
Submitted:
13 February 2024
Posted:
13 February 2024
You are already at the latest version
Abstract
Childhood glaucoma encompasses congenital and juvenile primary glaucoma, which are heterogeneous, uncommon, and irreversible optic neuropathies leading to visual impairment with poorly understood genetic basis. Our goal was to identify gene variants associated with these glaucoma types by assessing the mutational burden in 76 matrix metalloproteinase-related genes. We studied 101 childhood glaucoma patients with no identified monogenic alterations using next-generation sequencing. Gene expression was assessed through immunohistochemistry. Functional analysis of selected gene variants was conducted in cultured cells and in zebrafish. Patients presented a higher proportion of rare variants in four metalloproteinase-related genes, including CPAMD8 and ADAMTSL4, compared to controls. ADAMTSL4 protein expression was observed in the anterior segment of both the adult human and zebrafish larvae's eye, including tissues associated with glaucoma. In HEK-293T cells, expression of four ADAMTSL4 variants identified in this study showed that two variants (p.Arg774Trp and p.Arg98Trp) accumulated intracellularly, inducing endoplasmic reticulum stress. Additionally, overexpressing these ADAMTSL4 variants in zebrafish embryos confirmed partial loss-of-function effects for p.Ser719Leu and p.Arg1083His. Double heterozygous functional suppression of adamtsl4 and cpamd8 zebrafish orthologs resulted in reduced volume of both the anterior eye chamber and lens within the chamber, supporting a genetic interaction between these genes. Our findings suggest that accumulation of partial functional defects in matrix metalloproteinases-related genes may contribute to increased susceptibility to early-onset glaucoma and provide further evidence supporting the notion of a complex genetic inheritance pattern underlying the disease.
Keywords:
Glaucoma
; Matrix Metalloproteinases
; Multifactorial Inheritance.
1. Introduction
According to the World Glaucoma Association, childhood primary glaucoma presents before 16-18 years of age and encompasses both primary congenital glaucoma (PCG) and juvenile open-angle glaucoma (JOAG) [1]. These infrequent conditions can be viewed as part of a continuous disease spectrum [2] and represent a clinically diverse group of severe, progressive, and irreversible optic neuropathies characterized by apoptosis of retinal ganglion cells [3]. Untreated childhood glaucoma leads to irreversible visual impairment and blindness. PCG is the most common type of childhood glaucoma [4,5] with varying worldwide incidence, and JOAG has been described to account for approximately 4% of childhood glaucoma cases [6].
Developmental defects of the anterior chamber angle of the eye, known as goniodysgenesis, are believed to play a significant role in the pathogenesis of childhood glaucoma. In PCG, developmental arrest and malformation specifically affecting the trabecular meshwork (TM) and/or Schlemm's canal (SC) result in trabeculodysgenesis or goniodysgenesis [7,8]. JOAG patients present different forms of goniodysgenesis, including abnormally prominent iris processes, high iris insertion or a thick trabecular band that is due to the compact trabecular meshwork [9]. These structural alterations contribute to elevated intraocular pressure (IOP) and subsequent optic nerve damage.
The extracellular matrix (ECM) in the trabecular meshwork (TM) is abnormal in both PCG [8,10,11,12] and JOAG [13]. Physiological regulation of IOP and aqueous humor outflow involve continuous remodeling of the ECM in the TM, which is mediated by various matrix metalloproteinases (MMPs), [14] and the closely related ADAMs (A Disintegrin and Metalloproteinases) and ADAMTSs (A Disintegrin and Metalloproteinase with Thrombospondin Motif) [15]. ADAMTSL-like (ADAMTSL) is a second family of proteins that lack catalytic activity, are secreted and are part of the ECM, [16] with a member implicated in complex alterations that include congenital glaucoma [17].
Childhood glaucoma exhibits primarily a monogenic pattern, although some studies have indicated a complex genetic architecture in a significant proportion of patients [12,18,19,20,21]. Autosomal recessive inheritance is common in primary congenital glaucoma [21,22], while autosomal dominant inheritance is observed in JOAG [23,24]. CYP1B1 is the major gene involved in recessive PCG [25,26], and it is functionally disrupted in 18%-48% of non-consanguineous European patients [21,27,28,29]. Other genes, such as LTBP2 [30], MYOC [31], FOXC1 [32,33], and TEK [34], have also been reported to play a role in this pathology. Although GPATCH3 [19] and GUCA1C [35] have been proposed as candidate genes in PCG, their involvement in the disease remains unconfirmed. MYOC is the main gene associated with JOAG glaucoma [36], accounting for 8% to 36% of affected pedigrees [37] and contributing to the disease in 15-20% of European Caucasian patients [38]. EFEMP1 rare variants have been identified as the cause of JOAG in Filipino [39] and African-American families [40]. This genetic heterogeneity challenges the diagnosis and treatment of patients.
Here, we show an increased frequency of aggregate rare variants in several metalloproteinase-related genes, including CPAMD8 and ADAMTSL4, among childhood glaucoma patients, suggesting that this mutational burden may contribute to disease pathogenicity, as well as the existence of a complex inheritance. In addition, our data show deleterious effects of some of the identified ADAMTSL4 variants and reveal a functional interaction between cpamd8 and adamtsl4 in zebrafish. Finally, these results can be extrapolated to support the idea that other unexplained rare ocular diseases may also involve complex inheritance mechanisms.
2. Methods
2.1. Subjects
The study included patients enrolled from Hospital Clínico San Carlos (Madrid, Spain). A total of 101 unrelated childhood glaucoma patients (86 PCG and 15 JOAG), who did not carry pathogenic variants in CYP1B1, FOXC1, or MYOC genes, were selected for exome sequencing. The patients were mostly Spaniards of European ancestry (96%) and 4% were Arab and Sub-Saharan immigrants. Glaucoma specialists conducted comprehensive clinical examinations on all patients, including slit-lamp biomicroscopy, gonioscopy, biometry, intraocular pressure (IOP) measurement, and ophthalmoscopy. The childhood glaucoma diagnoses were made based on previously established criteria [21,24]. The study received approval from the Ethics Committee for Human Research at Hospital Clínico San Carlos (reference numbers 13/388-E and OFT-GCP-2019-01). Prior to their inclusion in the study, all participants provided informed written consent. The study adhered to the Declaration of Helsinki principles.
2.2. Human Tissue Samples
The eye of a 45-year-old Caucasian female donor, without any known ocular pathology, was obtained from the USA National Disease Research Interchange within 24 hours after enucleation and processed as previously described [12]. For immunohistochemical analysis, histological microtome sections (3 µm) of the eye were used.
2.3. Animals
Wild-type AB zebrafish (Danio rerio) were maintained under a 14 h light/10 h dark cycle at a temperature of 28 °C and fed a standard diet following established protocols [41]. Detailed experimental conditions are provided as Supplementary Methods. All animal husbandry and experimental procedures were carried out in adherence to the guidelines set by the Animal Research Committee of Castilla-La Mancha University (approved protocol number: PR-2017-01-19).
2.4. Next-Generation Sequencing
Peripheral blood samples were collected from the subjects, and genomic DNA was extracted using the Quiagen QIAamp DNA Blood Mini Kit. Whole-exome sequencing (WES) was carried out by Macrogen using the Twist Core Exome kit (Agilent Technologies). The libraries were pooled and sequenced on an Illumina NovaSeq6000 System (Illumina) with 151-bp paired-end reads, ensuring that over 98% of the target bases had a minimum coverage of 20x. Additional experimental information is provided in Supplementary Materials and Methods.
2.5. Variant Prioritization
A total of 76 matrix metalloproteinase genes (Table S1) were selected for aggregate rare variant burden analysis in childhood glaucoma patients. A multistep filtering approach was implemented to identify potential disease-causing variants, as described in Supplementary Methods. The presence of variants with pathogenic genotypes (i.e., compound heterozygous or homozygous recessive variants or heterozygous dominant variants) in known glaucoma-related genes, such as LTBP2, TEK, PITX2, PAX6, GPATCH3 and GUCA1C, that may explain the disease in a monogenic fashion was ruled out in all patients included in the study.
2.6. Fluorescence Immunohistochemistry (FIHC)
In this study, two types of tissue sections were utilized: paraffin-embedded human eye sections and zebrafish embryo cryosections. Detailed experimental conditions are provided as Supplementary Methods.
2.7. ADAMTSL4 Cloning and Site-Directed Mutagenesis
The human ADAMTSL4 cDNA (gene ID: 54507), cloned in the pCR4-TOPO cloning vector (Bioscience, ref: IRCBp5005E0212Q), was amplified and subcloned into an expression vector as described in Supplementary Methods. The four missense mutations (p.Arg98Trp, p.Ser719Leu, p.Arg774Trp, and p.Arg1083His) were also generated as described in Supplementary Methods.
2.8. Expression of Recombinant Proteins in Human Cells in Culture and Analysis of Endoplasmic Reticulum Stress
Human embryonic kidney 293T cells were seeded on coverslips in 24-well plates until they reached 70-80% confluence. They were transiently transfected with the different ADAMTSL4-GFP cDNA constructs at a concentration of 1.0 μg using the Superfect Transfection Reagent (Qiagen) according to the manufacturer's instructions. Detailed experimental conditions are provided as Supplementary Methods.
2.9. Functional Analyses in Zebrafish
Microinjection was performed on one-cell stage zebrafish embryos (50–300 embryos per experiment) using a Femtojet 5247 microinjector (Eppendorf) under a Nikon SMZ18 stereomicroscope. A total volume of 3 nl containing the respective pcDNA3.1-hADAMTSL4-GFP constructs (7 ng/μl) was injected. As a negative control, embryos were injected with pcDNA3.1-GFP. To ensure robustness of the results, at least three independent experiments were conducted for each cDNA construct, utilizing different zebrafish progenitors in each experiment. Lethality was quantified 24 hours after microinjection and expressed as a weighted average value (%). Larval phenotypes and ADAMTSL4-GFP protein fluorescence were evaluated at 3 days post-fertilization (dpf) using a Nikon SMZ18 stereo microscope.
2.10. Morphological Characterization of Zebrafish Anterior Segment Phenotypes
Adult zebrafish eyes (2 months post-fertilization) were dorsally photographed using a Nikon SMZ18 stereo microscope. The volumes of the anterior chamber and the lens within the anterior chamber were measured based on the assumption that they are spherical caps as indicated in the Supplementary Methods.
3. Results
3.1. Mutation Burden Analysis
This study included 101 unrelated patients diagnosed with childhood glaucoma, for whom a monogenic alteration could not be identified. Among these patients, 86 had PCG, while 15 had JOAG. The investigation explored the potential combined disruption of 76 MMP-related genes in these patients (Table S1). These genes encode proteins that participate in extracellular matrix (ECM) remodeling of the trabecular meshwork (TM), a process believed to play a key role in the pathogenesis of this type of glaucoma. To that end we carried out a variant burden analysis, in which we filtered heterozygous rare variants (<0.01) with predicted high or moderate functional impact (frameshift, non-sense, missense and donor/acceptor splicing sites). Furthermore, variants with at least 50 reads were compared to controls of European ancestry obtained from the ESP6500 and gnomAD v2.1.1 databases. In total, 338 rare variants were identified in the patients, averaging 4.45 filtered variants per gene (Table S1).
