
Submitted:
16 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
The use of privileged scaffolds as starting point for the construction of libraries of bioactive compounds is a widely used strategy in drug discovery and development. Scaffold decoration, morphing and hopping are additional techniques that allow to modify the privileged framework chosen and better explore the chemical space around it. In this study, two series of highly functionalized pyrimidine and pyridine derivatives were synthesized by scaffold morphing approach of triazine compounds obtained previously as antiviral agents. Newly synthesized azines were evaluated against lymphoma, hepatocarcinoma, and colon epithelial carcinoma cells showing in five cases acceptable to good anticancer activity associated to low cytotoxicity on healthy fibroblasts. Finally, ADME in vitro studies were conducted on the best derivatives of the two series showing good passive permeability and resistance to metabolic degradation.
Keywords:
Privileged scaffold
; triazines
; pyrimidines
; pyridines
; scaffold morphing
; anticancer activity.
1. Introduction
The concept of privileged scaffold was introduced by Evans in 1988 [1] and represents a molecular framework able to bind more than one receptor and act as agonist or antagonist, when conveniently functionalize. Among all the drug discovery strategies to harness privileged scaffolds, decoration with opportune substituents is a good technique that allow the exploration of the biologically relevant chemical space around the chosen promising framework [2]. In addition, scaffold morphing [3] and hopping [4] are useful procedures to replace the central core structure of an active molecule with the aim of identify new bioactive entities with increased pharmacodynamic and pharmacokinetic properties.
Basic nuclei of great interest are azines, six membered aromatic heterocycles that contain at least one nitrogen atom. Therefore, triazines, pyrimidines and pyridines (Figure 1 panel a), can be considered privileged scaffolds [5] since their high prevalence in commercially available drugs [6] and biologically relevant biomolecules (e.g., enzymes cofactors, nucleobases) [7].
In the last few years, our research group has been involved in the synthesis and decoration of privileged scaffolds [8]. Concerning triazines, our team recently published a paper [9] focused on the synthesis and evaluation of compounds 1-9 against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) (Figure 1 panel b, and Table 1). Unfortunately, from this library of molecules only compound 1 emerged as the one able to inhibit SARS-CoV-2 replication with an IC50 (half-maximal compound concentration inhibiting the 50% of virus replication) value of 12.1 micromolar (μM).
Figure 1.
Chemical structure of three azine basic nuclei (panel a) and a focused library of triazine derivatives (1-9) synthesized in a previous work (panel b) [9].
Figure 1.
Chemical structure of three azine basic nuclei (panel a) and a focused library of triazine derivatives (1-9) synthesized in a previous work (panel b) [9].
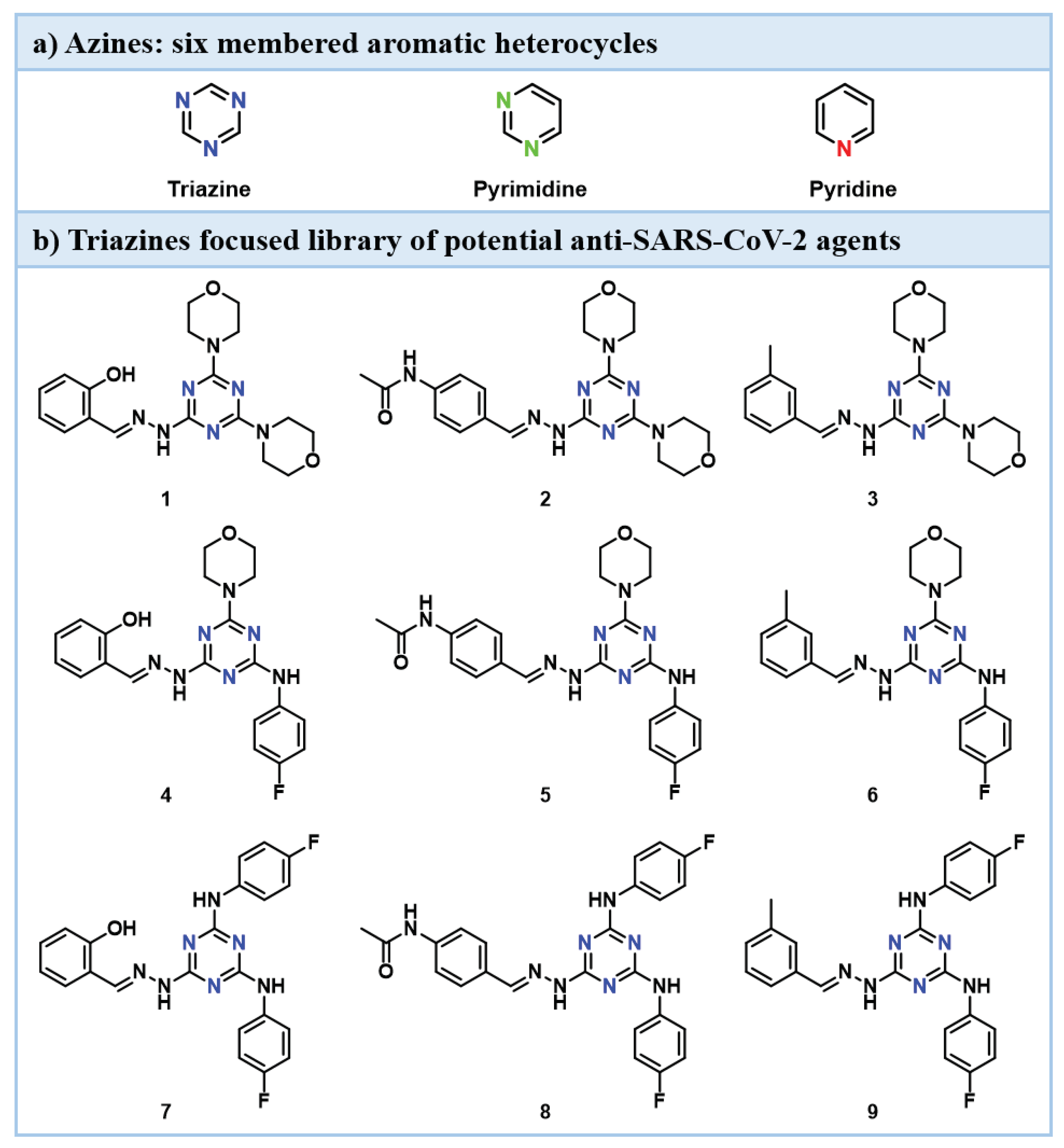
Additional studies aimed at addressing a potential antiviral activity of derivatives 1-9 were conducted on several pathogens, highlighting a low or absent effect on human immunodeficiency (HIV-1), Dengue (DENV) and West Nile viruses (WNV) (Table 1). The exception registered was compound 4 that showed potency on flaviviruses DENV and WNV in the low micromolar range (2.0 and 1.1 µM, respectively).
Surprisingly, viability assays conducted on uninfected lymphoma (H9), hepatocarcinoma (Huh7), and colon epithelial carcinoma (Caco-2) cells showed a cytotoxic effect of the synthesized molecules. This behavior prompts us to test compounds 1-9 on human normal fibroblasts FB789 and calculate the tumor selectivity indexes (TSIs) as the ratio between half-maximal compound cytotoxic concentration (CC50) on FB789 and in turn CC50 in H9, Huh7 and Caco-2. From all the evaluations made, derivatives 4 and 7 seemed to be interesting hit compounds, since they showed low micromolar antitumor activity on Huh7 (1.3 and 4.3 TSIs for derivatives 4 and 7, respectively) and H9 (5.7 and 3.0 TSIs for triazines 4 and 7, respectively) and submicromolar effect on Caco-2 for compound 7 (8.1 TSI) (Table 1, entries 4 and 7). Moreover, from the cytotoxicity on FB789 cells point of view, triazine 3 highlighted the best profile compared to other compounds of the series (Table 1, entry 3). For these reasons, we decided to begin a study based on these results serendipitously obtained with triazine molecules aimed at the identification of potential antineoplastic agents for lymphoma, hepatocarcinoma and colorectal cancer. Hence, we decided to apply a scaffold morphing approach to replace the triazine core with pyrimidine and pyridine ones and to decorate these azine nuclei with the substituents that showed the best pharmacological performance in the previous study [9]. In detail, morpholine and 4-fluoroaniline were used as the pharmacophoric functionalities directly linked to the central six membered heterocycle. Salicyl and 3-methylphenyl hydrazones were chosen to complete the panel of substituents for the novel highly functionalized pyrimidine and pyridine products, since characteristic of above-mentioned derivatives 3, 4 and 7. In the present study, in fact, two small libraries of totally twelve compounds were synthesized and were evaluated against H9, Huh7, and Caco-2 lines. Finally, the in vitro absorption, distribution, metabolism, and excretion (ADME) properties of selected derivatives were screened to define the most promising scaffold and the best highly functionalized derivative synthesized.

Table 1.
In vitro cytotoxicity and tumor selectivity index (TSI) of the synthesized compounds in a cell-based model.1
Table 1.
In vitro cytotoxicity and tumor selectivity index (TSI) of the synthesized compounds in a cell-based model.1
| FB7892 | H92 | Huh72 | Caco-22 | |||||
|---|---|---|---|---|---|---|---|---|
| Entry | CPD | CC503 | CC50 | TSI | CC50 µM | TSI | CC50 | TSI |
| 1 | 1 | 30.0±5.6 | >100 | <0.3 | >100 | <0.3 | >400 | <0.1 |
| 2 | 2 | 134.0±10.5 | >100 | <1.3 | >200 | <0.7 | >200 | <0.7 |
| 3 | 3 | 250.0±9.7 | 74.0±6.2 | 3.4 | 45.0±10 | 5.6 | 184.0±10.5 | 1.4 |
| 4 | 4 | 10.9±3.1 | 1.9±0.8 | 5.7 | 8.7±1.2 | 1.3 | 3±0.9 | 3.6 |
| 5 | 5 | 27.0±3.2 | 30.0±4.6 | 0.9 | 45.0±6.4 | 0.6 | 70.0±6.4 | 0.4 |
| 6 | 6 | 100.0±7.8 | >100 | <1.0 | 47.0±4.8 | 2.1 | 22.0±1.5 | 4.5 |
| 7 | 7 | 6.5±2.3 | 2.2±0.6 | 3.0 | 1.5±0.4 | 4.3 | 0.8±0.1 | 8.1 |
1 All experiments were conducted in duplicate in three independent experiments in FB789, H9, Huh7 and Caco-2 cells; 2 experiment read-out 48h; 3 CC50, half-maximal compound cytotoxic concentration, expressed in micromolar (µM) range, as determined by Cell-Titer Glo kit (Promega). TSI, tumor selectivity index (ratio between CC50 on FB789 and CC50 on cancer cells H9, Huh7 and Caco-2). CPD = compound.
2. Results and discussion
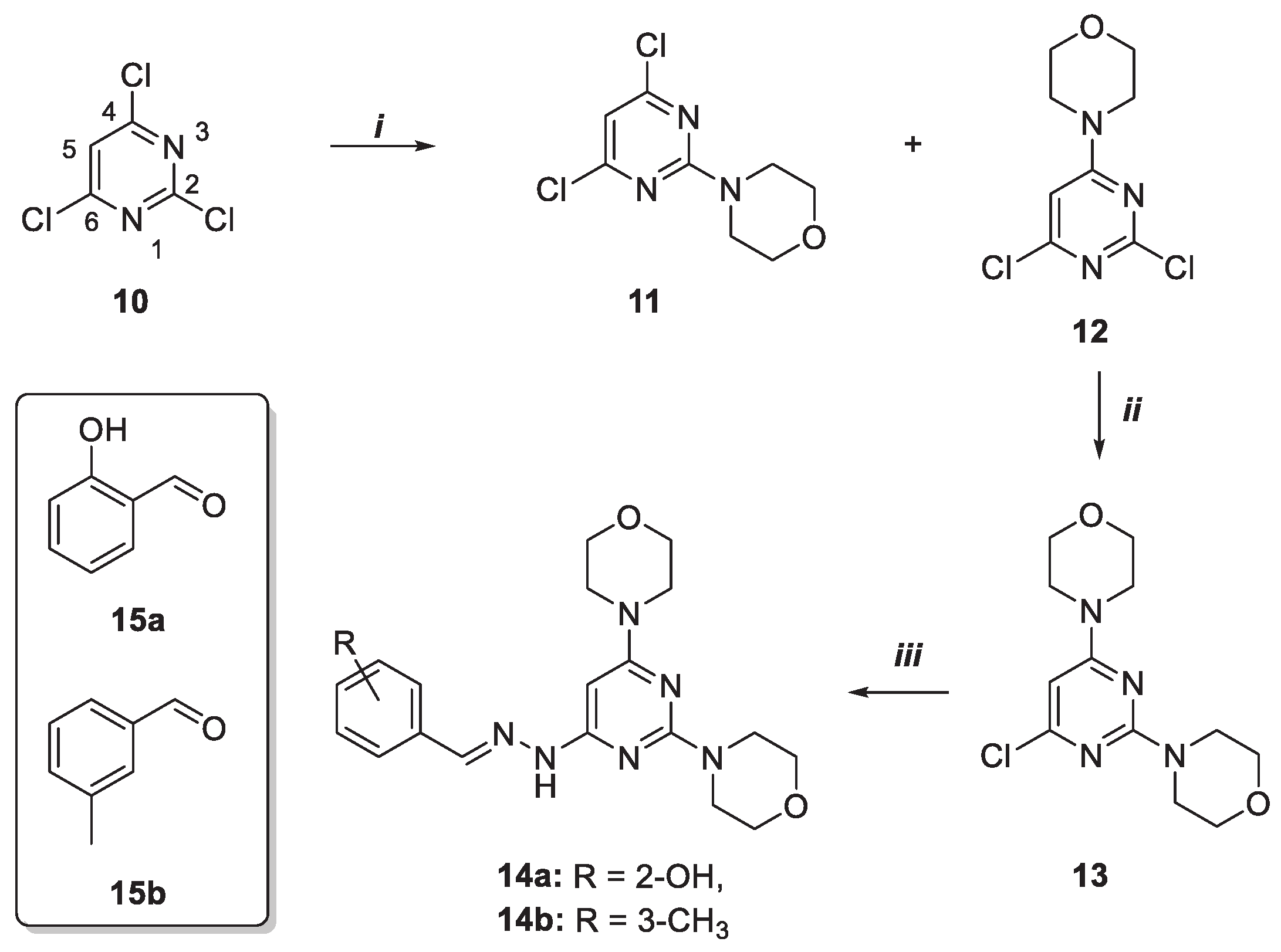
As depicted in Scheme 1, highly functionalized trisubstituted pyrimidine derivatives 15a–b were obtained in a three steps sequence starting from 2,4,6-trichloro pyrimidine 10. The latter was transformed into compounds 11 and 12 in 19 and 68% yield by reaction with morpholine in the presence of N,N-diisopropylethylamine (DIPEA) and at 0 °C. The higher yield of the C6-substituted 12, compared to the C-2 functionalized 11, is due to steric hinderance exerted by the lone pairs of the N1 and N3 atoms that discourage the access of the nucleophile in C2 position of the pyrimidine ring.
