
Submitted:
15 February 2024
Posted:
16 February 2024
You are already at the latest version
Abstract
Stimulation of the alpha 7 nicotinic acetylcholine receptor (α7nAChR) has shown beneficial effects in several acute inflammatory disease models. This study aims to examine whether treatment with the selective α7nAChR agonist PHA 568487 can dampen inflammation and thereby improve cardiac function after myocardial infarction in mice. The possible anti-inflammatory properties of α7nAChR agonist PHA 568487 were tested in vivo using the air pouch model and in a permanent occlusion model of acute myocardial infarction in mice. Hematology parameters and cytokine levels were determined. Infarct size and cardiac function were assessed via echocardiography 24 h and one week after the infarction. Treatment with α7nAChR agonist PHA 568487 decreased 12 (CCL27, CXCL5, IL6, CXCL10, CXCL11, CXCL1, CCL2, MIP1a, MIP2, CXCL16, CXCL12 and CCL25) out of 33 cytokines in the air pouch model of acute inflammation. However, α7nAChR agonist PHA 568487 did not alter infarct size, ejection fraction, cardiac output or stroke volume at 24 h nor at 7 days after the myocardial infarction compared with control mice. In conclusion, despite promising immunomodulatory effects in the acute inflammatory air pouch model, α7nAChR agonist PHA 568487 did not affect infarct size or cardiac function after a permanent occlusion model of acute myocardial infarction in mice. Consequently, this study does not strengthen the hypothesis that stimulation of the α7nAChR is a future treatment strategy for acute myocardial infarction when reperfusion is lacking. However, whether other agonists of the α7nAChR can have different effects remains to be investigated.
Keywords:
alpha 7 nicotinic acetylcholine receptor
; α7nAChR
; myocardial infarction
; inflammation
; α7nAChR agonist
1. Introduction
The rupture of an atherosclerotic plaque and the subsequent formation of a thrombus in a coronary artery represents the major mechanism of acute myocardial infarction [1,2]. The thrombotic occlusion will cause the cardiomyocytes in the ischemic myocardium to die, which results in an immense inflammatory reaction [3]. A balanced inflammatory process is crucial for the repair and recovery of the myocardium, but excessive inflammation can damage the heart and contribute to heart failure. Therefore, novel pharmacological interventions that can target specific components of the inflammatory response has been suggested [4−6]. The cholinergic anti-inflammatory pathway describes a connection between the autonomic nervous system and the immune system. Experimental studies have shown that stimulation of the Vagus nerve, the main component of the parasympathetic nervous system, can suppress the production of proinflammatory cytokines [7]. Hence, electrical and pharmacological stimulation of the vagal nerve has been investigated in several inflammatory diseases such as atherosclerosis, myocardial infarction and rheumatoid arthritis [8,9]. The exact mechanism whereby stimulation of the vagal nerve exerts its immunomodulatory effects remains unknown, but accumulating data shows that stimulation leads to the release of acetylcholine that binds to the alpha 7 nicotinic receptor (α7nAChR). The α7nAChR is expressed on immune cells, hence, several specific agonists have been developed to investigate whether stimulation of the α7nAChR can have immunomodulatory effects [10,11]. Our group have recently shown that stimulation of α7nAChR with the agonist AZ6983 decreases atherosclerosis via immunomodulatory effects [12]. Two previous myocardial infarction studies have shown that treatment with PNU-120596 (positive allosteric modulator of the α7nAChR) and PNU-282927 (selective α7nAChR agonist) can decrease both the infarct size and the production of proinflammatory cytokines after myocardial infarction in rats using a reperfusion injury model [13,14]. In line with this, α7nAChR knockout mice have been shown to get increased infarct size and impaired cardiac function after myocardial infarction [15]. However, whether stimulation of the α7nAChR with an agonist can affect the cardiac function following a myocardial infarction induced by permanent occlusion in mice remains to be investigated. Therefore, the aim of this study is to examine whether treatment with the selective α7nAChR agonist PHA 568487 can dampen inflammation and thereby reduce infarct size and improve cardiac function following a myocardial infarction.
2. Results
2.1. α7nAChRAgonist PHA 568487 Dampens Inflammation in the Air Pouch Model
To determine the dose and possible anti-inflammatory effect of α7nAChR agonist PHA 568487 we used the acute inflammatory air pouch model [16]. In the air pouch model, treatment with high dose PHA 568487 (50 mg/kg) significantly decreased the concentration of 12 cytokines compared to control mice (Figure 1A,B). After six hours of inflammation, IL6, CCL27, CXCL5, CXCL10 (IP-10), CXCL11, CXCL1 (GRO1), CCL2 (MCP-1), MIP1a, CXCL2 (MIP2), CXCL16, CXCL12 (SDF-1) and CCL25 were significantly decreased in the group receiving the high dose PHA 568487. Treatment with low dose PHA 568487 (5 mg/kg) did not affect the concentration of cytokines. There was no significant difference in immune cell count of the air pouch exudate (Figure 1C).
2.2. α7nAChR is Expressed in Primary Cardiomyocytes
To investigate a possible role for the α7nAChR in the heart, we first examined α7nAChR expression level in the heart. Using droplet digital PCR (ddPCR) gene expression of the gene coding for α7nAChR, Chrna7, in isolated primary cardiomyocytes was confirmed (Figure 1D).
