
Submitted:
22 February 2024
Posted:
23 February 2024
You are already at the latest version
Abstract
Nucleosomes are non-uniformly distributed across eukarytic genomes, with stretches of ‘open’ chromatin strongly associated with transcriptionally active promoters and enhancers. Understanding chromatin accessibility patterns in normal tissue and how they are altered in pathologies can provide critical insights to development and disease. With the advent of high-throughput sequencing, a variety of strategies have been devised to identify open regions across the genome, including DNase-seq, MNase-seq, FAIRE-seq, ATAC-seq, and NicE-seq. However, the broad application of such methods to FFPE (formalin-fixed paraffin embedded) tissues has been curtailed by the major technical challenges imposed by highly fixed and damaged genomic material. Here we review the most common approaches for mapping open chromatin regions, recent optimizations to overcome the challenges of working with FFPE tissue, and a brief overview of a typical data pipeline with analysis considerations.
Keywords:
Chromatin
; FFPE
; Nucleosome
; Nucleosome-free region
; Nucleosome-depleted region
Introduction
Alterations in chromatin structure and function are hallmark features of normal cell fate decisions, as well as pathological processes [1,2]. As such, understanding the epigenetic features that regulate chromatin states is essential to develop next-generation biomarkers and therapeutics. The chromatin landscape can be defined by the localization of histone post-translational modifications (PTMs) and chromatin associated proteins (CAPs), which together form a complex molecular language to govern genome transactions [3]. Indeed, gene expression patterns are controlled by the interplay of distinct genomic regions (e.g., promoters, enhancers, heterochromatin) marked by histone PTMs and engaged by chromatin regulatory complexes (e.g., nucleosome remodelers and modifiers) that in turn modulate local genome accessibility [4,5,6,7] (Figure 1).
Nucleosomes are the basic repeating unit of chromatin, comprising ~147bp of DNA wrapped around a histone octamer [8]. ‘Accessible’ or ‘open’ chromatin is conceptually defined as genomic regions containing stretches of free DNA longer than the average linker length between adjacent nucleosomes (~40 bp in human cells) [9,10,11]. These open chromatin regions are commonly referred to as nucleosome depleted/free regions (NDR/NFR; hereafter NDRs), reflecting dynamic nucleosome turnover and the spectrum of accessibility in population-based assays [12,13,14]. NDRs are characterized by relatively long free DNA stretches (~120-200 bp), over-represented in enhancers/promoters, bound by transcription factors (TFs), and positively correlated with transcriptional activity [15,16].
A variety of experimental strategies have been developed to map accessible chromatin at genome scale. Historically, nuclease DNAse I treatment of chromatin followed by primer extension identified hypersensitive cleavage sites [17,18], representing the NDRs (reviewed in [19,20,21]). Since commercial release, massively high-throughput sequencing technologies (aka. next generation sequencing (NGS): in 2005, Roche 454 pyrosequencing; in 2006, Illumina (formerly Solexa)) have revolutionized genome-scale studies by delivering ever-increasing amounts of sequence data at ever-decreasing cost. The first assay to take advantage of NGS for open chromatin mapping was DNase-seq in human CD4+ T cells [22], and shortly thereafter MNase-seq to map nucleosome positioning (an indirect approach: see below) in budding yeast [23] (Figure 2A,D). 2013 saw the first report of ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing), a Tn5-based assay that rapidly became established as the most frequently used chromatin profiling assay (Figure 2B,D).
Whole genome-scale chromatin accessibility assays have delivered breakthrough insights in diverse fields [6,24,25,26]. However, their ability to fully advance clinical research has been hampered by incompatibility with formalin-fixed paraffin-embedded (FFPE) tissue. FFPE is the routine method for preservation of clinical samples, with >20 million banked samples in the United States alone [27]. This material can be stored for years at ambient temperatures with minimal degradation of cytoarchitecture and proteomic content [28], making these banked tissues a potential goldmine for clinical research, especially for rare diseases and longitudinal studies. The first FFPE specimen was reported nearly 130 years ago [29], transforming the face of clinical research and enabling retrospective studies decades after initial tissue preservation [30]. Compared to the analysis of native cells, genomic mapping in FFPE tissue presents a number of unique challenges that require specific protocol modifications and considerations [31]. As an example, sample processing protocols must be implemented to extract biological material from the paraffin matrix and expose cross-linked chromatin epitopes. However the primary challenge is genome quality, as FFPE sample processing induces significant DNA adducts and fragmentation [32]. Further, DNA continues to degrade while in storage, increasing the challenge when analyzing older FFPE specimens.
In recent years, there has been increasing interest in chromatin accessibility studies of FFPE tissue, and thus mining this potentially rich data seam (Figure 2C,D). The goal of this review is to discuss the most common approaches for mapping NDRs (Figure 3), their suitability for profiling FFPE samples, and computational strategies to analyse the resulting data (Figure 4).
Genome-wide profiling of open chromatinThe most common approaches to map NDRs leverage nucleases, a transposase, a nickase, or the biochemical fractionation of chromatin (Figure 3). For the enzyme-based methods, their catalytic properties, molecular size, and potential steric hinderances influence the resulting open chromatin maps.
DNase I hypersensitivity mapping paved the way for genome-wide open chromatin profilingDeoxyribonuclease I (DNase I) endonuclease (31 kilodaltons, kDa) specifically degrades double- and single-stranded DNA to a 5’-phosphate and 3’-hydroxyl [33], and preferentially cleaves accessible chromatin in situ at eponymously named DNase I hypersensitive sites (DHSs). In a typical DNase-seq experiment (Figure 3A and Table 1), several million cells are digested to yield DHS subnucleosomal fragments, which are then identified by library preparation and NGS data analysis [19]. DNase I has proven an excellent molecular tool for studing chromatin structure for over three decades, most notably by the ENCODE consortium [15,21]. In a pioneering study, ~14,000 DHSs were mapped in primary CD4+ T cells, and ~90% shown to be shared across multiple cell types [22]. Although originally thought to lack intrinsic sequence bias, a recent systematic study showed DNAse I exhibits a C/G preference at the 5’-end of DHSs [34,35], and several DNase-seq data analysis pipelines now correct for this prejudice [36,37,38,39]. In an effort to map DHSs in FFPE tissue, a more sensitive DNase-seq strategy was developed using a circular carrier DNA-mediated sequencing method (Pico-Seq) [40,41]. However, despite a proof-of-concept study in human follicular thyroid carcinoma, the field has not adopted DNase-seq approaches for FFPE samples (a Pubmed search for “DNAse AND FFPE” returning only two related entries).
Micrococcal nuclease (MNase) digestion to decipher nucleosome positioning Staphylococcus aureus MNase has been used to study chromatin for nearly five decades [42], and employed in the NGS era to map genome-wide nucleosome positioning for multiple eukaryotes (e.g., yeast, worm, fly, mouse and human) [9,10,11,43,44]. The enzyme is a small (17 kDa), highly processive endo-exonuclease that degrades most types and forms of nucleic acids (e.g., supercoiled, linear and circular single-stranded and double-standed DNA and RNA) [33]. These properties enables it to thoroughly digest chromatin until protected by nucleosome structure, cleaving both NDRs and linker DNA. As such, MNase-Seq is distinct from other NDR mapping approaches since it enriches for protected DNA (i.e., nucleosome occupancy and position), and open chromatin is then inferred from low signal regions (Figure 3B and Table 1). MNase shows a strong sequence bias toward A/T rich sequences, and a correction factor is thus built into many data analysis pipelines [38,45]. Recent efforts using MNase to map chromatin accessibility have focused on limiting the MNase digestion [46,47,48], although these titration based variants have largely been superceded by competing direct NDR-mapping methods (Figure 3 & Table 1). Applying MNase-seq to FFPE tissue sections has met with minimal success, with a Pubmed search for “MNase AND FFPE” returning zero entries.