The variant burden analysis in patients across all individual genes revealed significant enrichment of rare variants with predicted high-moderate functional impact in four genes (ADAMTS2, ADAMTS18, ADAMTSL4, and CPAMD8), compared to at least one of the two control groups, after applying the extremely conservative Bonferroni correction for multiple testing (p<6.57x10-4) (Table 1 and Table S1). Additionally, the aggregate variant frequency in these four genes was significantly higher in patients compared to controls (29.20% vs. 9.13%-10.03% in controls, p<1x10-15, Table 1).
There was no significant enrichment of rare synonymous variants either across the individual genes (0.99%-4.46% vs. 0.97%-4.20% in controls, p=0.27-0.99, Table 1) or in the aggregate set of four genes (8.42% vs. 9.05%-9.83% in controls, p=0.57-0.63, Table 1). This finding indicates the specificity of the identified association. Therefore, patients with childhood glaucoma exhibited a higher likelihood of carrying rare variants in these MMP-related genes compared to the two control groups. This association was observed both when considering the genes individually, with odds ratios (OR) ranging from 1.77-2.75 for CPAMD8 to 4.49-4.70 for ADAMTS2 (Table 1), and when aggregating the variants (OR=4.06 (3.00-5.50), Table 1). In total, 59 variants were identified in these four genes, out of which 46 were unique, consisting of 45 missense nucleotide changes and one deletion of 4 nucleotides (Table S3). Twenty-three of the missense variants were predicted to be damaging or probably damaging by at least one pathogenicity algorithm (SIFT or PolyPhen2) (Table S3). The four nucleotides deletion (c.1899+1_1899+4delGTAG) was predicted to remove the complete consensus sequence of the splicing donor site in CPAMD8 intron 15. Therefore, this allele was classified as damaging. Fifty patients (49.5%) carried at least one filtered variant in one of the four significantly enriched MMP-related genes (Table S4). Seven cases (6.9%) carried more than two variants in these genes, of which, two patients (1.9%) presented 3 variants (Table 2). Segregation analyses by Sanger sequencing (Figure 1 and Figure S1) revealed that these multiple variants were not associated with a typical monogenic phenotype. This was evident as biallelic variants located on the same gene were also found in one of the progenitors who did not present glaucoma (i.e., PCG103 and PCG143), showing that these mutations were carried on the same chromosome (Figure 1). While ADAMTS2 variants appeared to segregate in an autosomal recessive compatible pattern in family PCG87, the absence of pathogenicity predicted by SIFT, PolyPhen2, and ClinVar classifications (Table S3) made it unlikely that these variants were the sole cause of the disease via a monogenic mechanism [42]. In addition, recessive mutations of this gene are associated with Ehlers-Danlos Syndrome, Dermatosparaxis Type (MIM # 225410). The main characteristics of this connective tissue disease, such as skin alterations, hernias, and mild to severe joint hypermobility [42], were not reported in the clinical records of this patient. This makes it unlikely that these mutations alone are the cause of the disease. Samples were not available for segregation analysis in one family with biallelic variants in the same gene (PCG291) and in one patient with two monoallelic variants in two genes (PCG219). Although we cannot completely dismiss the possibility that congenital glaucoma may be solely attributed to biallelic recessive ADAMTSL4 variants, we find it improbable. This is because only one of the variants (p.Val1073Ile) is classified as damaging, probably damaging or of unknown significant, while the other (p.Gly336Glu) is predicted to be benign of likely benign (Table S3). In addition, this patient did not have clinical records of typical alterations associated with ADAMTSL4 disruption (e.g., ectopia lentis or congenital cataract). The age at diagnosis for these patients with bilateral glaucoma ranged from 5 to 144 months. Except for PCG143, all of them required surgical treatment to correct IOP (Table 2). Five patients had European ancestry, while one patient had Arab ancestry (PCG291) and another one was a sub-Saharan immigrant (PCG219).
3.2. Genotype-Phenotype Correlation
We also investigated a potential relationship between disease severity and the presence of rare variants in MMP-related genes. To that end, we compared two clinical parameters, namely the age at diagnosis and the average number of surgeries per eye required to control IOP, between carriers and non-carriers of filtered variants in these genes. To provide additional context, from a previous study we selected PCG patients who carried null CYP1B1 genotypes associated with a severe phenotype characterized by early disease onset and a high number of ocular surgeries [21] (Table 3). The mean age at diagnosis did not exhibit a significant difference between carriers and non-carriers of filtered variants in MMP-related genes. However, the mean age at diagnosis for both carriers and non-carriers was higher than that of patients with null CYP1B1 genotypes (Table 3).
Interestingly, carriers of MMP-related genes demonstrated a twofold increase in the number of glaucoma surgeries per eye compared to non-carriers (1.98 surgeries vs. 0.88 surgeries), and both groups underwent fewer surgical interventions compared to individuals with null CYP1B1 genotypes (Table 3). There were no significant IOP differences among the three groups. Taken together, these findings suggest that patients without rare variants in MMP-related genes generally have a more favorable surgical prognosis compared to carriers of such variants, as well as those with null CYP1B1 genotypes.
3.3. Expression of ADAMTSL4 in Human and Zebrafish Ocular Tissues
We previously reported the expression of CPAMD8, one of the enriched genes, in both human and zebrafish eyes [12]. In this study, we selected another enriched gene, ADAMTSL4, to examine the presence of its encoded protein in glaucoma-related ocular tissues using confocal FIHC. Immunolabeling with anti-ADAMTSL4 antibodies revealed positive signals in various parts of the adult human anterior segment, including the lens epithelium and fibers (Figure 2A), corneal epithelium (Figure 2B) and endothelium (Figure 2D), as well as the stroma of the iris (Figure 2E) and ciliary processes (Figure 2F). Additionally, positive signals were observed in iris fibroblasts and iris sphincter muscle (Figure 2E). The retina was not stained with this antibody (Figure 2C). The specificity of these signals was confirmed by the absence of staining in the corresponding negative controls (Figure 2H-M). However, the negative controls revealed non-specific immunostaining in the lens capsule, corneal stroma and trabecular meshwork (Figure 2H, K, and N, asterisks).
We also used the same anti-ADAMTSL4 antibody to investigate the ocular expression of the ortholog protein in zebrafish larvae (6 dpf) using FIHC. Consistent with the human eye, positive signals were observed in the cornea and lens epithelium of wild type zebrafish (Figure 3A). Interestingly, ADAMTSL4 immunolabeling was also detected in the periocular mesenchyme, which plays a role in the morphogenesis of iris and anterior chamber angle structures (Figure 3A). Furthermore, the antibody decorated the two retinal plexiform layers, indicating the presence of the protein in these layers. These signals were completely absent in an adamtsl4 knock out zebrafish line and in ocular tissue sections incubated only with the secondary antibody (Figure 3B and C, respectively), which were used as negative controls, providing robust evidence for the specificity of the signals.
3.4. Functional Analysis of Selected ADAMTSL4 Variants in Cells in Culture and in Zebrafish Embryos
We selected four ADAMTSL4 variants predicted to be pathogenic (p.Arg98Trp, p.Ser719Leu, p.Arg774Trp and p.Arg1083His) for functional evaluation using both cells in culture and zebrafish. In addition, we included the wild type sequence as a control. The variants were cloned by site-directed mutagenesis and fused to GFP at their C-terminus as a reporter molecule. Upon transfection into HEK-293T cells, detection of GFP-fluorescence revealed the presence of the wild type protein in both the cytoplasm and nucleus (Figure 4A-D). In contrast, two mutant proteins (p.Arg98Trp and p.Arg774Trp) accumulated intracellularly in a granular pattern, probably within the ER, and were absent in the nucleus (Figure 4E-H and M-P, respectively). The remaining two variants (p.Ser719Leu and p.Arg1083His) displayed expression patterns similar to those of the wild type protein (Figure 4I-L and Q-T, respectively). In parallel, immunochemical detection of PDI, a marker of ER stress [43], was performed to evaluate protein misfolding induced by these variants. Cells expressing the wild type protein showed a diffuse cytoplasmic signal, indicative of the presence of endogenous PDI in the ER (Figure 4B-D).
In contrast, cells expressing p.Arg98Trp and p.Arg774Trp showed increased PDI immunofluorescence compared to cells expressing the wild type ADAMTSL4 (Figure 3F-H and N-P, respectively), suggesting the presence of induced ER stress. Cells expressing the other two variants (p.Ser719Leu and p.Arg1083His) showed PDI fluorescence levels similar to those of cells expressing the normal protein (Figure 4J-L and R-T, respectively).
The negative control indicated the specificity of the GFP signal (Figure 4U-X). To further evaluate ER stress associated with the expression of these variants, PDI was analyzed by western blotting using cell lysates of transfected cells. Detection of beta-actin and neomycin phosphotransferase II (NPTII) were used as loading and transfection controls, respectively. PDI levels were quantified by densitometry and normalized with beta-actin and NPTII. The analysis showed significantly increased PDI levels in cells expressing p.Arg98Trp and p.Arg774Trp variants compared to cells expressing the wild type protein (Figure 5). Expression of p.Ser719Leu and p.Arg1083His showed PDI levels, that were not statistically different from those associated with the wild type sequence (Figure 5).
Thus, the results support the pathogenicity of variants p.Arg98Trp and p.Arg774Trp due to ADAMTSL4 misfolding, while p.Ser719Leu and p.Arg1083His do not appear to significantly disrupt the protein's structure. Subsequently, we assessed the activity of these variants in zebrafish embryos microinjected with the different cDNA constructs. Expression of the control construct encoding wild type ADAMTSL4 resulted in early (24 hpf) lethal phenotypes in 20% of the embryos, which was two-times higher than the lethality observed in embryos microinjected with a control cDNA construct encoding GFP (Figure 6A).
Zebrafish embryos expressing variants p.Arg98Trp and p.Arg774Trp exhibited a three-fold increase in lethality compared to the GFP controls and approximately one and a half times higher lethality than embryos expressing wild type ADAMTSL4 (Fig 6A). Interestingly, mutants p.Ser719Leu and p.Arg1083His led to reduced lethality compared to the wild type protein (Figure 6A), indicating that these variants induce a partial loss-of-function (hypomorphic) effect on the protein.
Control zefrafish embryos (3 dpf) microinjected with the cDNA encoding GFP showed normal morphology and mosaic fluorescence, generally in the yolk sack (Figure 6B-D), but embryos expressing wild type ADAMTSL4 and variants p.Arg98Trp and p.Arg774Trp presented global maldevelopment and severe generalized edema, particularly noticeable in the pericardium and yolk sac (Figure 6E-G, H-J and N-P, respectively). The expression of variants p.Ser719Leu and p.Arg1083His was associated with less severe tissular alterations (Figure 6K-M and Q-S, respectively), such as jaw maldevelopment (Figure 6Q, arrowhead, and S) and diffuse appearance of the tissue in some areas of the body (Figure 6K, arrowhead, and M), suggesting an alteration of the organizational pattern. These results are in accordance with a hypomorphic, or partial loss-of-function (LoF), effect associated with these variants. The morphological and tissular changes correlated with the expression of the different ADAMTSL4 proteins (Figure 6K and M). Green fluorescence was absent in non-injected zebrafish embryos (Figure 6T-V), confirming the specificity of the signals. These findings support the notion that overexpression of these proteins disrupts normal embryonic development to varying degrees, likely by altering the extracellular matrix.
3.5. Functional Testing in Zebrafish of the Hypothesis on the Mutation Burden of MMP-Related Genes in Glaucoma
To investigate the influence of the mutation burden detected in glaucoma patients on the disease, we evaluated the ocular phenotypes resulting from the combination of monoallelic LoF mutations in some of the enriched genes. For simplicity, we selected adamtsl4 and cpamd8, which are evolutionarily conserved in zebrafish, and we took advantage of two unpublished knockout zebrafish lines that were generated in our laboratory. By crossing the adamtsl4 and cpamd8 knockout lines, we obtained double heterozygotes (adamtsl4/+ and cpamd8/+).
