
Submitted:
12 February 2024
Posted:
13 February 2024
You are already at the latest version
Abstract
The extracellular matrix (ECM) provides structural support for tissues and regulatory signals for resident cells. ECM requires a careful balance between protein accumulation and degradation for homeostasis. Disruption of this balance can lead to pathological processes such as fibrosis in organs across the body. Post-translational crosslinking modifications to ECM proteins such as collagens alter ECM structure and function. Dysregulation of crosslinking enzymes as well as changes in crosslinking composition are prevalent in fibrosis. Because of the crucial roles that ECM crosslinking pathways play in disease, the enzymes that govern crosslinking events are being explored as therapeutic targets for fibrosis. Here we review in-depth the molecular mechanisms underlying ECM crosslinking, how ECM crosslinking contributes to fibrosis, and therapeutic strategies being explored to target ECM crosslinking in fibrosis to restore normal tissue structure and function.
Keywords:
extracellular matrix
; crosslinking
; collagen
; fibrosis
1. Introduction
Fibrosis, or tissue “scarring”, is a complex, often lethal disease driven by different cell types and affecting multiple vital organs [1,2,3,4,5]. Even though fibrosis contributes to up to 45% of deaths in industrialized areas, there remains a limited understanding of how fibrosis develops over time and how the process could be reversed and resolved [2,6]. Available treatment options for fibrosis are currently minimal and lack disease-modifying efficacy [2]. One key cell type in fibrotic progression is the fibroblast. Upon activation by various local injury signals, fibroblasts transdifferentiate into myofibroblasts and produce excess extracellular matrix (ECM) [2,7,8]. Under normal, homeostatic circumstances, this process is necessary for repairing injured tissues. The deposited ECM can then be remodeled to restore normal tissue structure and function, and the transiently induced myofibroblasts can self-resolve. When the repair process is dysregulated or tissues are repeatedly injured and stimulated, however, overproduction of ECM by myofibroblasts leads to fibrosis [1,2,3,4,5,7]. This causes reduced levels of oxygen and promotes myofibroblast persistence and disease progression [1,3]. Tissue density, stiffness, and other biophysical features are also altered during increased ECM production which further activates myofibroblasts through mechano-signaling [2,3,5,9]. Overproduction and malformation of ECM is the signature characteristic of fibrosis and creates a positive feedback loop causing continued progression of fibrosis [3,9,10].
In fibrotic organs, ECM changes not only in abundance, but also in composition[8]. In a healthy state, the ECM is a complex 3D macromolecular network that fills the space between cells in solid tissues. It consists of approximately 300 different core matrisome proteins which includes collagens, proteoglycans and glycoproteins [11]. The ECM provides not only physical structural support to tissues but also biochemical and biomechanical signals to regulate numerous cellular functions during homeostasis and pathogenesis [11,12]. In fibrosis, activated myofibroblasts produce excess fibril-forming collagens I and III as well as fibronectin and elastin [8,10]. The buildup of these proteins can be balanced by ECM-remodeling enzymes including matrix metalloproteinases (MMPs), adamalysins, or meprins [13]. MMP and adamalysin enzyme activities are regulated by tissue inhibitor metalloproteinases (TIMP) family proteins which prevent over-degradation of the ECM[13]. The imbalance between ECM accumulation and remodeling has been highlighted as a recurring feature in fibrosis [10,14].
In addition to regulation by degradation enzymes, ECM proteins are subject to post-translational modifications (PTMs) which alter their structure and function [15,16]. The over-crosslinked, stiffened ECM is a defining feature in fibrotic disorders across various orans including lung, liver, and skin [15], as well as an important pathological driver of progressive fibrosis. This provides a unique opportunity for therapeutic strategies targeting ECM in fibrosis. Rather than depleting or degrading ECM as a whole, ECM crosslinking can be selectively targeted [15,16,17]. Here we will review the mechanisms underlying crosslinking modifications, the influence of crosslinking on fibrosis progression, and therapeutic approaches toward ECM crosslinking aimed at normalizing the diseased ECM and treating fibrotic diseases.
2. ECM Crosslinking Biochemical Pathways
Crosslinking serves as a powerful regulator of the biophysical properties of ECM. It creates strong connections between ECM molecules giving them increased stability and resistance to proteolysis [16,18,19]. Crosslinking modifications can be mediated by enzymatic or non-enzymatic reactions. Crosslinks formed via non-enzymatic reactions occur slowly and are associated with aging, making this type of crosslink challenging for therapeutic targeting[16,19]. Enzymatic crosslinking is mediated by multiple groups of enzymes including lysyl oxidase (LOX) and transglutaminase (TG) proteins [15,16]. While not directly contributing to crosslinking reactions, lysine hydroxylase (LH) enzymes also play a key role in LOX-mediated crosslinking as they modify amino acids that are subsequently acted on by the LOX family [15].
Since collagens are the most abundant fibrous proteins of interstitial tissue ECM and the major constituents of fibrotic ECM, we would like to put particular emphasis on pathways and mechanisms of collagen crosslinking in this section [20]. Collagen goes through a complex sequence of processing steps before reaching its final, mature form in the ECM. After being transcribed, collagen is translated into alpha chains containing N and C-terminal pro-peptides [21,22]. Proline hydroxylation of these alpha chains then allows for stable formation of a procollagen triple helix [23,24]. Procollagen can be made up of three of the same (homotrimeric), a combination of three different (heterotrimeric), or two of the same and one different (heterotrimeric) alpha chain. When these molecules are initially formed in the endoplasmic reticulum (ER), they still contain N and C-terminal pro-peptides which are cleaved after release into the ECM. After the cleavage, these tropocollagen triple helices contain three domains: N-terminal telopeptide, C-terminal telopeptide, and a helical domain in the center of the molecule. Finally, fibrillar collagen molecules can assemble into stable fibrils regulated by intra and intermolecular crosslinking (Figure 1) [16,22,23,25,26,27]. Next, we will discuss the enzyme families that mediate modification and crosslinking of ECM.
2.1. Transglutaminase (TG) Crosslinking
There are nine different TG genes of which eight produce catalytically active enzymes [28]. These enzymes regulate a crosslinking reaction between glutamine and lysine residues (Figure 2) [15,28]. Importantly, TG activity is not limited to crosslinking and TGs have a number of different substrates [15,29]. TGs also serve as scaffolding proteins important for basic cell functions including cell adhesion and signal transduction [28,30]. Such a diverse family of enzymes requires careful regulation for the maintenance of homeostasis. TG activity is calcium-dependent which can be advantageous for controlling enzyme activity. Homeostatic cells in the liver maintain inactive TG2 intracellularly but upon injury, release TG2 into the ECM where its activity increases due to higher calcium levels [31,32]. The activity of several TGs can also be activated by proteolytic cleavage or diminished through the binding of purine nucleotides [28].
Transglutaminase 2 (TG2) has been identified as the most broadly expressed TG and is dysregulated in a number of diseases including fibrosis [16,28,30]. It has been specifically studied in kidney, heart, lung, and liver fibrosis [33]. Beyond its functions in crosslinking, TG2 contributes to fibrosis through its protein binding capabilities. It binds to TGFβ, a key profibrotic molecule, which helps facilitate its conversion from a latent to active form [30,34]. Thus, TG2 could serve as a target for treating fibrosis, but drug delivery and specificity should be carefully considered due to the broad expression and variety of essential functions.
2.2. Lysine Hydroxylase (LH)
Three genes encode for three different lysine hydroxylase enzymes. While at the protein level these are referred to as LH1, LH2, and LH3, the genes are named PLOD1, PLOD2, and PLOD3 (procollagen-lysine, 2-oxoglutarate 5-dioxygenase), respectively [15,26]. The LH family does not catalyze crosslinks, but its activity is crucial for LOX-mediated crosslinking reactions. These enzymes contribute to crosslinking by converting lysine to hydroxylysine on collagens (Figure 3A). LOX enzymes can act on unmodified lysine, but a number of crosslinks are derived from the hydroxylysine generated by LH s[15,23].
LH modifications to collagen are substrate-specific. Hydroxylation of collagen telopeptides is conferred through LH2 which has two isoforms, LH2a and LH2b. LH2b, but not LH2a is necessary for downstream collagen crosslinking [15,24,26,35]. Helical regions of collagen are hydroxylated by LH1 and LH3 [15,26]. LH1 and LH3 play non-redundant crosslinking functions, because they have specificity for different types of collagens; LH1 plays a dominant role in helical region hydroxylation of collagen I and III, while LH3 plays a stronger role in modifying collagens IV and V [24]. LH3 additionally has a unique domain conferring galactosyltransferase activity [25]. All three LH enzymes predominantly localize to the endoplasmic reticulum within the cell. LH2 and LH3 have also been identified outside of the cell and have been shown to maintain catalytic activity [25,26,36]. LH enzyme activity is critical for modifying collagens before they are modified by LOX[ 23].
The differences between LH enzymes are crucial in considering crosslinking in fibrosis. For example, collagen I and III are key markers in lung fibrosis [9,37,38]. Thus, in pulmonary fibrosis, LH1 which preferentially modifies these collagens may be a more interesting contributor than LH3. However, this only accounts for helical region modifications to collagen; telopeptide region hydroxylation is also critical since it is required for the formation of mature crosslinks [26]. PLOD2, the gene encoding LH2, the protein responsible for telopeptide hydroxylation, is upregulated in samples from both idiopathic pulmonary fibrosis and systemic sclerosis patients [39,40,41]. Overall, these LH proteins create an important foundation for LOX-mediated crosslinking.
2.3. Lysyl Oxidase (LOX) Crosslinking
The LOX family includes LOX and four LOX-like proteins (LOXL1-4) [42]. Different LOX family proteins play unique roles in cytoskeleton organization and in transcriptional regulation through interactions with histones and transcription factors [42]. Perhaps of more interest to fibrosis, however, LOX proteins mediate crosslinking on collagen and elastin [17]. The crosslinks formed by LOX enzymes are both protein and region-specific. Collagen crosslinks differ from elastin crosslinks, and crosslinks on collagen vary between helical and telopeptide regions [17,23].
