
Submitted:
15 February 2024
Posted:
16 February 2024
You are already at the latest version
Abstract
Breast cancer develops upon sequential acquisition of driver mutations in mammary epithelial cells; however how these mutations collaborate to transform normal cells remains unclear in most cases. We aimed to reconstitute this process in a particular case. To this end, we combined the activated form of the PI 3-kinase harboring the H1047R mutation with the inactivation of the histone lysine methyl-transferase KMT2D in the non-tumorigenic human mammary epithelial cell line MCF10A. We found that PI 3-kinase activation promoted cell cycle progression, especially when growth signals were limiting, as well as cell migration, both in a collective monolayer and as single cells. Furthermore, we showed that KMT2D inactivation had relatively little influence on these processes, except for single cell migration that KMT2D inactivation promoted in synergy with PI 3-kinase activation. Combination of these two genetic alterations induced expression of the ARPC5L gene that encodes a subunit of the Arp2/3 complex. ARPC5L depletion fully abolished the enhanced migration persistence exhibited by double-mutant cells. Our reconstitution approach in MCF10A has thus revealed both the cell function, single cell migration, and the underlying Arp2/3-dependent mechanism, which are synergistically regulated when KMT2D inactivation is combined with the activation of the PI 3-kinase.
Keywords:
KMT2D
; MLL2
; MLL4
; PIK3CA
; Arp2/3
Introduction
Cancer develops when cells start proliferating independently from signals that normally regulate their growth in the body. A solid tumor, a mass that forms in one of our organs, represents the progeny of a single cancer cell and can ultimately interfere with proper functioning of that organ [1]. Most human cancers are derived from epithelial cells and are referred to as carcinomas, the most frequent of which are mammary carcinomas [2]. Breast cancer is not a fatal disease, as long as the tumor is restricted to this non-essential organ. Mammary carcinomas are typically removed by surgery. However, mammary carcinoma cells usually end up disseminating into the organism in the process of metastasis and seed secondary tumors in other organs, such as the brain, the liver, lungs and bones, where their surgical removal is much more difficult [3]. Moreover, secondary tumors can be numerous and scattered throughout the body. As a consequence, chemotherapy that delivers drugs throughout the body remains the only option to treat patients with metastases.
Tumor sequencing has been a major breakthrough in our understanding of the origin of cancers [4]. Indeed, it showed that some genes were frequently mutated in tumors, thus suggesting their critical role in cancer progression. One of such genes, PIK3CA, encodes the catalytic subunit of the PI 3-kinase α and is frequently mutated in various carcinomas [5], and especially in breast cancer, where it is mutated in about one third of the cases [6]. The vast majority of PIK3CA mutations are substitutions clustered at two well-defined positions. Such a profile of mutations in tumors is characteristic of oncogenes, which are activated by specific mutations. Since the mutated oncogene product is rendered constitutively active by the mutation, the mutated allele acts as a dominant allele. PI 3-kinase α is an atypical kinase that phosphorylates a lipid rather than proteins. Upon PI 3-kinase mediated phosphorylation, the membrane phospholipid phosphatidylinositol (4,5) bisphosphate (PIP2) is converted into phosphatidylinositol (3,4,5) trisphosphate (PIP3), which activates several downstream pathways, ultimately resulting in enhanced cell proliferation, survival and migration [7,8]. Because of its implication in tumorigenesis, the PI 3-kinase enzyme has been the subject of intense investigation to identify chemical inhibitors. One of these inhibitors, BYL719, also known as Alpelisib, is recommended for the treatment of mammary carcinomas, where PI 3-kinase activity is elevated due to PIK3CA mutations [9,10].
The histone methyl transferase encoding gene KMT2D, also known as MLL2 and MLL4, is also mutated in various cancers [11,12]. In breast cancer, the frequency of KMT2D mutations is about 4 to 5% [13,14]. KMT2D encodes a large enzyme composed of 5537 amino acids. KMT2D mutations are mostly substitutions scattered throughout the length of this long gene. 10 – 20% of mutations truncate the protein, removing in particular its C-terminal catalytic domain. This pattern of mutations is indicative of tumor suppressor genes, where the two alleles must be inactivated for tumors to arise. The tumor suppressor KMT2D, which incorporates into a large multiprotein complex of the COMPASS family, catalyzes the methylation of lysine 4 in histone 3, and thus regulates the expression of genes in an epigenetic manner [11,12].
We previously found KMT2D mutations associated with PIK3CA H1047R mutation in an invasive mammary carcinoma, where these two genes were the only cancer driving genes mutated in this particular tumor [15]. Association of driver mutations in these two cancer-associated genes is present in 1.5% of breast cancer patients in public databases [14]. To examine the potential synergy of these two cancer-associated genes in the transformation process, we combined the activated form of PIK3CA harboring the H1047R mutation with a biallelic knock-out of KMT2D in the non-tumorigenic mammary epithelial cell line MCF10A [16], and studied phenotypic consequences of the two genetic alterations when introduced in isolation or combined.
2. Materials and Methods
2.1. Cells and Drugs
MCF10A cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 20 ng/mL epidermal growth factor, 10 µg/mL insulin, 500 ng/mL hydrocortisone, and 100 ng/mL cholera toxin. PIK3CA H1047R/+ MCF10A clone (HD 101-011) were obtained from Horizon Discovery Ltd. (Cambridge, UK). KMT2D KO and double-mutant MCF10A clones were generated in the laboratory. Cells were incubated at 37°C in 5% CO2. All clones were routinely tested for mycoplasma and found to be negative. BYL719 was purchased from MedChemExpress (HY-15244), dissolved in DMSO and used at 5 µM, if not otherwise stated. The total amount of DMSO in treated or control cultures never exceeded 0.01%.
2.2. Antibodies, Western Blot and Immunofluorescence
Antibodies targeting GAPDH (#2597762) were purchased from Invitrogen, E-cadherin (#MABT26), Laminin-5 (#MAB19562), ARPC1A (HPA004334), ARPC1B (HPA004832) and alpha-tubulin (#T9026-2ML) from Sigma-Aldrich, ARPC5L (ab169763) from Abcam, ARPC5 (#305011, clone 323) from Synaptic Systems.
For Western blot analysis, MCF10A cells were lysed in RIPA buffer. SDS-PAGE was performed using NuPAGE 4-12% Bis-Tris gels (Life Technologies). Nitrocellulose membranes were incubated with HRP- conjugated secondary antibodies and developed with SuperSignal West Femto Maximum sensitivity substrate (Thermo Fisher Scientific).
For the immunofluorescencestaining of 3D structures, anti-mouse conjugated with Alexa Fluor 647 antibodies and anti-rat conjugated with Alexa Fluor 594 secondary antibodies were from Life Technologies. Actin was labeled using Acti-stainTM 488 phalloidin (#PHDG1-A, Tebubio).
2.3. Cas9-Mediated Knock-Out and siRNA-Mediated Knock-Down
To generate KMT2D knock-out (KO), parental MCF10A or PIK3CA H1047R cells were transfected with the gRNA that targets the CAGAGACCTCTCCCACATGT sequence in the second exon of KMT2D (ThermoFischer CRISPR1024276_CR). Cells were transfected with crRNA:tracrRNA duplex and the purified Cas9 protein using Lipofectamine CRISPRMAX™ Cas9 transfection reagent (all from Thermo Fisher Scientific). Cells were then diluted to 0.8 cells/well into 96-well plates. Genomic DNA of clones was screened by PCR using KMT2Dfor primer TCTCATGCGTCTGGTGAGTG and KMT2Drev primer GTAAGGCCCTCAGGGAAACC. Loss of the PciI restriction site in the PCR amplicon revealed the presence of Cas9-generated mutations. In candidate clones, PCR amplicons were sequenced using the Sanger method. In case of overlapping peaks indicating different alleles, PCR products were cloned using the Zero Blunt PCR Cloning Kit (Thermo Fisher Scientific) and resulting plasmids were sequenced to identify the two alleles.