In line with our hypothesis, we observed a reduction in the volume of both the anterior chamber of the eye and the portion of the lens inside the anterior chamber in the double heterozygotes compared to the wild type and single heterozygous adult zebrafish (Figure 7A-D). Quantitative analysis of these volumes confirmed their similarity in wild type zebrafish and single heterozygous adamstsl4 and cpamd8 zebrafish. However, in the double heterozygotes, these volumes were significantly reduced by approximately four-fold (Figure 7E and F).
These findings provide strong support for the existence of a genetic interaction between these two genes and offer evidence to suggest that their combined partial functional alterations may also lead to morphological changes in the anterior chamber, potentially contributing to childhood glaucoma.
4. Discussion
Despite the advances in identifying genetic alterations underlying childhood glaucoma, genomic analyses assuming a monogenic inheritance of the disease have failed to identify the causative genetic alterations in many patients. This fact, along with other lines of evidence, suggest a complex mechanism of transmission that involves the participation of multiple genes. Exceptions to Mendelian expectations are also increasingly recognized in other rare diseases, likely reflecting the action of modifier genes [44]. In the present study we investigate the possible role in childhood glaucoma of rare variants in MMP-related genes. Our mutational burden analysis applying a stringent statistical correction for multiple testing, suggests that partial functional alteration of four of these genes, combined with additional mild impairment in other genes, might contribute to the ECM anomalies present in the TM of patients affected by childhood glaucoma. Our filtering strategy selected uncommon variants potentially disrupting the function of proteins. Most of the unique variants (97.8%) were missense nucleotide changes, of which more than half were classified as damaging or probably damaging by at least one pathogenicity algorithm, providing a bioinformatic support for their functional impact. Most computational tools are developed to identify highly penetrant and clinically relevant missense variants, present in classic Mendelian diseases. However, our assumption is that the variants underlying the disease in our cohort of patients are mildly deleterious, and therefore, difficult to identify bioinformatically. Although highly time consuming functional assays are required to evaluate the functional impact of these variants, the evidence that most rare missense alleles are deleterious in humans [45], also indicate that this group of missense variants might contribute to the genetic basis of the disease in our cohort. We hypothesize that accumulation of mild or partial functional alterations in different genes involved in ECM formation may form part of a complex pathogenic mechanism resulting in development and progression of childhood glaucoma. The identification of an increased burden of rare variants in these metalloproteinase-related genes in patients with this type of glaucoma suggests the potential significance of analyzing genetic variants within these genes. Further investigations are necessary to determine whether such analysis could be valuable in identifying individuals at a higher risk for the disease and predicting the progression of the disease. Understanding the impact of these rare variants on disease pathogenicity could guide the development of targeted therapies aimed at modulating metalloproteinase-related pathways or compensating for their dysregulation. The four enriched genes are expressed in ocular tissues [46,47,48,49] and the presence of ADAMTSL4 and CPAMD8 proteins has been demonstrated in anterior segment structures of the eye involved in glaucoma [12,49]. In addition, these genes play relevant roles in physiology of the ocular ECM and their individual functional alteration are associated with different ocular pathologies, suggesting that the observed enrichment of rare variants in glaucoma patients may dysregulate MMP-related processes, contributing to glaucoma and other ocular pathologies. In this line, ADAMTS2 encodes a procollagen proteinase that plays a crucial role in the cleavage of fibrillar procollagens types I-III and type V [50], involved in the recessively inherited Ehlers-Danlos syndrome type VIIC, which manifest ocular alterations, including glaucoma in few patients [51,52]. ADAMTS18 participates in the morphology in various tissues and organs [53] and its LoF results in abnormal ocular development [54]. ADAMTSL4, participates in zonular formation and maintenance [48], and is involved in ectopia lentis [55]. CPAMD8 encodes a protein that is part of the A2M/C3 (alpha-2-macroglobulin/complement 3) protein family [56], which present a broad spectrum of endopeptidase inhibitor activity [57], and is associated with autosomal recessive anterior segment dysgenesis 8 (https://search.clinicalgenome.org/kb/genes/HGNC:23228, accessed 04/27/2023). It has been suggested that CPAMD8 loss-of-function may alter the regulation of extracellular proteinases involved in ECM remodeling [12].
The detection of the ADAMTSL4 protein in the ocular anterior segment of both adult human and zebrafish larvae, suggests that it has evolutionary conserved and specific roles in the physiology of these ocular tissues, as well as in glaucoma. In addition, the presence of the protein in the retina of zebrafish larvae could indicate additional functions or involvement in retinal physiology beyond the anterior segment.
The intracellular accumulation, along with increased PDI expression, in HEK-293T cells of two of the identified ADAMTSL4 variants (p.Arg774Trp and p.Arg98Trp), indicated the induction of ER stress, which is associated with various pathological conditions, including glaucoma [58]. These results provides a potential mechanism for the role of ADAMTSL4 variants in childhood glaucoma. The functional defect of the identified ADAMTSL4 variants was also supported by the overexpression studies in zebrafish embryos, which revealed two different effects caused by the four variants evaluated. Variants p.Ser719Leu and p.Arg1083His were found to be less harmful than the wild type protein, suggesting a partial loss-of-function effect. On the other hand, variants p.Arg774Trp and p.Arg98Trp showed increased lethality, indicating a gain-of-function effect.
Double heterozygous functional suppression of the zebrafish orthologs of ADAMTSL4 and CPAMD8, resulted in a reduction in the volume of the anterior chamber of the adult eye, demonstrating the collaborative role of these genes in the development of anterior segment ocular structures. Moreover, this result suggest that combined dysfunction of both genes could disrupt the normal development of the anterior segment of the eye and play a role in the pathogenesis of different ocular disorders, including glaucoma.
Limitations of this investigation include the relatively small number of patients available for the association study and the assumption that additional unidentified gene variants with subtle negative impacts, which are challenging to demonstrate, are also involved in the development of glaucoma. Additionally, zebrafish may not accurately represent the nature of human ocular development and the pathogenesis of glaucoma.
In summary, this study offers new insights into the role of ECM genes and its functional disruption on childhood glaucoma development. Further investigations are required to elucidate the precise mechanisms through which these variants contribute to disease pathogenesis and their relevance in clinical contexts. These findings may also have wider implications for understanding the molecular mechanisms involved in other inherited and unresolved eye pathologies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
"Instituto de Salud Carlos III/European Regional Development Fund (ERDF)", Grant/Award Numbers: RD16/0008/0019, OFTARED, PI19/00208, RD16/0008/0004, and PI19/01431; the “Consejería de Ciencia y Tecnología de la Junta de Comunidades de Castilla-La Mancha", Grant/Award Number: SBPLY/17/180501/000404; http://www.educa.jccm.es/idiuniv/es; the “Universidad de Castilla-La Mancha”, Grant/Award Numbers: 2022-GRIN-34136 and 2020-PREDUCLM-16605; the “Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana”, Grant/Award Number: UGP-21-227.
Acknowledgments
We express our sincere gratitude to the patients and their families for their invaluable cooperation in this study. Additionally, we would like to extend our thanks to Ms. María-José Cabañero for her exceptional technical assistance, and Ms. Ainhoa Colina Jareño for their outstanding support in the extraction and management of biological samples and clinical information. We also thanks Ms. Irene Llopis Torregrosa, nurse of the Ophthalmology Department, for her contribution extracting blood samples. The authors gratefully acknowledge the NHLBI GO Exome Sequencing Project and its continuous studies, which generated and supplied exome variant calls for comparative analysis: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Allen Beck TCPC, Sharon Freedman. Childhood Glaucoma. The 9th Consensus Report of the World Glaucoma Association. In: Grajewski AL, Papoudopoulos M, Grigg J, Freedman S, eds.Childhood Glaucoma. The 9th Consensus Report of the World Glaucoma Association, Ámsterdam, The Netherlands: Kugler Publications, 2013; 3-10.
- Selvan H, Gupta S, Wiggs JL, Gupta V. Juvenile-onset open-angle glaucoma - A clinical and genetic update. Surv Ophthalmol 2022; 67: 1099-117. [CrossRef]
- Quigley HA. Ganglion cell death in glaucoma: pathology recapitulates ontogeny. AustNZJOphthalmol 1995; 23: 85-91. [CrossRef]
- Taylor RH, Ainsworth JR, Evans AR, Levin AV. The epidemiology of pediatric glaucoma: the Toronto experience. J AAPOS 1999; 3: 308-15. [CrossRef]
- Chan TCP, Brookes J, Cavuoto K, Bitrian E, Grajewski AL. Primary congenital glaucoma and juvenile open-angle glaucoma. In: Weinreb RN, Grajewski AL, Papadopoulos M, Grigg J, Freedman S, eds.Primary congenital glaucoma and juvenile open-angle glaucoma, Ámsterdam, The Netherlands: Kluger Publications, 2013; 137-53. K: Netherlands.
- Fung DS, Roensch MA, Kooner KS, Cavanagh HD, Whitson JT. Epidemiology and characteristics of childhood glaucoma: results from the Dallas Glaucoma Registry. Clin Ophthalmol 2013; 7: 1739-46. [CrossRef]
- Gould DB, John SW. Anterior segment dysgenesis and the developmental glaucomas are complex traits. HumMolGenet 2002; 11: 1185-93. [CrossRef]
- Anderson DR. The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. TransAmOphthalmolSoc 1981; 79: 458-85.
- Gupta V, Srivastava RM, Rao A, Mittal M, Fingert J. Clinical correlates to the goniodysgensis among juvenile-onset primary open-angle glaucoma patients. Graefes Arch Clin Exp Ophthalmol 2013; 251: 1571-6. 10.1007/s00417-013-2262-2.
- García-Antón MT, Salazar JJ, de Hoz R, Rojas B, Ramírez AI, Triviño A, Aroca-Aguilar JD, García-Feijoo J, Escribano J, Ramírez JM. Goniodysgenesis variability and activity of CYP1B1 genotypes in primary congenital glaucoma. PLoS One 2017; 12: e0176386. [CrossRef]
- Hollander DA, Sarfarazi M, Stoilov I, Wood IS, Fredrick DR, Alvarado JA. Genotype and phenotype correlations in congenital glaucoma. TransAmOphthalmolSoc 2006; 104: 183-95.
- Bonet-Fernández JM, Aroca-Aguilar JD, Corton M, Ramírez AI, Alexandre-Moreno S, García-Antón MT, Salazar JJ, Ferre-Fernández JJ, Atienzar-Aroca R, Villaverde C, Iancu I, Tamayo A, Méndez-Hernández CD, Morales-Fernández L, Rojas B, Ayuso C, Coca-Prados M, Martinez-de-la-Casa JM, García-Feijoo J, Escribano J. CPAMD8 loss-of-function underlies non-dominant congenital glaucoma with variable anterior segment dysgenesis and abnormal extracellular matrix. Hum Genet 2020. [CrossRef]
- Tawara A, Inomata H. Developmental immaturity of the trabecular meshwork in juvenile glaucoma. Am J Ophthalmol 1984; 98: 82-97. [CrossRef]
- Weinreb RN, Robinson MR, Dibas M, Stamer WD. Matrix Metalloproteinases and Glaucoma Treatment. J Ocul Pharmacol Ther 2020; 36: 208-28. [CrossRef]
- Keller KE, Bradley JM, Acott TS. Differential effects of ADAMTS-1, -4, and -5 in the trabecular meshwork. Invest Ophthalmol Vis Sci 2009; 50: 5769-77.