While TGs are calcium-dependent, LOX enzymes are copper dependent [23,42]. Other regulatory mechanisms for LOX enzymes vary by protein. LOX and LOXL1 are activated by proteolytic cleavage of their N terminal domains [17,42]. This is done by bone morphogenetic protein-1 (BMP-1) after they are secreted [42]. Rather than an N-terminal pro-peptide, LOXL2, 3, and 4 have four SRCR repeats [42]. These structural differences provide some context as to the differing functions between these enzymes. Extensive work has provided a comprehensive view of the biochemical reactions underlying LOX crosslinking on both collagen and elastin.
2.4. LOX-Mediated Collagen Crosslinking
LOX-mediated collagen crosslinks can be intramolecular or intermolecular and can span across different types of collagens [17]. Unlike TGs, multiple subsequent LOX-mediated reactions are required to achieve crosslinked collagen. The initial chemical reaction triggered by LOX enzymes is oxidative deamination of a lysine or hydroxylysine resulting in an aldehyde (Figure 3B) [15]. These aldehydes go through condensation reactions with a lysine or a hydroxylysine [15]. If neither amino acid reacting was modified by an LH enzyme, a lysine aldehyde with a lysine, dehydro-lysino-norleucine (deH-LNL) is produced. When one or more of the reacting molecules was modified by LH, the product includes one or two hydroxyl groups. A lysine aldehyde with hydroxylysine produces dehydro-hydroxylysino-norleucine (deH-HLNL). Similarly, if a hydroxylysine aldehyde reacts with a lysine, the same product is produced, deH-HLNL. Lastly, if both reacting molecules were hydroxylated by LH, a hydroxylysine aldehyde and a hydroxylysine, dehydro-dihydroxylysino-norleucine (deH-DHLNL) is produced (Figure 4) [15,43]. In summary, crosslinks formed by LOX are deH-LNL, deH-HLNL, and deH-DHLNL where each product varies only in the number of hydroxyl groups, zero, one, or two, respectively.
deH-HLNL and deH-DHLNL are immature, divalent crosslinks and can go through an additional reaction to become mature and trivalent [23,44,45]. This reaction can create links between two to three different collagen molecules [23]. Reactions with various forms of lysine produce pyridinolines or pyrroles. Pyridinoline (Pyr) is formed by the reaction between deH-DHLNL and a hydroxylysine aldehyde. Deoxy-pyridinoline (DPyr) is formed with deH-HLNL and a hydroxylysine aldehyde. While Pyr and DPyr are formed with hydroxylysine aldehydes, pyrroles are formed with lysine aldehydes. deH-HLNL with a lysine aldehyde forms deoxy-pyrrole (DPrl) while deH-DHLNL with a lysine aldehyde forms pyrrole (Prl) (Figure 5) [23,44,46].
According to some studies, collagen crosslinks can also be formed with histidine residues. A crosslink can be formed between deH-HLNL and a histidine residue forming histidino-hydroxylysinonorleucine (HHL) [23,46]. Dehydro-histidino-hydroxymerodesmosine (HHMD) can also be formed through a series of reactions involving lysine aldehyde, hydroxylysine, and histidine [23,46]. In 2019, one group claimed that HHL was just an artifact; however, this claim was disputed by Yamauchi, Taga, and Terajima in a letter to the editor of the same journal [47,48]. This discrepancy may in part be due to a limitation in the methodology used [47]. Thus, it is important to consider methods available for answering specific questions about crosslink modifications. In summary, we have discussed nine unique crosslinking modifications to collagen facilitated by LOX: deH-LNL, deH-HLNL, deH-DHLNL, Pyr, DPyr, Prl, DPrl, HHL, and HHMD. Evaluation of variations in these crosslinking modifications can provide insights into ECM changes in fibrosis.
2.5. LOX-Mediated Elastin Crosslinking
LOX, LOXL1, and LOXL2 additionally facilitate crosslinking on elastin protein in the ECM [17,19]. As with collagen, the first step to this crosslinking is a LOX enzyme converting a lysine to a lysine aldehyde in the extracellular space. Subsequently, through a condensation reaction between a lysine aldehyde and a lysine, an immature deH-LNL crosslink is formed [17,19]. Since collagen also has deH-LNL crosslinks, assays measuring deH-LNL could identify these crosslinks from either collagen or elastin. On elastin, lysine aldehydes can also interact with one another to form an allysine aldol (AA). deH-LNL and AA are bifunctional, and as with immature collagen crosslinks, they go through additional reactions to form mature crosslinks. AA interacts with unmodified lysine to generate dehydromerodesmosine (deH-MDES) [19]. deH-MDES can also be formed through the interaction between a lysine aldehyde and deH-LNL. deH-MDES crosslinks are trifunctional. Elastin also has tetravalent mature crosslinks, desmosine (Des) and isodesmosine (IDes) which are formed through the reaction between deH-MDES and a lysine aldehyde [17,19]. These mature, tri-, and tetra-valent crosslinks are unique to elastin. Accumulation of crosslinks on elastin limits proteolytic degradation and stabilizes elastin for long periods of time [19].
2.6. LOX in Fibrosis
LOX activity is a critical element in fibrosis and is being investigated as a target for therapeutics [17,49,50]. Like TGs, LOX proteins bind to TGFβ, a key regulator of fibrosis [17]. LOX and LOXL2 are also regulated by hypoxia [17] which mechanistically explains the upregulation of LOX activity in fibrotic tissues where hypoxia is prevalent [1,3,51]. In a recent study by Brereton et al., this relationship between hypoxia and LOX activity was explored in depth. The group found that in addition to increased expression of LOXL2, collagen structure, tissue stiffness, and quantity of mature pyridinoline crosslinking were altered upon hypoxic induction in a lung fibrosis model [41].
Types of crosslinking dynamics observed in fibrosis vary by organ in part due to the tissue-specificity of crosslinking enzymes and modifications [25]. In skeletal tissues including bone and cartilage, LH2 drives formation of crosslinks from hydroxylysine aldehydes: HLNL, DHLNL, pyridinoline and deoxypyridinoline [17,22]. Pyrrole crosslinks are predominantly found in tendons and mineralizing tissue [17,23]. For this reason, pyrroles have less relevance for research in organ fibrosis. In skin and cornea, crosslinks from lysine aldehydes have been shown to be more prevalent [17,22]. Thus, maintaining appropriate crosslinking composition is necessary for homeostasis and basic organ functionality. When targeting crosslinking to treat fibrosis of one organ, the effect on crosslinking in other organs may also need to be considered if a drug is delivered systemically.
In fibrosis, there is not only an observed increase in LOX expression and overall crosslinking but also a change in proportions of different types of crosslinks [25]. Crosslinks derived from the hydroxylysine aldehyde pathway (deH-HLNL, deH-DHLNL, Pyr, and DPyr) have been shown to increase in fibrotic conditions [35,51,52,53]. These crosslinks not only alter ECM structure and biomechanical properties, but also change protein susceptibility to degradation by MMPs [16,25]. In summary, the crosslinking problem in fibrosis is created by a synergistic effect of overaccumulation of crosslinked ECM as well as increased stability of these crosslinked proteins.
3. ECM Crosslinking Analysis Methods
Methods to evaluate crosslinking are limited due to the complexity of modifications and the ECM structures formed. SDS-PAGE and size exclusion chromatography (SEC) can be used to detect the presence of crosslinks, but do not clearly decipher between specific types of crosslinks [54]. To quantify changes in certain crosslinks, some immunoblotting and ELISA assays have been developed. These are, however, limited by antibody specificity for crosslinking modifications. Antibodies can also fail to bind when ECM proteins are tightly aggregated or have altered structure [54,55]. Nevertheless, this method has served as an efficient way to evaluate crosslinking. Two different studies of lung fibrosis in recent years have shown changes in mature collagen crosslinks using an ELISA quantifying total Pyd and DPyd crosslinks [41,52]. An ELISA has also been developed to evaluate elastin Des/IDes crosslinks [56].
For more quantitative and specific analysis of crosslinking, liquid chromatography (LC) paired with mass spectrometry (MS) approaches have been developed. Typically, this approach requires proteins in the samples to be broken down to the amino acid level through hydrolysis. High-performance liquid chromatography (HPLC) or ultra-performance LC (UPLC) is then used for separation, and mass spectrometry is used for quantification [54,55]. This type of method can be used to simultaneously detect multiple crosslinking modifications, both immature and mature [51,57,58]. Effective identification of crosslinks with this method is dependent on the availability of standards [54]. Since the sample is hydrolyzed into individual amino acids, this method is also limited by its inability to identify the sites of crosslinks on proteins [54,55]. Overall, both ELISA and LC-MS strategies for quantifying crosslinking modifications are effective but still have limitations.
4. Dysregulation of ECM Crosslinking in Fibrotic Disease
The biochemical nature of ECM crosslinking and the enzymes that contribute to it are generally shared across organ systems. Expression patterns and crosslinking compositions, however, can vary [17]. Extensive work has been done to evaluate crosslinking enzymes and modifications in fibrosis of various organs. We will briefly touch on the nature of fibrosis in lung, liver, and skin as well as some of the work that has characterized crosslinking in these organs.
4.1. Lung Fibrosis
Idiopathic Pulmonary Fibrosis (IPF) is typically diagnosed in patients around the age of sixty-five and is more common in men. Upon diagnosis, the patients’ survival time is three to five years on average [59]. The two FDA-approved therapeutics, nintedanib and pirfenidone, only slow progression and do not significantly change prognosis [59]. The PLOD/LH family is dysregulated in IPF. PLOD genes are upregulated in IPF patient serum samples, and PLOD2 was the most upregulated family member [40]. Leveraging spatial transcriptomics data, PLOD2 gene expression was also shown to increase at active sites of fibrosis in IPF tissue [41]. Interestingly, research by Jones et al. assessed isoform-specific changes in LH2 in IPF tissue and did not demonstrate a significant increase in LH2b, the isoform responsible for facilitating collagen crosslinking [52]. Given isoform specific functions of LH2, this raises an important consideration that gene expression data alone may not be sufficient. Synthesis of information across studies, at both transcript and protein level is critical for identifying strong therapeutic targets.
