
Submitted:
20 February 2024
Posted:
20 February 2024
You are already at the latest version
Abstract
Polymer electrolyte was used as a medium for testing the performance of microband electrodes under conditions of linear diffusion. Cyclic voltammetry and chronoamperometry experiments were performed in such viscous medium, where diffusion rates are much slower than in fluid solutions. Confirmation of the existence of such conditions from the log i vs. log v or log i vs. log t relationships in the current equation always yielded values lower than the expected value of 0.5. This could indicate impure linear diffusion profile, i. e. some contribution from radial diffusion (edge effect). However, performing these tests in monomeric solvents of similar viscosities, such as glycerol or propylene glycol, gave the desired value of 0.5. These results lead to the conclusion that current equations which are based on Fick’s laws may not be applicable for polymer electrolytes, where various obstruction to diffusion make it a more complicated process than in monomeric solvents.
Keywords:
polymer electrolytes
; microband electrodes
; linear diffusion
; Fick’s equations
; diffusion in polymers
1. Introduction
Polymer electrolytes have been used in the past decades as media for electrochemical processes. In particular, polymers bearing polyether chains are capable to dissolve considerable amounts of electrolytes, such as lithium salts, which provide electrical conductivity to the system (1–6). Nevertheless, the conductivity in polymer electrolytes is still lower than in conventional electrolytic solutions, and therefore electrochemical experiments conducted in such media must employ microelectrodes, which are less sensitive to solution resistance than regular-size electrodes (7). A problem that can arise by the use of microelectrodes, such as microdisks or microsphere, is exceedingly low currents, especially in very viscous solutions where the diffusion rates are very slow. A practical way to overcome this difficulty is using microband electrodes. These electrodes have widths on the micrometic scale, which maintain the properties of microelectrodes, whereas their lengths are on the millimetric scale, which provide considerably higher currents. In the past we reported the fabrication procedures and testing of microband electrodes (8) and demonstrated their performance in viscous and resistive polymeric solutions (9).
In the course of characterization of microband electrodes, cyclic voltammetry (CV) experiments have been performed under conditions of radial and linear diffusion. Achieving conditions of linear diffusion at microbands on the width-scale of several micrometers requires a great suppression of the diffusion rate. This can be done by controlling the experimental parameters, such as the size and the shape of the diffusing probe (10), the temperature and the viscosity of the solution. Electrochemical measurements in polymer electrolytes have shown reduced values of the diffusion coefficients by several orders of magnitude, as compared to conventional fluid solvents (11-13). Therefore, it was an obvious choice to conduct these tests in polymeric solutions as one of the means to obtain pure linear diffusion, with only negligible contribution from edge effect.
An estimation of the diffusion geometry can be obtained using the dimensionless time parameter τ, defined as:
where r is the electrode’s radius, D is the diffusion coefficient and t is the electrolysis characteristic time. For cyclic voltammetry, τ equals to RT/nFv (14), where v is the potential scan-rate. When τ = 1, the diffusion distance of a solute is equal to the radius of the electrode. When τ >>1 the thickness of the diffusion layer δ is greater than r and the diffusion geometry is radial, whereas when τ <<1 the diffusion layer is thin relative to r, and thus the diffusion profile is linear ().
τ = 4Dt/r2
The approximate thickness of the diffusion layer at the electrode’s surface is given by (14):
δ = (2Dt)1/2
If, for the sake of argument, we define a limiting thickness which would not exceed one tenth of the electrode’s smallest dimension (i.e., δ = 0.1r), within which the diffusion geometry is pure linear, we get (after substituting the expression for t):
taking D = 10-8cm2/s and r = 3μm (the width of our microband electrode), then at ambient temperature, linear diffusion should be predominant at scan-rates greater than 0.7V/s. Substituting this value in Eq. 1 yield τ = 0.02, which is considerably smaller than 1, as should be for linear diffusion. At lower temperatures τ would be even smaller.
0.1r < (2DRT/nFv)1/2
An experimental criterion for the existence of conditions of linear diffusion is derived from equation [4] for the peak current in cyclic voltammetry (15).
where A is the electrode’s area and C is the concentration of the electroactive species. According to this equation, ip is a linear function of v½. Thus, conducting a series of CV experiments at a succession of potential scan-rates and plotting the results in the form of log ip vs. log v should yield, in the case of linear diffusion, a straight line with a slope of 0.5.
ip= 0.4463nFAC(nF/RT)½ D½ v½
In this paper we present results of such tests in polypropylene glycol (PPG). In spite of the expectations for pure linear diffusion, based on the above-mentioned calculation, slopes not higher than 0.4 were obtained under any experimental conditions. This was investigated and compared with results obtained in monomeric solvents of similarly high viscosities, and the different modes of diffusion in these systems are discussed.
2. Materials and Methods
Equipment: A locally built low current potentiostat were employed, controlled by a universal programmer (EG&G model PAR 175). All experiments were conducted inside a faraday cage.
Materials: Ferrocene-labeled monomethyl poly(ethylene glycol) [FcCO2PEG(350)CH3] (16) and a tetrathiafulvalene trimethoxy methyl [TTF-(OCH2)CH3] (17) were synthesized according to procedures described elsewhere. Polyethylene glycol 4000 and 10,000, glycerol and propylene glycol (Aldrich) were of analytical grade. LiSO3CF3 (Aldrich, analytical grade) was dried at 100 °C under vacuum and kept in a glove box.
Electrodes: Microband electrode was 2.7 µm wide and 0.7 cm long and included nearby, flanking parallel Ag band reference electrodes and Pt-wire counter electrodes. This structure was aimed at minimizing the effects of solution resistance and asymmetrical electric field. The preparation procedure is given in Ref. 18 (type D). Microdisk Pt electrodes of 25 and 50 µm radii with a nearby reference electrode were used. This preparation is also described in Ref. 18.
Solutions preparation: Polymer electrolyte solutions were prepared by adding weighted amounts of the electroactive probe and the electrolyte to a weighted amount of the polymer melt in a Schott tube, equipped with a vacuum outlet and a tightly-closed screw-cap with a rubber/Teflon septum. Dissolution of the solids was facilitated by adding a small amount of ethanol or acetonitrile, which were later removed by heating at ca. 70 °C under vacuum. The three electrodes were embedded in an Epon epoxy rod, which was inserted into the cell through the cap. Solutions in monomeric solvents (glycerol and propylene glycol) were prepared without adding a co-solvent.
3. Results
The performance of microband electrodes, manufactured in our laboratory, was tested in solution of polypropylene glycol of an average molecular weight of 4000 (PPG 4000). Addition of lithium salt (LiSO3CF3, 1.1M) as an electrolyte renders the polymer melt very viscous, due to the cross-linking of the polyether chains via coordinative bonds with the lithium ions. Such high viscosity is necessary for the suppression of the diffusion coefficient of the electroactive molecules, if pure linear diffusion profile is required at the very narrow microband. The higher the diffusion coefficient, the faster is the development of the diffusion layer in the vicinity of the electrode, resulting in growing contribution from the unwanted radial diffusion.
The linearity of the diffusion profile was judged from a plot of log ip vs. log v for a series of cyclic voltammograms at various scan-rates. Since equation 4 is valid only under conditions of linear diffusion, a slope of 0.5 would be indicative for the existence of such conditions. Examples for two of the voltammograms, recorded at room temperature at 1 and 200 mV/s, are shown in Figure 1:
Curve A was obtained at low scan-rate and represents a mixed diffusion profile, while curve B was recorded at faster scan-rate and shows higher contribution of linear diffusion. The plot of log i vs. log v is presented in Figure 2. Line A is drawn for a set of measurements carried out at room temperature; the slope of 0.4 may indicate that the diffusion profile is mainly linear, with some contribution from radial diffusion. We considered the possibly radial contribution to be a result of insufficient viscosity that could arise from either insufficiently-low temperature or from residual amounts of ethanol, which was used to facilitate the dissolution of the ferrocene probe in the polymer and was later removed by evaporation. That residual ethanol could act as a plasticizer and thus increase the diffusion rate in the polymer. These two possibilities were checked experimentally, as described herein.
Further removal of possible residues of ethanol was done by vacuum pumping under more drastic conditions: The polymeric solution was heated at a higher temperature (90 oC) during the evaporation, in order to increase its fluidity and enable any traces of ethanol to diffuse out more easily. Furthermore, the duration of the evaporation was longer (three days) and the vessel was held in the oil bath in a tilted position, so that the surface area of the polymeric solution was larger and its depth was smaller. Finally, some of the solutions were spread as a layer on the surface of the electrode-assembly rod and vacuum-pumped for three days. Nevertheless, no significant change in the value of the slope was observed.