- Apte SS. ADAMTS Proteins: Concepts, Challenges, and Prospects. Methods Mol Biol 2020; 2043: 1-12. [CrossRef]
- Hendee K, Wang LW, Reis LM, Rice GM, Apte SS, Semina EV. Identification and functional analysis of an ADAMTSL1 variant associated with a complex phenotype including congenital glaucoma, craniofacial, and other systemic features in a three-generation human pedigree. Hum Mutat 2017; 38: 1485-90. [CrossRef]
- Medina-Trillo C, Aroca-Aguilar JD, Ferre-Fernández JJ, Alexandre-Moreno S, Morales L, Méndez-Hernández CD, García-Feijoo J, Escribano J. Role of FOXC2 and PITX2 rare variants associated with mild functional alterations as modifier factors in congenital glaucoma. PLoS One 2019; 14: e0211029. [CrossRef]
- Ferre-Fernández JJ, Aroca-Aguilar JD, Medina-Trillo C, Bonet-Fernández JM, Méndez-Hernández CD, Morales-Fernández L, Corton M, Cabañero-Valera MJ, Gut M, Tonda R, Ayuso C, Coca-Prados M, García-Feijoo J, Escribano J. Whole-Exome Sequencing of Congenital Glaucoma Patients Reveals Hypermorphic Variants in GPATCH3, a New Gene Involved in Ocular and Craniofacial Development. Sci Rep 2017; 7: 46175. [CrossRef]
- Chakrabarti S, Pyatla G, Zhang W, Dixit R, Moreno L, Mandal AK, Senthil S, Bera S, Kumari S, Kabra M, Rathi S, Hameed S, Khanna SC, Kaur I, Khanna H. Complex Molecular Mechanisms Underlie Primary Congenital Glaucoma Due to the Involvement of Multiple Biochemical Pathways In.Complex Molecular Mechanisms Underlie Primary Congenital Glaucoma Due to the Involvement of Multiple Biochemical Pathways Vol. 61, Baltimore, MD, US: Investigative Ophthalmology & Visual Science 2020; 3516. I: MD, US.
- Lopez-Garrido MP, Medina-Trillo C, Morales-Fernandez L, Garcia-Feijoo J, Martinez-de-la-Casa JM, Garcia-Anton M, Escribano J. Null CYP1B1 genotypes in primary congenital and nondominant juvenile glaucoma. Ophthalmology 2013; 120: 716-23. [CrossRef]
- Sarfarazi M, Stoilov I. The Third Genetic Locus (GLC3C) for Primary Congenital Glaucoma (PCG) Maps to Chromosome 14q24.3. Am J HumGenet 2002; 71(4): (Supplement). A455.
- Stoilova D, Child A, Brice G, Desai T, Barsoum-Homsy M, Ozdemir N, Chevrette L, Adam MF, Garchon HJ, Pitts CR, Sarfarazi M. Novel TIGR/MYOC mutations in families with juvenile onset primary open angle glaucoma. JMedGenet 1998; 35: 989-92. [CrossRef]
- Campos-Mollo E, Sanchez-Sanchez F, Lopez-Garrido MP, Lopez-Sanchez E, Lopez-Martinez F, Escribano J. MYOC gene mutations in Spanish patients with autosomal dominant primary open-angle glaucoma: a founder effect in southeast Spain. Mol Vis 2007; 13: 1666-73.
- Sarfarazi M, Stoilov I. Molecular genetics of primary congenital glaucoma. Eye 2000; 14 ( Pt 3B): 422-8. [CrossRef]
- Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. HumMolGenet 1997; 6: 641-7.
- Weisschuh N, Wolf C, Wissinger B, Gramer E. A clinical and molecular genetic study of German patients with primary congenital glaucoma. AmJOphthalmol 2009; 147: 744-53.
- Colomb E, Kaplan J, Garchon HJ. Novel cytochrome P450 1B1 (CYP1B1) mutations in patients with primary congenital glaucoma in France. HumMutat 2003; 22: 496.
- Campos-Mollo E, Lopez-Garrido MP, Blanco-Marchite C, Garcia-Feijoo J, Peralta J, Belmonte-Martinez J, Ayuso C, Escribano J. CYP1B1 mutations in Spanish patients with primary congenital glaucoma: phenotypic and functional variability. Mol Vis 2009; 15: 417-31.
- Ali M, McKibbin M, Booth A, Parry DA, Jain P, Riazuddin SA, Hejtmancik JF, Khan SN, Firasat S, Shires M, Gilmour DF, Towns K, Murphy AL, Azmanov D, Tournev I, Cherninkova S, Jafri H, Raashid Y, Toomes C, Craig J, Mackey DA, Kalaydjieva L, Riazuddin S, Inglehearn CF. Null mutations in LTBP2 cause primary congenital glaucoma. AmJHumGenet 2009; 84: 664-71.
- Kaur K, Reddy AB, Mukhopadhyay A, Mandal AK, Hasnain SE, Ray K, Thomas R, Balasubramanian D, Chakrabarti S. Myocilin gene implicated in primary congenital glaucoma. ClinGenet 2005; 67: 335-40.
- Medina-Trillo C, Sánchez-Sánchez F, Aroca-Aguilar JD, Ferre-Fernández JJ, Morales L, Méndez-Hernández CD, Blanco-Kelly F, Ayuso C, García-Feijoo J, Escribano J. Hypo- and hypermorphic FOXC1 mutations in dominant glaucoma: transactivation and phenotypic variability. PLoS One 2015; 10: e0119272. [CrossRef]
- Medina-Trillo C, Aroca-Aguilar JD, Mendez-Hernandez CD, Morales L, Garcia-Anton M, Garcia-Feijoo J, Escribano J. Rare FOXC1 variants in congenital glaucoma: identification of translation regulatory sequences. Eur J Hum Genet 2016; 24: 672-80. [CrossRef]
- Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, Feng L, Limviphuvadh V, Whisenhunt KN, Maurer-Stroh S, Yanovitch TL, Kalaydjieva L, Azmanov DN, Finzi S, Mauri L, Javadiyan S, Souzeau E, Zhou T, Hewitt AW, Kloss B, Burdon KP, Mackey DA, Allen KF, Ruddle JB, Lim SH, Rozen S, Tran-Viet KN, Liu X, John S, Wiggs JL, Pasutto F, Craig JE, Jin J, Quaggin SE, Young TL. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest 2016; 126: 2575-87. [CrossRef]
- Morales-Camara S, Alexandre-Moreno S, Bonet-Fernandez JM, Atienzar-Aroca R, Aroca-Aguilar JD, Ferre-Fernandez JJ, Mendez CD, Morales L, Fernandez-Sanchez L, Cuenca N, Coca-Prados M, Martinez-de-la-Casa JM, Garcia-Feijoo J, Escribano J. Role of GUCA1C in Primary Congenital Glaucoma and in the Retina: Functional Evaluation in Zebrafish. Genes (Basel) 2020; 11. [CrossRef]
- Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, Nishimura D, Clark AF, Nystuen A, Nichols BE, Mackey DA, Ritch R, Kalenak JW, Craven ER, Sheffield VC. Identification of a gene that causes primary open angle glaucoma. Science 1997; 275: 668-70.
- Wiggs JL, Allingham RR, Vollrath D, Jones KH, De La PM, Kern J, Patterson K, Babb VL, Del Bono EA, Broomer BW, Pericak-Vance MA, Haines JL. Prevalence of mutations in TIGR/Myocilin in patients with adult and juvenile primary open-angle glaucoma. AmJHumGenet 1998; 63: 1549-52.
- Allen KF, Gaier ED, Wiggs JL. Genetics of Primary Inherited Disorders of the Optic Nerve: Clinical Applications. Cold Spring Harb Perspect Med 2015; 5: a017277. [CrossRef]
- Collantes ERA, Delfin MS, Fan B, Torregosa JMR, Siguan-Bell C, Vincent de Guzman Florcruz N, Martinez JMD, Joy Masna-Hidalgo B, Guzman VPT, Anotado-Flores JF, Levina FD, Hernandez SRC, Collantes AA, Sibulo MC, Rong S, Wiggs JL. EFEMP1 rare variants cause familial juvenile-onset open-angle glaucoma. Hum Mutat 2022; 43: 1343. [CrossRef]
- Mackay DS, Bennett TM, Shiels A. Exome Sequencing Identifies a Missense Variant in EFEMP1 Co-Segregating in a Family with Autosomal Dominant Primary Open-Angle Glaucoma. PLoS One 2015; 10: e0132529. [CrossRef]
- Monte W. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). 5th edn.: University of Oregon Press, Eugene, 2013.
- Colige A, Nuytinck L, Hausser I, van Essen AJ, Thiry M, Herens C, Ades LC, Malfait F, Paepe AD, Franck P, Wolff G, Oosterwijk JC, Smitt JH, Lapiere CM, Nusgens BV. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (Type VIIC) and common polymorphisms in the ADAMTS2 gene. J Invest Dermatol 2004; 123: 656-63.
- Okumura M, Kadokura H, Inaba K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic Biol Med 2015; 83: 314-22. [CrossRef]
- Chakravarti A. Magnitude of Mendelian versus complex inheritance of rare disorders. Am J Med Genet A 2021; 185: 3287-93. [CrossRef]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet 2007; 80: 727-39. [CrossRef]
- Aldahmesh MA, Khan AO, Mohamed JY, Alkuraya H, Ahmed H, Bobis S, Al-Mesfer S, Alkuraya FS. Identification of ADAMTS18 as a gene mutated in Knobloch syndrome. J Med Genet 2011; 48: 597-601. [CrossRef]
- Colige A, Li SW, Sieron AL, Nusgens BV, Prockop DJ, Lapiere CM. cDNA cloning and expression of bovine procollagen I N-proteinase: a new member of the superfamily of zinc-metalloproteinases with binding sites for cells and other matrix components. Proc Natl Acad Sci U S A 1997; 94: 2374-9. [CrossRef]
- Gabriel LA, Wang LW, Bader H, Ho JC, Majors AK, Hollyfield JG, Traboulsi EI, Apte SS. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci 2012; 53: 461-9. [CrossRef]
- Chandra A, Jones M, Cottrill P, Eastlake K, Limb GA, Charteris DG. Gene expression and protein distribution of ADAMTSL-4 in human iris, choroid and retina. Br J Ophthalmol 2013; 97: 1208-12. [CrossRef]
- Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, Byers PH, Lapiere CM, Prockop DJ, Nusgens BV. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet 1999; 65: 308-17. [CrossRef]
- Akar A, D Gl, Erdem Z, Sarathchandra P, Tysoe C, Pope M. Acrogeric Ehlers-Danlos syndrome type IV: report of a new patient with additional findings. Eur J Dermatol 2002; 12: 428-31.
