
Submitted:
11 April 2024
Posted:
12 April 2024
You are already at the latest version
Abstract
Background and Objectives Neurofibromatosis 1 (NF1 MIM # 162200), also known as von Recklinghausen disease, is among the most common autosomal dominantly transmitted disorders characterized mainly by neurocutaneous manifestations such as café au lait spots, intertriginous freckling, various types of neurofibromas (cutaneous neurofibromas or plexiform tumors), neurological manifestations, and bone abnormalities. It occurs with an incidence of 1:3000–1:4000 cases. Cone-rod dystrophies belong to the large group of retinal dystrophies, a highly heterogeneous group of disorders clinically manifested mainly by damage to cone and rod cells and have a broad spectrum of clinical manifestations. Many genes are involved in their etiology, with more than 100 currently identified, including the CRX gene responsible for the autosomal dominant form of cone-rod dystrophy (ad CORD). Materials and Methods: The authors present an intriguing case of a patient diagnosed with Neurofibromatosis type 1 (NF1) as an infant and ocular involvement (myopia and astigmatism onset at age 15), which yielded a surprising molecular testing result. Results: Molecular DNA analysis identified a rare occurrence of two mutational variants, including a pathogenic variant in the NF1 gene, c.3871-2A, and a likely pathogenic variant in the CRX gene c.119G>A. Conclusions: The authors present this case to highlight the surprises offered by molecular testing in a condition where the diagnosis is usually based on clinical criteria, thus providing patients with competent genetic advice.
Keywords:
neurofibromatosis type 1
; cone-rod dystrophy
; molecular diagnosis
; rare association
1. Introduction
Neurofibromatosis Type I (NF1) is one of the neurocutaneous syndromes along with von Hippel-Lindau syndrome, Sturge-Weber syndrome, and the tuberous sclerosis complex [1,2]. It is caused by pathogenic variants in the NF1 gene located on chromosome 17q11.2. The consequence of these mutations is overstimulation of the RAS/MAPK signaling pathway, leading to neurocutaneous manifestations and bone abnormalities [3]. The hallmark clinical findings include café-au-lait spots and neurofibromas, which can vary in size, number, and location [4,5]. Along with Gardner syndrome and Cowden syndrome, it is among the most common neoplastic hamartoma syndromes [6]. Penetrance is nearly 100%, and the expression of the disorder differs from one affected family to another and between individuals of the same family. NF1 is a disorder diagnosed in the vast majority of cases clinically, taking into account the clinical criteria proposed by the National Institutes of Health (NIH) in 1988 [7]. As the signs of the disorder appear gradually in evolution and are age-dependent, it is necessary to monitor these patients for several years before a clear diagnosis can be established. By the age of 8 years, approximately 97% of patients are diagnosed based on clinical criteria [8,9]. Genetic testing is an important criterion for confirming the diagnosis of the disorder and has been introduced in the new re-vised diagnostic criteria in 2021 [10,11].
Cone-rod dystrophy (CORD) is one of a large group of retinal dystrophies that have a broad spectrum of largely overlapping clinical manifestations; this makes diagnosis and therefore therapeutic approaches difficult [12]. Retinal dystrophies are characterized by an initial loss of central visual acuity and color vision, followed by a loss of peripheral vision. Although the onset of the disorder may occur in the first 10 years of life (usually during school years) with a decrease in visual acuity that does not improve after optical correction with glasses, most cases are diagnosed in adulthood [13]. Subsequently, the rod cells responsible for night vision are also affected. Photophobia and dyschromatopsia may also be associated. More than 90 genes and different patterns of inheritance associated with the condition have now been identified: autosomal dominant, autosomal recessive, or X-linked dominant. In autosomal dominant (adCORD) forms, more than 10 genes are identified, one of the most common being the Cone-Rod Homeobox gene (CRX, OMIM #602225). The encoded protein is a photoreceptor-specific transcription factor with a major role in photoreceptor differentiation and development [14]. Mutations in this gene cause different phenotypes, such as macular dystrophy, cone-rod dystrophy, and Leber’s congenital amaurosis [15]. The article aims to present a coincidental association of two mutational variants in two different genes in a patient initially diagnosed with NF1. Although NF1 is clinically diagnosed in most cases, molecular diagnosis can sometimes offer surprises, as in the case of our patient, who was identified with a likely pathogenic mutational variant in the CRX gene. Thus, we submit that a thorough clinical diagnosis combined with a molecular diagnosis provides an accurate diagnosis by identifying possible associated pathologies that would not otherwise have been suspected in the patient. A precise diagnosis is essential for competent, accurate genetic counseling and family screening.
2. Materials and Methods
2.1. Case Report
The authors present the case of a 19-year-old patient who was registered with the Bihor Regional Center for Medical Genetics and was referred by her family doctor during her infancy for genetic counseling.
2.2. Laboratory Investigations
The main lab tests performed involved carbohydrate, lipid (cholesterol, lipids, and triglycerides), protein (proteinemia, serum protein electrophoresis), mineral (calcemia, phosphatemia, and magnesemia) metabolisms, hormonal evaluation, and alpha-fetoprotein.
2.3. Imagistic Investigations
They targeted an MRI of the brain, and ocular computer tomography (OCT).
2.4. Molecular Investigations
Written informed consent was obtained from the mother before participation in the study. A next-generation sequencing technique was applied using the Illumina MiSeq platform, which allows the analysis of coding sequences in genomic DNA. Genomic DNA fragmentation was performed, followed by coding sequence amplification and library generation using the Illumina TruSight One Sequencing Panel kit at the Regional Genomic Centre Timisoara, Romania. End-to-end bioinformatics algorithms, including nitrogen base alignment, primary filtering of low-quality reads and likely artifacts, and variant annotation, were implemented using Isis (Analysis Software) 2.5.2.3; BWA (Aligner) 0.7.9a-isis-1.0.1; SAMtools 0.1.18 (r982:295); GATK (v1.6-23-gf0210b3)1.7. Data analysis was performed at the level of current knowledge using the following databases: UCSC Genome Browser, OMlM (Online Mendelian Inheritance in Man), and DGV (Database of Genomic Variants). All disease-causing variants reported in HGMD®, Clin-Var (class 1), and all variants with a minor allele frequency (MAF) less than 1% in the Ex-Ac database were considered. Only variants correlated with the phenotype for which the patient was referred were reported. Variants were interpreted according to the ACMG guidelines (1).
