
Submitted:
12 February 2024
Posted:
15 February 2024
You are already at the latest version
Abstract
In the early 2000s, matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) emerged as a performant and relevant tool for identifying micro-organisms. Since then, it has become practically essential for identifying bacteria in microbiological diagnostic laboratories. In the last decade, it was successfully applied for arthropod identification allowing to distinguish vectors from non-vectors of infectious diseases. However, identification failures are not rare, hampering it widely use. Failure is generally attributed either to the absence of respective counter species MS spectra in the database, or to the insufficient quality of query MS spectra (i.e. lower intensity and diversity of MS peaks detected). To avoid matching errors due to non-compliant spectra, the development of a strategy for detecting and excluding outlier MS profiles became compulsory. To this end, we created MSProfileR, an R package leading to a bioinformatics tool through a simple installation, integrating the control quality system of MS spectra and an analysis pipeline including peak detection and MS spectra comparisons. MSProfileR can also add metadata concerning the sample that the spectra are derived from. MSProfileR has been developed in the R environment and offers a user-friendly web interface using the R Shiny framework. It is available on Microsoft Windows as a web browser application by simply navigation using the link of the package on the Github. MSProfileR is therefore accessible to non-computer specialists and is freely available to the scientific community. We evaluated MSProfileR using two datasets including exclusively MS spectra from arthropods. In addition to coherent sample classification, outlier MS spectra were detected in each dataset confirming the value of MSProfileR.
Keywords:
R package
; quality control
; MALDI-TOF MS
; arthropod vectors
; Github
; rstudio
; Shiny interface.
Introduction
Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) tool, which was largely employed for protein identification, was later applied for rapid identification of whole bacteria based on comparison of spectral patterns [1]. The principle of MALDI-TOF MS profiling is to match MS spectra, resulting from laser ionization of the sample, with a library of reference MS spectra for microbial identification [2]. The cost-effective and rapidity of MALDI-TOF MS approach has revolutionized routine diagnostic identification of bacteria using simple and reliable procedures, substituting Gram staining and biochemical traditional methods. It was since been largely introduced in clinical laboratories for the identification of micro-organisms including bacteria, fungi and yeasts [3].
In the mid-2000s, this innovative tool was successfully applied for the identification of several arthropod families, vectors of infectious disease such as mosquitoes or ticks [4]. Correct classification of specimens requires that MS spectra are intra-species reproducible and inter-species specific. Unlike molecular assays, MS protein profiles from a same arthropod specimen could vary according to body part, developmental stages or sample preparation mode [5,6]. Then, to compare and to share results of MS profiling analyses, a standardization of the body parts selected for MS submission according to developmental stage and the conditions of sample homogenization for mosquitoes and ticks were established [7,8]. The legs and thorax for mosquitoes and the legs and half-idiosoma for ticks were the two body parts selected per specimen and submitted independently to MALDI-TOF MS for species identification [9,10,11]. The identification reliability was expressed with scores provided by algorithms matching MS spectra from the query sample with a library of reference MS spectra.
Despite the standardization of protocols, for some MS spectra, their query against the reference DB failed to reach the threshold score value established for reliable identification [12,13]. This failure was generally attributed either to the absence of respective counter species MS spectra in the database, or to the insufficient quality of queried MS spectra (i.e., lower intensity and diversity of MS peaks detected). The low quality of MS spectra could be attributed to several factors, such as the mode or duration of sample storing, sample preparation, sample loading onto the MALDI plate, the quality of the matrix buffer or the instrumental variations [12,14]. Currently, no pre-processing tests were integrated into the software proposed by manufacturers of MALDI-TOF MS profiling tools (e.g., Bruker Daltonics, Shimadzu) to assess quality of MS spectra. The classification of MS spectra as being of low quality relies essentially on visual inspection of their profile and only concerns those that did not reach the threshold score value established for reliable identification [12,13]. So, this classification depends on the reference DB and on the experimenter skill [15], which could be highly subjective. The classification of a MS spectrum as low quality or its exclusion should be based on rigorous criteria and a reproducible method.
Then, for a prospective adoption of this innovative method (i.e., MALDI-TOF MS profiling) for arthropod identification by the scientific entomologist community, it becomes imperative to provide integrated bio-informatics tools distinguishing conform from unacceptable MS spectra. In this way, the objective of the present study was to create a bio-informatics tool, using the R environment, that helps to determine the quality of MS spectra rigorously and before interrogation, and which integrates a complete MS spectra analysis pipeline including peak detection and MS spectra comparisons. In addition, it was essential to add metadata concerning the sample from which the spectra originate and to provide a user-friendly graphical interface using the R Shiny framework. It will be presented the advantages offered by this innovative bio-informatics tool called MSProfileR for analysing MS spectra of arthropods and discuss the future developments.
2. Details on MSProfileR Tool
2.1. General organization of the “MSProfileR_v1.0” Tool
The MSProfileR tool is a Shiny application assessing the conformity of mass spectra profiles for future analysis. It was developed in R programming language version 4.3.2 and in the RStudio environment version 2023.10.31 (R Core Team, 2020). The general workflow of the application was divided into five major steps: 1) the data loading tool that includes spectra import, 2) the preprocessing tool including successive steps to control the conformity and quality of spectra, 3) the processing tool including peak detection and MS spectra classification, 4) the spectra annotation tool allowing to add complementary metadata to each spectrum and finally 5) the output tool generating several files including a report (Figure 1). Each step of the MSProfileR was composed of several modules performing each one or more tasks. During the pipeline process, the user controls and adjusts the parameters of the MS spectra analysis. However, it is possible to realize the entire workflow without any intervention by using default parameters or loading parameters defined in a previous analysis. To make MSProfileR easier to use, a graphical user interface (GUI) was created using the Shiny R package version 1.7.2. A wrapping of existing R packages, available in Comprehensive R Archive Network (CRAN) repositories was done. Shiny R package allows to build an interactive web application straight from R. The different steps and modules from MSProfileR are described below.
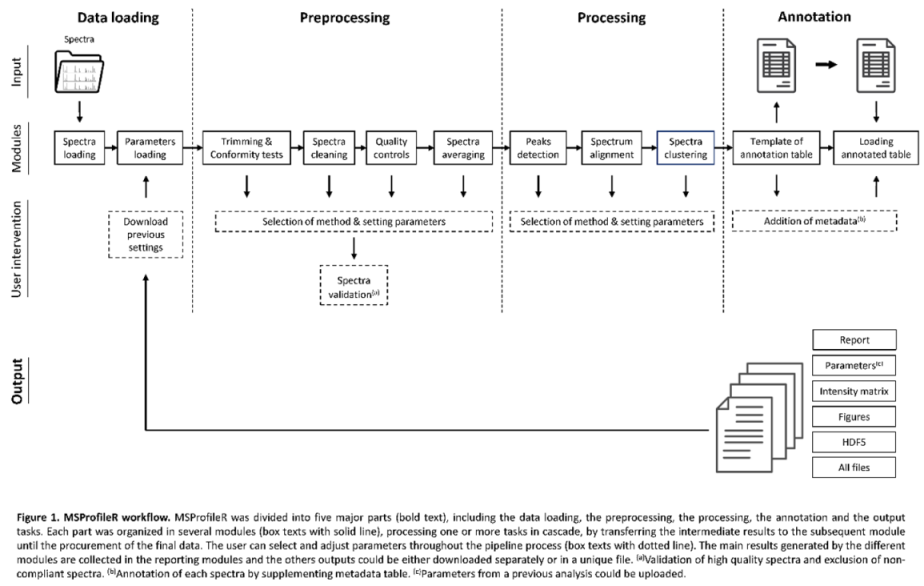
2.2. Data Loading
Data loading, which is the entry point to MSProfileR, contains a module for reading MS spectra, as well as an optional module for selecting and loading the setting parameters of a previous analysis.
2.2.1. Module of the MS Spectra Loading
Functions from the MALDIquant and MALDIquantForeign [16] packages were used for retrieving MALDI-TOF spectra on R system. These packages recognize MALDI-TOF mass spectra file types and load them automatically [16]. Different file formats (txt, tab, csv, fid, ciphergen, mzXML, mzML, imzML, analyze, cdf, msd) can be uploaded, in an automatic detection way, from a folder containing one or many spectra. However, as only MS spectra from Bruker instrument were available, the assessment of MSProfileR tool to these file format. The spectra files are loaded recursively, from which are retrieved, the m/z values and intensity values, in the x- and y-axis, respectively, plus several data related to each spectrum such as the spectrum name, the date of acquisition and the sample name. The sample name must be identical for the spectra of the biological replicates loaded by MSProfileR. Effectively, this parameter will be used during the preprocessing to generate automatically an average spectrum representative for the replicates originated from the same sample.
To download the spectra profiles correctly, the level number of each main folder path must be defined before loading the data. This path is the number of folders existing between the sampleName and the binary files (fid or aqu), which may change between a direct classification of spectra by Bruker Daltonics Software and a manual spectra folder classification by the user if the sample name was not indicated before spectra acquisition. By default, the level of the spectra folders set on the MSProfileR front is four but the user can change it. Moreover, an ID number was attributed to each spectrum which will be useful to follow and to display spectra during the whole pipeline analysis.
2.2.2. Module of Parameter Loading
This module is used to upload all the parameters saved from MSProfileR in a previous analysis. The user can submit distinct datasets to MSProfileR while applying the same parameters, enabling reproducibility.
2.3. Preprocessing
The preprocessing step of the workflow is composed of several modules aiming to evaluate the quality of the spectra and to exclude inconsistent MS profiles (Table 1). All these steps are designed with functions of the MALDIquant package [16], with the exception of the quality control step which is based on the MALDIrppa package [17]. MSProfileR tool allows assessing the MS spectra quality throughout four successive steps: 1) trimming and conformity tests, 2) spectra cleaning, 3) quality control and visualization of potentially outlier spectra, and 4) replicate averaging. All these tasks are using raw mass spectra data. Spectra passing these quality control steps are then selected for processing.
