
Submitted:
19 February 2024
Posted:
20 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The emergence of SARS-CoV-2 mutations pose significant challenges to diagnostic tests as these mutations can reduce the sensitivity of commonly used RT-PCR assays. Therefore, there is a need to design diagnostic assays with multiple targets to enhance sensitivity. In this study, we identified a novel diagnostic target, nsp10 gene, using nanopore sequencing. Firstly, we determined the analytical sensitivity and specificity of our COVID-19-nsp10 assay. The COVID-19-nsp10 assay had the limit of detection of 74 copies/mL (95% confidence interval: 48-299 copies/mL) and did not show cross-reactivity with other respiratory viruses. Next, we determined the diagnostic performance of the COVID-19-nsp10 assay using 261 respiratory specimens, including 147 SARS-CoV-2-positive specimens belonging to ancestral strain and Alpha, Beta, Gamma, Delta, Mu, Eta, Kappa, Theta and Omicron lineages. Using the LightMix E-gene RT-PCR assay as the reference method, the diagnostic sensitivity and specificity of the COVID-19-nsp10 assay were 100%. The median Cp values for the LightMix E-gene RT-PCR and our COVID-19-nsp10 RT-PCR were 22.48 (range: 12.95-36.60) and 25.94 (range 16.37-36.87), respectively. The Cp values of the COVID-19-nsp10 RT-PCR assay correlated well with those of the LightMix E-gene RT-PCR assay (Spearman’s ρ = 0.968; P<0.0001). In conclusion, nsp10 is a suitable target for SARS-CoV-2 RT-PCR assay.
Keywords:
SARS-CoV-2
; COVID-19
; nsp10
; real-time RT-PCR
; diagnostic
1. Introduction
In late December 2019, a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was first identified in patients with pneumonia in Wuhan, China [1]. At that time, most patients infected with SARS-CoV-2 typically experienced symptoms such as fever, cough, dyspnea, myalgia, and show radiological findings of ground-glass lung opacities consistent with atypical pneumonia [2,3,4]. However, there have also been documented cases of asymptomatic or mild presentations [5,6,7,8,9,10]. SARS-CoV-2 is highly contagious and can easily spread from person to person, leading to its rapid global dissemination [11,12,13,14,15]. This virus has caused the coronavirus disease 2019 (COVID-19) pandemic since 2020, resulting in a significant global morbidity and mortality, with nearly 7 million deaths reported [16]. Following the relaxation of the control measures, such as mask wearing and social distancing, the detection rates of common respiratory viruses, including SARS-CoV-2, have increased worldwide since 2023. RNA viruses, like SARS-CoV-2, are more prone to mutations compared to DNA viruses due to the lack of proofreading ability of their RNA-dependent-RNA-polymerase (RdRp) [17]. These factors will increase the likelihood of mutations in viral RNA genomes. Consequently, mismatches between virus genes and PCR primers/probe may occur frequently. As reported in various publications, mutations in the primer or probe binding sites can impact the sensitivity of RT-PCR assays, leading to the occurrence of false-negative results [18,19,20,21]. To minimize such occurrences, it is crucial to design molecular diagnostic assays with multiple gene targets and search for a novel real-time RT-PCR target located in a conserved region of the genome. A timely and accurate diagnosis can significantly aid in facilitating appropriate antiviral treatment and implementing effective infection control measures.
In our previous studies, we have successfully identified various abundantly expressed gene targets, including polymerase basic 2 (PB2) and nonstructural (NS) genes for influenza A virus, as well as nonstructural protein 1 (nsp1) and nsp8 genes for SARS-CoV-2 detection, using nanopore sequencing [22,23,24]. The diagnostic performance of these assays was comparable to other validated in-house or commercial RT-PCR assays. In the present study, we identified a novel highly expressed gene target, nsp10, using the same strategy for the development of a highly sensitive and specific RT-PCR assay for COVID-19 diagnosis. In contrast to our previous gene targets (nsp1 and nsp8) of SARS-CoV-2, nsp10 was found to be highly conserved when compared to other genes. A study published in 2023 examined the prevalence of the most common mutation in each gene within the SARS-CoV-2 genome. The finding revealed that the prevalence of the most common mutation in nsp10 was found to be 0.16%, while the prevalence of the most common mutations in nsp1 and nsp8 were 8.01% and 0.7% respectively [25]. Therefore, the conserved nature of the nsp10 gene reduces the likelihood of primer-binding site mutations. We describe the evaluation of the in-house developed COVID-19-nsp10 RT-PCR assay using clinical specimens and external quality assessment (EQA) samples.
2. Results
2.1. Design of a novel COVID-19 real-time RT-PCR assay targeting the nsp10 gene of SARS-CoV-2
In this study, we observed a high expression of the nsp10 gene of SARS-CoV-2 in 49 clinical specimens, as revealed by nanopore whole-genome sequencing with the nanopore protocol for PCR tiling of COVID-19 (PTC_9096_v109_revH_06Feb2020). Among all the genes in the SARS-CoV-2 genome, the nsp10 gene had the highest coverage (Figure 1). Also, despite the high expression of nsp10 in the clinical specimens, the large variances in the normalized coverages highlighted the importance of having different diagnostics targets for RT-PCR assays (Figure 1). Out of the 49 patients, 29 of them (59.2%) had a higher coverage in nsp10 compared to nsp1. The patient with the highest normalized nsp10 coverage had a coverage that was 56.8% higher than nsp1, while the patient with the lowest normalized nsp10 coverage had a coverage that was 63.7% lower than nsp1. As for nsp8, 42 out of 49 patients (85.7%) had a higher coverage in nsp10 compared to nsp8. The patient with the largest difference in coverage between nsp10 and nsp8 had a nsp10 coverage that was 3.7 times higher than the nsp8 coverage, while one of the patient’s nsp8 could not be sequenced. Therefore, we designed primers and probe targeting the nsp10 gene as a novel target for our in-house developed RT-PCR assay (Table 1). The multiple sequence alignment demonstrated that the target sites of our nsp10 primers and probe were well conserved among different variants (ancestral strain, Alpha, Beta, Gamma, Delta, Epsilon, Zeta, Eta, Theta, Iota, Kappa, Lambda, Mu and Omicron [including JN.1]) (Figure 2).
