
Submitted:
19 February 2024
Posted:
20 February 2024
Read the latest preprint version here
Abstract
The evolution of human society over the course of decades and centuries has led to an ever-increasingly complex modern life, one of the hallmark of which is the need for multi-tasking, both individually and collectively. Does this macroscopic evolution of the human societal dynamics have an empirical mapping to the microscopic evolution of the functional molecular units within cells and their manifold cross-talks? The relatively new wealth of knowledge coming from recent research on protein intrinsic disorder, fold-switching, domain shuffling, moonlighting, hub proteins etc. has given us new insights into the relationship between protein structure and function. This has led to a paradigm shift in protein science, clearly diverging from the traditional idea of 'one sequence – one structure – one function' to a more complex understanding of protein functionality. This paradigm shift has caused scientists to delve deeper into the subject, exploring the philosophical as well as the scientific basis of evolved protein multi-functionality, expressed by various evolutionary toys and tools to fit the cumulative multi-functional requirements within proteins. This commentary covers the different evolutionary arsenals to achieve the growing multi-functionality and attempts to connect intrinsic disorder in proteins as probably the sharpest weapon of all.
Keywords:
Protein multi-functionality
; Intrinsically Disordered Proteins
; Gene duplication
; Domain Shuffling
; Protein moonlighting
Protein Multi-Functionality from an Evolutionary Perspective
Proteins can serve multiple functions due to evolutionary pressures that lead to adaptations in their structure and function. The fundamental evolutionary basis of protein multi-functionality is rooted to an activity – stability trade-off between conservation of the core protein function (and corresponding key residue positions in the structure) and variation at other mutable peripheral positions allowing the scope for novel functions to evolve [1,2]. This trade-off can be attained by various evolutionary mechanisms. One mechanism is gene duplication – where redundant copies evolve novel functions [3]. Evolutionary changes like mutations or domain shuffling [4] can also increase a protein’s functional repertoire, enabling it to perform multiple roles crucial for an organism’s survival. Compared to prokaryotes, eukaryotic proteins have more intrinsic disorder with more than 30% of all eukaryotic proteins consisting of long disordered regions comprising of ≥ 50 consecutive amino acid residues [5]. In comparative genomics studies, it has been observed that bacteriophages and their host bacteria co-evolve on the chromosomal level [6]. Viruses can alter their host cells through short linear motifs in their intrinsically disordered regions (IDRs), which lead to functional binding promiscuity [7]. Understanding the evolutionary pathways of multi-functional proteins is crucial to unraveling their complexities and biological significance. This commentary presents a comprehensive comparison of different tools for evolved protein multi-functionality and highlights intrinsic disorder as a major means of achieving it.
Biophysical Basis of Multi-Functionality in IDPs
Intrinsically disordered proteins (IDPs) lack a stable fold and any recognizable domains therein. Unlike well-folded globular proteins with a deep energy-well (global minima), they map to rugged energy landscapes with many equivalent local minima. This imparts in them structural plasiticity (flexibility) for which they are represented as conformational ensembles rather than a single structure. This in turn, helps them inherit their characteristic binding promiscuity upon encountering different patterns to aid multi-functionality [8] without showing any overall preference in chaperone binding in vivo [9]. Their binding promiscuity stems from a phenomenon called ‘coupled folding and binding’ [10], where they undergo transition from disorder to different structured states upon binding to different partners. The root cause of this phenomenon lies in their non-disjoint flexible backbone trajectory that enables them to accommodate different combinations of side chain rotamers, consistent with different befitting surfaces of ordered protein partners [5]. Protein folding and binding (in globular proteins) are analogous processes and can be bridged by concepts like that of complementarity (in geometry and electrostatics) [11]. In case of IDPs, they keep on hovering across their rugged energy landscape in search for suitable intra and/or intermolecular interactions to stabilize them [12,13]. Eukaroytic IDPs stay disordered under normal physiological conditions and only fold into ordered structures when they come in contact with their ‘cellular targets’ [14,15,16,17]. It has been theorized that disordered proteins bind weakly and non-specifically to the target and aligns structurally to a befitting surface (and becomes structured) as it approaches the cognate binding site(s) [18]. In many cases, especially in signal transduction pathways, the bindings are essentially transient (meta-stable). IDPs can also escape protein degradation by undergoing a co-translational folding mechanism involving the ribosomal surface and molecular chaperons [19,20]. These adaptabilities enable IDPs to engage in numerous cellular processes, contributing to their multi-functionality despite lacking a defined structure. Importantly, IDRs of proteins also serves as promising (fuzzy) drug-targets [21,22], wherein a whole new approach for drug-development has lately been initiated, accounting for an acceptable representation of their conformational ensemble [22] as the receptor surface (in contrast to well demarcated drug-binding pockets of the folded proteins), thereby increasing the interacting cross-section for the ligands (drugs). The promiscuous binding nature of IDRs are also capitalized to make it potential drug-target, e.g., in case of castration resistant prostate cancer, the disordered N-terminal domain of androgen receptor are being targeted to overcome existing drug-resistance [23]. Formation of binding competent transient structures (conformational clusters) induced by molecular crowding in the close vicinity of IDPs / IDRs is another unique and idiosyncratic mechanisms to exhibit binding promiscuity and multi-functionality – as demonstrated in intrinsically disordered proteins of the Gab family (Gab1) during signal transduction [24]. It is presumed that these contribute to the spatial organization of complex components. These observations can show us the path which we can take to decipher and understand the mechanism of the assembly of very large and distinct signal transduction protein complexes (viz., ‘signalosomes’ that are stimulus specific) in response to certain stimuli in a short period of time [19].
