
Submitted:
19 February 2024
Posted:
20 February 2024
You are already at the latest version
Abstract
This review investigates the multifaceted role of the p66Shc adaptor protein and the gut microbiota in regulating mitochondrial function and oxidative stress, and their collective impact on the pathogenesis of chronic diseases. The study delves into molecular mechanisms by which p66Shc influences cellular stress responses through Rac1 activation, Forkhead-type transcription factors inactivation, and mitochondria-mediated apoptosis, alongside modulatory effects of gut microbiota-derived metabolites and endotoxins. Employing an integrative approach, the review synthesizes findings from a broad array of studies, including molecular biology techniques and analyses of microbial metabolites' impacts on host cellular pathways. The results underscore a complex interplay between microbial metabolites, p66Shc activation, and mitochondrial dysfunction, highlighting the significance of the gut microbiome in influencing disease outcomes through oxidative stress pathways. Conclusively, the review posits that targeting the gut microbiota-p66Shc-mitochondrial axis could offer novel therapeutic strategies for mitigating the development and progression of metabolic diseases. This underscores the potential of dietary interventions and microbiota modulation in managing oxidative stress and inflammation, pivotal factors in chronic disease etiology.
Keywords:
p66Shc
; adaptor protein
; oxidative stress
; gut microbiota
; inflammation
; mitochondrial dysfunction
1. Introduction
Mitochondria are membrane-bound organelles responsible for generating most of the chemical energy (in the form of adenosine triphosphate [ATP]) needed to support normal cellular function. Besides their familiar “powerhouse” trait, mitochondria are also critically involved in redox signaling and calcium homeostasis. Every human cell–except mature erythrocytes–contains mitochondria, with the proportion varying according to cell-specific metabolic demands. It is unsurprising, then, that mitochondrial dysfunction is implicated in the etiology of several diseases, including highly prevalent noncommunicable diseases such as type 2 diabetes and cardiovascular diseases [1,2].
The adaptor protein p66Shc is a redox sensor and oxidoreductase that plays a role in apoptosis and reactive oxygen species (ROS) production [3]. In particular, its localization in the mitochondria, which is increased during oxidative stress conditions, is a major driver of mitochondrial dysfunction. Since the recognition of p66Shc as a “lifespan regulator” [4], many studies have delved into elucidating its mechanistic pathways and cellular interactions. However, to date, there has been only one attempt to integrate the influence of the gut microbiota on p66Shc activation [5]. The understanding of how the gut microbiota and its metabolites are integrated into human health has inaugurated a more integrative approach to physiology [6,7,8]. Importantly, microbial metabolites are involved in redox signaling and oxidative stress response [9,10], which, in turn, influences p66Shc activation. Therefore, this review aims to describe and explore oxidative stress as the link between gut microbiota, p66Shc, and mitochondrial dysfunction and related diseases. Understanding this link can open new strategies to utilize non-pharmacological interventions (for instance, diet modification) to modulate p66Shc activity and decrease mitochondrial dysfunction.
2. Overview of the ShcA Protein Family
The Shc family comprises adaptor proteins that are critically involved in cellular signaling. In mammals, the ShcA protein sub-family is encoded by a single gene locus, ShcA. Using alternative splicing and different start codons, this locus encodes for three protein isoforms–p46Shc, p52Shc, and p66Shc. These proteins contain the same three functional domains: a phosphotyrosine-binding (PTB), a central collagen homology (CH1), and a C-terminal Src-homology (SH2) domain. The shortest isoform, p46Shc, has the PTB domain as N-terminal, whereas longer isoforms contain additional cytochrome C-binding domains. In addition, the longest isoform, p66Shc, has a second N-terminal collagen homology (CH2) domain [11,12] (Figure 1).
The functional domains of ShcA proteins undergo different post-translational modifications that modulate their activity. In p52Shc and p46Shc, tyrosine residues in the CH1 domain are phosphorylated in response to receptor tyrosine kinase activation. This event starts a signaling cascade resulting in the promotion of cellular survival, migration, and proliferation. Briefly, phosphorylated p52Shc or p46Shc recruited to an activated receptor tyrosine kinase binds to another adaptor protein, Grb2, which is constitutively associated with Sos1, a guanine nucleotide exchange factor for Ras. Recruitment of the Grb2-Sos1 complex to the plasma membrane induces Ras activation, which subsequently stimulates signaling via the mitogen-activated protein kinase (MAPK) pathway [12,13].
Although p66Shc also binds to activated receptor tyrosine kinases, this interaction does not induce the MAPK signaling cascade. Instead, p66Shc activation induces ROS production and apoptotic pathways (discussed in further detail in the following section) [13]. P66Shc is also antagonistic to p46/p52Shc proteins. ShcA proteins are encoded by a common gene locus; thus, an increase in p66Shc expression begets a decrease in p46/p52Shc expression. Additionally, post-translational modifications in the extra CH2 domain of p66Shc are associated with increased cellular ROS levels [14,15,16], setting it firmly apart from the other ShcA proteins (Figure 1). Particularly, phosphorylation of Ser36 in response to oxidative stress signals increases p66Shc translocation into mitochondria, an early event leading to excessive mitochondrial ROS production and cell death [14]. Thus, p66Shc plays a crucial role in mitochondrial dysfunction and related pathologies (Table 1), with several reviews exploring the connections between p66Shc and specific diseases [17,18,19,20].
3. P66Shc and Oxidative Stress
As abovementioned, p66Shc is associated with ROS production and apoptosis. This protein achieves these effects via three distinct mechanisms that are illustrated in Figure 2 and discussed below. However, before delving into these mechanisms, it is important to highlight that p66Shc is a redox sensor and its associated cellular outcomes are dependent on the local environment. The mechanisms discussed below are relevant when sustained and heightened (i.e., pathological) stress levels are present. Under normal physiological conditions, p66Shc responds to transient stress signals in a pro-survival and -proliferation manner [18,39,40].
3.1. Rac1 Activation
Under oxidative stress conditions, p66Shc is phosphorylated at the Ser36 residue in the CH2 domain by kinases such as protein kinase C-β (PKC-β) and c-Jun N-terminal kinase (JNK) [41]. This activated p66Shc competes with p52/p46Shc for binding to Grb2, causing its disassociation from Sos1. Sos1 is then free to associate with Eps8 and E3b1, forming a complex that activates Rac1 instead of Ras [42]. In this way, p52/p46Shc function is effectively inhibited and Ras-mediated activation of MAPK pathway is disrupted. Further, active Rac1 increases p66Shc stability and activity while decreasing proteasomal degradation [43]. Finally, active Rac1 also promotes the activation of membrane-bound NADPH oxidase, thereby increasing intracellular ROS levels [44].
3.2. Forkhead-Type Transcription Factors Inactivation
Forkhead-type transcription factors, particularly those in the O subgroup (FoxO), are involved in cellular stress response by modulating the expression of antioxidant enzymes such as catalase and superoxide dismutase [45,46]. FoxO proteins are negatively regulated by serine/threonine protein kinase Akt. When activated, Akt phosphorylates FoxO proteins, which causes them to be exported out of the nucleus, precluding their transcriptional activity [46]. Akt is activated by various stimuli such as insulin and growth factors. Under oxidative stress stimulus, activated p66Shc mediates Akt activation and subsequent FoxO sequestration from the nucleus [47,48]. P66Shc may also inactivate FoxO proteins in an Akt-independent manner via complexation with βPix [49,50]. In this way, p66Shc decreases the availability of endogenous antioxidants.
3.3. Mitochondria-Mediated Apoptosis
Under normal physiological conditions, p66Shc is distributed in the cytoplasm (32%), endoplasmic reticulum (24%), and mitochondria (44%) [51]. P66Shc in the cytoplasm remains inactive until stress signals induce p66Shc activation and increase trafficking into the mitochondria. Briefly, prolyl isomerase 1 (Pin1) interacts with active p66Shc, mediating a cis-trans isomerization. This change in conformation allows protein phosphatase 2A (PP2A) to interact with p66Shc and dephosphorylate the Ser36 residue. These consecutive changes facilitated by Pin1 and PP2A are necessary for p66Shc to interact with the outer membrane translocase and reach the intermembrane space [14]. In addition, stress signals also mediate the translocation of the small fraction of p66Shc normally present in the matrix into the intermembrane space via interaction with the intermembrane translocase [51,52]. This excessive p66Shc translocation into the mitochondrial intermembrane space increases local ROS levels and starts a cascade of signals leading to apoptosis.