The effect of temperature was tested by performing two sets of measurements at 12.6 and 2.6 °C, and their analyses are presented too in Figure 2 (lines B and C, respectively). Surprisingly, the trend in these results was in the opposite direction than expected: the slope values were decreasing with decreasing the temperature, rather than approach the value of 0.5. A possible explanation to this discrepancy could be the increased uncompensated solution resistance (iRu) at lower temperatures, which causes flattening of the peaks and thus lower measured currents. This effect was first tested by digital simulation of CV experiments, which compared two systems that were different from each other by the magnitude of Ru only. It was found that decreasing the magnitude of Ru from 500 MΩ to 100 MΩ results in an increase in the log i-log v slope from 0.40 to 0.46, in agreement with the assumption above. Experimental verification of this effect was done by taking three different steps: a) Decreasing the solution resistance by raising the electrolyte concentration. b) Decreasing the iR product by using lower concentration of the electroactive species, to get lower currents. C) Applying positive feedback for iR compensation. None of these steps seemed to cause a meaningful change in the magnitude of the slopes, which stayed within the range of 0.34-0.43. In testing steps (a) and (b) a similar trend in the slopes was observed with decreasing the temperature, although to a smaller extent. Testing the effect of positive feedback in two solutions with different electrolyte concentrations, 1.1M and 0.7M, yielded no change in the slope in either case, as shown in Figure 3. This is in spite of the remarkable reduction of the peak-to-peak separation (ΔEp) which indicates smaller iR drop. It was thus concluded that solution resistance is not the factor responsible for the decreasing slopes at lower temperatures, or for the unachieved target slope of 0.5.
Another means of increasing the solution viscosity was using polypropylene glycol of higher molecular weight, e.g., PPG 10,000, as the solvent. Upon dissolving the same concentration of electrolyte (O/Li =16), a very viscous melt was obtained, in which well-shaped voltammograms were recorded in the scan-rate range of 1-500 mV/s (Figure 4A). Plotting the data as log i vs. log v (Figure 4B) revealed another unexpected result: a slope of 0.26, which is considerably smaller than the slopes obtained in PPG 4000. Repeating the experiment in this solution with a wider microband (w = 4.6 μm) and with two microdisk electrodes of 25 and 50 μm radii, yielded higher slope-values of 0.35, 0.38 and 0.40, respectively. These results seem to indicate, on one hand, the existence of some radial contribution to the diffusion profile, since these slopes were getting closer to 0.5 as the ratios between the perimeters of the electrodes to their surface areas were getting smaller, i.e., the electrodes are less sensitive to edge effect. On the other hand, the fact that even with the 50 μm-radius microdisk electrode a value not higher than 0.4 was obtained, points to the possibility that such slopes cannot be obtained in viscous polymeric solvents at all.
The rationale of this conclusion may be found in the basic difference between the diffusion profiles in conventional monomeric solvents, such as water, alcohols, nitriles etc. and polymeric solvents. In monomeric solvents, free diffusion of the solute occurs with actually no physical restriction. Polymeric solvents, however, can be entangled, cross-linked or contain local aggregates or orderings, which can all introduce complications and constrains to the diffusion paths. The outcome of these is that equations which are derived from Fick’s laws, that have been developed for conditions of free diffusion (i.e., in monomeric solvents) may not be applicable in polymeric solvents, especially where such restrictions exist. Referring to our case, the reason for not obtaining slopes of 0.5, even when it was clear that the diffusion profile is nearly pure-linear, is that the current equation (Eq. 4) cannot be used in its present form. One of the changes should be the function of v; its power in such cases should be smaller than 0.5.
Experimental verification of this idea was conducted in two monomeric solvents at low temperatures, propylene glycol and glycerol, which are rather viscous already at room temperature. In propylene glycol, voltammograms at various scan-rates were recorded at three different temperatures, using TTF-(OCH2)3CH3 as the electroactive probe. A representative voltammogram, recorded at -23 °C, is shown in Figure 5 (inset); it has two well-resolved couples, and the currents of the first oxidation peaks were used for the log i vs. log v plot, which is shown in Figure 5. At -23.7 °C, a well-defined straight line was obtained, which exhibits the desired slope of exactly 0.5. This is actually the first time that pure linear diffusion has been observed at a microband electrode, and, perhaps more significant, this result is in accordance with the new concept of different diffusion rules in the two kinds of solvents. At higher temperatures, -12 °C and 1 °C, a slope of 0.5 was obtained at faster scan-rates, which were required in order to compete with the growing contribution from radial diffusion.
In glycerol, the composition of the solution, except the solvent, was similar to that of the PPG 4000 solution in which the first experiments were performed [10mM FcCO2PEG(350)CH3, 1M LiClO4]. The measurements were carried out at -27 oC, at scan-rates ranging from 1 to 200 mV/s, all yielding peak currents which are aligned on a straight line with a slope of 0.49 (Figure 6). Under these conditions it was possible to obtain pure linear diffusion at a 2.7 μm-wide microband at scan-rate as low as 1mV/s (Figure 6 inset).
In comparison with the results obtained in propylene glycol, a solution of the same probe in polypropylene glycol was prepared, and voltammograms were recorded at room temperatures, keeping all other experimental parameters unchanged. A typical voltammogram is shown in Figure 7 inset, having two main features: a) The first oxidation peak is less resolved than in propylene glycol but still distinguishable. b) The other peaks have sharp and symmetrical shapes, which are typical to surface-immobilized systems such as monolayers or thin-layer cells. This indicates that these bulky probe molecules are trapped in the polymer, and their freedom to diffuse is so limited that only molecules in the near vicinity of the electrode can undergo redox process within the time scale of the experiments. A plot of log i vs. log v for the first oxidation peak (that appear as a shoulder in the voltammogram) is linear with a slope of 0.35, which is close to the values that were previously measured in PPG 4000. More interestingly, such a plot for the first reduction peaks, which are clearly of a thin-layer shape and thus should show a linear dependence of i on v (8), has a slope of 0.32. This apparent paradox may be regarded as a situation of quasi-immobilization, i.e., the molecules are not covalently bound to the electrode surface or trapped in a thin layer but in principle can undergo diffusion, although in a restricted manner.
The unusual diffusional behavior in polymeric solvents was tested also by an independent electrochemical technique - chronoamperometry. Since the solution was dried under vacuum and the cell was kept tightly closed, the background curve could not be recorded in the same potential range prior to the dissolution of the substrate. Instead, it was done by stepping the potential from the start point to a value where no reaction occurs (point A on the inset CV, Figure 8) prior to recording the actual chronoamperogram at an equal potential step (point B). A typical response, obtained in the same solution as for the CV experiment described in Figure 1, is shown in Figure 8. The corresponding Cottrell plot (Figure 9A), based on the Cottrell equation:
is linear with zero intercept at the short time-segment after the charging spike. At times longer than 2 seconds a positive deviation is observed, which means that the rate of the current decay is slower than the Cottrell prediction. A similar deviation has been observed by Longmire et al. (9) in a system of ferrocene-labeled polyethylene glycol melt (which serves as both the electroactive moiety and electrolyte dissolving solvent), and the extra current was explained by the enhanced flux of material to the electrode by radial diffusion. Regarding the unusual results of our CV experiments, we checked the log-log relationship between i and t, which under conditions of regular diffusion should be linear with a slope of -0.5. The plot, shown in Figure 9A, has three linear segments: the first one, corresponds to time interval of 1 second after the charging spike (2 seconds total) has a slope of -0.49, in accordance with the Cottrell equation. This segment can be attribute to the solute molecules in the near vicinity of the electrode, which are capable to undergo relatively undisturbed diffusion. The third segment, corresponding to times longer than 3.6 seconds, has a slope of -0.32, which is close to the log i-log v slopes (0.33-0.40) obtained in the CV experiments that were performed in the same solutions.
i=nFAD1/2Cπ-1/2t-1/2
Chronoamperometry was performed also in the solution of PPG 10,000 (Figure 10). The Cottrell plot has a similar shape as that for the PPG 4000 solution, but the deviation from the Cottrell behavior starts after 0.6 second. Here, too, the plot of log i vs. log t includes three linear segments. The first, corresponding to a total time interval of 0.6 second, also has a slope of -0.49, while the third segment, corresponding to times longer than 2 seconds, has a lower value of -0.28. Here, too, the magnitude of the latter slope is very close to that of the log i-log v slope (0.26), obtained in the CV experiment in the same solution.
