
Submitted:
18 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
(1) Background: Sudden Infant Death Syndrome (SIDS) represents unexplained and sudden deaths during the sleep of infants under one year of age, despite thorough investigation. Screening for prolonged QTc or Long QT Syndrome (LQTS) should be conducted on all newborns to reduce the incidence of SIDS. Neonatal electrocardiogram (ECG) could identify congenital heart defects (CHD) early, especially those not detected at birth. Infants with prolonged QT intervals typically undergo genetic analysis for Long QT Syndrome; (2) Methods: The study involved infants aged 20-40 days, born clinically healthy, with initial ECG screening. Infants with prenatal diagnoses or signs/symptoms of CHD identified immediately after birth, as well as those who underwent ECG or echocardiogram for other clinical or historical indications, were excluded from the study. The data were analyzed using SPSS version 22.0; (3) Results: Of the 42,200 infants involved, 2,245 were enrolled, with 39.9% being males. Subsequently, 164 children (37.8% males) with prolonged QT intervals underwent further diagnostic investigations. Genetic mutations were identified in 11 out of 18 tested children. The most common mutations were LQT1 (54.5%), LQT2 (36.4%), and LQT3 (1 patient). Medications used included propranolol (39.8%), nadolol (22.2%), inderal (11.1%), metoprolol (11.1%), and no treatment (16.7%). The most common abnormalities were focal right bundle branch block (54.5%), left axis deviation (9.2%), and nonspecific ventricular repolarization abnormalities (7.1%). Multiple anomalies were found in 0.47% of children with focal right bundle branch block. Structural abnormalities were associated with specific features in 267 patients (11.9%), primarily isolated patent foramen ovale (PFO) at 61.4%; (4) Conclusions: The screening has proven effective in early diagnosing Long QT Syndrome and other cardiac rhythm anomalies, with additional benefits in detecting mutations and/or QTc prolongations in family members. Identifying other ECG abnormalities and congenital heart malformations further enhances the benefits of the screening.
Keywords:
children
; congenital heart disease
; Electrocardiogram
; Infant
; long QT syndrome
; neonatal screening
; sudden infant death syndrome.
1. Introduction
The term SIDS (sudden infant death syndrome) refers to the seemingly unexplained and sudden death during sleep of an infant under one year old after a thorough and careful investigation [1]. SIDS still stands as the leading cause of death in the first year of life in Western countries [2]. In 1995, the syndrome was documented at 5.6 cases per 10,000 births [3], varying between 2 and 5 per 10,000 live births in most countries [4]. A noteworthy observation is that fifty percent of deaths occur between 7.6 and 17.6 weeks from birth [2,3].
The cause of sudden death in some infants has long been theorized as multifactorial, involving interactions among various factors. These factors lead to death if expressed or experienced in combination with one or more others [5]. The primary exogenous risk factors include the prone sleeping position [6,7,8], exposure to cigarette smoke [9,10], bed-sharing [11], sleeping on soft surfaces [12], and covering the baby’s head under the sheets [13]. Among the various protective factors are breastfeeding [14], pacifier use [15], and placing the crib in the same room as the parents [2].
Possible endogenous causes may encompass systemic anomalies, neurological or autonomic dysfunctions [16], prematurity [3], and pregnancy-related factors [17]. In numerous SIDS cases, immune system activation [7] and down-regulation of the anti-inflammatory pathway have been noted, rendering the infant more susceptible to infections [18]. Metabolic disorders appear to account for 2% of SIDS cases [7,19].
Neonatal arrhythmias are categorized as either benign or non-benign. Non-benign arrhythmias include supraventricular tachycardia (SVT), ventricular tachycardia (VT), abnormalities in atrioventricular (AV) conduction, and genetic arrhythmias such as congenital long QT syndrome (LQTS) [20].
Specific genes have been linked to the development of arrhythmias that may be responsible for sudden infant death syndrome (SIDS) [21]. Notably, cardiac channelopathies are of significant concern [8]. Approximately 10% of SIDS cases have been attributed to cardiac channelopathies [8]. The scrutiny of all genes associated with SIDS underscores the importance of routine post-mortem genetic screening in suspected cases of SIDS, mainly focusing on genes related to cardiac arrhythmias [22]. This screening facilitates the identification of the most likely cause of death and identifies family members at risk [23].
A molecular association between long QT syndrome and SIDS has been documented [24]. According to the 2015 ESC guidelines [25], long QT syndrome is diagnosed when QTc ≥ 480 msec in repeated 12-lead ECGs or the Risk score for LQTS is > 3. Schwartz et al. found that infants who succumbed to SIDS exhibited a prolonged corrected QT interval (QTc) in comparison to survivors (435±45 vs. 400±20 msec, P<0.01) and infants who died from causes other than SIDS (393±24 msec, P<0.05) [26]. In 9.5% of SIDS cases, functionally significant genetic variants in Long QT Syndrome (LQTS) genes were identified [27]. LQTS is analyzed in the presence of a confirmed pathogenic mutation, irrespective of the duration of QTc [28]. Specifically, mutations in 14 genes encoding channels responsible for long QT syndrome have been recognized [29]. Roughly 75% of clinically defined LQTS cases result from mutations in three genes: KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) [30]. However, there is currently no comprehensive genetic test to identify infants at risk of SIDS, and the field is still grappling with methodological challenges [31].
Infants with inherited arrhythmias (ventricular tachycardia or fibrillation, long QT syndrome, or high-grade atrioventricular block) may face an elevated risk of congenital heart defects (CHD) [32]. CHD stands out as the most diagnosed genetic disorder in newborns. The prevalence of CHD at birth exhibits temporal and global variations. In Europe, the reported prevalence of CHD was 8.2 cases per 1,000 live births [95% CI: 8.1 to 8.3] [33]. The incidence of CHD has shown stability over the past three decades, indicating limited advancements in CHD prevention strategies [34].
Arrhythmogenic cardiomyopathy (ACM) represents a distinct hereditary form of cardiomyopathy characterized by prominent ventricular arrhythmias and often associated with sudden cardiac arrest. Conventional estimates of ACM typically suggest that between 1 in 2,000 and 1 in 5,000 people are affected [35].
To mitigate the incidence of SIDS, screening for prolonged QTc or Long QT Syndrome (LQTS) must possess sufficient sensitivity and be administered to all newborns [36]. Furthermore, the use of neonatal electrocardiogram (ECG) could aid in early identification of congenital heart defects (CHD), particularly those that may be undisclosed at birth [37,38].
Aims of the study
This study aims to evaluate whether an electrocardiogram (ECG) conducted in the neonatal period can significantly contribute to the early identification of children at risk of cardiovascular mortality and morbidity, encompassing arrhythmias, Long QT Syndrome (LQTS), and congenital heart defects (CHD) that may be undisclosed at birth.
2. Materials and Methods
Patients’ enrolment
This retrospective study was carried out at the Sleep-Related Respiratory Disorders clinic for ALTE/BRUE and SIDS at the University of Insubria and the Pediatric Cardiology Department of the Filippo Del Ponte Hospital in Varese, Italy.
All healthy infants underwent an ECG screening test primarily to investigate Long QT Syndrome (LQTS). Infants with prolonged QT intervals underwent follow-up and analysis for genetic mutations associated with Long QT Syndrome. The study aimed to identify other congenital heart defects (CHD) that may have gone unrecognized at birth.
The study included infants between 20 and 40 days who were born clinically healthy and had undergone initial routine ECG screening. Infants with prenatal diagnoses or signs/symptoms of CHD identified immediately after birth, as well as those who underwent ECG or echocardiogram for other clinical or historical indications, were excluded from the study.
Electrocardiographic screening
Analyzing paper medical records allowed us to identify infants with a prolongation of the QT interval observed in the first and/or subsequent ECG tracings taken at one- or two-week intervals. Twelve-lead ECGs were recorded on millimeter paper at 25 mm/sec speed using a Philips PageWriter TC50 electrocardiograph. Pediatric cardiologists manually analyzed the ECG tracings, following the European Society Guidelines for interpreting neonatal ECG [39]. QTc was considered prolonged when the QT interval, calculated with the Bazett formula, was ≥ 435 msec, rounding it up [40].
Children with prolonged QTc (≥ 435 msec) confirmed in multiple consecutive tracings (between three and four ECGs) were then subjected to a more in-depth screening.
