reported and labelled with the genus name. Dots are coloured by sample.
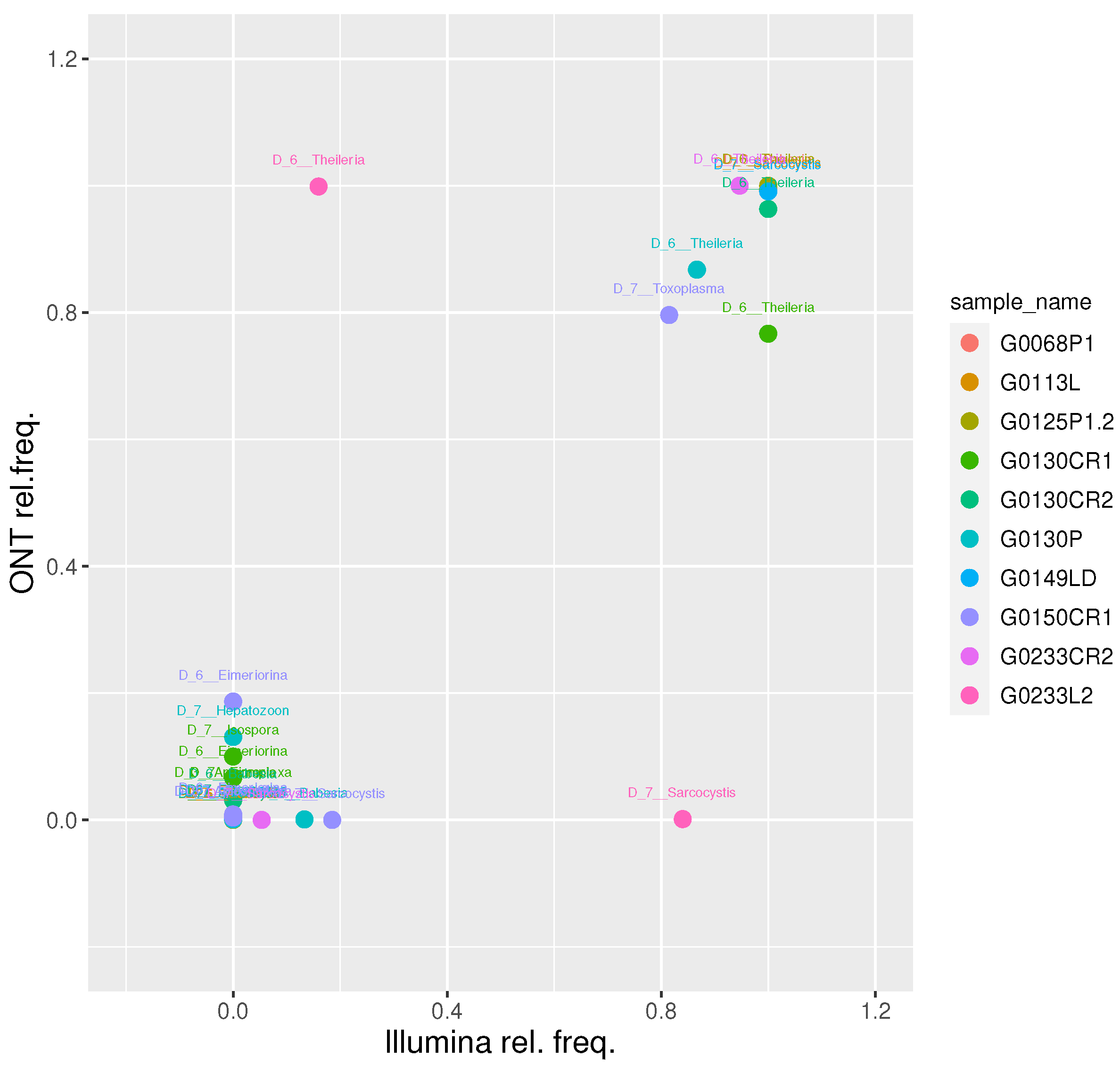

Table 1.
Distribution of animal species studied.
| Order Family | Genus species | Common name | Individual distribution |
| Rodentia Agoutidae | Cuniculus paca | Lowland paca, Paca | 1 |
| Rodentia Dasyproctidae | Dasyprocta leporina | Red-rumped agouti | 1 |
| Cingulata Dasypodidae | Dasypus sp. nov. | Nine-banded armadillo | 7 |
| Rodentia Caviidae | Hydrochoerus hydrochaeris | Capybara | 3 |
| Cetartiodactyla Cervidae | Mazama americana | Red brocket deer | 2 |
| Perissodactyla Tapiridae | Tapirus terrestris | Tapir | 1 |
| Total | 15 |
Table 2.
Demultiplexed sequences counts summary.
| Illumina reads | Nanopore reads | |
| Minimum | 71,565 | 18 |
| Median | 78,510 | 5,821 |
| Mean | 78,495.5 | 11,127.4 |
| Maximum | 91,032 | 42,449 |
| Total | 1,569,910 | 233,676 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