2.3.1. Module of Trimming and Conformity Check
The first task allows fixing the low and high limits of spectra m/z values using the trim function and inspects conformity of the spectra. Without specification, the limits of m/z values of default parameters correspond to a range from 2 to 20kDa. The inspection of data conformity checks whether errors occurred during MS spectra data acquisition. It assesses 3 criteria:
- (i)
- Completeness: Are there any empty spectra (i.e., no data to load)?
- (ii)
- Missing values: Are there any spectra with irregular m/z values? Normally, the interval between two successive m/z values should remain equal or increase uniformly (i.e., no missing point or aberrant values).
- (iii)
- Spectra range: Does the length (ie, m/z values range) of spectra differ?
Empty or irregular spectra, or spectra with too different an m/z range, can compromise the subsequent steps and must therefore be detected. These steps controlled the consistency and conformity of the spectra. The list of non-compliant spectra was removed before continuing the analysis.
2.3.2. Module of Spectra Cleaning
The MS spectra that have passed the previous tests are then subjected to a series of transformations in order to standardize the data: 1) intensity transformation, 2) smoothing, 3) baseline correction, and 4) normalization. The intensity transformation stabilizes the variance and improves the graphical visualization of spectra, by reducing the scale effect flattening the low-intensity peaks. By default, this transformation is performed using the square root method [18], but three log transformations (log, log2 and log10) are also proposed [19,20]. Mass spectra typically contain a mixture of noise and signal. To increase the signal-noise ratio, algorithms are used to improve the measure of peak m/z intensity values and facilitating the peak detection. By default, the Savitzki-Golay-Filter algorithm is used to smooth the spectra with a half window size of 10, but the Moving average is also available [21]. The noise altering the base level of mass spectra is then corrected with the Sensitive Nonlinear Iterative Peak (SNIP) algorithm (number of iterations, n=100) [22] or with the TopHat, Convex hull or Median methods. Subtracting the baseline from the spectra facilitates profile comparison. In order to compare intensities at each m/z value between spectra, normalization is required. Three normalizations are available, the Total-Ion-Current (TIC) [23], the Probabilistic Quotient Normalization (PQN) [24] or the median methods. The TIC is applied by default to each spectrum and corresponds to setting the sum of the relative abundances of all the ions (m/z peaks) to one. All these transformations are based on the mass spectra processing functions (transformIntensity, smoothIntensity, removeBaseline, calibrateIntensity) of the MALDIquant package.
2.3.3. Module of Quality Control
The subsequent steps consist in detecting and filtering MS spectra considered as no conform (i.e., outliers). To this end, the screenSpectra function from the MALDIrppa package is used. It computes for each spectrum an atypicality score (A score). The calculation of this score is based on robust estimators [25]. The A score pointed out MS spectra for which peak intensity profiles diverge from the rest of the dataset. An upper and lower tolerance limits are returned at the same time by the function. MS spectra classified above the upper limit correspond to profiles of low intensity and poor resolution, whereas those below the lower limit have high peak intensity profiles [17]. To avoid the deletion of profiles harbouring high peak intensity, the lower threshold limit is not considered by default. Nevertheless, the user could reactivate it.
Several parameters of the screenSpectra function are available to find the best estimator used in the calculation of the A score, the threshold and the method from which the cut-off is estimated. Two estimators are available, the median absolute deviation (MAD) and the Q. The MAD is defined as the median of the absolute deviations from the median of the intensities. It is a robust estimator for fairly symmetric distributions. The Q estimator is an improved version of the MAD and is more efficient and adequate for non-symmetric distributions [25]. Here, the Q estimator was selected as default parameters. Five methods (i.e., Rousseeuw & Croux (RC), Hampel, extreme studentized deviation (ESD), boxplot and adjusted boxplot) are available to compute the threshold limit for the detection of outliers [26]. By default, the RC method is selected.
The spectra with an A score outside the upper limit are regarded as potentially faulty spectra. By default, the spectra detected as outliers are automatically rejected from further analysis. However, at this step, a graphical interface was added giving to the user the possibility to visualize all MS spectra and notably the outliers. Based on their visualization, the user could refine the decision to exclude spectra near the A score limits or to keep some outliers. Generally, low-quality spectra are characterised by rippled and indented profiles due to low signal to noise intensities which could be absorbed by the background noise. Following this step, all mass spectra for which profiles were considered as atypically and confirmed by the user, are counted and excluded from the next analysis steps.
2.3.4. Module of Spectra Averaging
This module generates and proposes an archetype for MS spectra replicates. When available, the MS spectra replicates that passed the quality control steps were then averaged. An average spectrum is calculated for each sample by applying the mean, the sum or the median methods of its replicates, using the averageMassSpectra function from the MALDIquant R package. Then, one archetype spectrum of the replicates per sample is further analysed. The absence of replicates does not affect the workflow of the MSProfileR application. Spectra without replicates are the archetype profile of the sample. Samples with or without replicates could be downloaded and analysed concomitantly.
2.4. Processing
The processing tool consists in performing, successively, the peak detection, the spectra alignment until the spectra visualization, classification and the creation of an intensity matrix for further analyses.
2.4.1. Module of Peak Detection
This module aims to detect MS peaks excluding background noise for each spectrum taking into account homogeneity of the number of MS peaks detected in the entire dataset. The peak detection is performed on the average spectra using the detect Peaks function from MALDIquant package. It screens each spectrum with a sliding window in order to find the local maximums. A local maximum is detected as a peak whether its intensity is greater than the noise level, estimated using the MAD by default or super Smoother algorithms, multiplied by a signal-to-noise ratio (SNR), with a half window equal to 20, by default. Indeed, the spectra profiles are discretized and uniquely detected peaks are kept. Each spectrum profile can be regarded as a peak list. By default, the SNR value was set at 2 corresponding to a good compromise for detecting as many peaks as possible, without detecting too much parasite peaks which belong to the background noise. The SNR value is a parameter which could be adjusted by the user. To determine the optimal SNR value, the number of detected peaks and their standard deviation were measured for each SNR value tested, ranging from 2 to 7. A box-plot was generated showing the number of peaks detected per SNR value for the entire dataset. The SNR value for which the number of detected peaks was homogeneous and stable among dataset was considered as the optimal trade-off.
2.4.2. Module of Spectra Alignment
Prior to align detected peaks from averaged spectra, the determination of reference peaks is required. As no spectrum was selected as reference, the reference Peaks function from the MALDIquant package is used. In such conditions, all peaks from the dataset with an occurrence upper than the threshold (peak frequency) set by default at 0.9, are used as reference peaks for peak alignment, with a default tolerance of 0.002. The strict (default) or relaxed parameters allow to consider all peaks or uniquely the most highest.
The alignment of spectra was performed with alignSpectra function of the MALDIquant R package. All the peaks detected among the average spectra per sample are aligned and calibrated using one function of the correction phase (warping function), lowess, linear, quadratic or cubic. The lowess method was set as default.
Despite this alignment, very close detected peaks, but not identical, are binned into one, in accordance with an applied tolerance. This tolerance corresponds to the maximal relative deviation of a peak position (m/z value) to be considered as identical and is set to 0.002 by default. Thus, the peaks for which the difference of the positions (m/z values) is lower than this threshold get their mass values equalized forming one peak. Two binning functions are available, the strict one, for which bins never contain two or more peaks of the same sample, and the relaxed one, for which multiple peaks of the same sample were combined in one bin. By default, the strict binning function was selected.
The filtering step consists to remove peaks infrequently detected. A minimum frequency per m/z detected peak should be defined. For example, by setting this parameter to 1, uniquely peaks common to all spectra were filtered. By default, this value was set at 0.2, corresponding to an elimination of all peak positions found in less than 20% of the dataset. The number of peaks detected is indicated and then an adjustment of this frequency is possible by the user.
2.4.3. Module of Spectra Clustering
This module consists to visualize detected peaks and to ordinate samples according to their profiles. An intensity matrix is generated, where average spectra per sample are represented and was used for the classification. In this way, a hierarchical clustering algorithm was applied to generate a clustered heatmap using an unsupervised algorithm, the hclust function, and the pheatmap package [27]. This package computes bootstrap values to indicate for each query the positioning relevancy in the dendrogram. The columns correspond to the peak positions in m/z and the rows to the sample spectra. The peak intensity values are represented with a rainbow scale.
2.5. Spectra Annotation
Optionally, it is possible to annotate averaged spectra by the loading of an annotation table. The information introduced in this table from each sample allows to enrich metadata associated to each averaged spectra. The annotation table tool is composed of one module. The module allows to retrieve the list of sample names which passed the preprocessing and the processing tests and are then directly included in the first column headed “sampleName” of a downloadable table (.xlsx file format). At this step, as all spectra replicates were already averaged, a unique “sampleName” was associated to each averaged spectrum. In this way, the folder from each averaged spectrum could be easily paired with data from the annotation table using the “sampleName”. When the user considers the information added sufficient, the annotated table was imported into MSProfileR tool. The module controls whether all averaged spectra were annotated and also whether some annotations were not paired with an averaged spectra. Although the importation of the annotation table is not mandatory, these annotations could be helpful for future comparative results of mass spectra profiles. This module uses the readxl and writexl packages to read and write .xlsx files.
2.6. Output
The output module is created to guarantee traceability for the user about all the process steps, the methods and parameters selected, but also to download and to save data that can be useful for upcoming analyses.
