Submitted:
16 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
Separase is a key cysteine protease to separate sister chromatids through digestion of cohesion ring that inhibits chromosome segregation as trigger of metaphase–anaphase transition in eukaryotes and highly regulated its activity by binding with securin and cyclinB-CDK1 complex. Those bindings prevent proteolytic activity of separase until the onset of anaphase. Chromosome missegregation and aneuploidy are frequently observed in malignancies. However, there are some difficulties for biochemical examinations due to instability of separase in vitro and a few spatiotemporal resolution approaches monitoring live separase activity throughout mitotic processes. Here we have developed FRET-based molecular sensors including GFP variants with separase cleavable sequences as donor and covalently attached fluorescent dye as acceptor molecules applicable to conventional live cell imaging and flow cytometric analysis because of efficient live cell uptake. We investigated performance of equivalent molecular sensors localized or not localized inside nucleus under cell cycle control using flow cytometry. Synchronized cell cycle progression rendered significant separase activity detections in both molecular sensors. We obtained consistent outcomes with localized molecular sensor introduction and cell cycle control by fluorescent microscopic observations. We thus established live cell separase activity monitoring systems specifically or statistically that could lead to elucidate separase properties in detail.
Keywords:
separase
; cohesion
; securin
; CDK-1
; cyclin B1
; cell cycle
; chromosome segregation
; FRET
; live cell sensing
1. Introduction
Separase, a large eukaryotic endopeptidase (140-250kD) belonging to the cysteine protease family [1] that performs critical functions in the maintenance of genetic homeostasis. Specially, cells need to maintain genome stability during cell division to preserve and transmit their hereditary material intact to the next generation [2]. The cell cycle is a dynamic biological process that cause cell dividing into two daughter cells and most dramatic cellular phenomena involved in the cell cycle is the irreversible separation of sister chromatids, which happens during the mitotic phase specifically at the metaphase–anaphase transition in eukaryotes. This severely controlled processes are triggered by separase activation which is tightly regulated by its inhibitory chaperone, securin [3,4,5]. Securin stabilizes separase by co-translational binding to assist its correct folding [6,7,8]. The CDK-1 in vertebrates phosphorylates to connect phosphorylated separase and cyclin B1 to form the other controlling complex for separase activity [9,10,11].
Generally, separase has vital roles in many cellular functions, besides chromosome segregation during mitosis and meiosis [1,3,12], DNA damage repair [13,14,15], centrosome disengagement and duplication [16,17], spindle stabilization and elongation [18,19]. The proteins astrin and Aki1 function as inhibitors of centrosomal separase [20] and these combinations engages in proper spindle functions in mitosis [21]. Separase disintegrates the cohesion between sister chromatids by cleaving one of the subunits Scc1/Rad21/Mcd1 of the cohesin ring for chromosomal segregation [22]. Cohesin is the “glue” that binds the mother and daughter centrioles, and cleavage of this pool of cohesin by separase encourages centriole disengagement, according to subsequent biochemical investigations [23]. Interestingly, separase cleaves both the cohesin subunits Scc1/Rad21/Mcd1 and kendrin/pericentrin B at the centrosome [24]. But separase is well known to indicate minimal consensus motif as ExxR, as cleavage sites [1,4,22]. Separase activation is elicited by destruction of securin [25,26,27] and cyclin B1 [9] by proteasome degradation system after ubiquitination with anaphase promoting complex/cyclosome (APC/C) [28,29]. Additional mechanisms for separase activation process have been reported so that auto-cleavage of separase needed to promote mitosis, but not enhance protease activity [18,30,31,32]. These activation profiles are directed through phosphorylation states of separase including specific protein phosphatase 2A [33,34,35]. The molecular mechanisms underlying separase behaviors have been revealed by recent structural studies [36,37,38,39].
However, there still be remaining questions about separase activities and its regulation that can be clear with biochemical approaches. It is partially due to problems in defective expression system to obtain active separase unassisted with chaperone protein [7,40]. It is possible for anti-cancer medications to establish an index that will normalize excessive or insufficient separase activities. Therefore, in this investigation, we try to detect separase activity in mammalian live cells through FRET-based sensing by changing intramolecular FRET efficiencies upon enzymatic reactions. We have designed chimeric FRET-based molecular sensor that utilizes a green fluorescent protein (GFP) mutant (donor molecule) with separase cleavable sequences followed by unique cysteine for chemical modification with a fluorescent organic dye (acceptor molecule) as a simple conjugate (Figure 1) [41]. We could easily replace donor fluorescent protein, recognition sequences, and dye species to optimize assay system [42]. Furthermore, the molecular sensor can efficiently be delivered into live cells via endocytotic pathways. Here we have challenged to localize our molecular sensor for monitoring organelle specific event such as separase activations. We introduced microscopic observation to confirm our aim and flow cytometric analysis to check variety of activation extent for separase during cell cycle progression using molecular sensor localized inside nucleus and not localized one. We accomplished to detect separase activations upon pushing forward cell cycle in every case equally, demonstrating our robust molecular sensor functioned beyond approaches. Achievable flow cytometric separase assay would render the range of intracellular variation in separase activity or their alterations upon anti-cancer drug treatment in combination with other fluorescent markers or microscopic detailed observations. Very potentially and impressively, the versatile molecular sensor contributory systems we developed here not only monitor dynamics of separase in situ, but also can real-time assess the therapeutic efficacy of cancer treatment.
2. Materials and Methods
2.1. Plasmid Construction for Separase Activity Detection with or without Nuclear Localization Sequence (NLS)
Inverse polymerase chain reaction (iPCR) [43] was used to construct a plasmid containing the reporter genes for GFP and to detect separase activity. This plasmid was based on the pUV5casS52tag for caspase-3 activity sensing [44] and first manipulation was caried out to replace DEVD coded regions with DREIMRE around C- terminal to create pUV5Sep. Second manipulation included to insert GPKKKRKV (NLS) [45,46] around the N-terminal to obtain pUV5SepNLS. The genetic manipulations above were certified by usual gene sequencing. Coded amino acid sequences for GFP variants were shown in Figure S1.