Laboratory and instrumental tests
In some children, the following investigations were conducted to rule out the secondary causes of QT interval modification: blood tests, Holter ECG, and 12-lead ECG on family members (mainly the mother and/or father). Additionally, selected children underwent an echocardiogram to identify congenital heart defects not previously recognized. Specifically, infants with persistent prolongation of the QTc interval were evaluated for the possible onset of symptoms related to Long QT Syndrome, the results of the Holter ECG, and a therapeutic approach.
Genetic Testing
Genetic testing was performed using next-generation sequencing (NGS) to identify any of the pathogenic mutations in Long QT Syndrome (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, and KCNJ2) associated with SIDS and the patient’s family members. [22].
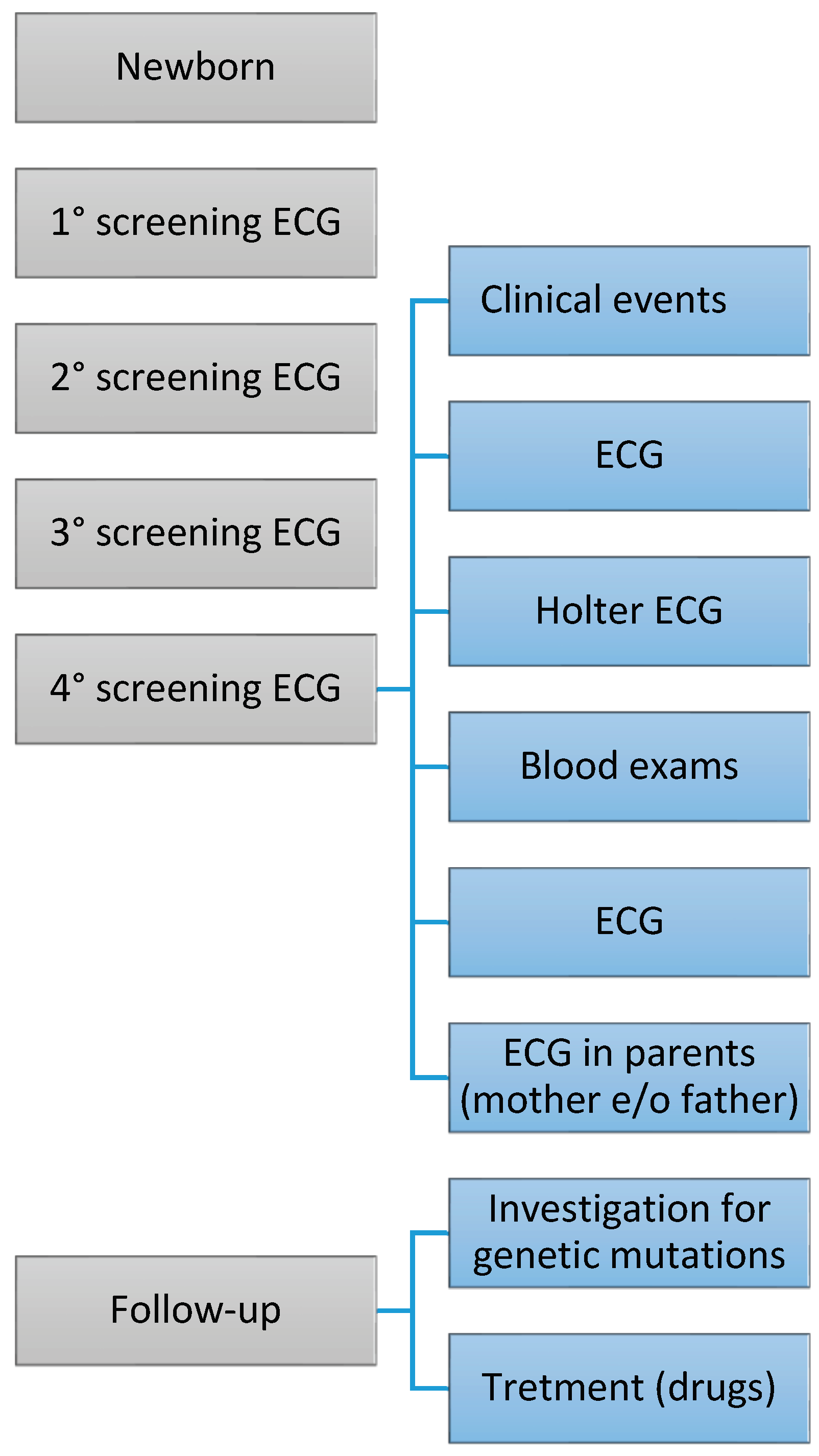
Figure 1.
The figure comprises a series of phases that unfold over time. The frequency of examinations may vary based on the severity of the congenital heart defect (CHD). The treatment for CHD can differ depending on the type of cardiac condition.
Figure 1.
The figure comprises a series of phases that unfold over time. The frequency of examinations may vary based on the severity of the congenital heart defect (CHD). The treatment for CHD can differ depending on the type of cardiac condition.
Statistical analysis
The data were recorded in a Microsoft® Excel® database for Windows 11 and statistically analyzed using SPSS version 22.0 for Windows (SPSS Inc., Chicago, IL). Categorical variables are presented as each category’s number (n) and percentage (%) of observations.
Continuous variables are reported with mean, standard deviation (SD), minimum, and maximum values. Group characteristics are expressed as mean, SD, standard error (SE), 95% confidence interval (95% C.I.), minimum value (Min), and maximum value (Max) for the variables of interest.
The Mann-Whitney U test was conducted to identify statistically significant differences between study groups.
3. Results
This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
Electrocardiographic screening
The study involved 42,200 infants (82.4% of those born from 2002 to 2017).
Table 1A presents the percentage of children with QTc values ≥ a predefined cut-off (msc) according to four different follow-up ECGs (ECG 1.0, ECG 2.0, ECG 3.0, and ECG 4.0), along with the percentage of males in each category. The baseline enrolees were 2,245 children (39.9% males).

Table 1.
A. Table 1 displays the percentage of children (percentage of males). Initially, 2,245 children were assessed (39.9% males). In subsequent control ECGs (from the 1st ECG to the 4th ECG), they exhibited prolonged QTc. Among these, 164 children were recruited (37.8% males) and underwent further diagnostic investigation. Data related to the ECGs, from the 1st repeat ECG to the 4th repeat ECG, are presented.
Table 1.
A. Table 1 displays the percentage of children (percentage of males). Initially, 2,245 children were assessed (39.9% males). In subsequent control ECGs (from the 1st ECG to the 4th ECG), they exhibited prolonged QTc. Among these, 164 children were recruited (37.8% males) and underwent further diagnostic investigation. Data related to the ECGs, from the 1st repeat ECG to the 4th repeat ECG, are presented.
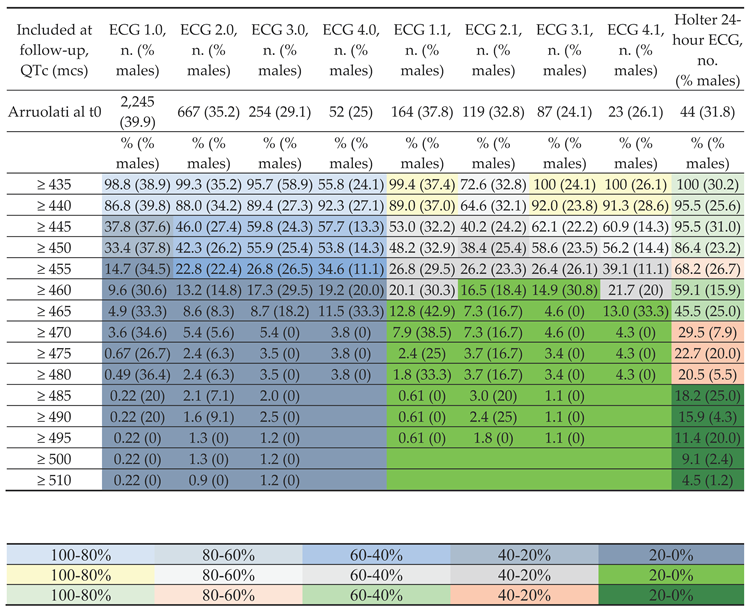 |
Legend: ECG, electrocardiogram; S.D., standard deviation.