3. Results
3.1. Personal Data
The patient is the second child in the family. Family history: young parents, no consanguineous relationship. The paternal grandfather, the father, and the father’s brother have numerous café au lait spots and skin neurofibromas. Past medical history: since birth, she has had six café au lait spots on her chest and abdomen, ranging in size from 7/4 cm to 2/1.5 cm, suggestive of neurofibromatosis. In evolution, the number of café au lait spots increased, and axillary freckles and posterior thoracic neurofibromas were reported. Since the age of 15, she has been registered with the ophthalmology service for myopia and astigmatism and later Lisch nodules. At the age of 16, she had a uni-grand mal seizure lasting about 30 seconds.
3.2. Clinical Evaluation of the Patient
Phenotypical traits at the age of 17: normal stature (166 cm) and weight (66 kg). The patient presents with multiple café au lait spots (more than 10) disseminated over the anterior and posterior thorax, neck, abdomen, upper and lower limbs, thoracic neurofibromas, and axillary freckles (Figure 1, Figure 2, Figure 3 and Figure 4).
3.3. Laboratory Investigations
Biochemical, hematological, and hormonal (thyroid hormones) tests were within normal limits, and alpha-fetoprotein, an important tumoral factor, was also normal.
3.4. Interdisciplinary Consultations and Imagistic Investigations
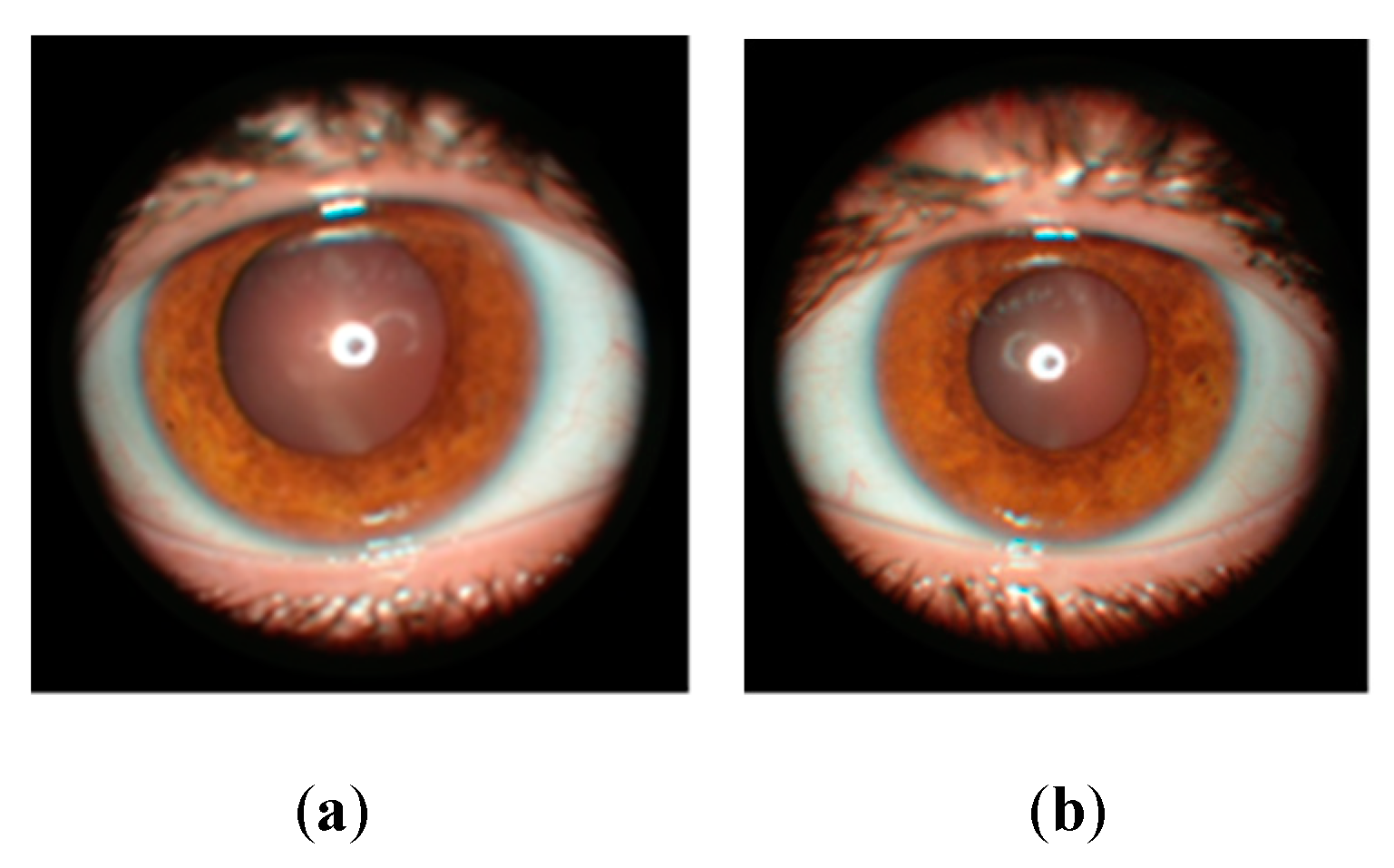
Ophthalmological examination revealed the presence of myopia and astigmatism. The visual acuity at the right eye was 0.8 with correction, and 1 with correction at the left eye. At the anterior pole, Lisch nodules are visualized (Figure 5) in both eyes. Fundus examination of the eye revealed well-contained optic nerve papillae and normal macular reflex. Optical coherence tomography (OCT Nidek DuoScan 3300) revealed a normal macular profile. The field of vision is within normal limits in both eyes.
3.5.
Brain MRI highlights several oval lesions in the hypersignal T2, FLAIR, and isosemnal T1, in the anterior region of the right mesencephalon (measuring approximately 5-7 mm), in the bilateral thalamus (measuring approximately 9 mm on the right and 7 mm on the left) and in the right capsulo-lenticular region (measuring approximately 5 mm, with the appearance of glioma). All lesions are relatively well delimited, without perilesional edema. The corpus callosum appears slightly hypertrophic but without changes in its signal. Electroencephalography (EEG) did not reveal pathological graphoelements.
3.6. Molecular Investigations
Using panel sequencing analysis two mutational variants have been detected: the first one with pathogenic significance in the NF1 gene, and the second in the CRX gene, with likely pathogenic significance.