Beyond the PLOD/LH family, LOX proteins have also been investigated in IPF. Jones et al. showed differential expression of LOXL2, LOXL3, and LOXL4, but not LOX and LOXL1 [52]. Brereton et al. and Ma et al., however, showed upregulation of all five LOX genes in IPF tissue [41,51]. Yet another study by Tjin et al. 2017 assessed two different IPF datasets and found that LOXL1 was upregulated in both, but LOXL2 was only upregulated in one [60]. This group followed up on the gene expression studies with an imaging approach and demonstrated that at the protein level LOXL1 and LOXL2 were upregulated in IPF tissue [60]. Strengthening these expression studies, LOX enzyme activity measured by amine oxidase also increased in IPF tissue [52]. Collectively these investigations suggest LOX is associated with fibrosis progression in lung.
Beyond changes in just LOX, recent work has put a spotlight on crosslinking changes in lung fibrosis. Jones et al. showed an increase in both immature (deH-HLNL, deH-DHLNL) and mature (Pyr, DPyr) crosslinks in IPF patient lung tissue. There was a higher ratio of deH-DHLNL to deH-HLNL in IPF tissue [52]. Consistent with this, Ma et al. evaluated crosslinking in the bleomycin mouse model of lung fibrosis. They observed a strong increase in deH-DHLNL. A subtle change in Pyr was seen but was not significant when normalized by total collagen [51]. In summary, both crosslink enzyme expression and crosslinking modifications are dysregulated in lung fibrosis.
4.2. Liver Fibrosis
The liver is subject to developing fibrosis, especially in cases of chronic wound repair. This wound repair process can be overactivated, particularly under circumstances involving viral infection and alcohol abuse [61,62]. Fibrosis of the liver is reversible and can be treated in early stages, but if left unchecked can progress to an irreversible state, cirrhosis [62]. Both collagen and elastin crosslinking have been shown to change in liver fibrosis. LOX genes expression increases in liver fibrosis and cirrhosis [63]. MMP family members and their inhibitors, TIMPs, also have dysregulated expression [61]. Along with enzyme expression changes, studies have highlighted changes to crosslinking modifications in liver fibrosis. In patient-derived liver samples, pyridinoline crosslinking was increased in both viral hepatitis and cirrhosis [64]. To model liver fibrosis, carbon tetrachloride (CCl4) is used to induce inflammation [61]. In one study using a fibrotic mouse CCl4 injection model, pyridinoline collagen crosslinking increased in fibrosis and cirrhosis. This study also showed an increase in elastin desmosine crosslinking in cirrhosis[63]. Lastly, recent work has highlighted non-enzymatic, advanced glycation end-product (AGE) mediated crosslinking in cirrhosis ECM. Targeting this diseased ECM showed promise for limiting fibrosis progression [65].
4.3. Skin Fibrosis
Systemic sclerosis (SSc) is an autoimmune, multi-organ fibrotic disease affecting connective tissues including the skin. Using autopsy samples from a SSc patient, one group showed an increase in Pyd crosslinks in skin, endocardium, fascia, and bladder [53]. With a bleomycin mouse model of skin fibrosis, however, another group failed to identify changes in Pyd, despite histological validation of the model. Upon removal of bleomycin, the fibrosis in this model is reversible [66]. Thus, it is likely that in this case the bleomycin mouse model did not fully recapitulate irreversible fibrotic changes that occur in human disease. As an alternative to a mouse model, Huang et al. developed self-assembling stromal tissues (SAS) and human skin equivalents (HSEs) using dermal fibroblasts derived from SSc patient skin or from normal healthy control donors [67]. They showed an increase in LOXL4 expression at the mRNA and protein levels. Using a CTX-I ELISA kit, they also saw an increase in crosslinked C-telopeptide Type I Collagen in SSc-derived cultures relative to normal healthy controls [67]. This is consistent with the increase in mature crosslinks observed in tissue from SSc patients [53]. Crosslinking has additionally been investigated in lipodermatosclerosis (LDS). As shown with SSc, an increase in crosslinking was observed in fibrotic skin from patients with LDS [68]. These studies collectively provide evidence of dysregulated crosslinking in fibrotic skin conditions.
5. ECM Crosslink-Based Therapeutic Strategies, Drug Targets, and Molecules
Due to strong evidence for dysregulated ECM crosslinking in promoting fibrosis and the key roles of ECM crosslinking enzymes, therapeutics are actively being investigated to target TG, LOX, and LH proteins, with the most advanced drug molecules in Phase I/II clinical trials (Table 1) [69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92]. LOX therapeutics vary by which LOX they target, and both small-molecule and antibody-based targeting approaches have been employed. One strategy has been to solely target LOXL2. For example, Simtuzumab is an antibody targeting LOXL2, but this has failed to improve fibrosis in clinical settings and has been discontinued [69,71,93]. The strategy of targeting LOXL2, however, continues with several small molecules currently in the clinic. PAT-1251 for example is now in a phase II clinical trial for myelofibrosis [76]. Another small molecule, GB2064, is also in phase II for myelofibrosis and results suggest disease-modifying activity [77]. These small-molecule LOXL2 inhibitors may also have the potential to treat other fibrotic diseases. This type of strategy, targeting only select LOX proteins, could be effective for ameliorating fibrosis progression without fully ablating functional collagen crosslinking. On the other hand, it is noteworthy that PXS-5505, a pan-LOX inhibitor, is also in Phase 1/2a trial for myelofibrosis. It was reported to be well-tolerated and showed preliminary signs of modification to disease [94]. It is too early to tell which LOX-targeting strategy would eventually work out for fibrotic diseases, but a variety of approaches are being explored.
TGs and LHs have been targeted to a lesser extent than LOX proteins. Of the TGs, TG2 has been the primary target of interest. Among the multiple small-molecule inhibitors of TG2, ZED1227 is the most advanced and has entered clinical development. While ZED1227 has been investigated for the treatment of celiac disease, it also entered clinical phase II in 2022 for treatment of non-alcoholic fatty liver disease (NAFLD) with significant fibrosis [82,83]. It is noteworthy that inhibitory antibodies have also been discovered to target TG2 [84,85].The most advanced anti-TG2 antibody, Zampilimab, is in clinical trials for adult kidney transplant patients with chronic allograft injury, with potential follow-up interest in fibrotic diseases (NCT04335578). For targeting LH2, small-molecule inhibitors have been discovered and proposed for use in fibrosis and cancer metastasis [90,91,92]. Overall, significant efforts are being made to develop novel therapeutics targeting crosslinking for fibrosis treatment. Continuing these efforts will be critical for determining if targeting collagen crosslinking can successfully treat fibrosis.
6. Discussion
Fibrosis across organ systems has proven to be extremely complex, dynamic, heterogeneous, and difficult to effectively target therapeutically. Finding consistencies across different types of fibrosis could serve as an effective strategy to build therapeutics applicable to multiple conditions [96]. One recurring similarity identified across organs is profibrotic adaptations of the ECM. Targeting and modifying the profibrotic ECM may offer a parallel approach to fibrosis therapy in addition to targeting disease-promoting cells in fibrotic tissues. With ECM targeting, we must account for both the cellular and molecular changes producing the ECM as well as the profibrotic ECM itself [5]. As for the profibrotic ECM, we then must consider both ECM protein expression profiles and the posttranslational crosslinking modifications that define its structure and function.
An important consideration with respect to fibrotic ECM is the progressive, heterogeneous nature of fibrotic disease [2,9]. Because of this, it would be interesting to explore spatial and temporal dynamics of crosslinking in organ fibrosis. To do this, the field will need to overcome the bottleneck of limited sample availability from diseased patients. Additionally, the advancement of spatially resolved methodology to assess collagen crosslinking will be required. Methods for assessing crosslinked, mature ECM are in part limited by the insolubility and complex three-dimensional structures formed[54]. Current LC-MS approaches are largely limited to quantification of crosslinking modification products from hydrolyzed samples where proteins are broken down into individual amino acids. This makes it impossible to determine the original site of crosslink modification [55]. Mapping out the three-dimensional ECM crosslink patterns and structures across organs and diseases is an underexplored field that will require breakthrough technologies.
Of almost equal importance as analysis of ECM crosslinking, is the modeling of ECM crosslink patterns associated with homeostatic and pathological processes to meet basic biology and drug discovery research needs. Limited sample availability from patients especially from early stages of fibrosis creates the requirement for laboratory-based fibrotic models to explore novel therapeutic targets. Many groups have utilized biochemical, cell culture, and animal models to characterize ECM crosslinking alterations associated with fibrosis and to support drug discovery programs [51,52,53,68]. However, the current biology models have significant limitations and do not support robust understanding and targeting of ECM for fibrotic diseases. For example, cell culture models, a popular backbone platform to many biomedical research fields, are often two-dimensional and lack the ability to generate a mature, three-dimensional ECM similar to that found in natural tissues [97,98,99]. Because of limitations to existing models, widespread efforts are being made to develop novel three-dimensional strategies incorporating more physiologically relevant ECM [98,100,101]. With the generation of new model platforms, cost and throughput may be a more limiting factor. Ultimately, improved modeling of ECM dynamics will advance our ability to develop and test fibrotic therapeutics.
Considering why therapeutics thus far have failed may help drive efforts moving forward. For example, Simtuzumab, an antibody targeting LOXL2, was discontinued from clinical trials due to lack of efficacy. It is possible that small molecule therapeutics with broader activity could be more efficacious. Drug distribution and penetration of different therapeutics could also play a role. If therapeutics continue to fail despite efforts to diversify therapeutic platforms, additional strategies could be considered. For instance, instead of targeting just the crosslinking enzymes, the diseased, accumulated ECM could be targeted. Delivery of ECM-degradation enzymes such as MMPs has been suggested and explored, but the approach can be limited by side effects, so more selective delivery approaches may be necessary [16]. Cellular therapy approaches based on cell types possessing ECM modifying and restoration capacities, such as mesenchymal stem cells and fibrolytic macrophages, may offer an additional strategy [10,95,102,103,104]. How to target and normalize diseased ECM and restore healthy tissue structure and function may be the biggest challenge of this research field. Ultimately, pairing therapies that can manage diseased ECM with therapies that can prevent further ECM build up may provide a promising strategy.
Author Contributions
Conceptualization, S.M.L. and Y.H.; writing—original draft preparation, S.M.L. and Y.H.; writing—review and editing, S.M.L. and Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank the following AbbVie Employees: Yu Tian, Lisa Hazelwood, and Qin Ji for their comments and suggestions on this manuscript. Diagrams were created with BioRender.com and exported under AbbVie’s paid subscription.