One of the ways p66Shc increases mitochondrial ROS levels is through its oxidoreductase activity, thereby being a source of superoxide anion [39]. This activity is mediated by intramolecular interactions between cysteine residues in the CH2 and PTB domains as well as a key tyrosine residue (Tyr10) in the CH2 domain [39]. However, the full mechanism of how p66Shc generates superoxide anion remains unknown. P66Shc also interacts with cytochrome C peroxidase to increase mitochondrial ROS levels. Cytochrome C peroxidase is part of the antioxidant machinery responsible for converting ROS into harmless products such as water. Although cytochrome C peroxidase activity is increased in response to escalating ROS levels, p66Shc inhibits its activity, thereby further blunting antioxidant defenses (see FoxO inactivation above). Prolonged oxidative stress causes cytochrome C disassociation, electron transport chain disruption, and permeability transition pore formation. Disassociated cytochrome C escapes into the cytoplasm through this pore, thus starting a caspase signaling cascade that leads to cell death [18].
4. Gut Microbiota and Oxidative Stress
The gut microbiota is a complex ecosystem of trillions of microorganisms including bacteria, viruses, yeast, protozoa, and fungi. These microorganisms reside in the gastrointestinal tract, primarily within the colon, and play an indispensable role in human physiology. The gut microbiota contributes to various processes such as digestion, immune response training and homeostasis, bioactive compound biosynthesis, toxin elimination, cell proliferation, among others [53,54]. Gut microbes influence their local environment as well as distal tissues and cells via microbial patterns (e.g., toll-like receptor [TLR] ligands) and a variety of metabolites. These microbial metabolites can be derived from dietary components (e.g., short-chain fatty acids [SCFA], tryptophan catabolites, trimethylamine-L-oxide [TMAO]) or from host metabolism (e.g., secondary bile acids), or synthesized de novo by the microbes themselves (e.g., branched-chain amino acids, bacterial vitamins) [8,55]. Thus, the metabolic output of the gut microbiota represents its main mode of communication with the host.
A diverse population of microorganisms primarily consisting of obligate anaerobes such as Firmicutes and Bacteroidetes allows for a health-promoting and functional environment primed for carrying out efficient physiologic processes. Conversely, conditions that promote the proliferation of facultative anaerobes decrease microbial diversity and increase local and systemic inflammation. This alteration in the gut microbiota community ultimately changes their metabolic output toward disease-promoting signals [54,56,57]. In fact, several pathophysiological conditions have been associated with the gut microbiota and its influence on inflammation and oxidative stress (Table 2).
In normal physiology, commensal bacteria stimulate transient ROS production in the gut, which is essential for cell proliferation and motility as well as inflammation and immune response [68,69,70]. They achieve this by shedding microbial patterns such as small formylated peptides that are recognized by pattern recognition receptors like formyl peptide receptors. These receptors increase the activity of NADPH oxidases [71], which generate ROS and activate redox sensor proteins and associated signal transduction pathways. For instance, Ubc12–a Nedd8 ligase involved in NF-κB activation–is inactivated in these conditions [72,73]. This mechanism likely mediates host immune tolerance to the gut microbiota. However, excessive and sustained ROS production can lead to detrimental immune suppression and downregulation of survival pathways.
Commensal bacteria also limit oxidative conditions in the gut. Butyrate-producing bacteria, such as those belonging to the Firmicutes phylum, stimulate the peroxisome proliferation activated receptor-gamma (PPAR-γ) pathway and β-oxidation in intestinal cells [74]. PPAR-γ signaling shifts cellular energy production toward oxidative phosphorylation, thereby stimulating oxygen consumption, and preventing it from translocating into the intestinal lumen. Depletion of SCFA-producing bacteria and subsequent down-regulation of the PPAR-γ pathway favors anaerobic glycolysis for energy production. Consequently, underutilized oxygen reaches the intestinal lumen, conferring a survival and proliferation advantage to facultative anaerobes such as Escherichia, Salmonella, and other genera of the Enterobacteriaceae family [75].
The proliferation of pathogenic members of the Enterobacteriaceae family is a driver of systemic inflammation and oxidative stress. Enterobacteriaceae are Gram-negative bacteria and, as such, they shed endotoxins termed lipopolysaccharides (LPS). LPS are components of Gram-negative bacterial outer membrane comprised of a hydrophobic domain (lipid A), a polysaccharide core, and an oligomeric polysaccharide tail (O-antigen) [76]. In a healthy gut, the intestinal barrier (formed by an intact epithelial cell layer and a mucus layer) will prevent LPS present in the luminal side from translocating into the basal side, where it can interact with pattern recognition receptors and induce inflammatory responses. However, alterations in the bacterial community and high levels of LPS disrupt intestinal barrier integrity, thus resulting in what is often termed a “leaky gut” [77,78]. This allows LPS to escape the intestinal lumen and enter systemic circulation.
While LPS shed by commensal bacteria can be beneficial to host metabolism [79], pathogenic bacteria cast off LPS that are associated with metabolic endotoxemia, a condition characterized by chronic low-grade inflammation. Strain-dependent variations in lipid A moiety dictate the immunologic activity of LPS based on how it interacts with pattern recognition receptors [80,81]. Briefly, LPS-binding proteins bind to circulating LPS and transport it to cluster of differentiation 14 (CD14), a co-receptor of LPS mainly expressed by macrophages and other cells involved in innate immune response. CD14 facilitates the transfer of LPS to the TLR-4–myeloid differentiation protein (MD)-2 complex. MD-2 is the main binding site of LPS; it contains a hydrophobic pocket that interacts with lipid A acyl chains [81]. Once LPS is inserted into the TLR-4–MD-2 complex, differences in acyl chain number and structure govern whether it will have an antagonistic or agonistic effect on TLR-4 [80]. Bacterial strains associated with metabolic endotoxemia produce agonistic LPS, which induce TLR-4 dimerization and subsequent activation. TLR-4 signaling cascades involve myeloid differentiation primary response 88 (MyD88)-dependent and -independent pathways [78,82]. These pathways culminate in the expression of pro-inflammatory mediators (e.g., interleukin [IL]-6, IL-18, TNF) through activation of NF-κB and interferon regulatory factor 3 (IRF-3). This inflammatory response is accompanied by an increase in ROS and oxidative stress [83], creating a forward-feeding mechanism of cellular damage. In this way, persistent metabolic endotoxemia promotes a state of chronic low-grade inflammation and oxidative stress, both of which are features of many pathophysiological conditions such as insulin resistance, type 2 diabetes, and obesity [84,85,86].
5. Oxidative Stress, Gut Microbiota, and p66Shc
The resilience of an organism is tied to its ability to adapt and respond to different stressors both endogenous and exogenous. Rheostasis is a feature of adaptation as the same biochemical processes or components govern different physiological outcomes through continuous regulation. ROS are a classic example of rheostatic activity, governing different cell fates depending on their type, level, and localization [87]. As mentioned in the previous section, transient ROS production in the gut stimulate cellular pathways leading to cell proliferation [70]. Here we contend that another rheostat, p66Shc, may play a role in this process.
Small formylated peptides shed by commensal bacteria and recognized by formyl peptide receptors induce an increase in the activity of NADPH oxidases, thereby increasing local ROS levels [70]. This inactivates certain redox sensitive proteins such as dual specific phosphatase-3 (DUSP3), a phosphatase involved in the regulation of MAPK/ERK proliferative pathways. When active, DUSP3 dephosphorylates MAPKs and downregulates MAPK/ERK pathways. ROS can oxidize cysteine residues in the DUSP3 catalytic site, rendering it inactive [88]. Thus, commensal bacteria can promote epithelial cell proliferation and gut barrier integrity through enzymatic ROS production.