4. Discussion
The attempts to find suitable experimental conditions for characterization of microband electrodes under linear diffusion profile have led us to consider the complicated manner of diffusion in polymeric solvent, in contrast to free diffusion in monomeric solvents. In the latter, the three dimensional random-walk model holds, on which the Fick’s laws are based. The expression for the peak current in cyclic voltammetry (Eq. 1) and for the time dependence of the current in chronoamperometry (Eq. 5, Cottrell equation) are both based on Fick’s laws and thus are valid only under conditions of Fickian diffusion. In polymer melts, however, a number of obstructions to diffusion exist, such as local entanglements, coordinative crosslinks, blocking ions or ionic aggregates and short-range local ordering, and therefore these equations may not be applicable in all cases. In solvent-swollen polymers wide voids or channels between polymer chains can permit nearly Fickian diffusion, so such constrains may not be very significant.
Masaro and Zhu, in their comprehensive review article on diffusion in polymers (19), indicated that this is indeed a complicated process and its rate depends on the concentration and the swelling degree of the polymers. They distinguished between Fickian diffusion (case 1) and two types of non-Fickian diffusion: Case 2 and anomalous diffusion. Fickian diffusion can be observed in polymers at temperatures considerably above the glass transition (Tg), or below Tg but with the addition of a plasticizer, such as water. In non-Fickian Case 2, the solvent diffusion rate is faster than the relaxation of the polymer chains, whereas in anomalous diffusion these rates are rather close. Fickian and Case 2 diffusions are considered as the two limiting types of transport, and the diffusion distance Mt is given by equation 6.
Mt = ktn
A supercase 2 was also defined (20) and these mechanisms were categorized based on the exponent n, as follows: Fickian: n<1/2, Anomalous ½<n<1, Case 2 n=1, Supercase 2: n>1.
Grinsted et al. (21) also noted that water has an effect of plasticizer on poly (methyl methacrylate) (PMMA). Therefore, diffusion of methanol in PMMA changes from Case 2 to Fickian as the water content increases.
Many studies and review articles were published on diffusion modes in polymers. Several diffusion models that are based on the obstruction effects were suggested, which consider the polymer chains stationary relative to the diffusing solvents or solutes due to their much smaller self-diffusion coefficient (19,). Modeling of entangled polymer diffusion was extensively review by Karatrantos et al. (22), concerning entangled (reptational) homopolymer diffusion in melts and nanocomposite. Diffusion in polymer electrolytes was widely discussed by Choo et al. (23) in their comprehensive review article. However it refers only to polymer-salt interactions at the segmental level and macroscopic ion transport, based on the system of poly(ethylene oxide) and lithium bis(trifluoromethanesulfonyl)imide (PEO/LiTFSI). No additional solute was included in this system as the diffusing molecules, unlike in our case,
In our polymeric solution the plasticizer was removed, so the polymer contained only the conductive electrolyte and the diffusing molecules. Due to the low temperatures and the high viscosity of the polymer electrolyte we can assume that the relaxation of the polymer chains is very slow. In such unswollen polymers the obstructions mentioned above may have a profound effect, especially when the diffusing molecules are large. Transport of the diffusant in such media will occur along paths of least resistance (PLR), which can be branched, or have dead ends. This PLR network can reorganize with time, as a result of polymer segmental motion, and create new paths for diffusion. In electrochemical measurements, insufficient reorganization rate, compared to the diffusivity of the electrophore, leads to “fractal diffusion”. Electrochemical current measurements would thus reflect a combination of the rate of reorganization together with the rate of diffusion of the electrophore within the existing or changing PLR network. The effect on the current equations is therefore reflected not only in the exponent of the diffusion coefficient, but also in the time-related terms (t or v). This can explain why the exponents of v or t in all of our experiments are smaller than 0.5, which is the expected value for linear profile of Fickian diffusion. The magnitudes of these exponents decrease as the experimental conditions turn more impedient to fast reorganization, such as lower temperatures or higher molecular weight of the polymer.
The last conclusion, which is based on empirical data, leads to generalization of the current equations for these voltammetric methods, in which the exponent of the time-related terms would be n/2, where n ≤ 1. The magnitude of n is a function of the nature of the diffusion environment, such as the reorganization time-constant of the polymer electrolyte or the longer diffusion paths, and should reflect the properties of the system. These are affected by factors such as the temperature, the concentration and the valency of the electrolyte, the structure of the polymer and its molecular weight. Their relative contributions yield complicated combinations which are specific to each system. Changes in the exponents of the time-related terms should imply additional changes in the current equations for CV and CA, to maintain the units of Amperes. Thus, the exponent of the diffusion coefficient, which is also a time-related term, should be changed accordingly. Evaluation of the current equation for polymer electrolytes is thus needed, which should reflect the combined contributions of both processes, i. e. polymer reorganization and solute diffusion, to mass transport.
5. Conclusions
1. Diffusion of molecules in polymer electrolyte melt is non-Fickian because of various perturbations to free random motion derived from concentrations gradients.
2. Therefore, the current equations for voltammetric measurements that were developed for fluid solutions are not valid in their regular form for such media.
3. These equations are valid, though, in very viscous solutions of monomeric solvents, where the high viscosity is reflected by low diffusion coefficients.
4. The modified current equations for voltammetric measurements in polymer electrolyte melt should include changes in the exponents of the time-related terms.
Acknowledgments
We thank Dr. Christopher S. Velazquez for his important ideas and contribution to this work.
References
- Watanabe, M.; Velazquez, C.S.; Porat, Z.; Haas, O.; Wooster, T.T.; Longmire, M.L.; Zhang, H.; Nishihara, H.; Murray, R.W. Electrochemical Properties of Lithium Ion Conducting Solid Polymer Electrolytes. In Proceedings of the Symposium on Advanced Lithium Batteries Case Western Reserve University, 1 July 1994. [Google Scholar]
- Di Noto, V.; Lavina, S.; Griffin, G.A.; Negro, E.; Scrosati, B. Polymer Electrolytes: Present, Past and Future. Electrochim. Acta, 2011, 5, 4–13. [Google Scholar] [CrossRef]
- Ngai, K.S.; Ramesh, S.; Juan, J.C. A Review of Polymer Electrolytes: Fundamental, Approaches and Applications. Ionics 2016, 22, 1259–1279. [Google Scholar] [CrossRef]
- Long, L.; Wang, S.; Xiao, M.; Meng, Y. Polymer Electrolytes for Lithium Polymer Batteries. J. Mater. Chem. A 2016, 26, 10038–10069. [Google Scholar] [CrossRef]
- Zhou, D.; Shanmukaraj, D.; Tkacheva, A.; Armand, M.; Wang, G. Polymer Electrolytes for Lithium-Based Batteries: Advances and Prospects. Chem. 2019, 5, 2326–2352. [Google Scholar] [CrossRef]
- Barbosa, J.C.; Gonçalves, R.; Costa, C.M.; Lanceros-Méndez, S. Toward Sustainable Solid Polymer Electrolytes for Lithium-Ion Batteries. Omega 2022, 7, 14457−14464–content content. [Google Scholar] [CrossRef] [PubMed]
- Longmire, M.L.; Watanabe, M.; Zhang, H.; Wooster, T.T.; Murray, R.W. Voltammetric Measurements of Ultraslow Diffusion Rates in Polymeric Media with Microdisk Electrodes. Anal. Chem. 1990, 62, 747–752. [Google Scholar] [CrossRef]
- Porat, Z.; Crooker, J.C.; Zhang, Y.; Le Mest, Y.; Murray, R.W. iRUNC Advantages and Real Geometrical Dimensions of Microband Electrodes. Anal. Chem. 1997, 69, 5073–5081. [Google Scholar] [CrossRef]
- Wilson Poupart, M.; Velazquez, C.S.; Hassett, K.; Porat, Z.; Haas, O.; Terrill, R.H.; Murray, R.W. Limit of Slow Diffusion to Electrodes: Molecule-Scale Diffusion Paths in a Redox Melt. J. Am. Chem. Soc. 1994, 116, 1165–1166. [Google Scholar] [CrossRef]
- Pinkerton, M.J.; Le Mest, Y.; Zhang, H.; Watanabe, M.; Murray, R.W. Solid-state voltammetry and self-diffusion dynamics of a linear monotagged redox polymer: omega-ferrocenecarboxamido-alpha-methoxypoly(ethylene oxide). J. Am. Chem. Soc. 1990, 112, 10, 3730–3736. [Google Scholar] [CrossRef]
- Geng, L.; Reed, R.A.; Longmire, M.L.; Murray, R.W. Solid-state linear sweep voltammetry: a probe of diffusion in thin films of polymer ion conductors on microdisk electrodes. J. Phys. Chem. 1987, 91(11), 2908–2914. [Google Scholar] [CrossRef]
- Watanabe, M.; Longmire, M.L.; Murray, R.W. A study of ferrocene diffusion dynamics in network poly (ethylene oxide) polymer electrolyte by solid-state voltammetry. J. Phys. Chem. 1990, 94, 2614–2619. [Google Scholar] [CrossRef]
- Wooster, T.T.; Longmire, M.L.; Watanabe, M.; Murray, R.W. Diffusion and Heterogeneous Electron-Transfer Rates in Acetonitrile and in Polyether Polymer Melts by Alternating Current Voltammetry at Microdisk Electrodes. J. Phys. Chem. 1991, 95, 5315. [Google Scholar] [CrossRef]
- “Electrochemical Methods - Fundamentals and Applications”, A. J. Bard and L. R. Faulkner: Wiley, NY 1980, P. 218.