2.3. No Effect of the α7nAChR Agonist PHA 568487 in the Permanent Occlusion Model of Myocardial Infarction
Next, we aimed to investigate whether the α7nAChR agonist PHA 568487 have a beneficial effect on cardiac function following a myocardial infarction. The mean mortality rate was 32% (37% in the PHA group, 40% in the Control group and 0% in the Sham group).
Cardiac function was assessed using echocardiography at 24 h and 7 days post myocardial infarction. No difference was seen in either myocardial infarct size, ejection fraction, cardiac output or left ventricular mass when comparing mice treated with PHA 568487 (50 mg/kg) with saline treated controls (Figure 2A,B). Furthermore, there was no difference between the groups in terms of heart rate, left ventricular volume, stroke volume, cardiac output, fractional shortening, fractional area change and left ventricular mass (Table 1). When investigating the cardiac parameters over time, from the 24 h time point to the 7 days time point, control mice recovered in terms of infarct area, as well as in cardiac output, an effect that was not seen in mice receiving PHA 568487 (Figure 2A). PHA568487 treated mice on the other hand increased in left ventricular weight (Figure 2A) assessed by echocardiography. In sham mice, there was no significant difference in cardiac function 24h post surgery (control vs PHA, ejection fraction: 70 ± 6.6 vs 60.3 ± 17.6, cardiac output 13.3 ± 1.3 vs 12.7 ± 2.5; left ventricular mass 82.5 ± 6.6 vs 99.3 ± 14.9, n = 3 in each group) nor at one week post sham surgery (control vs PHA, ejection fraction 71.7 ± 7.5 vs 58.4 ± 12:, cardiac output 16 ± 3.5 vs 12.6 ± 2.4; left ventricular mass 106.7 ± 25.7 vs 103.8 ± 15.3, n = 3 in each group).
3. Discussion
In this study we found a clear anti-inflammatory effect of the α7nAChR agonist PHA 568487 in the air pouch model of acute inflammation. However, despite this clear anti-inflammatory effect in the acute model, treatment with the α7nAChR agonist PHA 568487 did not alter infarct size or cardiac function at 24 h or one week post myocardial infarction.
Recent studies have shown that treatment with the α7nAChR agonists GTS-21 and PNU-282927, can acutely decrease infarct size and the production of serum proinflammatory cytokines after myocardial infarction in rats [17,18], where histological assessments revealed a decreased infarct area in the rats treated with GTS-21 and PNU-282987 1.5 and 2.5 h post infarction, respectively. Importantly, in these two studies the ischemia-reperfusion (I/R) model of myocardial infarction was used whereas in the current study we used a permanent occlusion model of myocardial infarction. The I/R method is considered to reflect the clinical setting of acute myocardial infarction, where restoration of blood flow plays a key role in the treatment strategy [19]. Importantly, permanent coronary occlusion is also a relevant animal model of acute myocardial infarction since it echoes patients who, due to contraindications or logistic issues, do not receive timely or successful reperfusion [20]. Hence, both models contribute with important information in different clinical settings. Nevertheless, taken together, these studies indicates that stimulation of the α7nAChR is beneficial following an ischemia reperfusion injury, however, not as favourable in the permanent occlusion model.
The effect of α7nAChR stimulation on cardiac function after myocardial infarction has been less studied. In a model of ischemic cardiomyopathy, Lin et al. [21] reported cardioprotective effects when rats received daily treatment with α7nAChR agonist PNU-292987 for four weeks, demonstrating an improved ejection fraction in rats receiving α7nAChR agonist PNU-292827. Furthermore, mice lacking the α7nAChR display an increased infarct size and decreased ejection fraction four weeks post acute myocardial infarction compared with control mice [15]. These studies are in contrast with the current study where we do not detect any difference in the functional parameters. Rather, control mice recover better, with decreased infarct size and improved CO one week after the myocardial infarction, an effect not seen in PHA 568487 treated mice. The discrepancy between these findings needs further evaluation but could potentially be due to the different models used as well as the different treatment strategies, both in terms of agonist used as well as the regimen of therapy.
The air pouch model is broadly used model of acute inflammation to identify potential anti-inflammatory drugs in inflammatory diseases [16,22,23,24]. Using the air pouch model we determined the dose of α7nAChR agonist PHA 568487. Interestingly, out of the 12 cytokines significantly decreased by the α7nAChR agonist PHA 568487, besides from IL-6, all of the other cytokines are involved in recruitment, i.e., acting chemotactic for neutrophils (CXCL1, CXCL2) [25], monocytes (CXCL10, CCL2, CCL25) as well as for T cells (CCL27, CXCL10) [26]. Despite the dampening effect of α7nAChR agonist PHA 568487 on several different chemokines, this did not alter the number of recruited cells to the air pouch.
Although, α7nAChR agonist PHA 568487 suppressed cytokine levels in the air pouch model, no differences were found in white blood cell count or in the concentrations of cytokines and chemokines when analysing the blood from the day of sacrifice. Possibly, the suppression of cytokines by PHA 568487 is more local, as seen in the air pouch experiments, and not a systemic effect detected in blood. Another possible explanation is the fact that the sacrifice took place six days after the final injection of the drugs and that the blood concentration of immune cells and cytokines could have time to go back to normal during this time. A more intense treatment protocol would perhaps show a different result.