Weaponry of Evolved Protein Multi-Functionality
Evolved protein multi-functionality harnesses several ammunitions (molecular evolutionary strategies) in its armoury (Figure 1).
Following is a comparative discussion of these evolutionary tools and strategies.
1. Gene Duplication & Functional Divergence: Gene duplications create redundant gene copies, allowing one copy to retain the original function while the paralog (often varying at their oligomric states [25]) accumulates mutations at a higher rate and is often fixed in the population by acquiring an adaptive function according to the classical model of divergence by neo-functionalization [31]. To that end, accelerated evolution in retained paralogs (e.g., Rck1/Rck2, Ptc2/Ptc3, Sim1/Sun4, Ktr5/Ktr6 [32] paralogs) have been observed through evolving post-translational regulation mechanisms (diversified short linear motif like sequences) [33]. At the other end, if we consider the model of sub-functionalization, after gene duplication and divergence, the biological functions of ancestor get partitioned between two paralogs. Sub-functionalization may be of two types: qualitative and quantitative. Qualitative subfunctionalization of the molecular functions that trade-off between each other in the ancestral gene. Each paralog may then evolve towards the optimization of the retained function. Alternatively, quantitative subfunctionalization occurs when neutral evolution results in complementary loss-of-function mutations between the paralogs. In this model, both duplicates become indispensable as they together provide the ancestral functional requirements [34,35,36].
2. Domain Shuffling: Reorganization of protein domains can create multi-functional proteins by combining existing functional units in new ways. It may come through horizontal gene transfer (e.g., from eukaryotes to prokaryotes) or by in-del mutations of genes, post duplication. One common way in which domain shuffling leads to novel functions is by the shuffling of exons (exon shuffling, analogous to alternative splicing at the m-RNA level) followed by in-del mutations. Usually this is established by a mapping of exons and domains (e.g., a single exon coding for a single complete domain) [4]. Insertion of a ‘nested’ domain may also interrupt the linear sequence of a structural domain. Such insertions often map to disordered loops in the parent structure. For example, this has been found in phospholipase Cγ wherein an insert of ~300 residues (comprising of one SH3 and two SH2 domains) separates one of its two Pleckstrin Homology (PH) domains [37]. Certain domains (e.g., the Xlink domain) of the protein aggrecan [26], the most abundant noncollagenous protein in cartilage, is also said to have been created by domain shuffling in ancestral vertebrates.
3. Protein Moonlighting: In contrast to gene-fusion, alternative splicing or functional peptides resulting from multiple proteolysis, protein moonlighting [28,38] refers to multi-functionality evolved in proteins (especially enzymes) without requiring any change in their primary sequence, typically expressed via alternative sites to that of the primary active site – which often maps to a pocket for catalysis [39,40]. In these proteins both classic and non-classic type protein functions co-exist wherein the former refers to enzymatic activities (i.e., involving covalent bond breaking and making) while the later refers to protein – protein interactions (via alternative part of the protein’s surface). However these alternative sites are different to that of allosteric regulations often found with enzymes like phosphofructokinase, hemoglobins etc. Heat shock proteins (HSPs) are classic examples of protein moonlighting.
4. Fold-switching Proteins: Fold-switching proteins [27], a newly emerging class of proteins undergo a distinct switching of their folds by remodelling their secondary structures upon change in environmental (physiological) conditions, for example, a change in pH [41]. Upon fold-switching, they respond to cellular stimuli enabling them to perform important alternative regulatory (e.g., transcriptional regulation) functions of the cell (demonstrated in proteins like RfaH, KaiB etc.) [42,43].
5. Adaptive Evolution: Environmental pressures, such as genetic drift, natural selection etc. drive proteins to adapt, acquiring new functions that enhance an organism’s survival fitness. Adaptive mutations are largely amino-acid substitutions that occur at the protein surface with a high degree of solvent accessibility for these exposed residues that make them most prone to mutations. Population genomics studies in model systems (Drosophila & Arabidopsis) surveyed a multitude of genomic, structural and functional descriptors and revealed that (i) the rate of adaptive substitutions are different for different functional classes (with the fastest rates of protein adaptation observed in proteins involved in translation, degradation and signalling) while (ii) intermolecular interactions (e.g., host-pathogen coevolution) is a major determinant for adaptive evolution [44]. Multifunctional viral proteins are classic examples of adaptive evolution [45]. The recent case is of course the Spike protein of the Coronavirus rapidly undergoing mutations (particularly at the solvent exposed disordered loop containing the crucial Furin like cleavage site or FLCSSpike [46,47]) from SARS-CoV-2 → omicron [48], deltacron [49] etc. Significant patterns of co-occurrence of adaptive events have also been identified in the RNA binding domains with functional overlapping of the HC-Pro of the potyvirus (established by covariation analyses) [45].