2.2. Analytical Sensitivity and Specificity of the COVID-19-nsp10 Real-Time RT-PCR Assay
To determine the analytical sensitivity of the in-house developed COVID-19-nsp10 RT-PCR assay, the limit of detection (LOD) was evaluated by using two-fold serial dilutions of a total nucleic acid (TNA) extracted from the SARS-CoV-2 Q control 01 and tested in quadruplicate in two independent runs. The LOD of the COVID-19-nsp10 RT-PCR assay was 74 copies/mL (95%CI 48 - 299 copies/mL) by probit analysis (Table 2).
To determine the analytical specificity of the COVID-19-nsp10 RT-PCR assay, we tested TNA of human coronaviruses SARS-CoV-1, Middle East Respiratory Syndrome coronavirus (MERS-CoV), HCoV-OC43, HCoV-HKU1, HCoV-229E, HCoV-NL63, and other common respiratory viruses including influenza A viruses ((H1N1)pdm09 and H3N2), influenza B virus, influenza C virus, respiratory syncytial virus, human metapneumovirus, parainfluenza virus types 1 – 4, rhinovirus and adenovirus. Our in-house developed COVID-19-nsp10 RT-PCR assay showed no cross reaction with these respiratory viruses.
2.3. Diagnostic Performance of the COVID-19-nsp10 Assay for SARS-CoV-2 Detection
To assess the diagnostic performance of our in-house developed COVID-19-nsp10 real-time RT-PCR assay, a total of 261 respiratory specimens from suspected COVID-19 patients were subjected to SARS-CoV-2 detection. Among these 261 clinical specimens collected during 2020-2023, 147 (56.3%) were tested positive for SARS-CoV-2 by both the in-house developed COVID-19-nsp10 RT-PCR (Cp range: 16.37 – 36.87) and the commercial LightMix E-gene RT-PCR (Cp range: 12.95 – 36.60). A good agreement in performance of the in-house COVID-19-nsp10 RT-PCR assay compared to the LightMix E-gene RT-PCR assay was revealed by a strong correlation (Spearman’s ρ = 0.968; P < 0.0001) (Figure 3). The median Cp value of the COVID-19-nsp10 RT-PCR assay (25.94) was significantly higher than that of the LightMix E-gene RT-PCR assay (22.48; P < 0.0001) (Figure 4). Using the LightMix E-gene RT-PCR assay as the reference method, the diagnostic sensitivity and specificity of the COVID-19-nsp10 RT-PCR assay were 100% (Table 3). The lineages of SARS-CoV-2 in the 147 positive specimens were identified by nanopore sequencing, sequencing results showed that the isolates belonged to a broad range of variants including ancestral strain, Alpha, Beta, Gamma, Delta, Mu, Eta, Kappa, Theta and Omicron variants. Among six EQA samples from the College of American Pathologists (CAP), both assays gave 100% correct results.
3. Discussion
In this study, we developed a real-time RT-PCR assay targeting the nsp10 gene of SARS-CoV-2 for COVID-19 diagnosis. Given the ongoing emergence of new variants with mutations scattered across the viral genome [26,27,28,29,30], it is advisable to design primers and probe that target different genes to ensure accurate detection of these variants. In December 2023, the World Health Organization classified the recently emerged JN.1 as variant of interest (VOI) following its rapid spread in multiple countries [31]. The identification of a newly emerged variant further highlights the need for robust diagnostic assays that can detect various SARS-CoV-2 strains. While the N gene of SARS-CoV-2 is commonly used as a target for COVID-19 nucleic acid amplification tests, instances of N gene target failure have been reported [32,33]. Hence, it is crucial to identify alternative conserved target genes for SARS-CoV-2 detection. The nsp10 gene was found to be highly expressed in clinical specimens and conserved among different SARS-CoV-2 variants and different genes [25], further supporting its potential as an attractive target for SARS-CoV-2 detection. Different from the nsp1 and N genes, the nsp10 gene is located near the center of the genome in the ORF1ab region. During the life cycle of the SARS-CoV-2 virus, nsp10 is essential for replicating and proofreading viral RNA [34]. The conservation of nsp10 is likely due to its importance in the viral life cycle and the need to maintain its interaction with RdRp. Notably, structural analysis by Wang et al showed that nsp10 has limited room for conformational changes, which also contributed to the conserved nature of the nsp10 protein [35]. In the present study, we also observed a high variability in the expression of different genes within the SARS-CoV-2 genome. This finding reemphasizes the importance of having multiple targets for real-time RT-PCR assays.
Our COVID-19-nsp10 assay has demonstrated high sensitivity and specificity in detecting all evaluated variants, including the recently emerged Omicron variants XBB and JN.1. The in-house developed COVID-19-nsp10 assay exhibits a high sensitivity, with the LOD below 100 copies/mL, and it does not show any cross-reactivity with other common respiratory viruses. When compared to the validated LightMix E-gene assay as the reference method, our COVID-19-nsp10 assay showed comparable diagnostic performance, with 100% diagnostic sensitivity and specificity using clinical specimens. The median Cp value of the in-house developed COVID-19-nsp10 assay was found to be significantly higher than the commercial LightMix E-gene assay. This difference in Cp values may be attributed to several factors, such as the lower volume of sample extract used in the in-house assay compared to the E-gene assay, as well as potential variations in PCR reagents and thermal cycling conditions between the two assays. However, despite this discrepancy, there was no significant difference in the diagnostic sensitivity between the two assays. In addition, the Cp values obtained from both the COVID-19-nsp10 and LightMix E-gene assays exhibited excellent correlation. We further evaluated the performance of our COVID-19-nsp10 assay and the LightMix E-gene assay using EQA samples from the CAP. Both assays performed well, reaffirming their accuracy and reliability in detecting SARS-CoV-2. These findings indicate that our in-house COVID-19-nsp10 assay, along with the LightMix E-gene assay, demonstrate excellent diagnostic performance for the detection of SARS-CoV-2 RNA.