Conflicts of Interest
S.M.L. and Y.H. are employees of AbbVie. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
References
- Lurje, I.; Gaisa, N.T.; Weiskirchen, R.; Tacke, F. Mechanisms of Organ Fibrosis: Emerging Concepts and Implications for Novel Treatment Strategies. Mol. Asp. Med. 2023, 92, 101191. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From Mechanisms to Medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and Distinct Mechanisms of Fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and Tissue Fibrosis: Molecular Signals, Cellular Mechanisms and Translational Implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The Big Five in Fibrosis: Macrophages, Myofibroblasts, Matrix, Mechanics, and Miscommunication. Matrix Biol. 2018, 68, 81–93. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic Disease and the TH1/TH2 Paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef]
- Talbott, H.E.; Mascharak, S.; Griffin, M.; Wan, D.C.; Longaker, M.T. Wound Healing, Fibroblast Heterogeneity, and Fibrosis. Cell Stem Cell 2022, 29, 1161–1180. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The Good and the Bad Collagens of Fibrosis – Their Role in Signaling and Organ Function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular Matrix as a Driver of Progressive Fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New Insights into Fibrosis from the ECM Degradation Perspective: The Macrophage-MMP-ECM Interaction. Cell Biosci. 2022, 12, 117. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From Mechanisms to Medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A. Collagen Cross-Linking Mediated by Lysyl Hydroxylase 2: An Enzymatic Battlefield to Combat Fibrosis. Essays Biochem. 2019, 63, 377–387. [Google Scholar] [CrossRef]
- Kong, W.; Lyu, C.; Liao, H.; Du, Y. Collagen Crosslinking: Effect on Structure, Mechanics and Fibrosis Progression. Biomed. Mater. 2021, 16, 062005. [Google Scholar] [CrossRef]
- Vallet, S.D.; Ricard-Blum, S. Lysyl Oxidases: From Enzyme Activity to Extracellular Matrix Cross-Links. Essays Biochem. 2019, 63, 349–364. [Google Scholar] [CrossRef]
- Beninati, S.; Piacentini, M. The Transglutaminase Family: An Overview: Minireview Article. Amino Acids 2004, 26, 367–372. [Google Scholar] [CrossRef]
- Schmelzer, C.E.H.; Hedtke, T.; Heinz, A. Unique Molecular Networks: Formation and Role of Elastin Cross-links. IUBMB Life 2020, 72, 842–854. [Google Scholar] [CrossRef]
- Muiznieks, L.D.; Keeley, F.W. Molecular Assembly and Mechanical Properties of the Extracellular Matrix: A Fibrous Protein Perspective. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2013, 1832, 866–875. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen Structure and Stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef]
- Huizen, N.A.; Ijzermans, J.N.M.; Burgers, P.C.; Luider, T.M. Collagen Analysis with Mass Spectrometry. Mass Spectrom. Rev. 2020, 39, 309–335. [Google Scholar] [CrossRef]
- Yamauchi, M.; Sricholpech, M. Lysine Post-Translational Modifications of Collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [CrossRef]
- Gjaltema, R.A.F.; Bank, R.A. Molecular Insights into Prolyl and Lysyl Hydroxylation of Fibrillar Collagens in Health and Disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef]
- Pehrsson, M.; Mortensen, J.H.; Manon-Jensen, T.; Bay-Jensen, A.-C.; Karsdal, M.A.; Davies, M.J. Enzymatic Cross-Linking of Collagens in Organ Fibrosis – Resolution and Assessment. Expert Rev. Mol. Diagn. 2021, 21, 1049–1064. [Google Scholar] [CrossRef]
- Salo, A.M.; Myllyharju, J. Prolyl and Lysyl Hydroxylases in Collagen Synthesis. Exp. Dermatol. 2021, 30, 38–49. [Google Scholar] [CrossRef]
- Lamande, S.R.; Bateman, J.F. Procollagen Folding and Assembly: The Role of Endoplasmic Reticulum Enzymes and Molecular Chaperones. Semin. Cell Dev. Biol. 1999, 10, 455–464. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.W.; Mehta, K. Transglutaminase Regulation of Cell Function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Takeuchi, T.; Shinoda, Y.; Hitomi, K. Identification and Characterization of Substrates Crosslinked by Transglutaminases in Liver and Kidney Fibrosis. Anal. Biochem. 2020, 604, 113629. [Google Scholar] [CrossRef]
- Benn, M.C.; Weber, W.; Klotzsch, E.; Vogel, V.; Pot, S.A. Tissue Transglutaminase in Fibrosis — More than an Extracellular Matrix Cross-Linker. Curr. Opin. Biomed. Eng. 2019, 10, 156–164. [Google Scholar] [CrossRef]
- Lee, Z.-W.; Kwon, S.-M.; Kim, S.-W.; Yi, S.-J.; Kim, Y.-M.; Ha, K.-S. Activation of in Situ Tissue Transglutaminase by Intracellular Reactive Oxygen Species. Biochem. Biophys. Res. Commun. 2003, 305, 633–640. [Google Scholar] [CrossRef]
- Siegel, M.; Strnad, P.; Watts, R.E.; Choi, K.; Jabri, B.; Omary, M.B.; Khosla, C. Extracellular Transglutaminase 2 Is Catalytically Inactive, but Is Transiently Activated upon Tissue Injury. PLoS ONE 2008, 3, e1861. [Google Scholar] [CrossRef]
- Soltani, F.; Kaartinen, M.T. Transglutaminases in Fibrosis—Overview and Recent Advances. Am. J. Physiol.-Cell Physiol. 2023, 325, C885–C894. [Google Scholar] [CrossRef]
- Troilo, H.; Steer, R.; Collins, R.F.; Kielty, C.M.; Baldock, C. Independent Multimerization of Latent TGFβ Binding Protein-1 Stabilized by Cross-Linking and Enhanced by Heparan Sulfate. Sci. Rep. 2016, 6, 34347. [Google Scholar] [CrossRef]
- Slot, A.J. van der; Zuurmond, A.-M.; Bogaerdt, A.J. van den; Ulrich, M.M.W.; Middelkoop, E.; Boers, W.; Ronday, H.K.; DeGroot, J.; Huizinga, T.W.J.; Bank, R.A. Increased Formation of Pyridinoline Cross-Links Due to Higher Telopeptide Lysyl Hydroxylase Levels Is a General Fibrotic Phenomenon. Matrix Biol. 2004, 23, 251–257. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, H.; Terajima, M.; Banerjee, P.; Liu, X.; Yu, J.; Momin, A.A.; Katayama, H.; Hanash, S.M.; Burns, A.R.; et al. Lysyl Hydroxylase 2 Is Secreted by Tumor Cells and Can Modify Collagen in the Extracellular Space*. J. Biol. Chem. 2016, 291, 25799–25808. [Google Scholar] [CrossRef]
- Jessen, H.; Hoyer, N.; Prior, T.S.; Frederiksen, P.; Karsdal, M.A.; Leeming, D.J.; Bendstrup, E.; Sand, J.M.B.; Shaker, S.B. Turnover of Type I and III Collagen Predicts Progression of Idiopathic Pulmonary Fibrosis. Respir. Res. 2021, 22, 205. [Google Scholar] [CrossRef]
- Agarwal, M.; Goheen, M.; Jia, S.; Ling, S.; White, E.S.; Kim, K.K. Type I Collagen Signaling Regulates Opposing Fibrotic Pathways through α 2 β 1 Integrin. Am. J. Respir. Cell Mol. Biol. 2020, 63, 613–622. [Google Scholar] [CrossRef]
- Slot, A.J. van der; Zuurmond, A.-M.; Bardoel, A.F.J.; Wijmenga, C.; Pruijs, H.E.H.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N.; et al. Identification of PLOD2 as Telopeptide Lysyl Hydroxylase, an Important Enzyme in Fibrosis*. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef]
- Shao, S.; Zhang, X.; Duan, L.; Fang, H.; Rao, S.; Liu, W.; Guo, B.; Zhang, X. Lysyl Hydroxylase Inhibition by Minoxidil Blocks Collagen Deposition and Prevents Pulmonary Fibrosis via TGF-Β1/Smad3 Signaling Pathway. Méd. Sci. Monit. : Int. Méd. J. Exp. Clin. Res. 2018, 24, 8592–8601. [Google Scholar] [CrossRef]
- Brereton, C.J.; Yao, L.; Davies, E.R.; Zhou, Y.; Vukmirovic, M.; Bell, J.A.; Wang, S.; Ridley, R.A.; Dean, L.S.; Andriotis, O.G.; et al. Pseudohypoxic HIF Pathway Activation Dysregulates Collagen Structure-Function in Human Lung Fibrosis. eLife 2022, 11, e69348. [Google Scholar] [CrossRef] [PubMed]
- Zaffryar-Eilot, S.; Hasson, P. Lysyl Oxidases: Orchestrators of Cellular Behavior and ECM Remodeling and Homeostasis. Int. J. Mol. Sci. 2022, 23, 11378. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The Fibrotic Tumor Stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Knott, L.; Bailey, A.J. Collagen Cross-Links in Mineralizing Tissues: A Review of Their Chemistry, Function, and Clinical Relevance. Bone 1998, 22, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Svensson, R.B.; Mulder, H.; Kovanen, V.; Magnusson, S.P. Fracture Mechanics of Collagen Fibrils: Influence of Natural Cross-Links. Biophys. J. 2013, 104, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.S.; Sharma, M.; Kabekkodu, S.P.; Kumar, N.A.; Satyamoorthy, K.; Radhakrishnan, R. Understanding the Molecular Mechanism Associated with Reversal of Oral Submucous Fibrosis Targeting Hydroxylysine Aldehyde-Derived Collagen Cross-Links. J. Carcinog. 2021, 20, 9. [Google Scholar] [CrossRef]
