
Submitted:
22 February 2024
Posted:
22 February 2024
You are already at the latest version
Abstract
Orthohepadnavirus causes chronic hepatitis in a broad range of mammals, including primates, cats, woodchucks, and bats. Hepatitis B virus (HBV) X protein inhibits type-I interferon (IFN) signaling, promoting HBV escape from the innate immune system to establish persistent infection. However, whether X proteins of Orthohepadnavirus viruses in the other species have similar inhibitory activity remains unknown. Here, we investigated the anti-IFN activity of 17 Orthohepadnavirus X proteins derived from various hosts. We observed conserved activity of Orthohepadnavirus X proteins in inhibiting TIR-domain-containing adaptor protein inducing IFN-β (TRIF)-mediated IFN-β signaling pathway through TRIF degradation. X proteins from domestic cat hepadnavirus (DCH), a novel member of Orthohepadnavirus, inhibited mitochondrial antiviral signaling protein (MAVS)-mediated IFNβ signaling pathway comparable with HBV X. These results indicate that inhibition of IFN signaling is conserved in Orthohepadnavirus X proteins.
Keywords:
Orthohepadnavirus
; X protein
; domestic cat hepadnavirus (DCH)
; hepatitis B virus (HBV)
; interferon-β signaling pathway
1. Introduction
Hepatitis B virus (HBV) affects >248 million individuals worldwide and is the major cause of chronic liver disease and liver cancer in humans [1]. HBV (family: Hepadnaviridae, genus: Orthohepadnavirus) infects mammals, including primates, cats, woodchucks, and bats [2]. Members of Orthohepadnavirus have a partially double-stranded DNA genome, ranging from 3.0–3.4 kb, that encodes four viral proteins, including core, polymerase, surface, and X [2]. HBV is a "stealth virus" because it triggers a minimal immune response, particularly the type-I interferon (IFN) response, during the initial stages of infection [3]. The failure to induce an innate immune response in infected hepatocytes can lead to incomplete clearance of infected hepatocytes and cause chronic infection [4].
The innate immune system acts as the first line of defense against viral infections by triggering pattern recognition receptors (PRRs), including retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) and toll-like receptors (TLRs) [5]. Several proteins are involved in the PRRs pathway, including mitochondrial antiviral signaling (MAVS) [6], cyclic GMP–AMP synthase (cGAS) [7], and stimulator of interferon genes (STING) [6]. In the TLR3/4-dependent pathway, TIR-domain-containing adaptor inducing interferon-beta (TRIF) is responsible for activating TANK-binding kinase 1 (TBK1), and subsequently, interferon regulatory factor 3 (IRF-3), contributing to IFN production [7]. Increasing evidence indicates that HBV suppresses IFN signaling [8,9]. Several HBV viral proteins, including polymerase (pol) [10], e antigen [11], core protein (HBcAG) [12], splice protein (HBSP) [13], and X protein (HBx) [14], can suppress cellular innate immunity [15]. HBx is a small protein (154 amino acids) that plays a crucial role in HBV replication [16] and infection [17]. In hepatocytes, HBx disrupted the structural maintenance of chromosome 5/6 complex (Smc5/6) in hepatocytes, which is a restriction factor that inhibits HBV transcription, leading to increased viral transcription and development of cellular transformation through impaired homologous recombination repair of DNA double-strand breaks (DSBs) [18]. HBx comprises two functional domains: N- and C-terminal domains [19,20]. The N-terminal is a negative regulatory domain (amino acid residues 1–50) that consists of a strongly conserved initial segment that exhibits transrepressor activity, specifically within residues 21–50 [21]. The functionality of HBx relies on the transactivation domain (C-terminal region), which contains the transactivation domain (amino acid residues 52–142) [22]. The stability of HBx is associated with 20 amino acids at the C-terminus (134–154) [23]. HBx inhibits IFN-β production by suppressing RIG-I, melanoma differentiation-associated protein (MDA5), and MAVS [24]. Residues Asn118 and Glu119 in HBx play a critical role in HBx-mediated inhibition of RIG-I–MAVS signaling [25]. Furthermore, HBx decreased TRIF expression through the proteasomal degradation of TRIF in hepatoma cells [26].
In 2018, domestic cat hepadnavirus (DCH), a novel member of the genus Orthohepadnavirus, was identified from a domestic cat in Australia [27]. Since then, the prevalence of DCH in cats has been investigated in Italy [28], Thailand [29], Malaysia [30], the United Kingdom [31], Japan [32], the USA [33], Hongkong [34], Taiwan [35], and Türkiye [36]. The prevalence of DCH infection ranged from 0.2% in the USA to 18.5% in Thailand [33,37]. DCH infection is associated with chronic hepatitis and hepatocellular carcinoma in cats, with increased levels of alanine transaminase (ALT) and aspartate transaminase (AST) [35,38], suggesting similarity with HBV infection in humans. We reported that DCH shares the sodium/bile acid cotransporter (NTCP) with HBV as a cellular entry receptor [39]. However, the functional conservation of DCH X proteins is unclear. We aimed to investigate the functional conservation of Orthohepadnavirus X protein from other animal species in inhibiting IFN-β signaling pathway.
In this study, we investigated the function of 17 Orthohepadnavirus X proteins on the IFN-β signaling pathway. We found that DCH X proteins and other Orthohepadnavirus X proteins tested inhibited TRIF-mediated IFN-β signaling. DCH X protein required 20 amino acids at the C-terminus for efficient expression and function, suggesting that this domain is required for protein stability and is conserved between HBV and DCH X proteins. Thus, the inhibitory function of TRIF-mediated IFN-β signaling pathway is conserved in Orthohepadnavirus X protein.
2. Results
2.1. Genetic characteristics of Orthohepadnavirus X proteins
We aligned 17 Orthohepadnavirus X proteins, including four strains of human HBV (genotype A, D, G, and H), four strains of DCH (KT-116, Rara, Sydney, and TR-SV15), three strains of bat HBV (pomona bat, horseshoe bat, and tent-making bat), one strain of woodchuck hepatitis virus, and one strain of HBV from the domestic donkey, Asian gray shrew, capuchin monkey, orangutan, and woolly monkey (Figure 1A).
Although X proteins derived from the four DCH strains consist of 145 amino acids (aa), HBV X protein comprises 154 aa. X protein from DCH (Rara) was distant from X proteins of other DCH strains (Figure 1B). X proteins from DCH (Rara) and DCH (Sydney: a reference strain of DCH) differed by 14 residues [35]. We observed that X protein of tent-making bat HBV was more distant from the X proteins of pomona bat HBV and horseshoe bat HBV. X proteins from bat HBV were not closely related, but were genetically similar to HBV strains from other animal species. This suggests viral transmission between bats and other animals (Figure 1B). In addition, X protein of orangutan HBV is genetically closer to X protein of HBV than X proteins of capuchin monkey HBV or woolly monkey HBV. This result suggests that X protein of ape HBV is more similar to X proteins of human HBVs than X proteins of New World monkey HBVs.
2.2. Both HBV (A) and DCH (KT-116) X proteins inhibited IFN-β signaling mediated by TRIF, MAVS, and IRF3
Considering the genetic diversity of Orthohepadnavirus X proteins across species and strains, we sought to investigate whether the inhibitory effect of HBx on IFN-β induction is conserved in other Orthohepadnavirus X proteins. First, we measured the expression level of HA-tagged Orthohepadnavirus X proteins in Lenti-X 293T cells with western blotting. We found comparable expression levels of X proteins in transfected Lenti-X 293T cells (Figure 2). Before testing the inhibitory effect of Orthohepadnavirus X proteins, we confirmed that co-transfection with the following plasmids: 1) IFN-β Luc and TRIF, 2) IgK-IFN Luc and MAVS, 3) IgK-IFN Luc and IKKe, 4) interferon-stimulated response element (ISRE) Luc and IRF3, and 5) ISRE Luc and TRIF, significantly induced Firefly luciferase (Figure S1), consistent with other studies [40,41,42].