In summary, treatment with the α7nAChR agonist PHA 568487 did not affect infarct size or cardiac function after a permanent occlusion model of acute myocardial infarction in mice. Consequently, this study does not strengthen the hypothesis that stimulation of the α7nAChR is a promising future treatment strategy for acute myocardial infarction when reperfusion is lacking. Possibly, targeting the α7nAChR in ischemia-reperfusion injury might have beneficial effects suggested by the previous acute studies, however, whether it also has an effect in a more long-term study needs to be further evaluated. In addition, we cannot rule out that other agonists of the α7nAChR can have different effects, hence, this remains to be investigated.
4. Materials and Methods
4.1. Isolation of Primary Cardiomyocytes and Droplet Digital pcr (ddpcr) Analysis
Isolation of primary cardiomyocytes from C57Bl/6N male mice (Taconic, Borup, Denmark) was carried out as previously described [27]. RNA from cardiomyocytes (n = 7) was extracted using the RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol. Reverse transcription was performed using the High-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA) according to the manufacturer’s protocol. The expression of the α7nAChR was determined with droplet digital pcr (ddpcr) utilizing EvaGreen (Bio-Rad Laboratories, CA, USA) as previously described [28]. Samples were analyzed in the QX200 Droplet Reader (Bio-Rad Laboratories, CA, USA) and the results, including positive and negative droplets, analyzed with QuantaSoft Analysis Pro™ (Bio-Rad Laboratories).
4.2. Air Pouch Model
To evaluate the possible immunomodulatory effects of PHA 568487 in vivo, the air pouch model of acute inflammation was used [12,24]. At 8 weeks of age, male C57BL/6JRj mice (Janvier Labs, Le Genest-Saint-Isle, France) were injected with 3 mL of sterile air between the scapulae under isoflurane anaesthesia. Three days later, the air pouch was re-filled with 3 mL of sterile air. On the sixth day, the mice received an intraperitoneal injection of either PBS (Thermo Fisher Scientific, Waltham, MA, USA) or PHA 568487 (Tocris Bioscience, Ellisville, MO, USA) at 12.4 μmol kg-1 (5 mg/kg) or 123.4 μmol kg-1 (50 mg/kg). 15 min later, all mice were injected with LPS (10 μg, List Biological Laboratories Inc., Campbell, USA) directly into the pouch. The mice were sacrificed six hours after the LPS-injection by an overdose of pentobarbital (Apoteket AB, Stockholm, Sweden). Finally, the pouch cavity was flushed with 2 mL of PBS with 1.6 mmol L-1 EDTA and the exudate was collected. White blood cell count of the exudate was analysed using the Vetscan HM5 Hematology Analyzer (Abaxis, Union City, USA) and thereafter centrifuged for 5 min at 400g. Supernatants were stored at -80 °C until Multiplex analysis.
4.3. Myocardial Infarction Study
A total of 47 mice were included in the myocardial infarction study, performed at two separate time points. Twelve-week-old, male C57BL/6JRj mice (n = 47) were purchased from Taconic (Denmark) and randomly assigned into two groups: Control (receiving PBS, n = 21) and PHA (receiving PHA 568487, n = 20). A permanent left anterior descending artery (LAD) occlusion was performed as previously described [29]. Each treatment group had a corresponding sham group (n = 3+3) undergoing the same surgical protocol with the exception of LAD ligation. Five minutes before the surgery, the mice received an intraperitoneal injection of either PBS (control group) or PHA 568487 (123.4 μmol kg-1, 50 mg/kg). The chest of each mouse was shaved. Further, the mice were anesthetized, orally intubated and connected to a small animal ventilator distributing a mixture of oxygen and 2-3% isoflurane. The mice were placed on a heating pad (Rodent Surgical Monitor, Scintica, London, Canada) where ECG was continuously measured. A rectal probe measuring temperature was applied and core temperature was maintained between 36.5-37.5 degrees Celsius. An incision was made between the 4th and 5th ribs to reveal the left ventricle. The pericardium was opened and myocardial infarction was induced by ligation of the left anterior descending coronary artery immediately after the bifurcation. Successful ligation was confirmed by characteristic ECG changes and akinesia of the ventricular wall. The lungs were hyperinflated, positive end expiratory pressure was applied and the chest was closed. The mice received an intraperitoneal injection of Temgesic (0.1 mg/kg) to relieve postoperative pain and were allowed to recover spontaneously after turning off the isoflurane administration. The mean mortality rate was 32% (37% in the PHA group, 40% in the Control group and 0% in the Sham group). The mice that did not survive the surgery all died within 10 min of chest closure. Twenty-four hours after the myocardial infarction, the mice received a second intraperitoneal injection of either PBS or PHA 568487 (123.4 μmol kg-1, 50 mg/kg). Mice were sacrificed 7 days after the myocardial infarction by an overdose of isoflurane. Heart tissue was perfused with saline, subsequently cooled in isopentane, snap-frozen in liquid nitrogen and stored at -80 °C until analysis. Blood was collected and analyzed using the Vetscan HM5 Hematology Analyzer (Abaxis, Union City, USA) and thereafter centrifuged for 5 min at 400g. Supernatants were stored at -80 °C until Multiplex analysis.