- ibid, p. 410.
- Barbour, C.J.; Murray, R.W.; Parcher, J.F. Evaluation of a Theory for the Sensitivity and Selectivity of a Polymer Diffusion-Plasticization Based Solid-State Electrochemical Gas Chromatography Detector. Anal. Chem. 1991, 63, 604–610. [Google Scholar] [CrossRef]
- C. S. Velazquez, R. W. Murray. Synthesis and voltammetry of a tetrathiafulvalene polymer electrolyte melt. J. Electroanal. Chem. 1995, 396(1), 349–357. [CrossRef]
- Porat, Z.; Crooker, J.C.; Zhang, Y.; . Le Mest, Y.; Murray, R.W. iRUNC Advantages and Real Geometrical Dimensions of Microband Electrodes Anal. Chem. 1997, 69, 5073–5081. [Google Scholar]
- Masaro, L.; Zhu, X.X. Physical models of diffusion for polymer solutions, gels and solids. Prog. Polym. Sci. 1999, 24, 31–75. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physical Polymer Science, Wiley Interscience, 2006.
- Grinsted, R.A.; Clark, L.; Koenig, J.L. Study of cyclic-desorption into poly (methyl methacrylate) rods using NMR imaging. Macromolecules 1992, 25, 125–1241. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Kröger, M.; Clarke, N. Modeling of EntanglN.ed Polymer Diffusion in Melts and Nanocomposites: A Review. Polymers 2019, 11, 8–905. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.; Halat, D.M.; Villaluenga, I. Timachova, K. P. Diffusion and Migration in Polymer Electrolytes 2020.
Figure 1.
Cyclic voltammograms for 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1M (O/Li=16:1). Working electrode: Pt microband, w=2.7µm. Ref. electrode: parallel Ag bands, d=3µm. A) v = 1mV/s, S=2nA. B) v = 200mV/s. S=10 nA.
Figure 1.
Cyclic voltammograms for 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1M (O/Li=16:1). Working electrode: Pt microband, w=2.7µm. Ref. electrode: parallel Ag bands, d=3µm. A) v = 1mV/s, S=2nA. B) v = 200mV/s. S=10 nA.
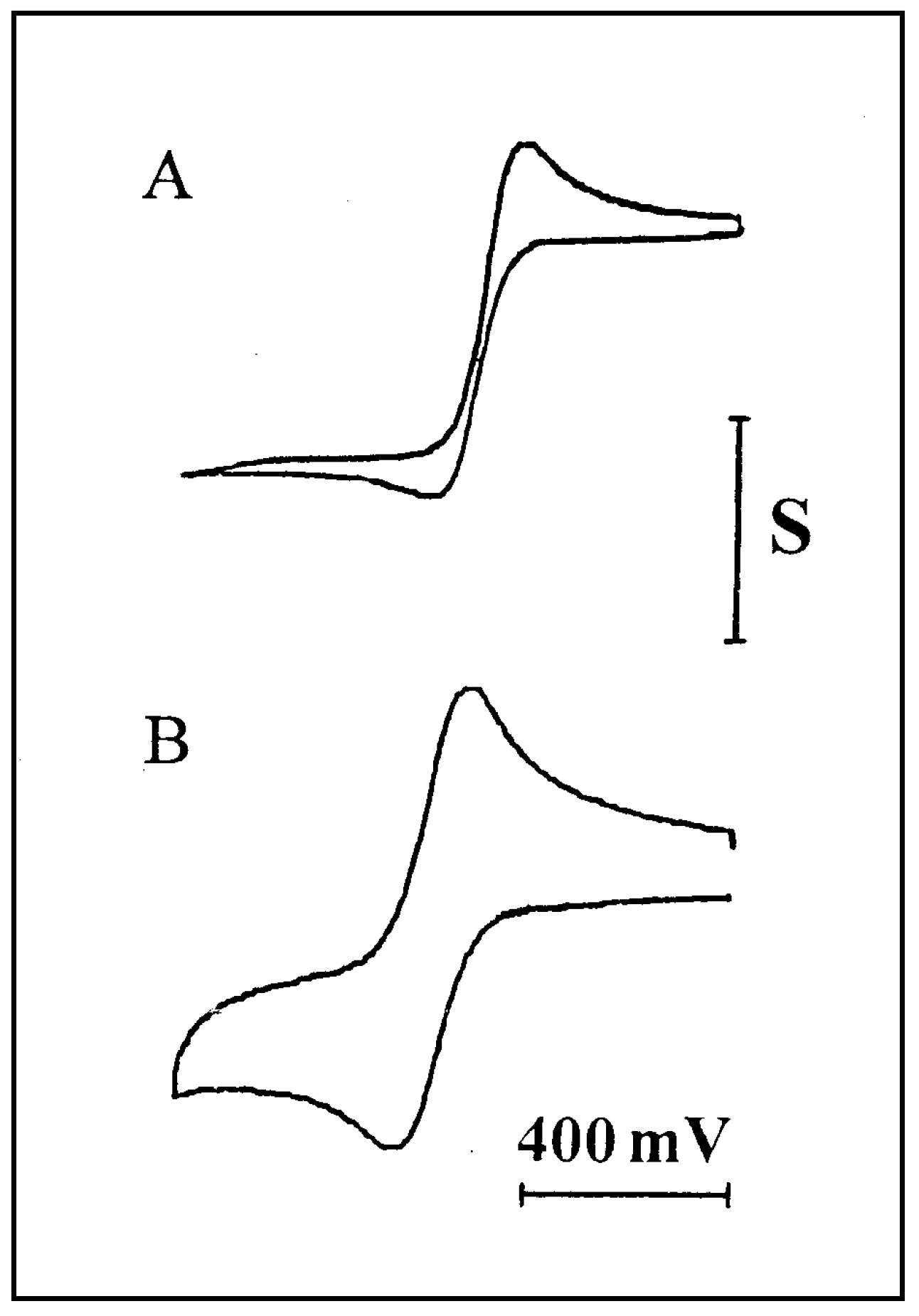
Figure 2.
Plot of log i vs. log v for three sets of CV experiments of 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1M (O/Li=16:1) at various temperatures. (●) t=22.8 oC, slope=0.40. (■) t=12.6 oC, slope=0.35. (▲) t=2.6 oC, slope=0.33.
Figure 2.
Plot of log i vs. log v for three sets of CV experiments of 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1M (O/Li=16:1) at various temperatures. (●) t=22.8 oC, slope=0.40. (■) t=12.6 oC, slope=0.35. (▲) t=2.6 oC, slope=0.33.