Next, we examined the inhibitory effect of HBV (genotype A) and DCH (KT-116) X proteins on IFN signaling pathways by co-transfection of Lenti-X 293T cells with plasmids expressing X proteins. Consistent with other findings [26], HBx inhibited the induction of Firefly luciferase in cells co-transfected with the following plasmids: IFN-β Luc and TRIF (Figure 3A), IgK-IFN Luc and MAVS (Figure 3B), and ISRE Luc and IRF3 (Figure 3D), but not IgK-IFN Luc and IKKe (Figure 3C). DCH (KT-116) X protein showed similar inhibitory effects with HBV (genotype A) X protein, suggesting functional conservation between HBV (genotype A) and DCH (KT-116) X proteins.
2.3. X proteins derived from a range of Orthohepadnavirus sp. inhibited TRIF-mediated IFN signaling
We observed a conserved inhibitory effect of HBV (genotype A) and DCH (KT-116) X proteins on IFN signaling (Figure 3A, 3B, and 3D). Notably, HBV (genotype A) and DCH (KT-116) X proteins potently inhibited TRIF-mediated IFN-β signaling (Figure 3A). To investigate whether the inhibitory effect of X proteins on TRIF-mediated IFN-β signaling is conserved across Orthohepadnavirus sp., Lenti-X 293T cells were co-transfected with plasmids expressing the 17 Orthohepadnavirus X proteins with IFN-β Luc and TRIF plasmids (Figure 4). We found that all Orthohepadnavirus X proteins significantly inhibited TRIF-mediated IFN-β signaling (Figure 4A and 4B), suggesting that the inhibitory effect on TRIF-mediated IFN signaling is conserved in X proteins across Orthohepadnavirus sp. To further test the inhibitory effect of Orthohepadnavirus X proteins, we co-transfected Lenti-X 293T cells with plasmids expressing IFN-β Luc, MAVS, and HBV strains (genotype A, D, G, and H) or DCH strains (KT-116, Rara, Sydney, and TR-SV15) X proteins. Although the inhibitory effect is smaller than on TRIF-mediated IFN-β signaling (Figure S2), Orthohepadnavirus X proteins target MAVS-mediated IFN-β signaling.
2.4 The C-terminus transactivation domain of X proteins plays an important role in stabilizing protein expression and function
We showed that the inhibitory effect on TRIF-mediated IFN-β signaling is conserved in Orthohepadnavirus X proteins (Figure 4A and 4B). We found genetic variations in Orthohepadnavirus X proteins, especially in the transactivation domain (Figure 1). Here, we aimed to identify domains in X proteins that determine inhibitory effects on TRIF-mediated IFN-β signaling. Therefore, we made a series of deletion mutants of HBV (genotype A) and DCH (KT-116) X proteins (Figure 5A and 5B), as described [43]. We made mutants with deletions in the 1) negative regulatory domain (amino acids (aa) 1-50: del(1-50), 2) the transactivation domain (aa 52-148: del(52-148)), 3) nuclear translocation domain (aa 120-140: del(120-140), 4) region that interacts with the host’s structural maintenance of chromosome 5/6 complex (Smc5/6 )(aa 45-140: del(45-140)), and 5) region that interacts with the host’s damaged DNA binding protein 1 (DDB1) (aa 88-100: del(88-100) (Figure 5B).
The deletion mutant plasmids of HBV (genotype A) and DCH (KT-116) X proteins were used to co-transfect Lenti-X 293T cells with IFN-β Luc and TRIF plasmids. We found significantly lower expression levels of del(52-148) and del(45-140) mutants of HBV (genotype A) X protein in Lenti-X 293T cells (Figure 5C). Although the similarity of the transactivation domains between HBV (genotype A) and DCH (KT-116) X proteins is low, del(52-148) and del(45-140) X protein mutants of DCH (KT-116) X protein shared phenotype with HBV (genotype A) del(52-148) and del(45-140) mutants.
Next, we examined the subcellular localization of X proteins in Lenti-X 293T cells. Wild-type (WT) X proteins of HBV (genotype A) and DCH (KT-116) primarily localized in the nucleus, but were also detected in the cytoplasm (Figure S3), which is consistent with another study [44]. Mutant X proteins with deletions had similar localization, suggesting that the deletion of N- and C-terminal amino acids of HBV (genotype A) and DCH (KT-116) X proteins minimally affected protein localization in Lenti-X 293T cells.
Consistent with the result of western blotting, we observed both del(52-148) and del(45-140) X protein mutants of both HBV (genotype A) and DCH (KT-116) failed to inhibit TRIF-mediated IFN-β signaling (Figure 5D and 5E). These results demonstrated that the transactivation domain at the C-terminus has an important role in stabilizing the expression and function of Orthohepadnavirus X proteins.
To elucidate the mechanism of the inhibitory effect on the TRIF-mediated IFN-β signaling pathway, we co-transfected Lenti-X 293T cells with plasmids expressing TRIF and X proteins from a broad range of species. We found that the expression of X proteins induced 13%–100% degradation of TRIF (Figure 6E). Although DCH (Rara) X protein degraded TRIF completely, TMBHBV X protein decreased TRIF expression by ~13% in Lenti-X 293T cells. These results suggest that X protein inhibit TRIF-mediated IFN-β signaling by degrading TRIF.