In addition to the analytical and diagnostic performance, the turnaround time (TAT) and cost are essential factors to consider when selecting diagnostic assays, particularly in the context of a pandemic where there is a high volume of clinical specimens from patients with suspected COVID-19. Regarding the TAT, both the COVID-19-nsp10 and E-gene assays had the same sample-to-extract time since the same extraction method was used, while the PCR running time of the E-gene assay (42 min) was shorter compared to our in-house COVID-19-nsp10 assay (72 min). In terms of cost, considering the expenses associated with PCR reagents and primers/probes, our in-house COVID-19-nsp10 assay (US $2 per reaction) proved to be significantly more cost-effective compared to the LightMix E-gene assay (US $10 per reaction). For clinical laboratories with the necessary expertise in the developing in-house assays, utilizing in-house developed tests can result in substantial cost savings, particularly when managing a large number of samples during a pandemic.
There is a limitation in this study. Due to the limited amount of clinical specimens for each patient, we were not able to classify each sample into groups of different expression level of different genes by sequencing and then compare the sensitivity of different RT-PCR assays. Therefore, an interesting direction for future projects would be to evaluate the sensitivity of different assays given different expression level of the genes.
In conclusion, a low-cost, sensitive, specific and reliable real-time RT-PCR assay targeting the nsp10 gene of SARS-CoV-2 was successfully developed for COVID-19 diagnosis. Next Generation Sequencing is a valuable tool for identifying suitable gene targets for molecular test development by analyzing the expression of each gene.
4. Materials and Methods
4.1. Viruses, clinical specimens and EQA samples for evaluation
For analytical sensitivity evaluation, two-fold serial dilutions of TNA extracted from the SARS-CoV-2 Q control 01 (Qnostics, Glasgow, UK) were used. For analytical specificity evaluation, TNA extracted from a clinical specimen positive for human coronavirus HCoV-HKU1 and 17 culture isolates of other human coronaviruses (SARS-CoV-1, MERS-CoV, HCoV-229E, HCoV-NL63, HCoV-OC43) and respiratory viruses (influenza A viruses – pdm09 H1N1 and H3, influenza B virus, influenza C virus, respiratory syncytial virus, human metapneumovirus, parainfluenza virus types 1 – 4, rhinovirus and adenovirus) were used [36]. For diagnostic performance evaluation, 261 clinical specimens including nasopharyngeal aspirate, nasopharyngeal swab, throat swab or posterior oropharyngeal saliva collected from hospitalized patients (male:female = 119:142; median age: 69 years; range: 3 months – 106 years) with suspected COVID-19 were included for SARS-CoV-2 detection. Among these 261 specimens collected between 2020 and 2023, 17 were from year 2020, 65 were from year 2021, 16 were from year 2022 and 163 were from year 2023. The specimens positive for SARS-CoV-2 were subjected to lineage identification by nanopore sequencing. In addition to clinical specimens, six EQA samples from the CAP in year 2023 with various concentrations of SARS-CoV-2 or negative for SARS-CoV-2 were evaluated.
4.2. Whole genome sequencing of SARS-CoV-2
Whole genome sequencing was performed using the Oxford Nanopore MinION device (Oxford Nanopore Technologies, Oxford, UK). Library preparation was performed following the Nanopore protocol - PCR tiling of COVID-19 (Version: PTC_9096_v109_revH_06Feb2020) according to the manufacturer’s instructions with minor modifications (Oxford Nanopore Technologies) as we described previously [37]. Briefly, extracted RNA was first reverse transcribed to cDNA using SuperScriptTM IV reverse transcriptase (ThermoFisher Scientific, MA, USA). PCR amplification was then performed using hCoV-2019/nCoV-2019 Version 3 Amplicon Set or hCoV-2019/nCoV-2019 Version 4.1 Amplicon Set (Integrated DNA Technologies, IA, USA) with the Q5® Hot Start High-Fidelity 2X Master Mix kit (New England Biolabs, MA, USA) according to the Nanopore protocol. PCR products were purified with 1x AMPure XP beads (Beckman Coulter, CA, USA) and quantified using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). The purified PCR products were then normalized for end-prep and native barcode ligation reactions according to the PCR tiling of COVID-19 virus protocol with Native Barcoding Expansion 96 (EXP-NBD196, Oxford Nanopore Technologies). Barcoded libraries were then pooled, purified with 0.4x AMPure XP beads and then quantified using Qubit dsDNA HS Assay Kit. Purified pooled libraries were ligated to sequencing adapters and sequenced with the Oxford Nanopore MinION device using R9.4.1 flow cells for 24-48 hours.
4.3. In-silico analysis
The raw reads from the sequenced 49 clinical specimens were quality checked. The sequencing primers were removed by trimming the extreme ends of each reads. They were then mapped to the SARS-CoV-2 ancestral strain (GenBank accession number: NC_045512.2) using the Medaka software [38]. The BAM files of the corresponding specimens were extracted and indexed for further analysis. The pysamstats module was used to extract the coverage of each position of each specimen across the whole SARS-CoV-2 genome [39]. The coverage of each gene was calculated by taking the average coverage of the bases located in different genes. The nucleotide coordinates of each gene were extracted from the National Center for Biotechnology Information (NCBI). The coverage of each gene was then normalized by the coverage of the nsp1 gene. Then, the normalized coverage of each gene is exported as a CSV file. The bar chart is created using PRISM v10.0.2.
Primers and probe targeting the nsp10 gene region of SARS-CoV-2 for in-house developed RT-PCR assay were designed and tested. Multiple sequence alignment was performed by ClustalX version 2.1 program using our primer/probe sequences and the nsp10 gene sequences of SARS-CoV-2 variants from different countries or geographical regions [40]. The SARS-CoV-2 ancestral strain sequence (GenBank accession number: NC_045512.2) and the sequences of other SARS-CoV-2 variants were retrieved from the Global Initiative on Sharing All Influenza Data (GISAID) EpiCoV database or the NCBI GenBank database.