To obtain ARPC5L knock-down, cells were transfected with ARPC5L siRNAs (ON-TARGET smart pool, Dharmacon, L-014690-02-0005) or control non-targeting siRNAs (D-001810-10-20), using lipofectamine RNAiMax (Thermo Fisher Scientific). Image acquisition started at 48 h post-transfection and finished at 72 h.
2.4. qRT-PCR
Total RNA from MCF10A cells was extracted using the NucleoSpin RNA Plus Mini kit for RNA purification (Macherey-Nagel, #740984.250). To generate cDNA, 1 μg of total RNA and Superscript II reverse transcriptase (Life Technologies, Inc., Gaithersburg, MD, #18064-071) were used. qRT-PCR was carried out using Power SYBR Green PCR master mix (Thermo Fisher Scientific, Waltham, MA, USA, #4368708) on the QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). The thermal cycling program was as follows: 95°C for 10 min for the initial denaturation, then 95°C for 15 sec and 65°C for 1 min repeated 50 times for template denaturation and primer annealing and extension respectively. A relative transcript level was calculated using the 2ΔΔCt method. The TBP gene was used as a reference gene for normalization. Nucleotide sequences of the primers used are listed in Table S1.
2.5. Cell Cycle
EdU assays were overall performed as previously described [17]. Briefly, cells were cultivated in the presence of 5% growth factor (GF) containing medium (supplemented with 5% horse serum and 20 ng/mL of the epidermal growth factor [EGF]) and seeded at a density of 104 cells per well in 24-well plates. After adhesion of the cells to the substratum, cells were starved from EGF and serum in the so-called 0% GF media for 32 h and then stimulated for 16 h with fresh medium containing either 0% or 1% GF. EdU was added 1h before cell fixation in 4% paraformaldehyde. Cells were permeabilized in 0.5% Triton X-100 and then labelled using the Alexa Fluor 488 Click-iT EdU Imaging kit (Thermo Fisher Scientific #C10337). The percentage of cells in S-phase was determined as the ratio between EdU-positive nuclei and DAPI-stained nuclei.
2.6.3. D Multicellular Structures
3D multicellular structures were obtained as we have previously described [15]. Briefly, MCF10A cells were seeded on top of gelified Matrigel (CB-40230C, Corning) in Millicell EZ SLIDE 8-well glass chamber slide (PEZGS0816, Millipore) in a medium containing 4 ng/mL EGF and 1% serum and 2% Matrigel. Medium was changed every 3 days for 21 days. Structures were then fixed in 2% PFA in PBS permeabilized with 0.5% Triton X-100, rinsed with PBS/glycine (130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 100mM glycine), blocked in IF buffer (130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 0.1% bovine serum albumin, 0.2% Triton X-100, 0.05% Tween-20) containing 10% fetal bovine serum (FBS) first and then with IF buffer supplemented with 10% FBS and 20 µg/mL goat anti-Rabbit Fc fragment (111-005-046, Jackson ImmunoResearch). Structures were incubated with primary antibodies in the latter buffer, washed with IF buffer and then incubated with secondary antibody in IF buffer + 10% FBS. Then structures were incubated with DAPI (Thermo Fisher Scientific), rinsed with IF buffer, and mounted with Mount Liquid Antifade (Abberior) and sealed with nail polish. Images of structures were obtained on a SP8ST-WS confocal microscope equipped with a HC PL APO 40x/1.10 W CS2 water immersion objective, a white light laser, HyD and PMT detectors. Image analysis was performed using the Fiji software [18]. Structures were manually outlined at their middle section; their size and aspect ratio were then analyzed. Overview images of multicellular structures at 11 days were acquired using an Olympus CKX53 microscope, equipped with a DP22 camera (Olympus) and DP2- SAL firmware (Olympus). Sizes of multicellular structures were calculated using the Fiji software after manually contouring structures.
2.7. Cell Migration Assays
All live imaging experiments were performed using an Axio Observer microscope (Zeiss) equipped with a Plan-Apochromat 10x/0.25 air objective, a Hamatsu camera C10600 OrcaR2 and a Pecon Zeiss incubator XL multi S1 RED LS.
For collective migration, 8.104 cells per insert (#80209 from Ibidi) were seeded in a 8-wells µ-Slide (#80826 Ibidi) in 1% GF medium (1% horse serum, 4 ng/mL EGF). Images were acquired every 10 min for 24 h after release from the insert. Edge progression was measured as the distance moved by the leading edge perpendicular to the initial edge, for each pixel along the wound. Analysis of time-lapse images was performed using Particle Image Velocimetry (PIV) analysis with a MATLAB PIV script [19].
For single cell migration, Ibidi 8-wells µ-Slide was coated with 20 μg/mL bovine fibronectin (Sigma) for 1 h at 37°C and 1000 cells were seeded per well in complete 5% GF medium. Images were acquired every 10 min for 24 h. Cell trajectories were obtained using MTrackJ plugin in the FIJI software only for cells that were freely migrating for 6 h or more. Migration persistence of random migration was based on the autocorrelation of angles between displacement vectors and is calculated using the DiPer program [20].
2.8. Statistical Analysis
Statistical analysis was conducted using GraphPad Prism software (v7.00). Two-way ANOVA followed by a post-hoc Tukey’s test was used when data followed a normal distribution. Otherwise, the non parametric Kruskal-Wallis followed by a post-hoc Dunn’s test was used. For migration persistence, statistics were performed using the R software, where directional autocorrelation over time was fit for each cell by an exponential decay with a plateau as follows:
where A is the autocorrelation, t the time interval, Amin the plateau and τ the time constant of decay [21]. The plateau value Amin is set to zero for cell lines in vitro, since they do not display overall directional movement. The time constant τ of exponential fits were then compared using one-way ANOVA on non-linear mixed-effect models for each condition. Four levels of statistical significance were distinguished: * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
3. Results
3.1. KMT2D KO does not Contribute to PI 3-Kinase Dependent Proliferation
We have created a set of four MCF10A cell lines to reconstitute the association between two genetic alterations found in breast cancer, the activation of PI 3-kinase by the H1047R mutation and the inactivation of KMT2D. We have first obtained the knock-in cell line where the H1047R mutation was introduced into one of the two alleles of the PIK3CA gene from a commercial source. We experimentally verified that this cell line indeed corresponds to what it is supposed to be by sequencing the two PIK3CA alleles (Figure S1). We then designed a gRNA that targets the second exon of KMT2D and obtained biallelic knock-outs (KOs) by CRISPR/Cas9 from both parental MCF10A and PIK3CA H1047R cell lines (Figure S1). This set of four cell lines with the parental MCF10A was subsequently used to investigate the consequences of each genetic alteration and their combination.
We first evaluated cell cycling in limiting amount of growth factors (GF), since GF-independent cell cycle progression is a hallmark of cell transformation in cancer. In 0% GF, MCF10A cells stopped proliferating. However, PIK3CA H1047R, but not KMT2D KO cells, were able to cycle in this condition (Figure 1, Table S2). The combination of KMT2D KO with PIK3CA H1047R did not affect the enhanced cycling due to PIK3CA H1047R alone. Interestingly, the cell lines that were cycling in 0% GF had no advantage over controls when 1% GF was present in the medium: double-mutant cells, in particular, were cycling significantly less than parental MCF10A cells in 1% GF. Since in 0% GF, PI 3-kinase activity appears to control the cell cycle in lines harboring the PIK3CA H1047R mutation, we studied cell cycling in the presence of 1 or 5 μM BYL719, which is the specific PI3-kinase α inhibitor approved for breast cancer treatment under the name of Alpelisib. BYL719 stopped cell cycling in the two cell lines harboring the PIK3CA H1047R mutation at 0% GF, and decreased cycling in all cell lines in the presence of 1% GF in a dose-dependent manner. We conclude from this first set of experiments that the ability of cells to cycle, and in particular when they should not, in the absence of GF, is determined by PI 3-kinase activity and that the inactivation of the histone methyl-transferase encoded by KMT2D has no effect in the proliferation of 2D cultures of MCF10A cells.