2.6. Variation in the C-terminus transactivation domain of DCH X protein determines the inhibitory effect on ISRE-mediated IFN-β signaling
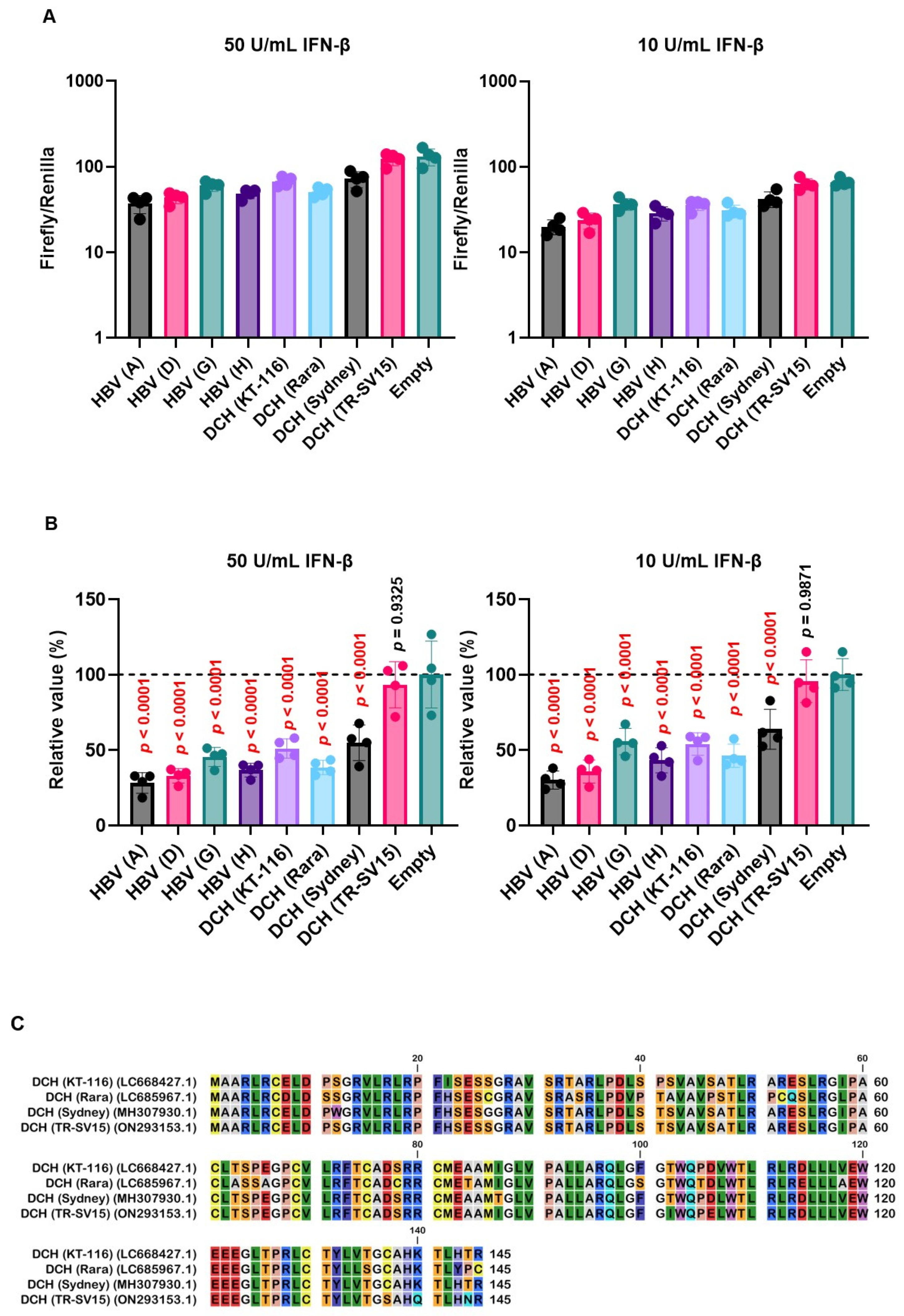
To evaluate the effect of Orthohepadnavirus X proteins in inhibiting ISRE-mediated IFN-β signaling, we used 293T-ISRE-luc2 cells. We confirmed that 293T-ISRE-luc2 cells could be stimulated with recombinant human IFN-β, leading to a significant induction of luciferase (Figure S4). Next, we transfected 293T-ISRE-luc2 cells with plasmids expressing Orthohepadnavirus X proteins to test the inhibitory effect of Orthohepadnavirus X proteins on ISRE-mediated IFN-β signaling. Most X proteins, except DCH (TR-SV15) X, suppressed ISRE-mediated IFN-β signaling (Figure 7A and 7B). Alignment of the X proteins of the four DCH strains showed that in DCH (TR-SV15) X protein, five residues (positions 102, 106, 137, 140, and 144) differed in the C-terminus transactivation domain compared with the other three strains (Figure 7C). This indicates that these five amino acids might be responsible for the impaired inhibition of ISRE-mediated IFN-β signaling.
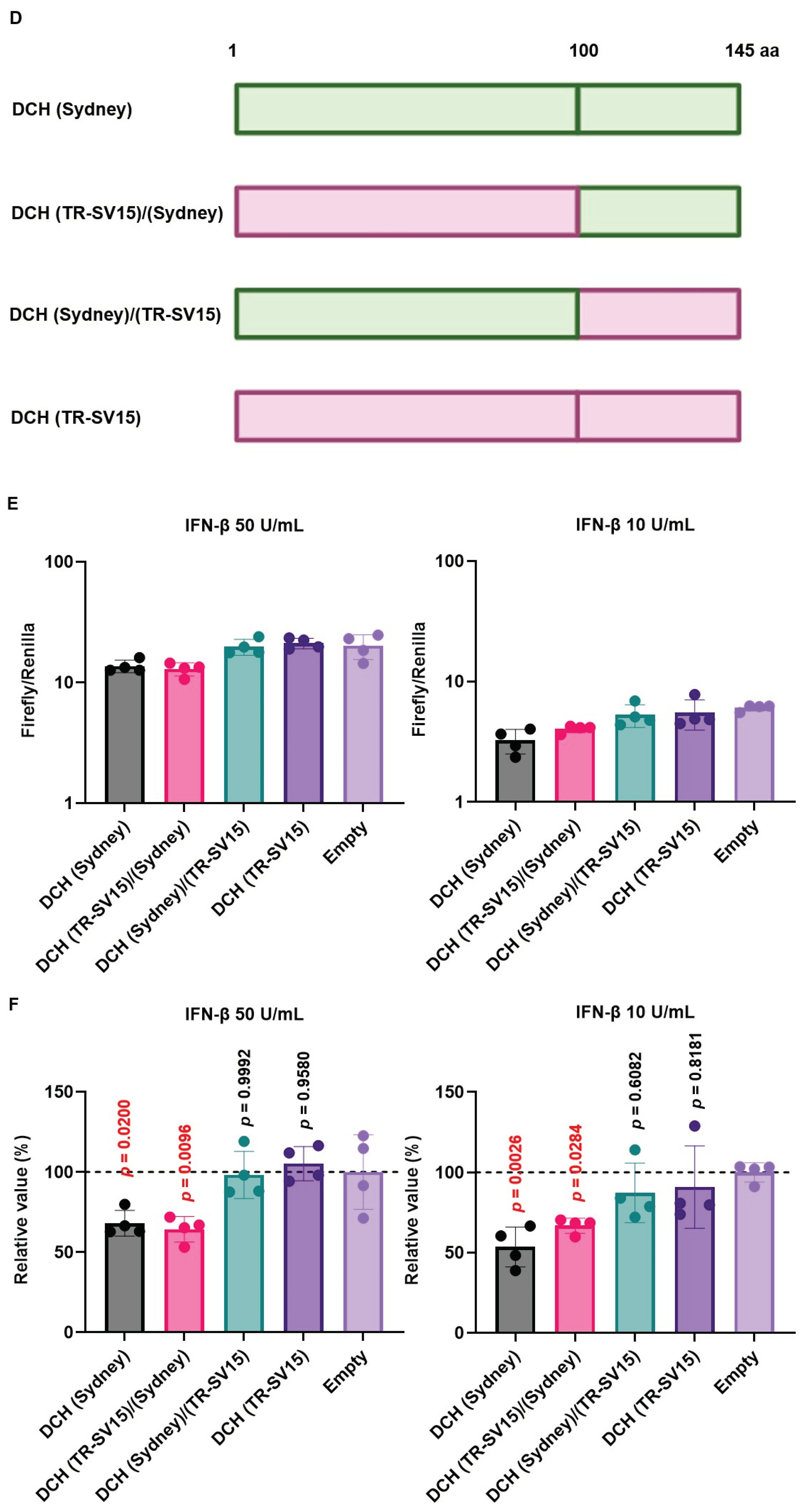
To address this possibility, we generated chimeric X proteins between DCH (Sydney) and DCH (TR-SV15) (Figure 7D). The chimeric DCH X proteins were efficiently expressed in 293T-ISRE-luc2 cells compared with WT proteins (Figure S5). Although DCH (Sydney)/(TR-SV15) X protein failed to inhibit ISRE-mediated IFN-β signaling, DCH (TR-SV15)/(Sydney) X protein showed potent inhibition of ISRE-mediated IFN-β signaling (Figure 7E and F). These results suggest that variations in the C-terminus transactivation domains of DCH X proteins determine the inhibitory effect on ISRE-mediated IFN-β signaling.
3. Discussion
In this study, we demonstrated that Orthohepadnavirus X protein inhibits TRIF-mediated IFN-β signaling, mainly through TRIF degradation, is conserved across Orthohepadnavirus X proteins. The C-terminus transactivation domains have an important role in stabilizing the expression and function of Orthohepadnavirus X protein to inhibit TRIF-mediated IFN-β signaling.