2.6.1. Reports
The report is implemented on R Markdown language [28], offering an interface to generate a downloadable document in pdf format including information about steps performed, methods selected and parameters applied. Moreover, the information about the spectra dataset, such as the name of the directory file, the number of spectra loaded and the results of pipeline analysis, and notably those about the conformity tests, the quality control steps with the list of excluded spectra (non-conform or outliers). In addition, all representations, such as tables, plots, gelviews and clustered heat map are included in the report.
2.6.2. Save Setting Parameters
This module allows, thanks to rjson package, to save in JSON format all methods chosen and associated setting parameters that could be easily downloaded for a future analysis.
2.6.3. Export Intensity Matrix
This module serves to export the data of the matrix intensity into a CSV file format. This matrix was extracted according to two criteria, on the ordinate are indicated the sample names, and on the abscissa the m/z values with respective.
2.6.4. Figure List (Plots, Graph, etc.)
This module exports all the figures generated during the analysis of the current dataset by the MSProfileR tool. All the figures are in SVG format and zipped into a unique file. This list includes ‘mass_bounds.svg’ for the defined extremities spectra in the trimming module, ‘screening.svg’ for the plot excluding the outliers in the quality control module, ‘peak_count_by_SNR.svg’ for the boxplot showing the number of peaks depending on the different SNR values in the peak detection module, ‘reference_peaks.svg’ for the generated peak list, ‘spectra_aligned.svg’ for the gelview displaying the alignment, ‘peaks_binned.svg’ for the gelview of the binned peaks, ‘peaks_filtering.svg’ for the gelview of the filtered peaks, the last four figures are illustrated in the spectra alignment module, ‘dendrogram.svg’ represents a clustering of sample spectra, ‘intensity_matrix_8x4.svg’,‘intensity_matrix_16x8.svg’ and ‘intensity_matrix_32x16.svg’, the figures of the intensity matrix can be loaded with three different dimension to improve the classification visualisation, which are illustrated in the clustering module.
2.6.5. Hierarchical Data Format Version 5 (HDF5) File
This module allows the storage in HDF5 file of the four main outputs of this analysis corresponding to the parameters selected, the averaged spectra, the intensity matrix and the annotation table. This HDF5 file is an open source format supporting a large, heterogeneous and complex data. The creation of this file is done using the hdf5r library, which generates a file format that can store and manage large, complex and heterogeneous data [29].
2.6.6. Module Downloading All Files
This last module was created in order that the user could download all files listed in the five preceding modules. It is then possible to download either some particular data or all the data generated by the analysis.
2.7. The User Interface (UI)
For unfamiliar users with the R language, a user-friendly interactive web interface was created making possible to exchange information following a user-machine interaction. The UI or graphical interface was based on the Shiny dashboard template (https://rstudio.github.io/shinydashboard/), using Shiny, a “package that makes it easy to build interactive web apps straight from R and Python”.(https://rdrr.io/cran/shiny/). The UI was compartmentalized in five tabs corresponding to the five steps of workflow, including 1) Data loading, 2) Preprocessing, 3) Processing 4) Annotation and 5) Output. The architecture of MSProfileR tool was organized in a modular way (Figure 2).
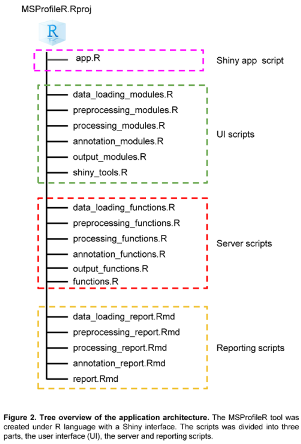
2.7.1. Web Interface Development Architecture
All R packages dependencies used for the creation of the MSProfileR tool are available in Bioconductor (https://www.bioconductor.org/) or Comprehensive R Archive Network (CRAN) repositories (https://cran.r-project.org/). The execution of MSProfileR tool modules is in cascade, with a specific modularity allowing the insertion of new functionalities and tasks without rewriting all R code. MSProfileR was made with the motivation to be used by everyone especially for individuals who have no programming skills. The installation of the R packages used for the tool building and the opening of the front of the interface, are obtained by installing the “MSProfileR” package on the R environment (version ≥ 4.3.0), which load all the needed dependencies for the launch of the application. The application launches into web browser.
2.7.2. UI of Data Loading Tab
The interface of the data loading tab was divided into two panels corresponding to the different modules from this tab (Supplementary Figure S1, Table 1). The MSProfileR tool starts with the loading of spectra. The level number of the binary files path sets at four by default should not be modified before spectra loading. The user selects the directory containing one or many spectra to analyse. The data related to each spectrum are gathered into a summarizing table, including the id spectra, a number generated by a programmatic way for each spectrum, the sample name, the replicate name and the acquisition date. The sample name will be used to detect sample replicates when available. The data from ten spectra are presented per table. Spectra are classified per loading order and the others are available by the page selection at the bottom of the table. The number of loaded spectra as well as the path are indicated at the top of the table. Thanks to the table, the user can control whether the sampleName and replicateName columns were correctly filled. If a shift was noticed, this problem could be rectified by adjusting the level number of the binary files path until the correct classification was obtained.
The second panel is optional. The user has the possibility at this step to upload setting parameters used in a previous analysis. This option allows to analyse the current spectrum dataset with the same parameters applied previously for another dataset.
2.7.3. UI of Preprocessing Tab
The preprocessing tool is represented on the interface by five panels corresponding to the four modules of this tab (Supplementary Figure S2, Table 1). It consisted of successive steps to evaluate the quality of the spectra to exclude inconsistent MS profiles. The quality control module was divided into two panels to distinct the automatic selection process from the optional manual validation of conform spectra. All these steps are interactive, the user could select a method and associated parameters when available, for dataset analysis.
The first panel consists to trim and to test the conformity of spectra (Supplementary Figure S2). The lower and upper limits of spectra range could be determined by the user and are plotted in a graduated scale of m/z (kiloDaltons, kDa). Following this step, conformity tests were automatically done and the results were presented in a table. The table rows are coloured in green if all spectra are compliant and in grey if some spectrum did not pass the test(s) successfully. Moreover, the number of spectra considered as non-conform per test was indicated and these spectra are automatically excluded from the rest of the analysis. Nevertheless, the user can keep these non-conform spectra by deselecting the checkbox untitled “Exclusion of non-conform MS spectra”.
The cleaning panel realizes successive steps to adjust spectra profiles including transformation, smoothing and normalization of intensity plus a baseline correction. The user can select different methods by radio button for each step and visualizes the result of treatment to each spectrum by a plot through a reactive window.
The panel of spectra quality control allows to visualize the results of the spectra screening according to the method selected, into a plot representing the atypical score (A score) of each spectrum indexed by a number corresponding to their loading order (i.e., id spectra). The dotted line represented the upper limit of the A score threshold above which spectra are considered as outliers. Numbers corresponding to atypical spectra are then coloured in red whereas those kept for the rest of the analysis are indicated in blue. Interestingly, it is also possible to detect spectra considered as outliers due to their low A score by deselecting the checkbox “Include spectra below the lower threshold”. In such condition, all spectra obtaining an A score outside the lower or upper limits will be considered as atypical. Nevertheless, based on our background, notably for MS spectra from entomological origin, protein profiles which obtained very low A score correspond generally to MS spectra with higher peak intensities. To avoid their classification as outliers, uniquely the upper limit is applied by default.
The validation of spectra classification as conform or atypical remains possible through the selection panel. This panel was composed of two boxes separating spectra according to their compliant status. One box lists the outliers (i.e., “Atypical spectra”), whereas the second box lists spectra kept for the next steps (i.e., “Selected spectra”). Spectra in each box are identified by their id spectra, sample name and A score. Using these box lists, the user can choose a spectrum which is plotted below. By default, atypical spectra are automatically removed from the rest of the analysis. However, after visual spectra checking, the user can decide to keep some “atypical spectra” or to remove some “selected spectra” by moving them from one box to another by using arrow buttons.
The last panel of this tab consists to average spectra replicates which succeeded in passing all the preprocessing steps. The user can select one of the three methods to perform the spectra averaging. Averaged spectra are presented in a table containing one representative spectrum per replicated sample, classified by the sample name. The number of averaged spectra was indicated at the top of the table. This averaging reduces the number of spectra to analyse in the processing part.
2.7.4. UI of Processing Tab
The interface of the processing tab is separated into five panels (Supplementary Figure S3). The first panel concerns the peak detection. The user can select one method of noise estimator and visualize the results of peak detection on a boxplot graph showing the variance of peak number detected per spectra in the dataset for each SNR value from 2 to 7. Based on this result, the user chooses the optimal SNR value for the current analysis using a graduated slider. A visualization of the peaks detected for each averaged spectrum is available. On the graph, the detected peaks are indexed in ascending order from highest to lowest intensities.
The next three panels correspond to the spectrum alignment modules, which were split in the interface to visualize and to control the outcomes of each step. Firstly, to realize spectrum alignment the requirement of reference peaks is compulsory. In this way, the user should select a method to obtain reference peaks among the spectra dataset based on two parameters, the minimum of peak occurrence (i.e., frequency) and the alignment tolerance. The number of peaks used as reference and their distribution in the range of m/z values are presented on a plot. Before to apply spectra alignment, an adjustment of parameters remains possible by the user to increase the number of the reference peaks. Then, the user launches the alignment process, by choosing one of the four methods of warping functions. The visualization of the alignment is presented by a gelview where abscissa and ordinate correspond respectively to peak position (m/z) and averaged spectra. Peak intensity is represented by a grey scale. Once the peaks are aligned, the next two panels concern the peak binning and peak filtering. For these two steps, different methods and parameters are offered to the user to reduce the number of peaks which will be retained for the creation of the matrix of peak intensities. The results of the binning and the filtering steps are illustrated by gelviews in the UI, and the total number of peaks (m/z values) from the dataset was also indicated at each step. Based on these information the user could decide either to adjust parameters or to continue the analysis.