To explore this point in greater depth, we challenged cells by cultivating them in 3D Matrigel for up to 3 weeks (in the presence of 1% GF). In these settings, cells develop as multicellular spherical structures. Spheres had various sizes and morphologies among different cultures. Cell lines harboring PIK3CA H1047R either alone or in combination with KMT2D KO were growing into structures that were significantly bigger and had less regular morphology than controls at 11 days (Figure 2A). In addition, we noticed the presence of protrusions invading into Matrigel in these two cell lines, but not with MCF10A or KMT2D KO cell lines. After 21 days, however, the effect of PIK3CA H1047R on structure size and shape regularity was less pronounced when KMT2D was also inactivated (Figure 2B), suggesting that the combination of these two driver mutations in double-mutant cells attenuated cell transformation induced by strong PI 3-kinase activity. BYL719 treatment decreased the aberrant growth and shape of the multicellular structures formed by the PIK3CA H1047R cell line.
3.2. Synergistic Activation of Migration Persistence by PIK3CA H1047R and KMT2D Inactivation
Migratory behavior is another feature of tranformed cells in addition to aberrant proliferation and growth. Therefore we examined collective cell migration of the mammary epithelial cell lines we had generated. To this end, we seeded the four cell lines at confluence in inserts and lifted the insert to study how the different epithelial monolayers migrated into the free space. Displacement vectors were obtained using Particle Image Velocimetry (PIV). The PIK3CA H1047R and the double-mutant cell lines showed significantly enhanced velocity of the edge compared with parental MCF10A and KMT2D KO alone (Figure 3, Video S1-4, Table S2). The PIV method allows examining how the information about free space sensed by the first row of cells is transmitted backwards to the follower cells in the monolayer. Velocity and the order parameter, which indicates the coordinated directional migration toward the free space, were transmitted further backwards in the two cell lines expressing active PI 3-kinase compared with controls. Basal and enhanced backward transmission of velocity and order parameter were in all cases abolished upon treatment with BYL719. In this collective migration assay, as well as in previous proliferation assays, KMT2D KO either alone or in combination with PIK3CA H1047R exerted little, if any, influence.
We reasoned that the histone methyl-transferase KMT2D should modulate gene expression in MCF10A cells. Indeed, the list of genes deregulated by KMT2D mutations was previously established [22]. Similarly, PIK3CA H1047R affects gene expression and similar lists were also established [23,24]. We selected in these lists of genes that were deregulated by either KMT2D mutation or PIK3CA H1047R mutation the genes that might be responsible for the phenotypes of proliferation and migration/invasion that we had observed in the four cell lines. We examined the expression of 12 selected genes using qRT-PCR in the presence or absence of the PI 3-kinase inhibitor BYL719 (Figure S2). Out of 12, one gene, ZEB1, was not detectable in MCF10A cells. Only two out of the 11 remaining genes showed significant variations in the set of four cell lines (Figure 4). These two genes were PLAUR that encodes the urokinase plasminogen activator receptor (uPAR), a glycolipid anchored protein of the cell surface involved in migration and invasion, and ARPC5L, that encodes one of the two paralogous ARPC5 subunits of the cytosolic Arp2/3 complex that polymerizes branched actin, which is instrumental in cell migration and intracellular trafficking. These two genes displayed the same pattern of regulation. Both showed a significantly higher expression level in the double-mutant than in parental MCF10A cells. These two genes, which were previously reported to be upregulated in cells expressing PIK3CA H1047R, were indeed upregulated in the PIK3CA H1047R MCF10A cell line, even if the difference did not reach statistical significance, but not in the KMT2D KO cell line. Deregulated expression of these two genes was the first evidence of a potential synergy between PIK3CA H1047R and KMT2D KO in our MCF10A cell system.
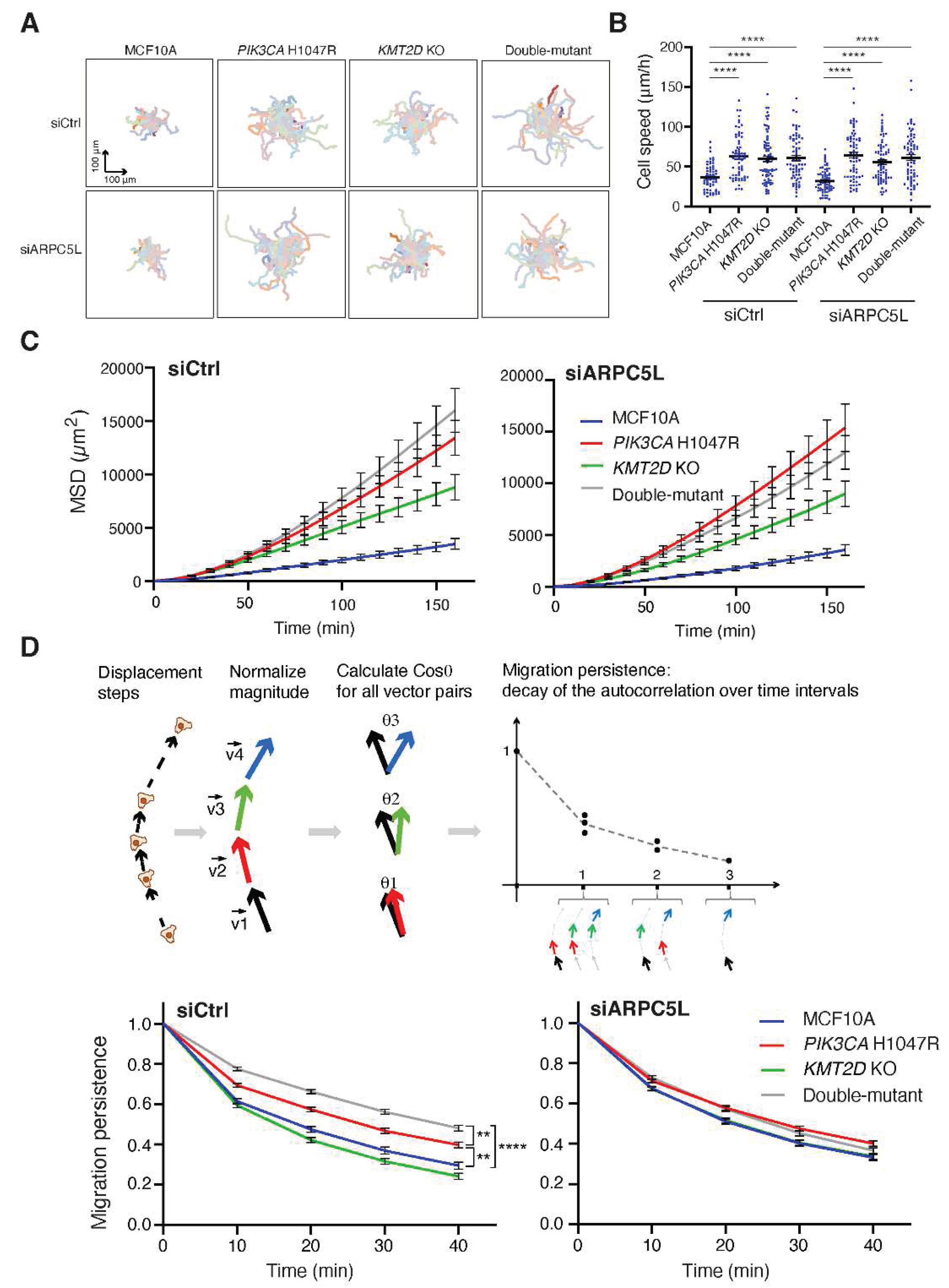
We next focused on ARPC5L, because the role of the Arp2/3 complex in cell migration has been well established in MCF10A cells [17,21,25]. We depleted ARPC5L from the four cell lines using pools of siRNAs and evaluated collective migration in these cells using the previous assay utilizing PIV. Surprisingly, ARPC5L depletion did not affect collective migration neither in terms of speed nor the directionality of migration that is transmitted backward from the free edge of the monolayer (Figure 5). However, our group has previously showed that Arp2/3 activity was implicated in the migration persistence of single MCF10A cells [17,21,25]. So we decided to assay the random migration of single cells in our four cell lines. In this assay, we compared the effect of ARPC5L depletion to that of depletion of its paralog ARPC5 (Video S5). Both were efficiently depleted (Figure S3). All cell lines that harbored a cancer associated genetic alteration either alone or in combination displayed increased speed of cell migration, but this increased speed was not affected by the depletion of ARPC5L or ARPC5 (Figure 6, Figure S4). In contrast, when the persistence of directional migration, was examined, PIK3CA H1047R cell line displayed increased persistence, which was even stronger when the PIK3CA mutation was combined with KMT2D KO. Importantly, the increased migration persistence in the double-mutant cell line was abolished when ARPC5L, but not ARPC5, was depleted (Figure 6D, Figure S4). Moreover, ARPC5L depletion did not affect the basal persistence of parental MCF10A cells. Mean Square Displacement (MSD), which is the surface area covered by migrating cells over time, depends on both speed and directional persistence and reflects the ability of cells to disseminate. As a result, MSD showed a stronger increase in the double-mutant cell line, which was no longer the case when ARPC5L was depleted (Figure 6). These data thus indicate that the synergistic enhancement of migration persistence by PIK3CA H1047R and KMT2D KO can be attributed to ARPC5L, whose expression is also enhanced by the two genetic alterations.