2.2. Chimeric FRET-Based Molecular Sensor Preparation
Two variant green fluorescent proteins (GFP) were isolated initially. Two plasmids (pUV5Sep and pUV5SepNLS) described above were transfected into Escherichia coli BL21 cells (DE3) respectively. The transfected bacteria were cultured in Luria Bertani (LB) medium containing 75 µg/ml of ampicillin; the expression of the GFP derivatives were induced by isopropyl-β-D-thiogalactopyranoside (IPTG). The bacteria were harvested and lysed with enough amount of bacterial protein extraction reagent, B-PER II (Thermo Scientific Pierce, Rockford, IL, USA). Each target protein was isolated from the lysate by centrifugation for separation from cell debris and purified using Ni2+-NTA (nitrilotriacetic acid) affinity chromatography through (Histidine)6 tag sequence fused with the GFP derivative. The remaining reagents were removed with gel filtration column and recovered proteins were then reconstituted in phosphate buffered saline (PBS) separately. Isolated fluorescent proteins (FP) were chemically modified with fluorescent dyes namely Alexa Fluor-546 (Life Technologies, Carlsbad, CA, USA) (Thermo Scientific Pierce). 10 µM of GFP dissolved in PBS was reduced with 1mM of dithiothreitol (DTT). Excess DTT was removed by gel filtration equilibrated with Phosphate Buffered Saline (PBS) (NICK column, GE Healthcare, Buckinghamshire, UK), and an aliquot of the eluate was immediately incubated with the corresponding fluorescent dye at the molar ratio of 1:10 at 37°C for 4 h followed to stand for at 4°C for 6 h. Ultrafiltration (EMD, Millipore, Billerica, MA, USA) was used to remove unreacted dye and concentrate the solution to an adequate volume for use in future studies. The FRET efficiencies were assessed to check emission profiles excited at 488 nm by fluorescence spectroscopy using a Shimadzu RF-5300PC spectrophotometer (Shimadzu, Kyoto, Japan).
2.3. Introduction of Chimeric FRET-Based Molecular Sensor into Cell Cycle Controlled HeLa Cells
Subcultured HeLa cells were seeded into a culture dish filled with Dulbecco’s Modified Eagle Medium (DMEM) supplied with 10% fetal bovine serum (FBS) at 37 °C under an atmosphere containing 5% CO2. After confirmations of proper growth, culture medium was replaced with DMEM without FBS to suppress cell cycle progression and incubated for 24 hr at 37 °C under an atmosphere containing 5% CO2. The culture medium was replaced with DMEM without FBS containing 5 μM corresponding FRET-based molecular sensor for 12 h incubation at 37 °C under an atmosphere containing 5% CO2. The residual FRET-based molecular sensor was removed by washing the cells with culture medium without FBS and subjected to further experiments.
2.4. Flow Cytometry
FRET-based molecular sensor introduced HeLa cells and further experimentally processed cells were rinsed with 250 µl of 0.2 mM EDTA. The chelating solution was removed to stock and the remaining cells were treated with 250 µl of Accutase and Accumax cell detachment solutions (Innovative Cell technologies, San Diego, CA, USA) for 5 minutes at 37°C. The detached cell mixtures were recovered and combined with stocked solutions. Remained cells were completely detached with 250 µl of 0.25% trypsin solution. The resulting cell mixtures were added to all combined solutions up to the points. The culture plate was washed with 250 µl of PBS to be transferred to whole recovered solutions described above. The resulting solutions were filtered before being subjected to flow cytometry analysis using a SONY FACS machine (LE-SH800 SONY Co., Ltd. Tokyo Japan). The fluorescence intensity and other optical data by the harvested cell population (30,000 cells) were monitored by excitation using a 488-nm semiconductor laser. FlowJo software (Becton, Dickinson and Company, San Jose, CA, USA) was used to process the data collected.
2.5. Verification of Cell Cycle Contorol by Serum Starvation through DNA Amount Estimation
Harvested subconfluent cells were centrifuged at 15000 rpm and resuspended in 1 ml of DMEM supplied with 10% FBS and 10% DMSO. The suspension was frozen for one hour at -20 °C. The resulting materials were thawed and centrifuged at 15000 rpm at 4°C to remove supernatant. Collected cells were resuspended to adjust recommended cell density as 1 x 106 cells per milliliter in 1ml of ice-cold Hoechst staining buffer and incubated at 4°C for 15 minutes in the dark. Ethidium bromide was added to 1g/ml solution to stand for 15 minutes at 4°C in the dark as well. Whole solutions were filtrated and DNA contents of recovered cell solutions were determined flow cytometric analysis described above. Data analysis was performed with Flow Jo software correspondingly.
2.6. Separase Activations through Cell Cycle Progression
FRET-based molecular sensor introduced HeLa cells described in the Section 2.3 were divided into two populations. One population was harvested to be applied to flow cytometric analysis directly. The other populations were continuously exposed to DMEM supplied with 10% FBS at 37 °C under an atmosphere containing 5% CO2 for 24 hr. Processed cells were then collected and analyzed by flow cytometry mentioned above.
2.7. Fluorescence Microscopic Observation of Bioprobes Localization Inside Cells
FRET-based molecular sensor introduced HeLa cells described in the Section 2.3 were divided into two populations. One was observed immediately as the population before cell cycle restarting and the other was observed after 24 h-exposure to DMEM supplied with 10% FBS as the population after cell cycle progression. Fluorescence imaging was performed at room temperature using an FV-1000 confocal microscope (Olympus, Tokyo, Japan) with a × 60 oil immersion objective lens. FRET-based molecular sensor was excited at 488 nm from an Ar laser through a dichroic mirror (DM405/488/559). Fluorescence was separated at 560 nm by a dichroic mirror and detected by two photomultipliers through suitable band path filters: 500–545 nm for GFP and 570–670 nm for Alexa 546.
2.8. Imaging Analysis
The acquired images were analyzed with FV10-ASW software (Olympus). Regions of interest (ROIs) were placed on the nucleus region on the sensor-introduced cells, and the average fluorescence intensity of each ROI was calculated. After the average fluorescence intensity of the ROI at the region without cells was subtracted as background, fluorescence ratio (GFP/Alexa 546) was calculated.
2.9. General Image Acquisitions for FRET-Based Molecular Sensor Localizations in HeLa Cells
Approximate examination of molecular sensor localization was carried out at room temperature using an ECLIPSE TE 2000U (Nikon Solutions Co. Ltd. Tokyo Japan) with a x 40 water immersion objective lens. FRET-based molecular sensor was excited with ultra-high pressure mercury lamp through adequate combination of dichroic mirror and filter to detect GFP and Alexa 546 emission and show bright images each other. The acquired images were processed with ImageJ supported by NIH.
2.10. Western Blot Analysis
Expression profiles of separase for cell populations in cell cycle arrest or its continuation were examined by western blotting. HeLa cells cultured with DMEM supplied with or without 10% FBS for 24 hr were collected individually by centrifugation at 15000 rpm following trypsinization. The recovered cells were rinsed with aliquots of PBS by centrifugation in the same way and lysed using M-PER™, Mammalian Protein Extraction Reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to standard procedures instructed by supplier Protein concentrations for recovered extracts were determined by spectrophotometer at 280 nm wavelength absorption. Denatured samples were applied to SDSPAGE and transferred to PVDF membrane to carry out western blotting using primary antibody, anti- Separase rabbit polyclonal antibody (abcam plc. Cambridge, UK) followed by detecting the 233 kDa full length separase with Goat Anti-Rabbit IgG HL (Alexa FluorR 680) as secondary antibody. Protein extraction of cell lysates was verified to adjust by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) detection with GAPDH rabbit polyclonal antibody (Proteintech Group Inc. Tokyo, Japan). Band images were taken by fluorescent imager (Tyhoon FLA 9500, GE Healthcare Japan, Tokyo Japan).