The last panel consists to classify averaged spectra according to their profiles. In this way, a hierarchical clustering was performed and represented by a heatmap linked to a dendrogram. At this stage of the application, an intensity matrix for all the detected peaks is generated for future analysis. Each peak does not necessarily exist in all the spectra. By default, when peaks are missing, intensity values of the spectra are used to fill the intensity matrix. A checkbox allows this filling to be deactivated. In this case, when peaks are missing, NA values are inserted into the intensity matrix. In the heatmap, missing values appear in grey whereas the detected peaks were coloured in a rainbow scale according to the intensity values.
2.7.5. UI of Annotation Tab
This tab divided into three panels, aims to add sample information (metadata) to each averaged spectrum (Supplementary Figure S4). An annotation table (.xlsx) generated automatically by MSProfileR is downloadable in the first panel. Uniquely the first column “sampleName” of the annotation table is filled with the names of the samples which passed all previous tests. In the subsequent columns, the information of interest could be listed. The body part used, the arthropod family, its geographical origin, the method and mode of storing are some examples of added information which could be useful for the next steps of the analysis. Once the annotation table is enriched with metadata, it could be uploaded using the second panel of this Table Finally, in the annotation processing panel, the success of the annotation and pairing with averaged MS spectra could be controlled in a summary table generated automatically.
2.7.6. UI of Output Tab
The output tab was constituted of six panels (Supplementary Figure S5). The first one serves to download the report generated during the analysis. The next four panels allow users to download, respectively, the parameters applied, the intensity matrix, the figures and the HDF5 files, including parameters, averaged spectra, intensity matrix and the annotated table. All these outputs can be downloaded as a single archive in the last panel named “All files”.
3. Assessment of MSProfileR Tool
Although MSProfileR can be applied beyond the entomological domain, its performance was assessed here with two datasets from arthropods. The two datasets of MS spectra were very different from each other, both in terms of sample diversity, but also in terms of the number of spectra tested. The first dataset was intentionally heterogeneous and included several distinct arthropod families. The second consisted in a single mosquito genera but included 13 species with a larger dataset.
3.1. Use Case N°1: Arthropod Families
This first dataset is composed of several arthropod species all laboratory reared. It comprises three arthropod families, mosquitoes (Culicidae), ticks (Ixodidae) and fleas (Pulicidae), including six species at two developmental stages for some of them. According to family and developmental stages, different body parts were submitted to MS analysis. Several recent papers established standardized operational protocols for sample preparation of these arthropod families [7,8,11]. Details about the arthropod species used for MS spectra analysis are summarized in Table 2. The breeding conditions and sample preparations are also indicated in this table, with respective references. This first dataset consisted in a total of 192 MS spectra coming from 48 samples, analysed in quadruplicate. These spectra come from our home-made reference spectra DB [30] and are then considered as high quality (Supplementary file S1).
The 192 MS spectra were uploaded in MSProfileR. The number of files uploaded and the name of samples can be controlled on the “data loading” tab (Supplementary Figure S1). As it is the first dataset of spectra analysed with this tool, no file containing MSProfileR parameters set in a previous analysis was available. Then, all the parameters were selected in the present analysis.
In the preprocessing tab (Supplementary Figure S2A), MS spectra were trimmed in the range of 2-20 kDa and were submitted to conformity tests (Supplementary Figure S2B and S2C). All spectra passed these tests. The parameters selected in the spectra cleaning panel were: square root method for transforming intensity, Savitzky-Golay method with a half-window size of 10 for smoothing intensity, SNIP method with 100 iterations for removing baseline and TIC method for normalizing the intensity of MS spectra. The result of the cleaning steps can be visualized on a plot for each spectrum (Supplementary Figure S2D). The Q estimator and the RC method with a threshold of 3 were computed for detecting of outliers in the quality control panel (Supplementary Figure S2E). In the present dataset, an upper atypical score (A score) limit of 0.59 revealed six spectra among 192 as outliers (3.1%). Interestingly, unticking the “Include spectra below the lower threshold” button did not exclude additional spectra. This underlines that none of the spectra with a low A score were considered outlier. All spectra classified as outliers are automatically removed from the next steps of the analysis. However, it remains possible to visualize all outliers and keep them (Supplementary Figure S2F). Using MSProfileR, all outlier spectra were compared with their respective replicates (Supplementary Figure S6). These spectra with higher A score (i.e., upper the limit) were clearly confirmed to be of lower quality, particularly because they presented a higher background noise. These spectra came from 5 distinct samples, two replicates from one Ae. albopictus at larval stage, two spectra from legs of two Ae. aegypti specimens and two spectra from cephalothoraxes of two Ct. felis specimens. As no sample had four replicates considered outliers, removing the six outlier spectra did not suppress any sample, which was confirmed in the averaging panel (Supplementary Figure S2G). The averaging of selected replicates was done with the mean methods revealing that 48 samples were available for further analysis.
In the processing tab (Supplementary Figure S3A), the MAD method with a half-window of 20 was applied for peak detection. Peak detection is directly linked to the signal-to-noise (SNR) value selected. In this dataset, when the SNR increases from 2 to 7, the mean number of peaks detected per spectrum decreases from about 144 to 39, respectively (Supplementary Figure S3B). The choice of the optimal SNR value could be decisive for the next steps of the analysis. To determine the most appropriate SNR value, a comparison of the peak list detected per arthropod family and per body part was carried out based on the SNR value from two to five (Supplementary Figure S7). It was noticed that at SNR equal to two, numerous peaks were detected between 2 to 8 kDa, among which several are unspecific and correspond to background noise. At SNR equal to three, the majority of these peaks of very low intensity were deleted. Moving to SNR equal to four, regardless of the sample types, some peaks which do not correspond to background noise were no longer detected. In such condition, some information about the protein profile of the sample was lost. To avoid this phenomenon, a SNR value of three was selected for this dataset. At this SNR value, the number of peaks detected per spectrum was 103.3 ± 11.5 (mean ± standard deviation (SD)).
After peak detection, the alignment of spectra was done (Supplementary Figure S3C). The strict method with a minimum frequency of 50% and a tolerance of 0.002 were set for selecting reference peaks. A total of 33 peaks met these criteria and were used to align spectra from the dataset by applying lowess method for spectra warping. For the binning step, the strict method with a tolerance of 0.002 was set and the parameter for peak filtering was a prevalence of at least 20% for a m/z value to be conserved (Supplementary Figure S3D and S3E). Among the 516 peak positions obtained after the binning step, the filtering reduced this number to 202. Finally, these 202 peaks were used to classify each sample based on their averaged MS profiles using a hierarchical clustering (Supplementary Figure S3F). The clustering confirmed that all averaged spectra from the same sample type were grouped per species and body parts. Interestingly, the first criterion of classification is the body part followed by the species (Supplementary Figure S8).
In the annotation tab (Supplementary Figure S4A), the list of the sample name of the 48 averaged spectra can be downloaded and was completed with several metadata from each sample prior to be uploaded (Supplementary Figure S4B and S4C). The annotation processing table allows to verify the correspondence of the averaged spectra with the metadata (Supplementary Figure S4D). Here, as the number of averaged spectra and annotated samples are identical (n=48) and as none of them was not paired, then all averaged spectra from the dataset N°1 were annotated (Supplementary fileS1).
In the output tab (Supplementary Figure S5A and S5(a)), several kinds of files could be downloaded. The report file detailed modules, methods and parameters applied, plus information about the dataset N°1 and the results (tables, plots, boxplot, gelviews and clustered heatmap) of the preprocessing and processing parts (Supplementary Figure S5B and S5(b)). The parameters could be uploaded as a JSON file and used for analysing a new dataset (Supplementary Figure S5C and S5(c)). This also ensures the traceability and the reproducibility of the analysis. The plots generated by the application during dataset N°1 analysis can also be exported (Supplementary Figure S5E and S5(e)), as well as the intensity matrix (Supplementary Figure S5D and S5(d)) and the HDF5 files (Supplementary Figure S5F and S5(f)). All the outputs from this dataset are downloadable in one zipped file which is available in the Supplementary file S2.
3.2. Use Case N°2: Culex Genus
This second dataset included MS spectra from Neotropical Culex mosquitoes (Diptera: Culicidae) coming from a work recently published [31]. The methods and protocols used for mosquito collection, for their morphological and/or molecular identification, for sample preparation and MS submission were detailed in this previous study [31]. Briefly, this dataset was composed of MS spectra from legs and thoraxes of 13 distinct Culex species collected in the field, in French Guiana. The number of specimens per species varied according to the availability of the sample, ranging from 1 to 34 (Supplementary file S3).
The dataset N°2 has interesting characteristics for the evaluation of MSProfileR. First, it includes cryptic species, morphologically undistinguishable. Secondly, specimens were collected in the field and then higher inter-sample variations could occurred among specimens from the same species compared to laboratory reared specimens. Thirdly, it encompasses a large number of samples, 169 mosquito specimens submitted to MALDI-TOF MS on two distinct body parts (legs and thoraxes) and loaded in quadruplicates, corresponding to a total of 1352 spectra (169 specimens × two body parts × four replicates). Finally, in the previous work using the same dataset [31], some spectra were excluded by the authors based on their visual inspection which were considered as low quality. The MSProfileR tool offers now the opportunity to detect and visualize all spectra considered as outliers and can then automatically exclude them from the dataset. The comparison of the spectra list excluded by the experimenters on the same dataset in the previous work will allow to assess the relevance of MSProfileR tool.