4.4. Echocardiographic Measurements in Mice
Mice were anesthetized with isoflurane (1.2%) and underwent echocardiographic examination at two different time points: 24 h and 7 days after the myocardial infarction. Echocardiography was performed using VEVO 2100 (VisualSonics Ontario, Canada), including an integrated rail system for consistent positioning of the ultrasound probe as previously described [29]. In brief: The chest of each mouse was shaved, the mice were placed on a heating pad and their paws connected to electrocardiographic (ECG) electrodes. A 45-MHz linear transducer (RMV 704) was used. An optimal parasternal long-axis cine loop was acquired to image the mitral and aortic valves and the maximum distance between the aortic valve and the cardiac apex. Parasternal short-axis cine loops were acquired at 1, 3, and 5 mm below the mitral annulus. End-diastolic and end- systolic left ventricular volumes and ejection fraction were calculated with the biplane Simpson’s formula using the 3 parasternal short-axis views and the parasternal long-axis view. For M-mode measurements (at the 3-mm level), the leading-edge method was used. End-diastole was defined at the onset of the QRS complex, and end-systole was defined as the time of peak inward motion of the interventricular septum. Infarct size was assessed based on wall motion score index (WMSI) [30] 24 h and 1 week after myocardial infarction by a 16-segments model on 3 short axis images, as 0 for normal, 0.5 for reduced wall thickening and excursion in a segment and 1 for no wall thickening and excursion in a segment. WMSI was calculated as the sum of scores divided by the total number of segments. The stored data was evaluated offline in a blinded fashion with Vevo Lab software (VisualSonics).
4.5. Histological Analysis of Myocardial Infarction Size
Frozen hearts were embedded in OCT and cryosectioned in 10µm sections. Sectioning begun from the apex and sections were collected every 100 µm up until 700 µm. Sections were stained with Masson-Goldner staining kit followed by Weigerts hematoxylin (both from Sigma-Aldrich, Merck KgaA, Darmstadt, Germany) according to the manufacturers protocol. Infarction area was calculated as percentage of cardiac tissue for each level and presented as the average infarction area per heart.
4.6. Multiplex Analysis
The Bio-Plex ProTM Mouse Chemokine Panel 33-plex (#12002231, Bio Rad Laboratories, Inc. Hercules, USA) and the Bio-Plex Pro Mouse Cytokine 23-plex Assay (#M60009RDPD, Bio Rad Laboratories) was used to measure cytokines in the exudate from the air pouch model and serum cytokine levels in mice from the myocardial infarction study, respectively. The assays were performed according to the manufacturers’ protocol as previously described [12].
4.7. Ethical Considerations
All procedures involving mice were approved by the Regional Animal Ethics Committee at the University of Gothenburg, in accordance with the European Communities Council Directives of 22 September 2010 (2010/63/EU).
4.8. Statistics
Data were tested for normality with Shapiro-Wilk’s test and statistical methods chosen according to the outcome. The data from the air pouch experiment, where control group and two different concentrations of PHA 568487 were compared, were analysed with the Kruskal-Wallis test followed by Holm- Sidak’s multiple comparison test. The data from the myocardial infarction study (echocardiographic measurements, multiplex assays, white blood cell count) were analysed with Friedmans test followed by Dunn’s multiple comparison test when comparing 24h and 1 week and with Mann-whitneys test when comparing Controls with the PHA group. Data is expressed as mean ± SEM unless otherwise stated and p < 0.05 was considered statistically significant.
Author Contributions
Conceptualization, FM, ML and MEJ.; Methodology, AM, ML, FM, MEJ.; Validation, AM, ML.; Formal Analysis, FM, AM.; Investigation, FM, AM.; Resources, ML, MEJ.; Data Curation, FM.; Writing—Original Draft Preparation, FM, MEJ.; Writing—Review & Editing, FM, AM, ML and MEJ.; Visualization, FM.; Supervision, MEJ.; Project Administration, FM.; Funding Acquisition, MEJ.
Funding
This work was supported by the Swedish Research Council, the Swedish Heart-Lung Foundation, Mary von Sydow foundation, Stiftelsen Gamla tjänarinnor, Stiftelsen Tornspiran, Dr. Felix Neuberghs Foundation, Wilhelm and Martina Lundgren foundation and Grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (ALF GBG-723131).