3. Results and Discussion
3.1. FRET-Based Molecular Sensor Preparations and Their Fluorescence Properties
We initially produced molecular sensors to target separase protease, and it could be straightforward to utilize Fluorescence Resonance Energy Transfer (FRET) system as the sensing mechanism for proteolysis. We have established chimeric FRET based molecular sensors for varied cysteine proteases as caspase-1, 3, 9, and 14. [47,48,49]. Though we have already demonstrated broad range of Alexa dyes (Alexa 532, 546, 555, 568 and 594) combined with GFP variants could perform emission spectrum changes upon proteolytic monitoring, there were preferable dyes for individual cysteine proteases. Structural basis for separase belonging to cysteine protease family confirmed to be highly homologous to caspase-9 [36] so that we introduced Alexa 546 for our new targeted FRET based molecular sensors because of optimal dye for caspase-9.
Next, we examined fluorescence emission patterns obtained on attachment of the dyes to two GFP variants with or without Nuclear Localization Signal (NLS) namely NLS based and WNLS (without NLS) based, to evaluate molecular sensors regarding FRET-based sensing by FRET efficiencies of the corresponding molecular sensors (amino acid sequences are shown in Figure S1). For each combination, significant decreases in donor fluorescence intensities and appearances of acceptor emissions were observed in Figure 2. The emission ratios were estimated as approximate FRET efficiency = donor emission intensity maximum / acceptor emission intensity maximum. Calculated FRET efficiencies of NLS based and WNLS were 0.164 and 0.200 respectively. In case of commercially available protease, we can assess sensing performances as emission spectrum changes with protease treatments to obtain exchanges for FRET efficiencies, however inaccessibility for isolated separase due to unstable behavior without chaperone protein prevent such operations.
3.2. Introduction of Chimeric FRET-Based Molecular Sensors into Cell Cycle-Controlled HeLa Cells and Flow Cytometry
After successfully preparation of molecular sensors, we next assessed molecular sensors uptake efficiencies into HeLa cells using flow cytometry. The cell sets were subjected to donor and acceptor emission dot-plot analysis for untreated cells, NLS GFP-Alexa 546 and WNLS GFP-Alexa 546 treated cells. By gating of untreated cell populations for auto fluorescent estimation, we confirmed that more than 90% cell populations could uptake significant amount of the fluorescent proteins by applying 5µM concentration of molecular sensors. Moreover, there was no big differences among the two GFP variants originated molecular sensors analysis shown in Figure 3A–C.
3.3. Checking Cell Cycle Control/Synchronization by DNA Amount Quantify
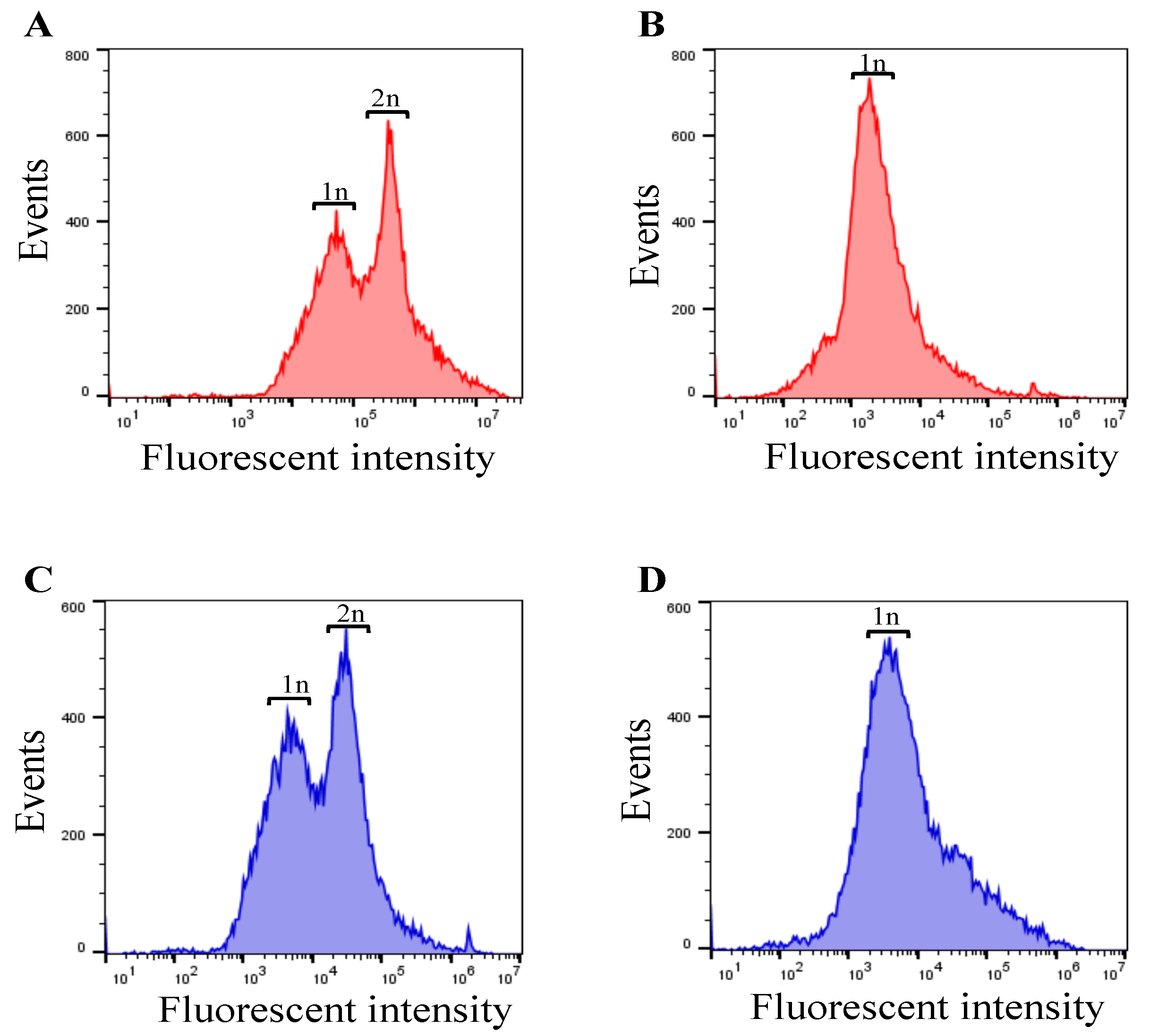
As separase activities in cells and proliferation rates of the cell population should correlate with DNA amount in the cells, we examined influences of cell cycle synchronization by serum starvation [50] on DNA amount contained in the cells. HeLa cells after 48 hr serum deprivation were stained with ethidium bromide and propidium iodide to compare with not serum starved HeLa cell population and DNA content was measured using a flow cytometer, SONY FACS machine. In case of serum starvation condition, only haploid cells were found, whereas cells with serum supplement condition both haploid and diploid cells were observed. We thus proved cell cycle arrest emerged as cells with haploid DNA content. Accordingly, we could collect separase activated cells through re-feeding with serum for cells.