For analyzing the dataset N°2, the parameters applied for the dataset N°1 were uploaded. Uniquely the modified parameters compared to the dataset N°1 were specified. In the preprocessing part, all the 1352 spectra passed the conformity tests. However, the quality control steps revealed that 65 spectra (4.8%) exceeded the upper limit of the atypical threshold (A score = 0.72) and were considered as outliers (Figure 3D). Interestingly, all spectra classified as outliers had leg origin (Figure 3A, Supplementary file S4).
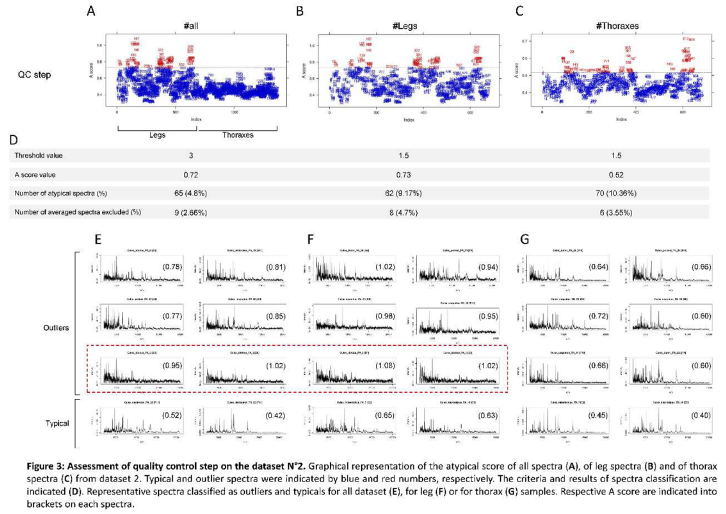
Several studies reported a higher diversity of spectra between body parts than between species [9,32]. Moreover, spectra from thoraxes generally had more numerous and higher intensity peaks than the paired leg spectra [11,31]. These two factors could likely explain why numerous spectra originating from legs were classified as outliers. To avoid this bias of exclusion, dataset N°2 was analyzed by splitting the list of spectra per body part, legs or thoraxes.
By applying the same parameters, “A” score thresholds of 0.93 and 0.59 were obtained for leg and thorax spectra, respectively (See report in Supplementary file S5 and Supplementary file S6). Nine and 20 spectra from leg and thorax, respectively, were classified as outliers. The inspection of these spectra confirmed the low quality of these outlier profiles compared to typical spectra. However, some spectra with a low-quality profile now scored below the threshold. (Figure 3E). Then, the threshold parameter of the quality control step was adjusted at 1.5 in order to exclude these spectra of low quality. “A” score thresholds of 0.73 and 0.52 were obtained for leg (Figure 3B) and thorax (Figure 3C) spectra, respectively. Details of the spectra listed as outliers are available in the reports of leg (Supplementary file S7) and thorax datasets (Supplementary file S8). Although 62 (9.2%) and 70 (10.4%) spectra from leg and thorax samples were classified as outliers (Figure 3D), the number of samples excluded remains modest, less than 5%. Effectively, the four spectra replicates from leg and thorax of eight and six specimens were excluded from the analysis. It is interesting to note that the eight specimens for which leg spectra were classified as outliers, encompassed those from the two Culex idottus which were excluded by the authors due to the low intensity and inter-sample heterogeneity of MS profiles in the previous work [31]. The inspection of the other spectra for which the four replicates were classified as outliers from legs or thoraxes confirmed the lower quality of these protein profiles. These samples were then excluded from the analysis. A total of 161 and 163 averaged spectra from legs or thoraxes, respectively, passed the preprocessing steps.
To check whether MSProfileR tool succeeded in classifying spectra according to species for each body part, averaged spectra were submitted to the peak detection steps by applying the parameters used in the dataset N°1. The filtering step revealed that 204 and 194 peaks for legs (Supplementary file S7) and thoraxes (Supplementary file S8), respectively, were retained for averaged spectra classification. Hierarchical clustering revealed that thorax averaged spectra were grouped per species. Solely two Cx. usquatus (#TH_24 and #TH_82) were not clustered with the other spectra from the same species; and the thorax averaged spectra from the unique Cx. adamesi (#TH_2) was classified inside the Cx. dunni group (Supplementary figure S10A). The clustering of leg averaged spectra appeared less efficient, with several samples intertwining with other species (Supplementary figure S10B). The lower classification of the leg averaged spectra compared to thorax was attributed, in part, to their lower spectra intensity. Effectively, as shown in the present study and in concordance with previous works, leg spectra were generally less intense than those of thoraxes from the same specimens [11,31].
For thoraxes, among the seven samples, from the previous analysis [31], which did not reach the threshold value (LSV>1.8) for relevant identification, for five of them, two or more of their spectra replicates were considered as outliers, leading to the exclusion of three samples, for which all replicates overtaken the “A” score threshold. The two other thorax samples (#TH_233 and #TH_234) from Cx. dunni conserved by the quality control step, obtained nearly relevant identification scores, 1.78 and 1.74, respectively, in the previous study [31]. The clustering of these last two thorax averaged spectra (#TH_233 and #TH_234) with the other samples from the same species support the conservation of their respective spectra by quality control steps.
Interestingly, the previous work [31] reported that the selection of the top ten of mass peak list per Culex species and per body part appeared sufficient to discriminate these 13 Culex species with a correct classification upper than 90%. Here, during the filtering step, all peaks with a frequency lower than 20 % (i.e., 0.2) across the dataset were excluded from the analysis. In the dataset N°2, for five species, one to five specimens were available which represents less than 1 to 4% of the number of averaged spectra of the dataset N°2 (161 for legs and 163 for thoraxes). It is then possible that some peaks specific to these species were not included in the intensity matrix due to their low representation (i.e., too few specimens from the same species to reach filtering threshold), which could explain the imperfect clustering of the samples per Culex species, notably for legs.
The decrease of the peak filtering from 20% to 0.5% (i.e., 0.005) led to the inclusion of 878 peaks in the intensity matrix without improving drastically the classification of leg averaged spectra. Among the peaks added in the intensity matrix, about 81.7% (n=717), some of them are heterogeneous between averaged spectra from the same species which should perturb clustering. MSProfileR then appears well adapted for the detection of atypical spectra, but also for their classification at condition that a subgroup is not too under-representative, which could alter the classification. Moreover, spectra of high intensity remain essential for relevant classification.
The duration of computational analyse is another parameter to take into account. Generally, the quickness of the analysis is directly linked to the power of the computer used. In the present work, the analysis was done on a laptop with classical configuration (Processor: Intel (R) Core(TM) i7-10510U CPU @ 1.80 GHz, Hard disk: RAM: 16 GiB, Graphics card: 00:02.0 VGA compatible controller: Intel Corporation UHD Graphics (rev 02), Operating System: Ubuntu 20.04.). The complete pipeline process for analysing the 1352 spectra of dataset N°2 took less than four minutes, by applying default parameters. This processing speed allows to the user to compare/adjust methods and parameters throughout the workflow without a loss of time.
Discussion
MALDI-TOF MS is capable of rapidly producing large volumes of extremely rich information. It is currently used in microbiology routine diagnosis laboratory for the identification of microorganisms, including bacteria, fungi, yeasts, filamentous and, more recently, for the identification of clinical tick samples collected on human hosts [33,34,35]. In microbiology, microorganisms are generally cultivated using standardized procedures prior to identification by MALDI-TOF MS [36]. For medical entomology studies, prior to arthropod specimen identification by MS, several factors can alter the quality of MS spectra, such as sample storing duration, storage conditions (temperature, with or without a buffer…) or sample preparation mode [5,37]. To overcome these limitations, the establishment of a standardised protocol and the development of a reproducible data scientific workflow to treat this kind of spectra appeared compulsory.
In the last decade, in order to improve the reproducibility and increase the noise-signal ratio of arthropod intra-species MS spectra, several guidelines have been proposed for sample preparation prior to MS analysis, notably for mosquitoes [6,7] and a consensus strategy seems to have emerged [11]. Some informatics tools were developed by private companies (eg, MALDI Biotyper from Bruker, Saramis from Biomerieux) for MALDI-TOF MS spectra investigation [38,39]. They are suitable for spectra analysis, based on spectral matching with a database of reference spectra, but some functionalities that seem essential, such as quality control or annotation of spectra, are missing, or, when available, the methods and parameters applied remain a black box [40]. As the identification success is essentially linked to the quality of sample MS spectra, the exclusion of spectra considered as non-conform should make it possible to save time and improve the relevance of classification. Some freely available packages were created by computational specialists to import and pre-process spectra raw data [16], or to filter of low-quality spectra among a dataset [17]. However, as they use R language, computer knowledge is required.
In the present study, the MSProfileR tool was created to perform the preprocessing including the filtering of non-compliant spectra, the classification and the annotation of MS spectra by the use of the R language and packages notably MALDIquant [16] and MALDIrppa [17]. Thanks to the R Shiny framework, which enables user-friendly interfaces to be built for the R environment, the MSProfileR tool offers rapid analysis and ease of use. The pipeline of analysis offered by MSProfileR consists in successive modules, and the tasks of each module offer several methods. Users can select the optimum method for their process and can also adjust its parameters to obtain the finest result. The consequences of each adjustment are presented by plots, graphics, tables or other illustrations on the Shiny interface. Throughout the workflow, users can then visualise each task of the pipeline and keep the control of all processes by adjusting parameters or changing methods. By recording the methods used and their parameters at the end of the analysis, they can be traced and reused. In this way, the same methods and parameters can be applied to a new dataset, reducing the experimenter’s intervention time and ensuring consistency in the analyses.