6.Intrinsically Disordered Proteins: IDPs are biological soft matters [30] that are highly dynamic and biologically active [50]. Unlike globular proteins, they do not have enough hydrophobic residues to trigger a hydrophobic collapse. Instead, they have high amounts of polar and charged residues [50,51,52,53] which contribute to less sequence complexity in the absence of folding [50,53]. This results in partial temporal order by hydrogen bonding, water mediated contacts (indirect readouts) [54] and formation of transient interchangeable salt-bridges [52]. IDPs do not have a characteristic deep well in their energy landscapes like globular proteins, which means they do not conform to a lone sTable 3D structure under physiological conditions. They have an affinity to undergo transition from disorder to order and back to disorder [51,52]. This makes them highly flexible and adaptable. Partially disordered proteins have intrinsically disordered regions that may be present in varying degrees. These regions often map to hybrid proteins that have both ordered and disordered regions [50,51]. A classic example of this is p53 [55].
Recurrent salt bridges (especially, those with short-range contact orders) impart local temporal structural rigidity in IDPs. However, it is crucial to maintain a balance between the number of salt bridges that allow flexibility and prevent complete rigidity, as seen in globular proteins [52]. Studies [30,46,52] have demonstrated that salt bridges in IDPs are typically not stable (or, persistant) and tend to dissolve and reform frequently with various interchangeable counter-ionic partners. This phenomenon is referred to as ‘transient salt bridge dynamics’. This is necessary to accommodate the high occurrence of oppositely charged residues and to allow for sampling of different conformations, leading to an ensemble. These conformations are not random but revolve around a finite number of structurally degenerate conformational clusters [30]. Phase transitions among these clusters are often triggered by switching of transient salt bridges, demonstrating critical behavior similar to a sand-pile model. The presence of these transient or flitting salt bridges may stabilize the IDP in a conformationally dependent manner, locked by befitting surfaces of its globular counterparts [30]. This is especially relevant in the case of cell signalling, such as suppressors of cytokine signalling (SOCs) [30], where IDRs in eukaryotic transcription factors [53] are evolving with high sequence heterogeneity and demonstrated dynamic multi-functionality through their binding promiscuity.
IDPs, lacking a fixed structure or folding code, exist as highly dynamic ‘dancing protein clouds’ [50] that can adopt different shapes depending on their local environment. When IDPs interact with ordered proteins, their binding contributes to at least partial folding, which depends on the binding partner. Different binding partners can induce different folds [14,50], making them highly adaptable. Additionally, IDPs exhibit fractals and heterogeneity, meaning that they neither converge to a steady state nor diverge to infinity, but rather stay within a defined and chaotic region.
7. Hub Proteins: Hub proteins [29] are well-structured proteins with a (hub-like) high degree in a PPI network. They can interact with multiple partners, even those associated with very different protein networks, leading to diverse biological processes. What sets hub proteins apart is their tendency to interact with disordered partners (IDPs/IDRs) [56], allowing them to interact with structurally diverse partners [51]. In simpler terms, hub proteins can fold into different ordered conformations when they bind to different Molecular Recognition Features (MoRFs) of their preferentially disordered binding partners. This feature makes them highly adaptable and valuable in a wide range of biological processes.
Necessity of IDPs as SOS (Ad Hoc) Tools for Multi-Functionality in Higher Organisms
The oversimplified ‘one gene - one enzyme’ hypothesis [57] has long been outdated with an evolving definition of “gene” [58], and, perhaps even more so, with the growing knowledge of IDPs in recent times. In a human cell, there are approximately 104 genes that code for proteins, while the number of different proteins is around 106. This suggests that there must be mechanisms that cause originally coded proteins to change biophysically, allowing them to acquire multiple functions [51,59]. One such mechanism is the diverse functionality exhibited by IDPs [60,61], which are more common in eukaryotes than in prokaryotes. The acquired multi-functionality in IDPs is supported by their fluid like flexibility, conformational dynamics [30,52] and binding promiscuity [62]. Eukaryotic proteins are mostly intrinsically disordered, with around 23-28% [63,64] showing this characteristic, while about 70% of signalling proteins seem to possess intrinsic disorder. IDPs are highly flexible and can undergo conformational changes to fit the surfaces of their binding partners. They exhibit high binding plasticity and a low affinity – high specificity trade-off as their conformational flexibility enables them to become almost tailor made for its partner(s) in each event of binding [51]. IDPs and IDRs thus complement the repertoire of ordered proteins (i.e., globular, fibrous, membrane proteins) by providing multi-functionality while the ordered proteins with one distinct native global minima structure carry out their routine (primarily, housekeeping) functions [50]. In particular, IDPs are essential for cell signalling pathways, as they allow for high specificity, transitory (switch-like) and reversible [65] interactions (formation of meta-stable ‘fuzzy complexes’ upon binding to suitable partners) that are not possible with ordered proteins. Ordered and disordered proteins work together to bring about cellular functions efficiently, with the presence of long IDRs being more common in eukaryotes than in Archaea and Bacteria [65]. The complexity of an organism is directly proportional to the demand for IDRs, as higher organisms often require more cellular signalling that relies on these proteins.