To distinguish between the two regioisomers and for the final structure assignment, 2D NMR Nuclear Overhauser Effect Spectroscopy (NOESY) experiments were conducted [supplementary materials S#1]. In derivative 12 was possible to identify the interaction between proton in position 4 of the pyrimidine ring and protons on the carbon atom adjacent to morpholine nitrogen, absent in 11.
Then, 12 was subjected to a second substitution reaction always in the presence of morpholine at 110 °C under microwave (MW) irradiation to obtain compound 13. Finally, disubstituted pyrimidine 13 was reacted with hydrazine (NH2NH2) and subsequently with the opportune aldehyde, salicyl- and 3-methylphenyl-aldehydes 15a-b, to give desired products 14a and 14b with 55 and 65% yield, respectively.
Scheme 1.
Synthesis of pyrimidine derivatives 14a-b. Reagents and conditions: i) dichloromethane (CH2Cl2), morpholine, N,N-Diisopropylethylamine (DIPEA), 0 °C, 5 h; ii) 1,4-dioxane, morpholine, HCl, MW, 110 °C, 40 min.; iii) 1. 1,4-dioxane, NH2NH2, MW, 100 °C, 4 h, 2. methanol (MeOH), 15a–b, acetic acid, 25 °C, 24 h.
Scheme 1.
Synthesis of pyrimidine derivatives 14a-b. Reagents and conditions: i) dichloromethane (CH2Cl2), morpholine, N,N-Diisopropylethylamine (DIPEA), 0 °C, 5 h; ii) 1,4-dioxane, morpholine, HCl, MW, 110 °C, 40 min.; iii) 1. 1,4-dioxane, NH2NH2, MW, 100 °C, 4 h, 2. methanol (MeOH), 15a–b, acetic acid, 25 °C, 24 h.
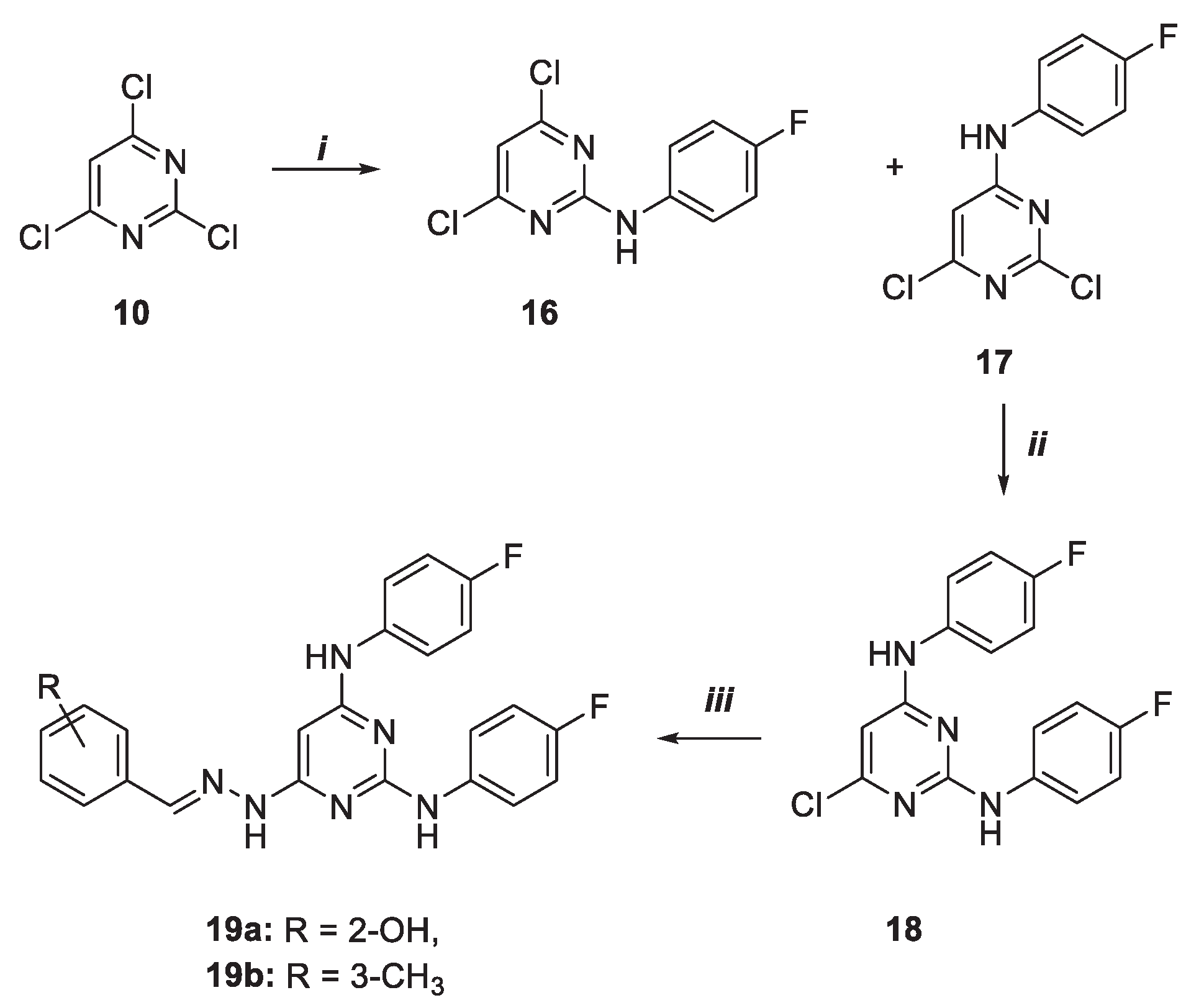
Highly functionalized pyrimidine 19a-b were synthesized with a pathway similar to the above-described one (Scheme 2). First, 2,4,6-trichloro pyrimidine 10 was reacted with 4-flouroaniline to give a mixture of two regioisomers 16 and 17 with 14 and 75% yield. The latter compound was further functionalized with a second 4-fluoroaniline by reaction at 110 °C in the presence of a catalytic amount of HCl as pyrimidine N-protonating/activating agent. The final hydrazone derivatives 19a-b were obtained by, again, NH2NH2 introduction and reaction with aldehydes 15a-b.
The last two desired pyrimidines 21a–b were afforded starting from the monosubstituted intermediate 17 previously obtained (Scheme 3). The latter was easily turned into final products by nucleophilic substitution with morpholine and the hydrazone two steps sequence installation.
Pyridine derivatives were synthesized as follows. Starting form 2,4,6-trichloropyridine 22, by nucleophilic substitution with morpholine in tetrahydrofuran (THF) and triethyl amine (Et3N) at 65 °C, a mixture of the two regioisomers 23 and 24, bearing the amine substituent on C-4 and C-2 respectively, was obtained (Scheme 4). Structural assignment was made based on 1H NMR experiments, being 23 symmetric and showing only one peak for C3-C5 protons in the recorded spectrum (S#2). From compound 24, two regioisomeric pyridines, compounds 25 and 28, were synthesized by morpholine nucleophilic substitution at 120°C. Then, many attempts to directly introduce NH2NH2 on the heterocyclic ring were made, such as the use of high temperature, MW irradiation, high concentration of NH2NH2, but the desired pyridine carrying the hydrazine functionality on C-4 was never isolated. Finally, with tert-butoxy carbonyl (Boc) protected hydrazine, namely tert-butyl carbazate (BocNHNH2), was possible to decorate the C-4 position of the azine core. Compound 26 so obtained, was turned into the final trisubstituted products 27 a-b with 39 and 18% yield by acidic Boc removal in the presence of trifluoroacetic acid (TFA) and reaction with the opportune aldehydes 15a-b in a mixture of MeOH and few dops of acetic acid at 25 °C.
As depicted in Scheme 5, the introduction of 4-fluoroaniline on 22 was obtained by palladium catalyzed coupling giving compounds 29 and 30. The latter was subjected to a second round of functionalization with 4-fluoroaniline that led to the recovery of the regioisomeric derivatives 31 and 34. The 2,6-disubstituited pyridine 31 was subjected to reaction with BocNHNH2 under MW irradiation at 160 °C in the presence of Pd2(dba)3, DPPF, and Cs2CO3. The boc-protected hydrazine product 32 obtained was converted into the desired highly decorated pyridines 33a-b by acid deprotection and reaction with aldehydes 15a-b, respectively.
Finally, compounds 38a-b were obtained in 37% and 33% yield with a three steps sequence starting from monosubstituted pyridine 30 (Scheme 6). The latter was converted into the desired products by reaction with morpholine at 160 °C, BocNHNH2 insertion, deprotection and hydrazone formation in the presence of aldehydes 15a-b.
All synthesized compounds were evaluated for their anticancer activity against lymphoma, hepatocarcinoma, and colon epithelial carcinoma on H9, Huh7 and Caco-2 cells respectively (Table 2). Healthy FB789 fibroblasts were used as a reference for cytotoxicity determinations. TSIs were calculated as the ratio of CC50 values obtained with FB789 cells and, in turn, H9, Huh7 and Caco-2.
In detail, cancer and normal cells were incubated in presence of increasing concentration of pyrimidine and pyridine derivatives for 48 h. As reported in Table 2, derivatives 14a, 14b and 21b are characterized by a cytotoxicity of the same order of magnitude of the antineoplastic activity in all cell line evaluated, as expressed by a TSI < 1 (Table 2, entries 1, 2 and 6). Pyrimidine 21a had a different behavior, showing moderate antiproliferative activity on the Huh7 (TSI = 3.3) and Caco-2 (TSI = 4.9) cell lines, but no effect on H9 cell line (Table 2, entries 5). Similarly, compounds 19a-b displayed high antiproliferative activity on Huh7 (13.6 and 10.2 TSIs for 19a and 19b respectively) and Caco-2 (6.8 and 15.3 TSIs for 19a and 19b respectively) cell lines (Table 2, entries 3 and 4), and low effects on H9 cell line (0.4 and 1.3 TSIs for 19a and 19b respectively). The difference observed between H9 and Caco-2/Huh7 could be imputable to the different nature and origin of these cell lines, considering that H9 are suspension cell line derived by a lymphoma, while Caco-2 and Huh7 are both adherent epithelial cell line derived by a carcinoma.
Regarding pyridines, derivatives 27a-b showed a low anticancer activity (TSI < 3) on H9, Huh7 and Caco-2 cells (Table 2, entries 7 and 8). Compounds 33a and 38a are characterized by a high cytotoxicity on FB789 (Table 2, entries 9 and 11). Pyridines 33b and 38b, instead showed high TSI values in all cells evaluated, due to a very low cytotoxic effect on healthy fibroblasts (Table 2, entries 10 and 12), associated to an antiproliferative activity that ranged from 22.7 μM, in the worst case, to 8.1 μM, in the best case.
Table 2.
Antiproliferative activity (µM) of highly functionalized pyrimidine and pyridine compounds against H9, Huh7 and Caco-2 cell lines.1
Table 2.