- Yamauchi, M.; Taga, Y.; Terajima, M. Analyses of Lysine Aldehyde Cross-Linking in Collagen Reveal That the Mature Cross-Link Histidinohydroxylysinonorleucine Is an Artifact. J. Biol. Chem. 2019, 294, 14163. [Google Scholar] [CrossRef]
- Eyre, D.R.; Weis, M.; Rai, J. Analyses of Lysine Aldehyde Cross-Linking in Collagen Reveal That the Mature Cross-Link Histidinohydroxylysinonorleucine Is an Artifact. J. Biol. Chem. 2019, 294, 6578–6590. [Google Scholar] [CrossRef]
- Puente, A.; Fortea, J.I.; Cabezas, J.; Loste, M.T.A.; Iruzubieta, P.; Llerena, S.; Huelin, P.; Fábrega, E.; Crespo, J. LOXL2—A New Target in Antifibrogenic Therapy? Int. J. Mol. Sci. 2019, 20, 1634. [Google Scholar] [CrossRef]
- Rosenbloom, J.; Ren, S.; Macarak, E. New Frontiers in Fibrotic Disease Therapies: The Focus of the Joan and Joel Rosenbloom Center for Fibrotic Diseases at Thomas Jefferson University. Matrix Biol. 2016, 51, 14–25. [Google Scholar] [CrossRef]
- Ma, H.-Y.; Li, Q.; Wong, W.R.; N’Diaye, E.-N.; Caplazi, P.; Bender, H.; Huang, Z.; Arlantico, A.; Jeet, S.; Wong, A.; et al. LOXL4, but Not LOXL2, Is the Critical Determinant of Pathological Collagen Cross-Linking and Fibrosis in the Lung. Sci. Adv. 2023, 9, eadf0133. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.G.; Andriotis, O.G.; Roberts, J.J.; Lunn, K.; Tear, V.J.; Cao, L.; Ask, K.; Smart, D.E.; Bonfanti, A.; Johnson, P.; et al. Nanoscale Dysregulation of Collagen Structure-Function Disrupts Mechano-Homeostasis and Mediates Pulmonary Fibrosis. eLife 2018, 7, e36354. [Google Scholar] [CrossRef]
- Istok, R.; Bely, M.; Stancikova, M.; Rovensky, J. Evidence for Increased Pyridinoline Concentration in Fibrotic Tissues in Diffuse Systemic Sclerosis. Experimental Dermatology 2001, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Lemus, E.; Hägglund, P.; López-Alarcón, C.; Davies, M.J. Oxidative Crosslinking of Peptides and Proteins: Mechanisms of Formation, Detection, Characterization and Quantification. Molecules 2021, 27, 15. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Detection, Identification, and Quantification of Oxidative Protein Modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, T.; Seyama, Y.; Yamashita, S. Comparison of ELISA and HPLC for the Determination of Desmosine or Isodesmosine in Aortic Tissue Elastin. Journal of Clinical Laboratory Analysis 1995, 9, 293–296. [Google Scholar] [CrossRef]
- Gineyts, E.; Borel, O.; Chapurlat, R.; Garnero, P. Quantification of Immature and Mature Collagen Crosslinks by Liquid Chromatography–Electrospray Ionization Mass Spectrometry in Connective Tissues. J. Chromatogr. B 2010, 878, 1449–1454. [Google Scholar] [CrossRef]
- Zork, N.M.; Myers, K.M.; Yoshida, K.; Cremers, S.; Jiang, H.; Ananth, C.V.; Wapner, R.J.; Kitajewski, J.; Vink, J. A Systematic Evaluation of Collagen Cross-Links in the Human Cervix. Am. J. Obstet. Gynecol. 2015, 212, 321.e1–321.e8. [Google Scholar] [CrossRef]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic Pulmonary Fibrosis: Current and Future Treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef]
- Tjin, G.; White, E.S.; Faiz, A.; Sicard, D.; Tschumperlin, D.J.; Mahar, A.; Kable, E.P.W.; Burgess, J.K. Lysyl Oxidases Regulate Fibrillar Collagen Remodelling in Idiopathic Pulmonary Fibrosis. Dis. Model. Mech. 2017, 10, 1301–1312. [Google Scholar] [CrossRef]
- Afratis, N.A.; Klepfish, M.; Karamanos, N.K.; Sagi, I. The Apparent Competitive Action of ECM Proteases and Cross-Linking Enzymes during Fibrosis: Applications to Drug Discovery. Adv. Drug Deliv. Rev. 2018, 129, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Stalnikowitz, D.K.; Weissbrod, A.B. Liver Fibrosis and Inflammation. A Review. Ann. Hepatol. 2003, 2, 159–163. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, A.; Chen, W.; Wang, P.; Liu, T.; Cong, M.; Xu, A.; Yan, X.; Jia, J.; You, H. Inhibition of Lysyl Oxidase-like 1 (LOXL1) Expression Arrests Liver Fibrosis Progression in Cirrhosis by Reducing Elastin Crosslinking. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2018, 1864, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, A.; Ilda, S.; Suzuki, N.; Kondo, F.; Miyazaki, M.; Yonemitsu, H. Pyridinoline Collagen Cross-Links in Patients with Chronic Viral Hepatitis and Cirrhosis. J. Hepatol. 1996, 24, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Lyu, C.; Kong, W.; Liu, Z.; Wang, S.; Zhao, P.; Liang, K.; Niu, Y.; Yang, W.; Xiang, C.; Hu, X.; et al. Advanced Glycation End-Products as Mediators of the Aberrant Crosslinking of Extracellular Matrix in Scarred Liver Tissue. Nature Biomedical Engineering 2023, 7, 1437–1454. [Google Scholar] [CrossRef] [PubMed]
- Slot-Verhoeven, A.J. van der; Dura, E.A. van; Attema, J.; Blauw, B.; DeGroot, J.; Huizinga, T.W.J.; Zuurmond, A.-M.; Bank, R.A. The Type of Collagen Cross-Link Determines the Reversibility of Experimental Skin Fibrosis. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2005, 1740, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Cai, G.; Baugh, L.M.; Liu, Z.; Smith, A.; Watson, M.; Popovich, D.; Zhang, T.; Stawski, L.S.; Trojanowska, M.; et al. Systemic Sclerosis Dermal Fibroblasts Induce Cutaneous Fibrosis Through Lysyl Oxidase–like 4: New Evidence From Three-Dimensional Skin-like Tissues. Arthritis Rheumatol. 2020, 72, 791–801. [Google Scholar] [CrossRef]
- Brinckmann, J.; Açil, Y.; Tronnier, M.; Notbohm, H.; Bätge, B.; Schmeller, W.; Koch, M.H.J.; Müller, P.K.; Wolff, H.H. Altered X-Ray Diffraction Pattern Is Accompanied by a Change in the Mode of Cross-Link Formation in Lipodermatosclerosis. J. Investig. Dermatol. 1996, 107, 589–592. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef]
- Muir, A.J.; Levy, C.; Janssen, H.L.A.; Montano-Loza, A.J.; Shiffman, M.L.; Caldwell, S.; Luketic, V.; Ding, D.; Jia, C.; McColgan, B.J.; et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results With Insights on the Natural History of the Disease. Hepatology 2019, 69, 684–698. [Google Scholar] [CrossRef]
- Verstovsek, S.; Savona, M.R.; Mesa, R.A.; Dong, H.; Maltzman, J.D.; Sharma, S.; Silverman, J.; Oh, S.T.; Gotlib, J. A Phase 2 Study of Simtuzumab in Patients with Primary, Post-polycythaemia Vera or Post-essential Thrombocythaemia Myelofibrosis. Br. J. Haematol. 2017, 176, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of Simtuzumab versus Placebo in Patients with Idiopathic Pulmonary Fibrosis: A Randomised, Double-Blind, Controlled, Phase 2 Trial. The Lancet Respiratory Medicine 2017, 5, 22–32. [Google Scholar] [CrossRef]
- Chitty, J.L.; Yam, M.; Perryman, L.; Parker, A.L.; Skhinas, J.N.; Setargew, Y.F.I.; Mok, E.T.Y.; Tran, E.; Grant, R.D.; Latham, S.L.; et al. A First-in-Class Pan-Lysyl Oxidase Inhibitor Impairs Stromal Remodeling and Enhances Gemcitabine Response and Survival in Pancreatic Cancer. Nat. Cancer 2023, 4, 1326–1344. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Findlay, A.; Stolp, J.; Rayner, B.; Ask, K.; Jarolimek, W. Pan-Lysyl Oxidase Inhibitor PXS-5505 Ameliorates Multiple-Organ Fibrosis by Inhibiting Collagen Crosslinks in Rodent Models of Systemic Sclerosis. Int. J. Mol. Sci. 2022, 23, 5533. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Saad, S.; Shi, Y.; Wang, R.; Chou, A.S.Y.; Gill, A.; Yao, Y.; Jarolimek, W.; Pollock, C.A. Lysyl Oxidase Inhibitors Attenuate Cyclosporin A-Induced Nephropathy in Mouse. Sci. Rep. 2021, 11, 12437. [Google Scholar] [CrossRef]
- Rowbottom, M.W.; Bain, G.; Calderon, I.; Lasof, T.; Lonergan, D.; Lai, A.; Huang, F.; Darlington, J.; Prodanovich, P.; Santini, A.M.; et al. Identification of 4-(Aminomethyl)-6-(Trifluoromethyl)-2-(Phenoxy)Pyridine Derivatives as Potent, Selective, and Orally Efficacious Inhibitors of the Copper-Dependent Amine Oxidase, Lysyl Oxidase-Like 2 (LOXL2). J. Med. Chem. 2017, 60, 4403–4423. [Google Scholar] [CrossRef]
- Harrison, C.; Mascarenhas, J.; Cilloni, D.; Schlenk, R.; Jacoby, B.; Slack, R.J.; Aslanis, V.; Singh, B.; Lindmark, B.; Verstovsek, S.; et al. P1024: MYLOX-1: A PHASE II STUDY EVALUATING THE SAFETY, TOLERABILITY, PHARMACOKINETICS AND PHARMACODYNAMICS OF ORAL LOXL2 INHIBITOR GB2064 (WITH FOCUS ON BONE MARROW COLLAGEN) IN PATIENTS WITH MYELOFIBROSIS. HemaSphere 2023, 7, e87048d4. [Google Scholar] [CrossRef]