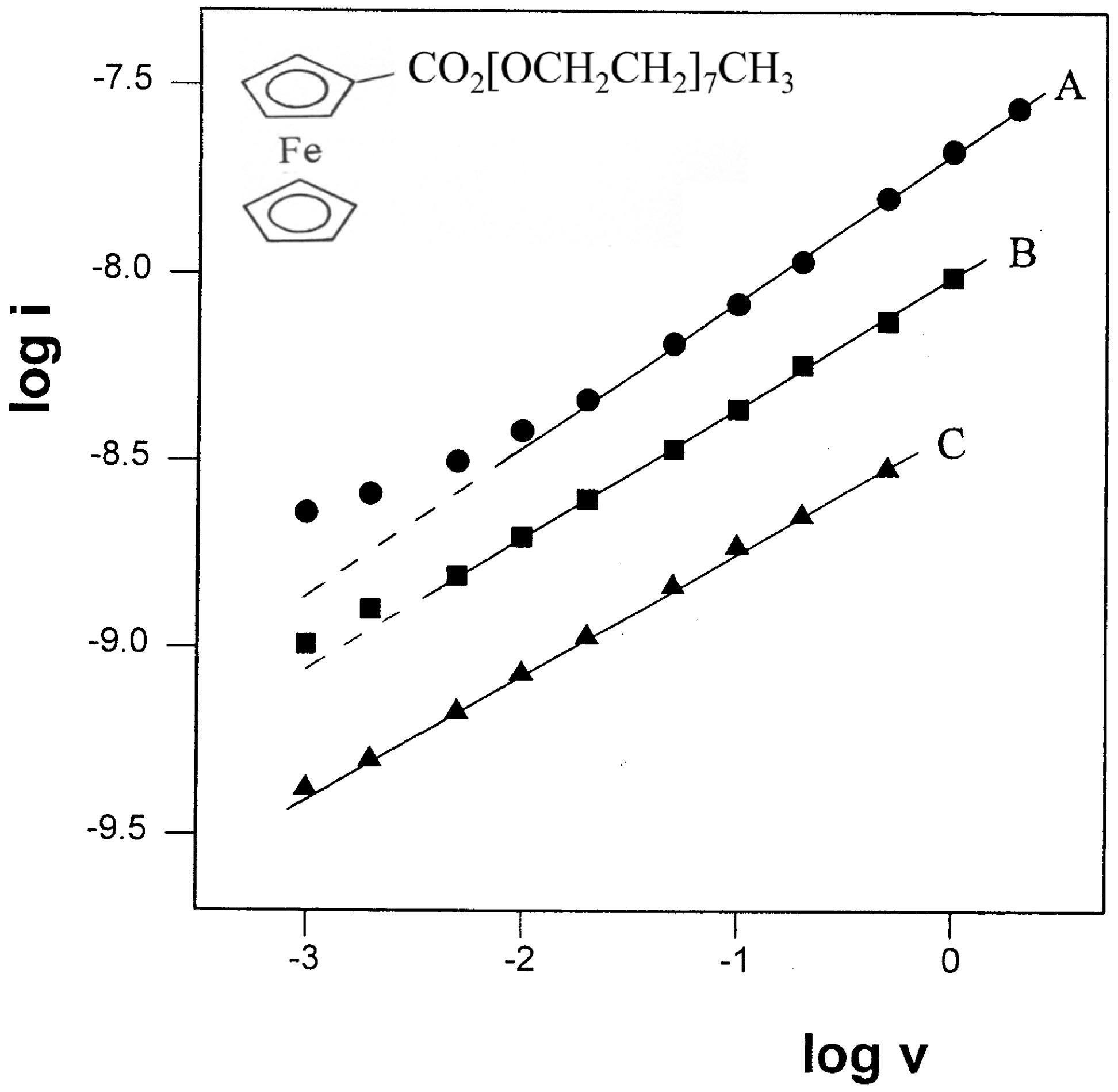
Figure 3.
Plot of log i vs. log v for two sets of CV of 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1 M, performed with (■) and without (●) applied positive feedback at room temperature. Slope = 0.33. Inset: representative voltammograms, recorded at 500 mV/s. A) With applied positive feedback, ΔEp=156mV. B) No applied positive feedback, ΔEp=218mV.
Figure 3.
Plot of log i vs. log v for two sets of CV of 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1 M, performed with (■) and without (●) applied positive feedback at room temperature. Slope = 0.33. Inset: representative voltammograms, recorded at 500 mV/s. A) With applied positive feedback, ΔEp=156mV. B) No applied positive feedback, ΔEp=218mV.
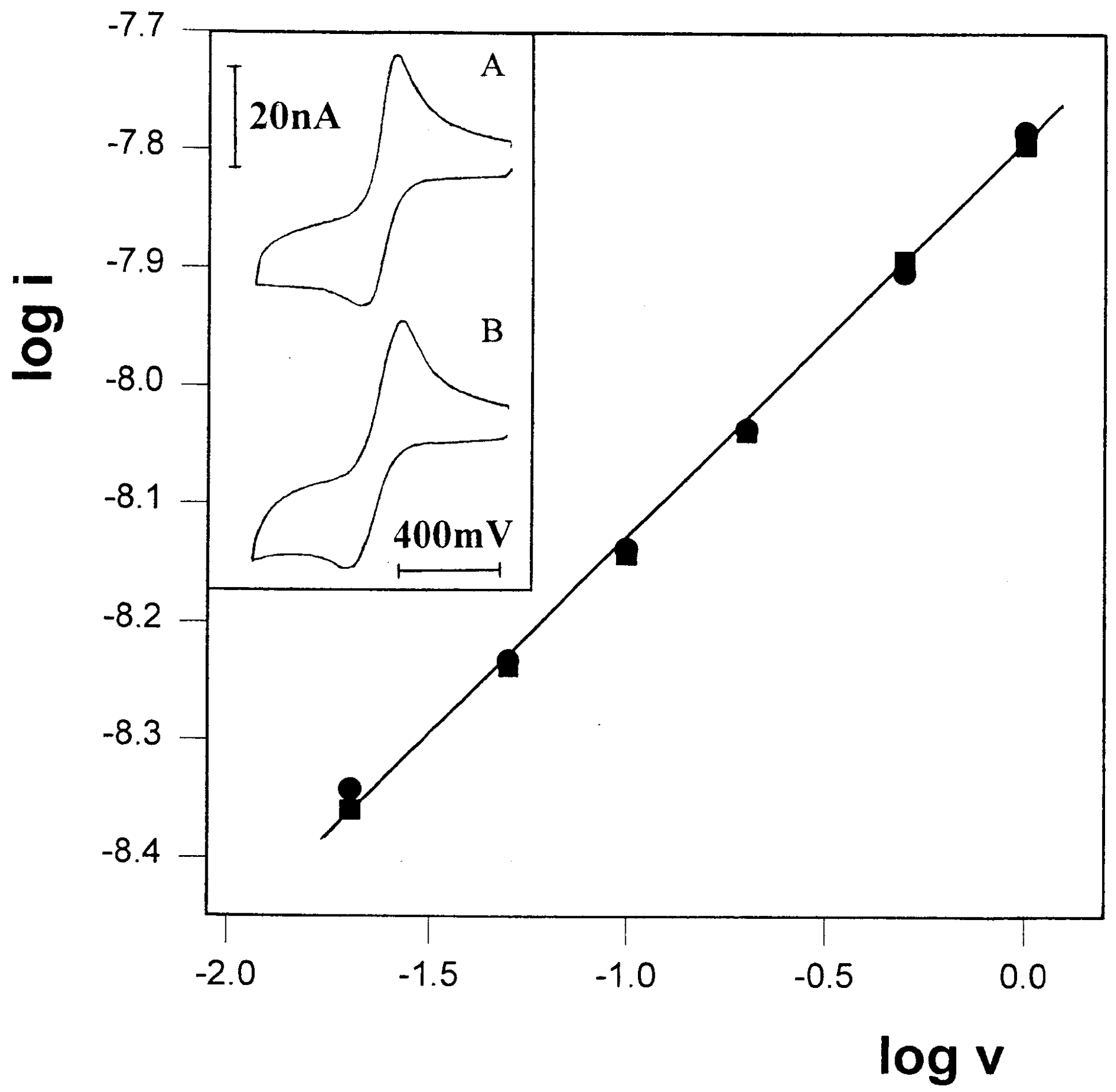
Figure 4.
A: Three of the cyclic voltammograms of 10 mM FcCO2PEG(350)CH3 out of a series recorded in PPG10,000/LiSO4CF3 1.1M at 23 °C. a) 1mV/s, S=2nA. b) 20mV/s, S=4nA. c) 500mV/s. S=8nA. B: The corresponding plot of log i vs. log v. Slope=0.26.
Figure 4.
A: Three of the cyclic voltammograms of 10 mM FcCO2PEG(350)CH3 out of a series recorded in PPG10,000/LiSO4CF3 1.1M at 23 °C. a) 1mV/s, S=2nA. b) 20mV/s, S=4nA. c) 500mV/s. S=8nA. B: The corresponding plot of log i vs. log v. Slope=0.26.

Figure 5.
Plot of log i vs. log v for three sets of cyclic voltammograms of 12mM TTF-(OCH2)3CH3 in propylene glycol/0.1 M LiClO4. (●) t=-23.7 oC. (■) t=-12.0 oC. (▲) t=1.0 oC. All lines are drawn with slopes of 0.5. Inset: A representative voltammogram recorded at scan-rate of 20 mV/s.
Figure 5.
Plot of log i vs. log v for three sets of cyclic voltammograms of 12mM TTF-(OCH2)3CH3 in propylene glycol/0.1 M LiClO4. (●) t=-23.7 oC. (■) t=-12.0 oC. (▲) t=1.0 oC. All lines are drawn with slopes of 0.5. Inset: A representative voltammogram recorded at scan-rate of 20 mV/s.