Antiproliferative activity (µM) of highly functionalized pyrimidine and pyridine compounds against H9, Huh7 and Caco-2 cell lines.1
| FB7892 | H92 | Huh72 | Caco-22 | |||||
|---|---|---|---|---|---|---|---|---|
| Entry | CPD | CC503 | CC50 µM | TSI | CC50 | TSI | CC50 | TSI |
| 1 | 14a | 30.2±5.9 | >100 | <0.3 | 82.3±6.1 | 0.4 | 84.0±6.5 | 0.4 |
| 2 | 14b | 9.2±1.3 | 14.0±7 | 0.7 | 8.2±4.5 | 1.1 | 11.0±1.8 | 0.8 |
| 3 | 19a | 13.6±2.8 | 30.5±7.8 | 0.4 | 1.0±0.3 | 13.6 | 2.0±0.5 | 6.8 |
| 4 | 19b | 61.3±9.3 | 47.5±3.5 | 1.3 | 6.0±1.1 | 10.2 | 4.0±1.0 | 15.3 |
| 5 | 21a | 9.8±5.4 | 11.0±2.1 | 0.9 | 3.0±0.9 | 3.3 | 2.0±0.4 | 4.9 |
| 6 | 21b | 95.1±0.9 | >100 | <1.0 | 67.0±23.0 | 1.4 | 113.0±4.1 | 0.8 |
| 7 | 27a | 10.9±3.4 | 76.0±4.0 | 0.1 | 44.0 ±5.1 | 0.2 | 29.0±1.8 | 0.4 |
| 8 | 27b | 80.9±2.3 | 50.0±4.3 | 1.6 | 39.0±3.4 | 2.1 | 30.0±1.8 | 2.7 |
| 9 | 33a | 0.3±0.3 | 42.6±9.1 | 0.01 | 17.6±4.9 | 0.02 | 12.2±3.8 | 0.02 |
| 10 | 33b | 390.3±29.8 | 22.7±7.1 | 17.2 | 20.9±12.2 | 18.7 | 11.3±8.6 | 34.5 |
| 11 | 38a | 2.8±0.9 | 37.4±1.8 | 0.1 | 24.3±14.0 | 0.1 | 18.8±0.1 | 0.1 |
| 12 | 38b | 90.3±8.9 | 18.1±3.7 | 5.0 | 9.0±5.7 | 10.0 | 8.1±2.8 | 11.1 |
1 Values are the means ± SD of experiments run in triplicate; 2 experiments read out at 48 h; 3 CC50, half-maximal compound cytotoxic concentration, expressed in micromolar (µM) unit. TSI, tumor selectivity index (ratio between CC50 on FB789 and CC50 on cancer cells H9, Huh7 and Caco-2). CPD = compound.
Based on results obtained by cellular evaluations, further studies aimed at investigating the in vitro ADME properties were conducted either on compounds that showed the highest TSI values, such as 19a, 19b and 21a, or the lowest cytotoxic effect on normal cells, products 33b and 38b (Figure 2).
Firstly, the kinetic aqueous solubility of all five derivatives was studied diluting a DMSO stock solution of these compounds with Mill-Q H2O and letting incubate for 3 h at room temperature (RT). As reported in Table 3, all pyrimidine derivatives 19a, 19b, and 21a showed suboptimal values of water solubility, less than 0.1 µg/mL (limit of detection, LOD).
Table 3.
Kinetic water solubility of tested compounds.
| CPD | µg/mL | LogS1 | Predicted LogS2 |
|---|---|---|---|
| 19a | <0.1 | - | -7.50 |
| 19b | <0.1 | - | -7.82 |
| 21a | <0.1 | - | -6.69 |
| 33b | <0.1 | - | -7.87 |
| 38b | 0.54 | -5.87 | -5.92 |
1 Log of Solubility reported as mol/L. 2 LogS data predicted with SwissADME.
Figure 2.
Structure of compounds 19a, 19b, 21a, 33b, 38b.
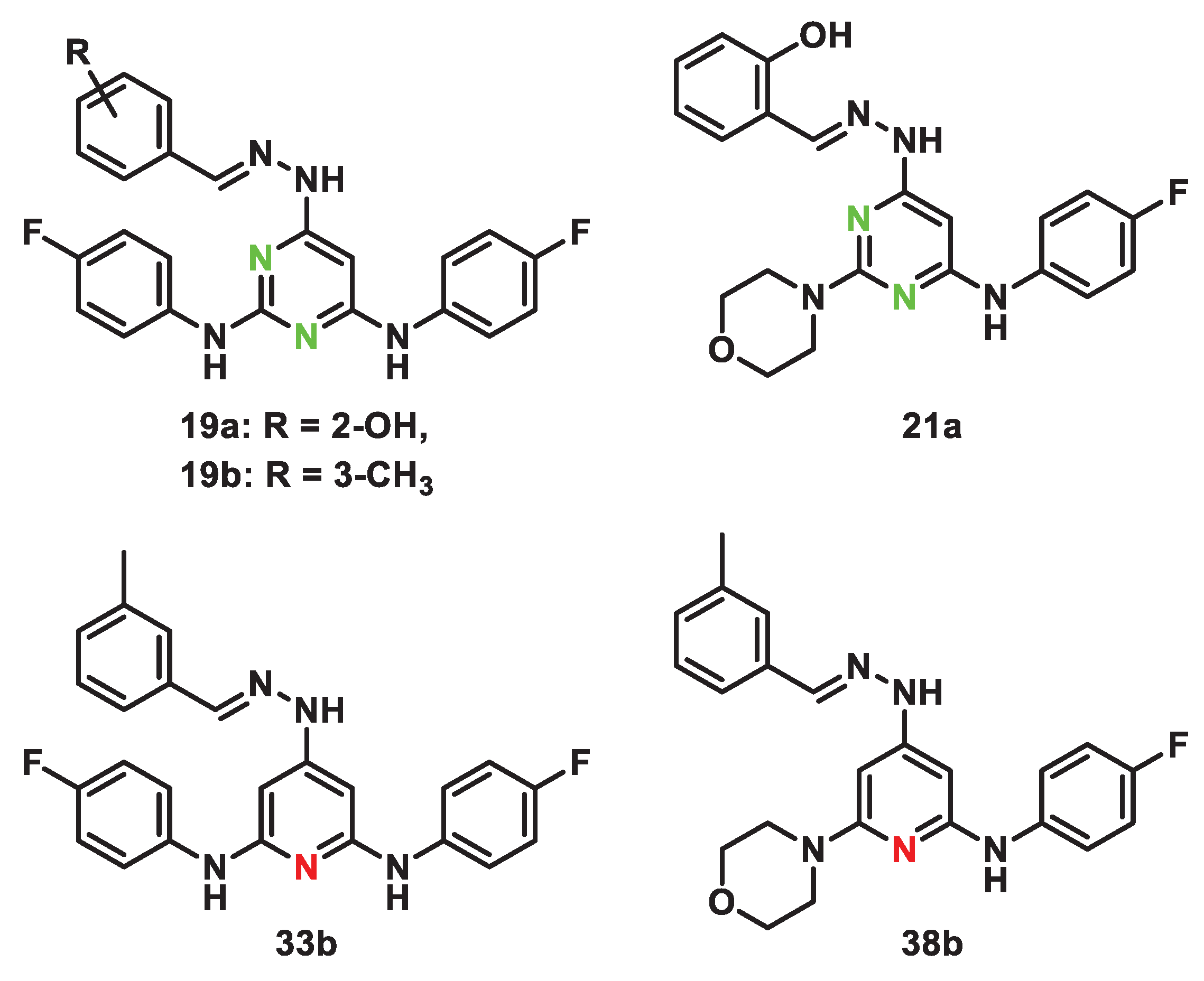
Indeed, the presence of the morpholine and phenol moieties in 21a, did not gave significant improvement to the water solubility in comparison to 19a (with a phenol group and two para fluorophenyl motifs) or 19b (with two para fluorophenyl groups and 3-methylphenyl residue). The replacement of the pyrimidine scaffold with a pyridine one, slight enhances the solubility only when one fluorophenyl group was substituted with a morpholine. In fact, if 38b showed a LogS value of -5.87, 33b confirmed the trend seen for 19a, 19b, and 21a. The aqueous solubility of selected compounds was also predicted using the online tool SwissADME; moving from data obtained for 38b, the predicted value resulted quite similar to experimental one suggesting the reliability of the prediction. Predicted values obtained for 19a, 19b, 21a, and 33b confirmed the suboptimal water solubility of tested derivatives with LogS values between -6.69 and -7.87.
The parallel artificial membrane permeability assay (PAMPA) underlined a general tendency to efficiently cross membranes. As reported in Table 4, the pyrimidine derivatives 19a, 19b, and 21a showed good apparent permeability values probably due to the scaffold that reduce the aqueous solubility and promotes the interaction with phospholipid bilayer. Indeed, 19a and 19b, both characterized by two para fluorophenyl groups, resulted in good apparent permeability values and higher percentages of membrane retention (20.99% and 27.42%, respectively) than 21a endowed with a good passive permeability and a very low membrane retention due to the presence of a morpholine moiety (Papp 4.80 x 10-6 cm/s, MR 25.9%).
Among the pyridine derivatives, the two para fluorophenyl moieties of 33b significantly increased the membrane retention up to 46.84% if compared to 38b whose percentage remained around 20.99%. In terms of apparent permeability, the high interaction of 33b with lipidic bilayer influenced its ability to cross membrane; indeed, the Papp value was noticeably reduced to 1.38 x 10-6 cm/s in comparison to 38b whose Papp value was 6.35 x 10-6 cm/s.
Regarding the stability of tested compounds in presence of human liver microsomes, the five derivatives showed percentages of metabolic stability never lower than 97%, suggesting a general high resistance to the transformations induced by liver microsomes (Table 5). 19a, 19b, and 21a resulted in very stable compounds (>99.9%), while 33b and 38b underwent to a slight metabolization leading to the formation of oxidized derivative M1 (1.68% and 2.06%, respectively).
Finally, the stability in human plasma was investigated incubating for 24 h a DMSO solution of compounds in presence of HEPES buffer and human plasma. According to Table 5, all tested derivatives undergone to a slight metabolization leading to percentages of plasmatic stabilities never lower than 81% after 24 h of incubation.
3. Materials and Methods
3.1. Chemistry – General part
All reactions were performed in flame-dried glassware under a nitrogen atmosphere. Reagents were obtained from commercial suppliers (Merck Srl, Milan, Italy) and used without further purification. TLC chromatography was performed on precoated aluminum silica gel SIL G/UV254 plates (Macherey-Nagel & Co.). The detection occurred via fluorescence quenching or development in a ninhydrin solution (0.2 g of ninhydrin in 99.5 ml ethanol and 0.5 ml acetic acid.). Merck silica gel 60 was used for flash chromatography (23-400 mesh). 1H NMR and 13C NMR spectra were measured on a Bruker Avance DRX400 (400 MHz/100 MHz) spectrometer. Chemical shifts for protons are reported in parts per million (ppm, δ scale) and internally referenced to the deuterated dimethyl sulfoxide (DMSO-d6), methanol (CD3OD) or chloroform (CDCl3) signal at δ 2.50, 3.33 and 7.28 ppm, respectively. 1H-NMR spectra are reported in this order: multiplicity and number of protons; signals were characterized as: s (singlet), d (doublet), dd (doublet of doublets), ddd (doublet of doublet of doublets), t (triplet), m (multiplet), bs (broad signal). 2,4,6-trichlorotriazine, 2,4,6-trichloropyrimidine and 2,4,6-trifluoropyridine have been bought from Merck.
Microwave irradiation experiments were conducted using a CEM Discover Synthesis Unit (CEM Corp., Matthews, NC, USA). The apparatus consists of a continuous focused microwave power delivery system with operator-selectable power output from 0 to 300 W. The temperature of the contents of the tube was monitored using a calibrated IR temperature control mounted under the reaction tube. All experiments were performed using a stirring option where the contents of the tube are stirred by means of a rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar in the tube.
Mass detention of compounds was performed using an Agilent 1260 Infinity HPLC-DAD system connected to an Agilent MSD 6130 system (Agilent Technologies, Palo Alto, CA, USA) as described in the UV/LC-MS method section below.
3.2. Chemistry – Experimental procedures and Compound characterization
Procedure for the synthesis of monosubstituted pyrimidine derivatives 11 and 12:
To a solution of 2,4,6-trichloropyrimidine 10 (1 equiv.) in CH2Cl2 (10 mL) at 0 °C, morpholine (1 equiv.) was slowly added, followed by DIPEA (1 equiv.). The reaction mixture was stirred at 0 °C for 5h, and then warmed up to 25 °C. The mixture was washed with H2O and brine. The organic phase was dried over anhydrous Na2SO4 and evaporated to dryness. The resulting residue was purified by column chromatography using a mixture of petroleum ether (PET)/ethyl acetate (EtOAc) 4:1 to give the desired products 11 and 12 with 19% and 68% yield, respectively.
Compound 11
Rf= 0.29 (PET/EtOAc 4:1); 1H NMR (400 MHz, CDCl3) δ 6.57 (s, 1H), 3.83 (d, J=5.2 Hz, 4H), 3.76 (d, J=4.8 Hz, 4H) ppm. 13C NMR (100 MHz, CDCl3) δ 161.8, 160.6, 108.3, 66.6, 44.4 ppm. MS m/z for C8H10Cl2N3O, (ESI+) m/z: 234.0 [M+H]+.
Compound 12
Rf= 0.26 (PET/EtOAc 4:1); 1H NMR (400 MHz, CDCl3) δ 6.41 (s, 1H), 3.79-3.77 (m, 4H), 3.65 (s, 4H) ppm. 13C NMR (100 MHz, CDCl3) δ 163.3, 160.7, 159.9, 99.7, 66.2, 44.6 ppm. MS m/z for C8H10Cl2N3O, (ESI+) m/z: 234.0 [M+H]+.