FAIRE-seq identifies accessible chromatin regions through principles of biochemical separation and solubilityIn contrast to nuclease based methods to map chromatin accessibility, FAIRE (formaldehyde assisted isolation of regulatory elements) identifies NDRs by building on the observation that transcriptionally active chromatin displays differential biochemical solubility after formaldehyde fixation [49]. In brief, cells are treated with formaldehyde to crosslink chromatin, sheared by sonication, and phenol-chloroform extracted, where the aqueous phase contains DNA fragments associated with NDRs (Figure 3B) [50]. While FAIRE-seq does not have the sequence-specific cleavage bias of nucleases [34,45], it is highly dependent on crosslinking efficiency, and often undermined by poor signal-to-noise [50,51], false positives [52], and the challenge posed by low cell numbers [53] (Table 1). Nevertheless, FAIRE-seq has been widely applied to model systems and cell lines [50,51,53,54,55], particularly as part of ENCODE efforts to systemically identify active regulatory elements [15]. A recent report mapping open chromatin by FAIRE-seq in Drosophila cleverly circumvented the challenge that pupa cuticles present to in situ enzyme-based methods, thus providing higher quality data than ATAC-seq for this particular tissue type [56]. Despite this, over the last decade ATAC-seq has clearly emerged as the preferred assay for mapping chromatin accessibility while FAIRE has declined in use (Figure 2A,B).
Tn5 transposon tagmentation of accessible chromatin (ATAC-seq)
Tn5 transposase was first discovered in the 1970s based on the kanamycin resistance it conferred to host bacteria [57,58]. In addition to providing a mechanistic model for transposases, Tn5 (106 kDa active dimer) has proven an invaluable molecular tool [59]. Most recently it has been leveraged to identify NDRs via ATAC-seq, and to map histone PTMs via CUT&Tag [60,61]. ATAC-seq is currently the most widely used open chromatin mapping assay (Figure 2B) due to its relative speed, efficiency, and sensitity (Table 1). The approach employs a genetically engineered hyperactive Tn5 transposase to insert loaded DNA adapters preferentially at accessible DNA in situ (i.e., tagmentation) for direct PCR amplification and NGS [60,62] (Figure 3D). Tn5 displays an enzymatic sequence bias which, while more complex than that of the nucleases used for NDR mapping, can also be compensated at data analysis [38,63]. With deep enough sequencing, TF binding footprints may also be inferred from protected fragments within the NDRs [39,64]. Early versions of the ATAC-seq protocol were hampered by high read duplications and contaminating mitochondrial DNA, which together consumed a majority of the sequencing bandwith. These issues were largely circumvented by the development of Omni-ATAC, wherein nuclei were isolated with a cocktail of detergents to remove contaminating mitochondria, increasing library complexity and signal-to-noise [65,66].
Beyond the application of ATAC to interrogate the epigenomes of model organisms and cell lines, recent efforts have sought to enable clicinal studies from FFPE tissue sections [67,68,69,70,71]. To prepare amplicons from Tn5-based approaches, two independent tagmentation events are required in opposing orientation and in close proximity (<~700 bp). This inherently reduces library efficiency and effective yields, which is exacerbated by the highly damaged and fragmented DNA in FFPE material. To address this complication, a recent approach combined Tn5 tagmentation with an in vitro transcription (IVT) step, such that a single insertion event can be amplified by T7 RNA polymerase [68,69,70,71]. While standard ATAC-seq yielded some success using nuclei isolated from mouse FFPE liver and kidney, the Tn5-IVT-modified approach improved library complexity, signal-to-noise, and other key metrics. However, the approach is limited by its complex, five day procedure that relies on harsh chemical, mechanical, and enzymatic methods (e.g., xylene, needle shearing, and a collagenase/hyaluronidase cocktail) to extract nuclei from FFPE tissue. Indeed, studies have shown that such exacting preparation methods contribute to genome fragmentation [72,73].
Noting these observations, Henikoff and colleagues instead used gentle heating and permeabilization, similar to how FFPE sections are routinely deparaffinized for histological analysis, to prepare samples for CUT&Tag [67]. NDRs facilitate access to DNA by the transcriptional machinery, so the CUTAC (Cleavage Under Targeted Accessible Chromatin) protocol was developed to target Tn5 to active chromatin via RNA Pol II, and yields short tagmentation fragments (~60bp) to reduce the impact of DNA damaged samples. Of note, FFPE-CUTAC yielded higher quality data than FFPE-ATAC from mouse brain [67], suggesting the potential of innovative Tn5-based approaches to map the epigenome of clinically relevant FFPE samples.
Nicking enzyme assisted accessible chromatin sequencing (NicE-seq)
NicE-seq is a recent approach (Figure 2A,B) that excels at NDR profiling from heavily fixed cells, including FFPE [74,75,76,77]. In contrast to nuclease or Tn5 transposase double-strand cleavage (as above), Chlorella virus Nt.CviPII (63 kDa) is a nicking endonuclease that cuts only one strand of double-stranded DNA at CCD sites (D=A/G/T), which occur by chance every ~21 bases [78]. In the latest version of the protocol (One-pot UniNicE-seq), Nt.CviPII nicks at NDRs are filled in using an NTP mix containing biotinylated- and 5-methyl- dCTP triphosphates (to respectively label and prevent further nicking), the genome is enzymatically sheared, biotin-labelled DNA is captured on streptavidin beads, and libraries are prepared on the matrix by PCR (Figure 3E). The method is a fast, simple, and robust one-tube workflow, although it is incompatible with native cells and yields larger DNA fragment sizes than ATAC-seq, which limits resolution. NicE-seq has now been applied to a wide variety of mouse and human cell lines, primary tissues, and FFPE sections [75,76]. Central to the theme of this review, the approach identified NDRs in human lung and liver FFPE samples from as few as five thousand cells (Table 1) [75,76]. Similar to FFPE-CUTAC, NicE-seq can be performed on permeabilized and minimally disrupted FFPE sections in situ, obviating the need for nuclei purification by harsh methods that damage genomic DNA. As such, the approach shows great potential for broad adoption to map open chromatin in clinical FFPE material.
Data analysisATAC-seq is the most popular open chromatin profiling approach (Figure 2), and several excellent papers describe methods to analyse the resulting data [79,80,81]. Instead this review aims to provide a brief overview of key considerations and data analysis pipelines broadly applicable to ATAC-seq, FAIRE-seq and NicE-seq (Figure 4). Because of their distinct data structure, DNase-seq and MNase-seq require application specific pipelines [79]. For example, open chromatin from MNase-seq data is inferred from nucleosome-centric maps, and so central considerations are to identify the nucleosome dyad, account for MNase sequence cleavage bias, and to quantify nucleosome occupancy and ‘fuzziness’ [23,46,48]. The major features of a typical ATAC-seq pipeline involve: 1) read pre-processing and quality control (QC); 2) primary analysis (read alignment and filtering); and 3) secondary analysis (peak-calling, visualization, reproducibility and differential accessibility). Paired end (PE) sequencing is highly recommended because it provides the DNA fragment length: an important metric for assay success and interpretation. A sequencing depth of 30-50M PE reads is usually sufficient for good genome coverage, but will depend on how much bandwidth is consumed by mitochondrial DNA contamination and read duplicates.
