
Submitted:
17 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
Molecular analysis of growing teratoma syndrome has not been extensively studied. Here, we report a 14-year-old boy with a growing mass during treatment for a mixed germ cell tumor of the pineal region. Tumor markers were negative; thus, growing teratoma syndrome was suspected. A radical resection via the occipital transtentorial approach was performed, and histopathological examination revealed a teratoma with malignant features. Methylation classifier analysis confirmed the diagnosis of teratoma, and DMRT1 loss and 12p gain were identified by copy number variation analysis, potentially elucidating the cause of growth and malignant transformation of the teratoma. The patient remains in remission after intense chemoradiation treatment as a high-risk germ cell tumor.
Keywords:
growing teratoma syndrome
; multimodal treatment
; methylation classifier
; copy number variation analysis
1. Introduction
Growing teratoma syndrome is a rare condition in which paradoxical growth of a mature teratoma during treatment for mixed malignant germ cell tumor despite normalization of tumor markers. It was first described in 1982 by Logothetis et al. [1]. Molecular studies of growing teratoma syndrome have not been extensively reported, and its pathophysiology remains largely obscure. We describe a 14-year-old boy with a growing mass during treatment for a mixed germ cell tumor of the pineal region. A radical resection was performed, and histopathological examination revealed a teratoma with malignant features. DMRT1 loss and 12p gain were identified by copy number variation analysis, potentially elucidating the cause of the formation and malignant phenotype of the teratoma. Intensive chemoradiation treatment was performed because morphological and molecular evidence of malignancy was observed.
2. Case Presentation
A 14-year-old boy without a notable history presented with worsening headaches, blurry vision, and vomiting. An MR scan taken at a neurosurgery clinic revealed a mass at the pineal region and accompanying obstructive hydrocephalus. He was referred to the Department of Neurosurgery, Niigata University, for further evaluation and treatment. At the initial presentation, he had a severe headache, and mild impairment of consciousness was observed (Glasgow Coma Scale 15 E4V5M6, but with mild disorientation). Extraocular movement was objectively normal, but the patient complained of blurry vision. No other neurological symptoms were observed. A head CT scan showed a slightly high-dense tumor mass, approximately 3.3 cm in diameter, at the pineal region with marked enlargement of bilateral lateral and third ventricles (Figure 1(a)). The tumor showed homogeneous enhancement, except for a small area at the left anterior part of the tumor, which was isodense on pre-contrast CT and did not enhance (Figure 1(b)). MR images also depicted tumor regions with different intensities. The main part of the tumor was isointense on T1-weighted images (T1WI) and slightly hyperintense on T2-weighted (T2WI) and diffusion-weighted images (DWI) with moderate enhancement. In retrospect, these intensities were thought to reflect calcification, although plain CT was not high dense. On the other hand, the left anterior portion of the tumor was hyperintense on T1WI, mixed hyper- and hypointense on T2WI, hypointense on DWI and showed heterogeneous enhancement (Figure 1(c)-(f)). A mixed type of germ cell tumor was suspected. However, at presentation, serum tumor markers were negative (AFP 5 ng/ml and β-HCG 6 mIU/ml).
An endoscopic biopsy and a third ventriculostomy were performed using a flexible endoscope. Both components of the tumor were sampled. The former was whitish and relatively soft; the latter component was obviously harder. The first component was histologically a germinoma component (Figure 2(a)), consisting of large, round tumor cells staining for placental alkaline phosphatase (PLAP) (Figure 2(d)) and mixed with small lymphocytes and an embryonal carcinoma-like component (Figure 2(b), (c)) which showed epithelial features and calcification (Figure 2(b)), as well as immunoreactivity for cytokeratin and partly for AFP, (Figure 2(e)) and CD30 (Figure 2(f)). Intraoperatively, cerebrospinal fluid (CSF) was obtained from inside the ventricle. CSF PLAP (1280 pg/ml) and β-HCG (15 mIU/ml) were elevated, but surprisingly, AFP was not elevated (5 ng/ml).
The mixed germ cell tumor was determined to be of intermediate risk according to the Matsutani classification [2,3], and a chemotherapeutic regimen followed by irradiation was planned. First, a regimen of carboplatin (450 mg/m2) and etoposide (150 mg/m2 x 3 days) was commenced. An elevation of serum AFP to 81 ng/ml was noted after the commencement of chemotherapy, and enlargement of the tumor and cystic component was observed on MRI (Figure 3(a)). After two courses of chemotherapy, serum AFP had normalized. However, the size of the tumor did not decrease (Figure 3(b)), so we decided to remove the tumor under the suspicion of growing teratoma syndrome. The pineal tumor was completely removed via the occipital transtentorial approach [4] (Figure 3(c)). The recurrent tumor was very hard and was resected en-bloc after the puncture of the cyst.
Pathological examination revealed a teratomatous tumor with components of cartilage, bone, and fibroblasts (Figure 4(a)). However, upon closer examination, striking malignant features such as cartilage consisting of chondrocytes with nuclear atypia and frequent double nuclei (Figure 4(b)) and immature mesenchymal tissue with ossification (Figure 4(a), (c)) were revealed. Although no embryonic neuroepithelial element was observed, there were GFAP-positive small cells with several mitotic figures (Figure 4(e)), probably representing neuroectodermal elements. Also, MIB-1 staining was diffusely elevated (Figure 4 (d), (g)), which is not consistent with the traditional definition of growing teratoma syndrome.