Procedure for the synthesis of disubstituted pyrimidine derivative 13
Compound 12 (1 equiv.) was dissolved in 1,4-dioxane (10 mL) and morpholine (2.5 equiv.) was added. Then, were slowly added few drops of HCl. The reaction was conducted under microwave irradiation for 40 minutes at 110 °C. After cooling to 25 °C, the mixture was washed with H2O and brine. The organic phase was dried over anhydrous Na2SO4 and evaporated to dryness. The solid residue obtained was purified by column chromatography using diethyl ether (Et2O)/PET 2:1 as eluant to give the disubstituted intermediate 13 with 90% yield.
Rf = 0.26 (Et2O/PET); 1H NMR (400 MHz, CDCl3) δ 5.87 (s, 1H), 3.76-3.73 (m, 8H), 3.56-3.54 (m, 8H) ppm. 13C NMR (100 MHz, CDCl3) δ 163.5, 160.8, 160.6, 91.1, 66.8, 66.5, 44.4, 44.3 ppm. MS m/z for C12H18ClN4O2, (ESI+) m/z: 285.1 [M+H]+.
General Procedure for the synthesis of trisubstituted pyrimidine derivatives 14a-b
Compound 13 (1 equiv.) was dissolved in 1,4-dioxane (1 mL) and hydrazine hydrate (20 equiv.) was added. The reaction was stirred under microwave irradiation for 4 h at 100 °C. After this time, the reaction mixture was diluted with EtOAc and then was washed with H2O and brine, dried over anhydrous Na2SO4 and evaporated to dryness. The crude residue was enough pure to be subjected to the next step.
To a solution of hydrazine derivative (1 equiv.) in MeOH (10 mL), the opportune aldehyde (1 equiv.) and a drop of acetic acid were added. The mixture was stirred for 24 h at 25 °C. The precipitate formed was filtered-off and dried under high vacuum giving the desired products 14a-b with 55 and 65% yield, respectively.
Compound 14a
Rf = 0.19 (hexane (Hex)/EtOAc 1:1). 1H-NMR (400 MHz, CDCl3) δ 10.54 (s, 1H), 8.40 (s,1H), 8.06 (s 1H), 7.32 (d, J=7.2 Hz, 1H), 7.23 (d, J=7.6 Hz, 1H), 7.00-6.93 (m, 2H), 5.55 (s 1H), 3.79 (d, J=8.4 Hz, 8H), 3.63 (s, 8H) ppm. 13C NMR (100 MHz, CDCl3) δ 164.0, 160.6, 157.3, 143.9, 130.9, 130.0, 119.7, 118.0, 116.6, 72.8, 66.8, 66.6, 44.5, 44.5 ppm. MS m/z for C19H25N6O3, (ESI+) m/z: 385.2 [M+H]+.
Compound 14b
Rf = 0.25 (Hex/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.68 (s, 1H), 7.47 (d, J=7.2 Hz, 2H), 7.30-7.26 (m, 2H), 7.17 (d, J=7.2 Hz, 1H), 5.92 (s, 1H), 3.81-3.79 (m, 4H), 3.74-3.72 (m, 8H), 3.63 (d, J=5.2 Hz, 4H), 2.39 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 164.2, 162.5, 161.1, 140.5, 138.3, 134.5, 130.0, 128.5, 127.2, 123.9, 73.8, 66.9, 66.7, 44.6, 44.4, 21.4 ppm. MS m/z for C20H27N6O2, (ESI+) m/z: 383.2 [M+H]+.
Procedure for the Synthesis of monosubstituted pyrimidine intermediate 16 and 17
To a solution of 2,4,6-trichloropyrimidine 10 (1 equiv.) in CH2Cl2 (10 mL) at 0 °C, 4-fluoroaniline (1 equiv.) was slowly added, followed by DIPEA (1 equiv.). The obtained reaction mixture was stirred at 0 °C for 5h, and then warmed up to 25 °C. The mixture was washed with water, dried over N2SO4 and the solvent removed under reduced pressure. The crude residue was purified by column chromatography PET/EtOAc 4:1 to give the desired products 16 and 17 with 14 and 75% yield, respectively.
Compound 16
Rf= 0.29 (PET/EtOAc 4:1). 1H NMR (400 MHz, CDCl3) δ 7.53-7.50 (m, 2H), 7.09-7.04 (m, 2H), 6.80 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 162.0, 159.8 (d, J= 149.0 Hz), 158.1, 133.7, 121.9 (d, J= 7.9 Hz), 115.8 (d, J= 22.5 Hz), 115.7, 111.3 ppm. MS m/z for C10H7Cl2FN3, (ESI+) m/z: 258.0 [M+H]+.
Compound 17
Rf= 0.22 (PET/EtOAc 4:1).1H NMR (400 MHz, CDCl3) δ 7.30-7.27 (m, 2H), 7.17-7.13 (m, 2H), 6.44 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 163.6, 161.8 (d, J= 116.0 Hz), 160.2, 160.0, 132.1, 126.4 (d, J= 7.1 Hz), 116.9 (d, J= 22.7 Hz), 100.7 ppm. MS m/z for C10H7Cl2FN3, (ESI+) m/z: 258.0 [M+H]+.
Procedure for the synthesis of disubstitutedpyrimidine intermediate 18
Compound 17 was dissolved in 1,4-dioxane (10 mL) and 4-fluoroaniline (2.5 eq) was added. Then few drops of HCl were slowly added to the mixture. The reaction was conducted under microwave irradiation for 40 minutes at 110 °C. The mixture was washed with water, brine and dried over N2SO4.The residue was purified by column chromatography with CH2Cl2 as eluant. The purified material was dried in vacuo to afford the desired product with 62% yield.
Rf = 0.20 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.49-7.46 (m, 2H), 7.31-7.27 (m, 2H), 7.11-7.06 (m, 2H), 7.03-6.99 (m, 2H), 6.98 (s, 1H), 6.06 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 162.7, 159.9 (d, J= 100.1 Hz), 157.6, 134.8, 133.5, 125.4 (d, J= 8.0 Hz), 121.8 (d, J= 7.7 Hz), 116.3 (d, J= 22.6 Hz), 115.5 (d, J= 22.3 Hz), 94.4 ppm. MS m/z for C16H12ClF2N4, (ESI+) m/z: 333.1 [M+H]+.
General procedure for the synthesis of trisubstituted pyrimidine derivatives 19a-b
Compound 18 (1 equiv.) was dissolved in 1,4-dioxane (1 mL) and hydrazine hydrate (20 equiv.) was added. The reaction was stirred under microwave irradiation for 4 h at 100 °C. After this time, the reaction mixture was diluted with EtOAc and then washed with H2O and brine, and dried over N2SO4. The crude residue was enough pure to be subjected to the next step.
To a solution of hydrazine derivative (1 equiv.) in MeOH (10 mL), the opportune aldehyde (1 equiv.) and a drop acetic acid were added. The mixture was stirred for 48 h at 25 °C. The precipitate formed was filtered-off and dried under high vacuum giving the desired products 19a-b with 53 and 61% yield, respectively.
Compound 19a
Rf = 0.42 (Hex/EtOAc 1:1) 1H-NMR (400 MHz, CD3OD) δ 8.11 (s, 1H), 7.69-7.65 (m, 3H), 7.56-7.53 (m, 3H), 7.29-7.28 (d, J=6.4 Hz, 1H), 7.27-7.23 (d, 1H), 7.04-7.00 (m, 4H), 6.98-6.91 (m, 4H), 5.76 (s, 1H) ppm. 13C NMR (100 MHz, CD3OD) δ 162.4, 160.5 (d, J= 145.2 Hz), 159.1, 157.1, 156.7, 143.5, 137.0, 136.6, 130.0, 129.4, 122.4 (d, J= 7.6 Hz), 120.9 (d, J= 7.4 Hz), 119.2, 118.8, 115.9, 114.7 (d, J= 22.3 Hz), 114.3 (d, J= 22.2 Hz), 76.1 ppm. MS m/z for C23H19F2N6O, (ESI+) m/z: 433.2 [M+H]+.
Compound 19b
Rf = 0.30 (Hex/EtOAc 1:1). 1H NMR (400 MHz, CD3OD) δ 7.87 (s, 1H),7.67-7.64 (m, 2H), 7.57-7.54 (m, 2H), 7.51 (s, 1H), 7.44 (d, J=7.6 Hz, 1H), 7.27 (d, J=7.6 Hz, 1H), 7.16 (d, J= 7.6, 2H), 7.04-6.97 (m, 4H), 2.34 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 162.3, 160.9 (d, J= 257.2 Hz), 159.1, 157.2, 156.7, 140.9, 138.0, 135.3, 129.4, 128.1, 126.5, 123.5, 122.3 (d, J= 7.5 Hz), 121.0 (d, J= 7.4 Hz), 114.6 (d, J= 22.3 Hz), 114.3 (d, J= 22.2 Hz), 76.9, 20.0 ppm. MS m/z for C24H21F2N6, (ESI+) m/z: 431.2 [M+H]+.
Procedure for the synthesis of disubstituted pyrimidine 20
To a solution of compound 17 (1 equiv.) in EtOH (10 mL), morpholine (1 equiv.) and triethyl amine (1.5 equiv.) were added. The reaction mixture was stirred at 80 °C for 18 h. After this time, the solvent was evaporated under reduced pressure and the residue was washed with water, brine and dried over Na2SO4. The crude obtained was purified by column chromatography using CH2Cl2/MeOH (10 mL:100 μL) as eluant. The purified material was dried in vacuo to afford the desired product with 62% yield.
Rf = 0.20 (CH2Cl2/MeOH 10mL:100 μL). 1H NMR (400 MHz, CDCl3) δ 7.29-7.26 (m, 2H), 7.08-7.04 (m, 2H), 6.55 (s,1H), 5.89 (s, 1H), 3.75 (d, J=4 Hz, 8H) ppm. 13C NMR (100 MHz, CDCl3) δ 164.3, 161.6 (d, J=200.2 Hz), 158.0, 135.5, 124.2 (d, J=7.1 Hz), 115.9 (d, J=22.4 Hz), 73.9, 67.0, 66.6, 44.6, 44.5 ppm. MS m/z for C14H15ClFN4O, (ESI+) m/z: 309.1 [M+H]+.
Procedure for the synthesis of disubstituted pyrimidine 21a-b
Compound 20 (1 equiv.) was dissolved in 1,4-dioxane (1 mL) and hydrazine hydrate (20 equiv.) was added. The reaction was stirred under microwave irradiation for 6 h at 100 °C. After this time, the reaction mixture was diluted with EtOAc and then washed with H2O and brine, and dried over N2SO4. The crude residue was enough pure to be subjected to the next step.
To a solution of hydrazine derivative (1 equiv.) in MeOH (10 mL), the opportune aldehyde (1 equiv.) and a drop acetic acid were added. The mixture was stirred for 48 h at 25 °C. The precipitate formed was filtered-off and dried under high vacuum giving the desired products 21a-b with 46% and 45% yield, respectively.
Compound 21a
Rf = 0.18 (CH2Cl2/MeOH 10:0,2). 1H-NMR (400 MHz, CDCl3) δ 10.48 (s, 1H), 7.88 (s, 1H), 7.36-7.33 (m, 2 H), 7.33-7.27 (m, 2H), 7.17 (d, J=7.6 Hz, 1H), 7.10-7.05 (m, 2 H), 7.00-6.98 (m, 1H), 6.94-6.92 (m, 1 H), 6.44 (s, 1H), 5.66 (s, 1H), 3.76 (s, 8H) ppm. 13C-NMR (100 MHz, CDCl3) δ 175.4, 162.3, 161.0, 159.3 (d, J= 251.4 Hz), 157.4, 144.2, 135.0, 131.0, 130.1, 123.6 (d, J= 8.0 Hz), 119.6, 117.9, 116.8, 115.9 (d, J= 22.4 Hz), 74.6, 66.9, 44.6 ppm. MS m/z for C21H22FN6O2, (ESI+) m/z: 409.2 [M+H]+.
Compound 21b
Rf: 0.29 (CH2Cl2/MeOH 10:0,2) 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.44 (s, 1H), 7.41 (s, 1H), 7.39-7.35 (m, 2H), 7.29 (s, 1H), 7.18 (d, J=7.6 Hz, 1H), 7.09-7.04 (m, 2H), 6.39 (s, 1H), 6.02 (s, 1H), 3.75 (s, 8H), 2.38 (3H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 162.4, 161.8, 160.0 (d, J= 309.8 Hz), 156.1, 140.8, 138.4, 138.0, 135.6, 130.0, 129.1, 126.8, 123.9, 121.1 (d, J= 7.4 Hz), 115.5 (d, J= 21.8 Hz), 76.5, 66.6, 44.7, 21.5 ppm. MS m/z for C22H24FN6O, (ESI+) m/z: 407.2 [M+H]+.
Procedure for the synthesis of monosubstituted pyridine derivatives 23 and 24
To a solution of morpholine (1 equiv.) and Et3N (2.5 equiv.) in dry THF (5 mL) was added a solution of 2,4,6-trichlorpyridine 22 (1 equiv.) in THF (2 mL). The reaction mixture was stirred 24 h at 65 °C. After cooling to 25 °C, the reaction was concentrated to remove THF and then diluted with Et2O and washed with water and brine, dried over anhydrous Na2SO4, filtered and evaporated in vacuo. The crude mixture was purified by column chromatography using Hex/EtOAc 5:1 as eluant to give 23 and 24 with 15 and 30 % yield, respectively.