- Findlay, A.; Turner, C.; Schilter, H.; Deodhar, M.; Zhou, W.; Perryman, L.; Foot, J.; Zahoor, A.; Yao, Y.; Hamilton, R.; et al. An Activity-based Bioprobe Differentiates a Novel Small Molecule Inhibitor from a LOXL2 Antibody and Provides Renewed Promise for Anti-fibrotic Therapeutic Strategies. Clin. Transl. Med. 2021, 11, e572. [Google Scholar] [CrossRef]
- Chaudhari, N.; Findlay, A.D.; Stevenson, A.W.; Clemons, T.D.; Yao, Y.; Joshi, A.; Sayyar, S.; Wallace, G.; Rea, S.; Toshniwal, P.; et al. Topical Application of an Irreversible Small Molecule Inhibitor of Lysyl Oxidases Ameliorates Skin Scarring and Fibrosis. Nat. Commun. 2022, 13, 5555. [Google Scholar] [CrossRef]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The Lysyl Oxidase like 2/3 Enzymatic Inhibitor, PXS-5153A, Reduces Crosslinks and Ameliorates Fibrosis. J. Cell. Mol. Med. 2019, 23, 1759–1770. [Google Scholar] [CrossRef]
- Leiva, O.; Ng, S.K.; Matsuura, S.; Chitalia, V.; Lucero, H.; Findlay, A.; Turner, C.; Jarolimek, W.; Ravid, K. Novel Lysyl Oxidase Inhibitors Attenuate Hallmarks of Primary Myelofibrosis in Mice. Int. J. Hematol. 2019, 110, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Büchold, C.; Hils, M.; Gerlach, U.; Weber, J.; Pelzer, C.; Heil, A.; Aeschlimann, D.; Pasternack, R. Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells 2022, 11, 1667. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Mäki, M.; Lundin, K.E.A.; Isola, J.; Friesing-Sosnik, T.; Taavela, J.; Popp, A.; Koskenpato, J.; Langhorst, J.; Hovde, Ø.; et al. A Randomized Trial of Transglutaminase 2 Inhibitor for Celiac Disease. The New England Journal of Medicine 2021. [Google Scholar] [CrossRef] [PubMed]
- Maamra, M.; Benayad, O.M.; Matthews, D.; Kettleborough, C.; Atkinson, J.; Cain, K.; Bon, H.; Brand, H.; Parkinson, M.; Watson, P.F.; et al. Transglutaminase 2: Development of Therapeutic Antibodies Reveals Four Inhibitory Epitopes and Confirms Extracellular Function in Fibrotic Remodelling. Br. J. Pharmacol. 2022, 179, 2697–2712. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Trinh-Minh, T.; Matei, A.; Györfi, A.; Hong, X.; Bergmann, C.; Schett, G.; Atkinson, J.; Bowcutt, R.; Patel, J.; et al. Amelioration of Fibrotic Remodeling of Human 3-Dimensional Full-Thickness Skin by Transglutamase 2 Inhibition. Arthritis Rheumatol. 2023, 75, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Fell, S.; Wang, Z.; Blanchard, A.; Nanthakumar, C.; Griffin, M. Transglutaminase 2: A Novel Therapeutic Target for Idiopathic Pulmonary Fibrosis Using Selective Small Molecule Inhibitors. Amino Acids 2021, 53, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Stuckey, D.J.; Murdoch, C.E.; Camelliti, P.; Lip, G.Y.H.; Griffin, M. Cardiac Fibrosis Can Be Attenuated by Blocking the Activity of Transglutaminase 2 Using a Selective Small-Molecule Inhibitor. Cell Death Dis. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zu, C.; Zhang, Y.; Wang, X.; Huan, X.; Wang, L. Blocking TG2 Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice through Inhibiting EMT. Respir. Physiol. Neurobiol. 2020, 276, 103402. [Google Scholar] [CrossRef]
- Badarau, E.; Wang, Z.; Rathbone, D.L.; Costanzi, A.; Thibault, T.; Murdoch, C.E.; Alaoui, S.E.; Bartkeviciute, M.; Griffin, M. Development of Potent and Selective Tissue Transglutaminase Inhibitors: Their Effect on TG2 Function and Application in Pathological Conditions. Chem. Biol. 2015, 22, 1347–1361. [Google Scholar] [CrossRef]
- Lee, J.; Guo, H.; Wang, S.; Maghsoud, Y.; Vázquez-Montelongo, E.A.; Jing, Z.; Sammons, R.M.; Cho, E.J.; Ren, P.; Cisneros, G.A.; et al. Unleashing the Potential of 1,3-Diketone Analogues as Selective LH2 Inhibitors. ACS Med. Chem. Lett. 2023, 14, 1396–1403. [Google Scholar] [CrossRef]
- Maghsoud, Y.; Vázquez-Montelongo, E.A.; Yang, X.; Liu, C.; Jing, Z.; Lee, J.; Harger, M.; Smith, A.K.; Espinoza, M.; Guo, H.-F.; et al. Computational Investigation of a Series of Small Molecules as Potential Compounds for Lysyl Hydroxylase-2 (LH2) Inhibition. J. Chem. Inf. Model. 2023, 63, 986–1001. [Google Scholar] [CrossRef] [PubMed]
- Devkota, A.K.; Veloria, J.R.; Guo, H.-F.; Kurie, J.M.; Cho, E.J.; Dalby, K.N. Development of a High-Throughput Lysyl Hydroxylase (LH) Assay and Identification of Small-Molecule Inhibitors against LH2. SLAS Discov. 2018, 24, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Meissner, E.G.; McLaughlin, M.; Matthews, L.; Gharib, A.M.; Wood, B.J.; Levy, E.; Sinkus, R.; Virtaneva, K.; Sturdevant, D.; Martens, C.; et al. Simtuzumab Treatment of Advanced Liver Fibrosis in HIV and HCV-infected Adults: Results of a 6-month Open-label Safety Trial. Liver Int. 2016, 36, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Vachhani, P.; Baskar, J.; Charlton, B.; Cheung, S.; Jarolimek, W.; Lee, S.-E.; Tan, P.; Watson, A.M.; Wu, S.-J. PXS5505-MF-101: A Phase 1/2a Study to Evaluate Safety, Pharmacokinetics and Pharmacodynamics of Pxs-5505 in Patients with Primary, Post-Polycythemia Vera or Post-Essential Thrombocythemia Myelofibrosis. Blood 2023, 142, 625. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, T.; Lyu, C.; Liang, K.; Du, Y. Cell Mediated ECM-Degradation as an Emerging Tool for Anti-Fibrotic Strategy. Cell Regen. 2023, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.W.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and Distinct Mechanisms of Fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The Extracellular Matrix in Development and Morphogenesis: A Dynamic View. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef]
- Phogat, S.; Thiam, F.; Yazeedi, S.A.; Abokor, F.A.; Osei, E.T. 3D in Vitro Hydrogel Models to Study the Human Lung Extracellular Matrix and Fibroblast Function. Respir. Res. 2023, 24, 242. [Google Scholar] [CrossRef]
- Talbott, H.E.; Mascharak, S.; Griffin, M.; Wan, D.C.; Longaker, M.T. Wound Healing, Fibroblast Heterogeneity, and Fibrosis. Cell Stem Cell 2022, 29, 1161–1180. [Google Scholar] [CrossRef]
- Urciuolo, F.; Imparato, G.; Netti, P.A. In Vitro Strategies for Mimicking Dynamic Cell–ECM Reciprocity in 3D Culture Models. Front. Bioeng. Biotechnol. 2023, 11, 1197075. [Google Scholar] [CrossRef]
- Perez-Castillejos, R. Replication of the 3D Architecture of Tissues. Mater. Today 2010, 13, 32–41. [Google Scholar] [CrossRef]
- Qin, L.; Liu, N.; Bao, C.; Yang, D.; Ma, G.; Yi, W.; Xiao, G.; Cao, H. Mesenchymal Stem Cells in Fibrotic Diseases—the Two Sides of the Same Coin. Acta Pharmacol. Sin. 2023, 44, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.N.; MacMillan, M.; Manship, T.; Moroni, F.; Glover, A.; Graham, C.; Semple, S.; Morris, D.M.; Fraser, A.R.; Pass, C.; et al. Study Protocol: A Multicentre, Open-Label, Parallel-Group, Phase 2, Randomised Controlled Trial of Autologous Macrophage Therapy for Liver Cirrhosis (MATCH). BMJ Open 2021, 11, e053190. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety Profile of Autologous Macrophage Therapy for Liver Cirrhosis. Nat. Med. 2019, 25, 1560–1565. [Google Scholar] [CrossRef]
Figure 1.
Collagen processing pathway, from transcription to crosslinked fibril formation. After transcription in the nucleus and subsequent translation, three alpha chains assemble into a procollagen triple helix. Upon export into the extracellular space, N and C-terminal pro-peptides are removed from procollagen converting it to tropocollagen. For fibrillar collagens, these tropocollagens then form fibrils which are subject to LOX-mediated crosslinking. LH hydroxylation precedes LOX activity and has been reported to occur both intra and extracellularly. Created with BioRender.com.
Figure 1.
Collagen processing pathway, from transcription to crosslinked fibril formation. After transcription in the nucleus and subsequent translation, three alpha chains assemble into a procollagen triple helix. Upon export into the extracellular space, N and C-terminal pro-peptides are removed from procollagen converting it to tropocollagen. For fibrillar collagens, these tropocollagens then form fibrils which are subject to LOX-mediated crosslinking. LH hydroxylation precedes LOX activity and has been reported to occur both intra and extracellularly. Created with BioRender.com.