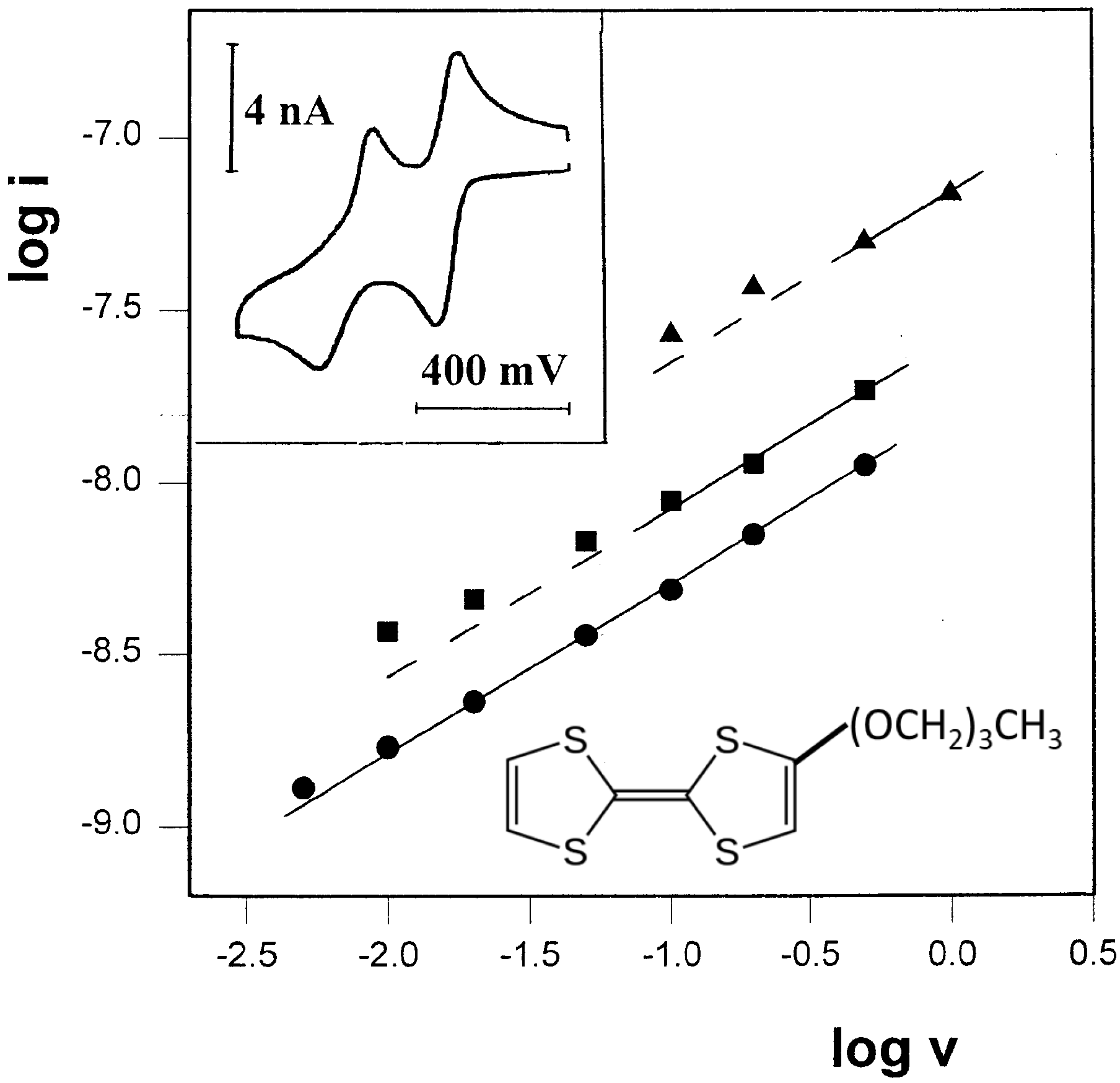
Figure 6.
Plot of log i vs. log v for the oxidation peak currents of 10 mM FcCO2PEG(350)CH3 in glycerol/LiSO3CF3, 1 M, at -27 oC. Slope=0.49. Inset: A representative voltammograms, recorded at scan-rate of 1 mV/s.
Figure 6.
Plot of log i vs. log v for the oxidation peak currents of 10 mM FcCO2PEG(350)CH3 in glycerol/LiSO3CF3, 1 M, at -27 oC. Slope=0.49. Inset: A representative voltammograms, recorded at scan-rate of 1 mV/s.
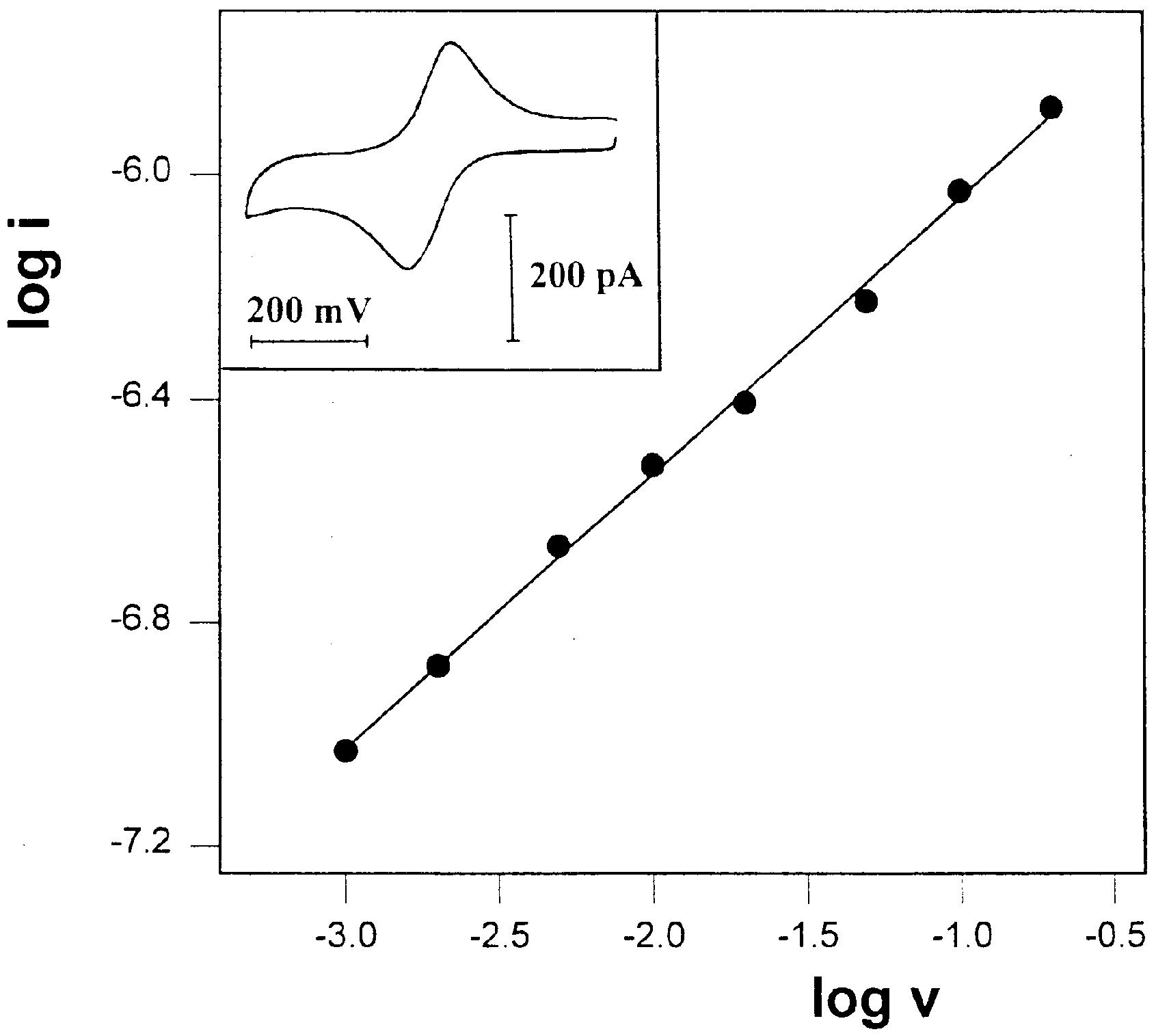
Figure 7.
Plot of log i vs. log v for the first oxidation peaks (●) and the first reduction peaks on the reverse scan (■) in the CV of 12mM TTF-(OCH2)3CH3 in propylene glycol/0.1M LiClO4, recorded at room temperature. Inset: A representative voltammograms, recorded at scan-rate 10 mV/s.