4. Discussion
PIK3CA acts as a major oncogene when activated by the H1047R mutation. We observed that PIK3CA H1047R MCF10A cells were still cycling in the absence of growth factors, as previously reported [7,26]. These cells were also shown to form enlarged multicellular structures in 3D Matrigel, with aberrant shapes and signs of invasion, a well-established hallmark of cell transformation [27]. We have performed detailed analysis of the migratory behavior using two protocols for collective monolayers and single cells. In both cases, PI3-kinase activity dramatically enhanced cell migration. In Matrigel-coated Boyden chamber assays, enhanced migration and invasion of cancer cells transformed with PIK3CA mutations was shown to depend on AKT1-mediated phosphorylation of cortactin [8].
In contrast, KMT2D appeared as a weak tumor suppressor gene. KMT2D KO cells did not behave differently from parental MCF10A cells in proliferation and most of the migration assays. The only migration parameter that was affected by KMT2D KO was the speed of single migrating cells, which was enhanced by KMT2D KO to the same level as that of PIK3CA H1047R. Surprisingly, in double-mutant cells, the speed of single migrating cells was not increased further. The only synergy between PIK3CA H1047R and KMT2D KO was found on the directional persistence of individually migrating cells. PIK3CA H1047R, but not KMT2D KO, already enhanced migration persistence. However, in double-mutant cells, persistence was further increased, suggesting that KMT2D controls this parameter even if this was not apparent in the single KMT2D KO.
Studies reporting the combined actions of driver mutations are indeed less numerous and relatively recent compared to those reporting the phenotypes induced by single driver mutations. Mutations of PIK3CA were shown to cooperate with KRAS mutations in MCF10A cells to induce anchorage-independent growth in soft agar [28]. Results of these combinations are sometimes counter-intuitive. For example, the PIK3CA H1047R mutation antagonizes lung metastases in a mouse model of ErbB2-induced breast cancer [29]. Toska et al. had previously studied the role of KMT2D inactivation in cells transformed by active PI 3-kinase and found that KMT2D was mediating the resistance to BYL719 in these cells in a mechanism that depended on the transcription of the estrogen receptor [30]. These results emphasize the importance of studying the synergy between oncogenes and tumor suppressor genes not only to understand cancer progression, but also to develop the most appropriate therapy.
The synergy we reported here between PIK3CA H1047R and KMT2D KO on the persistence of single cell migration was associated with enhanced expression of ARPC5L, a gene encoding an Arp2/3 subunit. The PIP3-dependent protein kinase AKT1 was shown to phosphorylate KMT2D and thereby decrease its ability to enhance gene expression [30]. But this phosphorylation event cannot account for the synergy of KMT2D KO with PIK3CA H1047R, since KMT2D should thereby be inactivated when PIK3CA harbors the H1047R mutation. This association of enhanced migration persistence with enhanced ARPC5L expression immediately attracted our attention, since we had previously shown that the Arp2/3 complex specifically controls the migration persistence of single cells [17,25,31]. The Arp2/3 complex is the only machinery that polymerizes branched actin. Polymerization of branched actin networks generates the pushing force driving membrane protrusions at the leading edge of migrating cells [32]. The Arp2/3 complex is composed of seven subunits. ARPC5 and ARPC5L are paralogous proteins, and as a consequence, ARPC5-containing Arp2/3 complexes co-exist with ARPC5L-containing Arp2/3 complexes [33].
ARPC5L-containing Arp2/3 complexes are more active in polymerizing branched actin networks, and stabilized more by cortactin against coronin1-mediated debranching than those containing ARPC5 [34]. The two ARPC5 paralogs were recently shown to have opposite effects on the lamellipodial width of B16-F1 cells, suggesting isoform-specific functions [35]. In MCF10A cells, we repeatedly found that migration persistence of single cells was the best read-out of Arp2/3 activity [17,21,25]. The enhanced migration persistence of double-mutant cells depended on ARPC5L and not on ARPC5. Surprisingly, collective migration of MCF10A cells did not depend on ARPC5L, even though cells in the first row made lamellipodia and those further in the monolayer probably made cryptic lamellipodia below the cells in front of them [36]. We have recently generated “super-migrator” cells with enhanced Arp2/3 activity at the cortex and these genetic manipulations similarly increased migration persistence of single cells, but not collective migration of MCF10A cells [25].
Our reconstitution approach of generating genetic alterations and combining them is a straightforward approach to study the potential cooperation of cancer-associated genes, which are altered in the same tumors. Here we have identified the major parameter that is synergistically potentialized by PIK3CA H1047R and KMT2D KO, migration persistence of single cells, and found a gene, possibly the gene, ARPC5L, whose expression is upregulated by these two genetic alterations and that appears responsible for the synergistic phenotype.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, AMG, JG, KDR. KDR and JG performed together the bulk of the work. Matrigel assays were performed by KDR, EdU assays were performed by JG, collective migration assays and analyses were performed by KDR, JG and AF, single cell migration assays and analyses were performed by KDR and JG. qRT-PCR measurements were performed by SV and LC. RT, AB, CM, MJ, MCNH and EG were rotating students from Ecole polytechnique, who isolated genome edited cell lines and genetically characterized them. Supervision, AMG, IB, AF. Writing, AMG. Funding acquisition, AMG and KDR. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from Agence Nationale de la Recherche (ANR-20-CE13-0016 to AMG and ANR-22-CE13-0041 to AMG), Fondation ARC pour la Recherche sur le Cancer (ARC PJA 2021 060003815 to AMG, ARCPDF12020080002578 to KDR), from Institut National du Cancer (INCA_11508 for AMG and IB, INCA_16712 for AMG). This work benefited from the support of the Personalized reconstitution of the tumoral process program led by l’X, Ecole polytechnique and the Fondation de l’Ecole polytechnique, sponsored by Servier. We thank the Polytechnique Bioimaging Facility for confocal microscopy partly supported by Région Ile-de-France (interDIM) and Agence Nationale de la Recherche (ANR-11-EQPX-0029 Morphoscope2, ANR-10-INBS-04 France BioImaging).