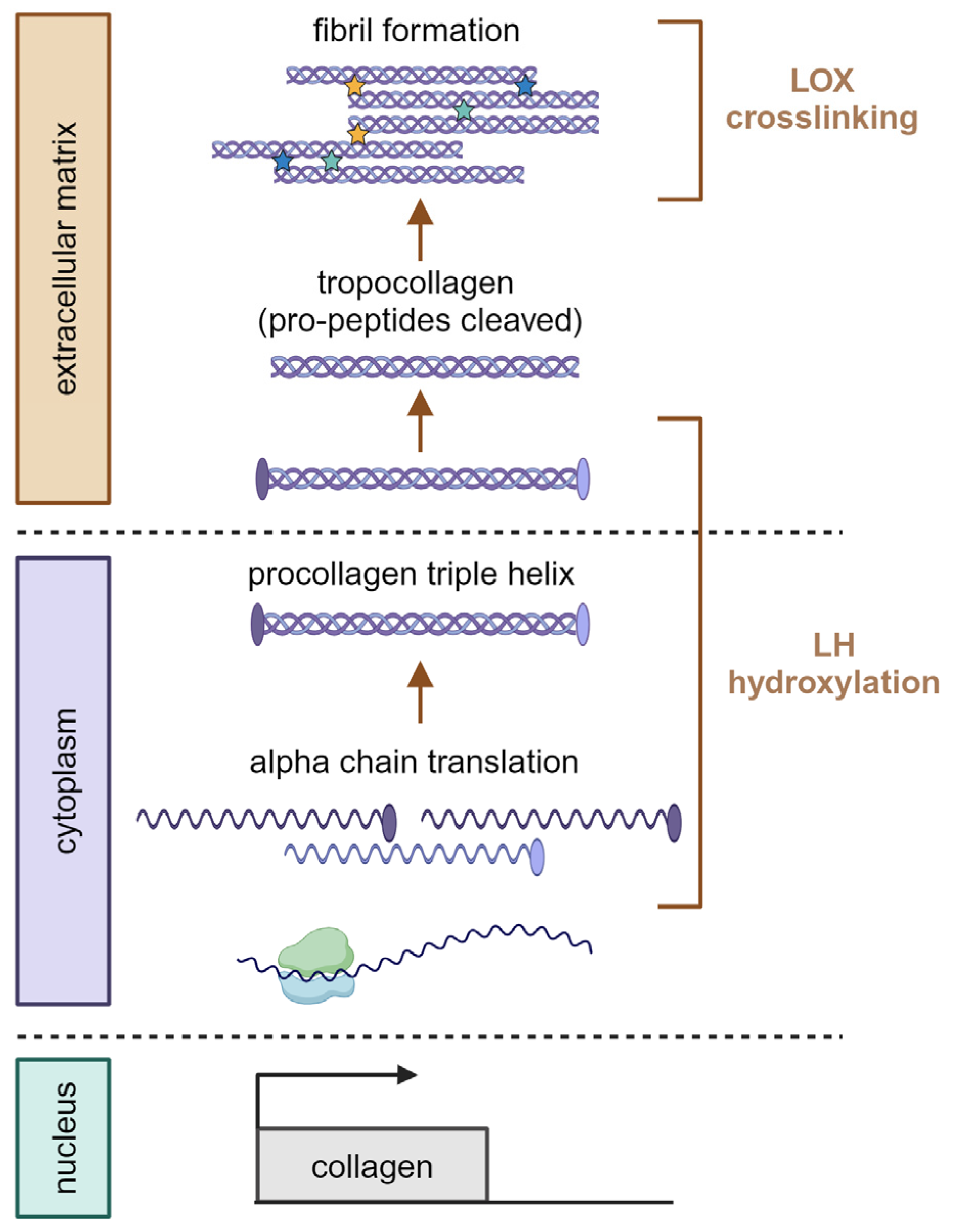
Figure 2.
Transglutaminase crosslinking. Transglutaminases (TGs) mediate a crosslinking reaction between lysine and glutamine residues. Created with BioRender.com.
Figure 2.
Transglutaminase crosslinking. Transglutaminases (TGs) mediate a crosslinking reaction between lysine and glutamine residues. Created with BioRender.com.
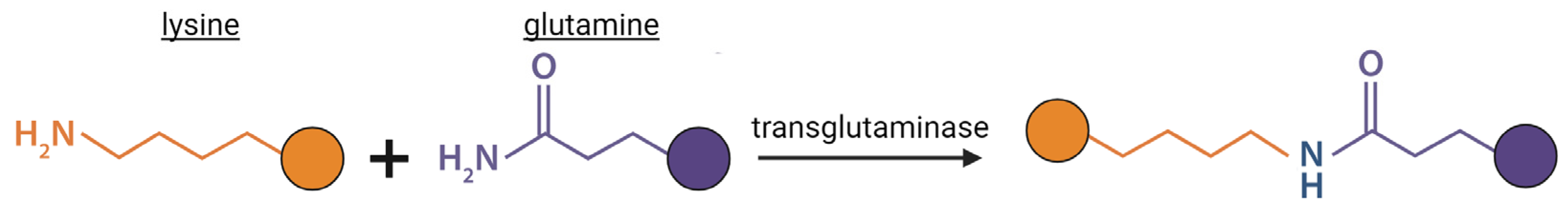
Figure 3.
Enzymatic reactions driven by lysine hydroxylase (LH) and lysyl oxidase (LOX) enzymes. A) LHs convert lysine to hydroxylysine through hydroxylation reaction. B) LOX’s convert hydroxylysine or lysine into aldehyde form via oxidative deamination. Created with BioRender.com.
Figure 3.
Enzymatic reactions driven by lysine hydroxylase (LH) and lysyl oxidase (LOX) enzymes. A) LHs convert lysine to hydroxylysine through hydroxylation reaction. B) LOX’s convert hydroxylysine or lysine into aldehyde form via oxidative deamination. Created with BioRender.com.
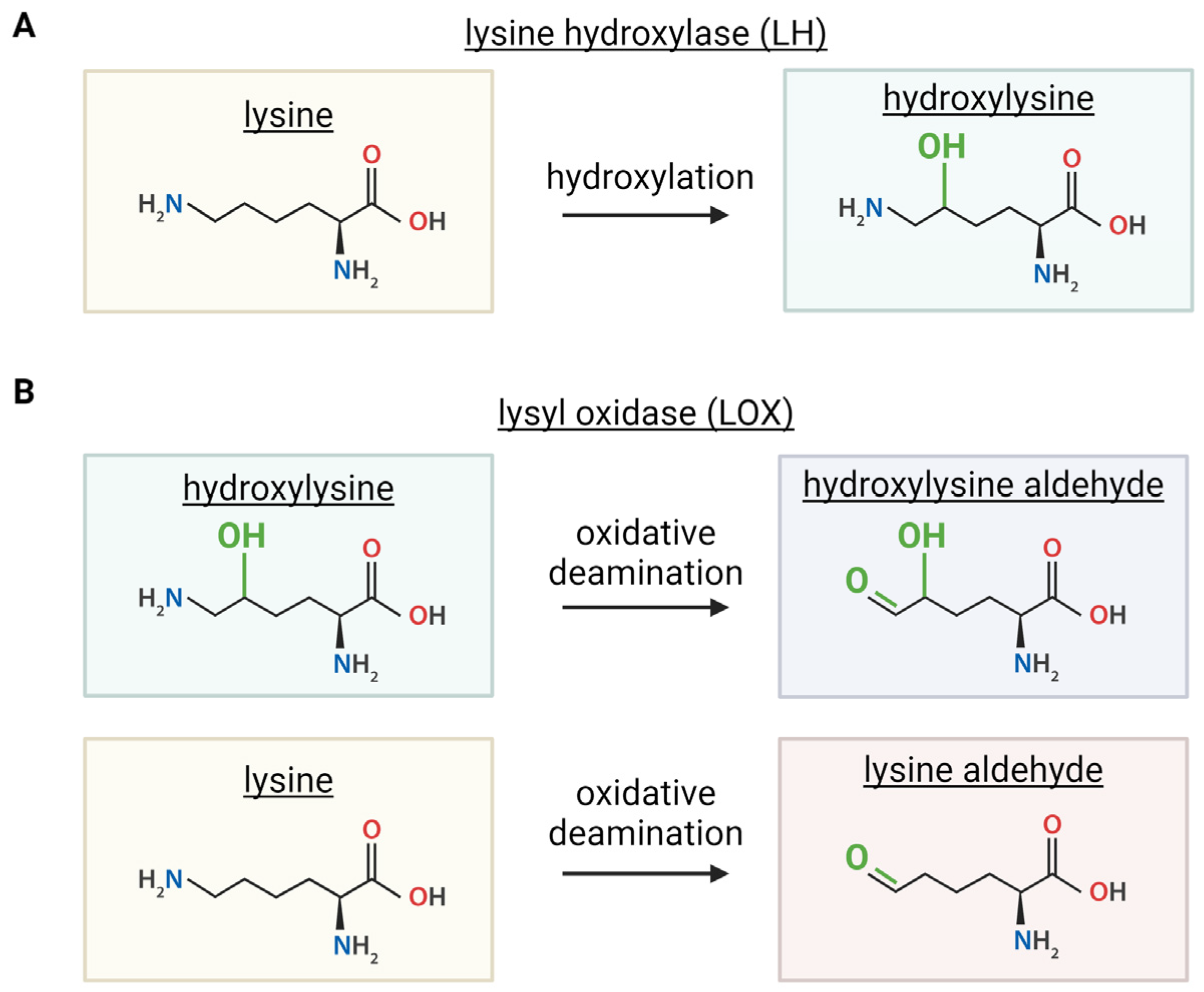
Figure 4.
Condensation reactions drive formation of immature collagen crosslinks. A lysine or hydroxylysine aldehyde from a LOX modification reacts with unmodified lysine or a hydroxylysine derived from an LH modification to form three different immature crosslinks. Immature collagen crosslinks vary by number of OH groups (green). Created with BioRender.com.
Figure 4.
Condensation reactions drive formation of immature collagen crosslinks. A lysine or hydroxylysine aldehyde from a LOX modification reacts with unmodified lysine or a hydroxylysine derived from an LH modification to form three different immature crosslinks. Immature collagen crosslinks vary by number of OH groups (green). Created with BioRender.com.