4.4. Nucleic acid extraction
MagNA Pure 96 extraction system (Roche, Basel, Switzerland) was used for total nucleic acid extraction according to manufacturer’s instructions as we described previously [24]. In brief, 200 μL of each sample was mixed with 250 μL of MagNA Pure External Lysis Buffer (Roche), which contains a chaotropic salt and a detergent to lyse cells and release nucleic acid. The lysed sample was then loaded onto the MagNA Pure 96 instrument, which automatically performs the extraction protocol, including binding, washing, and elution steps. The extracted TNA was then recovered in 50 μL of elution buffer and stored at -80 °C until required for further use.
4.5. Real-time RT-PCR assays for SARS-CoV-2 detection
The in-house developed COVID-19-nsp10 real-time RT-PCR assay for SARS-CoV-2 RNA detection was performed using the QuantiNova Probe RT-PCR Kit (QIAGEN, Hilden, Germany). Each 20 μL reaction mixture contained 10 μL of 2× QuantiNova Probe RT-PCR Master Mix, 1.2 μL of nuclease-free water, 0.2 μL of QN Probe RT-Mix, 1.6 μL of each 10 μM forward and reverse primer, 0.4 μL of 10 μM probe (Table 1), and 5 μL of TNA. RT-PCR was performed using LightCycler 480 II Real-Time PCR System (Roche). The thermal cycling condition consisted of 45°C for 10 min and 95°C for 5 min, followed by 45 cycles of 95°C for 5 sec and 55°C for 30 sec. Positive and negative controls were included in all runs to monitor assay performance.
The commercial LightMix E-gene kit for SARS-CoV-2 detection (TIB Molbiol, Berlin, Germany), along with LightCycler Multiplex RNA Virus Master (Roche), was used as the reference assay in this study. Briefly, each 20 μL reaction mixture contained 5.4 μL of nuclease-free water, 4 μL of Roche RT-qPCR Reaction Mix, 0.5 μL of TIB Molbiol primer/probe reagent mix, 0.1 μL of Roche RT Enzyme Solution, and 10 μL of TNA. The E-gene RT-PCR was performed using LightCycler 480 II real-time PCR system (Roche). The thermal cycling condition consisted of 55 °C for 3 min and 95°C for 30 sec, followed by 45 cycles of 95 °C for 3 sec and 60°C for 12 sec [24].
4.6. Statistical analysis
The kappa statistic was used to determine the agreement between the in-house developed RT-PCR assay and the reference RT-PCR assay. McNemar’s test was used to compare the performance of the in-house RT-PCR assay with the reference RT-PCR assay. Spearman’s correlation analysis was used to assess the relationship between the Cp values of the two real-time RT-PCR assays performed in this study. The Cp values obtained from the two real-time RT-PCR assays were compared using Wilcoxon signed-rank test. P < 0.05 was considered statistically significant. All statistical analyses were performed using the software GraphPad Prism 10 or IBM SPSS Statistics 27.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Acknowledgement of the originating and submitting laboratories who contributed sequences to GISAID.
Author Contributions
Conceptualization, C.C.-Y.Y., K.-Y.Y. and K.K.-W.T.; methodology, C.C.-Y.Y. and K.K.-W.T.; validation, C.C.-Y.Y., J.H.-C.P. and K.-H.L.; formal analysis, C.C.-Y.Y. and K.K.-W.T.; investigation, C.C.-Y.Y., J.H.-C.P., K.-H.L., W.-M.C., J.D.I. and A.W.-H.C.; resources, V.C.-C.C.; writing—original draft preparation, C.C.-Y.Y., W.-M.C., J.D.I. and K.K.-W.T.; writing—review and editing, V.C.-C.C. and K.-Y.Y.; supervision, K.-Y.Y. and K.K.-W.T.; funding acquisition, K.-Y.Y. and K.K.-W.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Health and Medical Research Fund (HMRF) Commissioned Research on Control of Infectious Disease (Phase IV), CID-HKU1-2. The funding source had no role in the design and conduct of the study, in the collection, analysis and interpretation of data, or in the preparation, review or approval of the manuscript.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HA HKW IRB) (UW 22-764). Written informed consent was waived due to using archived specimens.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy/ethical restriction.
Acknowledgments
We gratefully acknowledge the originating and submitting laboratories who contributed sequences to GISAID (Table S1).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Al-Sadeq, D.W.; Nasrallah, G.K. The incidence of the novel coronavirus SARS-CoV-2 among asymptomatic patients: A systematic review. Int. J. Infect. Dis. 2020, 98, 372–380. [Google Scholar] [CrossRef]
- Miyamae, Y.; Hayashi, T.; Yonezawa, H.; Fujihara, J.; Matsumoto, Y.; Ito, T.; Tsubota, T.; Ishii, K. Duration of viral shedding in asymptomatic or mild cases of novel coronavirus disease 2019 (COVID-19) from a cruise ship: A single-hospital experience in Tokyo, Japan. Int. J. Infect. Dis. 2020, 97, 293–295. [Google Scholar] [CrossRef]
- Wei, M.; Yuan, J.; Liu, Y.; Fu, T.; Yu, X.; Zhang, Z.J. Novel Coronavirus Infection in Hospitalized Infants Under 1 Year of Age in China. JAMA. 2020, 323, 1313–1314. [Google Scholar] [CrossRef]
- Tshokey, T.; Choden, J.; Dorjee, K.; Pempa, P.; Yangzom, P.; Gyeltshen, W.; Wangchuk, S.; Dorji, T.; Wangmo, D. Limited Secondary Transmission of the Novel Coronavirus (SARS-CoV-2) by Asymptomatic and Mild COVID-19 Patients in Bhutan. Am. J. Trop. Med. Hyg. 2020, 104, 490–495. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, B.; Mao, S.; Ye, Q. Assessment of global asymptomatic SARS-CoV-2 infection and management practices from China. Int. J. Biol. Sci. 2021, 17, 1119–1124. [Google Scholar] [CrossRef]
- Jefferson, T.; Spencer, E.A.; Brassey, J.; Onakpoya, I.J.; Rosca, E.C.; Plüddemann, A.; Evans, D.H.; Conly, J.M.; Heneghan, C.J. Transmission of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) from pre and asymptomatic infected individuals: a systematic review. Clin. Microbiol. Infect. 2022, 28, 178–189. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Phan, L.T.; Nguyen, T.V.; Luong, Q.C.; Nguyen, T.V.; Nguyen, H.T.; Le, H.Q.; Nguyen, T.T.; Cao, T.M.; Pham, Q.D. Importation and Human-to-Human Transmission of a Novel Coronavirus in Vietnam. N. Engl. J. Med. 2020, 382, 872–874. [Google Scholar] [CrossRef]
- Riou, J.; Althaus, C.L. Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus (2019-nCoV), December 2019 to January 2020. Euro. Surveill. 2020, 25, 2000058. [Google Scholar] [CrossRef]
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science. 2020, 368, 489–493. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature. 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Coronavirus diseases (COVID-19) pandemic by World Health Organization. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 31 January 2024).