Compound 23
Rf = 0.17 (Hex/EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ 6.57 (s, 2H), 3.81 (t, J=4 Hz, 4H), 3.30 (t, J=4 Hz, 4H) ppm. 13C NMR (100 MHz, CDCl3) δ 158.0, 151.2, 106.2, 66.0, 46.1 ppm. MS m/z for C9H11Cl2N2O, (ESI+) m/z: 233.0 [M+H]+.
Compound 24
Rf = 0.40 (Hex/EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ = 6.66 (d, J=1.2 Hz, 1H), 6.47 (d, J=4 Hz, 1H), 3.79 (t, J=4.8 Hz, 4H), 3.53 (t, J=4 Hz, 4H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.2, 150.2, 146.3, 112.6, 104.4, 66.4, 45.1 ppm. MS m/z for C9H11Cl2N2O, (ESI+) m/z: 233.0 [M+H]+.
Procedure for the Synthesis of disubstituted pyridine derivatives 25 and 28
To a solution of monosubstituted pyridine 24 (1 equiv.) in DMF (2 ml) was added morpholine (10 equiv.) and DIPEA (3 equiv.). The mixture was warmed at 120 °C for 48 h. After cooling to 25 °C, the reaction mixture was diluted with CH2Cl2 and washed with aqueous solution of 3% LiCl. The organic phase was dried over anhydrous Na2SO4, filtered and evaporated in vacuo to afford a crude mixture of two regioisomers. Products 25 and 28 can be separated by column chromatography using a mixture of Hex/EtOAc 2:1, and recovered with 58 and 18% yield, respectively.
Compound 25
Rf = 0.45 (Hex/EtOAc 2:1)] 1H NMR (400 MHz, CDCl3) δ 6.0 (s, 2H), 3.79 (d, J=4 Hz, 8H), 3.47 (d, J=8 Hz, 8H) ppm. 13C NMR (100 MHz, CDCl3) δ 158.2, 146.8, 97.0, 66.7, 49.3 ppm. MS m/z for C13H19ClN3O2, (ESI+) m/z: 284.1 [M+H]+.
Compound 28
Rf = 0.13 (Hex/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ 6.20 (d, J=4 Hz, 1H), 5.77 (d, J=1,2 Hz, 1H), 3.82-3.78 (m, 8H), 3.46 (t, J=4 Hz, 4H), 3.25 (t, J=4 Hz, 4H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.2, 150.2, 146.3, 112.6, 104.4, 66.4, 45.1 ppm. MS m/z for C13H19ClN3O2, (ESI+) m/z: 284.1 [M+H]+.
Procedure for the synthesis of Boc-protected trisubstituted pyridine carbazate 26
In a microwave tube were added under argon compound 25 (1 equiv.), tert-butyl carbazate (BocNHNH2, 10 equiv.), Pd2(dba)3 (0,04 equiv.), DPPF (0,12 equiv.), Cs2CO3 (2 equiv.) and dry toluene (1 mL). The reaction mixture was stirred at 150°C for 10 minutes under microwave irradiation. The mixture was filtered through celite, rinsed with EtOAc and concentrated. The crude was purified by column chromatography with CH2Cl2/EtOAc 4:1 as eluant to give the product 26 with 65% yield.
Rf= 0.43 (Hex/EtOAc 2:1)] 1H NMR (400 MHz, CDCl3) δ 6.2 (s, 2H), 3.98-3.85 (m, 8H), 3.81 (t, J=4.8 Hz, 4H), 3.50 (t, J=4.4 Hz, 4H), 1.49 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.0, 154.4, 153.2, 89.9, 82.6, 66.9, 46.0, 28.3 ppm. MS m/z for C18H30N5O4, (ESI+) m/z: 380.2 [M+H]+.
Procedure for the synthesis of trisubstituted pyridine derivatives 27a-b
Boc-protected pyridine 26 (1 equiv.) was dissolved in anhydrous CH2Cl2 (430µL) and cooled to 0°C. TFA (290µL) was added and the reaction mixture was warmed to 25 °C and stirred for 1h. After this time, the mixture was concentrated at reduced pressure. The residue obtained was enough pure to be subjected to the next step.
To a solution of hydrazine derivative (1 equiv.) in MeOH (10 mL), the opportune aldehyde (1 equiv.) and few drops of acetic acid were added. The mixture was stirred for 24 h at 25 °C and concentrated under vacuum. The residue was purified by column chromatography (Hex/EtOAc 2:1) furnishing the desired products 27a-b with 39% and 18% yield, respectively.
Compound 27a
Rf = 0.40 (Hex/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 10.92 (s, 1H), 8.39 (s, 1H), 7.86 (s, 1H), 7.27 (d, J=12.0 Hz, 1H), 7.16-7,14 (dd, J=1.6 Hz, J=7.6 Hz, 1H), 7.00 (d, J=8.0 Hz, 1H), 6.91 (s, 1H), 5.96 (d, J=1.6 Hz, 1H), 5.60 (d, J=1.2 Hz, 1H), 3.85-3.78 (m, 8H), 3.44 (t, J=4.0 Hz, 4H), 3.31 (t, J=4.0 Hz, 4H) ppm. 13C NMR (CDCl3, 100 Hz) δ 160.1, 159.6, 157.1, 155.3, 141.7, 130.1, 129.5, 119.5, 118.5, 116.5, 84.5, 82.1, 66.8, 47.0, 46.2 ppm. MS m/z for C20H26N5O3, (ESI+) m/z: 384.2 [M+H]+.
Compound 27b
Rf = 0.55 (Hex/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ = 8.11 (s, 1H), 7.66 (s, 1H), 7.47 (d, J=8.0 Hz, 2H), 7.27 (t, J=8.0 Hz, 1H), 7.14 (d, J=8.0 Hz, 1H), 6.31(d, J=4.0 Hz, 1H), 5.57 (d, J=2.0 Hz, 1H), 3.86-3.80 (m, 8H), 3.42 (t, J=8.0 Hz, 4H), 3.33 (t, J=8.0 Hz, 4H), 2.39 (s, 3H) ppm. 13CNMR (CDCl3, 100 Hz) δ 159.1, 138.4, 134.2, 130.6, 128.2, 127.9, 124.1, 82.7, 81.1, 66.2, 48.2, 46.3, 31.2, 29.7, 21.3 ppm. MS m/z for C21H28N5O2, (ESI+) m/z: 382.2 [M+H]+.
Procedure for the synthesis of monosubstituted pyridine derivatives 29 and 30
In a two necks flask equipped with molecular sieves, 2,4,6-tricloropyridine 22 (1 equiv.), Cs2CO3 (1 equiv.), Pd(PPh3)4 (0,02 equiv.) and dry toluene (5 mL) were added. Subsequently, 4-fluoroaniline (1,6 equiv.) was added under argon atmosphere and the reaction mixture was stirred for 24h at 150 °C. After this time, the mixture was filtered through celite, rinsed with EtOAc and concentrated. The residue so obtained was diluted with EtOAc, washed with a saturated aqueous solution of NaHCO3 and brine, dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by column chromatography with Pet/EtOAc 4:1 as eluant to give the products 29 and 30 with 19 and 50% yield, respectively.
Compound 29
Rf = 0.50 (Pet/EtOAc 4:1). 1H NMR (400 MHz, CDCl3) δ 7.27 (m, 2H), 7.12 (t, J=8.4 Hz, 2H), 7.00 (s, 1H), 6.7 (s, 2H) ppm. 13C (100 MHz, CDCl3) δ 159.3 (d, J=241.9 Hz), 156.2, 146.3, 135.6, 123.8 (d, J=6.9 Hz), 116.0 (d, J=22.3 Hz), 98.0 ppm. MS m/z for C11H8Cl2FN2, (ESI+) m/z: 257.0 [M+H]+.
Compound 30
Rf = 0.65 (Pet/EtOAc 4:1). 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 2H), 7.09 (t, J=8.8 Hz, 2H), 7.00 (s, 1H), 6.73 (d, J=1.2 Hz, 1H), 6.54 (d, J=1.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 160.0 (d, J=240.3 Hz), 157.2, 150.5, 146.6, 134.5, 124.8 (d, J=8.1 Hz), 116.4 (d, J=22.6 Hz), 114.2, 104.9 ppm. MS m/z for C11H8Cl2FN2, (ESI+) m/z: 257.0 [M+H]+.
Procedure for the synthesis of disubstituted pyridine derivatives 31 and 34
Pd(OAc)2 (0.02 equiv.) and BINAP (0.02 equiv.) were dissolved in anhydrous toluene (1 mL) and the mixture was kept under argon for 10 minutes. At the same time, in a Schenck tube were added compound 30 (1 equiv.), anhydrous toluene (1 mL), Cs2CO3 (3.5 equiv.), and 4-fluoroaniline (1.5 equiv.) and then the solution of Pd(OAc)2 and BINAP above described. The reaction mixture was warmed at 160°C and left under stirring for 4 h. After this time, the mixture was filtered through celite, rinsed with EtOAc and concentrated. The crude was purified by column chromatography with Pet/EtOAc 3:1 as eluant to give the products 31 and 34 with 36 and 13% yield, respectively.
Compound 31
Rf = 0.63 (Pet/EtOAc 3:1). 1H NMR (400 MHz, CDCl3) δ 7.29 (m, 4H), 7.06 (m, 4H), 6.65 (s, 1H), 6.13 (s, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.2 (d, J=260.2 Hz), 156.0, 146.6, 135.4, 123.9 (d, J=7.9 Hz), 116.1 (d, J=22.4 Hz), 98.0 ppm. MS m/z for C17H13ClF2N3, (ESI+) m/z: 332.1 [M+H]+.
Compound 34
Rf = 0.44 (Pet/EtOAc 3:1). 1H NMR (400 MHz, CDCl3) δ 7.21 (m, 4H), 7.11 (m, 4H), 6.45 (s, 1H), 6.24 (d, J=1.6 Hz, 1H), 6.0 (d, J=1.6 Hz, 1H), 5.9 (s, 1H) ppm. 13C (100 MHz, CDCl3) δ 159.2 (d, J=270.4 Hz), 156.8, 148.1, 141.5, 136.5, 123.4 (d, J=7.4 Hz), 116.3 (d, J=22.4 Hz), 100.0, 94.0 ppm. MS m/z for C17H13ClF2N3, (ESI+) m/z: 332.1 [M+H]+.
Procedure for the synthesis of Boc-protected trisubstituted pyridine carbazate 32
In a microwave tube were added under argon compound 31 (1 equiv.), BocNHNH2 (10 equiv.), Pd2(dba)3 (0,04 equiv.), DPPF (0,12 equiv.), Cs2CO3 (2 equiv.) and dry toluene (1 mL). The reaction mixture was stirred at 150°C for 10 minutes under microwave irradiation. The mixture was filtered through celite, rinsed with EtOAc and concentrated. The crude was purified by column chromatography with Hex/EtOAc 2:1 as eluant to give the product 32 with 59% yield.
Rf=0.37 (Hex/EtOAc 2:1). 1H NMR (400 MHz, CDCl3): δ 7.31 (m, 4H), 7.03 (m, 4H), 6.61 (s, 2H), 5.32 (s, 1H), 4.24 (s, 1H), 1.49 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 158.8 (d, J=240.2 Hz), 155.6, 154.2, 153.3, 136.5, 123.0 (d, J=7.7 Hz), 115. (d, J=22.2 Hz), 90.9, 83.1, 28.2 ppm. MS m/z for C22H24F2N5O2, (ESI+) m/z: 428.2 [M+H]+.
Procedure for the synthesis of trisubstituted pyridine derivativities 33a-b
Boc-protected pyridine 32 (1 equiv.) was dissolved in anhydrous CH2Cl2 (430µL) and cooled to 0°C. TFA (290µL) was added and the reaction mixture was warmed to 25 °C and stirred for 1h. After this time, the mixture was concentrated at reduced pressure. The residue obtained was enough pure to be subjected to the next step.
To a solution of hydrazine derivative (1 equiv.) in MeOH (10 mL), the opportune aldehyde (1 equiv.) and few drops of acetic acid were added. The mixture was stirred for 24 h at 25 °C and concentrated under vacuum. The residue was purified by column chromatography (Hex/EtOAc 1:1) furnishing the desired products 33a-b with 29% and 21% yield, respectively.
Compound 33a
Rf = 0.37 (Hex/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 10.47 (s, 1H), 7.81 (s, 1H), 7.30 (m, 4H), 7.06 (m, 6H), 6.93 (m, 2H), 6.36 (s, 2H), 5.80 (s, 2H), 5.63 (s, 1H) ppm. 13C (100 MHz, CDCl3) δ 162.5, 158.8 (d, J=240.2 Hz) 157.1, 155.3, 143.0, 140.3, 130.2, 129.6, 122.6 (d, J= 7.7 Hz), 119.2, 117.3, 116.5, 116.2 (d, J= 22.0 Hz), 90.9 ppm. MS m/z for C24H20F2N5O, (ESI+) m/z: 432.2 [M+H]+.