Mitochondrial dynamics are integral to cellular proliferation, with mitochondrial biogenesis and the replication of mitochondrial DNA (mtDNA) being essential for ensuring adequate energy supply and metabolic function in daughter cells [89,90], and the adaptor protein p66Shc has been implicated in this process. Studies by Trinei and colleagues [91] have demonstrated that p66Shc can upregulate mtDNA replication independently of its established roles in reactive oxygen species (ROS) generation and apoptosis. Remarkably, the absence of p66Shc was correlated with a substantial reduction in mtDNA content by 40–50%, suggesting an unanticipated role for p66Shc in mitochondrial maintenance [91]. This discovery aligns with observations made by Blank and collaborators [92], who found that increased mtDNA replication can trigger nuclear DNA replication and cellular proliferation in yeast, indicating a conserved mechanism potentially applicable to mammalian cells [92]. These insights position p66Shc as a potential regulator of mitochondrial distribution and function during cell division, warranting further investigation into its role in mtDNA replication and the consequent phenotypic effects. The implications of p66Shc's involvement in these processes could offer a novel perspective on the regulation of gut epithelial cell proliferation, an area ripe for exploration considering the critical importance of gut homeostasis in health and disease.
On the other side of the rheostatic spectrum, p66Shc may also exacerbate gut microbiota-derived signals that are associated with pathophysiological conditions. Locally, SCFAs contribute to colonic homeostasis by stimulating PPAR-γ signaling, inducing regulatory T cell maturation, and providing fuel for mitochondrial beta-oxidation [74,93]. Specifically, SCFAs bind to G protein-coupled receptor 43 on the surface of colonic T cells, thus inducing the maturation and expansion of regulatory T cells [94], which control inflammatory responses in mucosal tissues such as the colon. Consequently, when levels of SCFAs are insufficient, the colonic environment undergoes a shift in metabolism that favors oxidative and inflammatory conditions. PPAR-γ signaling downregulation increases local oxygen concentration and regulatory T cell depletion induces intestinal inflammation. This combination is a one-two punch to mitochondria as evidenced by a decreased oxygen consumption despite increased local oxygen bioavailability [74]. The oxidative and inflammatory environment described above favor p66Shc’s pro-apoptotic functions; therefore, it is plausible that the electron transport chain disruption observed in these conditions is mediated, at least in part, by p66Shc.
The relationship between the gut microbiota and p66Shc likely goes beyond local effects. As detailed in the previous section, gut microbiota-induced metabolic endotoxemia promotes low-grade chronic inflammation and oxidative stress through activation of NF-κB and IRF-3 pathways mediated by TLR4 activation [78]. This process is associated with the onset of many chronic diseases such as obesity and type 2 diabetes [61,84,85,86,95,96]. As inflammation and oxidative stress are associated with mitochondrial dysfunction, p66Shc has also been implicated in the onset and progression of these diseases (Table 1) [19,97,98,99]. However, as detailed in a recent review by Ciciliot and Fadini [19], the evidence for this association is conflicting.
This seemingly contradictory evidence may be parsed out by considering a missing confounder in these studies: the gut microbiota. To date, only one study has explored the differences between the gut microbiota composition of animals with and without p66Shc ablation [5]; the authors reported that p66Shc knockout altered gut microbiota composition and metabolic output in mice, and that this alteration modulated their phenotypic response to a high-fat diet. However, this study did not utilize a humanized gnotobiotic mouse model, which limits the interpretation of the results in the human context [100,101]. Thus, the complexity of the interaction between the metabolic output of gut microbes and p66Shc remains underexplored.
Herein, we hypothesize that adipose tissue is a major site for the interaction between microbial signals–namely, LPS–and p66Shc. Firstly, LPS receptor TLR4 and p66Shc are both highly expressed in adipocytes [102]. Further, LPS-induced TLR4 activation in macrophages leads to adipose tissue infiltration, ultimately leading to secretion of pro-inflammatory cytokines and other inflammatory signals to surrounding tissues [103]. Secondly, available evidence supports a complementary role of LPS and p66Shc in insulin-dependent signaling pathways regulating adipose tissue metabolism. As discussed previously, LPS-induced TLR4 activation stimulates NF-kB signaling, which leads to an increase in the expression of pro-inflammatory cytokines. TNF-alpha is one of these cytokines, and it plays a major role in the onset of insulin resistance [104]. Briefly, TNF-alpha secreted by stressed adipocytes induces phosphorylation of insulin receptor substrate 1 (IRS-1)–a critical regulator of insulin signaling–in muscle cells. This phosphorylation inactivates IRS-1 and impairs insulin-dependent downstream cascades [105,106]. Consequently, anabolic nutrient-sensing pathways such as those mediated by insulin and mTORC1 are inhibited.
In addition, LPS-induced inflammation can promote p66Shc activation due to the associated increase in oxidative stress. Ranieri and colleagues reported that p66Shc can inactivate IRS-1 in adipose tissue via interaction with insulin effector kinase 1 (S6K) [97,107], thereby contributing to the onset of insulin resistance. These observations were made in the context of high fat-induced obesity, and dietary fatty acids can stimulate TLR4 similarly to LPS [108]. However, because high-fat diets induce changes in gut microbiota that lead to metabolic endotoxemia [109,110], it is likely that dietary fatty acids affect TLR4 signaling via increased circulating LPS. In sum, LPS and p66Shc can work together to initiate insulin resistance and deregulate nutrient-sensing pathways. This deregulation has important implications for cellular adaptive stress responses such as endogenous antioxidant production and autophagy (including mitophagy), which ultimately lead to phenotypic manifestations and the development of chronic diseases.
6. Linking Oxidative Stress, Gut Microbiota, and p66Shc to Pathophysiological Outcomes
The previous sections outlined how the gut microbiota and p66Shc can exacerbate oxidative stress and inflammation. In this section, we will explore the pathophysiological outcomes of these processes and how they can contribute to the onset and progression of chronic diseases. Chronic diseases such as type 2 diabetes and cardiovascular diseases have common risk factors, including hyperglycemia, dyslipidemia, and endothelial dysfunction. These risk factors, in turn, have common etiologies, with the most prominent of them being insulin resistance [111].
Insulin is an essential endocrine hormone involved in glucose homeostasis and anabolic metabolism. Secreted by pancreatic beta cells upon nutrient availability signaling (e.g., exogenous glucose from a meal), insulin increases anabolic pathways while decreasing catabolic pathways. Insulin has systemic effects, with direct and indirect action on important organs and tissues, namely the liver, skeletal muscle, and adipose tissue, among others [112]. Insulin resistance is defined as an impaired response of these targets to insulin stimulation, which leads to hyperglycemia due to decreased glucose utilization and hyperinsulinemia due to compensatory insulin production [111]. These processes start a metabolic derangement that serves as a foundation to many chronic diseases.
Insulin resistance is most commonly initiated by modifiable lifestyle-related risk factors that lead to chronic overnutrition and obesity, such as physical inactivity and poor dietary habits [111]. In particular, diets high in saturated fat and simple carbohydrates (commonly referred to as a Western pattern diet) are most associated with an increased risk of developing insulin resistance and associated diseases [113]. Chronic overnutrition combined with a Western pattern diet induce changes in the microbial metabolic output that favor pro-oxidant and -inflammatory processes and disrupt energy metabolism; the most well-described changes are decreases in SCFAs and secondary bile acids and increases in LPS and branched-chain amino acids [114,115]. In sections 4 and 5, we described how SCFA depletion and LPS-mediated endotoxemia lead to an increase in inflammation via NF-kB signaling and subsequent IRS-1 inactivation, thereby dampening insulin-sensitive metabolism. In white adipose tissues, this dampening of metabolic responses to insulin means that lipogenesis is suppressed while lipolysis continues to be stimulated even in conditions of nutrient abundance, supplying non-adipose tissues with excess nonesterified fatty acids (NEFAs) and impairing lipid storage in adipocytes [116]. Circulating NEFAs are captured by the liver and skeletal muscle, where they can be stored in lipid droplets or utilized as an acetyl-CoA precursor for oxidative phosphorylation in the mitochondria. However, as the supply of NEFAs outpace the demand for ATP, excessive β oxidation leads to ROS overproduction [117,118] and mitochondrial dysfunction [119], thus perpetuating and amplifying stress-mediated IRS inactivation and insulin resistance.