Regarding the first mutation, a splice site null variant in heterozygous status c.3871-2A>T was found in the NF1 gene, with an 89X coverage in the gene’s area. Due to splice site localization is a loss of function variant. This variant is not present in populational databases (without ExAC and GnomAD frequency) thus suggesting that it is not a benign variation in the general population, and is not currently identified in people with NF1. In silico models predicting the effect of DNA modification on the protein estimate a pathogenic effect (6 pathogen predictions from BayesDel_addAF, DANN, EIGEN, FATHMM-MKL, MutationTaster and scSNV-Splicing versus 0 benign prediction). For these reasons, this variant has been classified with pathogenic significance according to the ACMG guide (1).
The second mutation variant, that of the CRX genes is a null missense variant in heterozygous status c.119G>A, with a 224X coverage in the variant region. It is not present in population databases (without frequency ExAC and GnomAD) indicating that it is not a benign variation in the population. In silico models predicting the effect of DNA modification on the protein estimate a pathogenic effect (12 pathogen predictions from BayesDel_addAF, DANN, DEOGEN2, EIGEN, FATHMM-MKL, LIST-S2, M-CAP, MVP, MutationAssessor, mutationTaster, PrimateAI, and SIFT versus 0 benign predicts. Thus, for the reasons described above, this variant has been classified with likely pathogenic significance according to the ACMG guide (1) and must be correlated with the patient’s phenotype.
4. Discussion
4.1. NF1 and CRX Genes
4.1.1. NF1 Gene
The NF1 gene is located on the long arm of chromosome 17 (17q11.2). It is a large gene of about 350 kb in length and contains 55 constitutive exons and 5 alternatively spliced exons [16]. NF1 genes encode neurofibromin, a large protein that consists of six functional domains with a role in regulating intracellular signaling pathways.
Neurofibromin is a regulator of the RAS/MAPK signaling pathway. If it loses its function, out-of-control RAS activity occurs, leading to uncontrolled cell division and the triggering of tumorigenesis by disrupting the cell cycle [17].
The Human Gene Mutation Database (HGMD; http://www.hgmd.org/) lists more than 3,600 pathogenic NF1 variants that are found throughout the length of the gene at both the exon and intron levels, affecting the splice region. http://www.hgmd.org/) [18]. These include microdeletions that may cover the entire NF1 gene; CNVs (copy number variants); frameshift, nonsense, and missense; and splice-site [19]. The prevalence of pathogenic variants in the NF1 gene is over 0.5% (31 different pathogenic variants), but the percentage of extremely rare or privately encountered variants is over 46% [20,21]. On the other hand, 90% of mutations occur in the intragenic region, with less than 10% being deletions that span the entire gene or are located in lacking genomic regions [22].
4.1.2. CRX Gene
The CRX gene is located on chromosome 19 (19q13.33); it belongs to the homeobox gene family and plays a major role in the development of the brain, cerebellum, eyes, and pineal gland. CRX encodes a cone-rod homeobox protein transcription factor that is a critical K50 homeodomain transcription factor with an important role in the differentiation and proper function of photoreceptors. This protein contains 299 amino acids [23].
The CRX gene is a small 25-kb stretch gene containing four exons [24]. Commonly encountered pathogenic variants are missense and in-frame deletions. More than 45 mutations with retinal damage are currently reported in the literature [12,25]. Most CRX mutations cause the autosomal dominant form of CORD, accounting for 5–10% of all dominant forms of CORD [26,27]. Mutations in CRX genes cause a wide range of retinopathies, including retinitis pigmentosa, cone-rod dystrophy, and the dominant form of Leber congenital amaurosis, a disorder that is usually autosomal recessive [28,29].
4.2. Neurofibromatosis
4.2.1. Clinical Aspects
Neurofibromatosis is a multisystem disorder with neurocutaneous involvement. It is characterized by skin changes such as café au lait spots, intertriginous freckles, and the appearance of various neurofibromas in both the central nervous system and the peripheral nervous system There are several known types of neurofibromatosis: neurofibromatosis type 1, the most common type, present in 90% of cases; neurofibromatosis type 2 or NF2-related schwannomatosis (NF2, MIM #101000), found in 3% of patients; SMARCB1-related schwannomatosis (MIM #162091), and LZTR1-related schwannomatosis (MIM #615670), which occurs much less frequently, less than 1% [30]. Along with Noonan, Costello, Legius, and Cardiofaciocutaneous syndromes, it is part of a large group of RASopathies and is the first condition identified as belonging to this signaling pathway. [10,31]. Neurofibromatosis type 1 occurs as a result of pathogenic variant in the NF1 gene and is autosomal dominant inherited. In 50% of patients, the disorder occurs spontaneously due to de novo mutations. Penetrance is almost 100%. It is a progressive disorder, with the life expectancy of these patients decreasing by 15% compared to the general population. Our patient meets the criteria for NF1 by presenting multiple café au lait spots, axillary freckles, cutaneous neurofibromas, and cortical gliomas. The molecular diagnosis has detected a pathogenic variant in the NF1 gene.
4.2.2. Cutaneous manifestations in NF1
Café au Lait Spots and Freckling
The first sign that draws attention to the disorder is the presence of café au lait spots since birth. They multiply gradually, become evident in early childhood, and grow in proportion to the body. Another major sign of NF1 is the presence of axillar or inguinal freckling. There are small cutaneous tumors, usually measuring between 2 and 4 mm, in small groups. Compared with café au lait spots, the freckling develops later in evolution, usually by the age of 3-5 years. These skin changes are seen in about 70% of the affected individuals.
Neurofibromas
Neurofibromas are tumors of the peripheral nerves that cause small, soft papules in the color of the skin or as small subcutaneous nodules [32]. They can be classified into cutaneous neurofibromas and plexiform neurofibromas. Cutaneous neurofibromas appear as soft papules between 1 and 2 cm in size; they usually appear at puberty and then increase in number and size until about 20 years of age. Unlike cutaneous neurofibromas, plexiform tumors pathognomonic for NF1 occur from birth, in about 30% of patients, and have an increased risk of malignant transformation (malignant peripheral nerve sheath tumor, MPNST) [33].
Our patient presents with multiple cafe au lait spots disseminated on the neck, anterior and posterior thorax, abdomen, buttocks, and lower limbs. Axillary freckles were observed at the age of 11 years when the first cutaneous neurofibroma appeared.