After thorough deliberation in a multi-disciplinary group, including pathologists, pediatric oncologists, radiation oncologists, and neurosurgeons, we decided to treat the patient as a high-risk patient further, considering that a teratoma with malignant features had enlarged even after treatment as an intermediate-risk mixed germ cell tumor. The patient subsequently received craniospinal irradiation (CSI) 30.6 Gy (17 fractions (fr)) and boost (23.4 Gy/) to the tumor removal cavity followed by 6 cycles of ifosfamide (900 mg/m2 x 5 days), cisplatin (20 mg/m2 x 5 days) and etoposide (60 mg/m2 x 5 days) (ICE) with autologous stem cell transplantation. The patient has remained relapse-free for over three years since the initial presentation.
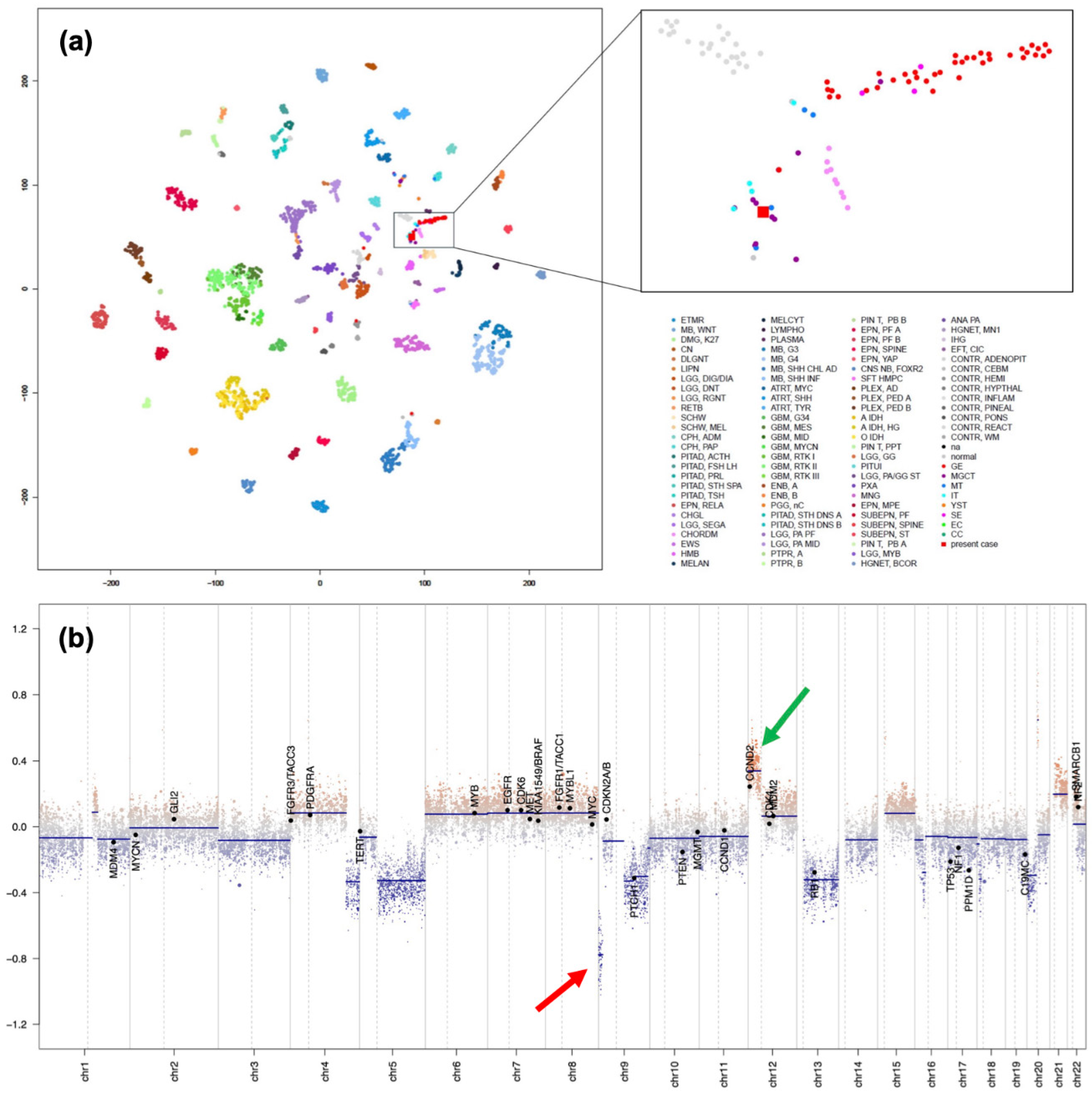
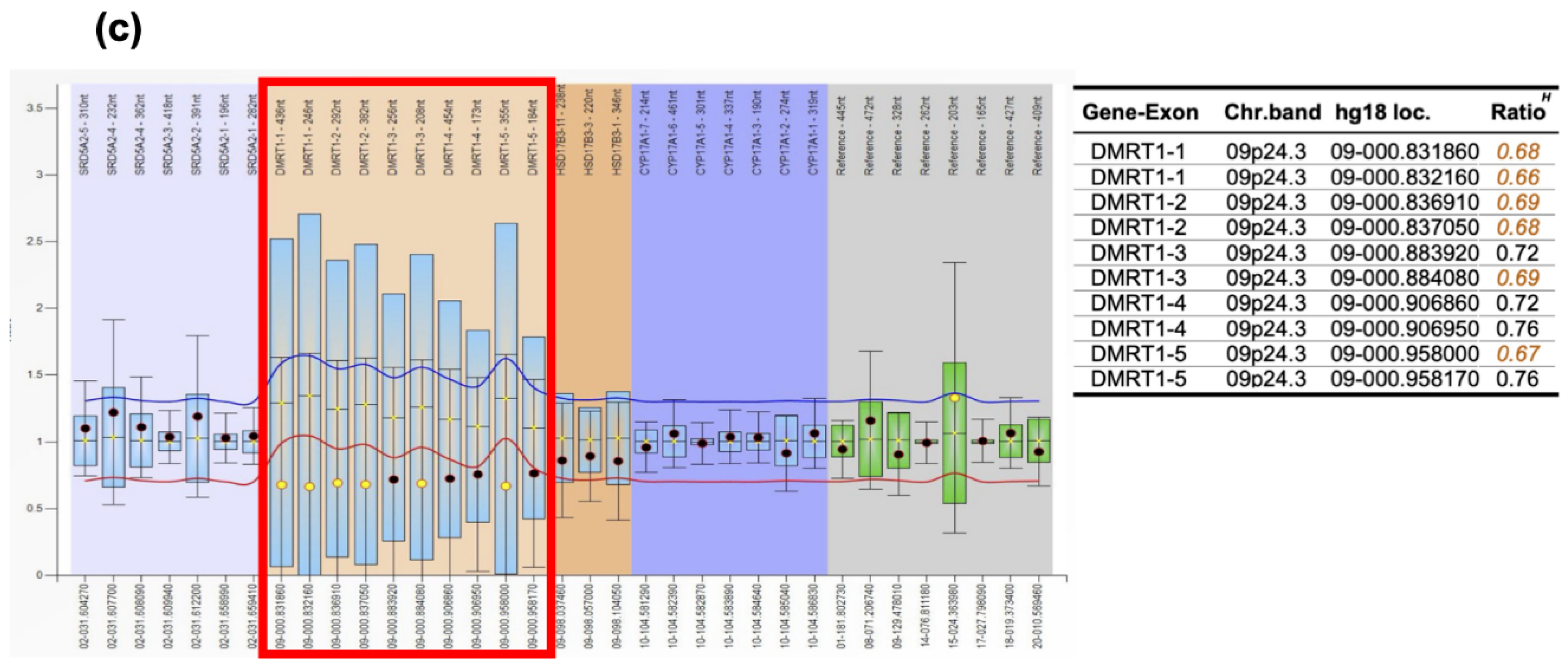
To confirm the pathological diagnosis, methylation classifier analysis ((The DKFZ classifier, developed and supplied by the German Cancer Research Center [Deutsches Krebsforschungszentrum, DKFZ] ver. 12.8 (https://www.molecularneuropathology.org)) was performed after approval by the Institutional Review Board of Niigata University (#G2022-0012) and obtaining written consent from the patient and family. The tumor matched to methylation class Teratoma with a high calibrated score of 0.99. Furthermore, t-distributed stochastic neighborhood embedding (t-SNE) analysis using relevant data provided by Capper et al. [5] and The Intracranial Germ Cell Tumor Genome Analysis (iGCT) Consortium [6] revealed that interestingly, this tumor fell in the vicinity of mixed germ cell tumors (MGCT), mature teratomas (MT) and immature teratomas (IT) (Figure 5(a)). Furthermore, analysis of copy number variation analysis showed DMRT1 loss at chromosome 9, possibly the trigger of teratoma formation, and 12p gain, which we have previously reported to be a marker of aggressive non-germinomatous germ cell tumors of the central nervous system and seen in approximately 30% of cases [6] (Figure 5(b)). However, 3p25.3 gain, a recently discovered independent predictor of poor prognosis in testicular, mediastinal [7], and central nervous system germ cell tumors [8], was not identified in the present case. We verified DMRT1 loss by MLPA analysis (Figure 5(c)) [9,10,11,12,13] using the SALSA MLPA P334-A3 Gonadal kit (MARC Holland, Amsterdam, the Netherlands).
4. Discussion