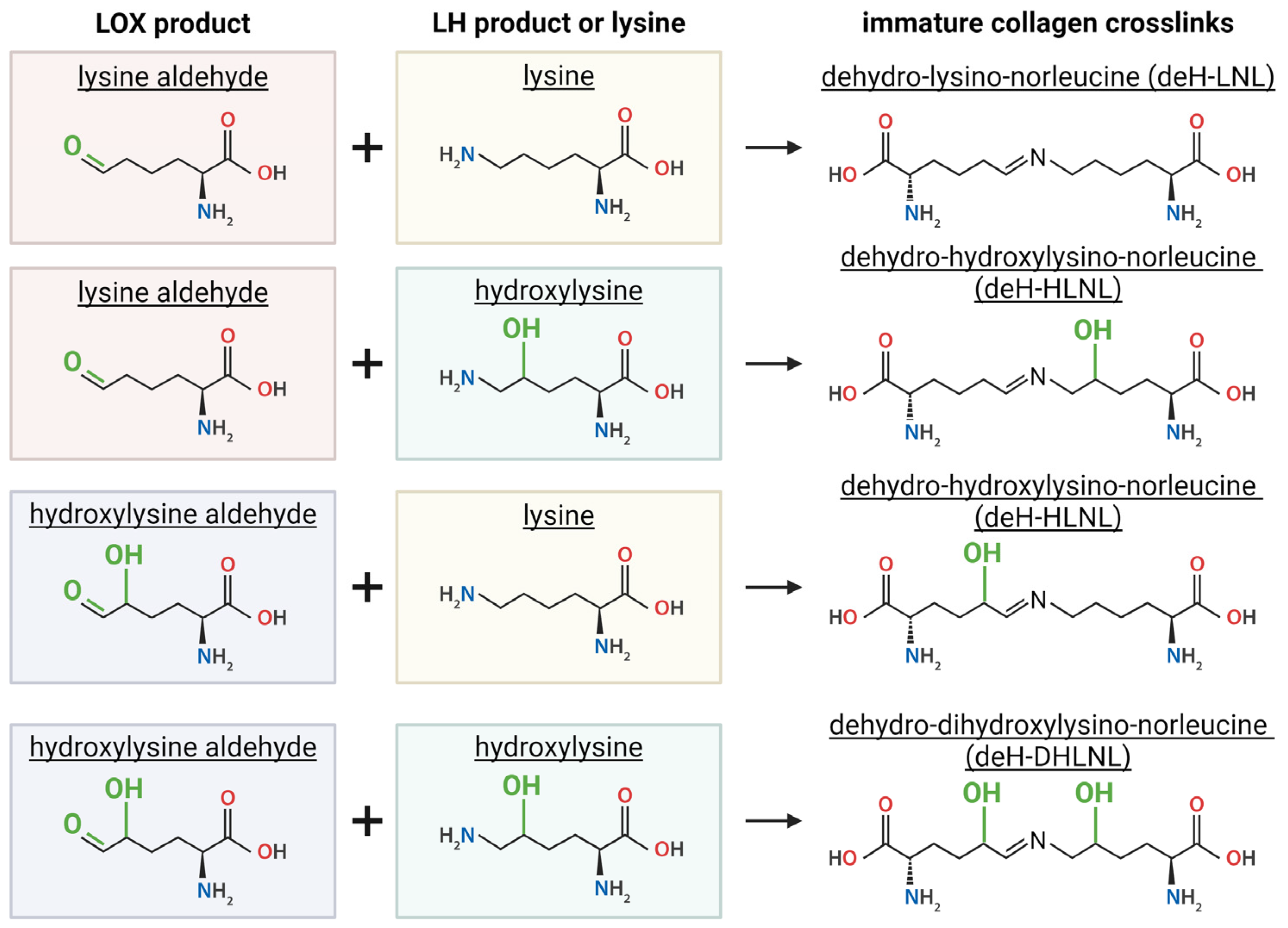
Figure 5.
Immature collagen crosslinks go through additional reactions to form mature crosslinks. Pyrroles are formed through a reaction between deHydro-Hydroxy-lysinenorleucine (deH-HLNL) or deHydro-Dihydroxy-lysinenorleucine (deH-DHLNL) with a lysine aldehyde. Pyridinolines are formed through a reaction between deH-HLNL or deH-DHLNL with a hydroxylysine aldehyde. Reactions with deH-HLNL produce Deoxy forms, Deoxy-pyridinoline and Deoxy-Pyrrole. Created with BioRender.com.
Figure 5.
Immature collagen crosslinks go through additional reactions to form mature crosslinks. Pyrroles are formed through a reaction between deHydro-Hydroxy-lysinenorleucine (deH-HLNL) or deHydro-Dihydroxy-lysinenorleucine (deH-DHLNL) with a lysine aldehyde. Pyridinolines are formed through a reaction between deH-HLNL or deH-DHLNL with a hydroxylysine aldehyde. Reactions with deH-HLNL produce Deoxy forms, Deoxy-pyridinoline and Deoxy-Pyrrole. Created with BioRender.com.
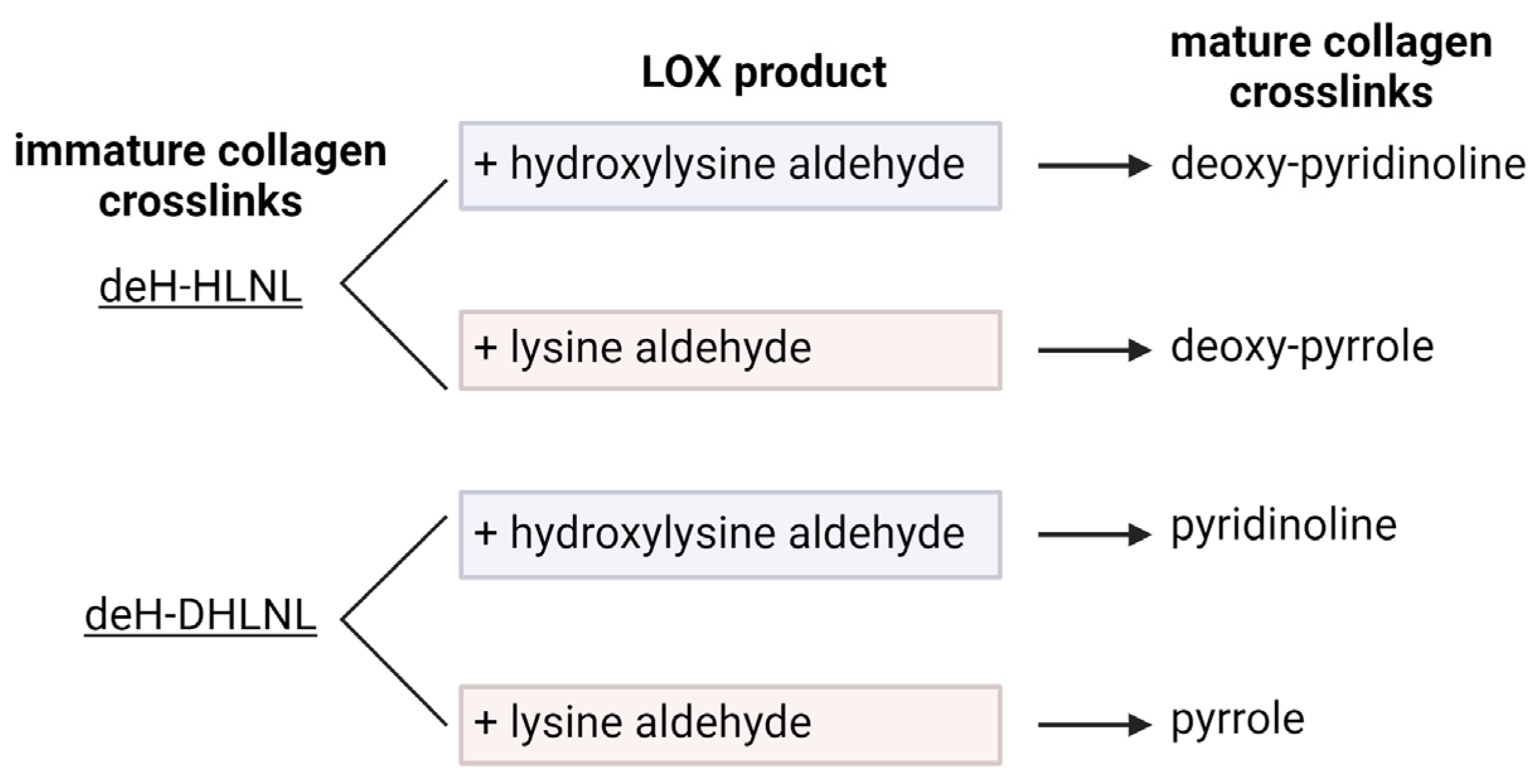

Table 1.
Therapeutic approaches to targeting ECM crosslinking for fibrotic diseases.
| Target | Target | Drug molecule | Drug type | Indication | Status | Reference |
|---|---|---|---|---|---|---|
| LOX | LOXL2 | Simtuzumab/GS-6624/AB0024 | Antibody | Cancer, IPF and liver fibrosis, myelofibrosis, PSC, NASH | Phase II - Discontinued | [69,70,71,72] |
| Pan-LOX | PXS-5505/SNT-5505 | small molecule | Myelofibrosis, Liver and pancreatic cancer | Phase II | [73,74,75] NCT05109052, NCT04676529 |
|
| LOXL2 | PAT-1251 | small molecule | IPF and other fibrotic diseases, Myelofibrosis | Phase II | [76], NCT04054245, NCT02852551 | |
| LOXL2 | GB2064 | small molecule | Myelofibrosis | Phase II | [77], NCT04679870 | |
| LOXL2 | PXS-5338 | small molecule | NASH, IPF, liver and kidney fibrosis | Phase I | [78] | |
| LOXL2 | PXS-5382/SNT-5382 | small molecule | Anti fibrotic IPF /CKD / NASH | Phase I | [75] | |
| Pan-LOX | PXS-6302 | small molecule | Anti scarring; Burns, established scars | Phase I | [79] | |
| LOXL2/LOXL3 | PXS-5153A | small molecule | liver fibrosis, myocardial infarction | Preclinical | [80] | |
| LOX | PXS-LOX_1 and PXS-LOX_2 | small molecule | Primary myelofibrosis (PMF) | Preclinical | [81] | |
| TG | TG2 | ZED1227/TAK-227 | small molecule | NAFLD with significant fibrosis | Phase II | [82,83], NCT05305599 |
| TG2 | Zampilimab | Antibody | Adult Kidney Transplant Recipients With Chronic Allograft Injury | Phase I/II | NCT04705350, NCT04335578 | |
| TG2 | AB1, DC1, and BB7 | Antibody | fibrosis and auto-immune disease | Preclinical | [84,95] | |
| TG2 | 1–155 | small molecule | IPF, Cardiac fibrosis | Preclinical | [86,87] | |
| TG2 | R281 | small molecule | IPF | Preclinical | [86] | |
| TG2 | GK921 | small molecule | Pulmonary fibrosis | Preclinical | [88] | |
| TG2 | compound 3h | small molecule | hypertensive nephrosclerosis | Preclinical | [89] | |
| LH | LH2 | 1,3-diketone analogues | small molecule | cancer metastasis | Preclinical | [90,91,92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