p53: Example of a Unique Idiosyncratic Multi-Functional Hybrid Protein with Functionally Crucial IDRs
Hybrid proteins contain structured regions that are connected by disordered loops (i.e., IDRs). IDRs are directly correlated with sequence diversity, making them robust for their regulatory functions. A prime example of this is p53, a protein found in both vertebrates and invertebrates, which has a unique structure-functional mapping. Its primary function is to suppress tumors by regulating cell cycle and control. However, it also has many other related non-enzymatic biological functions, such as PPI and DNA-binding. It can form different biologically active multimers like homo-tetramers and isoform-based hetero-tetramers. Additionally, it undergoes alternative splicing and has many preferentially localized pre- and post-translational modifications that lead to various isoforms known as ‘proteoforms’. These combinations, along with the presence of multiple disorder-based protein binding sites, allow p53 to adopt meta-stable states upon interacting with many binding partners in a switch-like transient manner, characteristic of signal transducers and eukaryotic transcription factors [53,59]. This is possible due to the flexibility and sequence diversity offered by its IDRs. While acting as a tumor suppressor, it binds to DNA via its highly-conserved, well-structured DNA-binding domains. The flanking and interconnecting IDRs often promote these bindings to different partners transiently [55]. These IDRs situated amidst structured domains in hybrid proteins have high amino acid substitution rates, leading to high sequence heterogeneity. The resultant expressed structural heterogeneity can be categorized into foldons, inducible foldons, semi-foldons, non-foldons, and unfoldons [59,66], underscoring their promiscuous binding capacity and their significance in PPI networks and signaling pathways. With over 1000 binding partners, p53’s intrinsic disorder is essential for its functionality. This intricate interplay between protein variation, intrinsic disorder, and functionality underscores the complexity of the biological machinery, with implications for understanding disease pathogenesis and the regulation of cellular processes.
Conclusion
The long-established model of protein structure-function relationship by ‘induced fit’ and/ ‘lock-n-key’ model was questioned by the rising evidence of IDRs of proteins which are often seen as functional complementary partners of structurally ordered proteins. The IDR is coined as an umbrella term to indicate either the full protein or any part of it which may adopt complex and heterogeneous spaciotemporal structure under various suitable conditions. Genetic drift and positive selection contribute to the rapid evolution and diversification of IDR/IDPs which could be functionally classified as chaperones, recognizers, protein assemblers, scavengers, signal transducers, effectors etc. IDPs are highly populated with structure-breaking residues like Gly and Pro and other polar residues and lack hydrophobic and aromatic residues which are the core components of a properly folded globular protein. Different structural mosaic patterns found in IDPs render the proteins multi-functionality and promiscuous binding nature. These two aspects are two sides of the coin, examining both advantages and disadvantages at the same time. The IDP/IDRs energy efficiency in the cellular milieu stems from its multifunctional nature, which causes it to transiently bind to a vast spectrum of interacting partners. But this apparent energy-efficient strategy to perform various physiological functions often gets backfired by high specificity – low affinity binding characteristic leading to neurodegenerative diseases, cardiovascular disease, and cancer. The promiscuous binding nature of IDR/IDP comes into play to alleviate the pathological conditions where the protein-protein interactions are being targeted as potential therapeutic measures to develop interacting partner mimicking drug molecules. The rapid advancements in bioinformatics tools are crucial for deciphering the complicated world of IDR/IDPs and utilizing it to our advantages.
References
- T. Sikosek, H.S. Chan, Biophysics of protein evolution and evolutionary protein biophysics, J R Soc Interface 11 (2014) 20140419. [CrossRef]
- N. Tokuriki, F. Stricher, L. Serrano, D.S. Tawfik, How Protein Stability and New Functions Trade Off, PLOS Comput Biol 4 (2008) e1000002. [CrossRef]
- Espinosa-Cantú, D. Ascencio, F. Barona-Gómez, A. DeLuna, Gene duplication and the evolution of moonlighting proteins, Frontiers in Genetics 6 (2015). https://www.frontiersin.org/articles/10.3389/fgene.2015.00227 (accessed February 1, 2024).
- Domain Shuffling - an overview | ScienceDirect Topics, (n.d.). https://www.sciencedirect.com/topics/neuroscience/domain-shuffling (accessed February 1, 2024).