Figure 4.
DNA amount in FBS supplied (100% FBS) and starved (0% FBS) HeLa cells stained with ethidium bromide (A-B) and propidium iodide (C-D).
Figure 4.
DNA amount in FBS supplied (100% FBS) and starved (0% FBS) HeLa cells stained with ethidium bromide (A-B) and propidium iodide (C-D).
3.4. Separase Activations through Cell Cycle Progression
Combined with the results of molecular sensors uptake efficiency and cell cycle synchronization by serum supplement conditions, we next tried to monitor separase activities with molecular sensors localized inside nucleus or dispersed in the whole cell upon separase activated cell collection. First, we observed localizations for two types of molecular sensors inside HeLa cells through microscopic observations mentioned in the Section 2.7. As shown in Figure S2, we obtained different distribution patterns for each molecular sensor. As for not localized type of molecular sensor (WNLS based), it seemed to stay at endosome around nucleus to disperse into cytosol. On the other hand, nucleus localized type of molecular sensor (NLS based) accumulated inside nucleus.
After confirmation of different localizations in cells for NLS and WNLS based molecular sensors, we measured separase activities upon cell cycle control shown in Figure 5A. We then checked distributions of FRET efficiencies for molecular sensors inside HeLa cells as histograms derived from populations in dot plot analysis (Figure 5B). Both Cell populations demonstrated FRET efficiencies increases after 24-hour re-feeding with serum. We calculated increasing ratios for 0 hr to 24 hr (FRET efficiencies at 24 hr / FRET efficiencies at 0 hr and calculated ratios for NLS based molecular sensor (1.40) was nearly the same as WNLS based one (1.42). (Figure 5C). This indicates that these molecular sensors would be adequately sensitive to detect separase activity inside cells and enough amount of not localized molecular sensor might spread to presence inside nucleus.
3.5. Imaging Analysis
We then attempt to validate our NLS based molecular sensor function using microscopic observations in detail upon corresponding cell cycle control. As shown in Figure 6A, it was demonstrated that NLS based molecular sensor almost existed inside nucleus normally. Some molecular sensor uptake cells changed their shape to spherical but indicated equivalent localization patterns shown in Figure S3. Representative emission profiles of them were shown in Figure 6B. A small but steady increase in GFP fluorescence was observed, indicating disappearance of FRET between GFP and Alexa 546 upon digestion of the inserted separase recognition sequence. We also calculated and compared average FRET efficiencies for 0 h and 24 h processed cells (Figure 6C). FRET efficiency (GFP/Alexa 546) increased after cell cycle progression. These observations almost fitted with our flow cytometric analysis, i.e., our chimeric FRET-based molecular sensor could appropriately detect separase activities upon cell cycle progression.
3.6. Western Blot Analysis
We analyzed separase occurrences for cells under cell cycle control as 0 hr and 24 hr processing by western blotting. As shown in Figure 7, separase occurrences in both cell populations were found. However, more clear presence of separase were shown in 0 hr processed cell lysate derived sample. On the other hands, 24 hr treatment indicated not only faint presence of separase but also several digested bands. These findings might be consistent with separase occurrences bound with chaperone protein securin and CDK1-cyclin B1 complex to inhibit their activity before cell cycle progression and separase activation through autocleavage [51].
4. Conclusions
We have established live cell monitoring systems for separase activity through microscopic observation and flow cytometric analysis that could allow quantitative assay for separase activation profiles during cell cycle with spatiotemporal resolution and statistic considerations owing to consistent outcomes from both approaches. Our monitoring systems could facilely be applied to other cell types [42]. It could render new insight for separase behaviors in live cells under cell cycle arrest or progress conditions in detail. Furthermore, our simple strategy to fabricate chimeric FRET-based molecular sensors for live cell proteolysis monitoring enable us to improve sensitivities, replace target proteases, alter localization inside cells and change fluorescent properties by protein engineering and introductions of other combinations for fluorescent proteins and dyes. It might expand sensing targets simultaneously. Already demonstrated combinatorial use of molecular sensors for caspase-9 and -3 might straightforwardly be substituted by the sensor in this work that would give us advanced comprehension for separase involved in programmed cell death [52]. In this context we are confident that our assay will be applicable as a clinical diagnostic and prognostic tool to monitor separase proteolytic activity combined with another cellular event in human malignancies and therapeutic mechanism in future studies.
Supporting Information
The supporting information is available free of charge on MDPI Web site.
Author Contributions
M.S. conceptualized, supervised, supported general microscopic observations and prepared the manuscript. M.S.R. prepared all molecular sensors, performed flow cytometric analysis and committed to manuscript preparation. Y.S. conducted microscopic observations for live imaging in detail. K.O. integrated whole works and checked the manuscript on the point of our concept. W.I. contributed to examine protein expression. This manuscript has been unanimously approved by all authors.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data are available upon request from the corresponding author.