Read pre-processing and quality control Prior to alignment, several tools are used to assess the quality of the library and sequencing run. FastQC reports the base calling quality and overrepresented sequences, such as primer- and adapter-dimers. Low base calling scores (<20 Q-score) may be indicative of a poor quality library and/or sequencing run. Overrepresented primer- and adapter-dimers are not as problematic in Tn5-based libraries as in ligation-mediated PCR libraries. If the accessible DNA fragment length is not greater than twice the paired-end read legth, sequencing will read through to the Illumina adapter regions. This readthrough can negatively impact genome alignment, so read trimming tools (e.g., Trimmomatic [82]) are used to detect and prune Illumina adapter sequences.
Primary analysis pipelineGenome alignment is typically the most time consuming and computationally intensive step in the primary pipeline, so fast, memory-efficient aligners optimized for short paired-end reads have been developed (e.g., Bowtie2 [83]). The goal is to identy the unique genomic location that corresponds to each read pair. However, multi-aligned reads pairs are common, and must be flagged/removed from subsequent analysis since they would introduce ambiguity to the data. In addition to removing multi-aligned reads using Samtools [84], other utilities (e.g., Bedtools [85,86] and Picard (http://broadinstitute.github.io/picard)) are used for read processing and filtering to remove PCR duplicates, contaminating mitochontrial DNA, and artifactual exclusion list regions [87].
Tools for secondary analysisIdentifying and visualizing statistically enriched NDRs enables data interpretation and provides biological insights. Accessible chromatin occurs in relatively narrow regions that can be identified using peak-calling tools, such as MACS2 [88]. DeepTools2 [89] is an excellent suite of utilities to assess data reproducibility, generate signal heatmaps, and convert alignment files for visualization in genome browsers, such as Integrative Genomics Viewer [90]. When two conditions are compared, EdgeR is widely used to identify peak locations that display a statistically significant differential signal [91]. While many additional follow-up analyses can provide further insights to the patterns of open chromatin, they are outside the scope of this paper and we recommend one of the more comprehensive analysis reviews [79,80,81].
DiscussionIdentifying NDRs throughout the genome provides a window into transcriptionally active regions in normal and disease states. Chromatin accessibility profiling has had an enormous impact on basic and pre-clinical research, and is of extreme interest to be applied to clinical biopsy specimens in FFPE blocks. The goal of any useful genomics method is to yield the maximum amount of high quality DNA (or RNA) to QC defined metrics. However, major challenges associated with FFPE tissue have thus far slowed the application of epigenomic approaches to archived biopsy specimens.
There are three main areas of focus if users are to generate high quality data from FFPE tissues: 1) best practices during clinical tissue preparation and preservation; 2) improved methods of material preparation; and, 3) the optimization of genomic assays specifically for FFPE material. The first is largely outside the control of the end-user, though minimizing any delay between tissue harvesting and fixation and shorter storage times improve DNA integrity and assay yields [92]. For the second, two different strategies were used during FFPE tissue preparation for ATAC-seq and CUTAC: nuclei extraction or in situ permeabilization [67,69]. Improved FFPE extraction and preparation methods should balance increased yields without further compromising genomic integrity. Of note, FFPE repair kits are increasingly available [93], but their impact on data quality for open chromatin profiling remains to be seen. Finally, lessons may be learned from efforts to develop RNA-seq and ChIP-seq for archived FFPE material [72,92,94]. For example, the crosslinking reversal step is a major source of DNA fragmentation [92,93], but this can be mitigated by high concentrations of Tris, which improved yields by three-fold and resulted in longer DNA fragments [73],
It is a given that the direct analysis of primary tissue provides insights to the development of human disease. Indeed, histopathological analysis of FFPE brain samples has been essential to characterize mechanisms of normal and pathological aging [95,96]. The ability to perform comprehensive epigenomic analyses in such samples could provide further understanding of these processes and revolutionize clinical research.
Competing Interests
New England Biolabs (NEB) and EpiCypher are engaged in the commercial development of NicE-seq based approaches as described in this review. VUSK, BJV, MWC and MCK are employed by (and own shares in) EpiCypher. KR, SS, POE and SP are employed by (and own shares in) NEB. MCK is a board member of EpiCypher.
Acknowledgements
NicE-seq development at EpiCypher and NEB is supported by NIH grant R44HG011006.
Abbreviations
ATAC-seq, assay for transposase-accessible chromatin using sequencing; CAP, chromatin associated protein; DHS, DNAse I hypersensitive site; DNAme, DNA methylation; ENCODE, encyclopedia of DNA elements; FAIRE, formaldehyde assisted isolation of regulatory elements; FFPE, formalin-fixed paraffin-embedded; NDR, nucleosome depleted region; NFR, nucleosome free region; NGS, next-generation sequencing; PTM, post-translational modification; TF, transcription factor.
References
- Berson, A., Nativio, R., Berger, S.L., and Bonini, N.M. (2018). Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci 41, 587-598. [CrossRef]
- Pal, S., and Tyler, J.K. (2016). Epigenetics and aging. Sci Adv 2, e1600584. [CrossRef]
- Strahl, B.D., and Allis, C.D. (2000). The language of covalent histone modifications. Nature 403, 41-45. [CrossRef]
- Bhat, K.P., Umit Kaniskan, H., Jin, J., and Gozani, O. (2021). Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discov 20, 265-286. [CrossRef]