Acknowledgments
The authors would like to thank Zeinab Mohammed Ali for the help with sectioning and staining of the cardiac tissues.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; Mickley, H.; Crea, F.; Van De Werf, F.; et al. Fourth universal definition of myocardial infarction (2018). Eur. Heart J. 2019, 40, 237–269. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. New Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amelia, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, N.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Fields, W.C.; Rocha-Resende, C.; Resende, R.R.; Guatimosim, S.; Prado, V.F.; Gros, R.; Prado, M.A. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 5072–5082. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef]
- Ulleryd, M.A.; Mjornstedt, F.; Panagaki, D.; Yang, L.J.; Engevall, K.; Gutierrez, S.; Wang, Y.; Gan, L.M.; Nilsson, H.; Michaelsson, E.; et al. Stimulation of alpha 7 nicotinic acetylcholine receptor (alpha7nAChR) inhibits atherosclerosis via immunomodulatory effects on myeloid cells. Atherosclerosis 2019, 287, 122–133. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.Z.; Zhan, J.; He, X.H.; Song, X.M.; Wang, Y.L. Protective effect of PNU-120596, a selective alpha7 nicotinic acetylcholine receptor-positive allosteric modulator, on myocardial ischemia-reperfusion injury in rats. J. Cardiovasc. Pharmacol. 2012, 59, 507–513. [Google Scholar] [CrossRef]
- Xiong, J.; Yuan, Y.J.; Xue, F.S.; Wang, Q.; Cheng, Y.; Li, R.P.; Liao, X.; Liu, J.H. Postconditioning with alpha7nAChR agonist attenuates systemic inflammatory response to myocardial ischemia--reperfusion injury in rats. Inflammation 2012, 35, 1357–1364. [Google Scholar] [CrossRef]
- Fang, J.; Wang, J.; Chen, F.; Xu, Y.; Zhang, H.; Wang, Y. α7nAChR Deletion Aggravates Myocardial Infarction and Enhances Systemic Inflammatory Reaction via mTOR-Signaling-Related Autophagy. Inflammation 2019, 42, 1190–1202. [Google Scholar] [CrossRef]
- Smith, C.J.; Zhang, Y.; Koboldt, C.M.; Muhammad, J.; Zweifel, B.S.; Shaffer, A.; Talley, J.J.; Masferrer, J.L.; Seibert, K.; Isakson, P.C. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. United States Am. 1998, 95, 13313–13318. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Zhou, Y.; Yang, H.; Liu, Y.; Mao, X.; Qin, X.; Li, X.; Zhang, X.; Hu, Y. Alpha7 nicotinic acetylcholine receptor activation protects against myocardial reperfusion injury through modulation of autophagy. Biochem. Biophys. Res. Commun. 2018, 500, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, S.A.; Khan, N.S.; Levy, A.C.; Faliks, B.T.; Sison, C.P.; Pavlov, V.A.; Zhang, Y.; Ojamaa, K. Nicotinic acetylcholine receptor-mediated protection of the rat heart exposed to ischemia reperfusion. Mol. Med. 2017, 23, 120–133. [Google Scholar] [CrossRef]
- Xu, H.; Li, J.; Zhang, L.; Li, N.; Su, S.; Ye, Z.; Xu, Y. Decreased α7nAChR mRNA levels in peripheral blood monocytes are associated with enhanced inflammatory cytokine production in patients with lupus nephritis. Biomed. Pharmacother. 2019, 111, 359–366. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Bolli, R.; Canty, J.M.; Jr. ; Du, X.J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H812–h838. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.H.; Li, Y.C.; Wu, S.J.; Zheng, C.; Lin, Y.Z.; Lian, H.; Lin, W.Q.; Lin, J.F. Eliciting α7-nAChR exerts cardioprotective effects on ischemic cardiomyopathy via activation of AMPK signalling. J. Cell. Mol. Med. 2019, 23, 4746–4758. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Diomede, L.; Sironi, M.; Massimiliano, L.; Sottocorno, M.; Polentarutti, N.; Guglielmotti, A.; Albani, D.; Bruno, A.; Fruscella, P.; et al. Inhibition of monocyte chemotactic protein-1 sysnthesis by statins. Lab. Investig. 2000, 80, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Naime, D.; Ostad, E. The antiinflammatory mechanism of methotrexate: Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Investig. 1993, 92, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.; Sedgwick, A.D.; Willoughby, D.A. The formation of a structure with the features of synovial lining by subcutaneous injection of air: An in vivo tissue culture system. J. Pathol. 1981, 134, 147–156. [Google Scholar] [CrossRef]
- Sawant, K.V.; Sepuru, K.M.; Penaranda, B.; Lowry, E.; Garofalo, R.P.; Rajarathnam, K. Chemokine Cxcl1-Cxcl2 heterodimer is a potent neutrophil chemoattractant. J. Leukoc. Biol. 2023, 114, 666–671. [Google Scholar] [CrossRef]
- David, B.A.; Kubes, P. Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol. Rev. 2019, 289, 9–30. [Google Scholar] [CrossRef]
- Mardani, I.; Tomas Dalen, K.; Drevinge, C.; Miljanovic, A.; Ståhlman, M.; Klevstig, M.; Scharin Täng, M.; Fogelstrand, P.; Levin, M.; Ekstrand, M.; et al. Plin2-deficiency reduces lipophagy and results in increased lipid accumulation in the heart. Sci. Rep. 2019, 9, 6909. [Google Scholar] [CrossRef] [PubMed]
- Pattanaik, B.; Hammarlund, M.; Mjörnstedt, F.; Ulleryd, M.A.; Zhong, W.; Uhlén, M.; Gummesson, A.; Bergström, G.; Johansson, M.E. Polymorphisms in alpha 7 nicotinic acetylcholine receptor gene, CHRNA7, and its partially duplicated gene, CHRFAM7A, associate with increased inflammatory response in human peripheral mononuclear cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22271. [Google Scholar] [CrossRef]
- Drevinge, C.; Dalen, K.T.; Mannila, M.N.; Täng, M.S.; Ståhlman, M.; Klevstig, M.; Lundqvist, A.; Mardani, I.; Haugen, F.; Fogelstrand, P.; et al. Perilipin 5 is protective in the ischemic heart. Int. J. Cardiol. 2016, 219, 446–454. [Google Scholar] [CrossRef]
- Zhang, Y.; Takagawa, J.; Sievers, R.E.; Khan, M.F.; Viswanathan, M.N.; Springer, M.L.; Foster, E.; Yeghiazarians, Y. Validation of the wall motion score and myocardial performance indexes as novel techniques to assess cardiac function in mice after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1187–1192. [Google Scholar] [CrossRef]
Figure 1.