In 1991, the domains of HBx were characterized; the domain between residues 103 and 117 of the C-terminus transactivation domain was found to be important for a fully functional HBx [45]. Here, we aligned 17 Orthohepadnavirus X proteins and found high genetic diversity at the C-terminus transactivation domains (Figure 1A). The phylogenetic tree showed that DCH X proteins are more distant from HBx than X proteins from other species (Figure 1B). Nevertheless, DCH X proteins shared a function with HBx to inhibit IFN-β signaling (Figure 3A, 3B, and 3D). We recently reported that DCH shares the cell entry molecule, sodium/bile acid cotransporter, with HBV [39]. However, DCH preS1 is genetically closer to HBV preS1 than woodchuck hepatitis virus or Arctic ground squirrel HBV [39]. Understanding the similarities and differences between HBV and DCH is critical for using DCH as a model for HBV research.
DCH (KT-116) X protein modulated the innate immune response, including the TLR3 and RIG-I-like receptor response, by inhibiting TRIF (Figure 3A), MAVS (Figure 3B), and IRF-3 signaling (Figure 3D), similar to HBx. HBx was reported to inhibit MAVS activation by aggregation without affecting the expression of MAVS and RIG-I [25]. By contrast, TRIF activation was inhibited by inducing TRIF degradation [26]. Consistently, our results indicate that Orthohepadnavirus X protein inhibits TRIF-mediated IFN-β signaling by degrading TRIF (Figure 6B and D). Interestingly, X proteins derived from TMBHBV (13%) and HBV (genotype H) (50%) showed weaker TRIF degradation activity than other X proteins. Nevertheless, these X proteins inhibited TRIF-mediated IFN-β signaling (Figure 4B). Although DCH (KT-116) del(120-140) X protein decreased TRIF expression (~40%) (Figure 6E), it failed to inhibit TRIF-mediated IFN-β signaling (Figure 5D and E), suggesting a TRIF degradation-independent mechanism to block signaling.
HBx was reported to induce ubiquitin-mediated protein degradation [46] and TRIF degradation through a proteasomal pathway in hepatoma cells [26]. In this study, Orthohepadnavirus X proteins from broad range of species exhibited a conserved mechanism for TRIF degradation to perturb innate immunity. Our results increase our understanding of Orthohepadnavirus X protein-mediated suppression of TLR signaling. Consistently, HBV was reported to utilize X protein to avoid the innate immune system, establishing chronic infection in the host [14,24,47,48].
The 3D structure of Orthohepadnavirus X protein is unsolved. However, it has two functional domains, including an N-terminal negative regulatory and a C-terminal transactivation domain [20]. The N-terminal region (aa1-50) contains a highly conserved region (aa 1-20) and a Ser/Pro-rich region (aa 21-50) that is essential for negative regulatory effects and association of the regulatory domain [21]. C-terminal residues 58-119 are associated with signal transduction to the nucleus [49], residues 120-140 play an important role in nuclear transactivation, and the last 20 amino acids (aa 134-154) are involved in protein stability [22,23]. Consistently, we found that the deletion of residues 120-140 impaired the stability of X protein (Figure 5C) and degradation activity of TRIF (Figure 6D), leading to failed inhibition of IFN signaling (Figure 5D and E). Although the amino acid length of DCH X proteins is nine amino acids shorter than HBx and there is high genetic variation at position 120-140 (Figure 5A), our results suggest that the C-terminal domains of DCH X proteins have conserved function with HBx (regarding protein stability and inhibition of IFN signaling).
The localization of HBx was shown to depend on its expression level: low expression leads to nuclear localization [44]; high expression leads to cytoplasmic localization and abnormal mitochondrial distribution [44]. In this study, Orthohepadnavirus X proteins from various species showed conserved localization primarily in the nucleus, but also localized to the cytoplasm (Figure S3A). Deletion at the N- or C-terminal domain marginally affected the localization of HBV and DCH X proteins (Figure S3B). These observations suggest that the C-terminal domain is important for the inhibitory effect of X proteins but does not determine their cellular localization.
X proteins derived from HBV and DCH showed an inhibitory activity on ISRE-mediated IFN-β signaling, except DCH (TR-SV15) (Figure 7A and B). Alignment of X proteins derived from the four DCH strains showed that DCH (TR-SV15) had five different amino acids in the C-terminus transactivation domain (Figure 7C). Analysis using chimeric X proteins between DCH (Sydney) and DCH (TR-SV15) proteins suggested that genetic variations in the C-terminus of DCH X protein determine its inhibitory effect on ISRE-mediated IFN-β signaling (Figure 7E and F). Further analyses are required to identify specific residue(s) that determine inhibitory activity.
Several host and viral factors, such as host age and gender, viral load, and viral genotype, can influence the IFN response during infection and IFN treatment [50]. Among viral factors, X protein is one of the important factors in the IFN response, because it not only inhibits TLR3–TRIF and RIG-I signaling, but also suppresses IFN induction by suppressing the transcription of tripartite motif 22 in a mouse model, primary human hepatocytes, and human liver tissues [51]. The mechanism for the loss of inhibitory effect of DCH (TR-SV15) on ISRE-mediated IFN-β signaling is unclear. Therefore, we must elucidate the determinant(s) that can affect the IFN response in DCH-infected cats. Our results provide clues to improve the efficiency of IFN therapy in cats with chronic hepatitis.
A limitation of this study is that we used a luciferase reporter system to probe the interaction between Orthohepadnavirus X protein and human-derived molecules involved in IFN signaling. We must investigate whether 1) Orthohepadnavirus X proteins a show similar effect on molecules derived from other species, and 2) our results can be reproduced in primary cells. Last, we must investigate the immunopathogenesis induced by Orthohepadnavirus X proteins, especially DCH. These findings will contribute to developing therapeutic guidelines to counteract DCH infection effectively.
In conclusion, our results revealed that DCH X proteins and other Orthohepadnavirus X proteins inhibit TRIF-mediated IFN-β signaling by degrading TRIF, suggesting that this mechanism is a conserved function of Orthohepadnavirus X proteins to perturb the host’s innate immune response. Our findings show that the C-terminus domains of HBV and DCH X proteins are important for protein stability and inhibitory function. Further investigations are required to understand the evasion strategy of viruses belonging to Orthohepadnavirus.
4. Materials and Methods
4.1. Plasmids
IFN-Beta_pGL3 was a gift from Nicolas Manel (Addgene plasmid # 102597; http://n2t.net/addgene:102597; RRID:Addgene_102597) [52]. pEF-Bos TRIF Flag was a gift from Kate Fitzgerald & Tom Maniatis (Addgene plasmid # 41550; http://n2t.net/addgene:41550; RRID:Addgene_41550) [53]. pMP31-1 (MAVS plasmid) was a gift from Harmit Malik (Addgene plasmid # 45905; http://n2t.net/addgene:45905; RRID:Addgene_45905) [54]. IgK-IFN-luc was a gift from David Baltimore (Addgene plasmid # 14886; http://n2t.net/addgene:14886; RRID:Addgene_14886) [55]. pcDNA3 IKKe Flag was a gift from Tom Maniatis (Addgene plasmid # 26201; http://n2t.net/addgene:26201; RRID:Addgene_26201) [53]. Human V5-IRF3-pcDNA3 was a gift from Saumen Sarkar (Addgene plasmid # 32713; http://n2t.net/addgene:32713; RRID:Addgene_32713) [56]. pRL-TK (Promega, Madison, WI, USA, Cat# E2241) and pGL4.45[luc2P/ISRE/Hygro] (Promega, Cat# E4141) are commercially available.
cDNA sequences of 17 Orthohepadnavirus X proteins with an N-terminal HA-tag were synthesized with codon optimization to human cells (Twist Bioscience). Synthesized DNA sequences are summarized in Supplementary Table 1. Inserts encoding cDNA were cloned into the pCAGGS vector [57], predigested with EcoRI-HF (New England Biolabs [NEB], Ipswich, MA, USA, Cat# R3101M) and NheI-HF (NEB, Cat# R3131M) using In-Fusion Snap Assembly Master Mix (TaKaRa, Kusatsu, Japan, Cat# Z8947N). Plasmids were amplified using NEB 5-alpha F Iq Competent E. coli (High Efficiency) (NEB, Cat# C2992H) and extracted with PureYield Plasmid Miniprep System (Promega, Cat# A1222). Sequences of all plasmids were verified using SupreDye v3.1 Cycle Sequencing Kit (M&S TechnoSystems, Osaka, Japan, Cat# 063001) with Spectrum Compact CE System (Promega).