- A.K. Dunker, J.D. Lawson, C.J. Brown, R.M. Williams, P. Romero, J.S. Oh, C.J. Oldfield, A.M. Campen, C.M. Ratliff, K.W. Hipps, J. Ausio, M.S. Nissen, R. Reeves, C. Kang, C.R. Kissinger, R.W. Bailey, M.D. Griswold, W. Chiu, E.C. Garner, Z. Obradovic, Intrinsically disordered protein, Journal of Molecular Graphics and Modelling 19 (2001) 26–59. [CrossRef]
- H. Brüssow, C. Canchaya, W.-D. Hardt, Phages and the Evolution of Bacterial Pathogens: from Genomic Rearrangements to Lysogenic Conversion, Microbiol Mol Biol Rev 68 (2004) 560–602. [CrossRef]
- N.E. Davey, G. Travé, T.J. Gibson, How viruses hijack cell regulation, Trends Biochem Sci 36 (2011) 159–169. [CrossRef]
- P.T. Fersht Alan, Structure and Function of Intrinsically Disordered Proteins, Chapman and Hall/CRC, New York, 2009. [CrossRef]
- H. Hegyi, P. Tompa, Intrinsically Disordered Proteins Display No Preference for Chaperone Binding In Vivo, PLOS Computational Biology 4 (2008) e1000017. [CrossRef]
- K. Sugase, H.J. Dyson, P.E. Wright, Mechanism of coupled folding and binding of an intrinsically disordered protein, Nature 447 (2007) 1021–1025. [CrossRef]
- S. Basu, D. Bhattacharyya, R. Banerjee, Self-Complementarity within Proteins: Bridging the Gap between Binding and Folding, Biophys J 102 (2012) 2605–2614. [CrossRef]
- C.J. Tsai, S. Kumar, B. Ma, R. Nussinov, Folding funnels, binding funnels, and protein function, Protein Sci 8 (1999) 1181–1190. [CrossRef]
- Y. Levy, S.S. Cho, J.N. Onuchic, P.G. Wolynes, A Survey of Flexible Protein Binding Mechanisms and their Transition States Using Native Topology Based Energy Landscapes, Journal of Molecular Biology 346 (2005) 1121–1145. [CrossRef]
- P.E. Wright, H.J. Dyson, Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm, J. Mol. Biol. 293 (1999) 321–331. [CrossRef]
- H.J. Dyson, P.E. Wright, Coupling of folding and binding for unstructured proteins, Curr Opin Struct Biol 12 (2002) 54–60. [CrossRef]
- H.J. Dyson, P.E. Wright, Intrinsically unstructured proteins and their functions, Nature Reviews Molecular Cell Biology 6 (2005) 197–208. [CrossRef]
- V.N. Uversky, Natively unfolded proteins: a point where biology waits for physics, Protein Sci 11 (2002) 739–756. [CrossRef]
- B.A. Shoemaker, J.J. Portman, P.G. Wolynes, Speeding molecular recognition by using the folding funnel: the fly-casting mechanism, Proc Natl Acad Sci U S A 97 (2000) 8868–8873. [CrossRef]
- P.C. Simister, F. Schaper, N. O’Reilly, S. McGowan, S.M. Feller, Self-Organization and Regulation of Intrinsically Disordered Proteins with Folded N-Termini, PLOS Biology 9 (2011) e1000591. [CrossRef]
- C.A. Waudby, C.M. Dobson, J. Christodoulou, Nature and Regulation of Protein Folding on the Ribosome, Trends in Biochemical Sciences 44 (2019) 914–926. [CrossRef]
- K. Kamagata, E. Mano, Y. Itoh, T. Wakamoto, R. Kitahara, S. Kanbayashi, H. Takahashi, A. Murata, T. Kameda, Rational design using sequence information only produces a peptide that binds to the intrinsically disordered region of p53, Sci Rep 9 (2019) 8584. [CrossRef]
- S. Saurabh, K. Nadendla, S.S. Purohit, P.M. Sivakumar, S. Cetinel, Fuzzy Drug Targets: Disordered Proteins in the Drug-Discovery Realm, ACS Omega 8 (2023) 9729–9747. [CrossRef]
- Q. Yi, W. Liu, J.H. Seo, J. Su, M.A. Alaoui-Jamali, J. Luo, R. Lin, J.H. Wu, Discovery of a Small-Molecule Inhibitor Targeting the Androgen Receptor N-Terminal Domain for Castration-Resistant Prostate Cancer, Molecular Cancer Therapeutics 22 (2023) 570–582. [CrossRef]
- T. Gruber, M. Lewitzky, L. Machner, U. Weininger, S.M. Feller, J. Balbach, Macromolecular Crowding Induces a Binding Competent Transient Structure in Intrinsically Disordered Gab1, Journal of Molecular Biology 434 (2022) 167407. [CrossRef]
- S. Mallik, D.S. Tawfik, E.D. Levy, How gene duplication diversifies the landscape of protein oligomeric state and function, Curr Opin Genet Dev 76 (2022) None. [CrossRef]
- T. Kawashima, S. Kawashima, C. Tanaka, M. Murai, M. Yoneda, N.H. Putnam, D.S. Rokhsar, M. Kanehisa, N. Satoh, H. Wada, Domain shuffling and the evolution of vertebrates, Genome Res 19 (2009) 1393–1403. [CrossRef]
- P.N. Bryan, J. Orban, Proteins that switch folds, Curr Opin Struct Biol 20 (2010) 482–488. [CrossRef]
- C.J. Jeffery, Protein moonlighting: what is it, and why is it important?, Philos Trans R Soc Lond B Biol Sci 373 (2018) 20160523. [CrossRef]
- M. Higurashi, T. Ishida, K. Kinoshita, Identification of transient hub proteins and the possible structural basis for their multiple interactions, Protein Sci 17 (2008) 72–78. [CrossRef]
- Bandyopadhyay, S. Basu, Criticality in the conformational phase transition among self-similar groups in intrinsically disordered proteins: Probed by salt-bridge dynamics, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1868 (2020) 140474. [CrossRef]
- Evolution by Gene Duplication | SpringerLink, (n.d.). https://link.springer.com/book/10.1007/978-3-642-86659-3 (accessed January 23, 2024).