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weinberg, R. The Biology of Cancer; Garland Science, Ed.; 2013.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Weigelt, B.; Peterse, J.L.; Veer, L.J. van’t Breast Cancer Metastasis: Markers and Models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Network, C.G.A.; Onofrio, R.C.; Ardlie, K.; Winckler, W.; Gabriel, S.B.; Meyerson, M.; Getz, G.; Saksena, G.; Park, P.J.; Chin, L.; et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Gustin, J.P.; Karakas, B.; Weiss, M.B.; Abukhdeir, A.M.; Lauring, J.; Garay, J.P.; Cosgrove, D.; Tamaki, A.; Konishi, H.; Konishi, Y.; et al. Knockin of Mutant PIK3CA Activates Multiple Oncogenic Pathways. Proc. Natl. Acad. Sci. 2009, 106, 2835–2840. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Renuse, S.; Sahasrabuddhe, N.A.; Zahari, M.S.; Chaerkady, R.; Kim, M.-S.; Nirujogi, R.S.; Mohseni, M.; Kumar, P.; Raju, R.; et al. Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations. Nature communications 2014, 5, 4961. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. The New England journal of medicine 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Prever, L.; Hirsch, E.; Gulluni, F. Targeting PI3K/AKT/MTOR Signaling Pathway in Breast Cancer. Cancers 2021, 13, 3517. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.J.; Dingwall, A.K. The Cancer COMPASS: Navigating the Functions of MLL Complexes in Cancer. Cancer Genet. 2015, 208, 178–191. [Google Scholar] [CrossRef]
- Dhar, S.S.; Lee, M.G. Cancer-Epigenetic Function of the Histone Methyltransferase KMT2D and Therapeutic Opportunities for the Treatment of KMT2D-Deficient Tumors. Oncotarget 2021, 12, 1296–1308. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Loo, P.V.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The Landscape of Cancer Genes and Mutational Processes in Breast Cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Bruijn, I. de; Kundra, R.; Mastrogiacomo, B.; Tran, T.Ngoc.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in CBioPortal. Cancer Res. 2023, 83, 3861–3867. [CrossRef]
- Dayoub, A.; Fokin, A.I.; Lomakina, M.E.; James, J.; Plays, M.; Jacquin, T.; Novikov, N.M.; Vorobyov, R.S.; Schegoleva, A.A.; Rysenkova, K.D.; et al. Inactivation of PTEN and ZFHX3 in Mammary Epithelial Cells Alters Patterns of Collective Cell Migration. Int. J. Mol. Sci. 2022, 24, 313. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and Characterization of a Spontaneously Immortalized Human Breast Epithelial Cell Line, MCF-10. Cancer Research 1990, 50, 6075–6086. [Google Scholar] [PubMed]
- Molinie, N.; Rubtsova, S.N.; Fokin, A.; Visweshwaran, S.P.; Rocques, N.; Polesskaya, A.; Schnitzler, A.; Vacher, S.; Denisov, E.V.; Tashireva, L.A.; et al. Cortical Branched Actin Determines Cell Cycle Progression. Cell Research 2019, 29, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Deforet, M.; Parrini, M.C.; Petitjean, L.; Biondini, M.; Buguin, A.; Camonis, J.; Silberzan, P. Automated Velocity Mapping of Migrating Cell Populations (AVeMap). Nature Methods 2012, 9, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, R.; Gautreau, A. Quantitative and Unbiased Analysis of Directional Persistence in Cell Migration. Nature Protocols 2014, 9, 1931–1943. [Google Scholar] [CrossRef]
- Polesskaya, A.; Boutillon, A.; Yang, S.; Wang, Y.; Romero, S.; Liu, Y.; Lavielle, M.; Molinie, N.; Rocques, N.; Fokin, A.; et al. Restrained Activation of CYFIP2-Containing WAVE Complexes Controls Membrane Protrusions and Cell Migration. Biorxiv 2022, 2020.07.02.184655. [CrossRef]
- Morcillo-Garcia, S.; Noblejas-Lopez, M. del M.; Nieto-Jimenez, C.; Perez-Peña, J.; Nuncia-Cantarero, M.; Győrffy, B.; Amir, E.; Pandiella, A.; Galan-Moya, E.M.; Ocana, A. Genetic Mutational Status of Genes Regulating Epigenetics: Role of the Histone Methyltransferase KMT2D in Triple Negative Breast Tumors. PLoS ONE 2019, 14, e0209134. [Google Scholar] [CrossRef]
- Wallin, J.J.; Guan, J.; Edgar, K.A.; Zhou, W.; Francis, R.; Torres, A.C.; Haverty, P.M.; Eastham-Anderson, J.; Arena, S.; Bardelli, A.; et al. Active PI3K Pathway Causes an Invasive Phenotype Which Can Be Reversed or Promoted by Blocking the Pathway at Divergent Nodes. PLoS ONE 2012, 7, e36402. [Google Scholar] [CrossRef]
- Young, C.D.; Zimmerman, L.J.; Hoshino, D.; Formisano, L.; Hanker, A.B.; Gatza, M.L.; Morrison, M.M.; Moore, P.D.; Whitwell, C.A.; Dave, B.; et al. Activating PIK3CA Mutations Induce an Epidermal Growth Factor Receptor (EGFR)/Extracellular Signal-Regulated Kinase (ERK) Paracrine Signaling Axis in Basal-like Breast Cancer*. Mol. Cell. Proteom. 2015, 14, 1959–1976. [Google Scholar] [CrossRef]
- Fokin, A.I.; Boutillon, A.; James, J.; Courtois, L.; Vacher, S.; Simanov, G.; Wang, Y.; Polesskaya, A.; Bièche, I.; David, N.B.; et al. Inactivating Negative Regulators of Cortical Branched Actin Enhances Persistence of Single Cell Migration. J. Cell Sci. 2024, 137. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; DeLong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA Promotes Cell Growth and Invasion of Human Cancer Cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Brugge, J.S. Modelling Glandular Epithelial Cancers in Three-Dimensional Cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.M.; Raffeiner, P.; Hart, J.R.; Vogt, P.K. PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9. Cancers 2019, 11, 1634. [Google Scholar] [CrossRef] [PubMed]
- Simond, A.M.; Bui, T.; Zuo, D.; Sanguin-Gendreau, V.; Rao, T.; Phillips, W.A.; Cardiff, R.D.; Muller, W.J. Physiological Expression of PI3K H1047R Mutation Reveals Its Anti-Metastatic Potential in ErbB2-Driven Breast Cancer. Oncogene 2022, 41, 3445–3451. [Google Scholar] [CrossRef]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K Pathway Regulates ER-Dependent Transcription in Breast Cancer through the Epigenetic Regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef]
- Dang, I.; Gorelik, R.; Sousa-Blin, C.; Derivery, E.; Guérin, C.; Linkner, J.; Nemethova, M.; Dumortier, J.G.; Giger, F.A.; Chipysheva, T.A.; et al. Inhibitory Signalling to the Arp2/3 Complex Steers Cell Migration. Nature 2013, 503, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Regulation of Actin Filament Assembly by Arp2/3 Complex and Formins. Annual Review of Biophysics and Biomolecular Structure 2007, 36, 451–477. [Google Scholar] [CrossRef]
- Pizarro-Cerdá, J.; Chorev, D.S.; Geiger, B.; Cossart, P. The Diverse Family of Arp2/3 Complexes. Trends in Cell Biology 2016, 1–8. [Google Scholar] [CrossRef]
- Abella, J.V.G.; Galloni, C.; Pernier, J.; Barry, D.J.; Kjær, S.; Carlier, M.-F.; Way, M. Isoform Diversity in the Arp2/3 Complex Determines Actin Filament Dynamics. Nature Cell Biology 2016, 18, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Fäßler, F.; Javoor, M.G.; Datler, J.; Döring, H.; Hofer, F.W.; Dimchev, G.; Hodirnau, V.-V.; Faix, J.; Rottner, K.; Schur, F.K.M. ArpC5 Isoforms Regulate Arp2/3 Complex–Dependent Protrusion through Differential Ena/VASP Positioning. Sci Adv 2023, 9, eadd6495. [Google Scholar] [CrossRef] [PubMed]
- Palamidessi, A.; Malinverno, C.; Frittoli, E.; Corallino, S.; Barbieri, E.; Sigismund, S.; Beznoussenko, G.V.; Martini, E.; Garré, M.; Ferrara, I.; et al. Unjamming Overcomes Kinetic and Proliferation Arrest in Terminally Differentiated Cells and Promotes Collective Motility of Carcinoma. Nature materials 2019, 80, 076601. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PI 3-kinase activity controls cell cycle progression of MCF10A cells. MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells are cultivated in media containing either 0 or 1 % of growth factors (GF, that is both serum and EGF) and incubated for 1 h in the presence of EdU that incorporates into DNA during S-phase. Cells are exposed to 1µM, 5µM BYL719 or DMSO as a control vehicle, for 16 h before incubation with EdU. Three independent experiments have been performed, six fields of view per sample are analyzed (n>1000 cells). Statistically significant differences with controls are represented by stars; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (two-way ANOVA followed by Tukey’s test).