- Isbel, L., Grand, R.S., and Schubeler, D. (2022). Generating specificity in genome regulation through transcription factor sensitivity to chromatin. Nat Rev Genet 23, 728-740. [CrossRef]
- Klemm, S.L., Shipony, Z., and Greenleaf, W.J. (2019). Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20, 207-220. [CrossRef]
- Shirvaliloo, M. (2022). The landscape of histone modifications in epigenomics since 2020. Epigenomics 14, 1465-1477. [CrossRef]
- Luger, K., Rechsteiner, T.J., Flaus, A.J., Waye, M.M., and Richmond, T.J. (1997). Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol 272, 301-311. [CrossRef]
- Ioshikhes, I.P., Albert, I., Zanton, S.J., and Pugh, B.F. (2006). Nucleosome positions predicted through comparative genomics. Nat Genet 38, 1210-1215. [CrossRef]
- Mavrich, T.N., Jiang, C., Ioshikhes, I.P., Li, X., Venters, B.J., Zanton, S.J., Tomsho, L.P., Qi, J., Glaser, R.L., Schuster, S.C., et al. (2008). Nucleosome organization in the Drosophila genome. Nature 453, 358-362. [CrossRef]
- Schones, D.E., Cui, K., Cuddapah, S., Roh, T.Y., Barski, A., Wang, Z., Wei, G., and Zhao, K. (2008). Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887-898. [CrossRef]
- Kraushaar, D.C., Jin, W., Maunakea, A., Abraham, B., Ha, M., and Zhao, K. (2013). Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol 14, R121. [CrossRef]
- Li, S., Wei, T., and Panchenko, A.R. (2023). Histone variant H2A.Z modulates nucleosome dynamics to promote DNA accessibility. Nat Commun 14, 769. [CrossRef]
- Weiner, A., Hsieh, T.H., Appleboim, A., Chen, H.V., Rahat, A., Amit, I., Rando, O.J., and Friedman, N. (2015). High-resolution chromatin dynamics during a yeast stress response. Mol Cell 58, 371-386. [CrossRef]
- Consortium, E.P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57-74. [CrossRef]
- Consortium, E.P., Moore, J.E., Purcaro, M.J., Pratt, H.E., Epstein, C.B., Shoresh, N., Adrian, J., Kawli, T., Davis, C.A., Dobin, A., et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699-710. [CrossRef]
- Shimizu, M., Roth, S.Y., Szent-Gyorgyi, C., and Simpson, R.T. (1991). Nucleosomes are positioned with base pair precision adjacent to the alpha 2 operator in Saccharomyces cerevisiae. EMBO J 10, 3033-3041. [CrossRef]
- Weintraub, H., and Groudine, M. (1976). Chromosomal subunits in active genes have an altered conformation. Science 193, 848-856. [CrossRef]
- Boyle, A.P., Davis, S., Shulha, H.P., Meltzer, P., Margulies, E.H., Weng, Z., Furey, T.S., and Crawford, G.E. (2008). High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311-322. [CrossRef]
- Simpson, R.T. (1998). Chromatin structure and analysis of mechanisms of activators and repressors. Methods 15, 283-294. [CrossRef]
- Vierstra, J., and Stamatoyannopoulos, J.A. (2016). Genomic footprinting. Nat Methods 13, 213-221. [CrossRef]
- Crawford, G.E., Holt, I.E., Whittle, J., Webb, B.D., Tai, D., Davis, S., Margulies, E.H., Chen, Y., Bernat, J.A., Ginsburg, D., et al. (2006). Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16, 123-131. [CrossRef]
- Albert, I., Mavrich, T.N., Tomsho, L.P., Qi, J., Zanton, S.J., Schuster, S.C., and Pugh, B.F. (2007). Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446, 572-576. [CrossRef]
- Tsompana, M., and Buck, M.J. (2014). Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7, 33. [CrossRef]
- Klein, D.C., and Hainer, S.J. (2020). Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res 28, 69-85. [CrossRef]
- Mansisidor, A.R., and Risca, V.I. (2022). Chromatin accessibility: methods, mechanisms, and biological insights. Nucleus 13, 236-276. [CrossRef]
- Waldron, L., Simpson, P., Parmigiani, G., and Huttenhower, C. (2012). Report on emerging technologies for translational bioinformatics: a symposium on gene expression profiling for archival tissues. BMC Cancer 12, 124. [CrossRef]
- Kokkat, T.J., Patel, M.S., McGarvey, D., LiVolsi, V.A., and Baloch, Z.W. (2013). Archived formalin-fixed paraffin-embedded (FFPE) blocks: A valuable underexploited resource for extraction of DNA, RNA, and protein. Biopreserv Biobank 11, 101-106. [CrossRef]
- Blum, F. (1894). Notiz über die Anwendung des Formaldehyds (Formol) als Härtungs-und Konservierungsmittel. Anat. Anz 9, 229-231.
- Donczo, B., and Guttman, A. (2018). Biomedical analysis of formalin-fixed, paraffin-embedded tissue samples: The Holy Grail for molecular diagnostics. J Pharm Biomed Anal 155, 125-134. [CrossRef]
- Steiert, T.A., Parra, G., Gut, M., Arnold, N., Trotta, J.R., Tonda, R., Moussy, A., Gerber, Z., Abuja, P.M., Zatloukal, K., et al. (2023). A critical spotlight on the paradigms of FFPE-DNA sequencing. Nucleic Acids Res 51, 7143-7162. [CrossRef]
- Guyard, A., Boyez, A., Pujals, A., Robe, C., Tran Van Nhieu, J., Allory, Y., Moroch, J., Georges, O., Fournet, J.C., Zafrani, E.S., and Leroy, K. (2017). DNA degrades during storage in formalin-fixed and paraffin-embedded tissue blocks. Virchows Arch 471, 491-500. [CrossRef]
- Nicieza, R.G., Huergo, J., Connolly, B.A., and Sanchez, J. (1999). Purification, characterization, and role of nucleases and serine proteases in Streptomyces differentiation. Analogies with the biochemical processes described in late steps of eukaryotic apoptosis. J Biol Chem 274, 20366-20375. [CrossRef]
- Koohy, H., Down, T.A., and Hubbard, T.J. (2013). Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS One 8, e69853. [CrossRef]
- Nordstrom, K.J.V., Schmidt, F., Gasparoni, N., Salhab, A., Gasparoni, G., Kattler, K., Muller, F., Ebert, P., Costa, I.G., consortium, D., et al. (2019). Unique and assay specific features of NOMe-, ATAC- and DNase I-seq data. Nucleic Acids Res 47, 10580-10596. [CrossRef]
- Yardimci, G.G., Frank, C.L., Crawford, G.E., and Ohler, U. (2014). Explicit DNase sequence bias modeling enables high-resolution transcription factor footprint detection. Nucleic Acids Res 42, 11865-11878. [CrossRef]
- Gusmao, E.G., Allhoff, M., Zenke, M., and Costa, I.G. (2016). Analysis of computational footprinting methods for DNase sequencing experiments. Nat Methods 13, 303-309. [CrossRef]
- Martins, A.L., Walavalkar, N.M., Anderson, W.D., Zang, C., and Guertin, M.J. (2018). Universal correction of enzymatic sequence bias reveals molecular signatures of protein/DNA interactions. Nucleic Acids Res 46, e9. [CrossRef]