PHA 568487 decreases the concentration of pro-inflammatory cytokines in the air pouch model of inflammation. Acute immunomodulatory properties of PHA 568487 were tested using the air pouch model of inflammation (A–C). Mice were treated with PHA 568487 12.4 μmol kg-1 (5 mg/kg) or PHA 568487 123.4 μmol kg-1 (50 mg/kg) i.p. and subsequently challenged with LPS into the pouch. Concentrations of cytokines in the air pouch-exudate was analysed after 6 h by Multiplex assay and presented as a heat map (A) and graphs representing cytokines that were significantly different compared to the controls (B). Immune cell count were measured with Vetscan HM5 Hematology Analyzer (C). Absolute quantification of Chrna7 mRNA expression using droplet digital PCR in primary cardiomyocytes from adult mice (n= 7). Kruskal-Wallis test was used and data expressed as mean ± SEM. *p < 0.05, **p < 0.01, n = 8–12 per group.
Figure 1.
PHA 568487 decreases the concentration of pro-inflammatory cytokines in the air pouch model of inflammation. Acute immunomodulatory properties of PHA 568487 were tested using the air pouch model of inflammation (A–C). Mice were treated with PHA 568487 12.4 μmol kg-1 (5 mg/kg) or PHA 568487 123.4 μmol kg-1 (50 mg/kg) i.p. and subsequently challenged with LPS into the pouch. Concentrations of cytokines in the air pouch-exudate was analysed after 6 h by Multiplex assay and presented as a heat map (A) and graphs representing cytokines that were significantly different compared to the controls (B). Immune cell count were measured with Vetscan HM5 Hematology Analyzer (C). Absolute quantification of Chrna7 mRNA expression using droplet digital PCR in primary cardiomyocytes from adult mice (n= 7). Kruskal-Wallis test was used and data expressed as mean ± SEM. *p < 0.05, **p < 0.01, n = 8–12 per group.
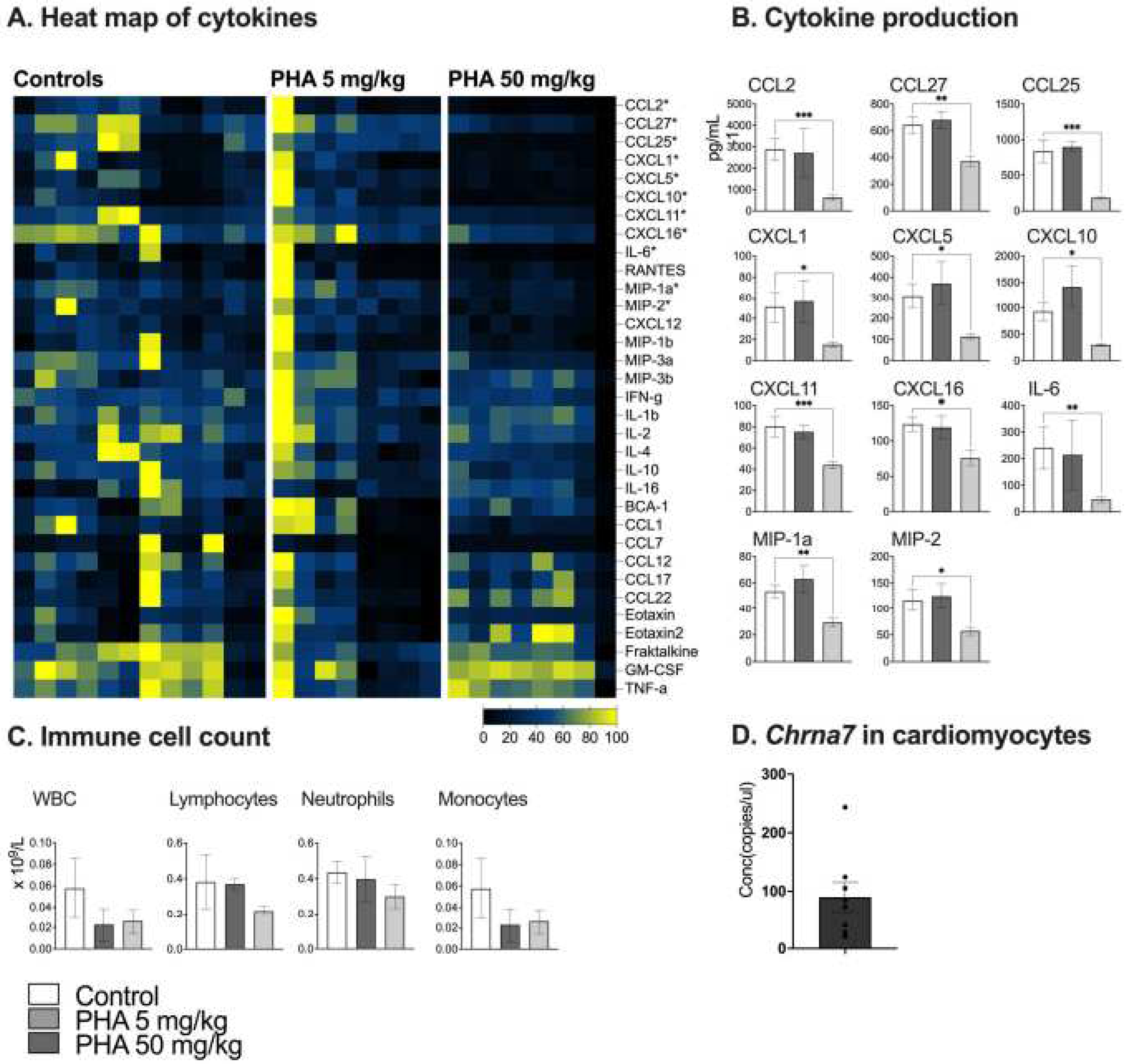
Figure 2.