For instance, at the ECG 1.0 follow-up, 37.8% (37.6%) of the subjects had a QTc ≥ 445 mcs, while at the ECG 4.0, this percentage had increased to 57.7% (13.3% males). Furthermore, Table 1A displays the same results regarding QTc values at the follow-up of children who were subsequently enrolled for advanced monitoring. These selective ECGs are denoted as ECG 1.1, ECG 2.1, ECG 3.1, and ECG 4.1. Specifically, the number of subjects enrolled at baseline was 164 (males 37.8%). At ECG 1.1, 53.0% (32.2% males) of the subjects had a QTc ≥ 445, while at the fourth follow-up ECG 4.1, this percentage had increased to 60.9% (14.3% males).
Finally, Table 1A presents the QTc values from the Holter ECG. The number of subjects enrolled at baseline was 44 (31.8% males). Notably, the percentage of subjects with QTc ≥ 445 on the Holter ECG was 95.5% (31% males), while for QTc ≥ 455 on the Holter ECG, it was 68.2% (26.7% males).
Table 1.
B. Table 1B displays milliseconds’ mean QTc values ± S.D. (min-max). Initially, 2,245 children were assessed (39.9% males). In subsequent follow-up ECGs, from ECG 1.0 to ECG 4.0, they exhibited prolonged QTc. Among these, 164 children were selected (37.8% males) and were then subjected to further diagnostic investigation.
Table 1.
B. Table 1B displays milliseconds’ mean QTc values ± S.D. (min-max). Initially, 2,245 children were assessed (39.9% males). In subsequent follow-up ECGs, from ECG 1.0 to ECG 4.0, they exhibited prolonged QTc. Among these, 164 children were selected (37.8% males) and were then subjected to further diagnostic investigation.
| Included at follow-up (QTc, mcs) | ECG 1.0,QTc mean ± S.D. (min-max) mcs | ECG 2.0,QTc mean ± S.D. (min-max) mcs | ECG 3.0,QTc mean ± S.D. (min-max) mcs | ECG 4.0,QTc mean ± S.D. (min-max) mcs | ECG 1.1,QTc mean ± S.D. (min-max) mcs | ECG 2.1,QTc mean ± S.D. (min-max) mcs | ECG 3.1,QTc mean ± S.D. (min-max) mcs | ECG 4.1,QTc mean ± S.D. (min-max) mcs | Holter 24-hour ECG, mean QTc ± S.D. (min-max) mcs |
| Arruolati al t0 | 445.9 ± 9.4 (392-513) | 448.1 ± 12.1 (427-513) | 450.2 ± 12.7 (428-520) | 450.5 ± 11.2 (435-482) | 449.6 ± 11.9(433-513) | 451.7 ± 14.8(435-513) | 450.4 ± 12.1(435-520) | 451.3 ± 11.6(435-482) | 465.7 ± 19.2(430-511) |
| ≥ 435 | 446.1 ± 9.2 | 448.3 ± 12.1 | 450.3 ± 12.6 | 449.9 ± 10.9 | 449.7 ± 11.9 | 451.7 ± 14.8 | 450.4 ± 12.1 | 451.3 ± 11.6 | 466.5 ± 18.6 |
| ≥ 440 | 447.5 ± 9.0 | 449.8 ± 11.9 | 452.0 ± 12.2 | 451.8 ± 10.6 | 451.3 ± 11.5 | 453.6 ± 14.6 | 451.7 ± 11.7 | 452.9 ± 11.0 | 467.1 ± 18.3 |
| ≥ 445 | 455.2 ± 8.7 | 457.5 ± 12.1 | 457.0 ± 12.0 | 458.0 ± 8.7 | 457.9 ± 10.7 | 460.9 ± 14.1 | 456.3 ± 11.7 | 458.5 ± 9.0 | 467.2 ± 18.3 |
| ≥ 450 | 456.3 ± 8.6 | 458.4 ± 12.2) | 457.7 ± 12.2 | 458.8 ± 8.5 | 459.0 ± 10.6 | 461.5 ± 14.1 | 456.9 ± 11.8 | 459.4 ± 8.7 | 469.4 ± 17.8 |
| ≥ 455 | 463.7 ± 8.3 | 465.2 ± 13.3 | 465.6 ± 13.7 | 463.1 ± 7.6 | 465.8 ± 9.8 | 466.7 ± 14.4 | 464.7 ± 14.1 | 463.1 ± 7.8 | 474.1 ± 17.2 |
| ≥ 460 | 467.0 ± 8.6 | 470.8 ± 15.1 | 470 ± 15.4 | 457.2 ± 8.2 | 468.5 ± 10.0 | 472.5 ± 15.7 | 470.4 ± 16.9 | 467.2 ± 8.7 | 476.9 ± 16.7 |
| ≥ 465 | 472.6 ± 8.9 | 479.2 ± 16.2 | 479.3 ± 17.4 | 471.3 ± 8.3 | 472.7 ± 10.3 | 484.7 ± 16.5 | 489.5 ± 20.7 | (466-482) | 481.6 ± 16.4 |
| ≥ 470 | 475.0 ± 9.3 | 483.7 ± 16.4 | 488.5 ± 17.4 | 482 | 476.9 ± 11.3 | 484.7 ± 16.5 | 489.5 ± 20.7 | 482 | 489.9 ± 14.4 |
| ≥ 475 | 488.4 ± 15.7 | 499.1 ± 13.3 | 495.4 ± 16.7 | 482 | 487.3 ± 17.3 | 497.5 ± 14.2 | (482-520) | 482 | 495.1 ± 12.2 |
| ≥ 480 | 492.9 ± 16.2 | 499.1 ± 13.3 | 495.4 ± 16.7 | 482 | 513 | 497.5 ± 14.2 | 520 | 482 | 497.3 ± 10.5 |
| ≥ 485 | 508.4 ± 10.3 | 501 ± 11.8 | 506 ± 15.2 | 513 | 501 ± 12.7 | 520 | 499.5 ± 8.8 | ||
| ≥ 490 | 508.4 ± 10.3 | 506.4 ± 8.6 | 506 ± 15.2 | 513 | 505 ± 10.4 | 520 | 501.6 ± 7.1 | ||
| ≥ 495 | 513 | 510 ± 3.1 | (511-520) | 513 | (506-513) | 520 | 504.6 ± 5.9 | ||
| ≥ 500 | 513 | 510 ± 3.1 | (511-520) | 506.0 ± 5.8 | |||||
| ≥ 510 | 513 | 512.0 ± 1.1 | (511-520) | 511 |
Legend: ECG, electrocardiogram; S.D., standard deviation.
Table 1B presents the mean values ± S.D. of QTc ranging from 435 mcs to 513 mcs (in increments of 5 mcs) during the four follow-up ECGs (ECG 1.0, ECG 2.0, ECG 3.0, and ECG 4.0). For instance, at the ECG 1.0 follow-up, the mean value ± S.D. for children with QTc ≥ 445 mcs was 455.2 ± 8.7 mcs, while at ECG 4.0, it was 458.0 ± 8.7 mcs. Additionally, Table 1B displays results related to QTc values for children enrolled for further diagnostic investigation (ECG 1.1 follow-up, ECG 2.1 follow-up, ECG 3.1 follow-up, and ECG 4.1). The mean value ± S.D. for subjects with QTc ≥ 445 mcs enrolled at baseline (t0) was 457.9 ± 10.7 mcs at ECG 1.1 and 458.5 ± 9.0 mcs at ECG 4.1. Finally, the table shows the mean values ± S.D. of QTc measured with Holter ECG ≥ 445 mcs as 467.2 ± 18.3 mcs
Genetic investigation
Table 2.
The QTc values from follow-up ECGs 1.1 to 4.1 and the QTc obtained from Holter ECG in 27 children (18.5% males) undergoing further diagnostic investigation are as follows.
Table 2.
The QTc values from follow-up ECGs 1.1 to 4.1 and the QTc obtained from Holter ECG in 27 children (18.5% males) undergoing further diagnostic investigation are as follows.
| N | Mean ± SD | Minimum – maximum | |
| ECG 1.1 QTc msec | 27 | 452,5 ±13,1 | 435 – 476 |
| ECG 2.1 QTc msec | 25 | 458,5 ±19,1 | 435 – 511 |
| ECG 3,1 QTc msec | 21 | 462,2 ±20,9 | 441 – 520 |
| ECG 4.1 QTc msec | 12 | 457,0 ±11,2 | 442 – 482 |
| ECG Holter, QTc msec | 18 | 452,9 ±26,2 | 417 – 538 |
Of the 27 children enrolled for further diagnostic investigation, 18 underwent genetic testing. Among the 18 children genetically examined, mutations were identified in 11, while no mutations were identified in the remaining 7 (see Table 3). The other 7 children spontaneously withdrew from the study and were lost to follow-up.