4.2. Construction of plasmids encoding Orthohepadnavirus X proteins with deletions
To construct pCAGGS vectors of Orthohepadnavirus X proteins with deletions, mutagenesis was performed with overlapping PCR using PrimeSTAR GXL DNA polymerase (TaKaRa, Cat# R050A). Primers are listed in Supplementary Table 2. The PCR protocol consisted of 35 cycles at 98°C for 10 s, 60°C for 15 s, and 68°C for 1 min, followed by 68°C for 7 min. Amplified PCR fragments encoding the deletion were cloned into the pCAGGS vector, as described in section 4.1. Plasmids were verified by sequencing.
4.3. Construction of plasmids encoding Myc-tagged TRIF
To construct the pCAGGS vector encoding Myc-tagged human TRIF, the insert encoding human TRIF was PCR amplified from the pEF-Bos TRIF Flag plasmid using PrimeSTAR GXL DNA polymerase. The primers are listed in Supplementary Table 3. The PCR protocol consisted of 35 cycles at 98°C for 10 s, 60°C for 15 s, and 68°C for 1 min, followed by 68°C for 7 min. Amplified PCR fragments encoding human TRIF were cloned into the pCAGGS vector, as described in section 4.1. Plasmids were verified by sequencing.
4.4. Construction of plasmids encoding chimeric DCH X proteins
To construct pCAGGS vectors encoding chimeric X proteins, mutagenesis was performed with overlapping PCR using PrimeSTAR GXL DNA polymerase. Primers are listed in Supplementary Table 4. The PCR protocol consisted of 35 cycles of 98°C for 10 s, 60°C for 15 s, and 68°C for 1 min, followed by 68°C for 7 min. Amplified PCR fragments encoding DCH (Sydney) and DCH (TR-SV15) X proteins were mixed and cloned into the pCAGGS vector using NEBuilder HiFi DNA Assembly Master Mix (NEB, Cat# E2621F). Plasmids were amplified as described in section 4.1. Plasmids were verified by sequencing.
4.5. Cell culture
Lenti-X 293T cells (TaKaRa, Cat# Z2180N) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Nacalai Tesque, Kyoto, Japan, Cat# 08458-16) supplemented with 10% fetal bovine serum (Cytiva, Shinjuku-Ku, Japan, Cat# SH30396) and 1 × penicillin–streptomycin (Nacalai Tesque, Cat# 09367-34).
4.6. Generation of 293T-ISRE-luc2 cells
Briefly, 293T cells (ATCC, Cat# CRL-3216) were transfected with 1 µg pGL4.45[luc2P/ISRE/Hygro] Vector with TransIT-LT1 Transfection Reagent (TaKaRa, Cat# V2304T) in Opti-MEM (Thermo Fisher Scientific, Minoto-Ku, Japan, Cat# 31985062). After 3 d, the cells were cultured in 250 µg/mL of hygromycin B (Nacalai Tesque, Cat# 09287-84) for 10 d. Single-cell cloning was then performed. After cell growth, we evaluated the induction of luciferase activity upon treatment with recombinant human IFN-β (PeproTech, Cranbury, NJ, USA, Cat# 300-02BC) in each clone.
4.7. Western blotting
To check the expression level of Orthohepadnavirus X proteins, pellets of Lenti-X 293T and 293T-ISRE-luc2 cells transfected with 500 ng pCAGGS plasmids encoding HA-tagged X protein were lysed with 2 × Bolt LDS sample buffer (Thermo Fisher Scientific, Cat# B0008) containing 2% β-mercaptoethanol (Bio-Rad, Hercules, CA, USA, Cat# 1610710) and incubated at 70°C for 10 min. Expression of HA-tagged X proteins was evaluated using Simple Western Abby (ProteinSimple, San Jose, CA, USA) with an anti-HA-Tag (6E2) mouse monoclonal antibody (CST, Danvers, MA, USA, Cat# 2367S, × 200) and Anti-Mouse Detection Module (ProteinSimple, Cat# DM-002). The amount of input protein was measured using Total Protein Detection Module (ProteinSimple, Cat# DM-TP01).
4.8. Luciferase reporter assay
4.8.1. IFN-β luciferase reporter assay
Lenti-X 293T cells were seeded in a 96-well plate (Fujifilm, Osaka, Japan, Cat# 635-28511) at 3 × 104 cells per well, cultured overnight, and transfected with 2.5 ng pIFN-β-Luc plasmid, 45 ng pRL-TK, 2.5 ng TRIF or MAVS plasmid, and 50 ng pCAGGS plasmid, encoding HA-tagged X protein or pCAGGS empty plasmid, using TransIT-LT1 Transfection Reagent in Opti-MEM. After 24 h, the cells were assayed for luciferase activity with Dual-Glo Luciferase Assay System (Promega, Cat# E2920). Firefly luciferase activity was normalized based on Renilla luciferase activity. Percent relative activity was calculated by comparing normalized luciferase data of Orthohepadnavirus X proteins plasmid transfected cells and empty plasmid transfected cells. The assays were repeated at least three times. The data shows mean values ± SD from one representative experiment.
4.8.2. IgK-IFN and ISRE luciferase reporter assay
Lenti-X 293T cells were seeded in a 96-well at 3 × 104 cells per well. After overnight incubation, cells were transfected with 5 ng IgK-IFN-Luc plasmid or ISRE Luc plasmid, 40 ng pRL-TK, 5 ng MAVS or IKKe or IRF-3 plasmid, and 50 ng plasmids encoding Orthohepadnavirus X proteins or empty plasmid. At 24-h after transfection, cells were assayed for luciferase activity with a Dual-Glo Luciferase Assay System as described above.
4.8.3. IFN-β bioassay in 293T-ISRE-luc2 cells
293T-ISRE-luc2 cells were seeded in a 96-well at 3 × 104 cells per well, cultured overnight, and transfected with 50 ng pRL-TK and 50 ng pCAGGS plasmid encoding Orthohepadnavirus X proteins, chimeric DCH X protein, or empty plasmid. At 24 h after transfection, the cells were treated with 50 or 10 U/mL recombinant human IFN-β. At 48 h after transfection, the cells were assayed for luciferase activity with Dual-Glo Luciferase Assay System.
4.9. TRIF-degradation assay
To investigate the effect of protein X-mediated degradation of TRIF, Lenti-X 293T cells were seeded in a 24-well plate (Fujifilm, Cat# 630-28441) at 1.25 × 105 cells per well. The cells were cultured overnight and co-transfected with 250 ng of pCAGGS plasmid encoding HA-tagged protein X and 250 ng of pCAGGS plasmid encoding Myc-tagged human TRIF. Cellular lysates were prepared as described above. The expression of Myc-tagged TRIF was measured using an anti-Myc (9B11) mouse monoclonal antibody (CST, Cat# 2276S, ×100) and Anti-Mouse Detection Module. The amount of input protein was measured using Total Protein Detection Module, as described above.