- Mitra A, Ramakrishnan R, Kader MA. Open angle glaucoma in a case of Type IV Ehler Danlos syndrome: a rarely reported association. Indian J Ophthalmol 2014; 62: 880-4. [CrossRef]
- Nie J, Zhang W. Secreted protease ADAMTS18 in development and disease. Gene 2023; 858: 147169. [CrossRef]
- Aldahmesh MA, Alshammari MJ, Khan AO, Mohamed JY, Alhabib FA, Alkuraya FS. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum Mutat 2013; 34: 1195-9. [CrossRef]
- Ahram D, Sato TS, Kohilan A, Tayeh M, Chen S, Leal S, Al-Salem M, El-Shanti H. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am J Hum Genet 2009; 84: 274-8. [CrossRef]
- Li ZF, Wu XH, Engvall E. Identification and characterization of CPAMD8, a novel member of the complement 3/alpha2-macroglobulin family with a C-terminal Kazal domain. Genomics 2004; 83: 1083-93. [CrossRef]
- Rehman AA, Ahsan H, Khan FH. α-2-Macroglobulin: a physiological guardian. J Cell Physiol 2013; 228: 1665-75. [CrossRef]
- Peters JC, Bhattacharya S, Clark AF, Zode GS. Increased Endoplasmic Reticulum Stress in Human Glaucomatous Trabecular Meshwork Cells and Tissues. Invest Ophthalmol Vis Sci 2015; 56: 3860-8. [CrossRef]
Figure 1.
Pedigree and segregations analysis in families of childhood glaucoma patients who carried more than one variant in MMP-related genes. In families PCG219 and PCG291, only proband DNA samples were available for the study. Black and grey symbols indicate childhood glaucoma and primary open-angle glaucoma, respectively. + : wildtype allele.
Figure 1.
Pedigree and segregations analysis in families of childhood glaucoma patients who carried more than one variant in MMP-related genes. In families PCG219 and PCG291, only proband DNA samples were available for the study. Black and grey symbols indicate childhood glaucoma and primary open-angle glaucoma, respectively. + : wildtype allele.
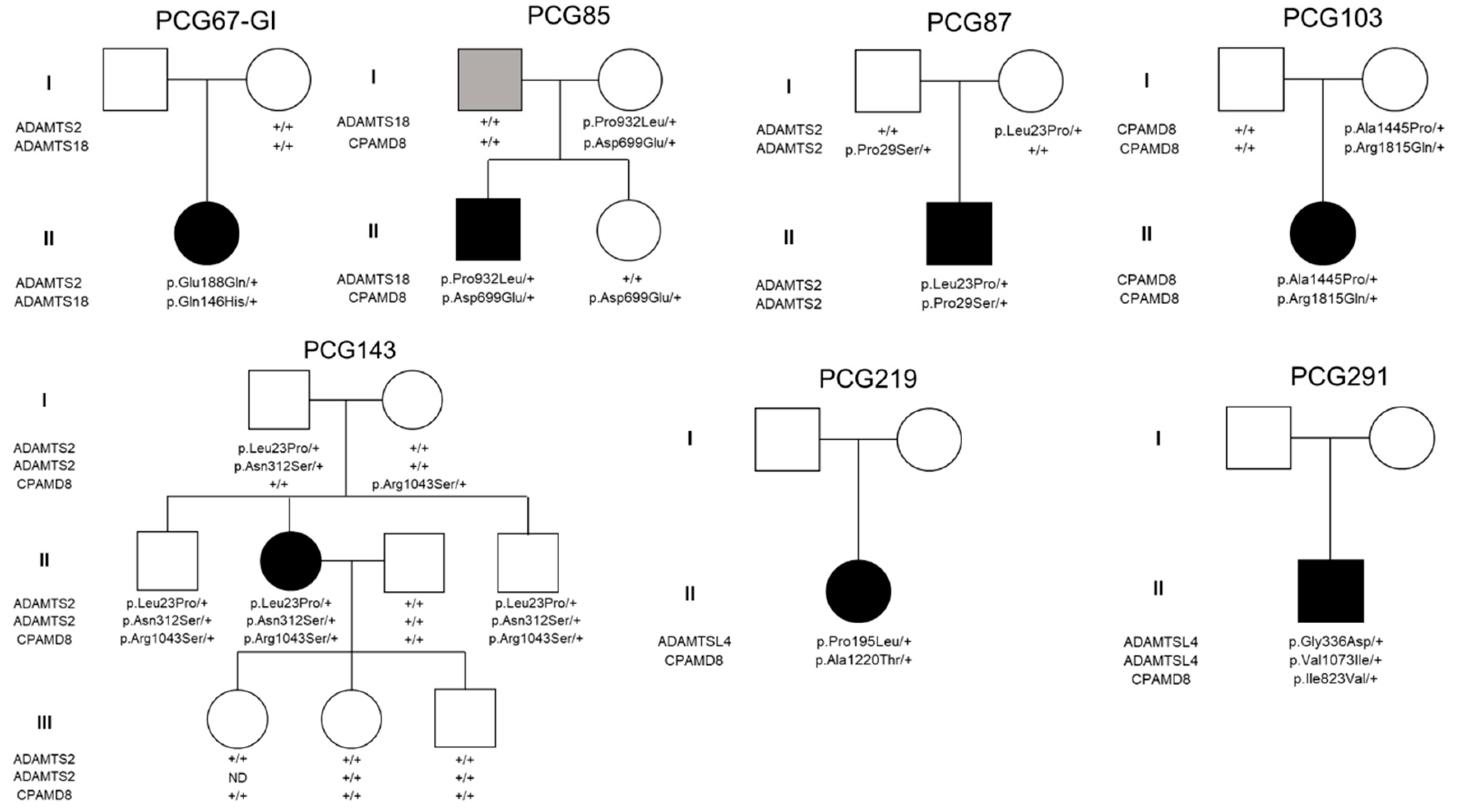
Figure 2.
Localization of ADAMTSL4 protein in adult human ocular tissues. Fluorescent immunohistochemistry was performed on 3 μm histological sections. The sections were incubated with a primary antibody, rabbit anti-ADAMTSL4 (1:250) (MBS716409, Quimigen), followed by incubation with a green-labeled secondary antibody (Cy2-conjugate donkey anti-rabbit) (1:1000). As a negative control, sections were incubated only with the secondary antibody (H-N). Confocal micrographs were captured to visualize the localization of ADAMTSL4 in different ocular tissues, including the lens (A), corneal epithelium (B), retina (C), corneal endothelium (D), iris (E), ciliary processes (F), and trabecular meshwork (G). The scale bars in panels (B-G) correspond to 50 μm, while in panel (A), it corresponds to 25 μm. CEN (corneal endothelium), CEP (corneal epithelium), CS (corneal stroma), GCL (ganglion cell layer), IF (iris fibroblasts), INL (inner nuclear layer), IPL (inner plexiform layer), IPE (iris pigment epithelium), IS (iris stroma), ISM (iris sphincter muscle), LC (lens capsule), LEP (lens epithelium), NPCE (non-pigmented epithelium), ONL (outer nuclear layer), OPL (outer plexiform layer), PCE (pigmented epithelium), PHL (photoreceptor layer), S (stroma), TM (trabecular meshwork). *: non-specific fluorescence.
Figure 2.
Localization of ADAMTSL4 protein in adult human ocular tissues. Fluorescent immunohistochemistry was performed on 3 μm histological sections. The sections were incubated with a primary antibody, rabbit anti-ADAMTSL4 (1:250) (MBS716409, Quimigen), followed by incubation with a green-labeled secondary antibody (Cy2-conjugate donkey anti-rabbit) (1:1000). As a negative control, sections were incubated only with the secondary antibody (H-N). Confocal micrographs were captured to visualize the localization of ADAMTSL4 in different ocular tissues, including the lens (A), corneal epithelium (B), retina (C), corneal endothelium (D), iris (E), ciliary processes (F), and trabecular meshwork (G). The scale bars in panels (B-G) correspond to 50 μm, while in panel (A), it corresponds to 25 μm. CEN (corneal endothelium), CEP (corneal epithelium), CS (corneal stroma), GCL (ganglion cell layer), IF (iris fibroblasts), INL (inner nuclear layer), IPL (inner plexiform layer), IPE (iris pigment epithelium), IS (iris stroma), ISM (iris sphincter muscle), LC (lens capsule), LEP (lens epithelium), NPCE (non-pigmented epithelium), ONL (outer nuclear layer), OPL (outer plexiform layer), PCE (pigmented epithelium), PHL (photoreceptor layer), S (stroma), TM (trabecular meshwork). *: non-specific fluorescence.
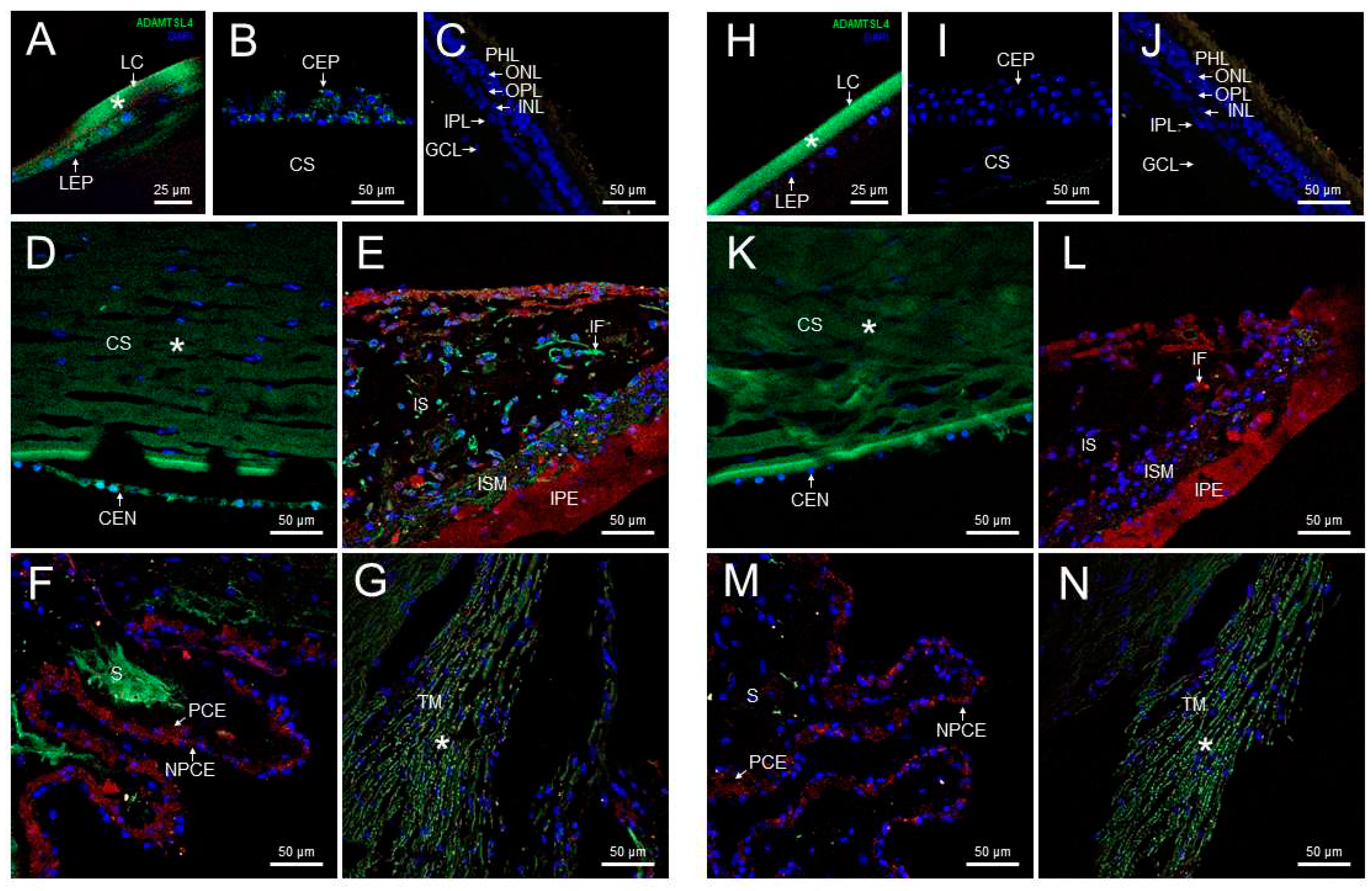
Figure 3.