Growing teratoma syndrome is a condition in which mature teratoma with negative tumor markers arises at the site of a treated malignant germ cell tumor. It was first described in 1982 by Logothetis et al. [1]. Several studies have documented growing teratoma syndrome, but mostly in adults. To date, only 98 pediatric growing teratoma syndrome patients, of which 53 (54%) were intracranial, have been reported [14]. Growing teratoma syndrome is considered rare, but a 5% frequency of intracranial growing teratoma syndrome has been reported [15]. Classically, growing teratoma syndrome is thought to be a histologically benign mature teratoma, and growth was mainly attributed to enlargement of the multicystic component of mature teratoma [16].
The genetic background of growing teratoma syndrome has yet to be extensively studied. A cancer predisposition syndrome caused by a PTEN gene variant was reported in a 2-year-old girl with a malignant ovarian germ cell tumor, whose treatment was complicated by growing teratoma syndrome [17]. In the present case, copy number variation analysis revealed DMRT1 loss and 12p gain. DMRT1, expressed in primordial germ cells, is known to drive the reprogramming and propagation of tumor cells, which have the capacity to induce pluripotent stem cells, leading to the development of cancer that resembles human germ cell tumors [18]. Dmrt1 acts as a dose-sensitive tumor suppressor gene in 129Sv mice, and loss of Dmrt1 has been shown to cause a high incidence of testicular teratomas in mice of the 129Sv strain [19]. DMRT1 loss was verified in the recurrent tumor by MLPA. Although further functional confirmation is needed, the results of copy number variation analysis provide clues as to the causes of the formation and malignant phenotype of growing teratoma. Due to the small sampling size of the tumor during the first surgery, we were unable to perform methylation classifiers and copy number variation analyses, which would have been of considerable interest.
A recent report longitudinally studied DNA methylation, microRNA expression, and secretome in two growing teratoma syndrome patients. They found the presence of pluripotency- and yolk-sac tumor-associated genes preceding the formation of yolk-sac tumor or somatic-type malignancy, suggesting that growing teratoma syndrome is a continuously growing teratoma that may harbor occult non-seminomatous components considerably reduced during chemotherapy but with the potential to outgrow over time [20]. This new data has potential clinical implications and stresses the importance of serial assessment of biological markers in growing teratoma syndrome.