Compound 33b
Rf = 0.49 (Hex/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ 7.62 (s, 1H), 7.43-7.21 (m, 8H), 7.05 (m, 4H), 6.33 (s, 1H), 5.96 (d, J=11.6 Hz, 2H), 2.39 (s, 3H) ppm. 13C (100 MHz, CDCl3) δ 161.6, 159.0 (d, J=241.0 Hz), 155.6, 154.0, 140.1, 138.4, 136.2, 134.3, 130.2, 128.6, 127.1, 123.5 (d, J= 7.8 Hz), 115.8 (d, J= 22.4 Hz), 106.9, 21.3 ppm. MS m/z for C25H22F2N5, (ESI+) m/z: 430.2 [M+H]+.
Procedure for the synthesis of disubstituted pyridine derivatives 35 and 36
In a microwave tube equipped with a stir bar were added the monosubstituted pyridine 30 (1 equiv.) dissolved in anhydrous 1,4-dioxane (1.5 mL), morpholine (5 equiv.) and DIPEA (1,5 equiv.). The reaction mixture was irradiated at 160°C for 10h in a microwave oven. Then, the mixture was extracted with EtOAc, washed with brine, dried over anhydrous Na2SO4 and concentrated under vacuum. The crude was purified by column chromatography using PET/EtOAc 5:1 as eluant to give products 35 and 36 with 19 and 46% yield, respectively.
Compound 35
Rf = 0.08 (PET/EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ = 7.21 (m, 2H), 7.11 (m, 2H), 6.24 (d, J=1.6 Hz, 1H), 6.0 (d, J=1.6 Hz, 1H), 5.9 (s, 1H), 3.82-3.78 (m, 4H), 3.46 (t, J=4 Hz, 2H), 3.25 (t, J=4 Hz, 2H) ppm. 13C (100 MHz, CDCl3) δ 161.1, 157.6 (d, J=197.8 Hz), 148.0, 134.8, 124.9 (d, J=6.6 Hz), 124.6, 116.4 (d, J=22.6 Hz), 100.0, 87.3, 68.8, 46.4 ppm. MS m/z for C15H16ClFN3O, (ESI+) m/z: 308.1 [M+H]+.
Compound 36
Rf = 0.37 (PET/EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ 7.27 (m, 2H), 7.05 (t, J=8.8 Hz, 2H), 6.24 (d, J=9.2 Hz, 1H), 6.08 (d, J=4.8 Hz, 1H), 3.81 (t, J=4.8 Hz, 2H), 3.49 (t, J=4Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.3, 159.1 (d, J=240.2 Hz), 155.7, 146.4, 135.9, 123.4 (d, J=6.7 Hz), 115.9 (d, J=22.4 Hz), 97.4, 96.9, 66.7, 45.4 ppm. MS m/z for C15H16ClFN3O, (ESI+) m/z: 308.1 [M+H]+.
Procedure for the synthesis of Boc-protected trisubstituted pyridine carbazate 37
In a microwave tube were added under argon compound 36 (1 equiv.), BocNHNH2 (10 equiv.), Pd2(dba)3 (0,04 equiv.), DPPF (0,12 equiv.), Cs2CO3 (2 equiv.) and dry toluene (1 mL). The reaction mixture was stirred at 150°C for 10 minutes under microwave irradiation. The mixture was filtered through celite, rinsed with EtOAc and concentrated. The crude was purified by column chromatography with Hex/EtOAc 2:1 as eluant to give the product 37 with 42% yield.
Rf=0.38 (Hex/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ 7.31 (m, 2H), 7.0 (t, J=8.4 Hz, 2H), 6.54 (s, 1H), 6.24 (s, 1H), 3.84 (t, J=4.8 Hz, 2H), 3.51 (t, J=4.8 Hz, 2H) ppm. 13C (100 MHz, CDCl3) δ 160.3, 159.6 (d, J= 240.2 Hz), 155.7, 154.2, 146.2, 135.8, 123.4 (d, J= 6.8 Hz), 115.9 (d, J= 22.2 Hz), 97.4, 96.8, 80.5, 66.9, 45.9, 28.3 ppm. MS m/z for C20H27FN5O3, (ESI+) m/z: 404.2 [M+H]+.
Procedure for the synthesis of trisubstituted pyridine derivativities 38a-b
Boc-protected pyridine 37 (1 equiv.) was dissolved in anhydrous CH2Cl2 (430µL) and cooled to 0°C. TFA (290µL) was added, and the reaction mixture was warmed to 25 °C and stirred for 1h. After this time, the mixture was concentrated at reduced pressure The residue obtained was enough pure to be subjected to the next step.
To a solution of hydrazine derivative (1 equiv.) in MeOH (10 mL), the opportune aldehyde (1 equiv.) and few drops of acetic acid were added. The mixture was stirred for 24 h at 25 °C and concentrated under vacuum. The residue was purified by column chromatography (Hex/EtOAc 1:1) furnishing the desired products 38a-b with 37% and 33% yield, respectively.
Compound 38a
Rf = 0.31 (Hex/EtOAc 1:1). 1H NMR (400 MHz, CDCl3). δ 10.63 (s, 1H), 7.84 (s, 1H), 7.31 (m, 2H), 7.03 (m, 4H), 6.94 (m, 2H), 5. 72 (s, 1H), 5.32 (s 1H), 3.79 (t, J=4.4 Hz, 2H), 3.49 (t, J=4.4 Hz, 2H) ppm. 13C (100 MHz, CDCl3) δ 160.3, 156.7 (d, J=113.7 Hz), 152.4, 142.5, 136.7, 130.7, 129.7, 123.9, 122.8 (d, J=7.5 Hz), 119.6, 118.0, 116.7, 116.5, 115.7 (d, J=22.3 Hz), 81.8, 81.7, 66.8, 45.7 ppm. MS m/z for C22H23FN5O2, (ESI+) m/z: 408.2 [M+H]+.
Compound 38b
Rf = 0.43 (Hex/EtOAc 1:1)]. 1H NMR (400 MHz, CDCl3) δ 7.65 (s, 1H), 7.44 (d, J=8.8 Hz, 2H), 7.30 (m, 4H), 7.18 (d, J=7.6 Hz, 1H), 7.03 (m, 2H), 6.0 (s, 2H), 3.84 (t, J=4.8 Hz, 2H), 3.51 (t, J=4.8 Hz, 2H), 2.42 (s, 3H) ppm. 13C (100 MHz, CDCl3) δ 162.5, 161.6, 159.2 (d, J=227.6 Hz), 155.6, 154.5, 148.9, 138.4, 134.2, 130.3, 128.6, 127.3, 123.9 (d, J=6.7 Hz), 123.7, 115.8 (d, J=22.3 Hz), 97.6, 97.0, 66.6, 46.4, 21.3 ppm. MS m/z for C23H25FN5O, (ESI+) m/z: 406.2 [M+H]+.
3.3. Biology – General part
All solvents, reagents and human plasma were purchased from Sigma-Aldrich Srl (Milan. Italy). Milli-Q quality water (Millipore. Milford. MA. USA), acetonitrile (ACN) and formic acid (FA) were used for the chromatographic analyses. Cell culture mediums, fetal bovine serum (FBS), L-glutamine, and penicillin–streptomycin were purchased from Euroclone S.p.A. (Milan, Italy). SOF (MCE® cat. HY-15005), REM (MCE® cat. HY-104077) and RAL (MCE® cat. HY-10353) used as reference compounds were purchased from MedChem Express (https://www.medchemexpress.com). RAL was dissolved in molecular grade water, SOF and REM were dissolved in 100% dimethyl sulfoxide (DMSO).
3.4. Biology – Cells
Cell culture
Cell-based assays were carried out on human normal fibroblast FB789 cell line (kindly provided by Elena Dell’Ambra from IRCCS Istituto Dermopatico dell’Immacolata, Rome), on Huh7 hepatocarcinoma cell line (kindly provided by Istituto Toscano Tumori, Core Research Laboratory, Siena, Italy), on Caco-2 adenocarcinoma colorectal cell line (ATCC catalog. n. HTB-37), on suspension H9 cell line (repository code ARP0001, NIBSC Centre for AIDS reagents) and on the adherent TZM-bl cell line (repository code ARP5011, NIBSC Centre for AIDS reagents).
FB789 were cultured in Dulbecco’s modified Eagle Medium (DMEM) and Ham’s F10, in a ratio of 50%, supplemented with 10% FBS, 2 mM L-glutamine, and 10,000 unit/mL penicillin/streptomycin.
Huh7 and Caco-2 were used to determine the cytotoxicity and the antiviral activity of candidate compounds against flaviviruses and SARS-CoV-2 respectively. H9 cells in combination with TZM-bl cells were used to evaluate the compounds against HIV-1, as described in the antiviral assays section.
High glucose Dulbecco's Modified Eagle's Medium with sodium pyruvate and L-glutamine (DMEM; Euroclone) was used to grow Huh-7 and TZM-bl. Minimum Essential Medium Eagle (EMEM; Euroclone) was used to propagate Caco-2. DMEM and EMEM were supplemented with 10% Fetal Bovine Serum (FBS; Euroclone) and 1% Penicillin/Streptomycin (Pen/Strep, Euroclone). The growth medium with a lower concentration of FBS (1%) was used for viral propagation, cytotoxic and antiviral experiments in adherent cell lines. Suspension cell lines were grown, propagated and infected in RPMI 1640 medium supplemented with 10% Fetal Bovine Serum (FBS; Euroclone), 2 mM L-glutamine and 1% Penicillin/Streptomycin (Pen/Strep, Euroclone). All cell lines were grown at 37°C in a 5% CO2 atmosphere in a humidified incubator.
Cytotoxicity Assay
The cytotoxicity of all investigated compounds was assayed in all cells lines as previously described [10, 11]. Briefly, confluent cells were treated with decreasing compound concentrations for 48h. The final DMSO concentration used was never greater than 0.5%v/v. Each experiment was performed in duplicated and repeated 2 independent experiments. Cell viability was calculated using the CellTiter Glo 2.0 kit (Promega) and luminescence values signal obtained from cells treated with serial dilution of the compounds were measured through the GloMax® Discover Multi mode Microplate Reader (Promega) and elaborated with the GraphPad PRISM software version 9.0 (La Jolla). The half-maximal cytotoxic concentration (CC50) was calculated using a non-linear regression analysis of the dose-response curves and the ECanything GraphPad function. A not toxic dose of each compound was used as the starting concentration in the antiviral activity assay.
3.5. Biology – Viruses
Viruses
The New Guinea C DENV serotype 2 and the WNV lineage 1 (Italy/2009) strains were kindly provided by the Istituto Superiore di Sanità (Rome, Italy) while the SARS-CoV-2 strain belonging to lineage B.1 (EPI_ISL_2472896) was kindly provided by the Department of Biomedical and Clinical Sciences Luigi Sacco, University of Milan (Italy). Once expanded in VERO E6 (African green monkey kidney cell line, ATCC catalog. n. CRL-1586), DENV, WNV and SARS-CoV-2 viral stocks were stored at -80 °C and titrated as previously described [10,11]. HIV-1 wild-type reference strain NL4-3 (catalog. n. ARP2006), was obtained through the NIH AIDS Reagent Program and viral titer was calculated in TZM- bl cells through the detection of β- galactosidase expression.
Antiviral Assays
Flaviviruses and SARS-CoV-2
To determine the antiviral activity of candidate compounds against DENV, WNV and SARS-CoV-2, a direct yield reduction assay, based on the infection of cells in the presence of serial drug dilutions was performed as previously described with minor modifications [10]. Briefly, Huh7 or Caco-2 cells, pre-seeded in 96-well format, were infected with DENV and WNV viral stocks at 0.005 multiplicity of infection (MOI) or with SARS-CoV-2 at 0.004 MOI. After 1h of adsorption of the virus at 37°C, viral inoculum was removed and serial dilutions of each tested compound, starting from the not-toxic dose, were added to the infected cells. After 48 h of incubation for DENV and WNV and 72h for SARS-CoV-2, the antiviral activity was measured on cell monolayer by immunodetection assay (IA), as previously described [10].
Absorbance was measured at 450 nm optical density (OD450) using the Absorbance Module of the GloMax® Discover Multimode Microplate Reader (Promega). In each plate the suitable reference compound, a mock control (uninfected cells) and a virus control were included. Each IA run was validated when the OD450 values of virus control showed an OD450>1. All drug concentrations were tested in duplicate in two independent experiments. In each plate, SOF and REM were used as reference compounds against flaviviruses and SARS-CoV-2, respectively. Infected and uninfected cells without drugs were used to calculate the 100% and 0% of viral replication, respectively. The half-maximal inhibitory concentration (IC50) was calculated through a non-linear regression analysis of the dose-response curves generated with GraphPad PRISM software version 9 (La Jolla, California, USA). The Selectivity Index (SI) of the compounds was calculated as the ratio between the CC50 and the IC50.
HIV-1
The antiviral activity of investigated compounds was evaluated by measuring the IC50 values against the HIV-1 wild-type reference strain NL4-3 in a TZM-bl cell line based phenotypic assay named BiCycle Assay [11,12]. The method includes a first round of infection in H9 cells at 0.08 MOI in presence of serial dilution of compounds in a 96-well plate. In each plate the reference compound, the mock control (uninfected cells) and the virus control were included. After 72 hours, 50 µlof supernatants from each well were used to infect TZM-bl cell line, which allow the quantitative analysis of HIV-1 infection by measuring the expression of the luciferase gene integrated in the genome of the cells under the control of HIV-1 LTR promoter. After 48 hours, dose-response curves were generated by measuring reporter gene expression in each well by using Bright-Glo Luciferase Assay (Promega) through the GloMax® Discover Multimode Microplate Reader (Promega). Relative luminescence units measured in each well were elaborated with the GraphPad PRISM software version 9 to calculate IC50 values.