4.2.3. Ocular Manifestations in NF1
NF1 presents a challenge to ophthalmologists in terms of early diagnosis, clinical picture, and treatment. The most representative brain tumor present in patients with NF1 is known to be low-grade optical glioma, which appears in 15% of patients during early childhood. [35]. In the last few years, considerable advances in multimodality imaging in ophthalmology have led to the description of new ocular manifestations in NF1, including choroidal abnormalities (CA), hyperpigmented spots, and retinal vascular abnormalities. The diagnostic criteria for NF1 have recently been revised to include the presence of two or more CAs as an alternative to the presence of Lisch nodules. Choroidal abnormalities are more common in adults (80–90%). In children, they occur at a much lower rate (60–78.6%), but compared to Lisch nodules, they are much more common [17,34,36]. The case presented by the authors shows ocular refractive disorders: myopia and astigmatism, respectively Lisch nodules.
4.2.4. Cerebral Manifestations in NF1
Cerebral gliomas are common and usually asymptomatic, with most of them being accidentally detected on an MRI [1]. Their incidence is between 2 and 5% and can occur at any age. The average age at diagnosis of these brain tumors is 13 years. Young adults with NF1 have a 10- to 50-fold increased risk of developing these deadly cancers [37]. The case presented by the authors has brain gliomas identified by brain MRI at the age of 15. The patient presented with a grand mal seizure episode at the age of 16 years. It is possible that the epilepsy was caused by the lesion of the corpus callosum, its hypertrophy may be due to small lesions that are still not visible on a brain MRI. Electroencephalography did not reveal pathological graphoelements.
4.2.5. Genotype-Phenotype Correlation in NF1
There is currently no clear genotype-phenotype correlation although specialized studies through the described mutational variants attempt to establish a clear genotype-phenotype relationship. In a retrospective study that included 38 patients with NF1, Well et al. demonstrated that large deletions encompassing the entire gene are associated with a much more severe clinical phenotype, a more severe tumor burden, and an accelerated tumor growth rate compared to patients who had atypical deletions. The authors recommend close monitoring of these patients to assess tumor progression and the risk of malignant transformation and, if appropriate, recommend drug treatment with MEK inhibitors [38]. Peduto et al. also demonstrated that large gene deletions correlate with a severe phenotype and that not all mutational variants have the same effects. Genotype-phenotype associations are on an upward curve at present, changing at a slower but pro-found pace the clinical and genetic approach to NF patients [3,22]. The splice site pathogenic variant identified in our patient has not been described in populational databases, so we cannot make an exact genotype-phenotype correlation.
4.2.6. Treatment
Surgical treatment is the main treatment option for neurofibromas, but the risk of tumor recurrence postoperatively is extremely high [30]. It is still considered the only curative treatment for patients with NF1. Surgical intervention takes into account the size of the NP, its location, growth rate, and radiological features, as well as the general health and well-being of the patient [39].
Drug Therapy
Targeted therapy can have a major impact on the course of the disorder. As such, Ras pathway inhibitors are therapeutic agents used in the treatment of inoperable plexiform tumors in children. In 2020 the US Food and Drug Administration approved the first monoclonal antibody, Selumetinib, an inhibitor of MEK1/2. This treatment represents a turning point in the treatment of inoperable plexiform tumors in children older than 2 years [40]. Another therapeutic agent is Rapamycin, an inhibitor of the mTOR pathway that plays a role in activating AKT, thus being another potential drug used in the treatment of plexiform tumors.
4.3. Cone Rod Dystrophies
4.3.1. Inheritance
The majority of current research regarding con/con-tying dystrophies focuses on elucidating the remaining causative genes and their molecular mechanisms, understanding the natural history of the disorder, and setting optimal clinical trial targets. More than 30 CORD-associated genes are currently reported in the RetNet database (http://www.sph.uth.tmc.edu/retnet/) [15]. They are monogenic disorders with autosomal dominant (adCORD), autosomal recessive (arCORD), or X-linked (xlCORD) inheritance. Related to adCORD, mutations occurring in 10 genes are described, including CRX gene [24]. For arCORD 13 genes are identified and there are other loci and chromosomal regions mapped: NF1, 18q21.1-q21.3, 1q12-q24, and 10q26. For the X-linked form, the mutations occur in the alternative terminal exon 15 (ORF15) of the RPGR gene located on the short arm of the X chromosome Xp21.1 [41].
4.3.2. Clinical Aspects
The main characteristic of CORD is retinal pigmentation by pigment deposition, especially in the macular area. While in retinitis pigmentosa (also called cone-rod dystrophy, RCD), rod cell receptors are lost first, in CORD, cone cell photoreceptors loss occurs first. This leads to a gradual decrease in visual acuity, disruption of color vision, and decreased sensitivity in the central visual field; later, there is a loss of peripheral vision and night blindness. Hammel et al. state that onset usually occurs in childhood, during the school years, although the diagnosis is usually established in adulthood [13]. In a study including 730 Japanese families with a hereditary retinal disorder, Fujinami-Yokokawa et al. identified 18 patients from 13 different families with retinal damage due to mutations in the CRX gene; in these patients, the mean age of onset was 45 years (range 15-77); in one patient, the onset of the disorder was at 15 years; late-onset was reported in one patient aged 45 years [42]. In the case presented by the authors, the patient has a likely pathogenic mutational variant that is currently not correlated with her phenotype; at this age, the patient has no suggestive symptomatology for cone-rod dystrophy. The patient has been under ophthalmological care since the age of 15 for myopia and astigmatism, with no clinical signs of cone-rod dystrophy. However, considering the potential late onset of the disease, proper ophthalmological monitoring is crucial.
4.3.3. Diagnosis of Cone-Rod Dystrophy
It is done based on ophthalmological consultation: fundus examination, OCT, and electroretinogram. The electroretinogram (ffERG) subdivides patients into 3 categories: patients with normal ffERG (MD), patients where cone cells are affected (CD), and patients who have both cone and rod cells affected (CORD) [43]. The case presented by the authors falls into the category of normal ffERG (MD).
4.3.4. Treatment
There is currently no curative therapeutic method to stop the progression of the disorder or restore lost vision. Identification of pathogenic variants in CRX genes is an extremely important step in the management of these patients, facilitating correct genetic counseling, providing prognostic data, and participating in clinical trials [44]. Treatment aims to slow the progression of the disorder, treat complications, and counsel patients to cope with the news of blindness [13]. Currently, the patient is undergoing ophthalmic correction for myopia and astigmatism, with no signs of cone-rod dystrophy. Given the likely pathogenic nature of the identified variant, close ophthalmological monitoring is essential for early detection if symptoms arise.