- Schmidt, F., Gasparoni, N., Gasparoni, G., Gianmoena, K., Cadenas, C., Polansky, J.K., Ebert, P., Nordstrom, K., Barann, M., Sinha, A., et al. (2017). Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res 45, 54-66. [CrossRef]
- Jin, W., Tang, Q., Wan, M., Cui, K., Zhang, Y., Ren, G., Ni, B., Sklar, J., Przytycka, T.M., Childs, R., et al. (2015). Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142-146. [CrossRef]
- Cooper, J., Ding, Y., Song, J., and Zhao, K. (2017). Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing. Nat Protoc 12, 2342-2354. [CrossRef]
- Axel, R. (1975). Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. Biochemistry 14, 2921-2925. [CrossRef]
- Valouev, A., Ichikawa, J., Tonthat, T., Stuart, J., Ranade, S., Peckham, H., Zeng, K., Malek, J.A., Costa, G., McKernan, K., et al. (2008). A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18, 1051-1063. [CrossRef]
- Li, Z., Schug, J., Tuteja, G., White, P., and Kaestner, K.H. (2011). The nucleosome map of the mammalian liver. Nat Struct Mol Biol 18, 742-746. [CrossRef]
- Gutierrez, G., Millan-Zambrano, G., Medina, D.A., Jordan-Pla, A., Perez-Ortin, J.E., Penate, X., and Chavez, S. (2017). Subtracting the sequence bias from partially digested MNase-seq data reveals a general contribution of TFIIS to nucleosome positioning. Epigenetics Chromatin 10, 58. [CrossRef]
- Mieczkowski, J., Cook, A., Bowman, S.K., Mueller, B., Alver, B.H., Kundu, S., Deaton, A.M., Urban, J.A., Larschan, E., Park, P.J., et al. (2016). MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 7, 11485. [CrossRef]
- Henikoff, J.G., Belsky, J.A., Krassovsky, K., MacAlpine, D.M., and Henikoff, S. (2011). Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A 108, 18318-18323. [CrossRef]
- Chereji, R.V., Bryson, T.D., and Henikoff, S. (2019). Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol 20, 198. [CrossRef]
- Nagy, P.L., Cleary, M.L., Brown, P.O., and Lieb, J.D. (2003). Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc Natl Acad Sci U S A 100, 6364-6369. [CrossRef]
- Giresi, P.G., Kim, J., McDaniell, R.M., Iyer, V.R., and Lieb, J.D. (2007). FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17, 877-885. [CrossRef]
- Simon, J.M., Giresi, P.G., Davis, I.J., and Lieb, J.D. (2012). Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc 7, 256-267. [CrossRef]
- Nagy, P.L., and Price, D.H. (2009). Formaldehyde-assisted isolation of regulatory elements. Wiley Interdiscip Rev Syst Biol Med 1, 400-406. [CrossRef]
- Zhou, W., Ji, Z., Fang, W., and Ji, H. (2019). Global prediction of chromatin accessibility using small-cell-number and single-cell RNA-seq. Nucleic Acids Res 47, e121. [CrossRef]
- Giresi, P.G., and Lieb, J.D. (2009). Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods 48, 233-239. [CrossRef]
- Gaulton, K.J., Nammo, T., Pasquali, L., Simon, J.M., Giresi, P.G., Fogarty, M.P., Panhuis, T.M., Mieczkowski, P., Secchi, A., Bosco, D., et al. (2010). A map of open chromatin in human pancreatic islets. Nat Genet 42, 255-259. [CrossRef]
- Buchert, E.M., Fogarty, E.A., Uyehara, C.M., McKay, D.J., and Buttitta, L.A. (2023). A tissue dissociation method for ATAC-seq and CUT&RUN in Drosophila pupal tissues. Fly (Austin) 17, 2209481. [CrossRef]
- Berg, D.E. (2017). Julian Davies and the discovery of kanamycin resistance transposon Tn5. J Antibiot (Tokyo) 70, 339-346. [CrossRef]
- Reznikoff, W.S. (2003). Tn5 as a model for understanding DNA transposition. Mol Microbiol 47, 1199-1206. [CrossRef]
- Li, N., Jin, K., Bai, Y., Fu, H., Liu, L., and Liu, B. (2020). Tn5 Transposase Applied in Genomics Research. Int J Mol Sci 21. [CrossRef]
- Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y., and Greenleaf, W.J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213-1218. [CrossRef]
- Kaya-Okur, H.S., Wu, S.J., Codomo, C.A., Pledger, E.S., Bryson, T.D., Henikoff, J.G., Ahmad, K., and Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930. [CrossRef]
- Buenrostro, J.D., Wu, B., Chang, H.Y., and Greenleaf, W.J. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21-21 29 29. [CrossRef]
- Wolpe, J.B., Martins, A.L., and Guertin, M.J. (2023). Correction of transposase sequence bias in ATAC-seq data with rule ensemble modeling. NAR Genom Bioinform 5, lqad054. [CrossRef]
- Li, Z., Schulz, M.H., Look, T., Begemann, M., Zenke, M., and Costa, I.G. (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol 20, 45. [CrossRef]
- Corces, M.R., Trevino, A.E., Hamilton, E.G., Greenside, P.G., Sinnott-Armstrong, N.A., Vesuna, S., Satpathy, A.T., Rubin, A.J., Montine, K.S., Wu, B., et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959-962. [CrossRef]
- Grandi, F.C., Modi, H., Kampman, L., and Corces, M.R. (2022). Chromatin accessibility profiling by ATAC-seq. Nat Protoc 17, 1518-1552. [CrossRef]
- Henikoff, S., Henikoff, J.G., Ahmad, K., Paranal, R.M., Janssens, D.H., Russell, Z.R., Szulzewsky, F., Kugel, S., and Holland, E.C. (2023). Epigenomic analysis of formalin-fixed paraffin-embedded samples by CUT&Tag. Nat Commun 14, 5930. [CrossRef]
- Yadav, R.P., Polavarapu, V.K., Xing, P., and Chen, X. (2022). FFPE-ATAC: A Highly Sensitive Method for Profiling Chromatin Accessibility in Formalin-Fixed Paraffin-Embedded Samples. Curr Protoc 2, e535. [CrossRef]
- Zhang, H., Polavarapu, V.K., Xing, P., Zhao, M., Mathot, L., Zhao, L., Rosen, G., Swartling, F.J., Sjoblom, T., and Chen, X. (2022). Profiling chromatin accessibility in formalin-fixed paraffin-embedded samples. Genome Res 32, 150-161. [CrossRef]
- Zhao, L., Polavarapu, V.K., Yadav, R.P., Xing, P., and Chen, X. (2022). A Highly Sensitive Method to Efficiently Profile the Histone Modifications of FFPE Samples. Bio Protoc 12. [CrossRef]
- Zhao, L., Xing, P., Polavarapu, V.K., Zhao, M., Valero-Martinez, B., Dang, Y., Maturi, N., Mathot, L., Neves, I., Yildirim, I., et al. (2021). FACT-seq: profiling histone modifications in formalin-fixed paraffin-embedded samples with low cell numbers. Nucleic Acids Res 49, e125. [CrossRef]
- Amatori, S., and Fanelli, M. (2022). The Current State of Chromatin Immunoprecipitation (ChIP) from FFPE Tissues. Int J Mol Sci 23. [CrossRef]