Localization of ADAMTSL4 protein in zebrafish eye larvae (6 dpf) by confocal fluorescence immunohistochemistry. Fluorescent immunohistochemistry was performed on 10 μm histological sections of zebrafish eyes from wild-type (+/+) (A) and adamtsl4 KO (-/-) (B) specimens. The sections were incubated with a rabbit anti-ADAMTSL4 primary antibody (MBS716409, Quimigen) at a dilution of 1:250, followed by a Cy2-conjugated donkey anti-rabbit secondary antibody at a dilution of 1:1000. As a negative control (C), a section was incubated only with the secondary antibody. In the resulting images, ADAMTSL4 immunoreactivity is represented by green signals, DAPI nuclear staining by blue signals, and tissue autofluorescence by red signals. Scale bars in the panels indicate 50 μm. The images are representative of the observed results in three zebrafish of each genotype. C: cornea. GCL: ganglion cell layer. INL: inner nuclear layer. IPL: inner plexiform layer. LEP: lens epithelium. ONL: outer nuclear layer. OPL: outer plexiform layer. PHL: photoreceptor layer. POM: periocular mesenchyme.
Figure 3.
Localization of ADAMTSL4 protein in zebrafish eye larvae (6 dpf) by confocal fluorescence immunohistochemistry. Fluorescent immunohistochemistry was performed on 10 μm histological sections of zebrafish eyes from wild-type (+/+) (A) and adamtsl4 KO (-/-) (B) specimens. The sections were incubated with a rabbit anti-ADAMTSL4 primary antibody (MBS716409, Quimigen) at a dilution of 1:250, followed by a Cy2-conjugated donkey anti-rabbit secondary antibody at a dilution of 1:1000. As a negative control (C), a section was incubated only with the secondary antibody. In the resulting images, ADAMTSL4 immunoreactivity is represented by green signals, DAPI nuclear staining by blue signals, and tissue autofluorescence by red signals. Scale bars in the panels indicate 50 μm. The images are representative of the observed results in three zebrafish of each genotype. C: cornea. GCL: ganglion cell layer. INL: inner nuclear layer. IPL: inner plexiform layer. LEP: lens epithelium. ONL: outer nuclear layer. OPL: outer plexiform layer. PHL: photoreceptor layer. POM: periocular mesenchyme.
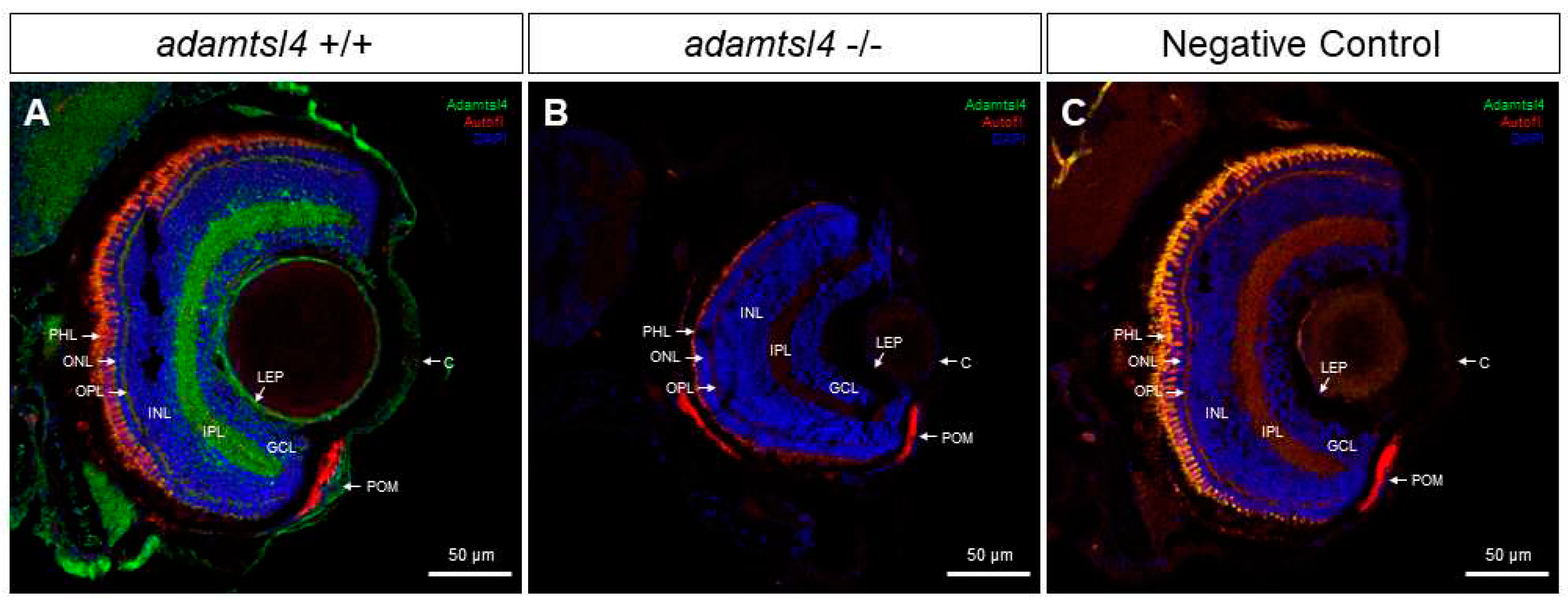
Figure 4.
Functional analysis of four rare ADAMTSL4 variants identified in childhood glaucoma patients, in HEK-293T cells. Subcellular distribution of the wild type protein (A) and variants R98W (E), S719L (I), R774W (M), and R1083H (Q) in HEK293T Cells (green signals). The cells were transfected with cDNA constructs encoding the different variants, fused to GFP at their C-terminal as a reporter protein and analyzed 48 h after transfection. (U) Non transfected cells were used as a negative control. (B, F, J N, R V) Immunocytochemistry was performed to detect PDI, a marker of ER stress (orange signals). (C, G, K, O, S, W) DAPI nuclear staining (blue signals). (D, H, L, P, T, X) Merged signals. The scale bars in the panels correspond to 20 μm. The white arrowheads indicate increased PDI signal colocalized with the granular expression pattern of the R98W and R774W variants. The inserts in panels (E) and (M) show granular deposits of recombinant protein, and dotted squares indicate the magnified areas. The images are representative of five fields per variant. R98W: p.Arg98Trp. S719L: p.Ser719Leu. R774W: p.Arg774Trp. R1083H: p.Arg1083His.
Figure 4.
Functional analysis of four rare ADAMTSL4 variants identified in childhood glaucoma patients, in HEK-293T cells. Subcellular distribution of the wild type protein (A) and variants R98W (E), S719L (I), R774W (M), and R1083H (Q) in HEK293T Cells (green signals). The cells were transfected with cDNA constructs encoding the different variants, fused to GFP at their C-terminal as a reporter protein and analyzed 48 h after transfection. (U) Non transfected cells were used as a negative control. (B, F, J N, R V) Immunocytochemistry was performed to detect PDI, a marker of ER stress (orange signals). (C, G, K, O, S, W) DAPI nuclear staining (blue signals). (D, H, L, P, T, X) Merged signals. The scale bars in the panels correspond to 20 μm. The white arrowheads indicate increased PDI signal colocalized with the granular expression pattern of the R98W and R774W variants. The inserts in panels (E) and (M) show granular deposits of recombinant protein, and dotted squares indicate the magnified areas. The images are representative of five fields per variant. R98W: p.Arg98Trp. S719L: p.Ser719Leu. R774W: p.Arg774Trp. R1083H: p.Arg1083His.
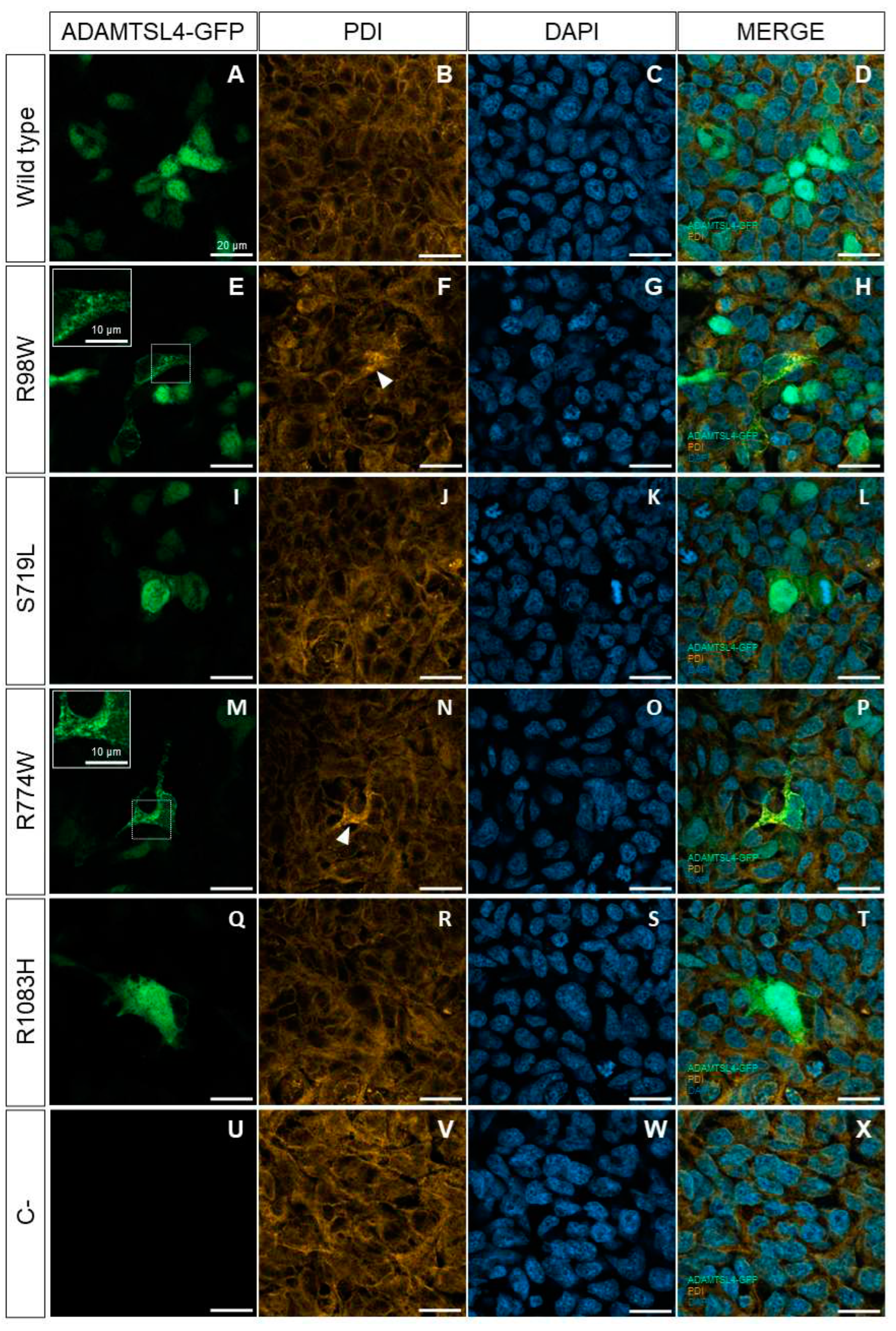
Figure 5.