4.3.5. Genotype-Phenotype Correlation in CORD
Genotype-phenotype correlations are complex: different mutations in the same gene can cause extremely complex and different phenotypes, and on the other hand, mutations in different genes cause relatively identical or similar phenotypes. Also, the symptoms in people with CORD with MD and people with retinitis pigmentosa (RP) sometimes overlap; as the disorder progresses, MD patients may switch to CORD [42,45,46]. The mutational variant identified in our patient is exceptionally rare and was described in a study by Carss et al., indicating its association with hereditary retinal disorders. Our case underscores the importance of molecular testing, which can reveal unexpected associations between genetic variants in different genes, such as NF1 and CRX, highlighting the necessity of individualized medical management and genetic counseling [47]. In 2022 Oh et al. identified the same missense type variant in heterozygous status c.119G>A, in two patients with pigmented paravenous retinochoroidal atrophy without associated NF1. This missense variant, identified in both our case and the cases presented by Oh, has previously been reported as pathogenic but with an unknown specific phenotype. Given that the allele occurs with extremely rare frequency in the general population, this demonstrates that this is not a common variant, and in silico tools predict the variant to be likely pathogenic [48]. The case described by us presents only myopia with astigmatism; the case presented by Oh et al. associates pigmented paravenous retinochoroidal atrophy, thus not being able to make an obvious genotype-phenotype correlation.
4.4. The Association of NF1 with Cone-Rod Dystrophy
It is extremely rarely reported in the literature. In 1993, Kylstra J. et al. reported the first case associating the two pathologies. In 2012, Zobor et al. described a case of NF1 associated with cone-rod dystrophy, but in an autosomal recessive form with a mutation in the CNNM4 gene and amelogenesis imperfecta [49,50]. The case presented by the authors represents a novel finding, being the first reported instance of the co-occurrence of a pathogenic NF1 variant and a likely pathogenic CRX variant. While the patient currently lacks symptoms of cone-rod dystrophy, the potential for its later onset cannot be ruled out. This underscores the complexity of genetic interactions and the necessity of comprehensive medical surveillance in such cases.
5. Conclusions
The diagnosis and management of neurofibromatosis often involve clinical evaluation by a complex medical team. Although the clinical diagnosis is very important and essential, it is important to look for other signs that are not consistent with the initial diagnosis. Molecular testing can unveil surprising associations, as evidenced in our case, which presents a rare combination of mutations in the NF1 and CRX genes. While the patient currently shows no symptoms of cone-rod dystrophy, ongoing ophthalmological monitoring is imperative for early detection. This case underscores the importance of personalized medicine, guided by genotypic features, in managing patients with complex genetic conditions like NF1 and cone-rod dystrophy.
Acknowledgments
We are extremely grateful to our colleagues from the Center of Genomic Medicine Timisoara for their sequencing services.
Conflict of interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Financial support
none.
Ethics statement
The study was conducted under the Declaration of Helsinki and approved by the Institutional Ethics Committee of Emergency Clinical County Hospital Bihor (protocol code 39652/15.11.2023).
References
- Friedman, J. M. Neurofibromatosis 1; University of Washington, Seattle 1993 updated, 2022, April. Book editor. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1109/.
- Jurca, C.M.; Kozma, K.; Petchesi, C.D.; Zaha, D.C.; Magyar, I.; Munteanu, M.; Faur, L.; Jurca, A.; Bembea, D.; Severin, E.; et al. Tuberous Sclerosis, Type II Diabetes Mellitus and the PI3K/AKT/mTOR Signaling Pathways—Case Report and Literature Review. Genes 2023, 14, 433. [Google Scholar] [CrossRef]
- Yoshida, Y. Neurofibromatosis 1 (von Recklinghausen Disease). Keio J. Med. 2023, No. 2023-0013-IR. [Google Scholar] [CrossRef] [PubMed]
- Adil, A.; Koritala, T.; Munakomi, S.; Singh, A. K. Neurofibromatosis Type 1; StatPearls Publishing, 2023.
- Farschtschi, S.; Mautner, V.-F.; McLean, A.C.L.; Schulz, A.; Friedrich, R.E.; Rosahl, S.K. The Neurofibromatoses. Dtsch. Aerzteblatt Online 2020, 117, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Jurca, C.M.; Frățilă, O.; Iliaș, T.; Jurca, A.; Cătana, A.; Moisa, C.; Jurca, A.D. A New Frameshift Mutation of PTEN Gene Associated with Cowden Syndrome—Case Report and Brief Review of the Literature. Genes 2023, 14, 1909. [Google Scholar] [CrossRef] [PubMed]
- Neurofibromatosis: Conference Statement. Arch. Neurol. 1988, 45, 575. [CrossRef]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the National Institutes of Health Criteria for Diagnosis of Neurofibromatosis 1 in Children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Gutmann, D.H. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, L.A.; Scanga, H.; Traboulsi, E.; Nischal, K.K. Novel clinical presentation of a CRX rod-cone dystrophy. BMJ Case Rep. 2021, 14, e233711. [Google Scholar] [CrossRef]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7–7. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a Novel otx-like Homeobox Gene, Shows Photoreceptor-Specific Expression and Regulates Photoreceptor Differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef]
- Wang, L.; Qi, A.; Pan, H.; Liu, B.; Feng, J.; Chen, W.; Wang, B. A novel CRX frameshift mutation causing cone-rod dystrophy in a Chinese family. Medicine 2018, 97, e11499. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Mallone, F.; Alisi, L.; Lucchino, L.; Di Martino, V.; Nebbioso, M.; Armentano, M.; Lambiase, A.; Moramarco, A. Insights into Novel Choroidal and Retinal Clinical Signs in Neurofibromatosis Type 1. Int. J. Mol. Sci. 2023, 24, 13481. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Tadini, G.; Brems, H.; Legius, E. Proposal of New Diagnostic Criteria. In Multidisciplinary Approach to Neurofibromatosis Type 1; Springer International Publishing: Cham, 2020: 309–313. Book chapter https://link.springer.com/chapter/10.1007/978-3-319-92450-2_21.