Available evidence indicates that p66Shc participates in the gut–mitochondria axis described above. Studies reported that p66Shc inhibits insulin-dependent anabolic metabolism [107,120,121] via IRS-1 inactivation, indicating that oxidative stress resulting from gut metabolite signaling can activate p66Shc and worsen insulin sensitivity. However, whether p66Shc antagonizes insulin through its effect on mitochondria or through its own enzymatic ROS production remains unclear. Further, in normal physiological conditions, Berniakovich and colleagues [98] reported that p66Shc couples insulin signaling to mitochondrial respiration in adipose tissue, increasing lipogenesis and decreasing fatty acid oxidation in adipocytes. As the p66Shc knockout mice in this study were reported to have less energy storage and increased energy expenditure than their counterparts, it is possible that the apparent longevity conferred to these mice by knocking out p66Shc would not occur in natural life scenarios, indicating an evolutionary adaptation. Interestingly, Ciciliot and colleagues [99] reported that p66Shc deletion had no protective effect on insulin resistance in mice fed a high-fat diet. In a complementary study, Ciciliot and collaborators [5] showed that the gut microbiota of p66Shc-knockout mice had a worse metabolite output than their wild-type counterparts even when fed a standard diet. This observation ties back to the argument described herein on the involvement of p66Shc in gut epithelial cell proliferation under normal physiological conditions (section 5), which is paramount for a health-promoting intestinal environment. Another possibility is that p66Shc deletion by itself cannot protect cells and tissues from the systemic and propagative injuries associated with insulin resistance (see [112] for an exhaustive review on the mechanisms of insulin resistance).
The events outlined in this section are just a part of the gut–mitochondria axis and illustrate how gut metabolites initiate a cascade of highly integrated pathways that lead to mitochondrial dysfunction and pathophysiological states. In the context of chronic diseases, gut and mitochondrial signals seem to converge in the adipose tissue, meriting further investigation. As a major endocrine organ, the pro-oxidant and -inflammatory signals transmitted by dysfunctional adipose tissue are amplified and contribute to the systemic low-grade inflammation that typifies obesity and insulin resistance.
7. Conclusions and Future Directions
This review elucidates p66Shc as a potential mediator for crosstalk between the gut microbiota and mitochondrial function, a relationship integral to cellular response to oxidative stress and pathogenesis of metabolic diseases. Evidence presented highlights the complexity of this interaction, where p66Shc modulates key signaling pathways implicated in both apoptotic initiation and inhibition, as well as regulation of antioxidative defenses. Insights garnered from this synthesis suggest a significant therapeutic potential in targeting the gut microbiota-p66Shc-mitochondrial axis, particularly through non-pharmacological interventions such as dietary modulation. These strategies may offer a promising avenue for mitigating deleterious effects of mitochondrial dysfunction in metabolic diseases. Further studies into molecular mechanisms underpinning microbial metabolite-mediated modulation of p66Shc activity are needed. Such investigations are vital for developing targeted interventions. In parallel, robust clinical studies are needed to establish causal relationships and support the translational potential of preclinical findings. Furthermore, embracing GM diversity across populations and individual genetic variability will be critical for advancing personalized medical approaches. As research progresses, the integration of advanced in vivo models and biotechnological innovations will be crucial for translating these complex biological interactions into tangible therapeutic modalities.
Author Contributions
Conceptualization, FAH; writing—original draft preparation, ACCPL, EM, and FAH.; writing—review and editing, ACCPL and FAH; visualization, ACCPL; supervision, FAH; project administration, ACCPL and FAH; funding acquisition, FAH. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health under the following award: National Institute of General Medical Sciences: R01GM118599 (FAH).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
MAPK - Mitogen-Activated Protein Kinase; PTB - Phosphotyrosine-Binding; CH - Collagen Homology; SH2 - Src Homology 2; PKC-β - Protein Kinase C-beta; JNK - c-Jun N-terminal Kinase; Grb2 - Growth Factor Receptor-Bound Protein 2; Sos1 - Son of Sevenless Homolog 1; Rac1 - Ras-related C3 Botulinum Toxin Substrate 1; FoxO - Forkhead Box O; Akt - Protein Kinase B; βPix - P21-Activated Kinase-Interactive Exchange Factor; PP2A - Protein Phosphatase 2A; TOM - Translocase of the Outer Mitochondrial Membrane; TIM - Translocase of the Inner Mitochondrial Membrane; IMM - Inner Mitochondrial Membrane; PTP - Permeability Transition Pore; Cyt c - Cytochrome c; SCFA - Short-Chain Fatty Acid; TMAO - Trimethylamine N-oxide; LPS - Lipopolysaccharides; TLR - Toll-Like Receptor; PPAR-γ - Peroxisome Proliferator-Activated Receptor Gamma; CD14 - Cluster of Differentiation 14; MD-2 - Myeloid Differentiation Factor 2; MyD88 - Myeloid Differentiation Primary Response 88; IRF-3 - Interferon Regulatory Factor 3; IL - Interleukin; TNF - Tumor Necrosis Factor; DUSP3 - Dual Specificity Phosphatase 3; mtDNA - Mitochondrial DNA; PPAR-γ - Peroxisome Proliferator-Activated Receptor Gamma; IRS-1 - Insulin Receptor Substrate 1; mTORC1 - Mechanistic Target of Rapamycin Complex 1; S6K - S6 Kinase; ESRD - End-Stage Renal Disease; CKD - Chronic Kidney Disease; CTEPH - Chronic Thromboembolic Pulmonary Hypertension; BMI - Body Mass Index; NEFA – non-esterified fatty acids.
References
- Zhunina, O.A.; Yabbarov, N.G.; Grechko, A.V.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Vascular Disease, Tumorigenesis, and Diabetes. Front Mol Biosci 2021, 8, 671908. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr Rev 2020, 41, bnaa005. [Google Scholar] [CrossRef]
- Mir, H.A.; Ali, R.; Mushtaq, U.; Khanday, F.A. Structure-functional implications of longevity protein p66Shc in health and disease. Ageing Res Rev 2020, 63, 101139. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef]
- Ciciliot, S.; Albiero, M.; Campanaro, S.; Poncina, N.; Tedesco, S.; Scattolini, V.; Dalla Costa, F.; Cignarella, A.; Vettore, M.; Di Gangi, I.M.; et al. Interplay between gut microbiota and p66Shc affects obesity-associated insulin resistance. FASEB J 2018, 32, 4004–4015. [Google Scholar] [CrossRef]
- Illiano, P.; Brambilla, R.; Parolini, C. The mutual interplay of gut microbiota, diet and human disease. FEBS J 2020, 287, 833–855. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front Cell Infect Microbiol 2021, 11, 625913. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Towarnicki, S.G. Mitochondria, the gut microbiome and ROS. Cell Signal 2020, 75, 109737. [Google Scholar] [CrossRef]
- Uchiyama, J.; Akiyama, M.; Hase, K.; Kumagai, Y.; Kim, Y.G. Gut microbiota reinforce host antioxidant capacity via the generation of reactive sulfur species. Cell Rep 2022, 38, 110479. [Google Scholar] [CrossRef]
- Galimov, E.R. The Role of p66shc in Oxidative Stress and Apoptosis. Acta Naturae 2010, 2, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef]
- Migliaccio, E.; Mele, S.; Salcini, A.E.; Pelicci, G.; Lai, K.M.; Superti-Furga, G.; Pawson, T.; Di Fiore, P.P.; Lanfrancone, L.; Pelicci, P.G. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J 1997, 16, 706–716. [Google Scholar] [CrossRef]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef]
- Kumar, S.; Kim, Y.R.; Vikram, A.; Naqvi, A.; Li, Q.; Kassan, M.; Kumar, V.; Bachschmid, M.M.; Jacobs, J.S.; Kumar, A.; et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc Natl Acad Sci U S A 2017, 114, 1714–1719. [Google Scholar] [CrossRef]
- Faisal, A.; el-Shemerly, M.; Hess, D.; Nagamine, Y. Serine/threonine phosphorylation of ShcA. Regulation of protein-tyrosine phosphatase-pest binding and involvement in insulin signaling. J Biol Chem 2002, 277, 30144–30152. [Google Scholar] [CrossRef]