PHA 568487 did not affect the cardiac function, myocardial infarct size nor the cytokine levels in plasma. Cardiac function and infarct size was assessed via echocardiography 24 hours and one week after the myocardial infarction (A). To verify the echocardiographic measurements, infarct size was also assessed histologically (B).Concentration of 23 cytokines in plasma were analysed at the day of sacrifice via multiplex assay (C). MI size: myocardial infarction size, EF: ejection fraction, CO: cardiac output, LV mass: left ventricular mass. In (A) Friedmans test was used followed by Dunn’s multiple comparison test. In (B) and (C) Mann-whitneys test was used. Data expressed as mean ± SEM. *p < 0.05, **p < 0.01, n = 13 per group.
Figure 2.
PHA 568487 did not affect the cardiac function, myocardial infarct size nor the cytokine levels in plasma. Cardiac function and infarct size was assessed via echocardiography 24 hours and one week after the myocardial infarction (A). To verify the echocardiographic measurements, infarct size was also assessed histologically (B).Concentration of 23 cytokines in plasma were analysed at the day of sacrifice via multiplex assay (C). MI size: myocardial infarction size, EF: ejection fraction, CO: cardiac output, LV mass: left ventricular mass. In (A) Friedmans test was used followed by Dunn’s multiple comparison test. In (B) and (C) Mann-whitneys test was used. Data expressed as mean ± SEM. *p < 0.05, **p < 0.01, n = 13 per group.
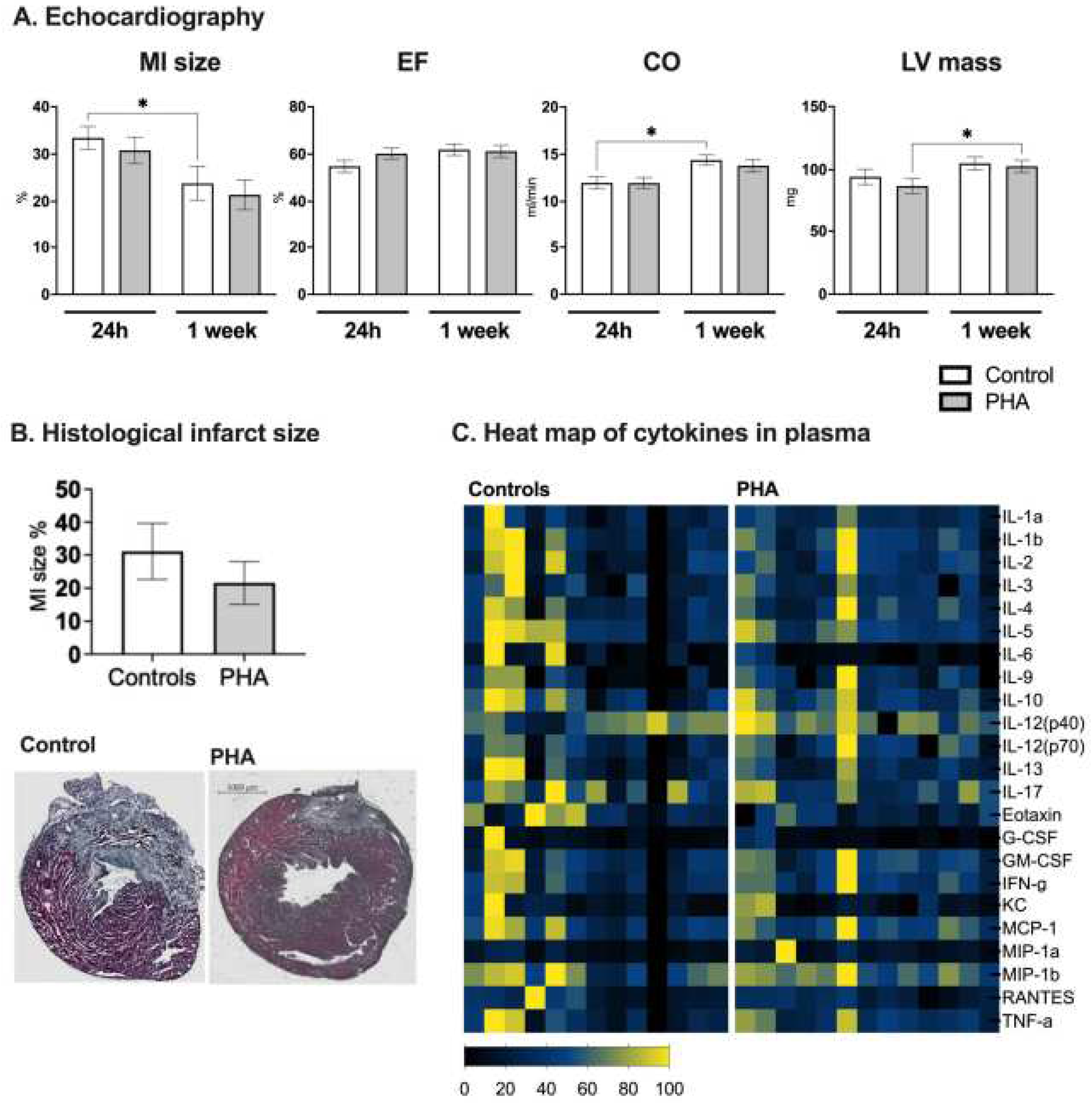

Table 1.