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.-C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2017, 102, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: genotype–phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Peduto, C.; Zanobio, M.; Nigro, V.; Perrotta, S.; Piluso, G.; Santoro, C. Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations. Cancers 2023, 15, 1217. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Senior Department of Ophthalmology, the Third Medical Center of PLA General Hospital, Beijing 100853, China; Yang, Q. -H.; Qu, L.-H.; Hou, B.-K.; Li, Z.-H.; Huang, H.-B.; Jin, X. Various phenotypes of autosomal dominant cone-rod dystrophy with cone-rod homeobox mutation in two Chinese families. Int. J. Ophthalmol. 2022, 15, 1915–1923. [Google Scholar] [CrossRef]
- Hodges, M.D.; Vieiraa, H.; Gregory-Evans, K.; Gregory-Evans, C.Y. Characterization of the Genomic and Transcriptional Structure of the CRX Gene: Substantial Differences between Human and Mouse. Genomics 2002, 80, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Arno, G.; Plagnol, V.; Chamney, S.; Russell-Eggitt, I.; Thompson, D.; Ramsden, S.C.; Black, G.C.M.; Robson, A.; Holder, G.E.; et al. The Phenotypic Variability of Retinal Dystrophies Associated With Mutations in CRX, With Report of a Novel Macular Dystrophy Phenotype. Investig. Opthalmology Vis. Sci. 2014, 55, 6934–6944. [Google Scholar] [CrossRef] [PubMed]
- Swain, P.K.; Chen, S.; Wang, Q.-L.; Affatigato, L.M.; Coats, C.L.; Brady, K.D.; A Fishman, G.; Jacobson, S.G.; Swaroop, A.; Stone, E.; et al. Mutations in the Cone-Rod Homeobox Gene Are Associated with the Cone-Rod Dystrophy Photoreceptor Degeneration. Neuron 1997, 19, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Clanor, P.B.; Buchholz, C.N.; Hayes, J.E.; Friedman, M.A.; White, A.M.; Enke, R.A.; Berndsen, C.E. Structural and functional analysis of the human cone-rod homeobox transcription factor. Proteins 2022, 90, 1584–1593. [Google Scholar] [CrossRef]
- Perrault, I.; Hanein, S.; Gerber, S.; Barbet, F.; Dufier, J.-L.; Munnich, A.; Rozet, J.-M.; Kaplan, J. Evidence of autosomal dominant Leber congenital amaurosis (LCA) underlain by a CRX heterozygous null allele. J. Med Genet. 2003, 40, 90e–90. [Google Scholar] [CrossRef]
- Tamura, R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int. J. Mol. Sci. 2021, 22, 5850. [Google Scholar] [CrossRef] [PubMed]
- Jurcă, M.C.; Iuhas, O.A.; Puiu, M.; Chiriţă-Emandi, A.; Andreescu, N.I.; Petcheşi, C.D.; Jurcă, A.D.; Magyar, I.; Jurcă, S.I.; Kozma, K.; et al. Cardiofaciocutaneous syndrome – a longitudinal study of a case over 33 years: case report and review of the literature. Romanian J. Morphol. Embryol. 2022, 62, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, L.; Krauland, K. Neurofibroma; StatPearls Publishing, 2023 book https://www.ncbi.nlm.nih.gov/books/NBK539707/.
- Ehara, Y.; Yamamoto, O.; Kosaki, K.; Yoshida, Y. Natural course and characteristics of cutaneous neurofibromas in neurofibromatosis 1. J. Dermatol. 2017, 45, 53–57. [Google Scholar] [CrossRef]
- Viola, F.; Villani, E.; Natacci, F.; Selicorni, A.; Melloni, G.; Vezzola, D.; Barteselli, G.; Mapelli, C.; Pirondini, C.; Ratiglia, R. Choroidal Abnormalities Detected by Near-Infrared Reflectance Imaging as a New Diagnostic Criterion for Neurofibromatosis 1. Ophthalmology 2011, 119, 369–375. [Google Scholar] [CrossRef]
- Pan, Y.; Hysinger, J.D.; Barron, T.; Schindler, N.F.; Cobb, O.; Guo, X.; Yalçın, B.; Anastasaki, C.; Mulinyawe, S.B.; Ponnuswami, A.; et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 2021, 594, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Vagge, A.; Camicione, P.; Capris, C.; Sburlati, C.; Panarello, S.; Calevo, M.G.; Traverso, C.E.; Capris, P. Choroidal abnormalities in neurofibromatosis type 1 detected by near-infrared reflectance imaging in paediatric population. Acta Ophthalmol. 2015, 93, e667–71. [Google Scholar] [CrossRef] [PubMed]
- Sellmer, L.; Farschtschi, S.; Marangoni, M.; Heran, M.K.S.; Birch, P.; Wenzel, R.; Friedman, J.M.; Mautner, V.-F. Non-optic glioma in adults and children with neurofibromatosis 1. Orphanet J. Rare Dis. 2017, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Well, L.; Döbel, K.; Kluwe, L.; Bannas, P.; Farschtschi, S.; Adam, G.; Mautner, V.-F.; Salamon, J. Genotype-phenotype correlation in neurofibromatosis type-1: NF1 whole gene deletions lead to high tumor-burden and increased tumor-growth. PLOS Genet. 2021, 17, e1009517. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Maitra, A.; Choudhury, S. Selumetinib: the first ever approved drug for neurofibromatosis-1 related inoperable plexiform neurofibroma. Curr. Med Res. Opin. 2021, 37, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.E.; Belzberg, A.J.; Crawford, J.R.; Hirbe, A.C.; Wang, Z.J. Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas. BMC Cancer 2023, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.-K.; Zhao, N.; Lv, Y.-S.; Gong, W.-K.; Wang, H.-Y.; Tong, Q.-H.; Lai, X.-M.; Liu, R.-R.; Fang, M.-Y.; Zhang, J.-G.; et al. A novel CRX mutation by whole-exome sequencing in an autosomal dominant cone-rod dystrophy pedigree. . 2015, 8, 1112–7. [Google Scholar] [CrossRef]
- Fujinami-Yokokawa, Y.; Fujinami, K.; Kuniyoshi, K.; Hayashi, T.; Ueno, S.; Mizota, A.; Shinoda, K.; Arno, G.; Pontikos, N.; Yang, L.; et al. Clinical and Genetic Characteristics of 18 Patients from 13 Japanese Families with CRX-associated retinal disorder: Identification of Genotype-phenotype Association. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Georgiou, M.; Fujinami, K.; Michaelides, M. Inherited retinal diseases: Therapeutics, clinical trials and end points—A review. Clin. Exp. Ophthalmol. 2021, 49, 270–288. [Google Scholar] [CrossRef]
- Boulanger-Scemama, E.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.-P.; Letexier, M.; Souied, E.; et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J. Rare Dis. 2015, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; Phan, T.M.L.; Zekveld-Vroon, R.C.; Leroy, B.P.; Born, L.I.v.D.; Hoyng, C.B.; Klaver, C.C.; Roosing, S.; Pott, J.-W.R.; van Schooneveld, M.J.; et al. Clinical Course, Genetic Etiology, and Visual Outcome in Cone and Cone–Rod Dystrophy. Ophthalmology 2012, 119, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Nuzbrokh, Y.; Lee, W.; de Carvalho, J.J.R.L.; Wang, N.K.; Sparrow, J.R.; Alliikmets, R.; Tsang, S.H. A mutation in CRX causing pigmented paravenous retinochoroidal atrophy. Eur. J. Ophthalmol. 2022, 32, NP235–NP239. [Google Scholar] [CrossRef] [PubMed]
- Zobor, D.; Kaufmann, D.H.; Weckerle, P.; Sauer, A.; Wissinger, B.; Wilhelm, H.; Kohl, S. Cone-rod dystrophy associated with amelogenesis imperfecta in a child with neurofibromatosis type 1. Ophthalmic Genet. 2012, 33, 34–38. [Google Scholar] [CrossRef]
- Kylstra, J. A.; Aylsworth, A. S. Cone-Rod Retinal Dystrophy in a Patient with Neurofibromatosis Type 1. Can. J. Ophthalmol. 1993, 28, 79–80. [Google Scholar]
Figure 1.