Figure 7.
Plot of log i vs. log v for the first oxidation peaks (●) and the first reduction peaks on the reverse scan (■) in the CV of 12mM TTF-(OCH2)3CH3 in propylene glycol/0.1M LiClO4, recorded at room temperature. Inset: A representative voltammograms, recorded at scan-rate 10 mV/s.
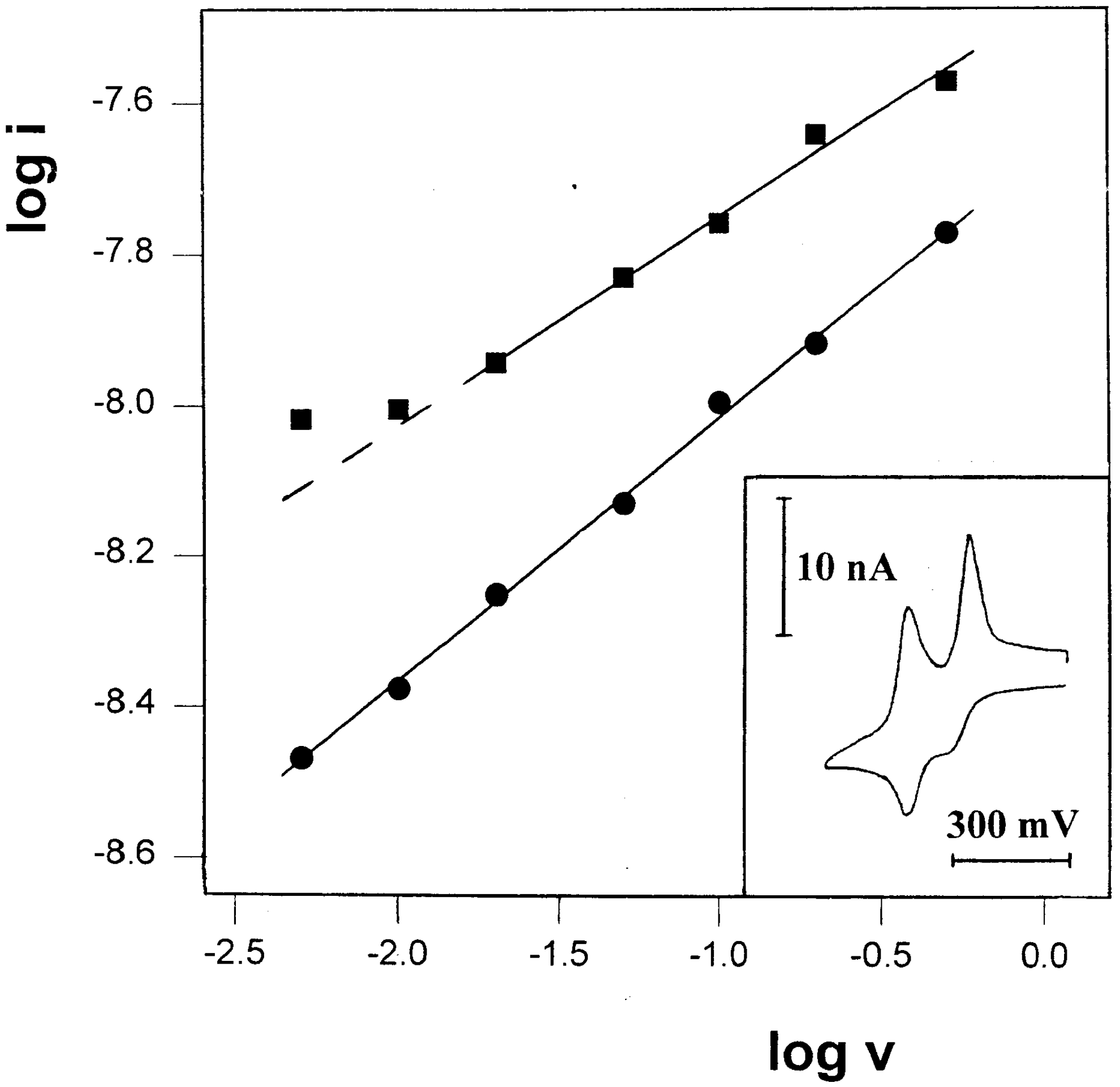
Figure 8.
Chronoamperometry of 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1M, recorded at room temperature. Inset: CV in the same system, showing the potential steps for the background curve (A) and for FcCO2PEG(350)CH3 (B).
Figure 8.
Chronoamperometry of 11.3 mM FcCO2PEG(350)CH3 in PPG 4000/LiSO4CF3, 1.1M, recorded at room temperature. Inset: CV in the same system, showing the potential steps for the background curve (A) and for FcCO2PEG(350)CH3 (B).
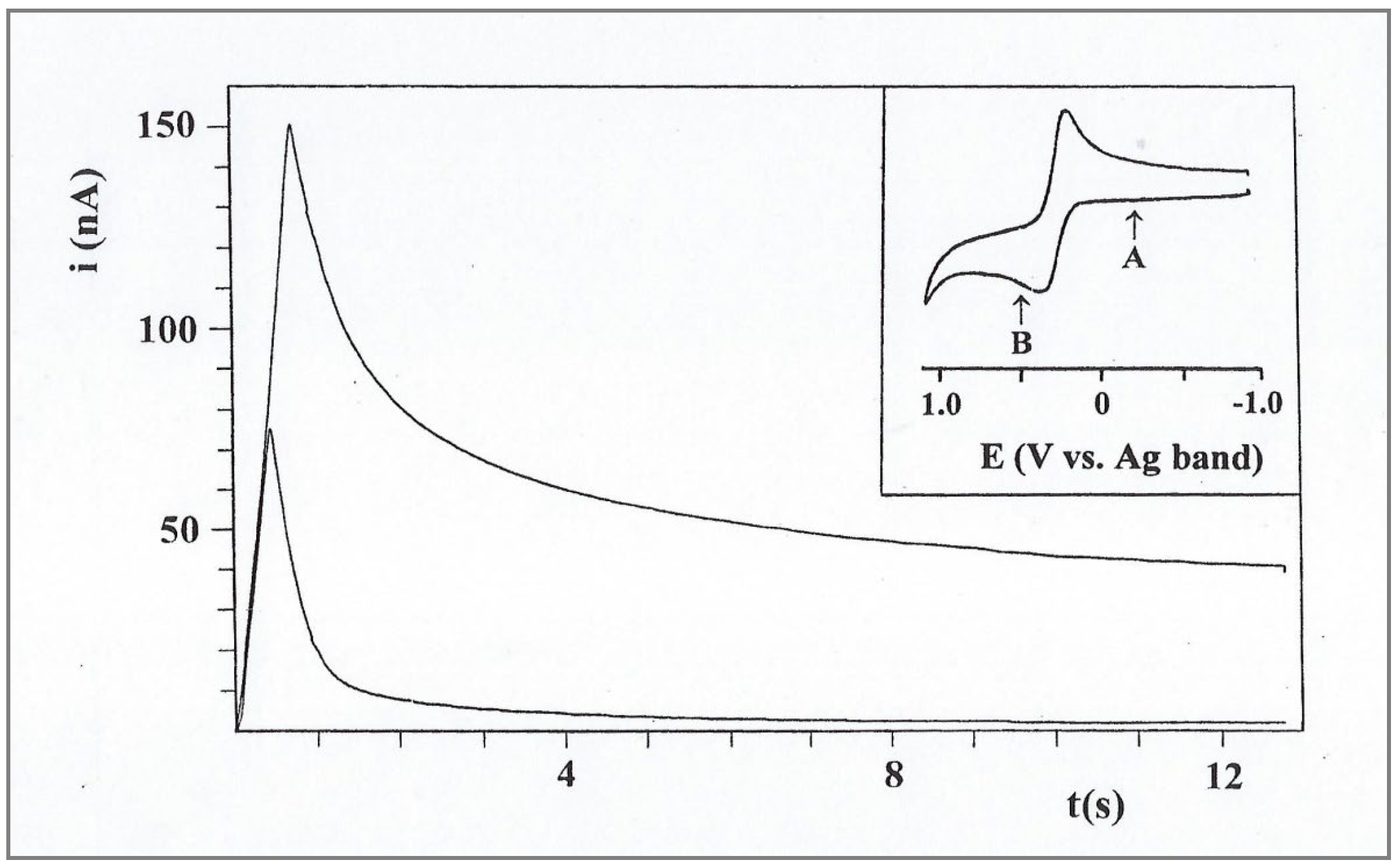