Western blot analysis of RE stress associated with the expression in HEK-293T cells of four rare ADAMTSL4 variants identified in childhood glaucoma patients. HEK-293T cells were transfected with various cDNA constructs as explained in the legend of Figure 3. Forty-eight hours after transfection, cells lysates were prepared and analyzed by Western immunoblot. PDI, beta-actin (protein loading control), and NPTII (plasmid expression control) immunosignals were quantified by densitometry. PDI levels were normalized using beta-actin and NPTII. We conducted two independent transfections per cDNA construct, each performed in triplicate. *: p<0.05. R98W: p.Arg98Trp. S719L: p.Ser719Leu. R774W: p.Arg774Trp. R1083H: p.Arg1083His.
Figure 5.
Western blot analysis of RE stress associated with the expression in HEK-293T cells of four rare ADAMTSL4 variants identified in childhood glaucoma patients. HEK-293T cells were transfected with various cDNA constructs as explained in the legend of Figure 3. Forty-eight hours after transfection, cells lysates were prepared and analyzed by Western immunoblot. PDI, beta-actin (protein loading control), and NPTII (plasmid expression control) immunosignals were quantified by densitometry. PDI levels were normalized using beta-actin and NPTII. We conducted two independent transfections per cDNA construct, each performed in triplicate. *: p<0.05. R98W: p.Arg98Trp. S719L: p.Ser719Leu. R774W: p.Arg774Trp. R1083H: p.Arg1083His.
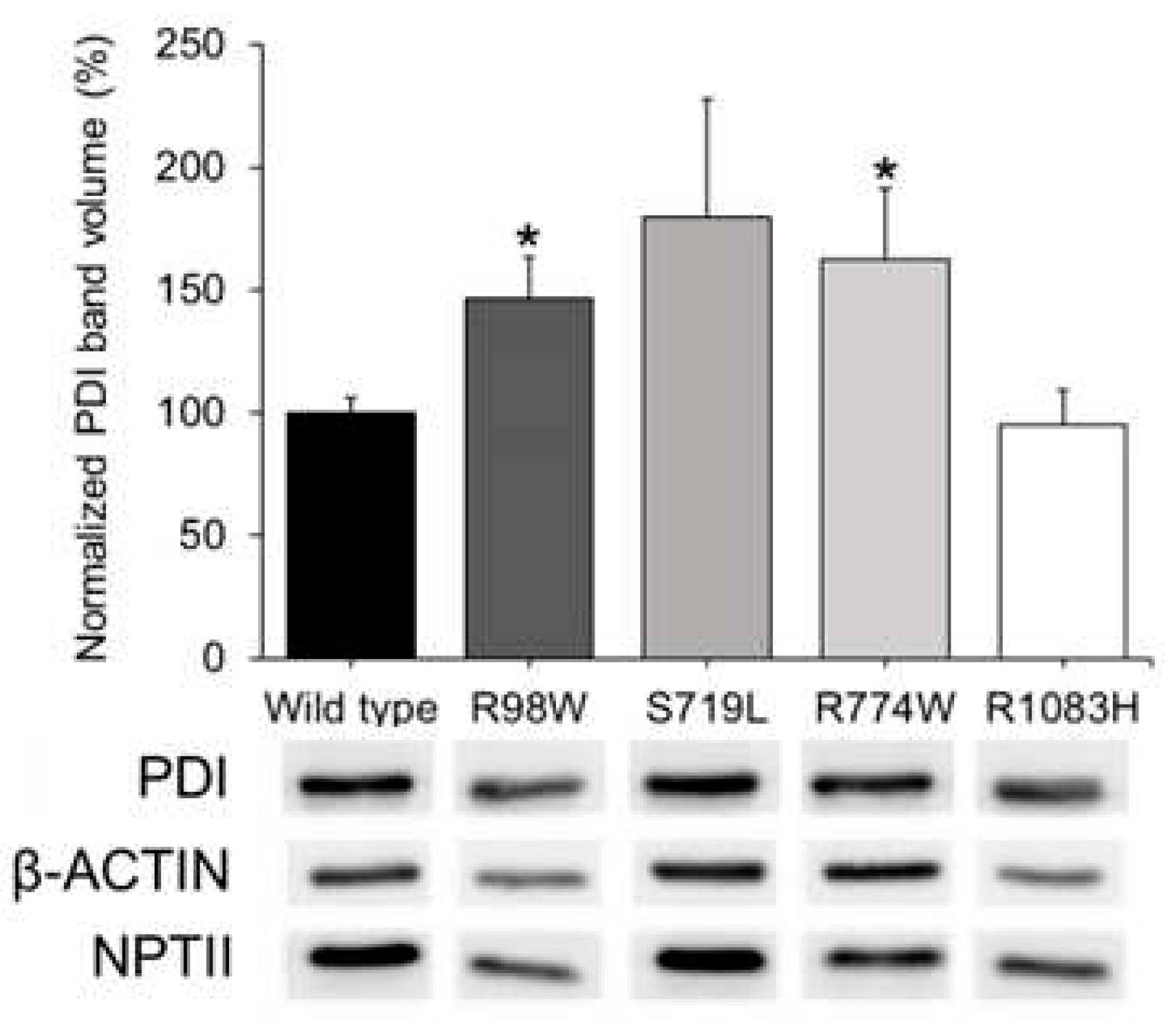
Figure 6.
Functional analysis of four rare ADAMTSL4 variants identified in childhood glaucoma patients, in zebrafish. One-cell zebrafish embryos were microinjected with pcDNA3.1-hADAMTSL4-GFP cDNA constructs encoding the four variants (R98W, n=374; S719L, n=381; R774W, n=477; and R1083H, n=485) and the wild type protein (n=1013). (A) Embryo lethality was quantified at 24 h after microinjection and (B-S) the phenotypes and recombinant protein expression were evaluated in larvae by bright field and fluorescence microscopy (3 dpf), respectively. (T-V) pcDNA3.1-GFP was microinjected as a control (n=784). *: p<0.05; ***: p<0.001. R98W: p.Arg98Trp. S719L: p.Ser719Leu. R774W: p.Arg774Trp. R1083H: p.Arg1083His.
Figure 6.
Functional analysis of four rare ADAMTSL4 variants identified in childhood glaucoma patients, in zebrafish. One-cell zebrafish embryos were microinjected with pcDNA3.1-hADAMTSL4-GFP cDNA constructs encoding the four variants (R98W, n=374; S719L, n=381; R774W, n=477; and R1083H, n=485) and the wild type protein (n=1013). (A) Embryo lethality was quantified at 24 h after microinjection and (B-S) the phenotypes and recombinant protein expression were evaluated in larvae by bright field and fluorescence microscopy (3 dpf), respectively. (T-V) pcDNA3.1-GFP was microinjected as a control (n=784). *: p<0.05; ***: p<0.001. R98W: p.Arg98Trp. S719L: p.Ser719Leu. R774W: p.Arg774Trp. R1083H: p.Arg1083His.
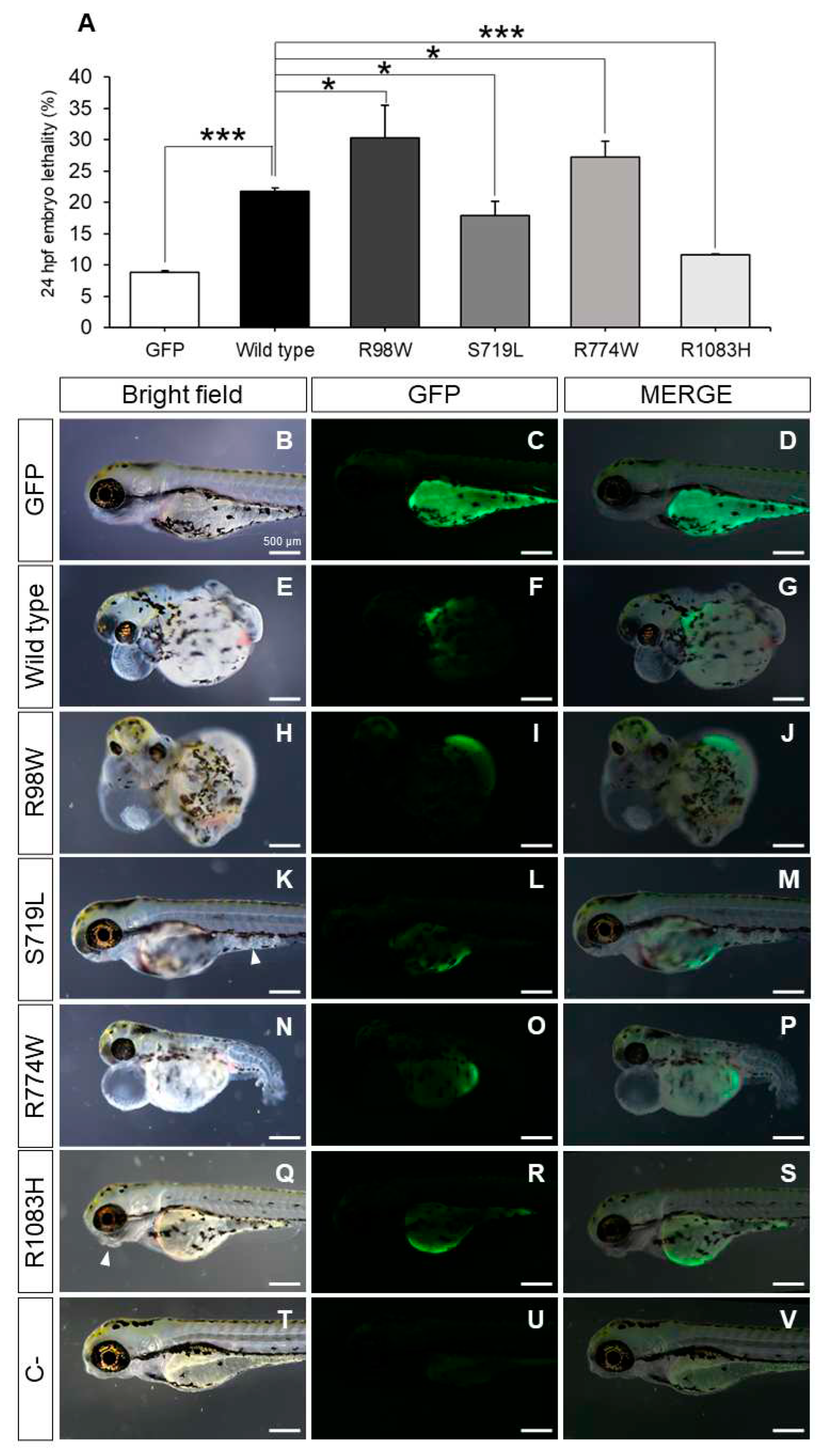
Figure 7.