Detecting aberrant spectra is an essential step in the analysis. The MSProfileR tool enables rapid and automatic detection, which is a considerable advantage for entomological studies. Until now, this detection was based on visual comparison of spectra and the application of certain criteria such as the diversity of MS profiles or the intensity of the most intense peak (> 3000 ua) [38,39]. This detection was highly dependent on the skills and experience of the experimenter. In the absence of standardisation, the reproducibility of this detection was not guaranteed between different experimenters. The use of MSProfileR on dataset N°1 revealed that six MS spectra were considered as non-conform. None of these had previously been detected as outliers by the experimenter. As these non-compliant spectra only concerned one repetition out of 4 per sample, the impact on the mean spectrum used as a reference in the DB remains negligible. However, in the future, excluding outliers before calculating the mean spectrum will improve the quality of the MS database and the relevance of the identifications.
Throughout the analysis workflow, MSProfileR offers interactive visualisations linked to the criteria (methods/parameters) chosen. This means that any changes can be quickly assessed, which was important for detecting non-compliant spectra in dataset N°2. Adjustment of the quality control parameters revealed that a single parameter setting was not possible because the spectral profiles between the mosquito’s leg and thorax differed in intensity and diversity. The dataset was therefore split beforehand according to body part, allowing good detection of non-compliant spectra. This data set highlighted the need to homogenise the origin of the samples in order to improve the quality control stage of the spectra. MSProfileR detected replicate MS spectra of the legs of two Culex samples as outliers, confirming the classification made in previous work [30]. In addition, new MS spectra were detected as outliers (n = 132 out of 1352). In the previous study [30], the selection was essentially based on the intensity of the most intense peak and the skill of the experimenter. In the previous study, this manual selection was complex and very time-consuming because this second data set included a total of 1352 spectra. Conversely, the user can now inspect the spectra classified as outliers by MSProfileR and validate or not this classification. Detecting new outlier spectra and validating them underlines the tool’s efficiency and saves time. By controlling the quality of spectra in a reproducible way and independently of the user, the analysis of spectra and the classification of species will improve.
A decisive parameter remains the peak detection threshold [41], which is linked to the selection of the optimal SNR value. A too low SNR value could conduct to the inclusion of peaks corresponding to background noise and inversely a too high SNR value could induce the miss-detection of true peaks, which could alter spectra classification in the next steps. The selection of the SNR value is rarely possible with the majority of commercial bio-informatics tools, and when it was possible the consequence of SNR value change on peak detection is not easy to judge. Often its choice is highly subjective. MSProfileR now makes it possible to directly see the effect of changing SNR value on the detected peaks. In a recent study assessing the COVID-19 diagnosis using human saliva MS spectra obtained by MALDI-TOF profiling, we reported the interest and importance of selecting the optimal SNR value [42].
An important original feature of MSProfileR is the ability to annotate all the spectra that are averaged after the validation stages. To our knowledge, no commercial or open source software allows information to be added to each spectrum. MSProfileR makes it possible to associate and store information about the sample analysed (origin, sample type, preservation, preparation protocol, etc.), which is essential for carrying out statistical analyses and storing these spectra as a reference in a database.
Finally, the correct spectra classification by the hierarchical clustering of the heterogeneous dataset N°1 and the highly homogeneous dataset N°2, underlined the widely useful of this tool for sample comparisons. As spectra from dataset N°2 were originated from field collection of arthropods without knowledge of the species population encountered, uniquely unsupervised classification is possible. The hierarchical clustering method is then an appropriate strategy for arthropod classification. For mosquitoes, two body parts, legs and thorax, could be submitted independently to improve specimen identification [9,31]. Here the comparison of dendrograms from paired samples could be informative to verify the concordance of classification. Although, the ordination of paired samples differs between dendrograms according of body part tested, the same sample list clusters on one branch per body part. To maintain the extensibility of the application, the HDF5 file panel was created by storing the averaged spectra of each dataset, their annotation, the parameters used and their intensity matrix. This module will be helpful for the future construction of a reference MS spectra database which will be used for specimen identification by spectral matching [43].
Recently, a web tool called GeenaR has been published [44]. Like MSProfileR, GeenaR offers complete workflow for analysis MALDI-TOF MS spectra with comparable functionalities. The main difference is that for GeenaR, all methods and parameters are defined in a unique web page before running the analysis. GeenaR presents the advantage of being very easy to use and also includes a quality control of MS spectra. Unfortunately, every time the user changes a method or a parameter, the whole application has to be re-run. The high heterogeneity of arthropod MS spectra according to numerous factors (e.g., family, body part, storing mode, duration of storing, sample preparation conditions, etc) requires an adjustment of software parameters to optimize spectra analysis [7,45]. The possibility to visualize rapidly each modification of method or setting parameters is essential for entomological spectra analyses, notably for the detection of the outliers and the determination of the cut-off value which could vary according to the body part tested for paired species-samples, as observed in the dataset N°2. MSProfileR make it possible to see the consequence of each parameter variation as soon as it is modified. MSProfileR is then completely well adapted for analysis MS spectra dataset including high diversity of protein profiles which occurred among arthropod species, from the distinct families (eg, dataset N°1), but also among species from the same family (eg, dataset N°2).
Conclusions
In developing the MSProfileR tool, we created a MALDI-TOF MS scientific workflow with functionalities adapted to investigate arthropod vector populations. Its main functionalities are the numerous quality control tests on the spectra, the classification of averaged spectra and their annotation. The advantage of MSProfileR resides in its semi-automatic processing allowing interventions of the analysis to improve the quality of results. Throughout the pipeline, the user can visualize and control all the tasks and quickly adjust each parameter. This easy-to-use Shiny graphical apps is accessible with a web browser application and is accessible on Windows, macOS and Linux, thanks to the MSProfileR R package. This package is available on the Github platform. MSProfileR is therefore a user-friendly tool that analyses spectral data, without need for programming expertise. MSProfileR seems to be a promising tool for analysing MS spectra from arthropods of public health importance like mosquito and tick vectors. MSProfileR is an open-source software that can be used by the scientific community, particularly entomologists.
List of Abbreviations
MALDI-TOF MS: Matrix Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry; DB: Database; GUI: Graphical User Interface; CRAN: Comprehensive R Archive Network; kDa: Kilo Daltons; m/z: mass/charge; SNIP: Sensitive Nonlinear Iterative Peak; TIC: Total-Ion-Current; PQN: Probabilistic Quotient Normalization; RC: Rousseeuw & Croux; ESD: Extreme Studentized Deviation; CQ: Control Quality; SNR: Signal-to-noise ratio; HDF5: Hierarchical Data Format version 5; UI: User Interface; T°: Temperature degree.
Supplementary Materials
Supplementary_Figure-S1.pptx; Supplementary_Figure-S2.pptx; Supplementary_Figure-S3.pptx; Supplementary_Figure-S4.pptx; Supplementary_Figure-S5.pptx; Supplementary_Figure-S6.pptx; Supplementary_Figure-S7.pptx; Supplementary_Figure-S8.pptx; Supplemetnary_File_S1; Supplemetnary_File_S2; Supplemetnary_File_S3; Supplementary_File_S4.zip; Supplementary_File_S5.zip; Supplementary_File_S6.zip; Supplementary_File_S7.zip; Supplementary_File_S8.zip.
Author Contributions
Conceptualization and methodology of the experiments: L.A.; B.E.; S.G.; K.H.; R.B.H.; experiments investigation and tools contribution: R.B.H.; A.C.; B.E.; K.H., S.G.; H.M.; J-M.L.; G.T.; data analysis: R.B.H., L.A., B.E.; writing—original draft preparation: L.A., R.B.H.; B.E.; S.G.; funding acquisition: L.A.. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by the Délégation Générale pour l’Armement (DGA, MS_ProfileR project, Grant no PDH-2-NRBC-2-B-2201).
Ethics Approval and Consent to Participate
Not applicable.
Consent for publication
Not applicable.
Availability of Data and Materials
The both spectra datasets (Dataset N°1 and N°2) used and/or analysed during the current study are available in the supplementary files as well as the other folders downloadable throughout the analysis process.
Acknowledgments
RBH was supported by the Institut Hospitalo-Universitaire (IHU) Méditerranée Infection throughout her PhD candidature.
Competing Interests
The authors declare that they have no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Availability
MSProfileR tool along with its source code and its associated existing R packages, MALDIquant and MALDIrppa, are freely available from the R archive CRAN (http://cran.r-project.org). The software with the R Shiny user interface, as a web browser tool, is available following request to the corresponding author. The package is distributed under the GNU General Public License (version 3 or later) and is accompanied by a tutorial.