- Oba, U., Kohashi, K., Sangatsuda, Y., Oda, Y., Sonoda, K.H., Ohga, S., Yoshimoto, K., Arai, Y., Yachida, S., Shibata, T., et al. (2022). An efficient procedure for the recovery of DNA from formalin-fixed paraffin-embedded tissue sections. Biol Methods Protoc 7, bpac014. [CrossRef]
- Ponnaluri, V.K.C., Zhang, G., Esteve, P.O., Spracklin, G., Sian, S., Xu, S.Y., Benoukraf, T., and Pradhan, S. (2017). NicE-seq: high resolution open chromatin profiling. Genome Biol 18, 122. [CrossRef]
- Chin, H.G., Sun, Z., Vishnu, U.S., Hao, P., Cejas, P., Spracklin, G., Esteve, P.O., Xu, S.Y., Long, H.W., and Pradhan, S. (2020). Universal NicE-seq for high-resolution accessible chromatin profiling for formaldehyde-fixed and FFPE tissues. Clin Epigenetics 12, 143. [CrossRef]
- Esteve, P.O., Vishnu, U.S., Chin, H.G., and Pradhan, S. (2020). Visualization and Sequencing of Accessible Chromatin Reveals Cell Cycle and Post-HDAC inhibitor Treatment Dynamics. J Mol Biol 432, 5304-5321. [CrossRef]
- Vishnu, U.S., Esteve, P.O., Chin, H.G., and Pradhan, S. (2021). One-pot universal NicE-seq: all enzymatic downstream processing of 4% formaldehyde crosslinked cells for chromatin accessibility genomics. Epigenetics Chromatin 14, 53. [CrossRef]
- Chan, S.H., Zhu, Z., Dunigan, D.D., Van Etten, J.L., and Xu, S.Y. (2006). Cloning of Nt.CviQII nicking endonuclease and its cognate methyltransferase: M.CviQII methylates AG sequences. Protein Expr Purif 49, 138-150. [CrossRef]
- Pranzatelli, T.J.F., Michael, D.G., and Chiorini, J.A. (2018). ATAC2GRN: optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference. BMC Genomics 19, 563. [CrossRef]
- Smith, J.P., and Sheffield, N.C. (2020). Analytical Approaches for ATAC-seq Data Analysis. Curr Protoc Hum Genet 106, e101. [CrossRef]
- Yan, F., Powell, D.R., Curtis, D.J., and Wong, N.C. (2020). From reads to insight: a hitchhiker's guide to ATAC-seq data analysis. Genome Biol 21, 22. [CrossRef]
- Bolger, A.M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114-2120. [CrossRef]
- Langmead, B., and Salzberg, S.L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357-359. [CrossRef]
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R., and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078-2079. [CrossRef]
- Quinlan, A.R. (2014). BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 47, 11 12 11-34. [CrossRef]
- Quinlan, A.R., and Hall, I.M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841-842. [CrossRef]
- Amemiya, H.M., Kundaje, A., and Boyle, A.P. (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. [CrossRef]
- Liu, T. (2014). Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 1150, 81-95. [CrossRef]
- Ramirez, F., Ryan, D.P., Gruning, B., Bhardwaj, V., Kilpert, F., Richter, A.S., Heyne, S., Dundar, F., and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160-165. [CrossRef]
- Robinson, J.T., Thorvaldsdottir, H., Turner, D., and Mesirov, J.P. (2023). igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 39. [CrossRef]
- Robinson, M.D., McCarthy, D.J., and Smyth, G.K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139-140. [CrossRef]
- Talebi, A., Thiery, J.P., and Kerachian, M.A. (2021). Fusion transcript discovery using RNA sequencing in formalin-fixed paraffin-embedded specimen. Crit Rev Oncol Hematol 160, 103303. [CrossRef]
- Siegel, E.M., Berglund, A.E., Riggs, B.M., Eschrich, S.A., Putney, R.M., Ajidahun, A.O., Coppola, D., and Shibata, D. (2014). Expanding epigenomics to archived FFPE tissues: an evaluation of DNA repair methodologies. Cancer Epidemiol Biomarkers Prev 23, 2622-2631. [CrossRef]
- Cejas, P., Li, L., O'Neill, N.K., Duarte, M., Rao, P., Bowden, M., Zhou, C.W., Mendiola, M., Burgos, E., Feliu, J., et al. (2016). Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat Med 22, 685-691. [CrossRef]
- DeTure, M.A., and Dickson, D.W. (2019). The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener 14, 32. [CrossRef]
- Willroider, M., Roeber, S., Horn, A.K.E., Arzberger, T., Scheifele, M., Respondek, G., Sabri, O., Barthel, H., Patt, M., Mishchenko, O., et al. (2021). Superiority of Formalin-Fixed Paraffin-Embedded Brain Tissue for in vitro Assessment of Progressive Supranuclear Palsy Tau Pathology With [(18)F]PI-2620. Front Neurol 12, 684523. [CrossRef]
- Clark, S.J., Statham, A., Stirzaker, C., Molloy, P.L., and Frommer, M. (2006). DNA methylation: bisulphite modification and analysis. Nat Protoc 1, 2353-2364. [CrossRef]
- Vaisvila, R., Ponnaluri, V.K.C., Sun, Z., Langhorst, B.W., Saleh, L., Guan, S., Dai, N., Campbell, M.A., Sexton, B.S., Marks, K., et al. (2021). Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res 31, 1280-1289. [CrossRef]
- Skene, P.J., and Henikoff, S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [CrossRef]
- Jiang, S., and Mortazavi, A. (2018). Integrating ChIP-seq with other functional genomics data. Brief Funct Genomics 17, 104-115. [CrossRef]
Figure 1.
Local features that define ‘open’ and ‘closed’ chromatin. Chromatin states (e.g., active transcriptional enhancers, repressed heterochromatin) can be functionally defined by integrating a range of data elements, including nucleosome depleted regions (NDRs; mapped by one of the methods discussed in this review), DNA methylation (DNAme (primarily 5-methylcytosine); mapped by bisulfite sequencing or EM-seq [97,98]), transcription factors (TFs) and histone post-translational modifications (PTMs; mapped by ChIP-seq or newer approaches like CUT&RUN or CUT&Tag [61,99]). Figure adapted from [100]. Missing nucleosome in active promoter shows NDR.
Figure 1.
Local features that define ‘open’ and ‘closed’ chromatin. Chromatin states (e.g., active transcriptional enhancers, repressed heterochromatin) can be functionally defined by integrating a range of data elements, including nucleosome depleted regions (NDRs; mapped by one of the methods discussed in this review), DNA methylation (DNAme (primarily 5-methylcytosine); mapped by bisulfite sequencing or EM-seq [97,98]), transcription factors (TFs) and histone post-translational modifications (PTMs; mapped by ChIP-seq or newer approaches like CUT&RUN or CUT&Tag [61,99]). Figure adapted from [100]. Missing nucleosome in active promoter shows NDR.
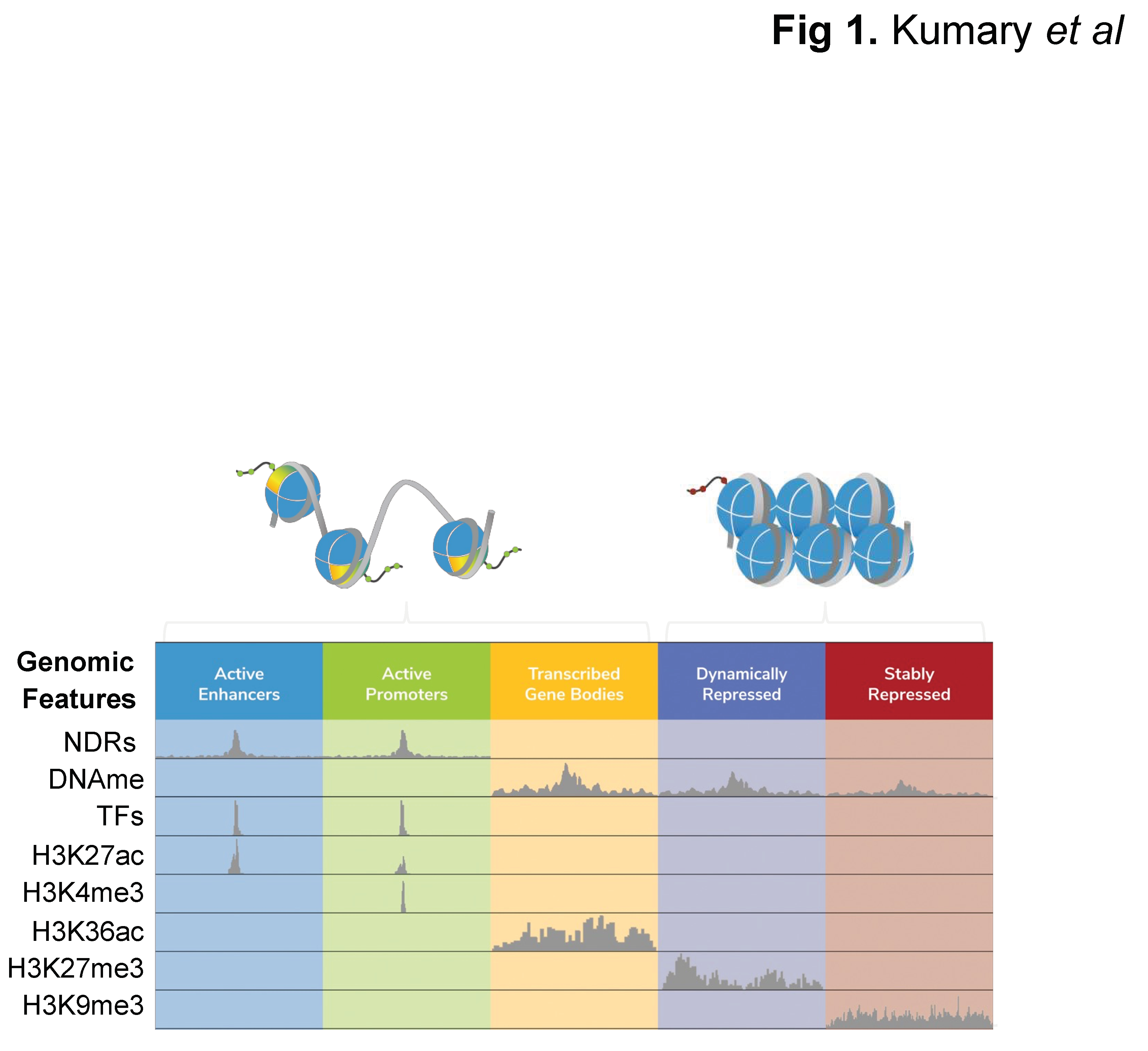
Figure 2.