4.10. Immunofluorescence assay
Lenti-X 293T cells were plated on collagen-coated, 24-well plates (IWAKI, Yoshida, Japan, Cat# 4860-010), cultured overnight, and transfected with 500 ng of pCAGGS vector encoding Orthohepadnavirus protein X or pCAGGS empty plasmid using TransIT-LT1 Transfection Reagent. At 24 h after transfection, the cells were fixed in 3% paraformaldehyde (Fujifilm, Cat# 163-20145) and permeabilized with 0.2% Triton X-100 (Sigma Aldrich, Meguro-Ku, Japan, Cat# 9002-93-1). HA-tagged Orthohepadnavirus protein X was probed with Alexa Flour 647 anti-HA.11, mouse IgG1 antibody (BioLegend, Cat# 682404, ×200). Nuclei were detected by staining with NucBlue Live ReadyProbes Reagent (Thermo Fisher Scientific, Cat# R37605). The localization of Orthohepadnavirus protein X was analyzed with EVOS M7000 Imaging System (Thermo Fisher Scientific).
4.11. Alignment of Orthohepadnavirus protein X and phylogenetic analysis
The amino acid sequences of protein X from 17 Orthohepadnavirus sp. were aligned using the MUSCLE algorithm in MEGA X (MEGA Software). A phylogenetic tree was constructed using the alignment of amino acid sequences from public databases, and evolutionary analysis was conducted using the maximum likelihood method and neighbor-joining method based on the Jones–Taylor–Thornton matrix-based model with 1000 bootstrap replicates.
4.12. Statistical analysis
The results are presented as the mean and standard deviation of four measurements from one assay, representing at least two or three independent experiments. Differences in relative values between Orthohepadnavirus X proteins and empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test. A p ≤ 0.05 was considered statistically significant. Analysis was performed using Prism 10 software v10.1.2 for Windows (GraphPad Software, Boston, Massachusetts, USA).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Induction of Firefly luciferase; Figure S2: Orthohepadnavirus X protein inhibits IFN-β induction mediated by TRIF and MAVS; Figure S3: Subcellular localization of Orthohepadnavirus X proteins in Lenti-X 293T cells; Figure S4: Induction of luciferase reporter upon stimulation of 293-ISRE-luc2 cells with human IFN-β; Figure S5: Expression levels of Orthohepadnavirus X proteins in 293T-ISRE-luc2 cells; Table S1: Synthesized DNAs for generating plasmids encoding X protein; Table S2: Primers used for generating plasmids encoding deletion mutant X proteins of HBV(A) and DCH (KT-116); Table S3: Primers used for generating a plasmid encoding Myc-tagged TRIF; Table S4: Primers used for generating plasmids encoding chimeric DCH X.
Author Contributions
Conceptualization, A.C., and A.S.; methodology, A.C., M.S., and A.S.; data analysis: A.C., M.S., A.S.; manuscript preparation: A.C., and A.S.; review and editing: M.S., T.O., and A.S.; supervision, A.S.; funding acquisition, A.S. All authors have read and agreed to the published version of this manuscript.
Funding
This work was supported by grants from the Japan Agency for Medical Research and Development (AMED) Research Program on HIV/AIDS JP23fk0410047, JP23fk0410056, and JP23fk0410058 (to A.S.); the AMED Research Program on Emerging and Re-emerging Infectious Diseases JP22fk0108511 and JP22fk0108506 (to A.S.); JSPS KAKENHI Grant-in-Aid for Scientific Research (B) 22H02500 (to A.S.) and 21H02361 (to T.O. and A.S.); the JSPS Fund for the Promotion of Joint International Research (International Leading Research) JP23K20041 (to A.S.); the G-7 Grant (to A.S.); and the Ito Foundation Research Grant R5 KEN77 (to A.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Ms. Yuki Shibatani, Ms. Tomoko Nishiuchi, and the staff of CADIC, University of Miyazaki, for their assistance.
Conflicts of Interest
The funders had no role in the design of this study; in the collection, analyses, or interpretation of data; in the writing of this manuscript; or in the decision to publish the results.
References
- World Health Organization WHO Guidelines on Hepatitis B and C Testing; World Health Organization: Geneva, 2017; ISBN 978-92-4-154998-1.
- Magnius, L.; Mason, W.S.; Taylor, J.; Kann, M.; Glebe, D.; Dény, P.; Sureau, C.; Norder, H. ; ICTV Report Consortium ICTV Virus Taxonomy Profile: Hepadnaviridae. J. Gen. Virol. 2020, 101, 571–572. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic Analysis of the Host Response to Hepatitis B Virus Infection. Proc. Natl. Acad. Sci. 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host–Virus Interactions in Hepatitis B Virus Infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Barber, G.N. STING Signaling and Host Defense against Microbial Infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Mani, S.K.K.; Andrisani, O. Interferon Signaling during Hepatitis B Virus (HBV) Infection and HBV-Associated Hepatocellular Carcinoma. Cytokine 2019, 124, 154518. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Song, H.; Xu, F.; Cheng, G. When Hepatitis B Virus Meets Interferons. Front. Microbiol. 2018, 9, 1611. [Google Scholar] [CrossRef]
- Wang, H.; Ryu, W.-S. Hepatitis B Virus Polymerase Blocks Pattern Recognition Receptor Signaling via Interaction with DDX3: Implications for Immune Evasion. PLoS Pathog. 2010, 6, e1000986. [Google Scholar] [CrossRef]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The Hepatitis B e Antigen (HBeAg) Targets and Suppresses Activation of the Toll-like Receptor Signaling Pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef]