Figure 1.
PI 3-kinase activity controls cell cycle progression of MCF10A cells. MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells are cultivated in media containing either 0 or 1 % of growth factors (GF, that is both serum and EGF) and incubated for 1 h in the presence of EdU that incorporates into DNA during S-phase. Cells are exposed to 1µM, 5µM BYL719 or DMSO as a control vehicle, for 16 h before incubation with EdU. Three independent experiments have been performed, six fields of view per sample are analyzed (n>1000 cells). Statistically significant differences with controls are represented by stars; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (two-way ANOVA followed by Tukey’s test).
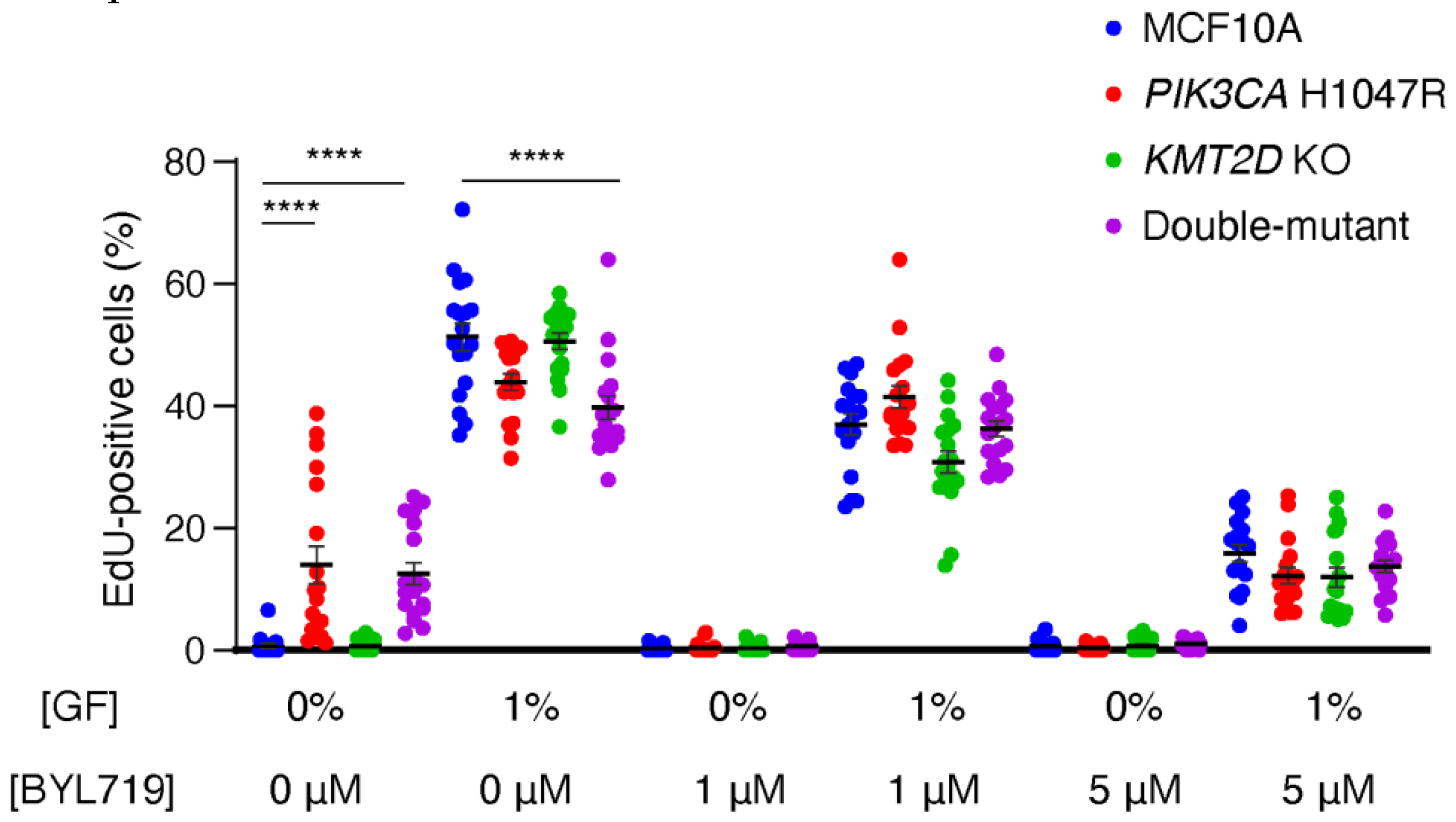
Figure 2.
KMT2D inactivation partially corrects the aberrant multicellular structures formed by PIK3CA H1047R expressing cells in Matrigel. (A) Morphologies of multicellular structures developed by MCF10A derivatives in Matrigel after 11 days of growth. PIK3CA H1047R and double-mutant structures are larger than controls and develop spiky protrusions that invade the Matrigel (arrowheads in high magnification views). (B) Multicellular structures were grown for 21 days in the presence of 1 µM BYL719 or DMSO vehicle. Structures are stained with DAPI (blue), phalloidin (green), E-cadherin (yellow), and Laminin-5 (red) antibodies and their size and shape are quantified using their major axis, which corresponds to the longest diameter of the middle z-section, and their roundness index, which corresponds to the ratio of the minor to the major axis of the same section. Four independent experiments (with DMSO) and two independent experiments (with BYL719) have been performed; more than 20 structures were included for each condition. Data from one representative experiment are displayed. Statistically significant differences with controls are represented by stars; *p<0.05, ** p< 0.01, *** p<0.001, ****p<0.0001 (two-way ANOVA followed by Tukey’s test).
Figure 2.
KMT2D inactivation partially corrects the aberrant multicellular structures formed by PIK3CA H1047R expressing cells in Matrigel. (A) Morphologies of multicellular structures developed by MCF10A derivatives in Matrigel after 11 days of growth. PIK3CA H1047R and double-mutant structures are larger than controls and develop spiky protrusions that invade the Matrigel (arrowheads in high magnification views). (B) Multicellular structures were grown for 21 days in the presence of 1 µM BYL719 or DMSO vehicle. Structures are stained with DAPI (blue), phalloidin (green), E-cadherin (yellow), and Laminin-5 (red) antibodies and their size and shape are quantified using their major axis, which corresponds to the longest diameter of the middle z-section, and their roundness index, which corresponds to the ratio of the minor to the major axis of the same section. Four independent experiments (with DMSO) and two independent experiments (with BYL719) have been performed; more than 20 structures were included for each condition. Data from one representative experiment are displayed. Statistically significant differences with controls are represented by stars; *p<0.05, ** p< 0.01, *** p<0.001, ****p<0.0001 (two-way ANOVA followed by Tukey’s test).