- Elena, S.F.; Sanjuán, R. Adaptive value of high mutation rates of RNA viruses: separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Euro. Surveill. 2020, 25, 2001650. [Google Scholar] [CrossRef]
- Ko, K.K.K.; Abdul Rahman, N.B.; Tan, S.Y.L.; Chan, K.X.L.; Goh, S.S.; Sim, J.H.C.; Lim, K.L.; Tan, W.L.; Chan, K.S.; Oon, L.L.E.; et al. SARS-CoV-2 N Gene G29195T Point Mutation May Affect Diagnostic Reverse Transcription-PCR Detection. Microbiol Spectr. 2022, 10, e0222321. [Google Scholar] [CrossRef]
- Wang, H.; Jean, S.; Wilson, S.A.; Lucyshyn, J.M.; McGrath, S.; Wilson, R.K.; Magrini, V.; Leber, A.L. A deletion in the N gene of SARS-CoV-2 may reduce test sensitivity for detection of SARS-CoV-2. Diagn. Microbiol. Infect. Dis. 2022, 102, 115631. [Google Scholar] [CrossRef] [PubMed]
- Mentes, A.; Papp, K.; Visontai, D.; Stéger, J.; VEO Technical Working Group; Csabai, I.; Medgyes-Horváth, A.; Pipek, O.A. Identification of mutations in SARS-CoV-2 PCR primer regions. Sci Rep. 2022, 12, 18651. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.C.; Chan, W.M.; Ip, J.D.; Seng, C.W.; Leung, K.H.; Poon, R.W.; Ng, A.C.; Wu, W.L.; Zhao, H.; Chan, K.H.; et al. Nanopore Sequencing Reveals Novel Targets for Detection and Surveillance of Human and Avian Influenza A Viruses. J. Clin. Microbiol. 2020, 58, e02127-19. [Google Scholar] [CrossRef]
- Chan, W.M.; Ip, J.D.; Chu, A.W.; Yip, C.C.; Lo, L.S.; Chan, K.H.; Ng, A.C.; Poon, R.W.; To, W.K.; Tsang, O.T.; et al. Identification of nsp1 gene as the target of SARS-CoV-2 real-time RT-PCR using nanopore whole-genome sequencing. J. Med. Virol. 2020, 92, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.C.; Sridhar, S.; Chan, W.M.; Ip, J.D.; Chu, A.W.; Leung, K.H.; Cheng, V.C.; Yuen, K.Y.; To, K.K. Development and Validation of a Novel COVID-19 nsp8 One-Tube RT-LAMP-CRISPR Assay for SARS-CoV-2 Diagnosis. Microbiol. Spectr. 2022, 10, e0196222. [Google Scholar] [CrossRef]
- Abbasian, M.H.; Mahmanzar, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Moradi, B.; Sisakht, M.M.; Deng, Y. Global landscape of SARS-CoV-2 mutations and conserved regions. J. Transl. Med. 2023, 21, 152. [Google Scholar] [CrossRef]
- Bui, N.N.; Lin, Y.T.; Huang, S.H.; Lin, C.W. The extent of molecular variation in novel SARS-CoV-2 after the six-month global spread. Infect. Genet. Evol. 2021, 91, 104800. [Google Scholar] [CrossRef]
- Flores-Vega, V.R.; Monroy-Molina, J.V.; Jiménez-Hernández, L.E.; Torres, A.G.; Santos-Preciado, J.I.; Rosales-Reyes, R. SARS-CoV-2: Evolution and Emergence of New Viral Variants. Viruses. 2022, 14, 653. [Google Scholar] [CrossRef] [PubMed]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science. 2022, 376, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Jeworowski, L.M.; Mühlemann, B.; Walper, F.; Schmidt, M.L.; Jansen, J.; Krumbholz, A.; Simon-Lorière, E.; Jones, T.C.; Corman, V.M.; Drosten, C. Humoral immune escape by current SARS-CoV-2 variants BA.2.86 and JN.1, December 2023. Euro. Surveill. 2024, 29, 2300740. [Google Scholar] [CrossRef]
- Initial Risk Evaluation of JN.1, 19 December 2023 by World Health Organization. Available online: https://www.who.int/docs/default-source/coronaviruse/18122023_jn.1_ire_clean.pdf?sfvrsn=6103754a_3 (accessed on 31 January 2024).