Table 3 presents the genetic mutations detected in 11 children. One girl had an SCN5A mutation (c.647C>T). In three girls, a mutation in the KCNH2 gene (LQT2) was found; one girl had a nucleotide substitution in the KCNH2 gene (c.1196C>T), and another girl had a nucleotide substitution in the KCNH2 gene (c.3367G>C). In one girl, a nucleotide substitution was detected in both KCNH2 (c.2560T>G) and SCN5A (c.5845G>A). Follow-up data were not available for this girl. Patients were assessed in follow-up as asymptomatic, except for one with a KCNQ1 mutation and polymorphisms in other genes (SCN5A-H558R, KCNH2-K897K, KCNE1-S38G). This patient had a syncopal episode. Five children (three males and two siblings) had heterozygous mutations in KCNQ1 (LQT1). In the other male child, the LQT1 mutation was known in the family history (mother and sister of the child). Six children (3 females and 3 males, including two siblings) reported positive family history. Eight out of 11 children had a therapeutic protocol. Data for a female carrier of KCNH2 (c.2560T>G), SCN5A (c.5845G>A), and a female carrier of KCNQ1 (LQT1) were incomplete. A female carrier of KCNH2 (c.1196C>T) was not undergoing any therapy. In summary, the KCNQ1 (LQT1) mutation was present in 54.5% of patients, the KCNH2 (LQT2) mutation in 36.4% of patients, and the SCN5A (LQT3) mutation in 1 patient.
Table 3 also provides information on the results of neonatal ECG screening for Long QT in 7 children where specific mutations were not identified (6 females and 1 male). QTc values were measured both through ECG and Holter ECG. QTc values varied among children (QTc: minimum 402 mcs - maximum 494 mcs; QTc Holter: minimum 440 mcs – maximum 537 mcs), indicating heterogeneity in QTc length. The prescribed therapy varies among patients and mainly uses beta-blockers such as propranolol, nadolol, and metoprolol. Propranolol is administered at 2 mg/kg dosages, with frequencies of 1 to 3 times daily, depending on the specific case. Nadolol is administered thrice daily at a dosage of ¾ tablet (40 mg). Metoprolol is administered at a dosage of 8 mg twice a day. In summary, the drugs used for the treatment of long QTc were Propranolol 39.8 % of children, Nadolol 22.2 % of children, Inderal 11.1 % of children, Metoprolol 11.1 % of children, and none 16.7 % of children.
Table 4 provides information on single ECG abnormalities found in children identified through neonatal screening for Long QT (total 4.3% of children). The most frequent were: patients with focal right bundle branch block were 54.5% of the total children with a single anomaly, patients with left axis deviation were 9.2% of the total single anomalies, patients with nonspecific ventricular repolarization abnormalities were 7.1% of the total single anomalies, patients with ventricular extrasystole were 4.6% of the total subjects with single anomalies, patients with supraventricular extrasystole were 4.5% of the total single anomalies, and patients with complete right bundle branch block were 3.7% of the total single anomalies. These results suggest a variety of electrical heart anomalies in children at risk of Long QT, with a significant prevalence of focal right bundle branch block.
Table 4 also provides information on multiple ECG abnormalities found in children with focal right bundle branch block identified through neonatal ECG screening for Long QT (0.47% of children). The most frequent were focal right bundle branch block associated with right ventricular prevalence was 61% of the total children with multiple anomalies, focal right bundle branch block plus left axis deviation was 26% of the total children with various anomalies, patients with focal right bundle branch block associated with other electrical abnormalities were 66.7% of the total children with multiple ECG anomalies.
Table 5 provides information on the frequency of structural heart abnormalities associated with specific ECG characteristics in 267 patients (11.9% of the total patients with structural heart abnormalities). The most frequent were: patients with isolated PFO were 164 (61.4%), patients with PFO associated with ASD OS (Atrial septal defect ostium secundum) were 25 (9.4%), patients with PFO plus Mitral insufficiency were 21 (7.9%), and patients with PFO plus PDA were 12 (4.5%). These results indicate that PFO is the main structural heart abnormality observed in patients undergoing ECG screening and is often associated with other anatomical conditions such as atrial septal defect, mitral insufficiency, and patent ductus arteriosus.
Table 5 also presents associations between positive screening for prolonged QTc interval, included in the follow-up, and structural heart abnormalities in 116 patients (5.17 %). In particular, the most frequent were: 62.1% had PFO associated with other structural abnormalities, 5.2% had PFO plus VSD, and 3.4% had PFO plus PDA. Finally, 10.3% of patients had ASD OS.
4. Discussion
This study analyzed the utility of neonatal ECG screening for diagnosing long QT syndrome, heart rhythm abnormalities, and structural heart conditions that could be responsible for neonatal and infant morbidity and mortality [41,42]. In particular, a retrospective analysis was conducted over 16 years of ECG screening performed on 42,200 newborns, highlighting changes over time, gender differences, and the importance of specific diagnostic investigations in certain subgroups of children undergoing neonatal ECG screening for long QTc syndrome. Gender differences emerged significantly, especially in the male subgroup. The results of the Holter ECG indicate high sensitivity in detecting anomalies. The study also highlights average variations in the QTc interval over time, while average values during follow-up can be useful in assessing cardiovascular risk in children with prolonged QTc beyond a specific predefined value. Delayed diagnosis of critical congenital heart disease poses a severe risk of mortality, morbidity, and disability. The utility of ECG screening as a universal method to identify affected newborns was emphasized by Schwartz et al. in 2002 [39].
The physiopathological mechanisms leading to sudden infant death syndrome (SIDS) in infants are not fully understood. It has been suggested that arrhythmias and cardiovascular changes may be responsible for death in infants affected by SIDS [43], including prolongation of the QTc interval in the early years of life [26]. However, there are significant challenges in establishing a cardiac cause for SIDS [44]. Some hypotheses overlap; for example, the conceptual failure in respiration could include apnea, prone sleep position, and asphyxia, which might be considered connected to co-sleeping [45]. However, a crucial step in understanding the complex pathophysiology of SIDS was the establishment of the Triple-Risk Model, which successfully conceptualized epidemiological, physiological, and neuropathological data associated with SIDS [46].
Prolonged QT syndrome can cause abnormal heartbeats and an increased risk of sudden cardiac arrest [36]. Its estimated prevalence is around 1 case per 2,000-2,500 individuals [47]. Although long QT syndrome is rare, it can be hazardous in infants and children as their cardiovascular system is still in the developmental stage. Moreover, an abnormal heart rhythm, such as ventricular tachycardia or ventricular arrhythmia, could lead to sudden cardiac arrest [48]. Finally, structural heart abnormalities are congenital conditions that can contribute to an increased risk of sudden cardiac arrest [42].
The QTc undergoes a physiological prolongation from the second month of life, returning to normal values at six months. Some deaths labelled as SIDS may be due to a mechanism similar or identical to that responsible for long QT syndrome [27]. The prevalence of long QT syndrome varies from 1 in 20,000 to 1 in 5,000 [37]. A prospective study demonstrated that in 50% of SIDS victims, the QTc was > 435 msec [40]. The QT interval was increased in SIDS victims (QTc ≥ 440 ms, exceeding the 97.5th percentile). The risk of SIDS in infants with QTc > 435 ms was calculated to be 41 times higher than that of children with a normal QTc [26]. Long QT syndrome and SIDS share similar phenotypes, such as prolonged QTc interval, reduced parasympathetic tone, and/or autonomic nervous system imbalance [49].
The prevalence of long QT syndrome (LQTS) identified in children undergoing ECG screening for long QT is 5.32%. The prevalence of long QTc is 1 in 2381 (0.42 ‰), similar to that found in the study by Schwartz et al. (1 in 2,534) [37]. However, our data might be underestimated because follow-up data for all children are unavailable.
Long QT syndrome (LQTS) refers to a group of "channelopathies," disorders affecting cardiac ion channels, contributing to approximately 12% of SIDS cases [36]. Screening in the neonatal period allows the identification of "at-risk" infants before the peak incidence of SIDS (2-6 months).