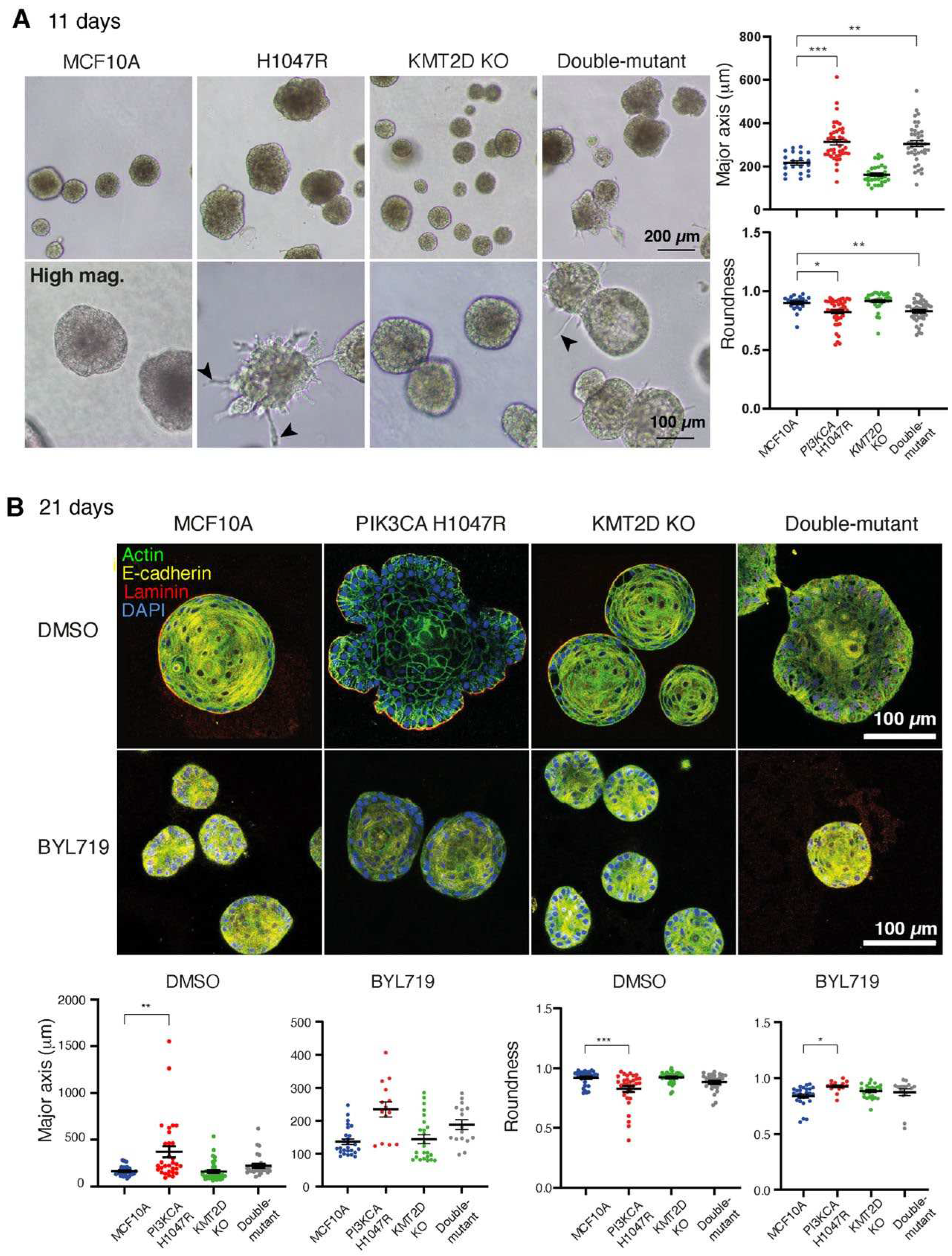
Figure 3.
PI 3-kinase activity is critical for collective cell migration. Confluent monolayers of parental, PIK3CA H1047R, KMT2D KO and double-mutant MCF10A cells are induced to migrate into the free space obtained by lifting a physical boundary. Displacement vectors within the monolayer are obtained using Particle Image Velocimetry ((A), PIV). These vectors are analyzed over space and time their position for their magnitude ((B), velocity) and directionality ((C), order parameter, i.e. the cosine of the angle made by the displacement vector with respect to the vector oriented toward the free space). These parameters are represented as heat maps. Three biological repeats with similar results, four fields of view in each condition. Plots show the average of all measurements. Each pixel of the heatmaps average all fields of view in all biological replicates over 42 µm and 40 min. These parameters are compared at a single time point or at a single distance from the edge using two-way ANOVA with Tukey’s test. *p<0.05, ** p<0.01, *** p<0.001.
Figure 3.
PI 3-kinase activity is critical for collective cell migration. Confluent monolayers of parental, PIK3CA H1047R, KMT2D KO and double-mutant MCF10A cells are induced to migrate into the free space obtained by lifting a physical boundary. Displacement vectors within the monolayer are obtained using Particle Image Velocimetry ((A), PIV). These vectors are analyzed over space and time their position for their magnitude ((B), velocity) and directionality ((C), order parameter, i.e. the cosine of the angle made by the displacement vector with respect to the vector oriented toward the free space). These parameters are represented as heat maps. Three biological repeats with similar results, four fields of view in each condition. Plots show the average of all measurements. Each pixel of the heatmaps average all fields of view in all biological replicates over 42 µm and 40 min. These parameters are compared at a single time point or at a single distance from the edge using two-way ANOVA with Tukey’s test. *p<0.05, ** p<0.01, *** p<0.001.
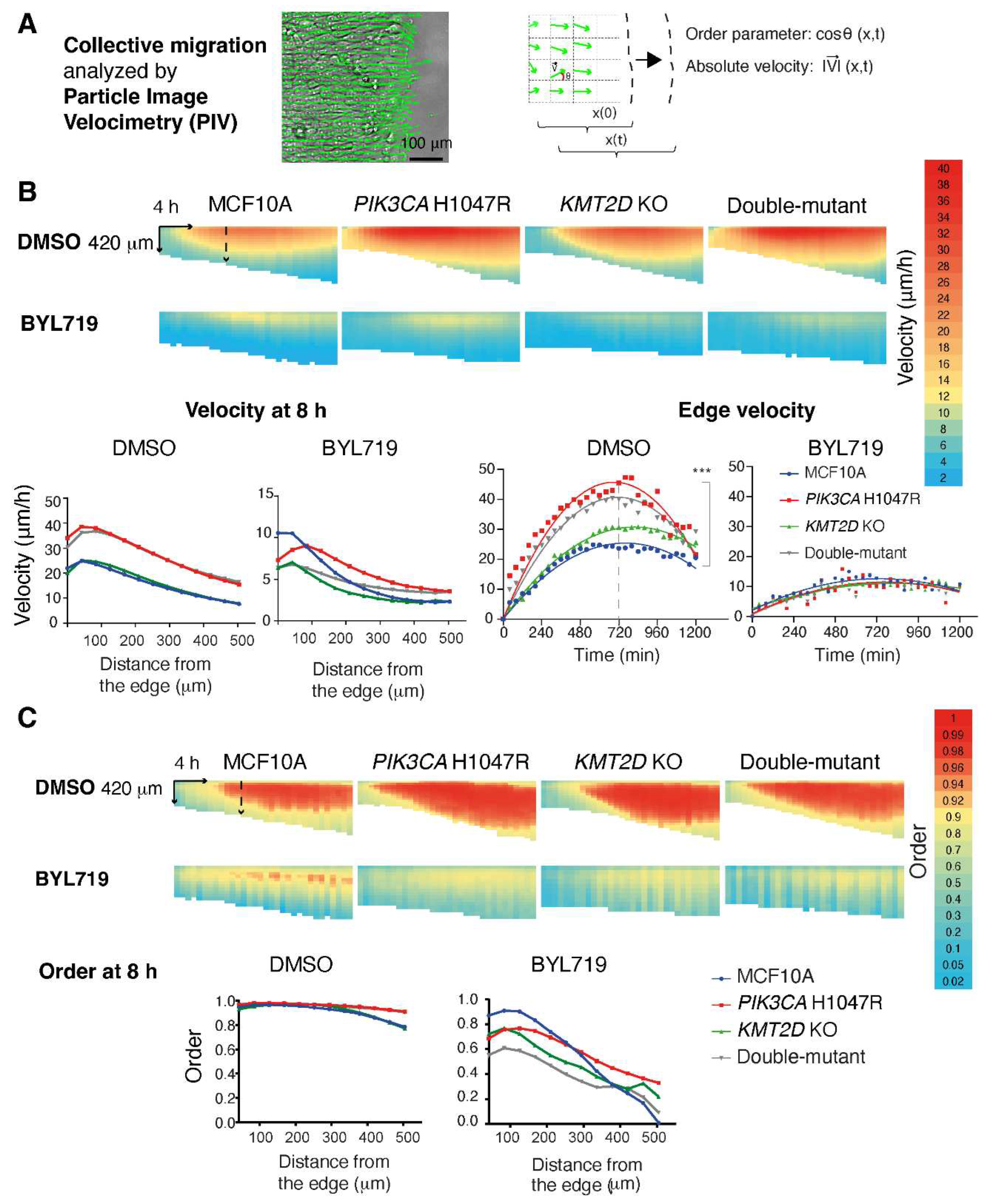
Figure 4.