- Boengler, K.; Bornbaum, J.; Schluter, K.D.; Schulz, R. P66shc and its role in ischemic cardiovascular diseases. Basic Res Cardiol 2019, 114, 29. [Google Scholar] [CrossRef]
- Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11, 1855. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Fadini, G.P. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66(Shc). Int J Mol Sci 2019, 20, 985. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.; Khazeei Tabari, M.A.; Bagheri, A.; Samieefar, N.; Shaterian, N.; Kelishadi, R. The Role of p66Shc in Diabetes: A Comprehensive Review from Bench to Bedside. J Diabetes Res 2022, 2022, 7703520. [Google Scholar] [CrossRef] [PubMed]
- Hughes, W.E.; Hockenberry, J.; Miller, B.; Sorokin, A.; Beyer, A.M. Modulation of p66Shc impairs cerebrovascular myogenic tone in low renin but not low nitric oxide models of systemic hypertension. Am J Physiol Heart Circ Physiol 2021, 321, H1096–H1102. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Gadi, I.; Nazir, S.; Al-Dabet, M.M.; Kohli, S.; Bock, F.; Breitenstein, L.; Ranjan, S.; Fuchs, T.; Halloul, Z.; et al. Activated protein C reverses epigenetically sustained p66(Shc) expression in plaque-associated macrophages in diabetes. Commun Biol 2018, 1, 104. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Luscher, T.F.; et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66(Shc). Int J Cardiol 2018, 268, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Vashistha, H.; Marrero, L.; Reiss, K.; Cohen, A.J.; Malhotra, A.; Javed, T.; Bradley, A.; Abbruscato, F.; Giusti, S.; Jimenez, A.; et al. Aging phenotype(s) in kidneys of diabetic mice are p66ShcA dependent. Am J Physiol Renal Physiol 2018, 315, F1833–F1842. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Krankel, N.; Cosentino, F.; Luscher, T.F. Reprogramming ageing and longevity genes restores paracrine angiogenic properties of early outgrowth cells. Eur Heart J 2016, 37, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Vono, R.; Fuoco, C.; Testa, S.; Pirro, S.; Maselli, D.; Ferland McCollough, D.; Sangalli, E.; Pintus, G.; Giordo, R.; Finzi, G.; et al. Activation of the Pro-Oxidant PKCbetaII-p66Shc Signaling Pathway Contributes to Pericyte Dysfunction in Skeletal Muscles of Patients With Diabetes With Critical Limb Ischemia. Diabetes 2016, 65, 3691–3704. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.; Montecucco, F.; Braunersreuther, V.; Camici, G.G.; Jakob, P.; Reiner, M.F.; Glanzmann, M.; Burger, F.; Paneni, F.; Galan, K.; et al. Genetic deletion of the adaptor protein p66Shc increases susceptibility to short-term ischaemic myocardial injury via intracellular salvage pathways. Eur Heart J 2015, 36, 516–526a. [Google Scholar] [CrossRef]
- Spescha, R.D.; Klohs, J.; Semerano, A.; Giacalone, G.; Derungs, R.S.; Reiner, M.F.; Rodriguez Gutierrez, D.; Mendez-Carmona, N.; Glanzmann, M.; Savarese, G.; et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur Heart J 2015, 36, 1590–1600. [Google Scholar] [CrossRef]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef]
- Shi, Y.; Savarese, G.; Perrone-Filardi, P.; Luscher, T.F.; Camici, G.G. Enhanced age-dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int J Cardiol 2014, 175, 446–450. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Cosentino, F. p66(Shc)-induced redox changes drive endothelial insulin resistance. Atherosclerosis 2014, 236, 426–429. [Google Scholar] [CrossRef]
- Spescha, R.D.; Glanzmann, M.; Simic, B.; Witassek, F.; Keller, S.; Akhmedov, A.; Tanner, F.C.; Luscher, T.F.; Camici, G.G. Adaptor protein p66(Shc) mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension 2014, 64, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Naqvi, A.; Hoffman, T.A.; Kumar, A.; Miller, F.J., Jr.; Kim, C.S.; Irani, K. Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arterioscler Thromb Vasc Biol 2014, 34, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, G.; Zhai, X.; Hu, Y.; Gao, D.; Ma, L.; Yao, J.; Tian, X. Selective inhibition of protein kinase C beta2 attenuates the adaptor P66 Shc-mediated intestinal ischemia-reperfusion injury. Cell Death Dis 2014, 5, e1164. [Google Scholar] [CrossRef]
- Bellisario, V.; Berry, A.; Capoccia, S.; Raggi, C.; Panetta, P.; Branchi, I.; Piccaro, G.; Giorgio, M.; Pelicci, P.G.; Cirulli, F. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66(Shc-/-) mouse. Front Behav Neurosci 2014, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Spescha, R.D.; Shi, Y.; Wegener, S.; Keller, S.; Weber, B.; Wyss, M.M.; Lauinger, N.; Tabatabai, G.; Paneni, F.; Cosentino, F.; et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur Heart J 2013, 34, 96–103. [Google Scholar] [CrossRef]
- Laviola, L.; Orlando, M.R.; Incalza, M.A.; Caccioppoli, C.; Melchiorre, M.; Leonardini, A.; Cignarelli, A.; Tortosa, F.; Labarbuta, R.; Martemucci, S.; et al. TNFalpha signals via p66(Shc) to induce E-Selectin, promote leukocyte transmigration and enhance permeability in human endothelial cells. PLoS One 2013, 8, e81930. [Google Scholar] [CrossRef]
- Bock, F.; Shahzad, K.; Wang, H.; Stoyanov, S.; Wolter, J.; Dong, W.; Pelicci, P.G.; Kashif, M.; Ranjan, S.; Schmidt, S.; et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A 2013, 110, 648–653. [Google Scholar] [CrossRef]
- Haslem, L.; Hays, J.M.; Schmitz, H.; Matsuzaki, S.; Sjoelund, V.; Byrum, S.D.; Humphries, K.M.; Frazer, J.K.; Demeler, B.; Benbrook, D.M.; et al. p66Shc is an apoptotic rheostat whose targeted ROS inhibition improves MI outcomes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bhat, S.S.; Anand, D.; Khanday, F.A. p66Shc as a switch in bringing about contrasting responses in cell growth: implications on cell proliferation and apoptosis. Mol Cancer 2015, 14, 76. [Google Scholar] [CrossRef]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Luscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler Thromb Vasc Biol 2011, 31, 2090–2097. [Google Scholar] [CrossRef]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J Cell Biol 2006, 172, 817–822. [Google Scholar] [CrossRef]
- Khanday, F.A.; Yamamori, T.; Mattagajasingh, I.; Zhang, Z.; Bugayenko, A.; Naqvi, A.; Santhanam, L.; Nabi, N.; Kasuno, K.; Day, B.W.; et al. Rac1 leads to phosphorylation-dependent increase in stability of the p66shc adaptor protein: role in Rac1-induced oxidative stress. Mol Biol Cell 2006, 17, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr Biol 2007, 17, R113–R114. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Izumiya, Y.; Walsh, K. Forkhead transcription factors and cardiovascular biology. Circ Res 2008, 102, 16–31. [Google Scholar] [CrossRef]
- Guo, J.; Gertsberg, Z.; Ozgen, N.; Steinberg, S.F. p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circ Res 2009, 104, 660–669. [Google Scholar] [CrossRef]
- Nemoto, S.; Finkel, T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002, 295, 2450–2452. [Google Scholar] [CrossRef]
- Chahdi, A.; Sorokin, A. Endothelin-1 couples betaPix to p66Shc: role of betaPix in cell proliferation through FOXO3a phosphorylation and p27kip1 down-regulation independently of Akt. Mol Biol Cell 2008, 19, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Chahdi, A.; Sorokin, A. Endothelin-1 induces p66Shc activation through EGF receptor transactivation: Role of beta(1)Pix/Galpha(i3) interaction. Cell Signal 2010, 22, 325–329. [Google Scholar] [CrossRef]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem 2004, 279, 25689–25695. [Google Scholar] [CrossRef]
- Orsini, F.; Moroni, M.; Contursi, C.; Yano, M.; Pelicci, P.; Giorgio, M.; Migliaccio, E. Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol Chem 2006, 387, 1405–1410. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N Engl J Med 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol Immunol 2021, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Hoyles, L. Human microbiome myths and misconceptions. Nat Microbiol 2023, 8, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct Target Ther 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Larke, J.A.; Bacalzo, N.; Castillo, J.J.; Couture, G.; Chen, Y.; Xue, Z.; Alkan, Z.; Kable, M.E.; Lebrilla, C.B.; Stephensen, C.B.; et al. Dietary Intake of Monosaccharides from Foods is Associated with Characteristics of the Gut Microbiota and Gastrointestinal Inflammation in Healthy US Adults. J Nutr 2023, 153, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Ikubo, Y.; Sanada, T.J.; Hosomi, K.; Park, J.; Naito, A.; Shoji, H.; Misawa, T.; Suda, R.; Sekine, A.; Sugiura, T.; et al. Altered gut microbiota and its association with inflammation in patients with chronic thromboembolic pulmonary hypertension: a single-center observational study in Japan. BMC Pulm Med 2022, 22, 138. [Google Scholar] [CrossRef]