- Fernández, M.; Quiroga, J.A.; Carreño, V. Hepatitis B Virus Downregulates the Human Interferon-Inducible MxA Promoter through Direct Interaction of Precore/Core Proteins. J. Gen. Virol. 2003, 84, 2073–2082. [Google Scholar] [CrossRef]
- Soussan, P.; Garreau, F.; Zylberberg, H.; Ferray, C.; Brechot, C.; Kremsdorf, D. In Vivo Expression of a New Hepatitis B Virus Protein Encoded by a Spliced RNA. J. Clin. Invest. 2000, 105, 55–60. [Google Scholar] [CrossRef]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The Hepatitis B Virus X Protein Disrupts Innate Immunity by Downregulating Mitochondrial Antiviral Signaling Protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef]
- Megahed, F.A.K.; Zhou, X.; Sun, P. The Interactions Between HBV and the Innate Immunity of Hepatocytes. Viruses 2020, 12, 285. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B Virus X Protein Is Essential to Initiate and Maintain Virus Replication after Infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Role of HBx in Hepatitis B Virus Persistence and Its Therapeutic Implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Funato, K.; Miyakawa, Y.; Tanaka, E.; Seimiya, T.; Yamagami, M.; Tsutsumi, T.; Okushin, K.; Miyakawa, K.; et al. HBx-Induced Degradation of Smc5/6 Complex Impairs Homologous Recombination-Mediated Repair of Damaged DNA. J. Hepatol. 2022, 76, 53–62. [Google Scholar] [CrossRef]
- Murakami, S.; Cheong, J.H.; Kaneko, S. Human Hepatitis Virus X Gene Encodes a Regulatory Domain That Represses Transactivation of X Protein. J. Biol. Chem. 1994, 269, 15118–15123. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular Functions and Biological Roles of Hepatitis B Virus x Protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef]
- Parashar Misra, K.; Mukherji, A.; Kumar, V. The Conserved Amino-Terminal Region (Amino Acids 1–20) of the Hepatitis B Virus X Protein Shows a Transrepression Function. Virus Res. 2004, 105, 157–165. [Google Scholar] [CrossRef]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A Truncated Mutant (Residues 58-140) of the Hepatitis B Virus X Protein Retains Transactivation Function. Proc. Natl. Acad. Sci. 1996, 93, 5647–5652. [Google Scholar] [CrossRef]
- Lizzano, R.A.; Yang, B.; Clippinger, A.J.; Bouchard, M.J. The C-Terminal Region of the Hepatitis B Virus X Protein Is Essential for Its Stability and Function. Virus Res. 2011, 155, 231–239. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, H. Mechanism of Inhibiting Type I Interferon Induction by Hepatitis B Virus X Protein. Protein Cell 2010, 1, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Shen, F.; Wang, Y.; Li, Z.; Chen, J.; Yuan, Z. Residues Asn118 and Glu119 of Hepatitis B Virus X Protein Are Critical for HBx-Mediated Inhibition of RIG-I-MAVS Signaling. Virology 2020, 539, 92–103. [Google Scholar] [CrossRef]
- Hong, Y.; Zhou, L.; Xie, H.; Zheng, S. Innate Immune Evasion by Hepatitis B Virus-Mediated Downregulation of TRIF. Biochem. Biophys. Res. Commun. 2015, 463, 719–725. [Google Scholar] [CrossRef]
- Aghazadeh, M.; Shi, M.; Barrs, V.; McLuckie, A.; Lindsay, S.; Jameson, B.; Hampson, B.; Holmes, E.; Beatty, J. A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia. Viruses 2018, 10, 269. [Google Scholar] [CrossRef]
- Lanave, G.; Capozza, P.; Diakoudi, G.; Catella, C.; Catucci, L.; Ghergo, P.; Stasi, F.; Barrs, V.; Beatty, J.; Decaro, N.; et al. Identification of Hepadnavirus in the Sera of Cats. Sci. Rep. 2019, 9, 10668. [Google Scholar] [CrossRef]
- Piewbang, C.; Wardhani, S.W.; Chaiyasak, S.; Yostawonkul, J.; Chai-in, P.; Boonrungsiman, S.; Kasantikul, T.; Techangamsuwan, S. Insights into the Genetic Diversity, Recombination, and Systemic Infections with Evidence of Intracellular Maturation of Hepadnavirus in Cats. PLOS ONE 2020, 15, e0241212. [Google Scholar] [CrossRef]
- Anpuanandam, K.; Selvarajah, G.T.; Choy, M.M.K.; Ng, S.W.; Kumar, K.; Ali, R.M.; Rajendran, S.K.; Ho, K.L.; Tan, W.S. Molecular Detection and Characterisation of Domestic Cat Hepadnavirus (DCH) from Blood and Liver Tissues of Cats in Malaysia. BMC Vet. Res. 2021, 17, 9. [Google Scholar] [CrossRef]
- Jeanes, E.C.; Wegg, M.L.; Mitchell, J.A.; Priestnall, S.L.; Fleming, L.; Dawson, C. Comparison of the Prevalence of Domestic Cat Hepadnavirus in a Population of Cats with Uveitis and in a Healthy Blood Donor Cat Population in the United Kingdom. Vet. Ophthalmol. 2022, 25, 165–172. [Google Scholar] [CrossRef]
- Takahashi, K.; Kaneko, Y.; Shibanai, A.; Yamamoto, S.; Katagiri, A.; Osuga, T.; Inoue, Y.; Kuroda, K.; Tanabe, M.; Okabayashi, T.; et al. Identification of Domestic Cat Hepadnavirus from a Cat Blood Sample in Japan. J. Vet. Med. Sci. 2022, 84, 648–652. [Google Scholar] [CrossRef]
- Stone, C.; Petch, R.; Gagne, R.B.; Nehring, M.; Tu, T.; Beatty, J.A.; VandeWoude, S. Prevalence and Genomic Sequence Analysis of Domestic Cat Hepadnavirus in the United States. Viruses 2022, 14, 2091. [Google Scholar] [CrossRef]
- Capozza, P.; Carrai, M.; Choi, Y.R.; Tu, T.; Nekouei, O.; Lanave, G.; Martella, V.; Beatty, J.A.; Barrs, V.R. Domestic Cat Hepadnavirus: Molecular Epidemiology and Phylogeny in Cats in Hong Kong. Viruses 2023, 15, 150. [Google Scholar] [CrossRef]
- Silva, B.B.I.; Chen, J.-Y.; Villanueva, B.H.A.; Lu, Z.-Y.; Hsing, H.-Z.; Montecillo, A.D.; Shofa, M.; Minh, H.; Chuang, J.-P.; Huang, H.-Y.; et al. Genetic Diversity of Domestic Cat Hepadnavirus in Southern Taiwan. Viruses 2023, 15, 2128. [Google Scholar] [CrossRef]
- Adıgüzel, E.; Erdem-Şahinkesen, E.; Koç, B.T.; Demirden, C.; Oğuzoğlu, T.Ç. The Detection and Full Genomic Characterization of Domestic Cat Orthohepadnaviruses from Türkiye. Vet. Med. Sci. 2023, 9, 1965–1972. [Google Scholar] [CrossRef] [PubMed]