Publication trends. A-B. Publication frequency for chromatin accessibility approaches using PubMed search terms “DNase-seq”, MNase-seq”, “FAIRE-seq”, “NicE-seq” and “ATAC-seq” (last the focus of (B) to accommodate its overwhelming field adoption rate). C. Publication frequency for PubMed search term “FFPE”. D. Accumulated publications/the first incidence of each search term on PubMed.
Figure 2.
Publication trends. A-B. Publication frequency for chromatin accessibility approaches using PubMed search terms “DNase-seq”, MNase-seq”, “FAIRE-seq”, “NicE-seq” and “ATAC-seq” (last the focus of (B) to accommodate its overwhelming field adoption rate). C. Publication frequency for PubMed search term “FFPE”. D. Accumulated publications/the first incidence of each search term on PubMed.
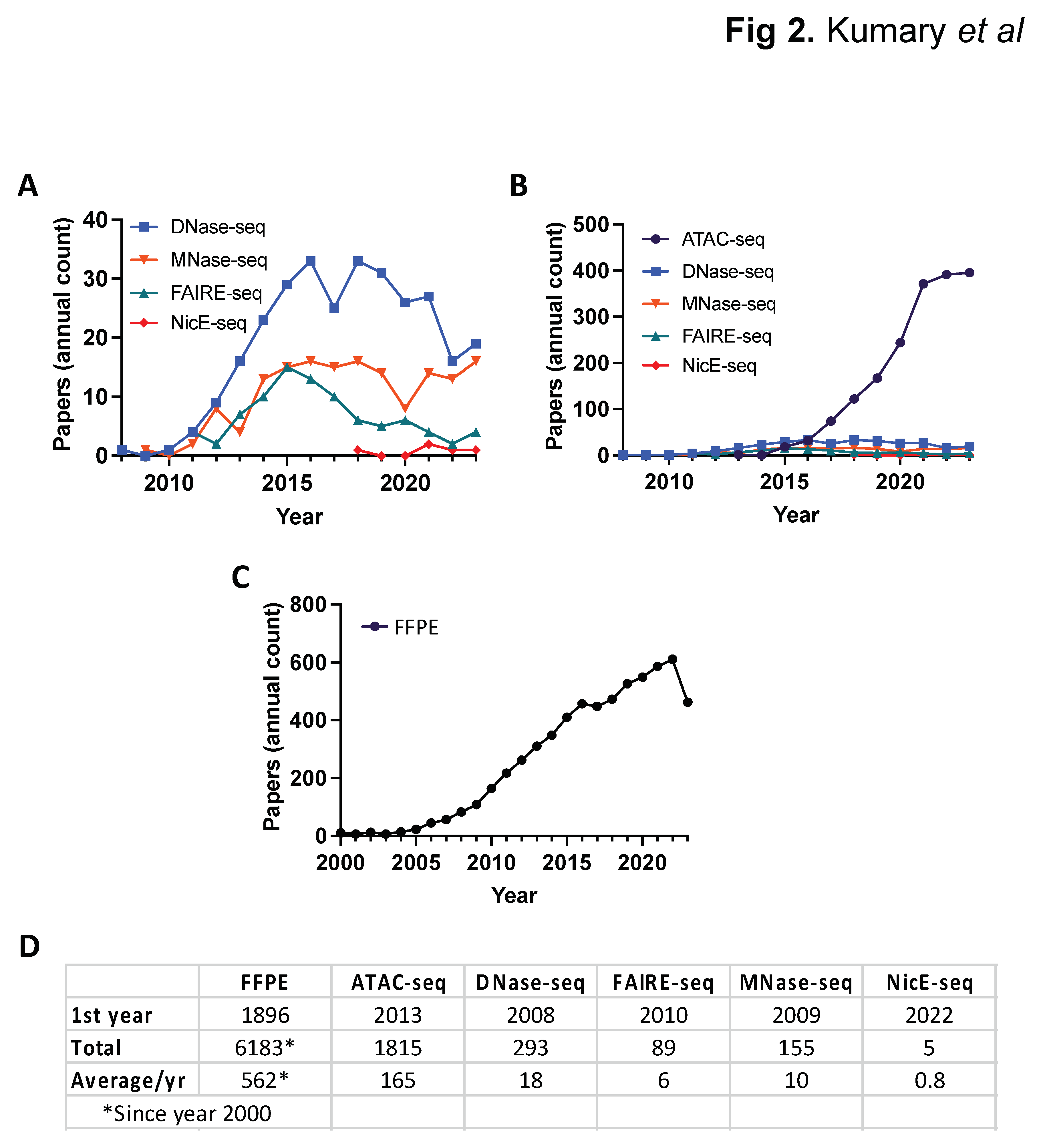
Figure 3.
Schematic of techniques used for chromatin accessibility profiling. DNase-seq. B. MNase-seq. C. FAIRE-seq. D. ATAC-seq. E. NicE-seq. Figure was created with BioRender.
Figure 3.
Schematic of techniques used for chromatin accessibility profiling. DNase-seq. B. MNase-seq. C. FAIRE-seq. D. ATAC-seq. E. NicE-seq. Figure was created with BioRender.
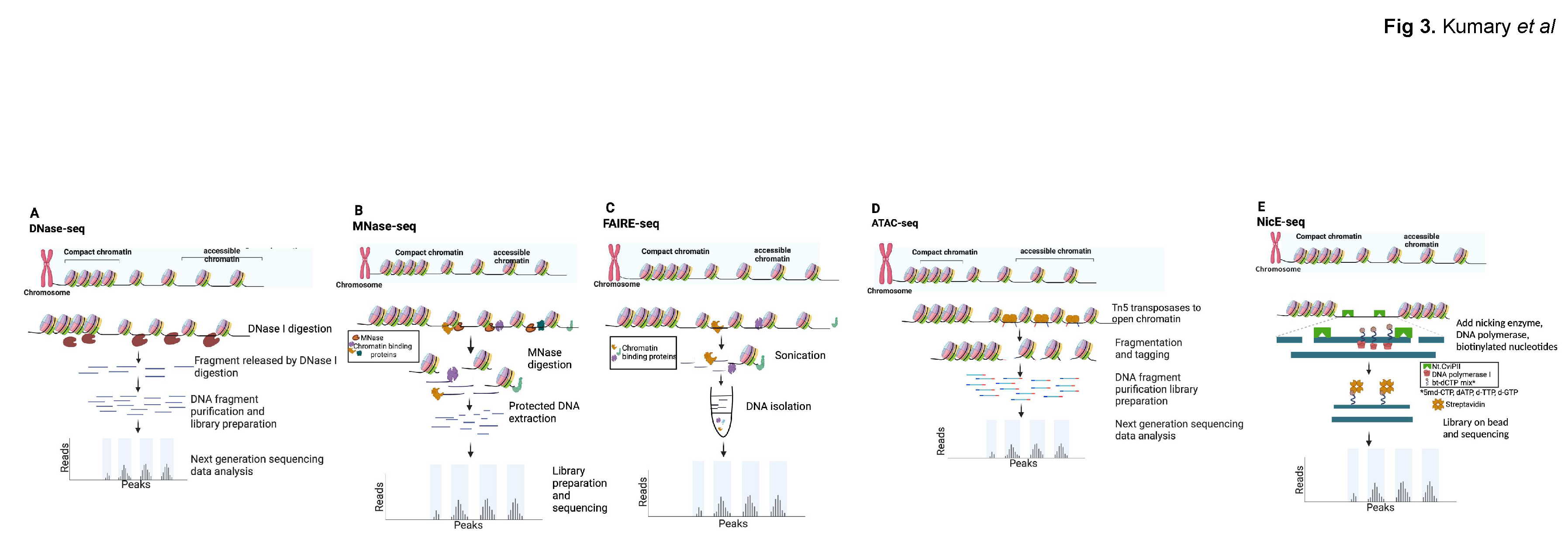
Figure 4.