- Walker, R.L.; Vlamakis, H.; Lee, J.W.J.; Besse, L.A.; Xanthakis, V.; Vasan, R.S.; Shaw, S.Y.; Xavier, R.J. Population study of the gut microbiome: associations with diet, lifestyle, and cardiometabolic disease. Genome Med 2021, 13, 188. [Google Scholar] [CrossRef]
- Rohm, T.V.; Fuchs, R.; Muller, R.L.; Keller, L.; Baumann, Z.; Bosch, A.J.T.; Schneider, R.; Labes, D.; Langer, I.; Pilz, J.B.; et al. Obesity in Humans Is Characterized by Gut Inflammation as Shown by Pro-Inflammatory Intestinal Macrophage Accumulation. Front Immunol 2021, 12, 668654. [Google Scholar] [CrossRef]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef]
- Wan, Y.; Yuan, J.; Li, J.; Li, H.; Yin, K.; Wang, F.; Li, D. Overweight and underweight status are linked to specific gut microbiota and intestinal tricarboxylic acid cycle intermediates. Clin Nutr 2020, 39, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Pinero, F.; Vazquez, M.; Bare, P.; Rohr, C.; Mendizabal, M.; Sciara, M.; Alonso, C.; Fay, F.; Silva, M. A different gut microbiome linked to inflammation found in cirrhotic patients with and without hepatocellular carcinoma. Ann Hepatol 2019, 18, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.; Cohen, N.A.; Shalev, V.; Uzan, A.; Koren, O.; Maharshak, N. Psoriatic patients have a distinct structural and functional fecal microbiota compared with controls. J Dermatol 2019, 46, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Troseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjorndal, B.; Halvorsen, B.; et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med 2015, 277, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: a cross-sectional study in stage 3-4 chronic kidney disease. Arch Med Res 2014, 45, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive oxygen production induced by the gut microbiota: pharmacotherapeutic implications. Curr Med Chem 2012, 19, 1519–1529. [Google Scholar] [CrossRef]
- Jones, R.M.; Luo, L.; Ardita, C.S.; Richardson, A.N.; Kwon, Y.M.; Mercante, J.W.; Alam, A.; Gates, C.L.; Wu, H.; Swanson, P.A.; et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J 2013, 32, 3017–3028. [Google Scholar] [CrossRef]
- Jones, R.M.; Neish, A.S. Redox signaling mediated by the gut microbiota. Free Radic Biol Med 2017, 105, 41–47. [Google Scholar] [CrossRef]
- Jeong, Y.S.; Bae, Y.S. Formyl peptide receptors in the mucosal immune system. Exp Mol Med 2020, 52, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Wu, H.; Collier-Hyams, L.S.; Hansen, J.M.; Li, T.; Yamoah, K.; Pan, Z.Q.; Jones, D.P.; Neish, A.S. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J 2007, 26, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J. Bacterial-modulated signaling pathways in gut homeostasis. Sci Signal 2008, 1, pe24. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Rivera-Chavez, F.; Zhang, L.F.; Faber, F.; Lopez, C.A.; Byndloss, M.X.; Olsan, E.E.; Xu, G.; Velazquez, E.M.; Lebrilla, C.B.; Winter, S.E.; et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 2016, 19, 443–454. [Google Scholar] [CrossRef]
- Farhana, A.; Khan, Y. Biochemistry, Lipopolysaccharide. In StatPearls; StatPearls Publishing: Treasure Island, FL, 2023. [Google Scholar]
- McGuckin, M.A.; Linden, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat Rev Microbiol 2011, 9, 265–278. [Google Scholar] [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front Immunol 2020, 11, 594150. [Google Scholar] [CrossRef]
- Anhe, F.F.; Barra, N.G.; Cavallari, J.F.; Henriksbo, B.D.; Schertzer, J.D. Metabolic endotoxemia is dictated by the type of lipopolysaccharide. Cell Rep 2021, 36, 109691. [Google Scholar] [CrossRef]
- Steimle, A.; Autenrieth, I.B.; Frick, J.S. Structure and function: Lipid A modifications in commensals and pathogens. Int J Med Microbiol 2016, 306, 290–301. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 2013, 45, e66. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Takeuchi, S.; Kubota, K.; Kobayashi, Y.; Kozakai, S.; Ukai, I.; Shichiku, A.; Okubo, M.; Numasaki, M.; Kanemitsu, Y.; et al. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKKϵ-IRF3 axis activation. J Biol Chem 2018, 293, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R., Jr. Metabolic endotoxemia: a molecular link between obesity and cardiovascular risk. J Mol Endocrinol 2013, 51, R51–R64. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol 2013, 23, 129–134. [Google Scholar] [CrossRef]
- Wentworth, C.C.; Alam, A.; Jones, R.M.; Nusrat, A.; Neish, A.S. Enteric commensal bacteria induce extracellular signal-regulated kinase pathway signaling via formyl peptide receptor-dependent redox modulation of dual specific phosphatase 3. J Biol Chem 2011, 286, 38448–38455. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Trinei, M.; Berniakovich, I.; Pelicci, P.G.; Giorgio, M. Mitochondrial DNA copy number is regulated by cellular proliferation: a role for Ras and p66(Shc). Biochim Biophys Acta 2006, 1757, 624–630. [Google Scholar] [CrossRef]
- Blank, H.M.; Li, C.; Mueller, J.E.; Bogomolnaya, L.M.; Bryk, M.; Polymenis, M. An increase in mitochondrial DNA promotes nuclear DNA replication in yeast. PLoS Genet 2008, 4, e1000047. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Baumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Luche, E.; Cousin, B.; Garidou, L.; Serino, M.; Waget, A.; Barreau, C.; Andre, M.; Valet, P.; Courtney, M.; Casteilla, L.; et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a CD14-dependent mechanism. Mol Metab 2013, 2, 281–291. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Oliva-Olivera, W.; Coin-Araguez, L.; Ramos-Molina, B.; Giraldez-Perez, R.M.; Lhamyani, S.; Alcaide-Torres, J.; Perez-Martinez, P.; El Bekay, R.; Cardona, F.; et al. Metabolic endotoxemia promotes adipose dysfunction and inflammation in human obesity. Am J Physiol Endocrinol Metab 2019, 316, E319–E332. [Google Scholar] [CrossRef]
- Ranieri, S.C.; Fusco, S.; Pani, G. p66(ShcA): linking mammalian longevity with obesity-induced insulin resistance. Vitam Horm 2013, 91, 219–241. [Google Scholar] [CrossRef]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. p66Shc-generated oxidative signal promotes fat accumulation. J Biol Chem 2008, 283, 34283–34293. [Google Scholar] [CrossRef]
- Ciciliot, S.; Albiero, M.; Menegazzo, L.; Poncina, N.; Scattolini, V.; Danesi, A.; Pagnin, E.; Marabita, M.; Blaauw, B.; Giorgio, M.; et al. p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia 2015, 58, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis Model Mech 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Im, S.H. Of men in mice: the development and application of a humanized gnotobiotic mouse model for microbiome therapeutics. Exp Mol Med 2020, 52, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Paz, K.; Hemi, R.; LeRoith, D.; Karasik, A.; Elhanany, E.; Kanety, H.; Zick, Y. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem 1997, 272, 29911–29918. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Ranieri, S.C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara, A.M.; Maulucci, G.; De Spirito, M.; Martorana, G.E.; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc Natl Acad Sci U S A 2010, 107, 13420–13425. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.P.; Texeira, T.F.; Ferreira, A.B.; Peluzio Mdo, C.; Alfenas Rde, C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 2012, 108, 801–809. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101. [Google Scholar] [CrossRef]
- Freeman, A.M.; Acevedo, L.A.; Pennings, A. Insulin Resistance. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507839/ (accessed on 14 February 2024).