Cutaneous neurofibroma.
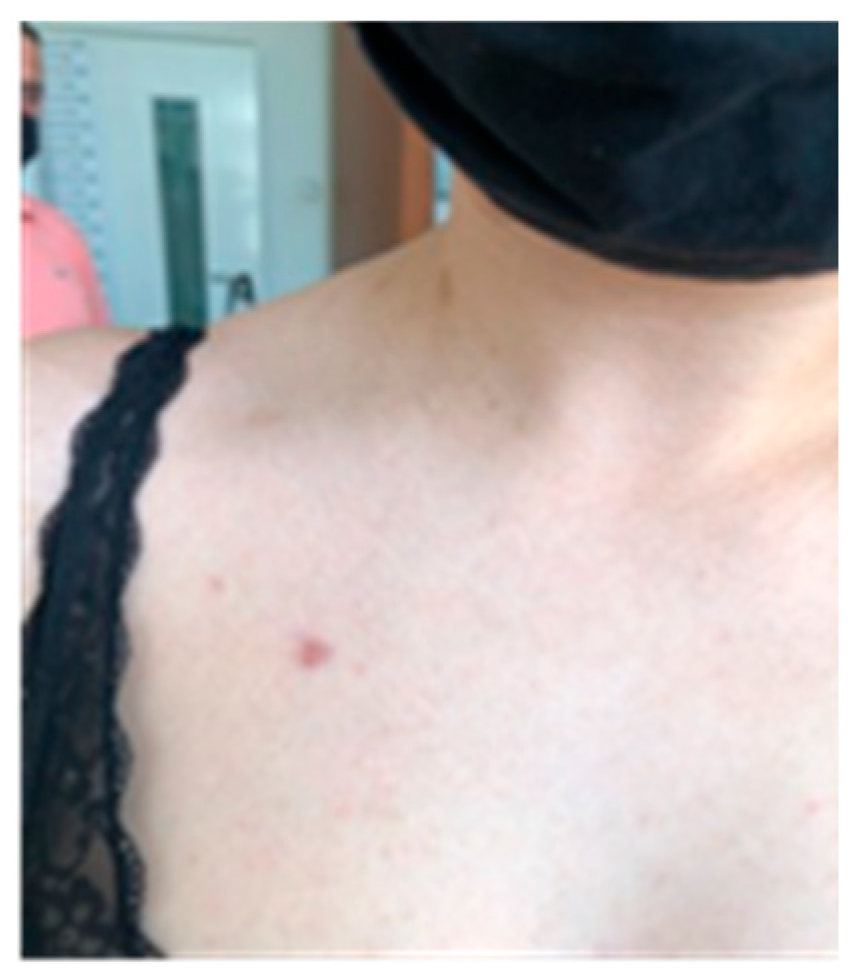
Figure 2.
Posterior thoracal café au lait spots.
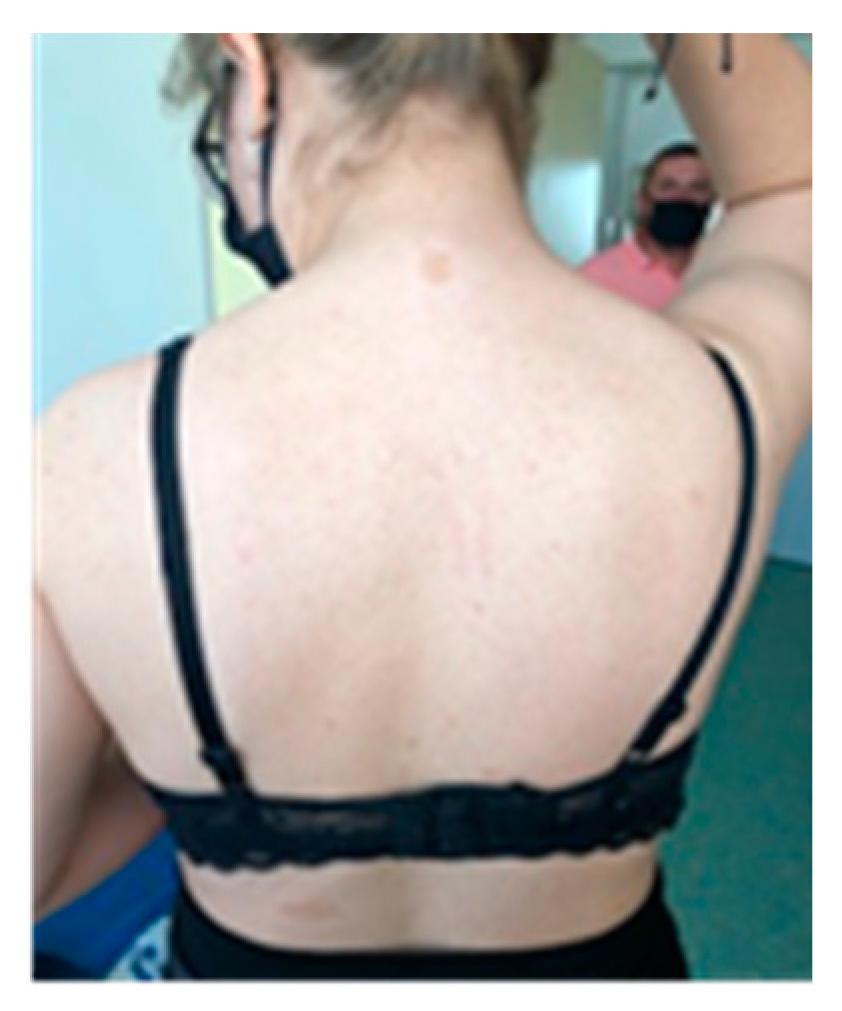
Figure 3.