We identified 11 children carrying pathogenic mutations of LQTS: 6 LQT1 (KCNQ1; 54.5%), 3 LQT2 (KCNH2; 27.3%), 1 LQT3 (SCN5A; 9.1%); 1 case (9.1%) presented a combined mutation of LQT1 (KCNH2) and LQT3 (SCN5A). About 10% of sudden infant deaths classified as SIDS are likely caused by LQTS [27]. Follow-up evaluation predominantly shows asymptomaticity, except for one case with KCNQ1 mutation that manifested a syncopal episode, emphasizing the importance of genotype-phenotype correlation. In the other children in our case series, the diagnosis of long QT syndrome was confirmed, but the genetic mutation was not identified.
Mutations in the LQT1 gene (KCNQ1) cause defects in potassium channels with an LQT1 phenotype, the most common form of Long QT Syndrome (LQTS). LQT1 carriers have a higher risk of ventricular arrhythmias and fatal events [50]. The distribution within the LQTS subgroups in our study confirms the prevalence of LQT1 compared to other variants, in line with literature data [37,51]. Genetic screening has allowed the identification and genetic characterization of affected family members with the syndrome. The nucleotide substitution in the LQT3 gene (SCN5A; c.647C>T) has been described in patients with LQTS, Brugada syndrome, atrial fibrillation, and SIDS [52,53]. Patients with LQT3 showed a rapid trajectory towards cardiac events, with 20% being fatal. In contrast, with one exception, both LQT1 and LQT2 patients had relatively benign clinical course and outcomes, with no deaths and only one heart transplant [54].
After the discovery of the first three Long QT Syndrome (LQTS) susceptibility genes (KCNQ1, KCNH2, and SCN5A), a total of 17 genes have been identified, with seven genes discovered by Ackerman et al. [30]. However, more than half of the genes reported as responsible for LQTS have limited or contested evidence supporting their causality in the disease [55].
Long QT Syndrome (LQTS) is typically inherited in an autosomal dominant manner. Most individuals diagnosed with LQTS have an affected parent, and the proportion of LQTS caused by a de novo pathogenic variant is small. The penetrance of the disorder can vary [56]. The study has demonstrated the potential of neonatal screening to identify families at risk of Long QT Syndrome. Therefore, the benefit of neonatal screening has been extended to the relatives of children with Long QT Syndrome. Neonatal ECG screening reveals variations in the length of the QTc interval, suggesting the need for comprehensive assessments even in the absence of specific genetic mutations.
The therapeutic management outlined in the children’s protocol emphasizes the importance of early identification and personalized treatment. The prescribed therapy, based on beta-blockers such as propranolol, nadolol, and metoprolol, reflects a joint approach with dosages tailored to individual needs, underscoring the significance of personalized treatment in Long QT management.
Neonatal ECG screening facilitated the identification of cardiac rhythm anomalies, such as right bundle branch block (2.33%), left axis deviation (0.39%), and nonspecific ventricular repolarization anomalies (0.30%) of the total screened neonates. The most common combined ECG anomalies were right bundle branch block associated with right ventricular prevalence (0.14%) and right bundle branch block associated with left axis deviation (0.06%). Therefore, echocardiographic assessment in our patients with prolonged QTc and/or other ECG abnormalities aided in identifying congenital heart defects (CHD), as reported in the literature [37].
Routine ECG screening is a valuable tool for identifying unrecognized congenital heart defects (CHD) not detected by prenatal ultrasound and neonatal examinations in asymptomatic patients [42]. A study reported that postnatal echocardiographic screening does not contribute to improving the detection rate of critical or severe heart defects in newborns without prenatal diagnosis or clinical signs. The study authors suggest conducting the examination a few months after birth, but it has a high rate of false positives [57].
In total, the structural heart abnormalities identified through neonatal ECG screening were 6.3‰, with Patent Foramen Ovale (PFO) alone (3.9‰) or associated with other cardiac anomalies (6.3‰) being the most frequent. Following this, mitral valve insufficiency was less frequently identified (0.28‰). Overall, the frequency of cardiac anomalies identified in children enrolled for serial neonatal ECG screening for suspected prolonged QTc was 5.57%, with PFO, either single (3.21%) or associated with other structural heart anomalies, being the most common (4.01%). Subsequently, isolated ostium secundum atrial septal defect (ASD) (5.3‰) or associated with other cardiac anomalies was less commonly observed (6.7‰). Echocardiographic evaluation in these patients with prolonged QTc and/or other ECG abnormalities facilitated the identification of congenital heart defects, as reported in the literature [37].
It has been reported that the overall incidence of Congenital Heart Defects (CHD) is approximately 6-9‰ [58]. However, neonatal clinical examination generally cannot detect all forms of CHD [59]. The incidence of congenital heart diseases in asymptomatic neonates born at high altitudes was 27.8%, with Ostium Secundum Atrial Septal Defect (ASD OS) (62.7%), Patent Ductus Arteriosus (PDA) (21.9%), Ventricular Septal Defect (VSD) (2.9%), and multiple defects (12.6%) being observed [60]. Almost all CHD cases were simple forms with left-to-right shunting, including ASD OS, PDA, and VSD [60].
Neonatal ECG screening aims to recognise cardiovascular morbidity and mortality risk-related cardiac anomalies. During the study, 5.3% of newborns undergoing neonatal ECG screening underwent cardiac follow-up with serial ECGs for prolonged QTc. At the end of the follow-up, 0.12% of the children had completed the follow-up. Of the enrolled children, 0.10% had undergone ECG Holter monitoring. Finally, 18 children had been diagnosed with prolonged QTc, of whom 11 had a identified genetic mutation responsible for long QT. The primary drug used to treat prolonged QT was propranolol.
Additionally, ECG screening identified cardiac rhythm abnormalities in 4.7% of the children. The most frequent isolated cardiac rhythm abnormality was right bundle branch block (54.5% of total isolated rhythm abnormalities), and right bundle branch block was associated with right ventricular prevalence (30.8% of total multiple cardiac rhythm abnormalities). Overall, right bundle branch block, associated with other cardiac rhythm anomalies, was found in 66.7% of total multiple cardiac rhythm abnormalities.
The echocardiographic examination identified structural heart anomalies in many children with ECG abnormalities. Specifically, the patent foramen ovale (PFO) was found in 61.4% of total cardiac anomalies. Furthermore, overall results from this study suggest that neonatal ECG screening is valuable in recognizing cardiac abnormalities associated with the risk of cardiovascular morbidity and mortality, and echocardiographic examination is an essential additional resource to confirm the presence of structural heart anomalies.
Expanded genetic investigations allowed the identification of mutations in family members, enabling them to access genetic counselling. Additionally, routine neonatal ECG screening facilitated the diagnosis of other arrhythmogenic or structural cardiovascular anomalies not identified in previous prenatal and neonatal examinations. Early diagnosis of these conditions in asymptomatic patients is encouraged to provide these infants with immediate access to medical or surgical therapy, thereby reducing mortality and morbidity.
Considering the lack of some follow-up data for children with prolonged QT, there appears to be a need to establish a data collection network to monitor children’s evolution more thoroughly with prolonged QTc.
5. Conclusions
The study demonstrates straightforward clinical utility, emphasizing the importance of neonatal ECG screening between 20 and 40 days of life. This screening has proven effective in diagnosing long QT syndrome and other cardiac rhythm anomalies early, enabling suspicions about structural heart pathologies. Identifying genetic mutations, particularly channelopathies, in over half of the children with prolonged QT, highlights the possibility of early interventions such as treatment with beta-blockers. Cost-benefit assessment indicates that screening saves newborns with long QT syndrome and provides additional benefits by identifying mutations and/or QTc prolongations in family members. Moreover, detecting other ECG abnormalities and congenital heart diseases further expands the number of children benefiting from screening.
Author Contributions
Conceptualization, L.N. and M.Ag.; methodology, A.G. and M.Ab.; software, M.Z.; validation, G.P., M.Ag., and C.L.; formal analysis, M.Z. and A.G.; investigation, A.G. and M.Ab.; data curation, L.N., M.Z, and C.L.; writing—original draft preparation, M.Z. and L.N.; writing—review and editing, L.N., C.L. and G.P.; supervision, G.P., and M.Ag.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. Permission from the Ethics Committee was not required for this study as our Research Center is the national reference center for Sudden Unexplained Infant Death, in accordance with the Italian Law n. 31/2006.