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Van Hul, M.; Cani, P.D. The gut microbiota in obesity and weight management: microbes as friends or foe? Nat Rev Endocrinol 2023, 19, 258–271. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front Immunol 2020, 11, 571731. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology (Bethesda) 2016, 31, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J Clin Invest 2018, 128, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Tomilov, A.A.; Ramsey, J.J.; Hagopian, K.; Giorgio, M.; Kim, K.M.; Lam, A.; Migliaccio, E.; Lloyd, K.C.; Berniakovich, I.; Prolla, T.A.; et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011, 10, 55–65. [Google Scholar] [CrossRef]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.W.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The adaptor protein p66Shc inhibits mTOR-dependent anabolic metabolism. Sci Signal 2014, 7, ra17. [Google Scholar] [CrossRef]
Figure 1.
Functional domains of the ShcA proteins: collagen homology (CH1 and CH2), cytochrome C-binding (CB), phosphotyrosine-binding (PTB), and Src-homology (SH2) domains. Post-translation modifications highlighted in the CH2 domain of p66Shc are involved in its activity affecting reactive oxygen species production and apoptosis. Ser36 phosphorylation increases p66Shc translocation into mitochondria; Ser54 phosphorylation decreases p66Sch degradation by the proteasome; Lys81 acetylation increases Ser36 phosphorylation. P: phosphorylation, Ac: acetylation. Created with BioRender.com.
Figure 1.
Functional domains of the ShcA proteins: collagen homology (CH1 and CH2), cytochrome C-binding (CB), phosphotyrosine-binding (PTB), and Src-homology (SH2) domains. Post-translation modifications highlighted in the CH2 domain of p66Shc are involved in its activity affecting reactive oxygen species production and apoptosis. Ser36 phosphorylation increases p66Shc translocation into mitochondria; Ser54 phosphorylation decreases p66Sch degradation by the proteasome; Lys81 acetylation increases Ser36 phosphorylation. P: phosphorylation, Ac: acetylation. Created with BioRender.com.
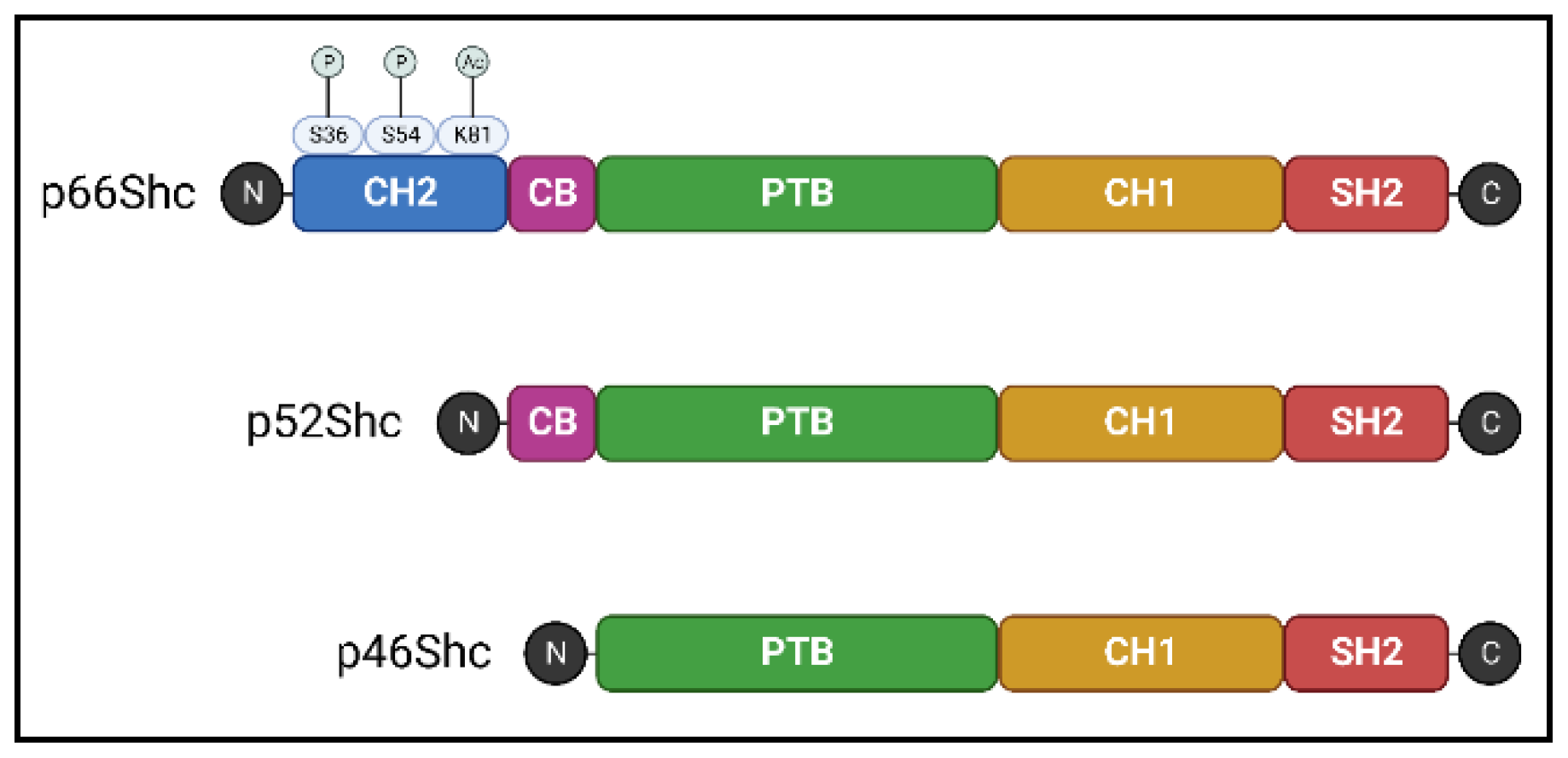
Figure 2.
Schematic representation of p66Shc-mediated signaling pathways and their role in apoptosis. (A) competitive inhibition of p52/p46Shc by p66Shc at the Grb2-SOS complex, resulting in the activation of Rac1 instead of Ras, leading to increased NADPH oxidase activity and subsequent reactive oxygen species (ROS) production, culminating in apoptosis. (B) Role of p66Shc in the inactivation of Forkhead-type transcription factors (FOXO3a) via Akt phosphorylation and βPix sequestration from the nucleus, which diminishes the expression of antioxidant enzymes such as MnSOD and GPx, further promoting apoptosis. (C) Role of p66Shc in mitochondrial apoptosis through its translocation into the intermembrane space mediated by Pin1 and PP2A, leading to increased mitochondrial ROS production and cyt c release, triggering the apoptotic cascade. Created with BioRender.com.
Figure 2.
Schematic representation of p66Shc-mediated signaling pathways and their role in apoptosis. (A) competitive inhibition of p52/p46Shc by p66Shc at the Grb2-SOS complex, resulting in the activation of Rac1 instead of Ras, leading to increased NADPH oxidase activity and subsequent reactive oxygen species (ROS) production, culminating in apoptosis. (B) Role of p66Shc in the inactivation of Forkhead-type transcription factors (FOXO3a) via Akt phosphorylation and βPix sequestration from the nucleus, which diminishes the expression of antioxidant enzymes such as MnSOD and GPx, further promoting apoptosis. (C) Role of p66Shc in mitochondrial apoptosis through its translocation into the intermembrane space mediated by Pin1 and PP2A, leading to increased mitochondrial ROS production and cyt c release, triggering the apoptotic cascade. Created with BioRender.com.