Acknowledgments
We gratefully acknowledge the work of past and present members of our laboratory.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Uhlmann, F.; Wernic, D.; Poupart, M.A.; Koonin, E.V.; Nasmyth, K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 2000, 103, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Ciosk, R.; Zachariae, W.; Michaelis, C.; Shevchenko, A.; Mann, M.; Nasmyth, K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 1998, 93, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Henschke, L.; Frese, M.; Hellmuth, S.; Marx, A.; Stemmann, O.; Mayer, T.U. Identification of Bioactive Small Molecule Inhibitors of Separase. ACS Chem. Biol. 2019, 14, 2155–2159. [Google Scholar] [CrossRef] [PubMed]
- Jallepalli, P.V.; Lengauer, C. Chromosome segregation and cancer:Cutting through the mystery. Nat. Rev. Cancer 2001, 1, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Hornig, N.C.; Knowles, P.P.; McDonald, N.Q.; Uhlmann, F. The dual mechanism of separase regulation by securin. Curr. Biol. 2002, 12, 973–982. [Google Scholar] [CrossRef]
- Waizenegger, I.; Giménez-Abián, J.F.; Wernic, D.; Peters, J.-M. Regulation of human separase by securin binding and autocleavage. Curr. Biol. 2002, 12, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Gorr, I.H.; Boos, D.; Stemmann, O. Mutual inhibition of separase and Cdk1 by two-step complex formation. Molecular Cell 2005, 19, 135–141. [Google Scholar] [CrossRef]
- Sanchez-Puig, N.; Veprintsev, D.B.; Fersht, A.R. Human full-length Securin is a natively unfolded protein. Protein Sci. 2005, 14, 1410–1418. [Google Scholar] [CrossRef]
- Csizmok, V.; Felli, I.C.; Tompa, P.; Banci, L.; Bertini, I. Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 16873–16879. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, S.B.C.; Clyne, R.K.; Fuchs, J.; Loidl, J.; Uhlmann, F.; Nasmyth, K. Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 2000, 103, 387–398. [Google Scholar] [CrossRef]
- Nagao, K.; Adachi, Y.; Yanagida, M. Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 2004, 430, 1044–1048. [Google Scholar] [CrossRef]
- McAleenan, A.; Clemente-Blanco, A.; Cordon-Preciado, V.; Sen, N.; Esteras, M.; Jarmuz, A.; Aragón, L. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 2013, 493, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, S.; Gutiérrez-Caballero, C.; Llano, E.; Pendás, A.M.; Stemmann, O. Local activation of mammalian separase in interphase promotes double-strand break repair and prevents oncogenic transformation. EMBO J. 2018, 37, e99184. [Google Scholar] [CrossRef] [PubMed]
- Tsou, M.F.; Wang, W.J.; George, K.A.; Uryu, K.; Stearns, T.; Jallepalli, P.V. Polo kinase and separase regulatethe mitotic licensing of centriole duplication in human cells. Dev. Cell 2009, 17, 344–354. [Google Scholar] [CrossRef]
- Lee, K.; Rhee, K. Separase-dependent cleavage of pericentrin B is necessary and sufficient for centriole disengagement during mitosis. Cell Cycle 2012, 11, 2476–2485. [Google Scholar] [CrossRef]
- Papi, M.; Berdougo, E.; Randall, C.L.; Ganguly, S.; Jallepalli, P.V. Multiple roles for separase auto-cleavage during the G2/M transi- tion. Nat. Cell Biol. 2005, 7, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, C.; Segal, M.; Reed, S.I. The protease activity of yeast separase (esp1) is required for anaphase spindle elongation independently of its role in cleavage of cohesin. Genetics 2008, 178, 2361–2372. [Google Scholar] [CrossRef]
- Thein, K.H.; Kleylein-Sohn, J.; Nigg, E.A.; Gruneberg, U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J. Cell Biol. 2007, 178, 345–354. [Google Scholar] [CrossRef]
- Nakamura, A.; Arai, H.; Fujita, N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J. Cell Biol. 2009, 187, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Hauf, S.; Waizenegger, I.C.; Peters, J.M. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Sci. 2001, 293, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Schockel, L.; Mockel, M.; Mayer, B.; Boos, D.; Stemmann, O. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat. Cell Biol. 2011, 13, 966–972. [Google Scholar] [CrossRef]
- Matsuo, K.; Ohsumi, K.; Iwabuchi, M.; Kawamata, T.; Ono, Y.; Takahashi, M. Kendrin is a novel substrate for separase involved in the licensing of centriole duplication. Current Biol. 2012, 22, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Funabiki, H.; Kumada, K.; Yanagida, M. Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO J. 1996, 15, 6617–6628. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Fix, O.; Peters, J.M.; Kirschner, M.W.; Koshland, D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996, 10, 3081–3093. [Google Scholar] [CrossRef]
- Rao, H.; Uhlmann, F.; Nasmyth, K.; Varshavsky, A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature 2001, 410, 955–959. [Google Scholar] [CrossRef]
- Zachariae, W.; Shin, T.H.; Galova, M.; Obermaier, B.; Nasmyth, K. Identification of subunits of the anaphasepromoting complex of Saccharomyces cerevisiae. Sci. 1996, 274, 1201–1204. [Google Scholar] [CrossRef]
- Peters, J.-M. The anaphase-promoting complex: Proteolysis in mitosis andbeyond. Mol. Cell 2002, 9, 931–943. [Google Scholar] [CrossRef]