Informed Consent Statement
This retrospective study utilized a dataset extracted from a database, which was anonymized and stripped of personally identifiable information. The study is grounded in pre-existing data, encompassing paper medical records securely stored in hospital cabinets with keys and electronic data recorded in password-protected computers accessible exclusively by authorized healthcare personnel. Attempting to contact and secure individual consent from each parent in a study of this magnitude proved impractical. In conclusion, the study delivers significant scientific benefits, with minimal risk to privacy.
Data Availability Statement
None.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Krous, H.F.; Byard, R.W.; Rognum, T.O. Pathology research into sudden infant death syndrome: where do we go from here? Pediatrics 2004, 114, 492–494. [Google Scholar] [CrossRef]
- Moon, R.Y. SIDS and Other Sleep-Related Infant Deaths: Evidence Base for 2016 Updated Recommendations for a Safe Infant Sleeping Environment. Pediatrics 2016, 138. [Google Scholar] [CrossRef] [PubMed]
- Hakeem, G.F.; Oddy, L.; Holcroft, C.A.; Abenhaim, H.A. Incidence and determinants of sudden infant death syndrome: a population-based study on 37 million births. World J Pediatr 2015, 11, 41–47. [Google Scholar] [CrossRef]
- Haas, E.A. Sudden Unexplained Death in Childhood: An Overview. In SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future, Duncan, J.R., Byard, R.W., Eds.; University of Adelaide Press © 2018 The Contributors, with the exception of which is by Federal United States employees and is therefore in the public domain.: Adelaide (AU), 2018.
- Hauck, F.R.; Hunt, C.E. Sudden infant death syndrome in 2000. Current Problems in Pediatrics 2000, 30, 237–261. [Google Scholar] [CrossRef]
- Garcia, A.J., 3rd; Koschnitzky, J.E.; Ramirez, J.M. The physiological determinants of sudden infant death syndrome. Respir Physiol Neurobiol 2013, 189, 288–300. [Google Scholar] [CrossRef]
- Opdal, S.H.; Rognum, T.O. Gene variants predisposing to SIDS: current knowledge. Forensic Sci Med Pathol 2011, 7, 26–36. [Google Scholar] [CrossRef]
- Tfelt-Hansen, J.; Winkel, B.G.; Grunnet, M.; Jespersen, T. Cardiac channelopathies and sudden infant death syndrome. Cardiology 2011, 119, 21–33. [Google Scholar] [CrossRef]
- Fifer, W.P.; Fingers, S.T.; Youngman, M.; Gomez-Gribben, E.; Myers, M.M. Effects of alcohol and smoking during pregnancy on infant autonomic control. Dev Psychobiol 2009, 51, 234–242. [Google Scholar] [CrossRef]
- Richardson, H.L.; Walker, A.M.; Horne, R.S. Maternal smoking impairs arousal patterns in sleeping infants. Sleep 2009, 32, 515–521. [Google Scholar] [CrossRef]
- American Academy of Pediatrics Task Force on Sudden Infant Death, S. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics 2005, 116, 1245-1255. [CrossRef]
- Carlin, R.F.; Moon, R.Y. Risk Factors, Protective Factors, and Current Recommendations to Reduce Sudden Infant Death Syndrome: A Review. JAMA Pediatr 2017, 171, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.S.; Mitchell, E.A.; Heckstall-Smith, E.M.A.; Fleming, P.J. Head covering - a major modifiable risk factor for sudden infant death syndrome: a systematic review. Archives of Disease in Childhood 2008, 93, 778–783. [Google Scholar] [CrossRef]
- Hauck, F.R.; Thompson, J.M.; Tanabe, K.O.; Moon, R.Y.; Vennemann, M.M. Breastfeeding and reduced risk of sudden infant death syndrome: a meta-analysis. Pediatrics 2011, 128, 103–110. [Google Scholar] [CrossRef]
- Yiallourou, S.R.; Poole, H.; Prathivadi, P.; Odoi, A.; Wong, F.Y.; Horne, R.S. The effects of dummy/pacifier use on infant blood pressure and autonomic activity during sleep. Sleep Med 2014, 15, 1508–1516. [Google Scholar] [CrossRef]
- Weese-Mayer, D.E.; Berry-Kravis, E.M. Genetics of congenital central hypoventilation syndrome: lessons from a seemingly orphan disease. Am J Respir Crit Care Med 2004, 170, 16–21. [Google Scholar] [CrossRef]
- Getahun, D.; Amre, D.; Rhoads, G.G.; Demissie, K. Maternal and obstetric risk factors for sudden infant death syndrome in the United States. Obstet Gynecol 2004, 103, 646–652. [Google Scholar] [CrossRef]
- Highet, A.R. An infectious aetiology of sudden infant death syndrome. Journal of Applied Microbiology 2008, 105, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Pryce, J.W.; Weber, M.A.; Heales, S.; Malone, M.; Sebire, N.J. Tandem mass spectrometry findings at autopsy for detection of metabolic disease in infant deaths: postmortem changes and confounding factors. Journal of Clinical Pathology 2011, 64, 1005–1009. [Google Scholar] [CrossRef]
- Jaeggi, E.; Öhman, A. Fetal and Neonatal Arrhythmias. Clin Perinatol 2016, 43, 99–112. [Google Scholar] [CrossRef]
- Mazzanti, A.; Kanthan, A.; Monteforte, N.; Memmi, M.; Bloise, R.; Novelli, V.; Miceli, C.; O’Rourke, S.; Borio, G.; Zienciuk-Krajka, A.; et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol 2014, 63, 1300–1308. [Google Scholar] [CrossRef]
- Liebrechts-Akkerman, G.; Liu, F.; van Marion, R.; Dinjens, W.N.M.; Kayser, M. Explaining sudden infant death with cardiac arrhythmias: Complete exon sequencing of nine cardiac arrhythmia genes in Dutch SIDS cases highlights new and known DNA variants. Forensic Sci Int Genet 2020, 46, 102266. [Google Scholar] [CrossRef]
- Sarquella-Brugada, G.; Campuzano, O.; Cesar, S.; Iglesias, A.; Fernandez, A.; Brugada, J.; Brugada, R. Sudden infant death syndrome caused by cardiac arrhythmias: only a matter of genes encoding ion channels? Int J Legal Med 2016, 130, 415–420. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Dumaine, R.; Napolitano, C.; Antzelevitch, C.; Stramba-Badiale, M.; Richard, T.A.; Berti, M.R.; Bloise, R. A molecular link between the sudden infant death syndrome and the long-QT syndrome. The New England Journal of Medicine 2000, 343, 262–267. [Google Scholar] [CrossRef]
- Aktaa, S.; Tzeis, S.; Gale, C.P.; Ackerman, M.J.; Arbelo, E.; Behr, E.R.; Crotti, L.; d’Avila, A.; de Chillou, C.; Deneke, T.; et al. European Society of Cardiology quality indicators for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace 2023, 25, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Segantini, A.; Austoni, P.; Bosi, G.; Giorgetti, R.; Grancini, F.; Marni, E.D.; Perticone, F.; Rosti, D.; et al. Prolongation of the QT interval and the sudden infant death syndrome. The New England Journal of Medicine 1998, 338, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Arnestad, M.; Crotti, L.; Rognum, T.O.; Insolia, R.; Pedrazzini, M.; Ferrandi, C.; Vege, A.; Wang, D.W.; Rhodes, T.E.; George, A.L., Jr.; et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation 2007, 115, 361–367. [Google Scholar] [CrossRef]
- Zareba, W. Genotype-specific ECG patterns in long QT syndrome. J Electrocardiol 2006, 39, S101–106. [Google Scholar] [CrossRef]
- Tester, D.J.; Benton, A.J.; Train, L.; Deal, B.; Baudhuin, L.M.; Ackerman, M.J. Prevalence and spectrum of large deletions or duplications in the major long QT syndrome-susceptibility genes and implications for long QT syndrome genetic testing. Am J Cardiol 2010, 106, 1124–1128. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef]
- Johannsen, E.B.; Baughn, L.B.; Sharma, N.; Zjacic, N.; Pirooznia, M.; Elhaik, E. The Genetics of Sudden Infant Death Syndrome-Towards a Gene Reference Resource. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Dai, Y.; Yin, R.; Yang, L.; Li, Z.H. Clinical and genetic spectrum of neonatal arrhythmia in a NICU. Transl Pediatr 2021, 10, 2432–2438. [Google Scholar] [CrossRef]