The classical definition of growing teratoma syndrome involves the histopathological confirmation of a benign, mature teratoma. In the present case, a teratoma with apparent malignant features was observed, and intense chemoradiation treatment was applied. In such cases, chemoradiation treatment may be necessary to eradicate the tumor, whereas, in classical growing teratoma cases, only prompt and complete surgical removal is necessary. Careful pathological examination is vital, and molecular analysis can be a helpful adjunct in understanding the biology of these recurrent tumors.
In treating the patient, we determined that the relapsed tumor was a teratoma with malignant features. Thus, we decided to treat with intensive chemoradiation and succeeded in eradicating the tumor. Close monitoring of potential relapse with serial MR imaging and analysis of tumor markers are being performed.
In conclusion, much remains unknown regarding the pathophysiology of growing teratoma syndrome. The presence of growing teratomas with malignant features, such as the case presented, can further confound this rare condition. Performing molecular analysis of individual cases may help elucidate the pathophysiology of this complex syndrome.
Author Contributions
Conceptualization, D.S., M.N., K.S., and M.Oi.; methodology, M.N., M.Ok., K.S., M.T.; formal analysis, M.N., K.S., T.M., and M.Ok.; investigation, M.N., M.T., T.S., K.K., H.T., Y.T., M.S., N.S., M.I., C.I., H.U., and A.K.; resources, H.T., S.F., K.I., R.N.; writing—original draft preparation, D.S., K.S., M.T., and M.N.; writing—review and editing, Y.T., H.T., and R.N.; supervision, M.Oi.; funding acquisition, M.N. and M.Oi. All authors have read and agreed to the published version of the manuscript.
Funding
This paper was funded in part by a Japanese Society for the Promotion of Science (JSPS) KAKENHI grants to M.N. (21KK0156) and M.Oi. (20K09384).
Institutional Review Board Statement
The study was conducted per the Declaration of Helsinki and approved by the Institutional Review Board of Niigata University (approval #G2022-0012 and date of approval: October 5, 2023).
Informed Consent Statement
Written informed consent was obtained from the patient and family to participate in the study and publish this paper.
Data Availability Statement
The data presented in this study are available upon reasonable request from the corresponding author.
Acknowledgments
The authors would like to acknowledge Akiko Yoshii and Tamami Murakami for their technical assistance and all participating centers of The Intracranial Germ Cell Tumor Genome Analysis (iGCT) Consortium.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Logothetis, C.J.; Samuels, M.L.; Trindade, A.; Johnson, D.E. The growing teratoma syndrome. Cancer 1982, 50, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Matsutani, M.; Sano, K.; Takakura, K.; Fujimaki, T.; Nakamura, O.; Funata, N.; Seto, T. Primary intracranial germ cell tumors: A clinical analysis of 153 histologically verified cases. J Neurosurg 1997, 86, 446–455. [Google Scholar] [CrossRef]
- Nakamura, H.; Takami, H.; Yanagisawa, T.; Kumabe, T.; Fujimaki, T.; Arakawa, Y.; Karasawa, K.; Terashima, K.; Yokoo, H.; Fukuoka, K.; et al. The Japan Society for Neuro-oncology Guideline on the diagnosis and treatment of central nervous system germ cell tumors. Neuro-Oncol 2021, 24, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Tsumanuma, I.; Tanaka, R.; Fujii, Y. (2009) Occipital transtentorial approach and combined treatments for pineal parenchymal tumors. In: Kobayashi, T. and Lunsford, L.D. eds. Pineal region tumors: Diagnosis and treatment options, Vol. 23: S.Karger AG. [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Satomi, K.; Takami, H.; Fukushima, S.; Yamashita, S.; Matsushita, Y.; Nakazato, Y.; Suzuki, T.; Tanaka, S.; Mukasa, A.; Saito, N.; et al. 12p gain is predominantly observed in non-germinomatous germ cell tumors and identifies an unfavorable subgroup of central nervous system germ cell tumors. Neuro-Oncol 2021, 24, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, D.M.; Eleveld, T.F.; Sriram, S.; Dorssers, L.C.J.; Gillis, A.J.M.; Schmidtova, S.; Kalavska, K.; van de Werken, H.J.G.; Oing, C.; Honecker, F.; et al. Chromosome 3p25.3 gain is associated with cisplatin resistance and is an independent predictor of poor outcome in male malignant germ cell tumors. J Clin Oncol 2022, 40, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Takami, H.; Satomi, K.; Fukuoka, K.; Nakamura, T.; Tanaka, S.; Mukasa, A.; Saito, N.; Suzuki, T.; Yanagisawa, T.; Sugiyama, K.; et al. Distinct patterns of copy number alterations may predict poor outcome in central nervous system germ cell tumors. Sci Rep 2023, 13, 15760. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, Y.; Natsumeda, M.; Okada, M.; Saito, R.; Kobayashi, D.; Eda, T.; Watanabe, J.; Saito, S.; Tsukamoto, Y.; Oishi, M.; et al. Dramatic response of BRAF V600E-mutant epithelioid glioblastoma to combination therapy with BRAF and MEK inhibitor: Establishment and xenograft of a cell line to predict clinical efficacy. Acta Neuropathol Commun 2019, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Okada, M.; Natsumeda, M.; Eda, T.; Abe, H.; Tsukamoto, Y.; Okamoto, K.; Oishi, M.; Takahashi, H.; Fujii, Y.; Kakita, A. EGFRvIII is expressed in cellular areas of tumor in a subset of glioblastoma. Neurol Med Chir (Tokyo) 2019, 59, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y.; Natsumeda, M.; Takahashi, H.; On, J.; Seto, H.; Saito, T.; Shibuya, K.; Ogura, R.; Ito, J.; Okada, M.; et al. Diffusely infiltrating gliomas with poor prognosis, TERT promotor mutations, and histological anaplastic pleomorphic xanthoastrocytoma-like appearance classify as mesenchymal type of glioblastoma, IDH-wildtype by methylation analysis. Neurosurgery Practice 2023, 4, e00040. [Google Scholar] [CrossRef]
- On, J.; Natsumeda, M.; Takahashi, H.; Koyama, A.; Shibuma, S.; Shibata, N.; Watanabe, J.; Saito, S.; Kanemaru, Y.; Tsukamoto, Y.; et al. Reliable detection of genetic alterations in cyst fluid DNA for the diagnosis of brain tumors. J Neurooncol 2024, 166, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Bertini, V.; Baldinotti, F.; Parma, P.; Tyutyusheva, N.; Sepich, M.; Bertolucci, G.; Rosano, C.; Caligo, M.A.; Peroni, D.; Valetto, A.; Bertelloni, S. In tandem intragenic duplication of doublesex and mab-3-related transcription factor 1 (DMRT1) in an sry-negative boy with a 46,xx disorder of sex development. Genes 2023, 14, 2067. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-Y.; Chen, H.-H.; Lee, C.-Y.; Hung, G.-Y.; Chang, T.-Y.; Chen, S.-H.; Lai, J.-Y.; Jaing, T.-H.; Cheng, C.-N.; Chen, J.-S.; et al. A case series and literature review on 98 pediatric patients of germ cell tumor developing growing teratoma syndrome. Cancer Med 2023, 12, 13256–13269. [Google Scholar] [CrossRef] [PubMed]
- Michaiel, G.; Strother, D.; Gottardo, N.; Bartels, U.; Coltin, H.; Hukin, J.; Wilson, B.; Zelcer, S.; Hansford, J.R.; Hassall, T.; et al. Intracranial growing teratoma syndrome (igts): An international case series and review of the literature. Journal of Neurooncol 2020, 147, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Tsuyuguchi, S.; Sugiyama, K.; Kinoshita, Y.; Kolakshyapati, M.; Takayasu, T.; Usui, S.; Takano, M.; Yonezawa, U.; Taguchi, A.; Amatya, V.J.; et al. Primary and recurrent growing teratoma syndrome in central nervous system nongerminomatous germ cell tumors: Case series and review of the literature. World Neurosurg 2020, 134, e360–e371. [Google Scholar] [CrossRef] [PubMed]
- Tullius, B.P.; Shankar, S.P.; Cole, S.; Triano, V.; Aradhya, S.; Huang, E.C.; Sanchez, T.; Pawar, A. Novel heterozygous mutation in the pten gene associated with ovarian germ cell tumor complicated by growing teratoma syndrome and overgrowth in a two-year-old female. Pediatr Blood Cancer 2019, 66, e27788. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, J.; Shibata, H.; Kabata, M.; Kato, M.; Fukuda, K.; Tanaka, A.; Ohta, S.; Ukai, T.; Mitsunaga, K.; Yamada, Y.; et al. DMRT1-mediated reprogramming drives development of cancer resembling human germ cell tumors with features of totipotency. Nature Commun 2021, 12, 5041. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.D.; Murphy, M.W.; Kim, S.; Cook, M.S.; Capel, B.; Zhu, R.; Matin, A.; Sarver, A.L.; Parker, K.L.; Griswold, M.D.; et al. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc Nat Acad Sci 2009, 106, 22323–22328. [Google Scholar] [CrossRef] [PubMed]
- Pongratanakul, P.; Bremmer, F.; Pauls, S.; Poschmann, G.; Kresbach, C.; Parmaksiz, F.; Skowron, M.A.; Fuß, J; Stephan, A.; Paffenholz, P.; et al. Assessing the risk to develop a growing teratoma syndrome based on molecular and epigenetic subtyping as well as novel secreted biomarkers. Cancer Lett 2024, 585, 216673. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pre-contrast (a) and post-contrast CT and T1WI (c), T2WI (d), DWI (e), and post-contrast MRI showing two components (small component indicated with yellow arrow).
Figure 1.
Pre-contrast (a) and post-contrast CT and T1WI (c), T2WI (d), DWI (e), and post-contrast MRI showing two components (small component indicated with yellow arrow).