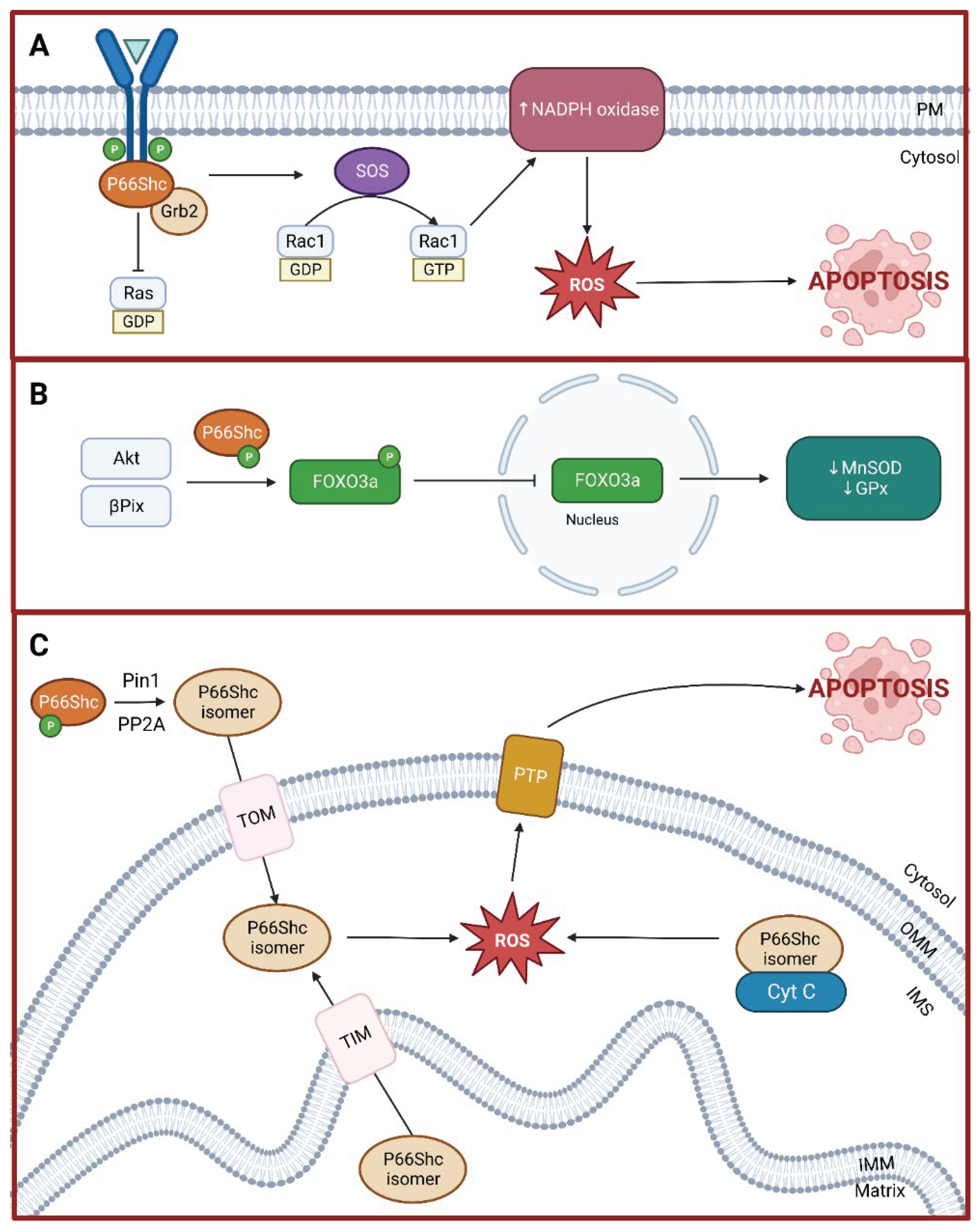

Table 1.
Selected studies published in the last 10 years investigating the role of p66Shc on different pathophysiological conditions.
Table 1.
Selected studies published in the last 10 years investigating the role of p66Shc on different pathophysiological conditions.
| First author | Year | Study population or model | Pathophysiological condition |
|---|---|---|---|
| W. E. Hughes [21] | 2021 | Animal (rats) | Hypertension |
| K. Shahzad [22] | 2018 | Animal (mice) | Hyperglycemia-induced atherosclerosis |
| S. Costantino [23] | 2018 | Animal (mice) Cell culture (human cardiomyocytes) |
Diabetes-related cardiomyopathy |
| H. Vashistha [24] | 2018 | Animal (mice) Cell culture (Sca-1+ mesenchymal stem cells) |
Diabetes-related renal dysfunction |
| F. Paneni [25] | 2016 | Cell culture (early outgrowth cells) | Age-related impaired vascular repair |
| R. Vono [26] | 2016 | Humans (patients with diabetes undergoing major limb amputation) | Diabetes-related critical limb ischemia |
| A. Akhmedov [27] | 2015 | Animal (mice) | Cardiac ischemia and reperfusion |
| R. D. Spescha [28] | 2015 | Animal (mice) Cell culture (primary HBMVECs) Human (acute ischemic stroke patients) |
Ischemia/reperfusion brain injury; stroke |
| A. Natalicchio [29] | 2015 | Animal (mice) Cell culture (rat INS-1E cells; murine, human, and mouse islets) |
Hyperglycemia |
| Y. Shi [30] | 2014 | Animal (mice) | Age-related cerebrovascular impairment |
| F. Paneni [31] | 2014 | Animal (mice) | Obesity-induced endothelial insulin resistance |
| R. D. Spescha [32] | 2014 | Cell culture (primary human AECs and rat AECs) | Hypertension |
| A. Vikram [33] | 2014 | Animal (mice) Cell culture (various) |
Endothelial dysfunction |
| Z. Chen [34] | 2014 | Animal (mice) Cell culture (Caco-2 cells) |
Ischemia/reperfusion intestinal injury |
| V. Bellisario [35] | 2014 | Animal (mice) | Detrimental developmental programming |
| R. D. Spescha [36] | 2013 | Animal (mice) | Ischemia/reperfusion brain injury; stroke |
| L. Laviola [37] | 2013 | Cell culture (HUVECs) | Endothelial dysfunction |
| F. Bock [38] | 2013 | Animal (mice) | Diabetes-related nephropathy |
AECs, aorta endothelial cells; HBMVECs, human brain microvascular endothelial cells; HUVECs, human umbilical vein endothelial cells.
Table 2.
Selected studies published in the last 10 years investigating associations between the gut microbiota and different pathophysiological conditions.
Table 2.
Selected studies published in the last 10 years investigating associations between the gut microbiota and different pathophysiological conditions.
| First author | Year | Study population | Pathophysiological condition |
|---|---|---|---|
| J. A. Larke [58] | 2023 | Healthy adults | Gastrointestinal inflammation |
| Y. Ikubo [59] | 2022 | Patients with CTEPH | Pulmonary hypertension |
| R. L. Walker [60] | 2021 | Framingham Heart Study cohort | Cardiometabolic diseases |
| T. V. Rohm [61] | 2021 | Obese and non-obese adults | Obesity |
| X. Wang [62] | 2020 | Patients with ESRD | Renal disease |
| Y. Wan [63] | 2020 | Adults in different BMI categories | Cardiometabolic diseases |
| F. Piñero [64] | 2019 | Patients with cirrhosis | Liver cancer |
| J. Shapiro [65] | 2019 | Patients with psoriasis | Autoimmune diseases |
| M. Trøseid [66] | 2015 | Patients with chronic heart failure | Cardiovascular diseases |
| M. Rossi [67] | 2014 | Patients with CKD | Renal disease |
CTEPH, chronic thromboembolic pulmonary hypertension; BMI, body mass index; ESRN, end-stage renal disease; CKD, chronic kidney disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