- van der Linde, D.; Konings, E.E.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 2011, 58, 2241–2247. [Google Scholar] [CrossRef]
- Wu, W.; He, J.; Shao, X. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990-2017. Medicine (Baltimore) 2020, 99, e20593. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Judge, D.P. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol 2021, 18, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Ioakeimidis, N.S.; Papamitsou, T.; Meditskou, S.; Iakovidou-Kritsi, Z. Sudden infant death syndrome due to long QT syndrome: a brief review of the genetic substrate and prevalence. J Biol Res (Thessalon) 2017, 24, 6. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Quaglini, S.; Rognoni, C.; Spazzolini, C.; Priori, S.G.; Mannarino, S.; Schwartz, P.J. Cost-effectiveness of neonatal ECG screening for the long QT syndrome. Eur Heart J 2006, 27, 1824–1832. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Garson, A.; Paul, T.; Stramba-Badiale, M.; Vetter, V.L.; Wren, C.; European Society of, C. Guidelines for the interpretation of the neonatal electrocardiogram. A task force of the European Society of Cardiology. European Heart Journal 2002, 23, 1329–1344. [Google Scholar] [CrossRef]
- Schwartz, A.R.; O’Donnell, C.P.; Baron, J.; Schubert, N.; Alam, D.; Samadi, S.D.; Smith, P.L. The hypotonic upper airway in obstructive sleep apnea: role of structures and neuromuscular activity. Am J Respir Crit Care Med 1998, 157, 1051–1057. [Google Scholar] [CrossRef]
- Ran, L.; Li, J.; Bao, L.; Chen, L. Association Between Neonatal Arrhythmia and Mortality and Recurrence: A Retrospective Study. Front Pediatr 2022, 10, 818164. [Google Scholar] [CrossRef]
- Mishra, V.; Zaidi, S.; Axiaq, A.; Harky, A. Sudden cardiac death in children with congenital heart disease: a critical review of the literature. Cardiol Young 2020, 30, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Guilleminault, C.; Ariagno, R.; Coons, S.; Winkle, R.; Korobkin, R.; Baldwin, R.; Souquet, M. Near-miss sudden infant death syndrome in eight infants with sleep apnea-related cardiac arrhythmias. Pediatrics 1985, 76, 236–242. [Google Scholar]
- Southall, D.P.; Richards, J.M.; Stebbens, V.; Wilson, A.J.; Taylor, V.; Alexander, J.R. Cardiorespiratory function in 16 full-term infants with sudden infant death syndrome. Pediatrics 1986, 78, 787–796. [Google Scholar] [CrossRef]
- Goldwater, P.N. A perspective on SIDS pathogenesis. the hypotheses: plausibility and evidence. BMC Med 2011, 9, 64. [Google Scholar] [CrossRef]
- Filiano, J.J.; Kinney, H.C. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biology of the Neonate 1994, 65, 194–197. [Google Scholar] [CrossRef]
- Schwartz, J.R.; Roth, T. Neurophysiology of sleep and wakefulness: basic science and clinical implications. Curr Neuropharmacol 2008, 6, 367–378. [Google Scholar] [CrossRef]
- Kline, J.; Costantini, O. Inherited Cardiac Arrhythmias and Channelopathies. Med Clin North Am 2019, 103, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; Groswasser, J.; Scaillet, S.; Lanquart, J.P.; Benatar, A.; Sastre, J.P.; Chevalier, P.; Kugener, B.; Kahn, A.; Lin, J.S. QT interval prolongation in future SIDS victims: a polysomnographic study. Sleep 2008, 31, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, K.H.; Leren, I.S. Prevalence, Clinical Presentation, and Management of Channelopathies and Cardiomyopathies, Long QT Syndrome, Brugada Syndrome, Arrhythmogenic Cardiomyopathy, and Hypertrophic Cardiomyopathy. Current Cardiovascular Risk Reports 2019, 13, 16 %U. [Google Scholar] [CrossRef]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk stratification in the long-QT syndrome. N Engl J Med 2003, 348, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Gosselin-Badaroudine, P.; Moreau, A.; Chahine, M. Nav 1.5 mutations linked to dilated cardiomyopathy phenotypes: Is the gating pore current the missing link? Channels (Austin) 2014, 8, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zaklyazminskaya, E.; Dzemeshkevich, S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta 2016, 1863, 1799–1805. [Google Scholar] [CrossRef]
- Moore, J.P.; Gallotti, R.G.; Shannon, K.M.; Bos, J.M.; Sadeghi, E.; Strasburger, J.F.; Wakai, R.T.; Horigome, H.; Clur, S.A.; Hill, A.C.; et al. Genotype Predicts Outcomes in Fetuses and Neonates With Severe Congenital Long QT Syndrome. JACC Clin Electrophysiol 2020, 6, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Alders, M.; Bikker, H.; Christiaans, I. Long QT Syndrome. In GeneReviews(®), Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle. Copyright © 1993-2023, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
- Kondo, M.; Ohishi, A.; Baba, T.; Fujita, T.; Iijima, S. Can echocardiographic screening in the early days of life detect critical congenital heart disease among apparently healthy newborns? BMC Pediatr 2018, 18, 359. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J Am Coll Cardiol 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Dawson, A.L.; Cassell, C.H.; Riehle-Colarusso, T.; Grosse, S.D.; Tanner, J.P.; Kirby, R.S.; Watkins, S.M.; Correia, J.A.; Olney, R.S. Factors associated with late detection of critical congenital heart disease in newborns. Pediatrics 2013, 132, e604–611. [Google Scholar] [CrossRef]
- Li, J.J.; Liu, Y.; Xie, S.Y.; Zhao, G.D.; Dai, T.; Chen, H.; Mu, L.F.; Qi, H.Y.; Li, J. Newborn screening for congenital heart disease using echocardiography and follow-up at high altitude in China. Int J Cardiol 2019, 274, 106–112. [Google Scholar] [CrossRef]
Table 3.
Mutations were identified, and therapy was initiated in 11 children, including 2 siblings, affected by Long QTc in the screening ECG and Holter ECG. Additionally, results of QTc in the ECG, Holter ECG, and the therapy undertaken by children with Long QTc in whom the effects of genetic investigations were negative.
Table 3.
Mutations were identified, and therapy was initiated in 11 children, including 2 siblings, affected by Long QTc in the screening ECG and Holter ECG. Additionally, results of QTc in the ECG, Holter ECG, and the therapy undertaken by children with Long QTc in whom the effects of genetic investigations were negative.
| SEX | MUTATION | Familiarity | QTc msc (ECG) | QTc msec (ECG Holter) | Therapy | Dosage | SEX | QTc msc (ECG) | QTc msec (ECG holter) | Therapy | Dosage |
| F | SCN5A (LQT3; c.647C>T) | No | 427 | 438 | Propranolol | 2 mg/kg, 3 vv/die | F | 468 | 460 | Propranolol | 2 mg/kg, 2 vv/die |
| F | KCNH2 (LQT2) | Father | 458 | 461 | Inderal | 20 mg | F | 402 | 441 | Propranolol | 2 mg/kg, 1 v /die |
| F | KCNH2 (LQT2; c.1196C>T) | No | 438 + PR corto | - | - | - | F | 413 | 449 | Propranolol | 2 mg/kg, 3 vv/die |
| F | KCNH2 (LQT2; c.3367G>C) | No | 452 | 462 | Propranolol | 3 mg/kg, 3 vv/die | M | 433 | 440 | Propranolol | 2 mg/kg, 3 vv/die |
| F | KCNH2 (LQT2; c.2560T>G), SCN5A (LQT3; c.5845G>A) | No | - | - | - | F | 494 | 537 | Nadolol | ¾ cp (40 mg), 3 vv/die | |
| F | KCNQ1 (polymorphism SCN5A-H558R, KCNH2-K897K, e KCNE1-S38G) | No | 448 | 465 | Propranolol | 2 mg/kg, 3 vv/die | F | EXTENDED | EXTENDED | Metoprolol | 8 mg, 2 vv/die |
| F | KCNQ1 (LQT1) | Mother | ND | ND | Nadolol | 1,5 mg/Kg/die | F | EXTENDED | EXTENDED | Metoprolol | 8 mg, 2 vv/die |
| F | KCNQ1 (LQT1) | Mom and maternal grandfather | ND | ND | - | - | |||||
| M | KCNQ1 (LQT1) | Mom and sister | 434 | 465 | Inderal | ¾ + ½ + ½ | |||||
| M* | KCNQ1 (LQT1) | Father (asymptomatic, QTc in the norm) | ND | ND | Nadolol | 1 mg/Kg, 1 v/die | |||||
| M* | KCNQ1 (LQT1) | Father (asymptomatic QTc in the norm) | ND | ND | Nadolol | 1 mg/Kg, 1 v/die |
* brothers. Legend: N.O., not available.