Genetic interaction of adamtsl4 and cpamd8 in zebrafish. (A-D) To obtain double heterozygotes (adamtsl4/+ and cpamd8/+), zebrafish progenitors with heterozygous mutations in both adamtsl4 and cpamd8 genes were crossed. The progeny was genotyped, and the ocular anterior segment was evaluated in adult specimens (2 months) using brightfield microscopy. Sibling wild type (+/+) and single heterozygous (+/-) animals were used as controls. (E, F) The volumes of the anterior segment and the lens within the anterior chamber were calculated as indicated in Methods section. These calculations assumed that the anterior chamber and lens can be represented as spherical caps. In panel (A), the base (r) and height (h) of the spherical caps for the anterior chamber and lens are depicted in yellow and red, respectively. These values were normalized to those of wild-type eyes. The following number of eyes were used: Wild-type: n=16; adamtsl4 +/ and cpamd8 +/-: n=16; adamtsl4 +/- cpamd8 +/-: n=158. **: p<0.01. ***: p<0.001.
Figure 7.
Genetic interaction of adamtsl4 and cpamd8 in zebrafish. (A-D) To obtain double heterozygotes (adamtsl4/+ and cpamd8/+), zebrafish progenitors with heterozygous mutations in both adamtsl4 and cpamd8 genes were crossed. The progeny was genotyped, and the ocular anterior segment was evaluated in adult specimens (2 months) using brightfield microscopy. Sibling wild type (+/+) and single heterozygous (+/-) animals were used as controls. (E, F) The volumes of the anterior segment and the lens within the anterior chamber were calculated as indicated in Methods section. These calculations assumed that the anterior chamber and lens can be represented as spherical caps. In panel (A), the base (r) and height (h) of the spherical caps for the anterior chamber and lens are depicted in yellow and red, respectively. These values were normalized to those of wild-type eyes. The following number of eyes were used: Wild-type: n=16; adamtsl4 +/ and cpamd8 +/-: n=16; adamtsl4 +/- cpamd8 +/-: n=158. **: p<0.01. ***: p<0.001.
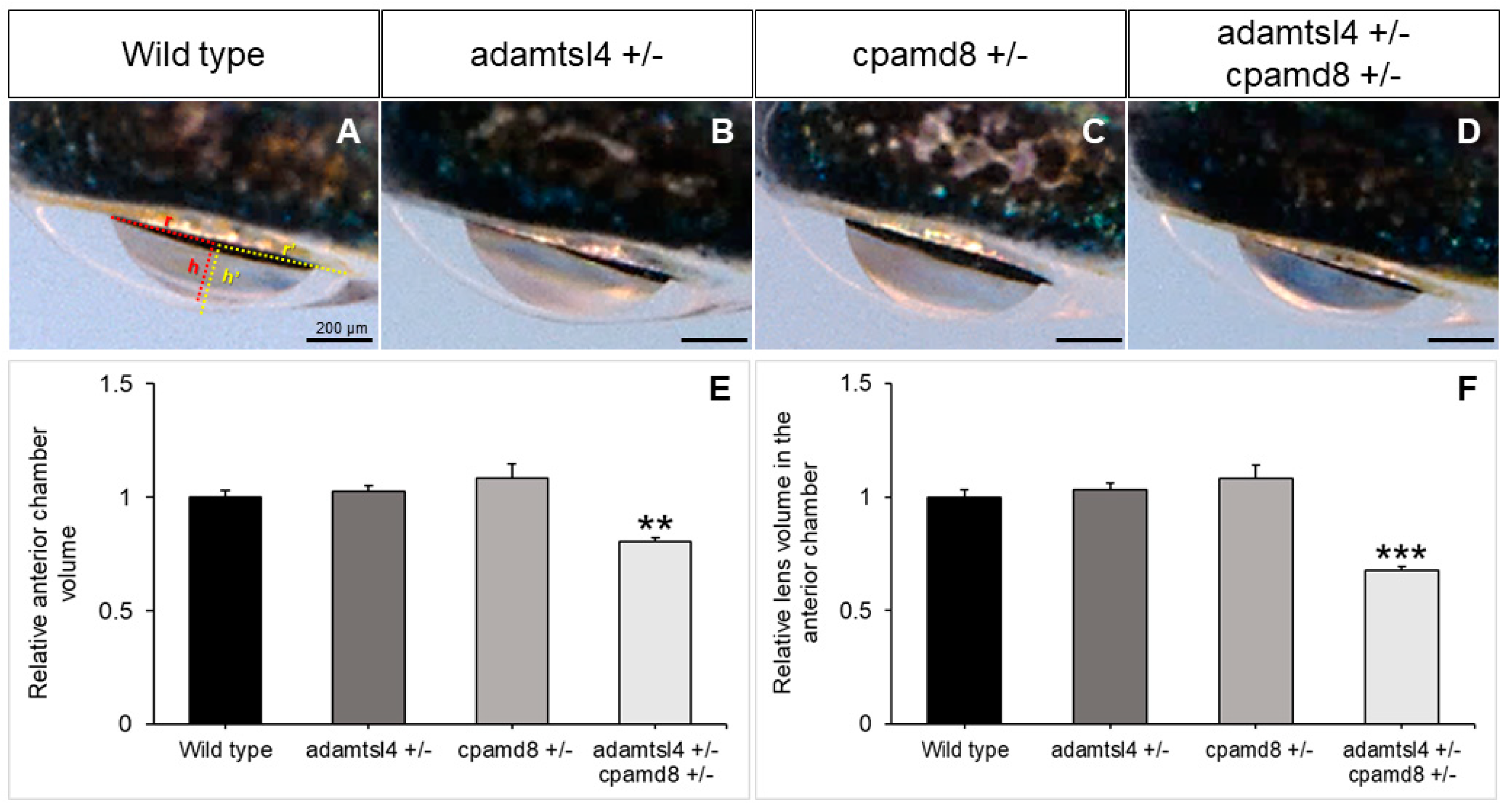

Table 1.
Rare variant burden analysis across 4 significantly enriched genes in childhood glaucoma vs. controls.
Table 1.
Rare variant burden analysis across 4 significantly enriched genes in childhood glaucoma vs. controls.
| Childhood glaucoma Alleles (%) [n=202] | ESP6500* Alleles (%) [n=8600] | gnomAD* v2.1.1 Alleles (%) [n=104400] | p-Value (Chi-square test) (CG vs. ESP6500) | p-Value (Chi-square test) (CG vs. gnomAD v2.1.1) | Odds Ratio (95% CI) (CG vs. ESP6500) | Odds Ratio (95% CI) (CG vs. gnomAD v2.1.1 | ||
|---|---|---|---|---|---|---|---|---|
| Aggregate genes | Rare frameshift, non-sense, missense and donor/acceptor splicing sites | 59 (29.20) | 854 (10.03) | 9489 (9.13) | <1E-15 | <1E-15 | 3.49 (2.56-4.77) | 4.06 (3.00-5.50) |
| Rare Synonymous | 17 (8.42) | 773 (9.05) | 10584 (9.83) | 6.39E-01 | 5.77E-01 | 0.86 (0.52-1.42) | 0.84 (0.51-1.39) | |
| ADAMTS2 | Rare frameshift, non-sense, missense and donor/acceptor splicing sites | 14 (6.93) | 140 (1.63) | 1611 (1.56) | 6.52E-08 | 5.00E-09 | 4.49 (2.55-7.93) | 4.70 (2.72-8.10) |
| Rare Synonymous | 2 (0.99) | 184 (2.15) | 2616 (2.43) | 3.79E-01 | 2.70E-01 | 0.46 (0.11-1.85) | 0.40 (0.10-1.61) | |
| ADAMTS18 | Rare frameshift, non-sense, missense and donor/acceptor splicing sites | 14 (6.93) | 178 (2.07) | 2953 (2.70) | 9.47E-06 | 5.01E-04 | 3.52 (2.01-6.18) | 2.68 (1.56-4.62) |
| Rare Synonymous | 3 (1.49) | 158 (1.84) | 2110 (1.93) | 9.17E-01 | 8.36E-01 | 0.81 (0.25-2.55) | 0.76 (0.24-2.39) | |
| ADAMTSL4 | Rare frameshift, non-sense, missense and donor/acceptor splicing sites | 16 (7.92) | 176 (2.05) | 2213 (2.06) | 6.97E-08 | 2.08E-08 | 4.10 (2.41-6.99) | 4.10 (2.45-6.84) |
| Rare Synonymous | 3 (1.49) | 83 (0.97) | 1545 (1.41) | 7.09E-01 | 8.31E-01 | 1.54 (0.48-4.91) | 1.05 (0.34-3.29) | |
| CPAMD8 | Rare frameshift, non-sense, missense and donor/acceptor splicing sites | 15 (7.43) | 360 (4.34) | 2712 (2.84) | 5.24E-02 | 2.13E-04 | 1.77 (1.04-3.03) | 2.75 (1.62-4.65) |
| Rare Synonymous | 9 (4.46) | 348 (4.19) | 4313 (4.20) | 9.92E-01 | 9.94E-01 | 1.07 (0.54-2.10) | 1.06 (0.55-2.08) |
*: individuals of European ancestry.
Table 2.
Genetic and clinical features of patients with at least two filtered variants in significantly enriched genes.
Table 2.
Genetic and clinical features of patients with at least two filtered variants in significantly enriched genes.
| Patient | ADAMTS2 | ADAMTS18 | ADAMTSL4 | CPAMD8 | Total number ofvariants per patient | Age at diagnosis (months) | IOP at diagnosis (mm Hg) (RE/LE) | Cup/discratio(RE/LE) | Surgical treatment (# surgeries) | Gender/laterality | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PCG67-GI | p.Glu188Gln | p.Gln146His | 2 | 144 | 47/37 | 0.4/0.3 | T (4), C/T (6), V, C | ||||||||||
| PCG85 | p.Pro932Leu | p.Asp746Glu | 2 | 10 | NA | NA | G/G (2), T (2) | M/B | |||||||||
| PCG87 | p.Pro29Serp.Leu23Pro | 2 | 16 | 48/42 | 0.8/0.8 | G/G | M/B | ||||||||||
| PCG103 | p.Arg1862Glnp.Ala1492Pro | 2 | 8 | NA | NA | G/G | F/B | ||||||||||
| PCG143 | p.Asn312Serp.Leu23Pro | p.Arg1090Ser | 3 | 60 | NA | NA | None | F/B | |||||||||
| PCG219 | p.Pro195Leu | p.Ala1267Thr | 2 | 5 | NA | NA | T/T | F/B | |||||||||
| PCG291 | p.Val1073Ilep.Gly336Asp | p.Ile823Val | 3 | 36 | NA | NA | G/G | M/B | |||||||||
AV: AHMED valve. B/U: bilateral/unilateral. F/M: female/male. G: goniotomy. NA: not available. RE/LE: right eye/left eye. T: trabeculectomy.
Table 3.
Clinical features of carriers of rare variants in MMP-related genes.
| Variable | MMP-related gene carriers (A) | MMP-related gene non-carriers (B) | PCGs with null CYP1B1 genotypes (n=37) (C) | P (A vs. B) | p (A vs. C) | p (B vs. C) |
|---|---|---|---|---|---|---|
| aAge at diagnosis (months)(mean±SD) | 17.1±37.4 (n=50) | 12.18±25.2 (n=28) | 1.9±5.2 | ns | 0.0075 | 0.0089 |
| Number of surgeries per eye(mean±SD) | 1.9±1.4 (n=47) | 0.8±1.40 (n=31) | 3.1±1.7 | 0.001 | 0.0014 | 0.00000019 |
| IOP at diagnosis (mm Hg) (mean±SD) | 30.0±1.4(n=48) | 30.5±9.3(n=30) | 28.0±5.5 | ns | ns | ns |
a Only patients with a clinical diagnosis of congenital glaucoma were considered for the analysis. ns: not significant.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