Expression levels of the PLAUR gene, encoding the urokinase plasminogen activator receptor (uPAR), and the ARPC5L gene, encoding one of the two paralogous ARPC5 subunits of the Arp2/3 complex, are significantly up-regulated in double-mutant cells. qRT-PCR analysis of gene expression is performed from MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells in the presence or absence of 5 µM BYL719. Mean ± SEM, n=3 biological repeats. Statistically significant differences with controls, using two-way ANOVA followed by Tukey’s test, are represented by stars; * p<0.05; ** p<0.01; *** p<0.001.
Figure 4.
Expression levels of the PLAUR gene, encoding the urokinase plasminogen activator receptor (uPAR), and the ARPC5L gene, encoding one of the two paralogous ARPC5 subunits of the Arp2/3 complex, are significantly up-regulated in double-mutant cells. qRT-PCR analysis of gene expression is performed from MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells in the presence or absence of 5 µM BYL719. Mean ± SEM, n=3 biological repeats. Statistically significant differences with controls, using two-way ANOVA followed by Tukey’s test, are represented by stars; * p<0.05; ** p<0.01; *** p<0.001.
Figure 5.
The Arp2/3 subunit ARPC5L does not control collective cell migration. (A) ARPC5L is depleted from MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells using siRNA-mediated knock-down. Collective migration is induced by lifting a physical boundary. Displacement vectors within monolayers are obtained using PIV and analyzed as in Figure 3. (B) Velocity. (C) Order. Two biological repeats with similar results, four fields of view in each condition. Plots show the average of all measurements. Each pixel of the heatmaps represents an average of all fields of view and all biological replicates over 42 µm and 40 min.
Figure 5.
The Arp2/3 subunit ARPC5L does not control collective cell migration. (A) ARPC5L is depleted from MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells using siRNA-mediated knock-down. Collective migration is induced by lifting a physical boundary. Displacement vectors within monolayers are obtained using PIV and analyzed as in Figure 3. (B) Velocity. (C) Order. Two biological repeats with similar results, four fields of view in each condition. Plots show the average of all measurements. Each pixel of the heatmaps represents an average of all fields of view and all biological replicates over 42 µm and 40 min.
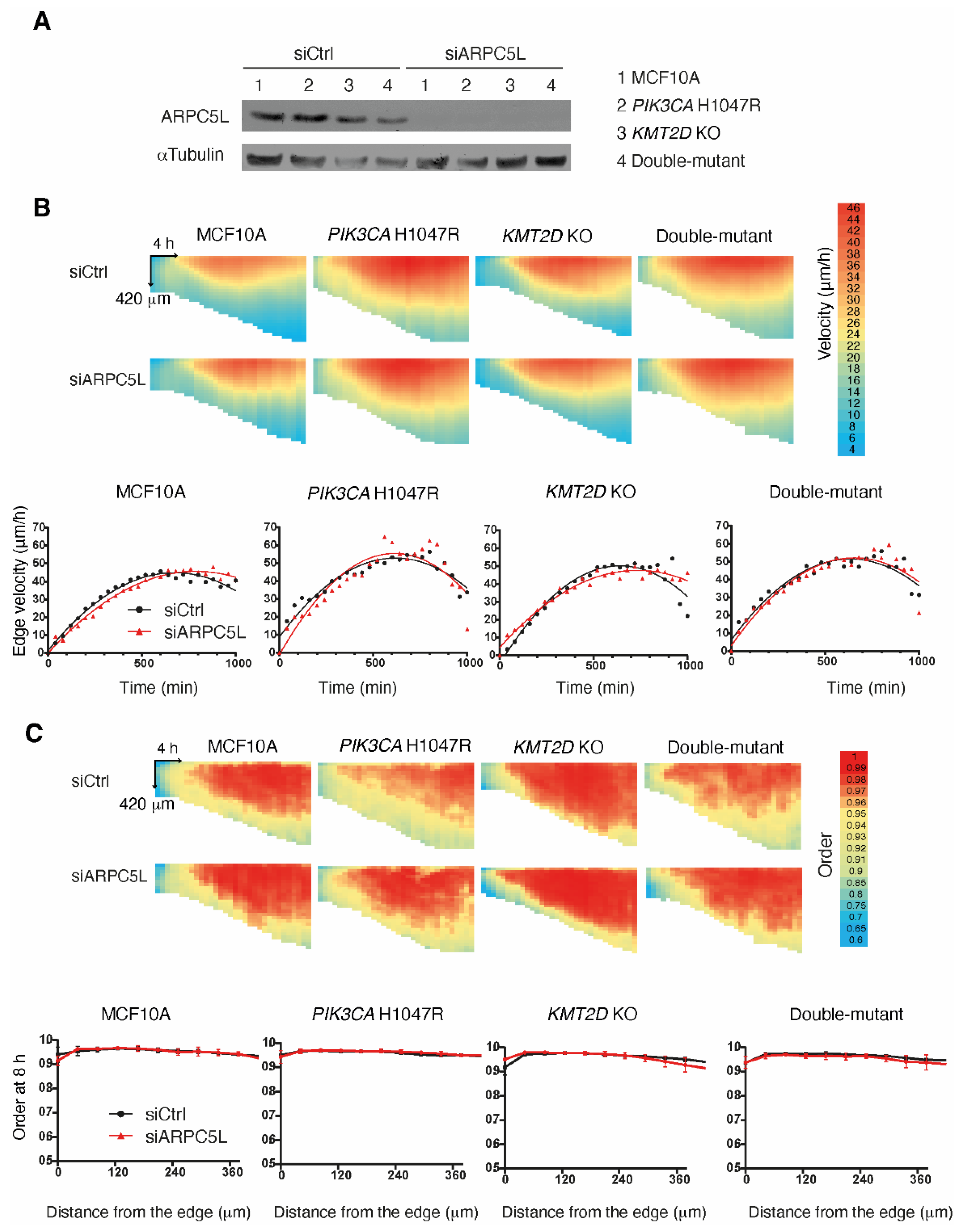
Figure 6.
ARPC5L stimulates migration persistence, but not speed, in double-mutant cells. Random migration of single cells is tracked for 6 h. (A) Trajectories of 56 MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells, depleted of ARPC5L or not. The origin of each track is registered to the center of the plot. (B) Cell speed (mean ± SEM). (C) Mean Square Displacement (MSD). (D) Migration persistence. Three independent experiments are performed, overall number of cells ranges from 56 to 78 for each condition. ** p<0.01, **** p<0.0001 (two-way ANOVA followed by Tukey’s test for speed, 1-way ANOVA on non linear mixed-effect models for persistence).
Figure 6.
ARPC5L stimulates migration persistence, but not speed, in double-mutant cells. Random migration of single cells is tracked for 6 h. (A) Trajectories of 56 MCF10A, PIK3CA H1047R, KMT2D KO and double-mutant cells, depleted of ARPC5L or not. The origin of each track is registered to the center of the plot. (B) Cell speed (mean ± SEM). (C) Mean Square Displacement (MSD). (D) Migration persistence. Three independent experiments are performed, overall number of cells ranges from 56 to 78 for each condition. ** p<0.01, **** p<0.0001 (two-way ANOVA followed by Tukey’s test for speed, 1-way ANOVA on non linear mixed-effect models for persistence).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