Table 4.
Frequency of ECG abnormalities in the population of 42,200 study subjects identified by newborn screening by ECG.
Table 4.
Frequency of ECG abnormalities in the population of 42,200 study subjects identified by newborn screening by ECG.
| SINGLE ECG ABNORMALITY | Patients, No. (Partial %) | MULTIPLE ECG ABNORMALITY | Patients, No. (Partial %) |
| Right bundle branch focal block | 983 (54.5) | Right Bundle Branch Focal Block + Right Ventricular Prevalence | 61 |
| Left axial deviation | 166 (9.2) | Right Bundle Branch Focal Block + Left Axial Deviation | 26 |
| Nonspecific abnormalities of ventricular repolarization | 128 (7.1) | Right bundle branch focal block + Non-specific alterations of repolarization | 8 |
| Ventricular extrasystole | 83 (4.6) | Right Bundle Branch Focal Block + Supraventricular Extrasystole | 6 |
| Supraventricular extrasystole | 82 (4.5) | Right Bundle Branch Focal Block + Ventricular Extrasystole | 6 |
| Complete right bundle branch block | 66 (3.7) | Right bundle branch focal block + High voltages of QRS | 5 |
| High Voltage QRS | 46 | Right Bundle Branch Focal Block + PQ at Upper Limits | 5 |
| Ventricular pre-excitation | 32 | Right bundle branch focal block + negative T wave | 2 |
| Increased P wave amplitude | 28 | Right Bundle Branch Focal Block + Inf Q Waves | 2 |
| FP at Upper Limits | 27 | Right Bundle Branch Focal Block + Right Ventricular Head + Left Axial Deviation | 2 |
| Low QRS voltages | 20 | Right bundle branch focal block + Sinus tachycardia | 2 |
| Tachycardia sinusale | 20 | Blocco focale di branca dx + Deviazione assiale sx + Extrasistolia sopraventricolare | 1 |
| Bradicardia sinusale relativa | 18 | Right Bundle Branch Focal Block + Supraventricular Extrasystole + Ventricular Extrasystole | 1 |
| FP at Lower Limits | 16 | Right bundle branch focal block + Ventricular parasystole | 1 |
| AV conduction at the sup limits | 10 | Right Bundle Branch Focal Block + Right Ventricular Prevalence + PQ at Lower Limits | 1 |
| Positive T | 10 | Right Branch Focal Block + Migrant Steplight | 1 |
| Ectopic atrial rhythm | 9 | Right bundle branch focal block + Right ventricular and atrial prevalence | 1 |
| Positive T-wave | 8 (97.1) | Right Bundle Branch Focal Block + Right Ventricular Prevalence + PQ at Upper Limits | 1 |
| Migrant step marker | 8 | TOTAL Right bundle branch focal block | 132 (66.7%) |
| Right Axial Deviation | 7 | Right ventricular head + left axial deviation | 17 |
| Left front hemiblock | 5 | Right Ventricular Head + Deep Q Waves | 7 |
| Delayed right intraventricular conduction | 4 | Right ventricular prevalence + PR at the experimental limits | 6 |
| Respiratory sinus arrhythmia | 3 | Right Ventricular Head + High QRS Voltages | 4 |
| ST Elevation | 3 | Right Ventricular Prevalence + Non-Specific Abnormalities of Ventricular Repolarization | 4 |
| Negative T | 3 | Right Ventricular Prevalence + Ventricular Extrasystole | 4 |
| Dextrocardia | 2 | Right Ventricular Head + Low QRS Voltages | 4 |
| QTc and PQ at the lower limits | 2 | Right Ventricular Prevalence + Nonspecific Abnormalities of Ventricular Repolarization | 3 |
| Ventricular hyperkinetic arrhythmia | 1 | Right Ventricular Prevalence + Supraventricular Extrasystole | 3 |
| Appearance S1-Q3 | 1 | Right ventricular prevalence + PQ at upper limits | 2 |
| A-V Dissociation | 1 | Right Ventricular Head + Appearance S1-Q3 | 1 |
| Right atrial engagement | 1 | Right Ventricular Prevalence + AV Conduction at Upper Limits | 1 |
| Increased amplitude P waves | 1 | TOTAL Right ventricular prevalence | 56 (28.3%) |
| Septal Q waves | 1 | Supraventricular Extrasystole + Ventricular Extrasystole | 7 |
| Flat T Waves | 1 | Ventricular Pre-Excitation + Right Bundle Branch Focal Block | 2 |
| AV Conduction Extension | 1 | Ventricular Pre-Excitation + Right Ventricular Head | 2 |
| qR in inferolateral site | 1 (2.5) | TOTAL Ventricular pre-excitation | 4 (2%) |
| Biventricular overload s. | 1 | Left Axial Deviation + Sinus Tachycardia | 1 |
| Signs of bi-atrial engagement | 1 | Left front hemiblock + Left axial deviation | 1 |
| Right Overload | 1 | Biventricular Hypertrophy + Supraventricular Extrasystole | 1 |
| Right ventricular overload | 1 | PQ at Lower Limits + Supraventricular Extrasystole | 1 |
| Diphasic T | 2 | FP at Lower Limits + Probable Junctional Rhythm in Migrant Stepper | 1 |
| Paroxysmal supraventricular tachycardia | 1 | Ventricular Pre-Excitation + Supraventricular Extrasystoa | 1 |
| Total | 1805 | Migrant Steplight + Supraventricular Extrasystole | 1 |
| TOTAL | 198 | ||
Table 5.
Frequency of congenital heart diseases and/or valvulopathies in the population with ECG abnormalities. Frequency of congenital heart diseases and/or valvulopathies in the studied population affected by prolonged QTc interval.
Table 5.
Frequency of congenital heart diseases and/or valvulopathies in the population with ECG abnormalities. Frequency of congenital heart diseases and/or valvulopathies in the studied population affected by prolonged QTc interval.
| Structural alteration of the heart associated with ECG abnormalities | Patients, n. (%) | Multiple structural alterations of the heart associated with a prolongation of the QTc interval | Patients, n. (%) |
| PFO | 164 (61.4) | PFO | 72 (62.1) |
| PFO / DIA OS | 25 (9.4) | PFO + DIV | 6 (5.2) |
| PFO + Mitral insufficiency | 21 (7.9) | PFO + PDA | 4 (3.4) |
| PFO + PDA | 12 (4.5) | PFO / DIA OS | 3 |
| PFO + DIV | 6 | PFO + Mitral insufficiency | 3 |
| PFO / DIA OS + Mitral insufficiency | 3 | PFO + Aortic insufficiency | 1 |
| PFO + PDA + Mitral insufficiency | 3 | PFO + Tricuspid insufficiency | 1 |
| PFO + Aortic insufficiency | 2 | DIA OS | 12 (10.3) |
| PFO / DIA OS + DIV | 1 | DIA OS + IM | 1 |
| PFO / DIA OS + PDA | 1 | DIA OS + Itric | 1 |
| PFO + Aortic Coarctation + Mitral Insufficiency | 1 | DIA OS + Aortic dysplasia | 1 |
| PFO + Tricuspid insufficiency | 1 | PDA | 4 |
| PFO + Flow acceleration at the level of the aortic isthmus without obstructive gradient + Mitral insufficiency | 2 | DIV | 3 |
| Mitral insufficiency | 12 (4.5) | Mitral insufficiency | 2 |
| DIA OS | 2 | Aortic Insufficiency | 1 |
| DIA OS + Tricuspid insufficiency | 2 | Pulmonary stenosis | 1 (11) |
| PDA | 2 | TOTAL | 125 |
| PDA + Mitral insufficiency | 2 | ||
| DIV | 1 | ||
| Pulmonary insufficiency | 1 | ||
| Tricuspid insufficiency | 1 | ||
| Mild aortic insufficiency in apparently tricuspid valve | 1 | ||
| Multiple ventricular echodense neoformations, referable in the first hypothesis to rhabdomyomas | 1 | ||
| TOTAL | 267 |
Legend: DIA, atrial defect; DIV, interventricular defect; PFO, patent foramen ovale; PDA, patency of the duct of Botallo.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