- A.L. Hughes, R. Friedman, Parallel Evolution by Gene Duplication in the Genomes of Two Unicellular Fungi, Genome Res 13 (2003) 794–799. [CrossRef]
- A.N. Nguyen Ba, B. Strome, J.J. Hua, J. Desmond, I. Gagnon-Arsenault, E.L. Weiss, C.R. Landry, A.M. Moses, Detecting Functional Divergence after Gene Duplication through Evolutionary Changes in Posttranslational Regulatory Sequences, PLoS Comput Biol 10 (2014) e1003977. [CrossRef]
- S.W. Fewell, J.L. Woolford, Ribosomal protein S14 of Saccharomyces cerevisiae regulates its expression by binding to RPS14B pre-mRNA and to 18S rRNA, Mol Cell Biol 19 (1999) 826–834. [CrossRef]
- M. Lynch, A. Force, The probability of duplicate gene preservation by subfunctionalization, Genetics 154 (2000) 459–473. [CrossRef]
- X. He, J. Zhang, Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution, Genetics 169 (2005) 1157–1164. [CrossRef]
- M. Fadri, A. Daquinag, S. Wang, T. Xue, J. Kunz, The Pleckstrin Homology Domain Proteins Slm1 and Slm2 Are Required for Actin Cytoskeleton Organization in Yeast and Bind Phosphatidylinositol-4,5-Bisphosphate and TORC2, Mol Biol Cell 16 (2005) 1883–1900. [CrossRef]
- C.J. Jeffery, Multifunctional proteins: examples of gene sharing, Ann Med 35 (2003) 28–35. [CrossRef]
- C.J. Jeffery, Moonlighting proteins, Trends Biochem Sci 24 (1999) 8–11. [CrossRef]
- J. Piatigorsky, Gene Sharing and Evolution: The Diversity of Protein Functions, in: Gene Sharing and Evolution, Harvard University Press, 2009. [CrossRef]
- Baruah, P. Biswas, Designing pH induced fold switch in proteins, J. Chem. Phys. 142 (2015) 185102. [CrossRef]
- A.K. Kim, L.L. Porter, Functional and Regulatory Roles of Fold-Switching Proteins, Structure 29 (2021) 6–14. [CrossRef]
- N.A. Bernhardt, U.H.E. Hansmann, Multifunnel Landscape of the Fold-Switching Protein RfaH-CTD, J Phys Chem B 122 (2018) 1600–1607. [CrossRef]
- A.F. Moutinho, F.F. Trancoso, J.Y. Dutheil, The Impact of Protein Architecture on Adaptive Evolution, Mol Biol Evol 36 (2019) 2013–2028. [CrossRef]
- Hasiów-Jaroszewska, M.A. Fares, S.F. Elena, Molecular evolution of viral multifunctional proteins: the case of potyvirus HC-Pro, J Mol Evol 78 (2014) 75–86. [CrossRef]
- S. Roy, P. Ghosh, A. Bandyopadhyay, S. Basu, Capturing a Crucial ‘Disorder-to-Order Transition’ at the Heart of the Coronavirus Molecular Pathology—Triggered by Highly Persistent, Interchangeable Salt-Bridges, Vaccines 10 (2022) 301. [CrossRef]
- P. Balaram, The murky origins of the coronavirus SARS-CoV-2, the causative agent of the COVID-19 pandemic, CURRENT SCIENCE 120 (2021) 4.