- Zou, H.; Stemman, O.; Anderson, J.S.; Mann, M.; Kirschner, M.W. Anaphase specific auto-cleavage of separase. FEBS Lett. 2002, 528, 246–250. [Google Scholar] [CrossRef]
- Herzig, A.; Lehner, C.F.; Heidmann, S. Proteolytic cleavage of the THR subunit during anaphase limits Drosophila separase function. Genes Dev. 2002, 16, 2443–54. [Google Scholar] [CrossRef] [PubMed]
- Chestukhin, A.; Pfeffer, C.; Milligan, S.; DeCaprio, J.A.; Pellman, D. Processing, localization, and requirement of human separase for normal anaphase progression. Proc. Natl. Acad. Sci. USA. 2003, 100, 4574–4579. [Google Scholar] [CrossRef]
- Holland, A.J.; Böttger, F.; Stemmann, O.; Taylor, S.S. Protein phosphatase 2A and separase form a complex regulated by separase autocleavage. J. Biol. Chem. 2007, 282, 24623–24632. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, S.; Böttger, F.; Pan, C.; Mann, M.; Stemmann, O. PP2A delays APC/C-dependent degradation of separase-associated but not free securin. EMBO J. 2014, 33, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Cohen-Fix, O. Phosphorylation of the mitotic regulator Pds1/securin by Cdc28 is required for efficient nuclear localization of Esp1/separase. Genes Dev. 2002, 16, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Luo, X.; Yu, H. Structural basis of cohesin cleavage by separase. Nature 2016, 532, 131–134. [Google Scholar] [CrossRef]
- Luo, S.; Tong, L. Molecular mechanism for the regulation of yeast separase by securin. Nature 2017, 542, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Boland, A.; Martin, T.G.; Zhang, Z.; Yang, J.; Bai, X.C.; Chang, L.; Scheres, S.H.; Barford, D. Cryo-EM structure of a metazoan separase-securin complex at near-atomic resolution. Nat. Struct. Mol. Biol. 2017, 24, 414–418. [Google Scholar] [CrossRef]
- Yu, J.; Raia, P.; Ghent, C.M.; Raisch, T.; Sadian, Y.; Cavadini, S.; Sabale, P.M.; Barford, D.; Raunser, S.; Morgan, D.O.; et al. Structural basis of human separase regulation by securin and CDK1-cyclin B1. Nature 2021, 596, 138–142. [Google Scholar] [CrossRef]
- Hellmuth, S.; Rata, S.; Brown, A.; Heidmann, S.; Novak, B.; Stemmann, O. Human chromosome segregation involves multi-layered regulation of separase by the peptidyl-prolyl-isomerase Pin1. Mol. Cell 2015, 58, 495–506. [Google Scholar] [CrossRef]
- Zhang, W.; Suzuki, M.; Ito, Y.; Douglas, K.T. A Chemically Modified Green-Fluorescent Protein that Responds to Cleavage of an Engineered Disulphide Bond by Fluorescence Resonance Energy Transfer (FRET)-Based Changes. Chemistry Letters 2005, 34, 766–767. [Google Scholar] [CrossRef]
- Suzuki, M.; Shindo, Y.; Yamanaka, R.; Oka, K. Live imaging of apoptotic signaling flow using tunable combinatorial FRET-based bioprobes for cell population analysis of caspase cascades. Sci. Rep. 2022, 12, 21160. [Google Scholar] [CrossRef]
- Imai, Y.; Matsushima, Y.; Sugimura, T.; Terada, M. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res. 1991, 19, 2785. [Google Scholar] [CrossRef] [PubMed]
- Alnemri, T.F.; Armstrong, R.C.; Krebs, J.; Srinivasula, S.M.; Wang, L.; Bullrich, F.; Fritz, L.C.; Trapani, J.A.; Tomaselli, K.J.; Litwack, G.; et al. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad. Sci. USA 1996, 93, 7464–7469. [Google Scholar] [CrossRef] [PubMed]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Kitamura, A.; Nakayama, Y.; Kinjo, M. Efficient and dynamic nuclear localization of green fluorescent protein via RNA binding. Biochem. Biophys. Res. Commun. 2015, 463, 401–6. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tanaka, S.; Ito, Y.; Inoue, M.; Sakai, T.; Nishigaki, K. Simple and tunable Förster resonance energy transfer-based bioprobes for high-throughput monitoring of caspase-3 activation in living cells by using flow cytometry. Biochim Biophys Acta. 2012, 1823, 215–26. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Shindo, Y.; Yamanaka, R.; Oka, K. Live imaging of apoptotic signaling flow using tunable combinatorial FRET-based bioprobes for cell population analysis of caspase cascades. Sci. Rep. 2022, 12, 21160. [Google Scholar] [CrossRef]
- Suzuki, M.; Taguma, K.; Bandaranayake, U.K.; Ikeda, W. Development of Ratiometric Fluorescence Sensing Molecule for Caspase-14, a Key Enzyme of Epidermis Metabolism. J. Biomed. Res. Environ. Sci. 2023, 4, 793–800. [Google Scholar] [CrossRef]
- Chen, M.; Huang, J.; Yang, X.; Liu, B.; Zhang, W.; Huang, L. Serum Starvation Induced Cell Cycle Synchronization Facilitates Human Somatic Cells Reprogramming. PLoS ONE 2012, 7, e28203. [Google Scholar] [CrossRef]
- Konecna, M.; Abbasi Sani, S.; Anger, M. Separase and Roads to Disengage Sister Chromatids during Anaphase. Int. J. Mol. Sci. 2023, 27, 24. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, S.; Stemman, O. Separase-triggered apoptosis enforces minimal length mitosis. Nature 2020, 580, 542–547. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sensing mechanism of FRET based separase molecular sensors.
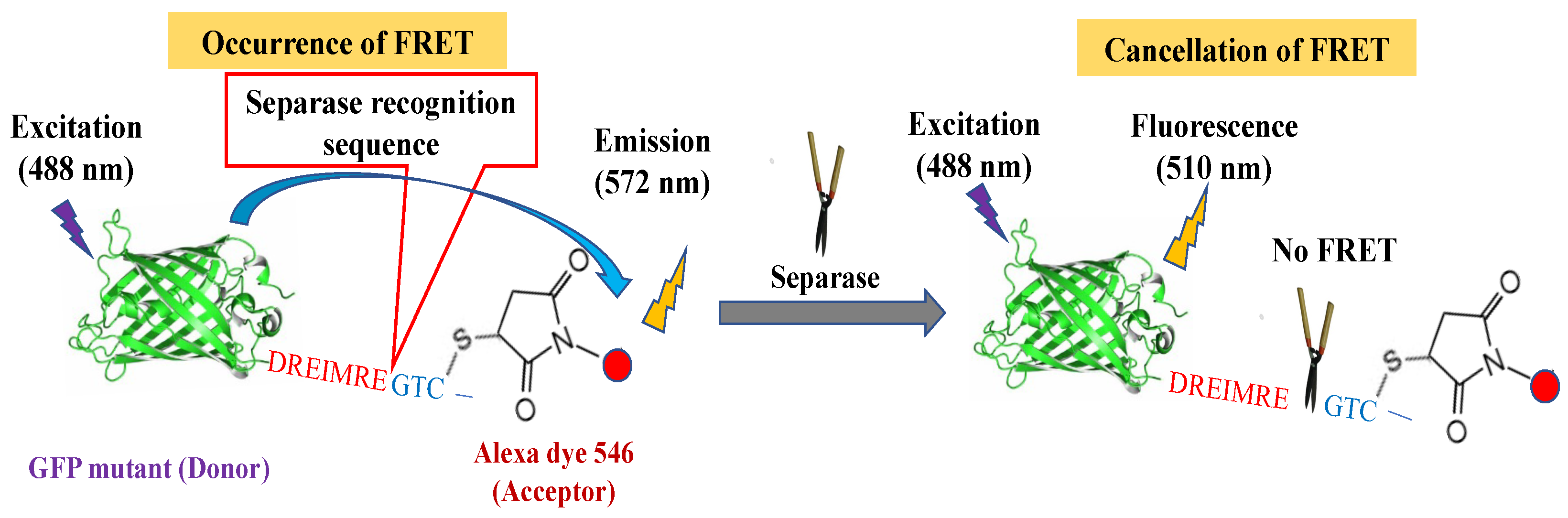
Figure 2.
Emission patterns of molecular sensors excited at 488 nm.
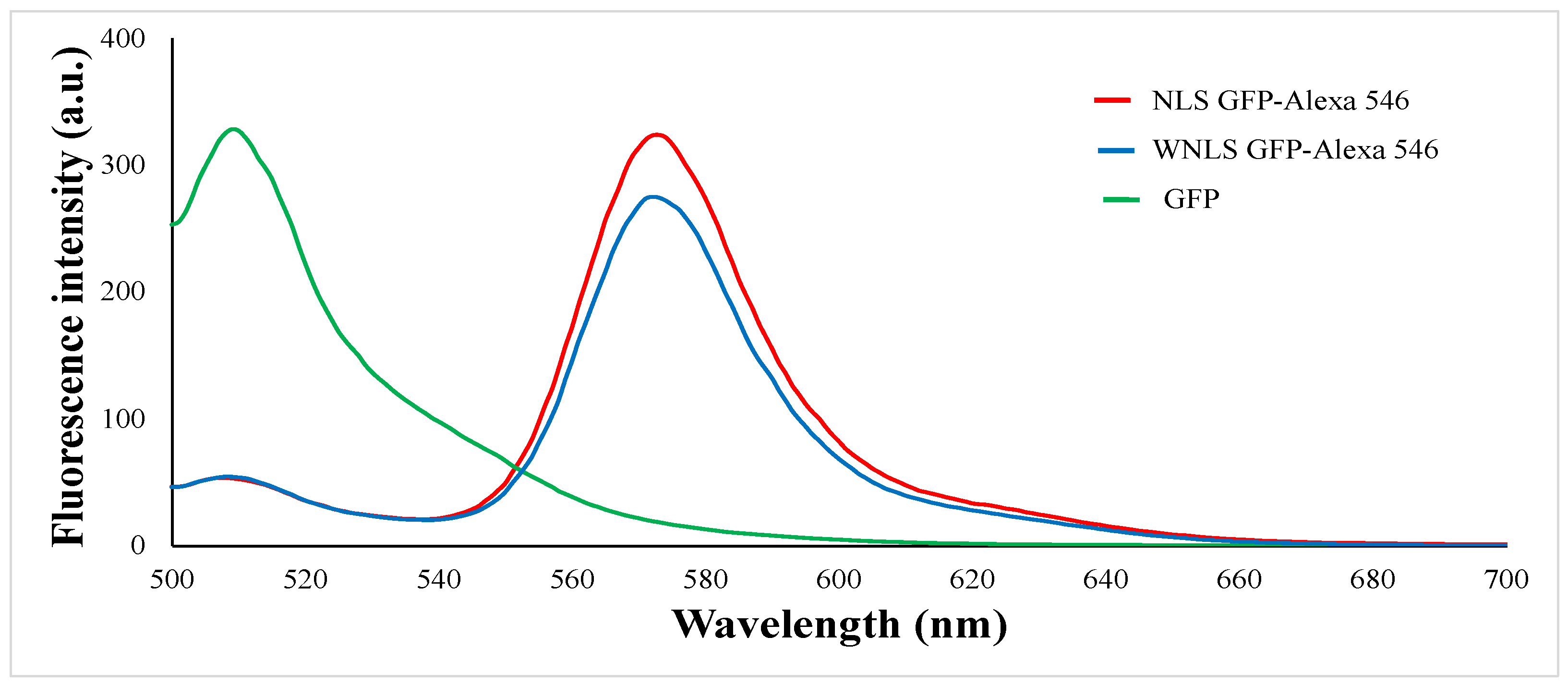
Figure 3.