References
- Sandrin, T.R.; Goldstein, J.E.; Schumaker, S. MALDI TOF MS Profiling of Bacteria at the Strain Level: A Review. Mass Spectrom. Rev. 2013, 32, 188–217. [Google Scholar] [CrossRef]
- Fenselau, C.; Demirev, P.A. Characterization of Intact Microorganisms by MALDI Mass Spectrometry. Mass Spectrom. Rev. 2001, 20, 157–171. [Google Scholar] [CrossRef]
- Seng, P.; Rolain, J.-M.; Fournier, P.E.; La Scola, B.; Drancourt, M.; Raoult, D. MALDI-TOF-Mass Spectrometry Applications in Clinical Microbiology. Future Microbiol. 2010, 5, 1733–1754. [Google Scholar] [CrossRef]
- Yssouf, A.; Almeras, L.; Raoult, D.; Parola, P. Emerging Tools for Identification of Arthropod Vectors. Future Microbiol. 2016, 11, 549–566. [Google Scholar] [CrossRef]
- Dieme, C.; Yssouf, A.; Vega-Rúa, A.; Berenger, J.-M.; Failloux, A.-B.; Raoult, D.; Parola, P.; Almeras, L. Accurate Identification of Culicidae at Aquatic Developmental Stages by MALDI-TOF MS Profiling. Parasit. Vectors 2014, 7, 544. [Google Scholar] [CrossRef]
- Nabet, C.; Kone, A.K.; Dia, A.K.; Sylla, M.; Gautier, M.; Yattara, M.; Thera, M.A.; Faye, O.; Braack, L.; Manguin, S.; et al. New Assessment of Anopheles Vector Species Identification Using MALDI-TOF MS. Malar. J. 2021, 20, 33. [Google Scholar] [CrossRef]
- Nebbak, A.; Willcox, A.C.; Bitam, I.; Raoult, D.; Parola, P.; Almeras, L. Standardization of Sample Homogenization for Mosquito Identification Using an Innovative Proteomic Tool Based on Protein Profiling. Proteomics 2016, 16, 3148–3160. [Google Scholar] [CrossRef] [PubMed]
- Nebbak, A.; El Hamzaoui, B.; Berenger, J.-M.; Bitam, I.; Raoult, D.; Almeras, L.; Parola, P. Comparative Analysis of Storage Conditions and Homogenization Methods for Tick and Flea Species for Identification by MALDI-TOF MS. Med. Vet. Entomol. 2017, 31, 438–448. [Google Scholar] [CrossRef]
- Vega-Rúa, A.; Pagès, N.; Fontaine, A.; Nuccio, C.; Hery, L.; Goindin, D.; Gustave, J.; Almeras, L. Improvement of Mosquito Identification by MALDI-TOF MS Biotyping Using Protein Signatures from Two Body Parts. Parasit. Vectors 2018, 11, 574. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.H.; Boulanger, N.; Nebbak, A.; Collin, E.; Jaulhac, B.; Almeras, L. Assessment of MALDI-TOF MS Biotyping for Borrelia Burgdorferi Sl Detection in Ixodes Ricinus. PloS One 2017, 12, e0185430. [Google Scholar] [CrossRef] [PubMed]
- Bamou, R.; Costa, M.M.; Diarra, A.Z.; Martins, A.J.; Parola, P.; Almeras, L. Enhanced Procedures for Mosquito Identification by MALDI-TOF MS. Parasit. Vectors 2022, 15, 240. [Google Scholar] [CrossRef]
- Yssouf, A.; Parola, P.; Lindström, A.; Lilja, T.; L’Ambert, G.; Bondesson, U.; Berenger, J.-M.; Raoult, D.; Almeras, L. Identification of European Mosquito Species by MALDI-TOF MS. Parasitol. Res. 2014, 113, 2375–2378. [Google Scholar] [CrossRef]
- Kumsa, B.; Laroche, M.; Almeras, L.; Mediannikov, O.; Raoult, D.; Parola, P. Morphological, Molecular and MALDI-TOF Mass Spectrometry Identification of Ixodid Tick Species Collected in Oromia, Ethiopia. Parasitol. Res. 2016, 115, 4199–4210. [Google Scholar] [CrossRef]
- Albrethsen, J. Reproducibility in Protein Profiling by MALDI-TOF Mass Spectrometry. Clin. Chem. 2007, 53, 852–858. [Google Scholar] [CrossRef]
- Diarra, A.Z.; Laroche, M.; Berger, F.; Parola, P. Use of MALDI-TOF MS for the Identification of Chad Mosquitoes and the Origin of Their Blood Meal. Am. J. Trop. Med. Hyg. 2019, 100, 47–53. [Google Scholar] [CrossRef]
- Gibb, S.; Strimmer, K. MALDIquant: A Versatile R Package for the Analysis of Mass Spectrometry Data. Bioinforma. Oxf. Engl. 2012, 28, 2270–2271. [Google Scholar] [CrossRef]
- Palarea-Albaladejo, J.; Mclean, K.; Wright, F.; Smith, D.G.E. MALDIrppa: Quality Control and Robust Analysis for Mass Spectrometry Data. Bioinforma. Oxf. Engl. 2018, 34, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Susmita, D.; Bart J. A., M. Statistical Analysis of Proteomics, Metabolomics, and Lipidomics Data Using Mass Spectrometry.
- Coombes, K.R.; Tsavachidis, S.; Morris, J.S.; Baggerly, K.A.; Hung, M.-C.; Kuerer, H.M. Improved Peak Detection and Quantification of Mass Spectrometry Data Acquired from Surface-Enhanced Laser Desorption and Ionization by Denoising Spectra with the Undecimated Discrete Wavelet Transform. Proteomics 2005, 5, 4107–4117. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Soltys, S.; Shi, G.; Koong, A.; Le, Q.-T. Sample Classification from Protein Mass Spectrometry, by “Peak Probability Contrasts. ” Bioinforma. Oxf. Engl. 2004, 20, 3034–3044. [Google Scholar] [CrossRef] [PubMed]
- Savitzky, Abraham.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [CrossRef]
- Ryan, C.G.; Clayton, E.; Griffin, W.L.; Sie, S.H.; Cousens, D.R. SNIP, a Statistics-Sensitive Background Treatment for the Quantitative Analysis of PIXE Spectra in Geoscience Applications. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1988, 34, 396–402. [Google Scholar] [CrossRef]
- Deininger, S.-O.; Cornett, D.S.; Paape, R.; Becker, M.; Pineau, C.; Rauser, S.; Walch, A.; Wolski, E. Normalization in MALDI-TOF Imaging Datasets of Proteins: Practical Considerations. Anal. Bioanal. Chem. 2011, 401, 167–181. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J.; Croux, C. Alternatives to the Median Absolute Deviation. J. Am. Stat. Assoc. 1993, 88, 1273–1283. [Google Scholar] [CrossRef]
- Rosner, B. Percentage Points for a Generalized ESD Many-Outlier Procedure. Technometrics 1983, 25, 165–172. [Google Scholar] [CrossRef]
- Zheng, X.; Ma, Y.; Bai, Y.; Huang, T.; Lv, X.; Deng, J.; Wang, Z.; Lian, W.; Tong, Y.; Zhang, X.; et al. Identification and Validation of Immunotherapy for Four Novel Clusters of Colorectal Cancer Based on the Tumor Microenvironment. Front. Immunol. 2022, 13, 984480. [Google Scholar] [CrossRef] [PubMed]
- Yihui, X.; J. J., A.; Garrett, G. Chapter 15 Parameterized Reports. In R Markdown: The Definitive Guide; 2023.
- Ingargiola, A.; Laurence, T.; Boutelle, R.; Weiss, S.; Michalet, X. Photon-HDF5: Open Data Format and Computational Tools for Timestamp-Based Single-Molecule Experiments. Proc. SPIE-- Int. Soc. Opt. Eng. 2016, 9714, 971405. [Google Scholar] [CrossRef] [PubMed]
- Fall, F.K.; Laroche, M.; Bossin, H.; Musso, D.; Parola, P. Performance of MALDI-TOF Mass Spectrometry to Determine the Sex of Mosquitoes and Identify Specific Colonies from French Polynesia. Am. J. Trop. Med. Hyg. 2021, 104, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.M.; Guidez, A.; Briolant, S.; Talaga, S.; Issaly, J.; Naroua, H.; Carinci, R.; Gaborit, P.; Lavergne, A.; Dusfour, I.; et al. Identification of Neotropical Culex Mosquitoes by MALDI-TOF MS Profiling. Trop. Med. Infect. Dis. 2023, 8, 168. [Google Scholar] [CrossRef]
- Briolant, S.; Costa, M.M.; Nguyen, C.; Dusfour, I.; Pommier de Santi, V.; Girod, R.; Almeras, L. Identification of French Guiana Anopheline Mosquitoes by MALDI-TOF MS Profiling Using Protein Signatures from Two Body Parts. PloS One 2020, 15, e0234098. [Google Scholar] [CrossRef]
- Sevestre, J.; Diarra, A.Z.; Laroche, M.; Almeras, L.; Parola, P. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry: An Emerging Tool for Studying the Vectors of Human Infectious Diseases. Future Microbiol. 2021, 16, 323–340. [Google Scholar] [CrossRef]
- Beltran, A.; Palomar, A.M.; Ercibengoa, M.; Goñi, P.; Benito, R.; Lopez, B.; Oteo, J.A. MALDI-TOF MS as a Tick Identification Tool in a Tertiary Hospital in Spain. Acta Trop. 2023, 242, 106868. [Google Scholar] [CrossRef] [PubMed]
- Jumpertz, M.; Sevestre, J.; Luciani, L.; Houhamdi, L.; Fournier, P.-E.; Parola, P. Bacterial Agents Detected in 418 Ticks Removed from Humans during 2014-2021, France. Emerg. Infect. Dis. 2023, 29, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Dingle, T.C.; Butler-Wu, S.M. Maldi-Tof Mass Spectrometry for Microorganism Identification. Clin. Lab. Med. 2013, 33, 589–609. [Google Scholar] [CrossRef] [PubMed]
- Diarra, A.Z.; Almeras, L.; Laroche, M.; Berenger, J.-M.; Koné, A.K.; Bocoum, Z.; Dabo, A.; Doumbo, O.; Raoult, D.; Parola, P. Molecular and MALDI-TOF Identification of Ticks and Tick-Associated Bacteria in Mali. PLoS Negl. Trop. Dis. 2017, 11, e0005762. [Google Scholar] [CrossRef]
- Ilina, E.N.; Borovskaya, A.D.; Malakhova, M.M.; Vereshchagin, V.A.; Kubanova, A.A.; Kruglov, A.N.; Svistunova, T.S.; Gazarian, A.O.; Maier, T.; Kostrzewa, M.; et al. Direct Bacterial Profiling by Matrix-Assisted Laser Desorption-Ionization Time-of-Flight Mass Spectrometry for Identification of Pathogenic Neisseria. J. Mol. Diagn. JMD 2009, 11, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, A.; Kasinathan, V.; Hyman, J.; Walsh, J.; Drake, T.; Wang, Y.F.W. Rapid Identification of Bacteria and Yeasts from Positive-Blood-Culture Bottles by Using a Lysis-Filtration Method and Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrum Analysis with the SARAMIS Database. J. Clin. Microbiol. 2013, 51, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Yssouf, A.; Socolovschi, C.; Leulmi, H.; Kernif, T.; Bitam, I.; Audoly, G.; Almeras, L.; Raoult, D.; Parola, P. Identification of Flea Species Using MALDI-TOF/MS. Comp. Immunol. Microbiol. Infect. Dis. 2014, 37, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Cramer, R.; Schuchhardt, J. Evaluation of Peak-Picking Algorithms for Protein Mass Spectrometry. Methods Mol. Biol. Clifton NJ 2011, 696, 341–352. [Google Scholar] [CrossRef]
- Costa, M.M.; Martin, H.; Estellon, B.; Dupé, F.-X.; Saby, F.; Benoit, N.; Tissot-Dupont, H.; Million, M.; Pradines, B.; Granjeaud, S.; et al. Exploratory Study on Application of MALDI-TOF-MS to Detect SARS-CoV-2 Infection in Human Saliva. J. Clin. Med. 2022, 11, 295. [Google Scholar] [CrossRef]
- Asare, P.T.; Lee, C.-H.; Hürlimann, V.; Teo, Y.; Cuénod, A.; Akduman, N.; Gekeler, C.; Afrizal, A.; Corthesy, M.; Kohout, C.; et al. A MALDI-TOF MS Library for Rapid Identification of Human Commensal Gut Bacteria from the Class Clostridia. Front. Microbiol. 2023, 14, 1104707. [Google Scholar] [CrossRef]
- Del Prete, E.; Facchiano, A.; Profumo, A.; Angelini, C.; Romano, P. GeenaR: A Web Tool for Reproducible MALDI-TOF Analysis. Front. Genet. 2021, 12, 635814. [Google Scholar] [CrossRef] [PubMed]
- Nebbak, A.; Almeras, L. Identification of Aedes Mosquitoes by MALDI-TOF MS Biotyping Using Protein Signatures from Larval and Pupal Exuviae. Parasit. Vectors 2020, 13, 161. [Google Scholar] [CrossRef]

Table 1.