- Y. Araf, F. Akter, Y.-D. Tang, R. Fatemi, M.S.A. Parvez, C. Zheng, M.G. Hossain, Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines, J Med Virol 94 (2022) 1825–1832. [CrossRef]
- S.Q. Maulud, D.A. Hasan, R.K. Ali, R.F. Rashid, A.A. Saied, M. Dhawan, Priyanka, O.P. Choudhary, Deltacron: Apprehending a new phase of the COVID-19 pandemic, Int J Surg 102 (2022) 106654. [CrossRef]
- V.N. Uversky, Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins, J Biol Chem 291 (2016) 6681–6688. [CrossRef]
- X. Sun, E.H.A. Rikkerink, W.T. Jones, V.N. Uversky, Multifarious Roles of Intrinsic Disorder in Proteins Illustrate Its Broad Impact on Plant Biology, The Plant Cell 25 (2013) 38–55. [CrossRef]
- S. Basu, P. Biswas, Salt-bridge dynamics in intrinsically disordered proteins: A trade-off between electrostatic interactions and structural flexibility, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1866 (2018) 624–641. [CrossRef]
- M. Már, K. Nitsenko, P.O. Heidarsson, Multifunctional Intrinsically Disordered Regions in Transcription Factors, Chemistry – A European Journal 29 (2023) e202203369. [CrossRef]
- K.M. Reid, H. Poudel, D.M. Leitner, Dynamics of Hydrogen Bonds between Water and Intrinsically Disordered and Structured Regions of Proteins, J Phys Chem B 127 (2023) 7839–7847. [CrossRef]
- Xue, C.J. Brown, A.K. Dunker, V.N. Uversky, Intrinsically disordered regions of p53 family are highly diversified in evolution, Biochim Biophys Acta 1834 (2013) 725–738. [CrossRef]
- E.A. Cino, R.C. Killoran, M. Karttunen, W.-Y. Choy, Binding of disordered proteins to a protein hub, Sci Rep 3 (2013) 2305. [CrossRef]
- G.W. Beadle, E.L. Tatum, Genetic Control of Biochemical Reactions in Neurospora*, Proceedings of the National Academy of Sciences 27 (1941) 499–506. [CrossRef]
- P. Portin, A. Wilkins, The Evolving Definition of the Term “Gene,” Genetics 205 (2017) 1353–1364. [CrossRef]
- V.N. Uversky, p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept, Int J Mol Sci 17 (2016) 1874. [CrossRef]
- S.A. Clark, N. Jespersen, C. Woodward, E. Barbar, Multivalent IDP assemblies: Unique properties of LC8-associated, IDP duplex scaffolds, FEBS Letters 589 (2015) 2543–2551. [CrossRef]
- M. Sickmeier, J.A. Hamilton, T. LeGall, V. Vacic, M.S. Cortese, A. Tantos, B. Szabo, P. Tompa, J. Chen, V.N. Uversky, Z. Obradovic, A.K. Dunker, DisProt: the Database of Disordered Proteins, Nucleic Acids Res 35 (2007) D786–D793. [CrossRef]
- Morris, J.H. Torpey, R.L. Isaacson, Intrinsically disordered proteins: modes of binding with emphasis on disordered domains, Open Biol 11 (n.d.) 210222. [CrossRef]
- W. Basile, M. Salvatore, C. Bassot, A. Elofsson, Why do eukaryotic proteins contain more intrinsically disordered regions?, PLoS Comput Biol 15 (2019) e1007186. [CrossRef]