3.6. In vitro ADME
UV/LC-MS method
For the quantitative analysis was used a UV/LC-MS system. LC analysis was performed by Agilent 1260 Infinity HPLC-DAD system (Agilent Technologies. Palo Alto. CA) constituted by a vacuum solvent degassing unit, a binary high-pressure gradient pump, an UV detector, connected to an Agilent MSD 6130 system (Agilent Technologies, Palo Alto, CA, USA). The Agilent 1260 series mass spectra detection (MSD) single-quadrupole instrument was equipped with the orthogonal spray API-ES (Agilent Technologies. Palo Alto. CA). Nitrogen was used as nebulizing and drying gas. Chromatographic separation was performed using a Phenomenex Kinetex EVO C18-100 Å (150 x 4.6 mm, 5 μm particle size) at 25 °C and gradient elution with a binary solution; eluent A was H2O, while eluent B consisted of ACN (both eluents were acidified with FA 0.1%v/v). The analysis started with 0% of B for 1 minute, then rapidly increased up to 80% of B in 15 minutes remaining until 19 minutes; finally, in one minute came back to the initial conditions of 100% of A. The analysis was performed at a flow rate of 0.6 mL/min. UV detection was monitored at 254 nm. Spectra were acquired over the scan range m/z 100-1500 in positive mode.
Kinetic Aqueous Solubility
DMSO-stock compounds’ solutions were diluted with Mill-Q H2O to a final concentration of 200 μM. The percentage of DMSO never exceed 2%v/v. Samples were incubated under gentle shaking at 25 °C (RT) conditions for 3 h. The suspensions were filtered using a 0.45 µm nylon filter (Acrodisc), and the amount of solubilized compound was determined with the HPLC-UV-MS method above reported. The quantification of the solubilized compound was made with the appropriate calibration curve realized with stock solutions in DMSO (0.1–100 µg/mL). The limit of detection (LOD) was quantified at 0.1 µg/mL.
Parallel Artificial Membrane Permeability Assay (PAMPA)
To evaluate the apparent permeability of tested compounds, a DMSO stock solution [1mM] of each derivative was prepared and then diluted 1:1v/v with phosphate buffer (PBS 25 mM, pH 7.4), in order to make the donor solutions. According to the protocol already published [9,13], the gastrointestinal (GI) phospholipidic bilayer was mimed by adding a 1%w/v dodecane solution of phosphatidylcholine (PC). The sandwich plates were assembled and incubated for 5 h at RT. At the end time point, the amount of compound passed through the phospholipid bilayer was measured by HPLC-UV/MS. Finally, apparent permeability (Papp) and membrane retention (MR%) were calculated as previously described [doi.org/10.3390/molecules27248829].
Metabolic Stability Assay
A DMSO solution of selected compounds was incubated in the presence of phosphate buffer (25 mM, pH 7.4), human liver microsomal protein (200 µg/mL), and an NADPH regenerating system in MgCl2 (48 mM) at a final concentration of 50 µM. The metabolic reactions were conducted for 1 h under shaking at 37 °C and then stopped by adding cold acetonitrile (ACN). Centrifuging the mixtures at 5000 rpm, the supernatant was separated, dried under nitrogen flow, and finally suspended in methanol (MeOH). The amount of parent drug and the metabolites were determined as previously described [13].
Plasma Stability Assay
A DMSO solution of each compound was incubated at a final concentration of 100 µM at 37°C under shaking with HEPES buffer (25 mM, 140 mM NaCl, pH 7.4) and human plasma. At time 0 and after 24 h, samples were collected from the reaction mixtures and treated with cold ACN. After centrifugation at 5000 rpm for 10 minutes, the supernatant was collected, and the amount of unmodified compound was determined using the HLPC-UV/MS method described above. Modified compounds were calculated using time zero as 100% of the unmodified compound.
4. Conclusions
In conclusion two small libraries of derivatives containing pyrimidine and pyridine privileged scaffolds were synthesized using a scaffold morphing approach, starting from previously obtained triazines. Newly synthesized compounds have been evaluated for their anticancer activity on lymphoma, hepatocarcinoma, and colon epithelial carcinoma cells Three pyrimidine derivatives, 19a, 19b, and 21a showed low micromolar antineoplastic activity associated to a low cytotoxic effect, especially in the case of 19b. Two pyridine compounds, 33b and 38b, highlighted the lowest cytotoxicity of both the libraries synthesized.
Finally, the above-mentioned five azines were evaluated with in vitro ADME protocols showing a general suboptimal aqueous solubility accompanied by good passive permeability and discrete percentages of membrane retention. From the metabolic point of view, neither in presence of liver microsomes nor with human plasma, tested compounds undergone massive degradation, thus letting us to state that most of them manage to reach the potential biological target unaltered. Further studies aimed at the identification of a specific biological target for the newly synthesized azines are ongoing in our laboratories.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Nuclear Overhauser Effect Spectroscopy (NOESY) of pyrimidine intermediates 11 and 12; Figure S2: 1H NMR of pyridine intermediates 23 and 24.
Author Contributions
Conceptualization, L.B., M.Z. and E.D.; methodology, S.C., I.V., P.F., F.S.; validation, E.M., L.F. and C.V.; formal analysis, E.D.M. and F.G.; investigation, B.M.B.; data curation, E.M., L.F., E.D.M., F.G., B.M.B. and C.V.; writing—original draft preparation, S.C., I.V., P.F., F.S.; writing—review and editing, L.B., M.Z. and E.D.; supervision, R.S.; funding acquisition, R.S., E.D, M.Z., L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministero dell’Istruzione, dell’Università della Ricerca Italiano (MIUR), PRIN 2017, project N. 2017BMK8JR, Title “ORIGINALE CHEMIAE in Antiviral Strategy— Origin and Modernization of Multi-Component Chemistry as a Source of Innovative Broad Spectrum Antiviral Strategy” (L.B., R.S., E.D. and M.Z.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
M.Z. reports consultancy for ViiV Healthcare, Gilead Sciences, GlaxoSmithKline, Janssen-Cilag, Theratechnologies, Merck Sharp, and Dohme, and grants for his institution from ViiV Healthcare, Theratechnologies, and Gilead Sciences outside the submitted work. All other authors: no conflicts to declare.
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; Lotti, V.J.; Cerino, D.J.; Chen, T.B.; Kling, P.J.; Kunkel, K.A.; Springer, J.P.; Hirshfieldt, J. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Park, S.B. Privileged structures: efficient chemical "navigators" toward unexplored biologically relevant chemical spaces. J Am Chem Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef] [PubMed]
- Kounde, C.S.; Yeo, H.Q.; Wang, Q.Y.; Wan, K.F.; Dong, H.; Karuna, R.; Dix, I.; Wagner, T.; Zou, B.; Simon, O.; Bonamy, G.M.C.; Yeung, B.K.S.; Yokokawa, F. Discovery of 2-oxopiperazine dengue inhibitors by scaffold morphing of a phenotypic high-throughput screening hit. Bioorg Med Chem Lett. 2017, 27, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.R.; Modh, R.P.; Chikhalia, K.H. Privileged s-triazines: Structure and pharmacological applications. Future Med. Chem. 2014, 6, 463–477; Celena M. Josephitis, Hillary M. H. Nguyen, Andrew McNally. Late-Stage C–H Functionalization of Azines. Chem. Rev. 2023, 123, 7655–7691. [Google Scholar]
- Baumann, M.; Baxendale, I. R. An Overview of the Synthetic Routes to the Best Selling Drugs Containing 6-Membered Heterocycles. Beilstein J. Org. Chem. 2013, 9, 2265–2319. [Google Scholar] [CrossRef] [PubMed]
- Chiacchio, M.A.; Iannazzo, D.; Romeo, R.; Giofrè, S.V.; Legnani, L. Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Biologically Active Agents. Curr Med Chem. 2019, 26, 7166–7195. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Botta, M. Practical one-pot two-step protocol for the microwave-assisted synthesis of highly functionalized rhodanine derivatives. J. Comb. Chem. 2010, 12, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, S.; Vicenti, I.; Poggialini, F.; Secchi, M.; Giammarino, F.; Varasi, I.; Lodola, C.; Zazzi, M.; Dreassi, E.; Maga, G.; Botta, L.; Saladino, R. Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity. Molecules 2022, 27, 8829. [Google Scholar] [CrossRef] [PubMed]
- Vicenti, I.; Martina, M.G.; Boccuto, A.; De Angelis, M.; Giavarini, G.; Dragoni, F.; Marchi, S.; Trombetta, C.M.; Crespan, E.; Maga, G.; Eydoux, C.; Decroly, E.; Montomoli, E.; Nencioni, L.; Zazzi, M.; Radi, M. System-oriented optimization of multi-target 2,6-diaminopurine derivatives: Easily accessible broad-spectrum antivirals active against flaviviruses, influenza virus and SARS-CoV-2. Eur J Med Chem. 2021, 224, 113683. [Google Scholar] [CrossRef] [PubMed]
- Sanna, C.; D'Abrosca, B.; Fiorentino, A.; Giammarino, F.; Vicenti, I.; Corona, A.; Caredda, A.; Tramontano, E.; Esposito, F. HIV-1 Integrase Inhibition Activity by Spiroketals Derived from Plagius flosculosus, an Endemic Plant of Sardinia (Italy) and Corsica (France). Pharmaceuticals 2023, 16, 1118. [Google Scholar] [CrossRef] [PubMed]
- Saladini, F.; Giannini, A.; Boccuto, A.; Vicenti, I.; Zazzi, M. Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors. J Clin Lab Anal. 2018, 32, e22206. [Google Scholar] [CrossRef] [PubMed]
- Poggialini, F.; Vagaggini, C.; Brai, A.; Pasqualini, C.; Crespan, E.; Maga, G.; Perini, C.; Cabella, N.; Botta, L.; Musumeci, F.; Frosini, M.; Schenone, S.; Dreassi, E. Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme. Pharmaceutics 2023, 15, 453. [Google Scholar] [CrossRef]
Scheme 2.
Synthesis of pyrimidine derivatives 19a-b. Reagents and conditions: i) CH2Cl2, 4-fluoroaniline, DIPEA, 0 °C, 5 h; ii) 1,4-dioxane, 4-fluoroaniline, HCl, MW, 110 °C, 40 min.; iii) 1. 1,4-dioxane, NH2NH2, MW, 100 °C, 4 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 48 h.
Scheme 2.
Synthesis of pyrimidine derivatives 19a-b. Reagents and conditions: i) CH2Cl2, 4-fluoroaniline, DIPEA, 0 °C, 5 h; ii) 1,4-dioxane, 4-fluoroaniline, HCl, MW, 110 °C, 40 min.; iii) 1. 1,4-dioxane, NH2NH2, MW, 100 °C, 4 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 48 h.
Scheme 3.
Synthesis of pyrimidine derivatives 21a, b. Reagents and conditions: i) ethanol (EtOH), morpholine, Et3N, 80 °C, 18 h; ii) 1. 1,4-dioxane, NH2NH2, MW, 100 °C, 6 h, 2. MeOH, 15a–b, acetic acid, 25 °C 48 h.
Scheme 3.
Synthesis of pyrimidine derivatives 21a, b. Reagents and conditions: i) ethanol (EtOH), morpholine, Et3N, 80 °C, 18 h; ii) 1. 1,4-dioxane, NH2NH2, MW, 100 °C, 6 h, 2. MeOH, 15a–b, acetic acid, 25 °C 48 h.
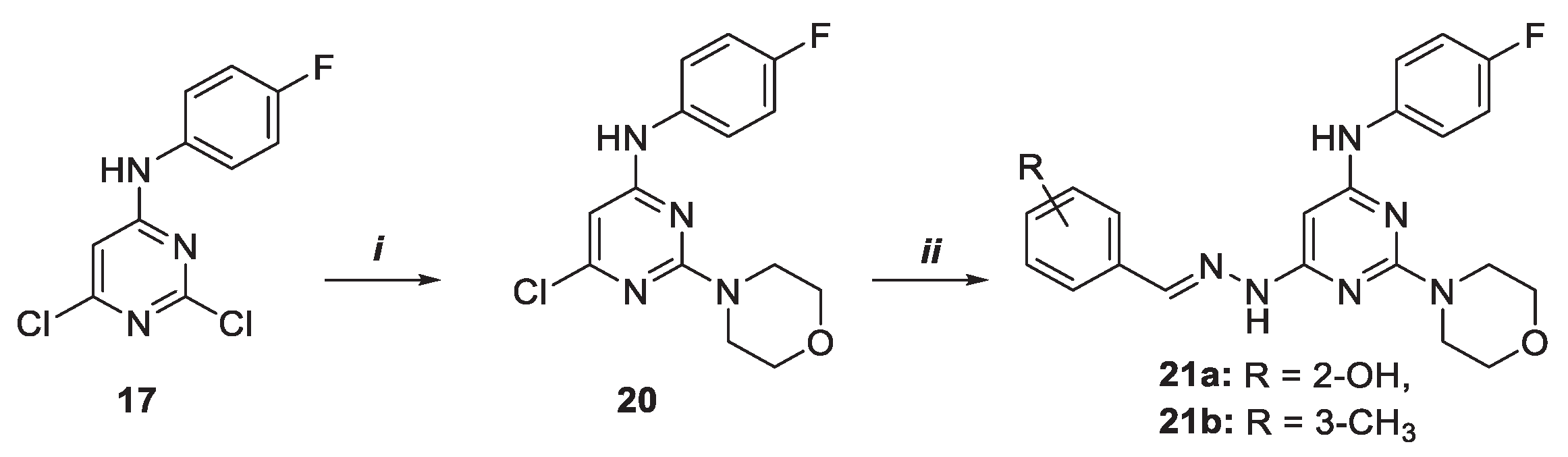
Scheme 4.