- Miller, S.; Lee, T.; Merritt, A.; Pryce, T.; Levy, A.; Speers, D. Single-Point Mutations in the N Gene of SARS-CoV-2 Adversely Impact Detection by a Commercial Dual Target Diagnostic Assay. Microbiol. Spectr. 2021, 9, e0149421. [Google Scholar] [CrossRef]
- Wollschläger, P.; Todt, D.; Gerlitz, N.; Pfaender, S.; Bollinger, T.; Sing, A.; Dangel, A.; Ackermann, N.; Korn, K.; Ensser, A.; et al. SARS-CoV-2 N gene dropout and N gene Ct value shift as indicator for the presence of B.1.1.7 lineage in a commercial multiplex PCR assay. Clin. Microbiol. Infect. 2021, 27, 1353.e1–1353.e5. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell. Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Wang, H.; Rizvi, S.R.A.; Dong, D.; Lou, J.; Wang, Q.; Sopipong, W.; Su, Y.; Najar, F.; Agarwal, P.K.; Kozielski, F.; et al. Emerging variants of SARS-CoV-2 NSP10 highlight strong functional conservation of its binding to two non-structural proteins, NSP14 and NSP16. Elife. 2023, 12, RP87884. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Yip, C.C.; To, K.K.; Tang, T.H.; Wong, S.C.; Leung, K.H.; Fung, A.Y.; Ng, A.C.; Zou, Z.; Tsoi, H.W.; et al. Improved Molecular Diagnosis of COVID-19 by the Novel, Highly Sensitive and Specific COVID-19-RdRp/Hel Real-Time Reverse Transcription-PCR Assay Validated In Vitro and with Clinical Specimens. J. Clin. Microbiol. 2020, 58, e00310-20. [Google Scholar] [CrossRef]
- To, K.K.; Chan, W.M.; Ip, J.D.; Chu, A.W.; Tam, A.R.; Liu, R.; Wu, A.K.; Lung, K.C.; Tsang, O.T.; Lau, D.P.; et al. Unique Clusters of Severe Acute Respiratory Syndrome Coronavirus 2 Causing a Large Coronavirus Disease 2019 Outbreak in Hong Kong. Clin. Infect. Dis. 2021, 73, 137–142. [Google Scholar] [CrossRef] [PubMed]
- nanoporetech/medaka. Available online: https://github.com/nanoporetech/medaka (accessed on 31 January 2024).
- alimanfoo/pysamstats. Available online: https://github.com/alimanfoo/pysamstats (accessed on 31 January 2024).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
Figure 1.
The normalized coverages of different genes in the SARS-CoV-2 genome from 49 whole genome sequenced clinical specimens. The coverage of each gene was calculated by averaging the number of reads mapped to each position of the same gene. The coverage of the NSP1 gene was used to normalize the coverages of the other genes and the desired region for comparison. The Y-axis represents the normalized coverage and the X-axis shows the genes.
Figure 1.
The normalized coverages of different genes in the SARS-CoV-2 genome from 49 whole genome sequenced clinical specimens. The coverage of each gene was calculated by averaging the number of reads mapped to each position of the same gene. The coverage of the NSP1 gene was used to normalize the coverages of the other genes and the desired region for comparison. The Y-axis represents the normalized coverage and the X-axis shows the genes.
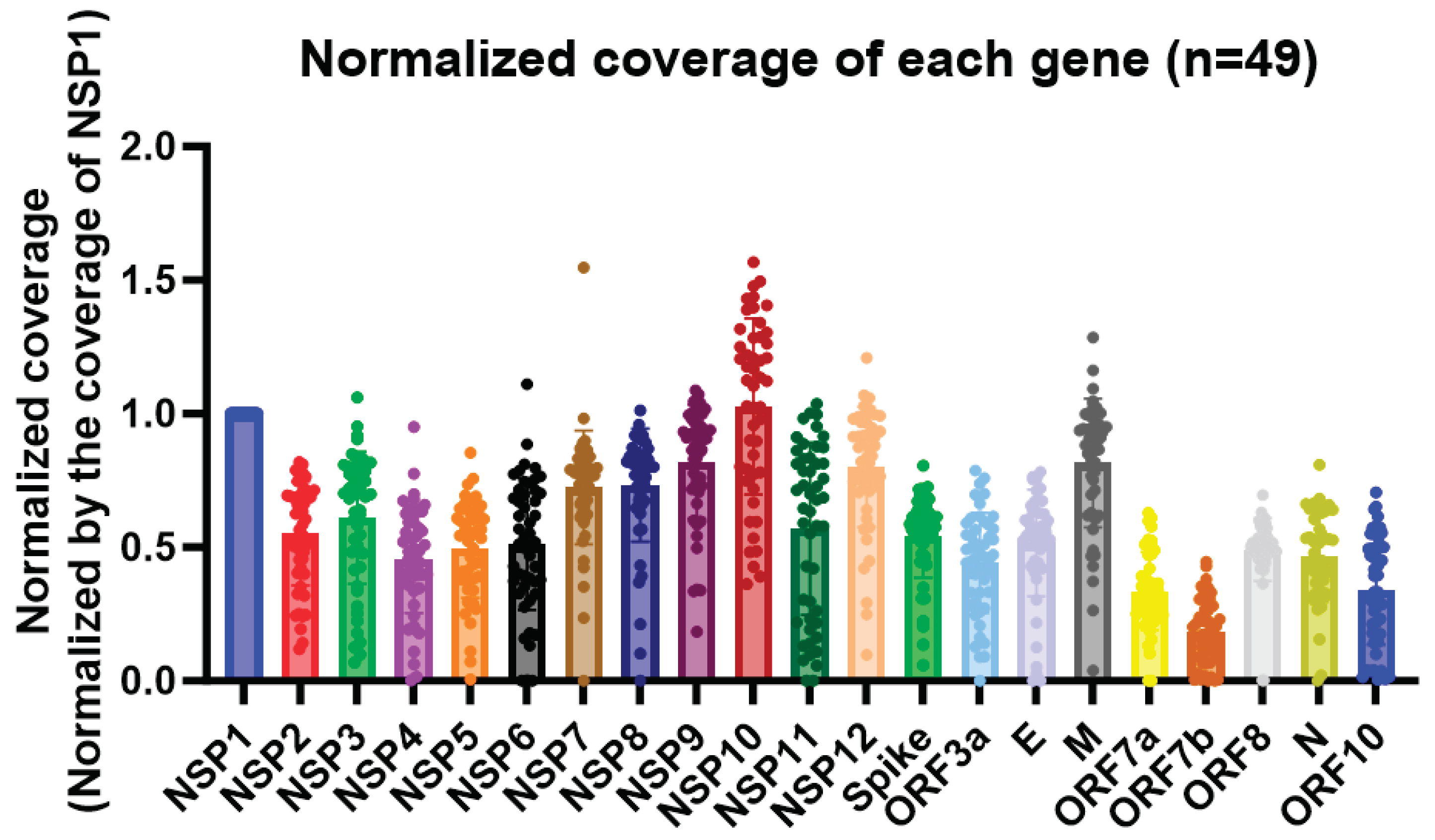
Figure 2.