- J.A. Zamora-Briseño, A. Pereira-Santana, S.J. Reyes-Hernández, D. Cerqueda-García, E. Castaño, L.C. Rodríguez-Zapata, Towards an understanding of the role of intrinsic protein disorder on plant adaptation to environmental challenges, Cell Stress and Chaperones 26 (2021) 141–150. [CrossRef]
- P.E. Wright, H.J. Dyson, Intrinsically Disordered Proteins in Cellular Signaling and Regulation, Nat Rev Mol Cell Biol 16 (2015) 18–29. [CrossRef]
- V.N. Uversky, Unusual biophysics of intrinsically disordered proteins, Biochim. Biophys. Acta 1834 (2013) 932–951. [CrossRef]
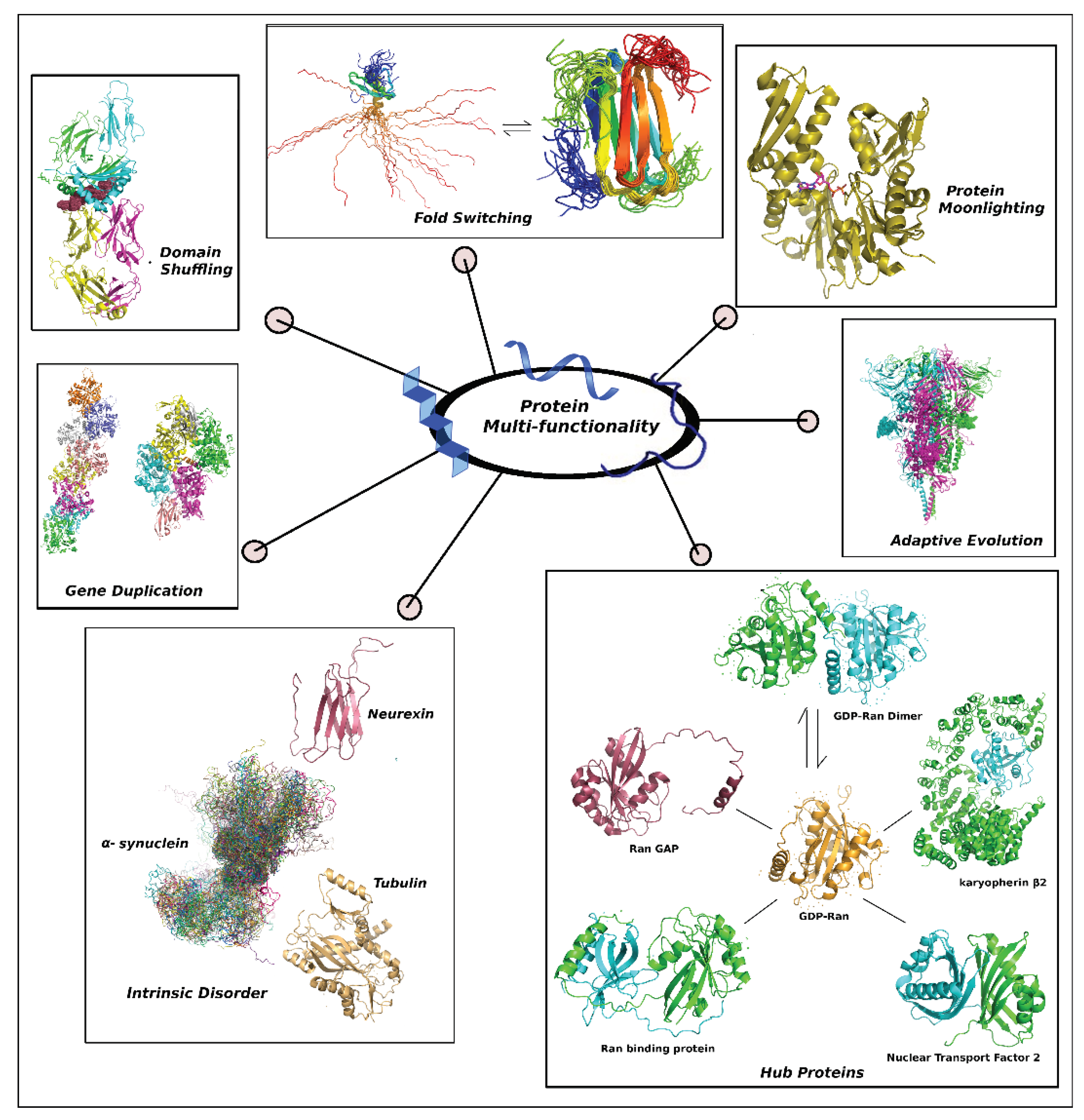
Figure 1.
The composite figure portrays different evolutionary tools to express protein multi-functionality. The represented repertoire includes (i) Gene Duplication: Actins seperated by two insertions - deletions (in-del) mutations (Q211→D231, D299→T365): ARP4 (PDB ID: 5NBM) where filament formation is inhibited and its heteromerized paralog ACT1 (6BNO) [25], (ii) Domain Shuffling: Aggrecan core peptide (highlighted as dots) presented by class II Major Histocompatibility Complex (7RDV) [26], (iii) Fold Switching: human Lymphotacin (1J8I ↔ 2JP1) [27], (iv) Protein Moonlighting: Yeast Heat shock protein Hsp70 bound with ADP (3QFU) [28], (v) Adaptive Evolution: the Spike protein with highly mutable FLCSSpike (highlighted as dots) from SARS-CoV-2 (6XR8), (vi) Hub Proteins: GDP-Ran (monomer: 1QG4 ↔ dimer: 1BYU) with its interactome (Ran binding protein: 1RRP, Ran GAP: 1K5D, karyopherin β2: 1QBK, Nuclear Transport Factor 2: 5BXQ) [29] and (iii) Intrinsic Disorder: alpha synuclein (a conformational ensemble picked up from its MD-simulated trajectory [30]) with its two cognate globular binding partners: Tubulin (4YRL) and β-neurexin 1 (3MW2).
Figure 1.
The composite figure portrays different evolutionary tools to express protein multi-functionality. The represented repertoire includes (i) Gene Duplication: Actins seperated by two insertions - deletions (in-del) mutations (Q211→D231, D299→T365): ARP4 (PDB ID: 5NBM) where filament formation is inhibited and its heteromerized paralog ACT1 (6BNO) [25], (ii) Domain Shuffling: Aggrecan core peptide (highlighted as dots) presented by class II Major Histocompatibility Complex (7RDV) [26], (iii) Fold Switching: human Lymphotacin (1J8I ↔ 2JP1) [27], (iv) Protein Moonlighting: Yeast Heat shock protein Hsp70 bound with ADP (3QFU) [28], (v) Adaptive Evolution: the Spike protein with highly mutable FLCSSpike (highlighted as dots) from SARS-CoV-2 (6XR8), (vi) Hub Proteins: GDP-Ran (monomer: 1QG4 ↔ dimer: 1BYU) with its interactome (Ran binding protein: 1RRP, Ran GAP: 1K5D, karyopherin β2: 1QBK, Nuclear Transport Factor 2: 5BXQ) [29] and (iii) Intrinsic Disorder: alpha synuclein (a conformational ensemble picked up from its MD-simulated trajectory [30]) with its two cognate globular binding partners: Tubulin (4YRL) and β-neurexin 1 (3MW2).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