Axillary freckles.
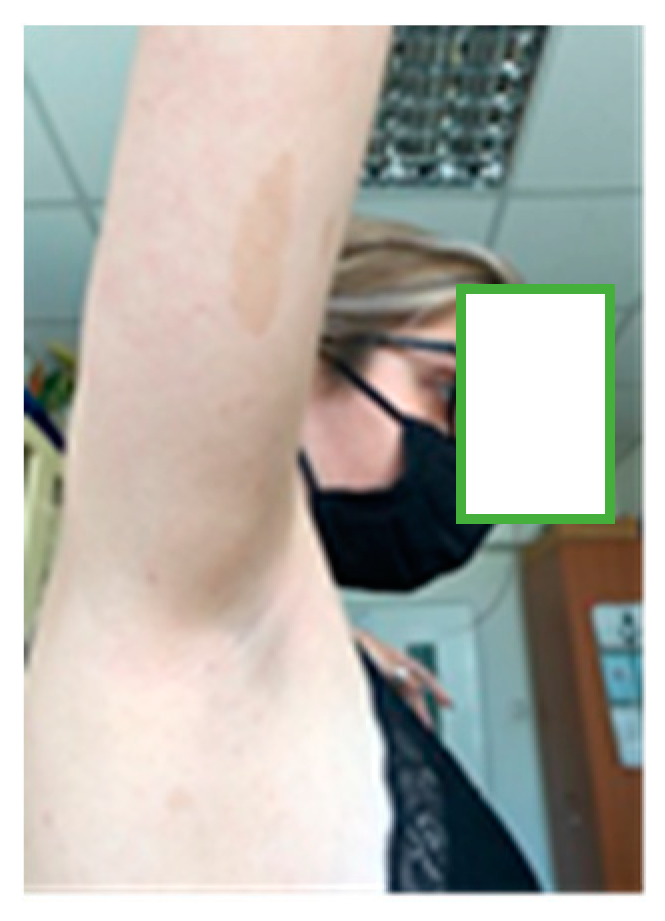
Figure 4.
Café au lait spots on the lower limbs.
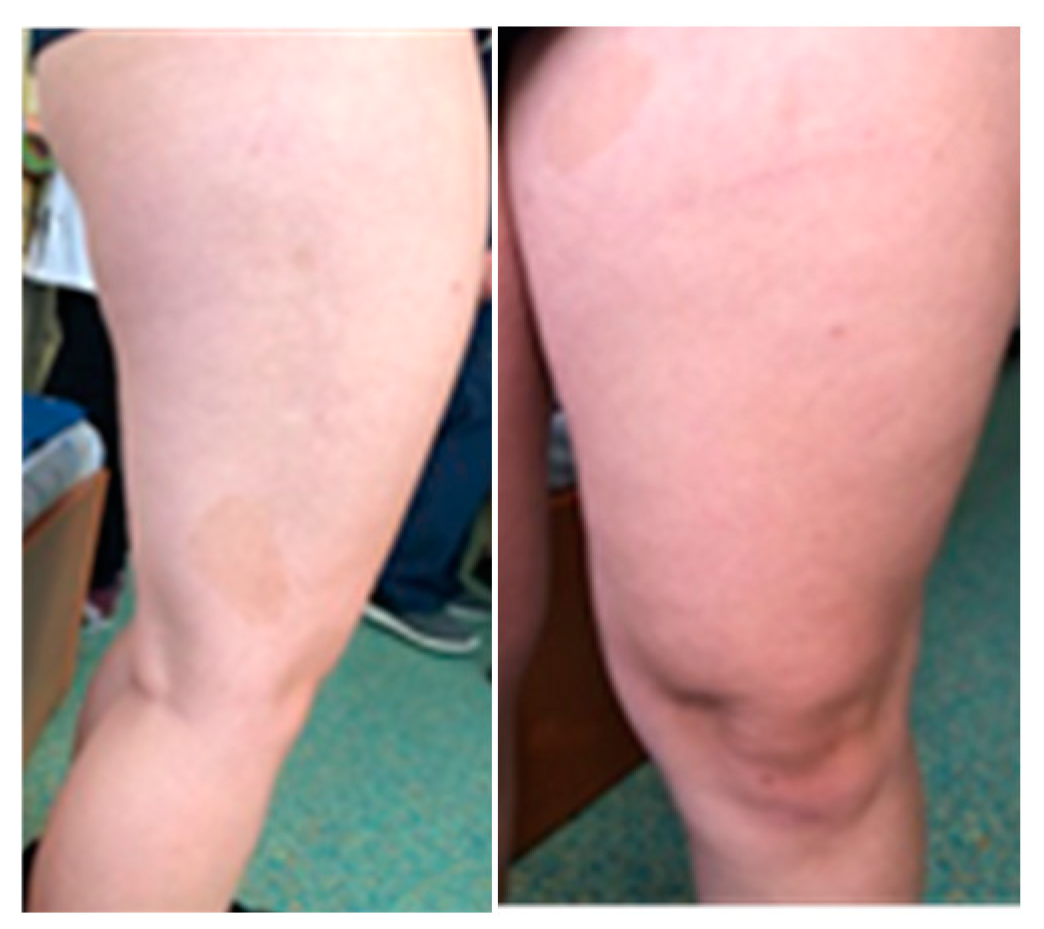
Figure 5.
Lisch nodules: (a) right eye; (b) left eye.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