Flow cytometric confirmation of molecular sensors for separase sensing uptake into HeLa cells by dot plot analysis.
Figure 3.
Flow cytometric confirmation of molecular sensors for separase sensing uptake into HeLa cells by dot plot analysis.
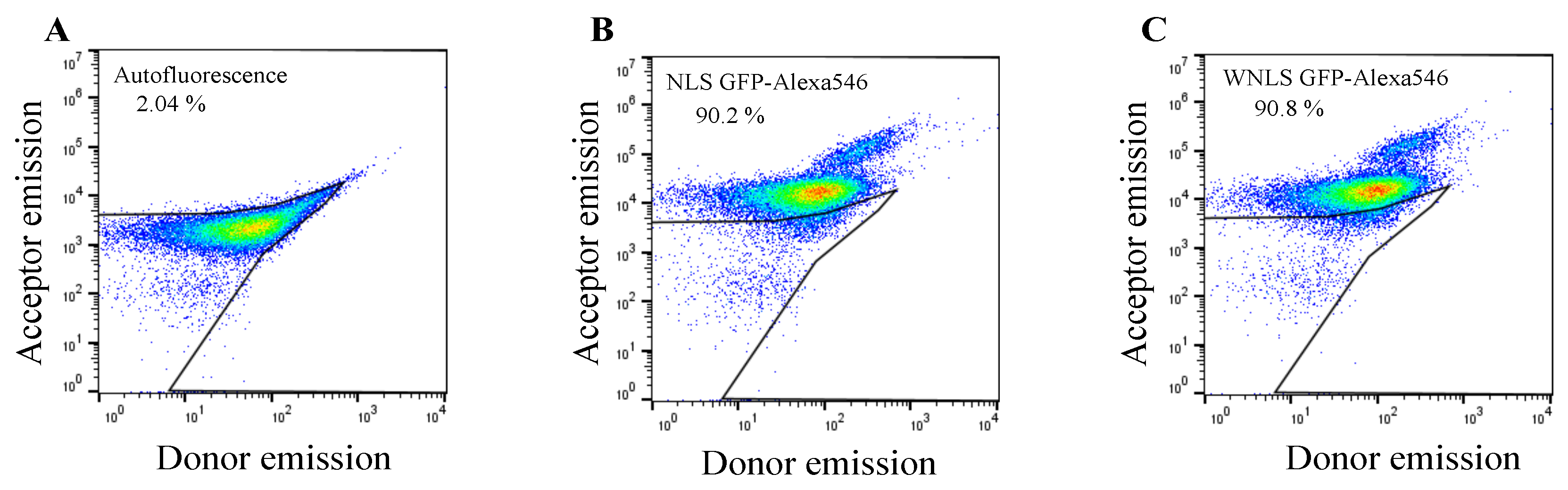
Figure 5.
Separase activity monitoring through cell cycle progression A. Cell cycle control experimental flow was schematically drawn. FRET efficiencies (after 0hr and 24hr incubation) were approximately estimated using median values from histograms for FRET efficiencies. C. Increasing ratios for FRET efficiencies of 0 hr and 24 hr were calculated for NLS and WNLS based molecular sensor introduced cell populations.
Figure 5.
Separase activity monitoring through cell cycle progression A. Cell cycle control experimental flow was schematically drawn. FRET efficiencies (after 0hr and 24hr incubation) were approximately estimated using median values from histograms for FRET efficiencies. C. Increasing ratios for FRET efficiencies of 0 hr and 24 hr were calculated for NLS and WNLS based molecular sensor introduced cell populations.