Bioinformatic pipeline for NGS data processing and analysis. Figure was created with BioRender.
Figure 4.
Bioinformatic pipeline for NGS data processing and analysis. Figure was created with BioRender.
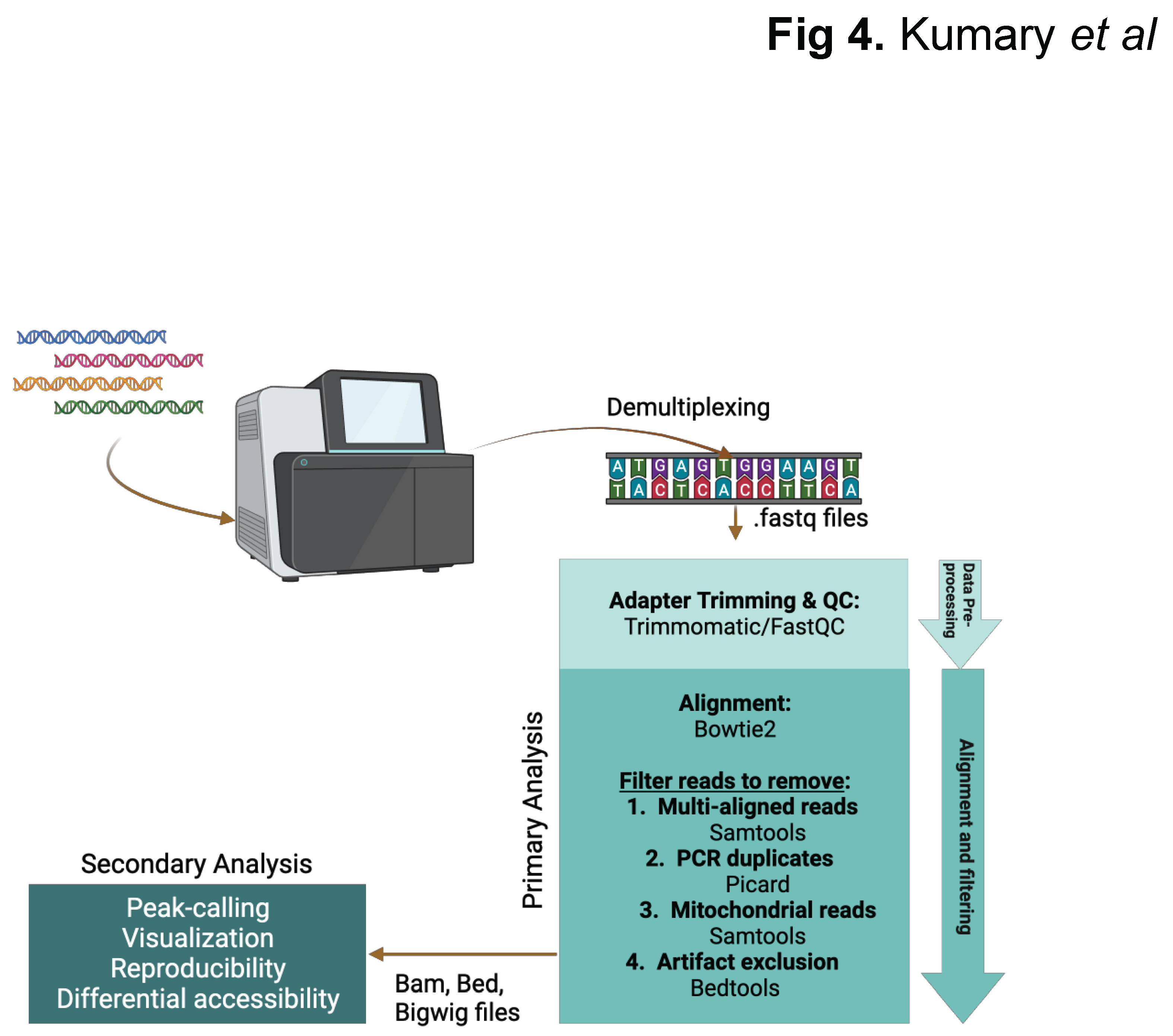

Table 1.
Commonly used approaches for chromatin accessibility profiling.
| DNase-seq | MNase-seq | FAIRE-seq | ATAC-seq | NicE-seq | |
|---|---|---|---|---|---|
| Type of input cells/tissue | Fresh/Formaldehyde cross-linked/FFPE (Formalin Fixed Paraffin Embedded) | Fresh/formaldehyde cross-linked | Formaldehyde cross-linked | Fresh/formaldehyde cross-linked (less efficient in fixed) | Formaldehyde cross-linked/FFPE |
| Application to FFPE (PubMed) | 1 | 0 | 1 | 2 | 2 |
| Number of input cells | 1M-10M | 10K-10M | 10K-10M | 1 cell – 50k | 25 cells – 100k |
| Approach | DNase I (endonuclease) cuts unprotected DNA | MNase (endo-exonuclease) digests unprotected DNA | Sonicate unprotected DNA in crosslinked material | Tn5 transposase tagments open region with DNA adapters | Nt-CviPII nickase cuts/labels CCD sites in unprotected DNA |
| Sequencing type | Single/Paired End | Single/Paired End | Single/Paired End | Single/Paired End | Single/Paired End |
| Target region | NDR | Linker DNA between Nucleosomes | NDR | NDR | NDR |
| Sequencing depth (human genome; ~3B bp) | 20-50 M mapped reads | 150-200 M mapped reads | 20-50 M mapped reads | 25-30 million (M) mapped non-mitochondrial (mito) reads | 20-30 M mapped reads |
| Cleavage bias | Yes | Yes | No | Yes | Yes |
|
Advantages / Disadvantages |
No prior knowledge of the sequence or binding protein is required / Time consuming. Requires laborious enzyme titrations and calibrations. Requires high sequencing depth. |
Nucleosome positioning can be inferred / Requires laborious enzyme titrations and calibrations. Requires high sequencing depth. Indirect profiling of open regions. |
No enzymes optimization or titration required / Low signal-to-noise. Relatively complex computational data analysis and interpretation. Results are highly fixation dependent. |
Simple, fast and sensitive approach. High signal-to-noise. / High mito DNA counts (unless nuclei isolated). Requires two independent tagmentation events in opposite orientation. Tn5 sequence bias and promoter-enrichment bias. |
Simple enzymatic approach. 5% mito DNA counts. Optimal in fixed or FFPE samples. Can be used in clinical settings. Efficiently profiles promoters and enhancers. / AT-rich sequences may be underrepresented. |
| References | [19] | [9–11,43,44] | [50] | [60,62] | [74–77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