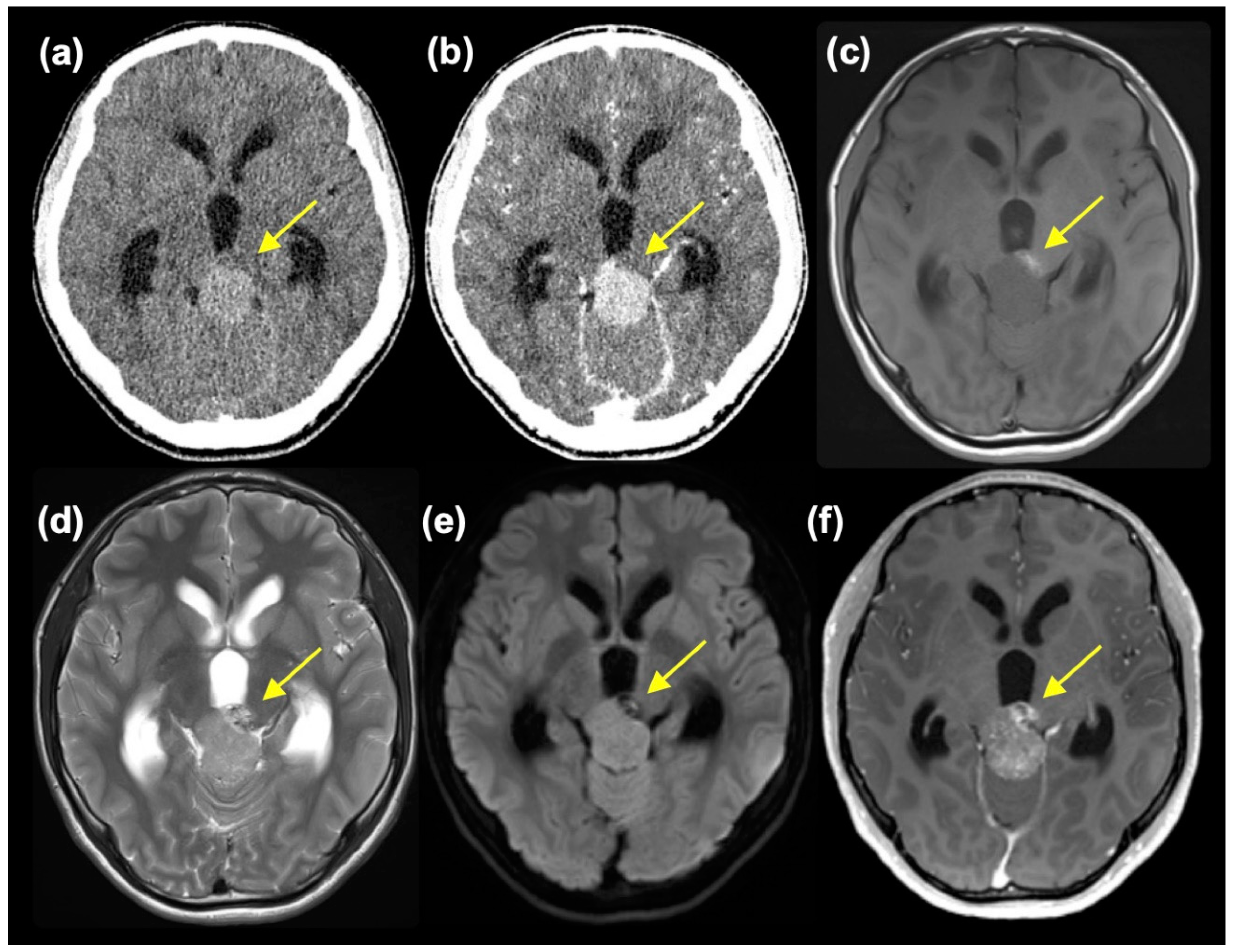
Figure 2.
Pathological findings of the initial surgery. The germinoma component showing a classic two-cell pattern (a) with PLAP-positive tumor cells (d) and small lymphoid cells. The AFP-positive component forms a thick epithelial cord (b), (e), with a small glandular structure (inset in (e)). Small calcification deposits are observed in this region (inset in (b)). CD30-positive component (c), (f). CD-30 labels large cells with severe nuclear atypia (arrows in (c)). Immunohistochemistry. ((d)-(f)). (d), PLAP; (e), AFP; (f), CD30. Bar = 50 μm for (a), (b), (d), (e), 25 μm for (c), (f), and 35 μm for insets in (b) and (e).
Figure 2.
Pathological findings of the initial surgery. The germinoma component showing a classic two-cell pattern (a) with PLAP-positive tumor