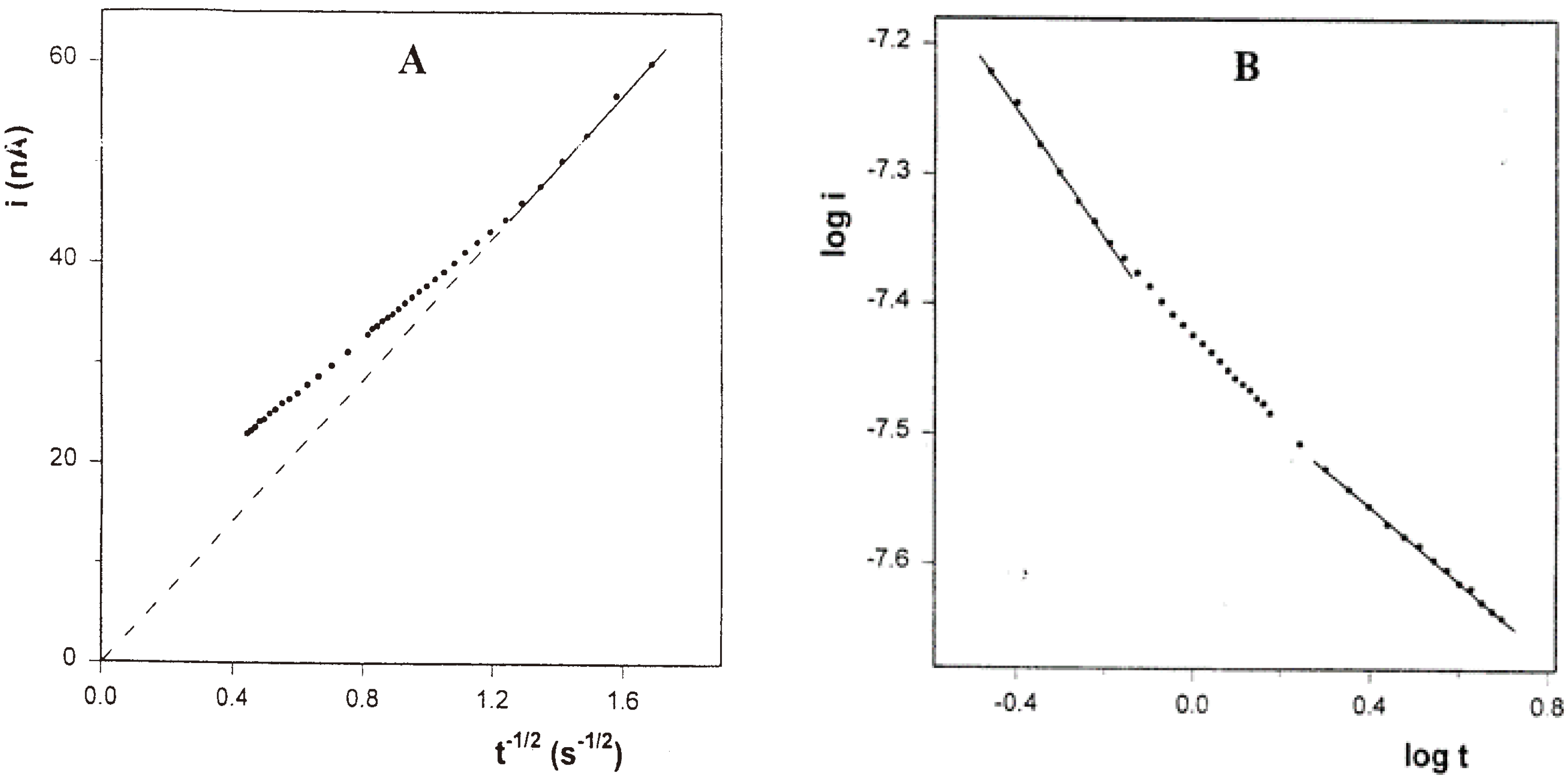
Figure 9.
A) Cottrell plot for the chronoamperogram in Figure 8. B) Plot of log i vs. log t corresponding to the same chronoamperogram.
Figure 9.
A) Cottrell plot for the chronoamperogram in Figure 8. B) Plot of log i vs. log t corresponding to the same chronoamperogram.
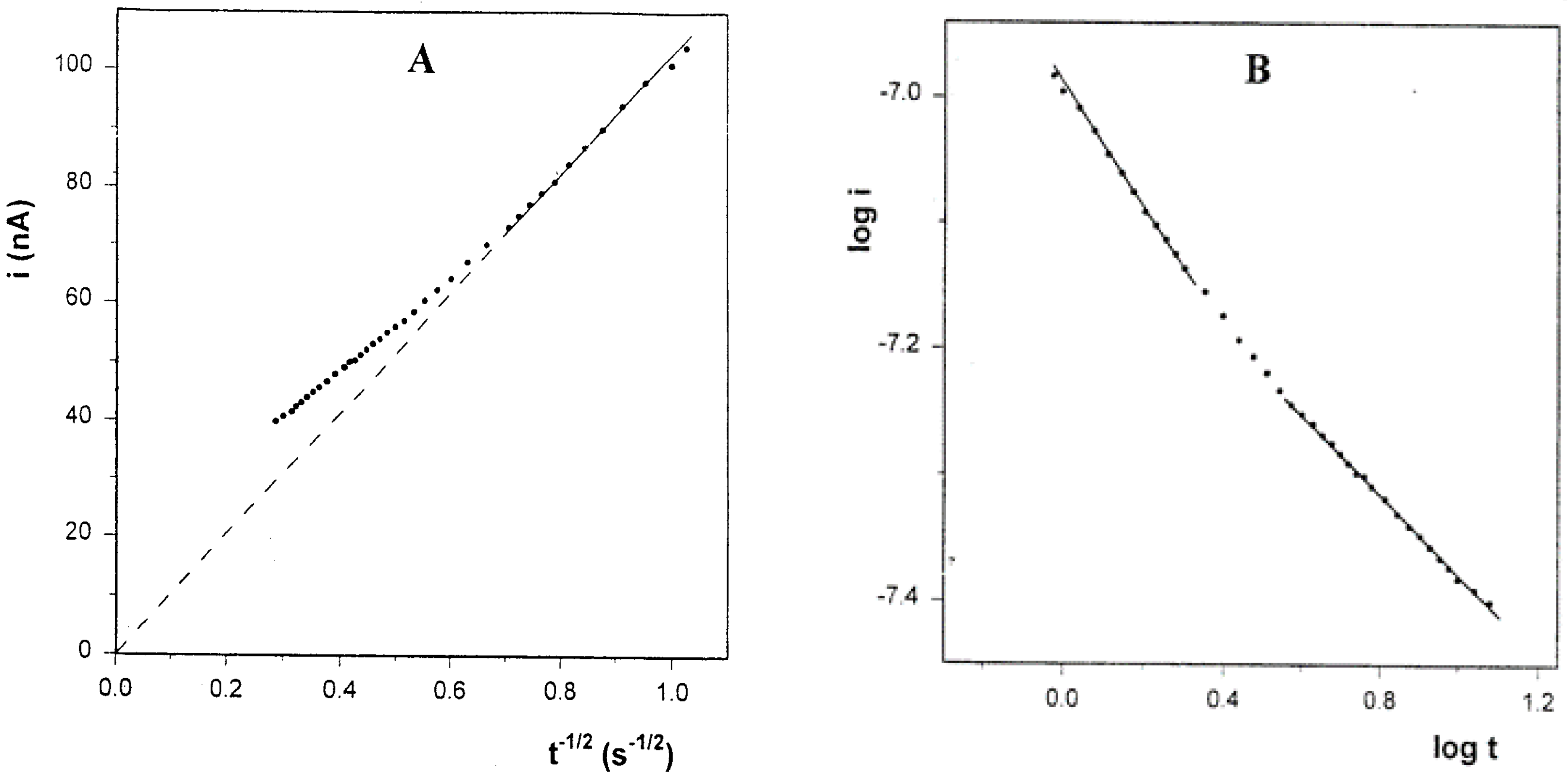
Figure 10.
A) Cottrell plot for the chronoamperogram of 10 mM FcCO2PEG(350)CH3 in PPG10,000/LiSO4CF3 1.1 M at room temperature. B) Plot of log i vs. log t corresponding to the same chronoamperogram.
Figure 10.
A) Cottrell plot for the chronoamperogram of 10 mM FcCO2PEG(350)CH3 in PPG10,000/LiSO4CF3 1.1 M at room temperature. B) Plot of log i vs. log t corresponding to the same chronoamperogram.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