Cardiac and haematological parameters 24 h and 7 days post MI.
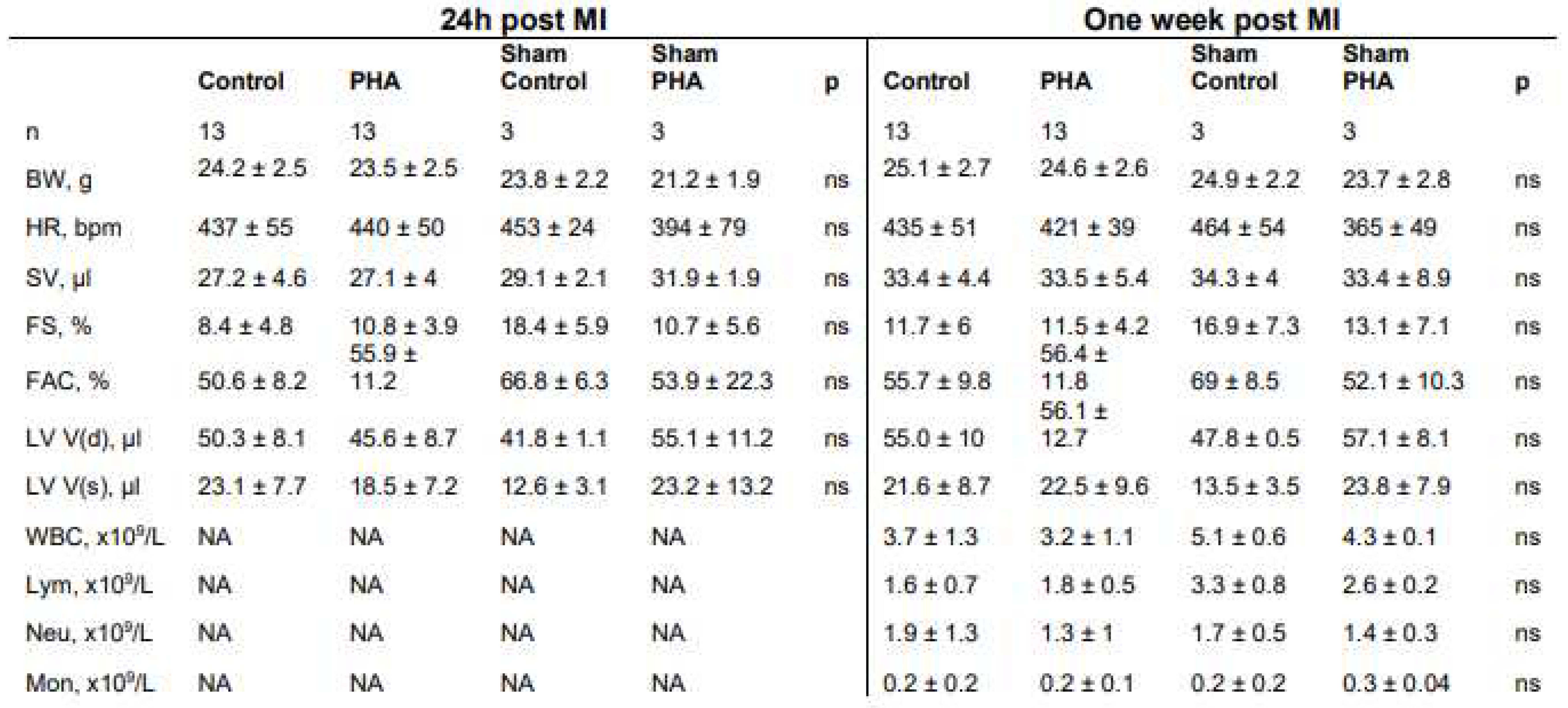 |
Table 1. Echocardiographic measurements and immune cell populations Treatment with PHA 568487 did not affect echocardiographic measurements of the heart 24 hours and 7 days after myocardial infarction. No difference in immune cell population in blood at the day of sacrifice. Body weight (BW), heart rate (HR), stroke volume (SV), fractional shortening (FS), fractional area change (FAC), left ventricular volume in diastole (LV V(d)), left ventricular volume in systole (LV V(s)), white blood cell count (WBC), lymphocyte count (Lym), neutrophil count (Neu), monocyte count (Mon). Mann whitney’s test was used to compare control and PHA group at the two different time points. Values represented as mean ± SEM.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