cells (d) and small lymphoid cells. The AFP-positive component forms a thick epithelial cord (b), (e), with a small glandular structure (inset in (e)). Small calcification deposits are observed in this region (inset in (b)). CD30-positive component (c), (f). CD-30 labels large cells with severe nuclear atypia (arrows in (c)). Immunohistochemistry. ((d)-(f)). (d), PLAP; (e), AFP; (f), CD30. Bar = 50 μm for (a), (b), (d), (e), 25 μm for (c), (f), and 35 μm for insets in (b) and (e).
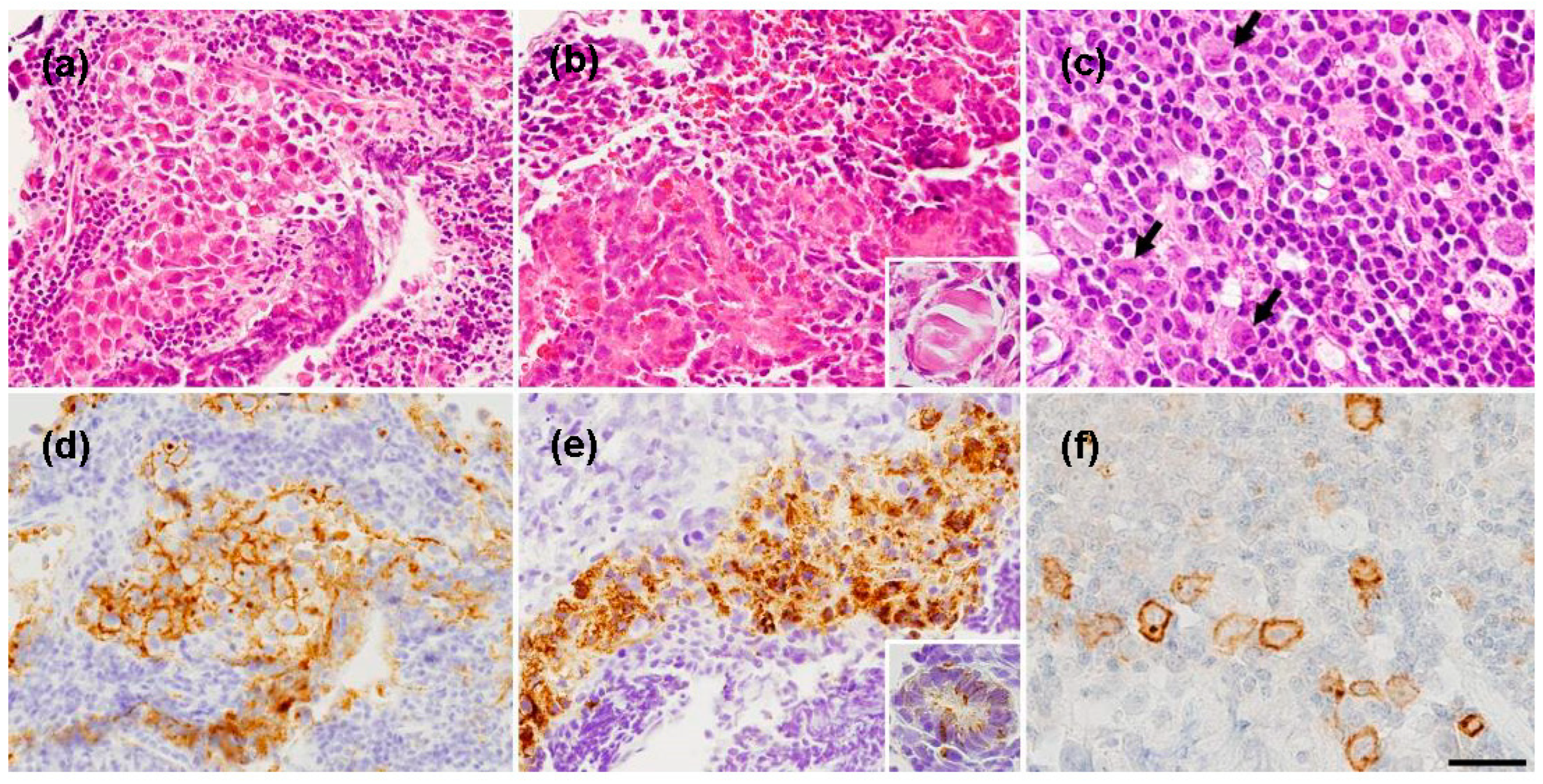
Figure 3.
Post-contrast MR images taken after one course (a), two courses (b) of chemotherapy, and after removal (c).
Figure 3.
Post-contrast MR images taken after one course (a), two courses (b) of chemotherapy, and after removal (c).
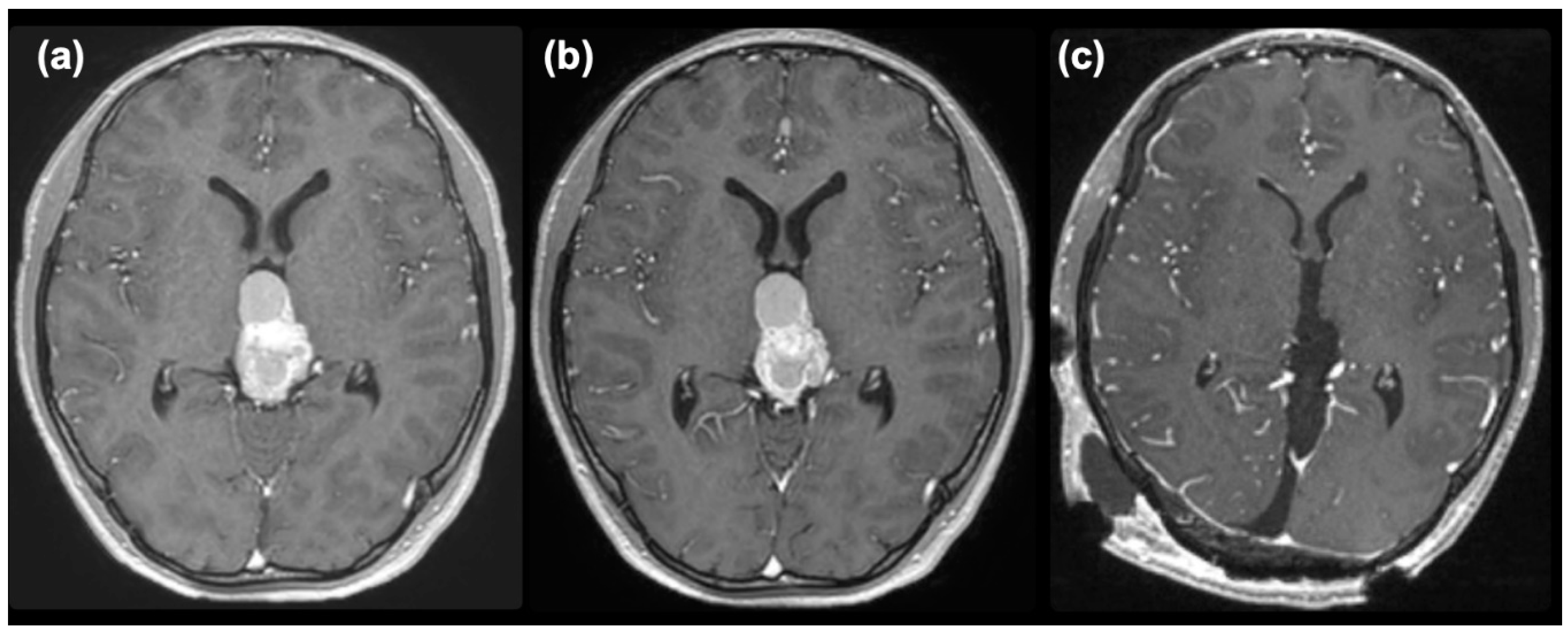
Figure 4.