Overview of the organization of each tabs of MSProfileR tool user’s interface.
| Tab (or Steps) | Modules | Tasks | Methods | Parameters (Range) | Default Setting | Graphical Interface |
|---|---|---|---|---|---|---|
|
Data loading |
Spectra loading |
Choose the level of spectra path issues from the MALDI-TOF MS directories | number of folders (1 to n) | 4 | ||
| Import of raw spectra data and spectra metadata | Table | |||||
| Setting parameters (optional) | Download previous settings | JSON file(a) | ||||
| Pre-processing | Trimming & Conformity tests | Trimming of spectra | Lower – upper limits (0.1-500 kDa) |
2-20 kDa | Plotting the limits | |
| Elimination of empty, irregular or non-compliant length spectra | Count and color code of spectra status (green, compliant; orange, non-compliant spectra) | |||||
| Cleaning spectra | Variance stabilization | Sqrt* Log Log 2 Log 10 |
||||
| Smoothing | Savitzky-Golay* Moving average |
Half Window Size (1- 100) | 10 | |||
| Baseline removal | SNIP* TopHat Convex hull Median |
Number of iterations (1-100) | 100 | |||
| Normalization | TIC* PQN Median |
Plotting of stabilized, smoothed, corrected and normalized spectra | ||||
| Quality control | Estimator | Q* MAD |
||||
| Associated method | RC* Hampel ESD Boxplot Adj.boxplot |
Threshold value (0.1- 10) | 3 (1.5 for boxplot rules, 3 for others methods) | |||
| Detection of outliers outside the upper and lower thresholds | Ascore | Spectra below the lower threshold included |
Plotting of spectra with respective Ascore, threshold indicated by dotted line(s) and outliers colored in red numbers. | |||
| Selection of spectra | User intervention | Listing of selected and excluded spectra Plotting of selected spectra |
||||
| Averaging | Average replicates | Mean* Median Sum |
Count of averaged spectra Table |
|||
| Processing | Peak detection | Estimator | MAD* Super Smoother |
Half window size (1-100) |
20 | |
| Background subtraction | SNR | SNR value (2-7) | 2 | Boxplot counting detected peak per SNR value Plotting of detected peak per selected spectra |
||
| Spectrum alignment | Reference peak detection | Strict* Relaxed |
min Frequency (0.1-1) Tolerance (0.0001-0.5) |
0.9 0.002 |
Plotting of reference peaks | |
| Warping | Lowess* Linear Quadratic Cubic |
Gel view of spectra from dataset | ||||
| Peak binning | Strict* Relaxed |
Tolerance (0.0001-0.5) | 0.002 | Peak counting Plotting of aligned spectra |
||
| Peak filtering | Frequency | min Frequency (0.1-1) | 0.2 | Plotting of filtered spectra | ||
| Clustering | Hierarchical clustering Creation of matrix |
Plot a clustred heatmap | ||||
| Spectra annotation | Loading template for annotation table | Table in .csv format | ||||
| Upload annotation table | Table in .csv format | |||||
| Output | Reporting | Pdf file listing the successive methods, parameters, outputs and results of spectra treatment applied | ||||
| Parameters | Json file Registration of methods and parameters selected during spectra analysis |
|||||
| Intensity matrix | Excel file Matrix table inventorying peak list and respective intensities |
|||||
| Figures | Svg files A zip file contain all the graphs during the process |
|||||
| HDF5 | HDF5 File(b) Registration of imported raw spectra, averaged spectra, annotated table and parameters |
|||||
| All files | Zipped file Contain all the previous outputs |
* Method used by default in MSProfileR tool. (a) A human-readable file format stores previous entered parameters by default. (b) HDF5 file is a special file with a database function to store all the raw data introduced during the analysis. ESD, extreme studentized deviation; HDF5, Hierarchical Data Format version 5; kDa, kilo Dalton; MAD, median absolute deviation; PQN, Probabilistic Quotient Normalization; RC, Rousseeuw and Croux; SNIP, Sensitive Nonlinear Iterative Peak; SNR, signal-to-noise ratio; min, minimum; Sqrt, square root method; TIC, Total-Ion-Current.
Table 2.
Overview of arthropod species selected from “dataset N°1” for MS spectra analysis, including reference list for breeding or samples preparation.
Table 2.
Overview of arthropod species selected from “dataset N°1” for MS spectra analysis, including reference list for breeding or samples preparation.
| Family | Species | Origin (country) | Developmental stage | Body part$ | Number of specimens* | Sample preparation | Rearing conditions |
|---|---|---|---|---|---|---|---|
| Culicidae | An. coluzzii | Dakar (Senegal) | Adult | Legs | 4 | (27862981) | |
| Thorax | 4 | (30390691) | (35773735) | ||||
| Larvae | Whole | 4 | (25442218) | ||||
| Ae. aegypti (Bora strain) | French Polynesia (France) | Adult | Legs | 4 | (27862981) | ||
| Thorax | 4 | (30390691) | (35773735) | ||||
| Larvae | Whole | 4 | (25442218) | ||||
| Ae. albopictus | Marseille (France) | Adult | Legs | 4 | (27862981) | ||
| Thorax | 4 | (30390691) | (35773735) | ||||
| Larvae | Whole | 4 | (25442218) | ||||
| Ixodidae | Rh. sanguineus | Southern France (France) |
Adult | Legs | 4 | (31622384) | (23040662) |
| Am. variegatum | (Senegal) | Adult | Legs | 4 | (31622384) | (26051210) | |
| Siphonaptera (Pulicidae) | Ct. felis | Bristol (UK) | Adult | Cephalothorax | 4 | (31622384) | (29451890) |
| Total# | 48 |
$For larval stage the totality of the specimen was submitted to MS analysis. *Number of specimens submitted to MS analysis. #As each specimen were loaded in quadruplicate on MS plate the total number is 192 MS spectra. Ae. aegypti; Am. amblyomma; An. anopheles; Ct. ctenocephalides; Rh. rhipicephalus.
Table 3.
Overview of Culex mosquito species selected to compose the “dataset N°2” of MS spectra*.
| Genus | Subgenera | Species | Number of specimens$ | Number of specimens included in the reference MS DB per body part (Thoraxes/Legs)§ |
|---|---|---|---|---|
| Culex (Cx.) | Culex (Cux.) | Culex declarator | 7 | 2 / 1 |
| Culex nigripalpus | 12 | 3 / 2 | ||
| Culex quinquefasciatus | 34 | 4 / 4 | ||
| Culex usquatus | 22 | 4 / 4 | ||
| Melanoconion (Mel). | Culex adamesi | 1 | 1 / 1 | |
| Culex dunni | 30 | 4 / 3 | ||
| Culex eastor | 3 | 1 / 1 | ||
| Culex idottus | 2 | 1 / 0 | ||
| Culex pedroi | 15 | 4 / 2 | ||
| Culex portesi | 28 | 4 / 1 | ||
| Culex rabanicolus | 5 | 2 / 2 | ||
| Culex spissipes | 9 | 3 / 2 | ||
| Culex. phlogistus | 1 | 1 / 1 | ||
| Total | 169# | 34/24 |
*Dataset obtained from Costa et al. (36977169). §Number of specimens used to create the reference MS database per body part according to Costa et al. (36977169). $Number of specimens submitted to MS analysis. #As two body parts (legs and thoraxes) from each specimen were loaded in quadruplicate on MS plate, the total number is 1352 MS spectra. Cx., Culex; Cux., Culex; Mel., Melanoconion.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