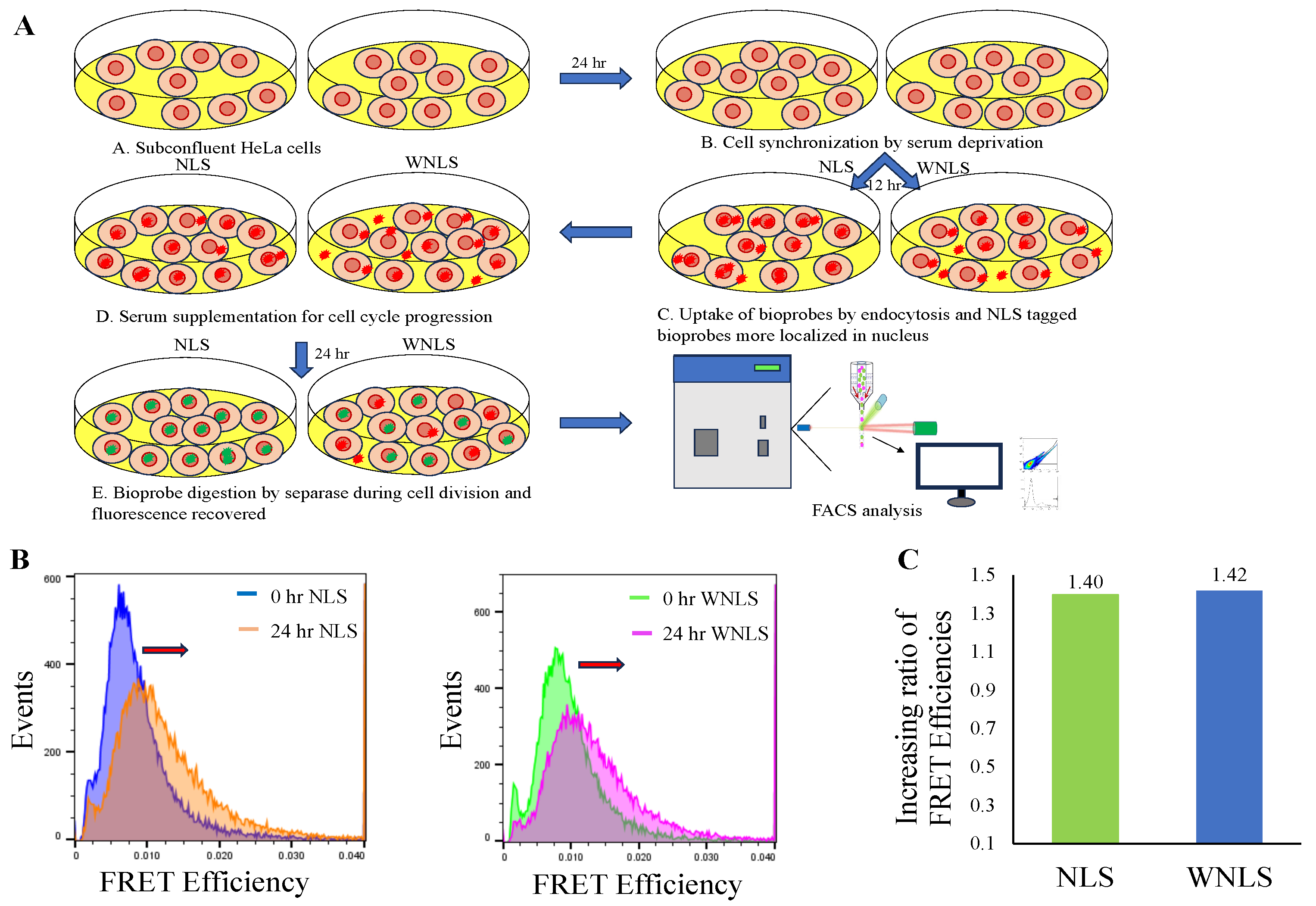
Figure 6.
Separase activity monitoring through cell cycle progression. A. Fluorescence microscopic observation of molecular sensors localization inside cells. B. The emission profiles in HeLa cell after 0 hr and 24 hr incubation in culture medium containing serum. Fluorescence intensity was normalized at isosbestic point. C. FRET efficiencies exchanges before and after cell cycle progression (Before: n = 55 cells from three different experiments, After: 34 cells from three different experiments). Error bars: S.E.M. *: p < 0.05 in Student’s t-test.
Figure 6.
Separase activity monitoring through cell cycle progression. A. Fluorescence microscopic observation of molecular sensors localization inside cells. B. The emission profiles in HeLa cell after 0 hr and 24 hr incubation in culture medium containing serum. Fluorescence intensity was normalized at isosbestic point. C. FRET efficiencies exchanges before and after cell cycle progression (Before: n = 55 cells from three different experiments, After: 34 cells from three different experiments). Error bars: S.E.M. *: p < 0.05 in Student’s t-test.
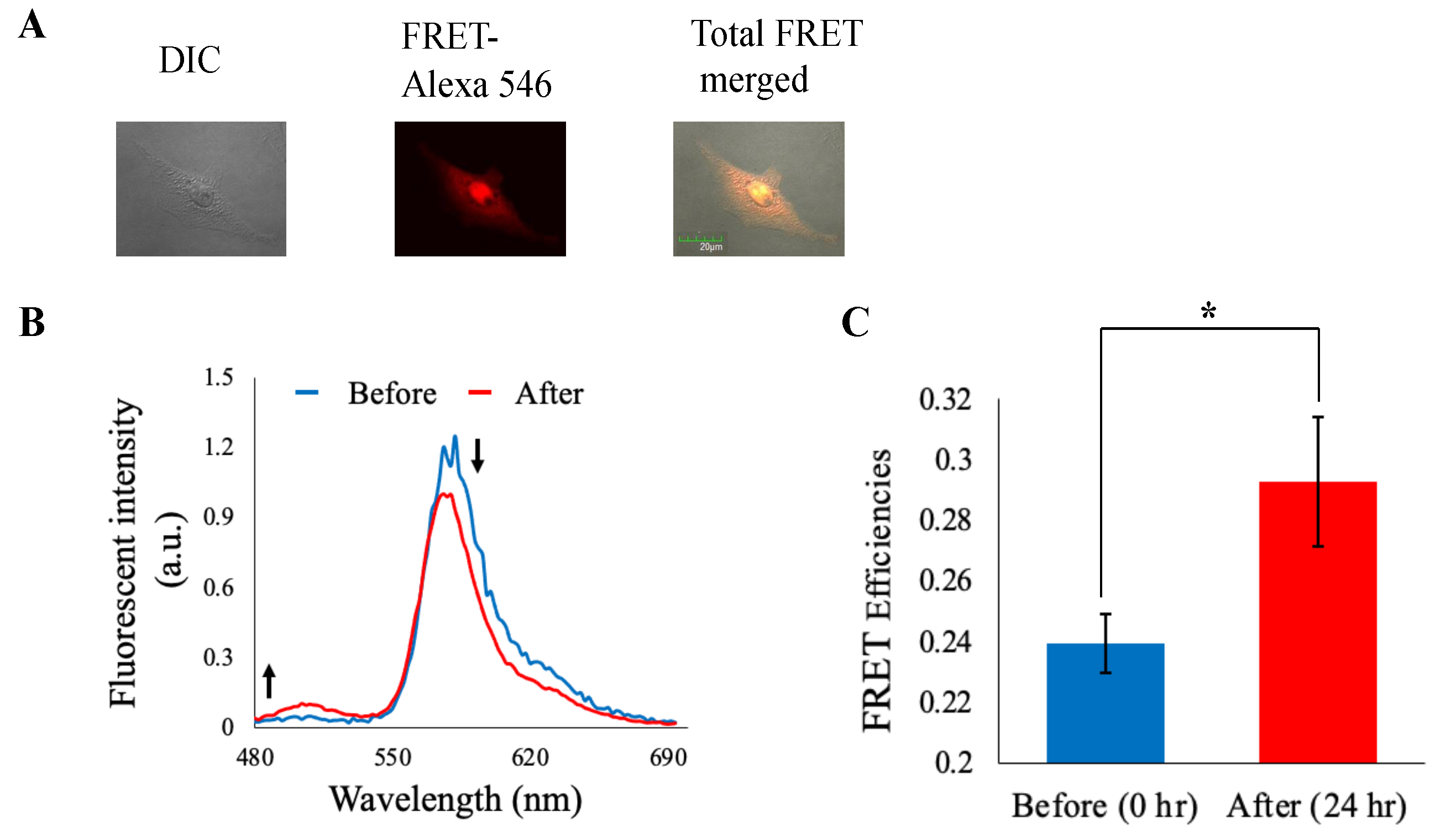
Figure 7.
Separase expression profiles of cell cycle synchronized 0 hr and 24 hr.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