Pathologic findings of the second surgery. Teratomatous tumor with cartilage, bone, fibroblasts, and mesenchymal tissue (a). Cartilage tissue with relatively high cellularity containing chondrocytes with nuclear atypia (arrowhead) and frequent double nuclei (arrows). (b) Immature mesenchymal tissue (c) with a high MIB-1 labeling index of 30%. Plump eosinophilic large cells (e) with with GFAP-positive small cells (f). MIB-1 labeling index in this area is 13% (g). Immunohistochemistry ((d), (f), (g)). (d), (g); MIB-1(f); GFAP. Bar = 120 µm for (a), 50 µm for (b)-(g), and 10 µm for insets in (c) and (e).
Figure 4.
Pathologic findings of the second surgery. Teratomatous tumor with cartilage, bone, fibroblasts, and mesenchymal tissue (a). Cartilage tissue with relatively high cellularity containing chondrocytes with nuclear atypia (arrowhead) and frequent double nuclei (arrows). (b) Immature mesenchymal tissue (c) with a high MIB-1 labeling index of 30%. Plump eosinophilic large cells (e) with with GFAP-positive small cells (f). MIB-1 labeling index in this area is 13% (g). Immunohistochemistry ((d), (f), (g)). (d), (g); MIB-1(f); GFAP. Bar = 120 µm for (a), 50 µm for (b)-(g), and 10 µm for insets in (c) and (e).
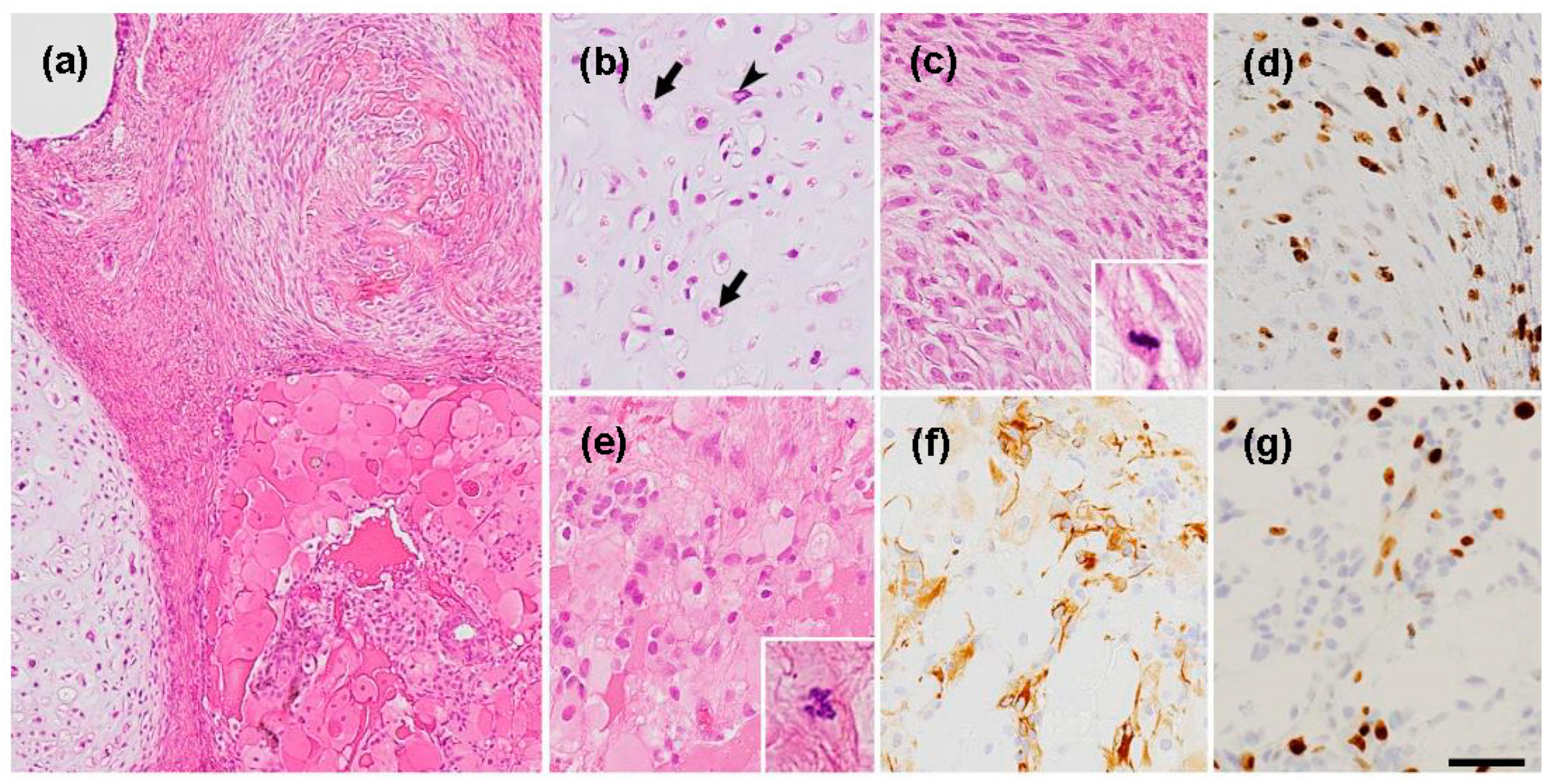
Figure 5.
(a) tSNE analysis of recurrent tumor in the present case (black arrow) (b) Copy number variation analysis revealing DMRT1 loss (red arrow) at chromosome 9 and 12p gain (green arrow). (c) MLPA analysis confirmed DMRT1 loss.
Figure 5.
(a) tSNE analysis of recurrent tumor in the present case (black arrow) (b) Copy number variation analysis revealing DMRT1 loss (red arrow) at chromosome 9 and 12p gain (green arrow). (c) MLPA analysis confirmed DMRT1 loss.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