Synthesis of pyridine derivatives 27a, b. Reagents and conditions: i) THF, morpholine, Et3N, 65 °C, 18 h; ii) dimethyl formamide (DMF), morpholine, DIPEA, 120 °C, 48 h; iii) toluene, BocNHNH2, tris(dibenzylideneacetone)dipalladium (0) [Pd2(dba)3], 1,1'-Bis(diphenylphosphino)ferrocene (DPPF), cesium carbonate (Cs2CO3), MW, 150 °C, 10 min.; iv) 1. CH2Cl2, TFA, 25 °C, 1 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 24 h.
Scheme 4.
Synthesis of pyridine derivatives 27a, b. Reagents and conditions: i) THF, morpholine, Et3N, 65 °C, 18 h; ii) dimethyl formamide (DMF), morpholine, DIPEA, 120 °C, 48 h; iii) toluene, BocNHNH2, tris(dibenzylideneacetone)dipalladium (0) [Pd2(dba)3], 1,1'-Bis(diphenylphosphino)ferrocene (DPPF), cesium carbonate (Cs2CO3), MW, 150 °C, 10 min.; iv) 1. CH2Cl2, TFA, 25 °C, 1 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 24 h.
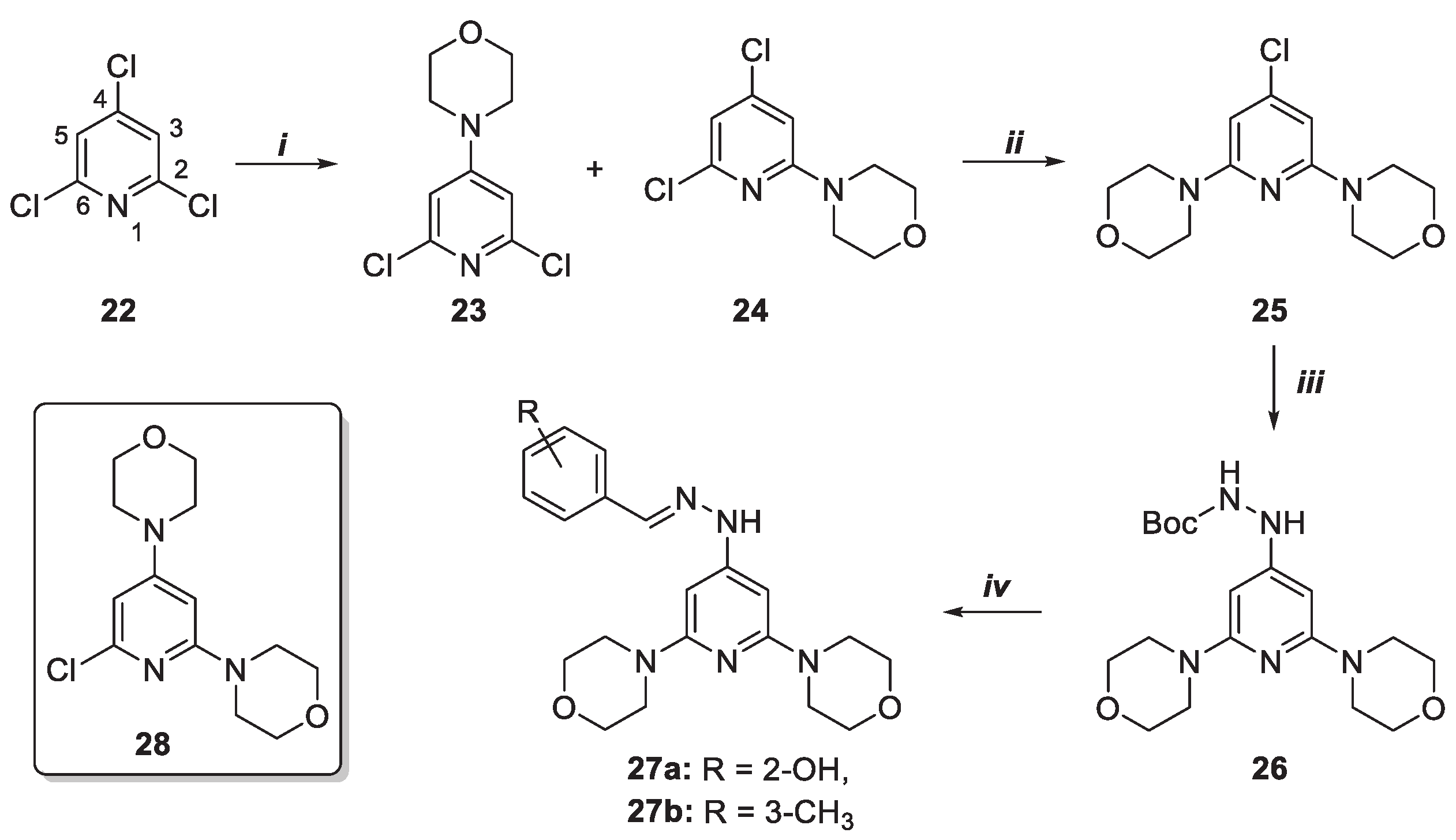
Scheme 5.
Synthesis of pyridine derivatives 33a, b. Reagents and conditions: i) toluene, tetrakis(triphenylphosphine)-palladium (0) [Pd(PPh3)4], 4-fluoroanilina, Cs2CO3, 150 °C, 24 h; ii) toluene, palladium(II) acetate [Pd(OAc)2], 2,2′-Bis(diphenylphosphino)-1,1′-binaphyl (BINAP), 4-fluoroaniline, Cs2CO3, 160 °C, 4 h; iii) toluene, BocNHNH2, Pd2(dba)3, DPPF, Cs2CO3 MW, 150 °C, 10 min.; iv) 1. CH2Cl2, TFA, 25 °C, 1 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 24 h.
Scheme 5.
Synthesis of pyridine derivatives 33a, b. Reagents and conditions: i) toluene, tetrakis(triphenylphosphine)-palladium (0) [Pd(PPh3)4], 4-fluoroanilina, Cs2CO3, 150 °C, 24 h; ii) toluene, palladium(II) acetate [Pd(OAc)2], 2,2′-Bis(diphenylphosphino)-1,1′-binaphyl (BINAP), 4-fluoroaniline, Cs2CO3, 160 °C, 4 h; iii) toluene, BocNHNH2, Pd2(dba)3, DPPF, Cs2CO3 MW, 150 °C, 10 min.; iv) 1. CH2Cl2, TFA, 25 °C, 1 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 24 h.
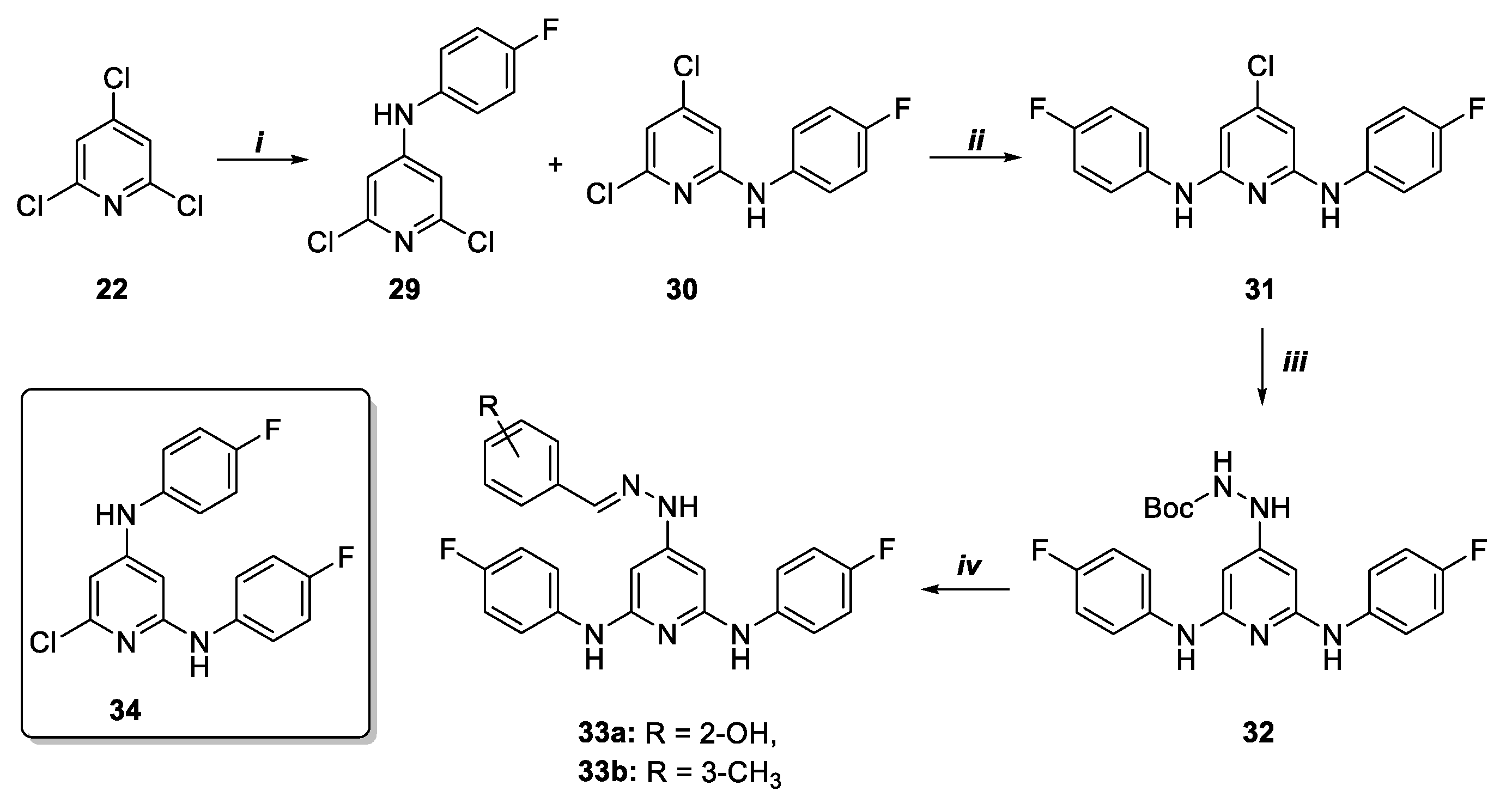
Scheme 6.
Synthesis of pyridine derivatives 38a, b. Reagents and conditions: i) 1,4-dioxane, morpholine, DIPEA, 160 °C, 10 h; ii) toluene, BocNHNH2, Pd2(dba)3, DPPF, Cs2CO3 MW, 150 °C, 10 min.; iv) 1. CH2Cl2, TFA, 25 °C, 1 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 24 h.
Scheme 6.
Synthesis of pyridine derivatives 38a, b. Reagents and conditions: i) 1,4-dioxane, morpholine, DIPEA, 160 °C, 10 h; ii) toluene, BocNHNH2, Pd2(dba)3, DPPF, Cs2CO3 MW, 150 °C, 10 min.; iv) 1. CH2Cl2, TFA, 25 °C, 1 h, 2. MeOH, 15a–b, acetic acid, 25 °C, 24 h.
Table 4.
In vitro PAMPA permeability studies of tested compounds.
| CPD | Papp1 | MR2 (%) |
|---|---|---|
| 19a | 3.59 ± 0.21 | 20.91 ± 1.75 |
| 19b | 5.64 ± 0.59 | 27.42 ± 1.22 |
| 21a | 4.80 ± 0.73 | 26.03 ± 6.17 |
| 33b | 1.38 ± 0.60 | 46.84 ± 4.99 |
| 38b | 6.35 ± 0.67 | 20.99 ± 1.69 |
1Apparent Permeability (Papp) reported in cm/s × 10−6. 2 Membrane retention %.
Table 5.
In vitro metabolic stability studies of tested compounds in presence of liver microsomes and plasma.
Table 5.
In vitro metabolic stability studies of tested compounds in presence of liver microsomes and plasma.
| CPD | Metabolic Stability (%) | Metabolite Formation (%)1 | Plasmatic Stability (%)2 |
|---|---|---|---|
| 19a | >99.9 | - | 86.64 ± 0.17 |
| 19b | 98.31 ± 0.11 | M1 = 1.68 ± 0.11 | >99.9 |
| 21a | >99.9 | - | 96.93 ± 2.73 |
| 33b | >99.9 | - | 81.01 ± 3.28 |
| 38b | 97.93 ± 0.46 | M1 =2.06 ± 0.46 | 87.13 ± 0.48 |
1 M1 = M + OH (+16). 2 Plasma stabilities after 24 h of incubation were calculated as percentages of Time 0 taken as 100%.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