Multiple sequence alignment using our primer and probe sequences and the nsp10 gene sequences of SARS-CoV-2 variants from different geographical regions.
Figure 2.
Multiple sequence alignment using our primer and probe sequences and the nsp10 gene sequences of SARS-CoV-2 variants from different geographical regions.

Figure 3.
Correlation of the Cp values of the samples found positive for SARS-CoV-2 by the in-house COVID-19-nsp10 assay and the LightMix E-gene assay.
Figure 3.
Correlation of the Cp values of the samples found positive for SARS-CoV-2 by the in-house COVID-19-nsp10 assay and the LightMix E-gene assay.
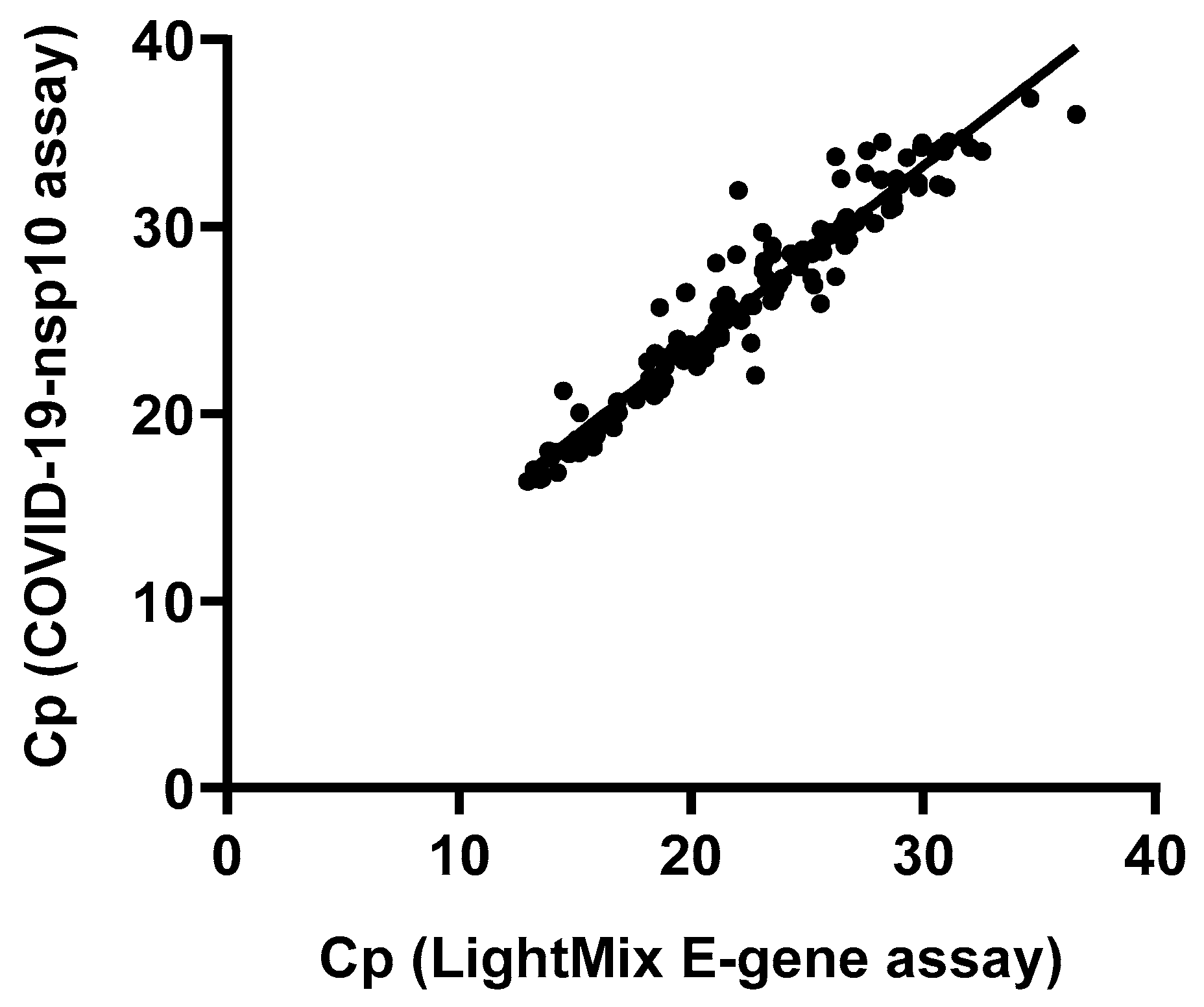
Figure 4.
Comparison of the Cp values of the COVID-19-nsp10 and LightMix E-gene assays. **** indicates P < 0.0001.
Figure 4.
Comparison of the Cp values of the COVID-19-nsp10 and LightMix E-gene assays. **** indicates P < 0.0001.
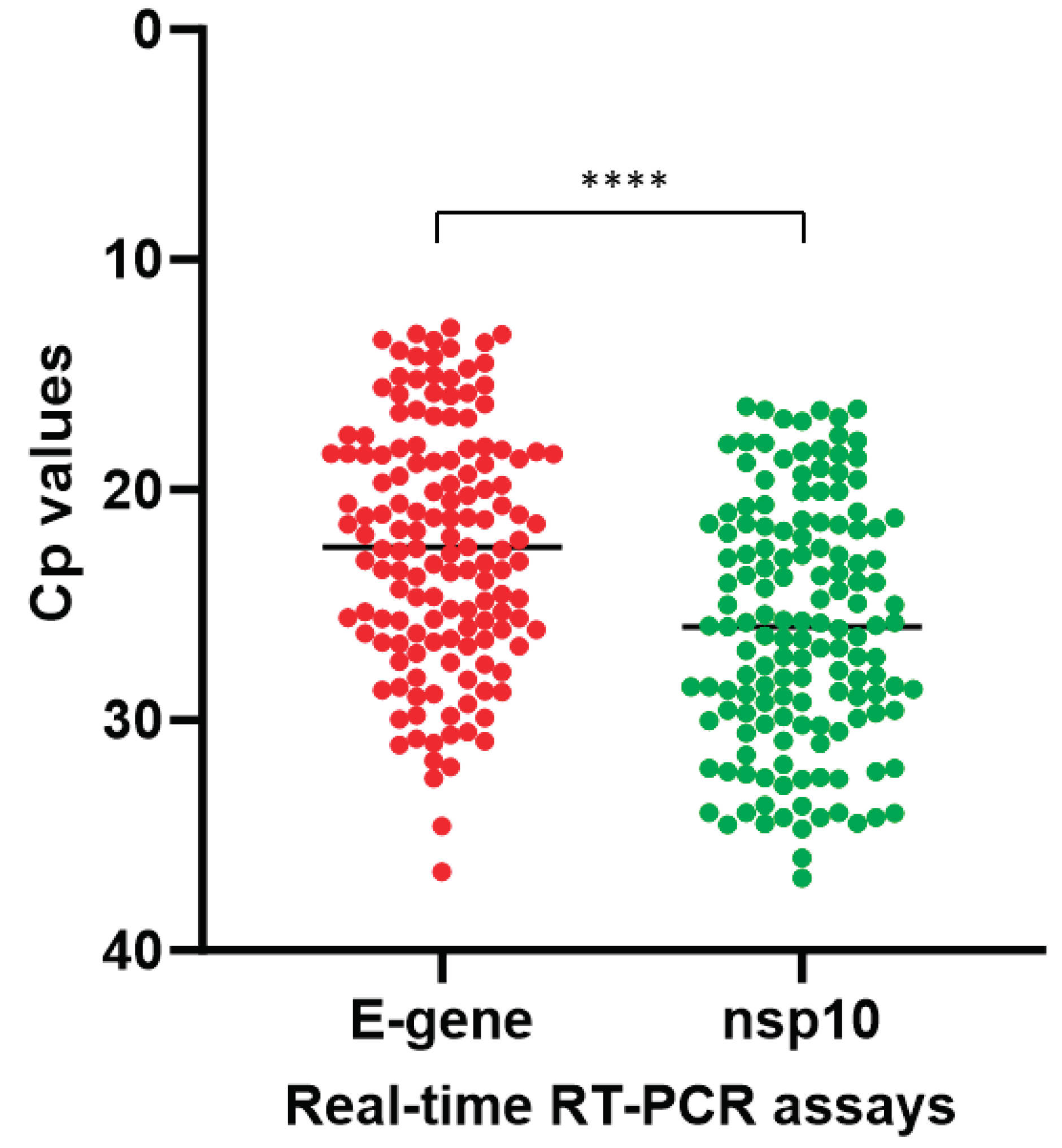

Table 1.
Primer and probe used in this study.
| Primer/probe 1 | Sequence (5’- 3’) | Position 2 |
|---|---|---|
| nCoV_nsp10-F | TGGTGCATCGTGTTGTCTGT | 13231-13250 |
| nCoV_nsp10-R | CCACAGGGTCATTAGCACAA | 13330-13349 |
| nCoV_nsp10-probe | FAM- CTGCCGTTGCCACATAGATCAT –IABkFQ | 13252-13273 |
1 F, forward primer; R, reverse primer. 2 Primer and probe position corresponding to the genome sequence of SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank: NC_045512.2).
Table 2.
Test results for determining the limit of detection of the in-house developed COVID-19-nsp10 RT-PCR assay with TNA extracted from a Qnostics SARS-CoV-2 Q Control 01.
Table 2.
Test results for determining the limit of detection of the in-house developed COVID-19-nsp10 RT-PCR assay with TNA extracted from a Qnostics SARS-CoV-2 Q Control 01.
| Concentration (copies/mL) | Cp (Intra-run) | Cp (Inter-run) | ||||||
|---|---|---|---|---|---|---|---|---|
| Test 1 | Test 2 | Test 3 | Test 4 | Test 1 | Test 2 | Test 3 | Test 4 | |
| 500 | 36.31 | 35.62 | 36.49 | 36.25 | 35.92 | 35.35 | 35.86 | 35.25 |
| 250 | 37.09 | 36.65 | 37.02 | 37.01 | 36.85 | 35.68 | 36.07 | 36.59 |
| 125 | 38.07 | 38.73 | 37.78 | 36.94 | 37.17 | 37.14 | 36.76 | 37.17 |
| 62.5 | 37.23 | 38.66 | 38.13 | - | 37.92 | 37.2 | 36.71 | 39.12 |
| 31.3 | - | 40 | - | 40 | 38.48 | 38.39 | - | 37.68 |
| 15.6 | - | - | - | - | 38.56 | - | - | - |
| NTC | - | - | - | - | - | - | - | - |
Cp, crossing point; NTC, no template control; -, not detected.
Table 3.
Diagnostic performance of the in-house developed COVID-19-nsp10 assay compared to the LightMix E-gene RT-PCR assay for SARS-CoV-2 detection using clinical specimens.
Table 3.
Diagnostic performance of the in-house developed COVID-19-nsp10 assay compared to the LightMix E-gene RT-PCR assay for SARS-CoV-2 detection using clinical specimens.
| Molecular assay | LightMix E-gene RT-PCR | ||||
|---|---|---|---|---|---|
| Positive | Negative | Kappa value (95% CI)1 | McNemar’s test | ||
| COVID-19-nsp10 real-time RT-PCR | |||||
| Positive | 147 | 0 | 1.00 (1.00-1.00) | P=1.000 | |
| Negative | 0 | 114 | |||
1 CI, confident interval.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