- Piewbang, C.; Dankaona, W.; Poonsin, P.; Yostawonkul, J.; Lacharoje, S.; Sirivisoot, S.; Kasantikul, T.; Tummaruk, P.; Techangamsuwan, S. Domestic Cat Hepadnavirus Associated with Hepatopathy in Cats: A Retrospective Study. J. Vet. Intern. Med. 2022, 36, 1648–1659. [Google Scholar] [CrossRef]
- Pesavento; Jackson; Hampson; Munday; Barrs; Beatty A Novel Hepadnavirus Is Associated with Chronic Hepatitis and Hepatocellular Carcinoma in Cats. Viruses 2019, 11, 969. [CrossRef]
- Shofa, M.; Ohkawa, A.; Kaneko, Y.; Saito, A. Conserved Use of the Sodium/Bile Acid Cotransporter (NTCP) as an Entry Receptor by Hepatitis B Virus and Domestic Cat Hepadnavirus. Antiviral Res. 2023, 217, 105695. [Google Scholar] [CrossRef]
- Luo, M.; Qu, X.; Pan, R.; Zhu, D.; Zhang, Y.; Wu, J.; Pan, Z. The Virus-Induced Signaling Adaptor Molecule Enhances DNA-Raised Immune Protection against H5N1 Influenza Virus Infection in Mice. Vaccine 2011, 29, 2561–2567. [Google Scholar] [CrossRef]
- Lui, W.-Y.; Bharti, A.; Wong, N.-H.M.; Jangra, S.; Botelho, M.G.; Yuen, K.-S.; Jin, D.-Y. Suppression of cGAS- and RIG-I-Mediated Innate Immune Signaling by Epstein-Barr Virus Deubiquitinase BPLF1. PLOS Pathog. 2023, 19, e1011186. [Google Scholar] [CrossRef]
- Han, K.-J.; Yang, Y.; Xu, L.-G.; Shu, H.-B. Analysis of a TIR-Less Splice Variant of TRIF Reveals an Unexpected Mechanism of TLR3-Mediated Signaling. J. Biol. Chem. 2010, 285, 12543–12550. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Xu, F.; Xiao, Q.; Tan, G. Hepatitis B Virus X Protein and Its Host Partners. Cell. Mol. Immunol. 2021, 18, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Henkler, F.; Hoare, J.; Waseem, N.; Goldin, R.D.; McGarvey, M.J.; Koshy, R.; King, I.A. Intracellular Localization of the Hepatitis B Virus HBx Protein. J. Gen. Virol. 2001, 82, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Balsano, C.; Avantaggiati, M.L.; Natoli, G.; De Marzio, E.; Will, H.; Perricaudet, M.; Levrero, M. Full-Length and Truncated Versions of the Hepatitis B Virus (HBV) X Protein (pX) Transactivate the cMYC Protooncogene at the Transcriptional Level. Biochem. Biophys. Res. Commun. 1991, 176, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.K.; Kwun, H.J.; Lee, J.-O.; Arora, P.; Jang, K.L. Hepatitis B Virus X Protein Differentially Affects the Ubiquitin-Mediated Proteasomal Degradation of β-Catenin Depending on the Status of Cellular P53. J. Gen. Virol. 2007, 88, 2144–2154. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.-R.; Oh, M.; Koh, S.S.; Malilas, W.; Srisuttee, R.; Jhun, B.H.; Pellegrini, S.; Fuchs, S.Y.; Chung, Y.-H. Hepatitis B Virus X Protein Inhibits Extracellular IFN-α-Mediated Signal Transduction by Downregulation of Type I IFN Receptor. Int. J. Mol. Med. 2012, 29, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Kuipery, A.; Gehring, A.J.; Isogawa, M. Mechanisms of HBV Immune Evasion. Antiviral Res. 2020, 179, 104816. [Google Scholar] [CrossRef]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Kumar, V.; Sarkar, D.P. An Internal Segment (Residues 58–119) of the Hepatitis B Virus X Protein Is Sufficient to Activate MAP Kinase Pathways in Mouse Liver. FEBS Lett. 2001, 504, 59–64. [Google Scholar] [CrossRef]
- Ye, J.; Chen, J. Interferon and Hepatitis B: Current and Future Perspectives. Front. Immunol. 2021, 12, 733364. [Google Scholar] [CrossRef]
- Lim, K.-H.; Park, E.-S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.-H.; Kim, C.W.; et al. Suppression of Interferon-Mediated Anti-HBV Response by Single CpG Methylation in the 5′-UTR of TRIM22. Gut 2018, 67, 166–178. [Google Scholar] [CrossRef]
- Gentili, M.; Kowal, J.; Tkach, M.; Satoh, T.; Lahaye, X.; Conrad, C.; Boyron, M.; Lombard, B.; Durand, S.; Kroemer, G.; et al. Transmission of Innate Immune Signaling by Packaging of cGAMP in Viral Particles. Science 2015, 349, 1232–1236. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 Are Essential Components of the IRF3 Signaling Pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Loo, Y.-M.; Horner, S.M.; Gale, M.; Malik, H.S. Convergent Evolution of Escape from Hepaciviral Antagonism in Primates. PLoS Biol. 2012, 10, e1001282. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.L.; Denny, E.M.; Baltimore, D. CARD11 Mediates Factor-Specific Activation of NF-κB by the T Cell Receptor Complex. EMBO J. 2002, 21, 5184–5194. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Smith, K.; Hsieh, P.N.; Mburu, Y.K.; Chattopadhyay, S.; Sen, G.C.; Sarkar, S.N. High-Throughput Screening for TLR3–IFN Regulatory Factor 3 Signaling Pathway Modulators Identifies Several Antipsychotic Drugs as TLR Inhibitors. J. Immunol. 2010, 184, 5768–5776. [Google Scholar] [CrossRef]
- Hitoshi, N.; Ken-ichi, Y.; Jun-ichi, M. Efficient Selection for High-Expression Transfectants with a Novel Eukaryotic Vector. Gene 1991, 108, 193–199. [Google Scholar] [CrossRef]
Figure 1.
The alignment and phylogenetic tree of Orthohepadnavirus X proteins. (A) Amino acid alignment of Orthohepadnavirus X proteins obtained from public databases. (B) Phylogenetic tree constructed with MEGA software.
Figure 1.
The alignment and phylogenetic tree of Orthohepadnavirus X proteins. (A) Amino acid alignment of Orthohepadnavirus X proteins obtained from public databases. (B) Phylogenetic tree constructed with MEGA software.
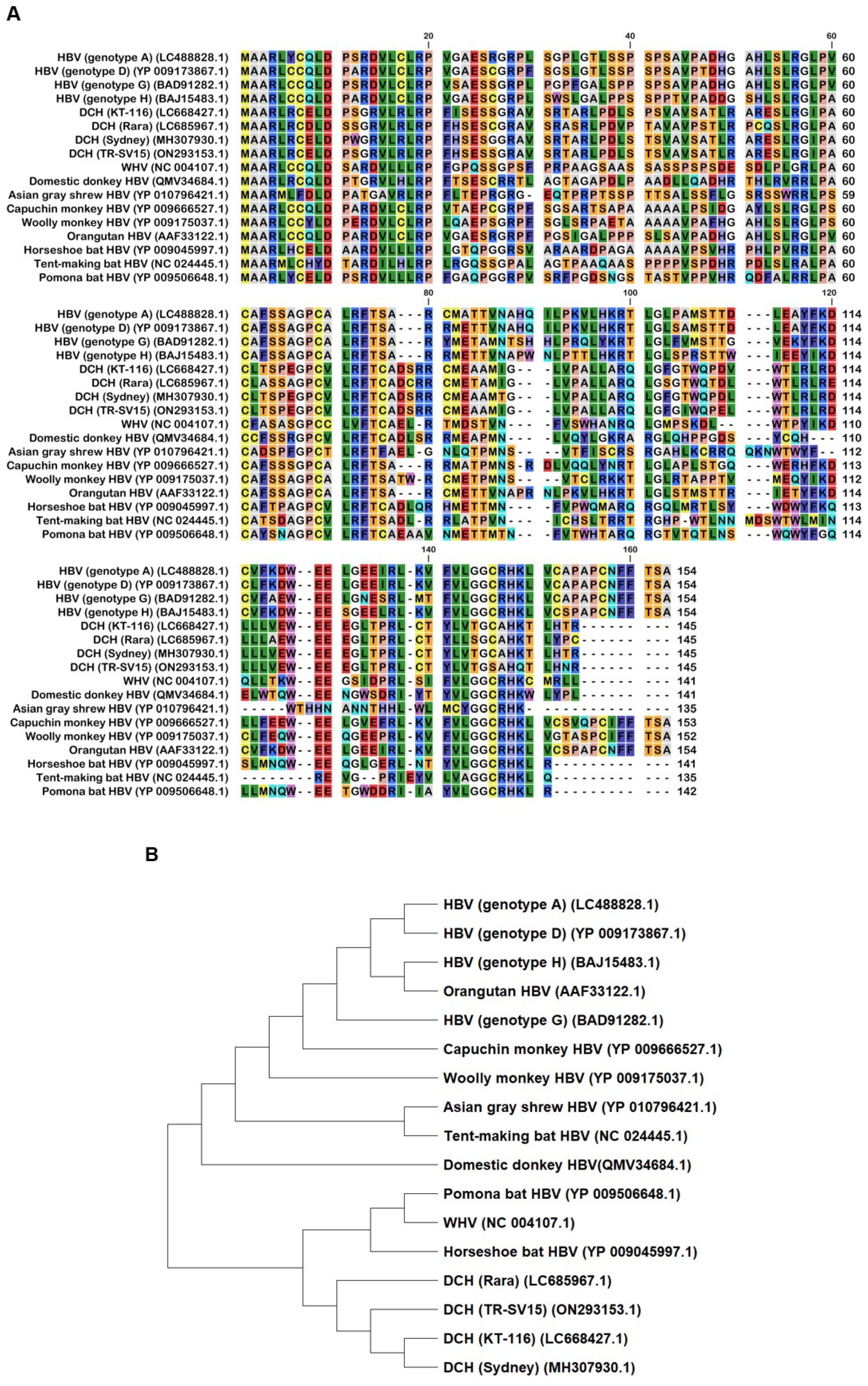
Figure 2.
Expression levels of Orthohepadnavirus X proteins in Lenti-X 293T cells.
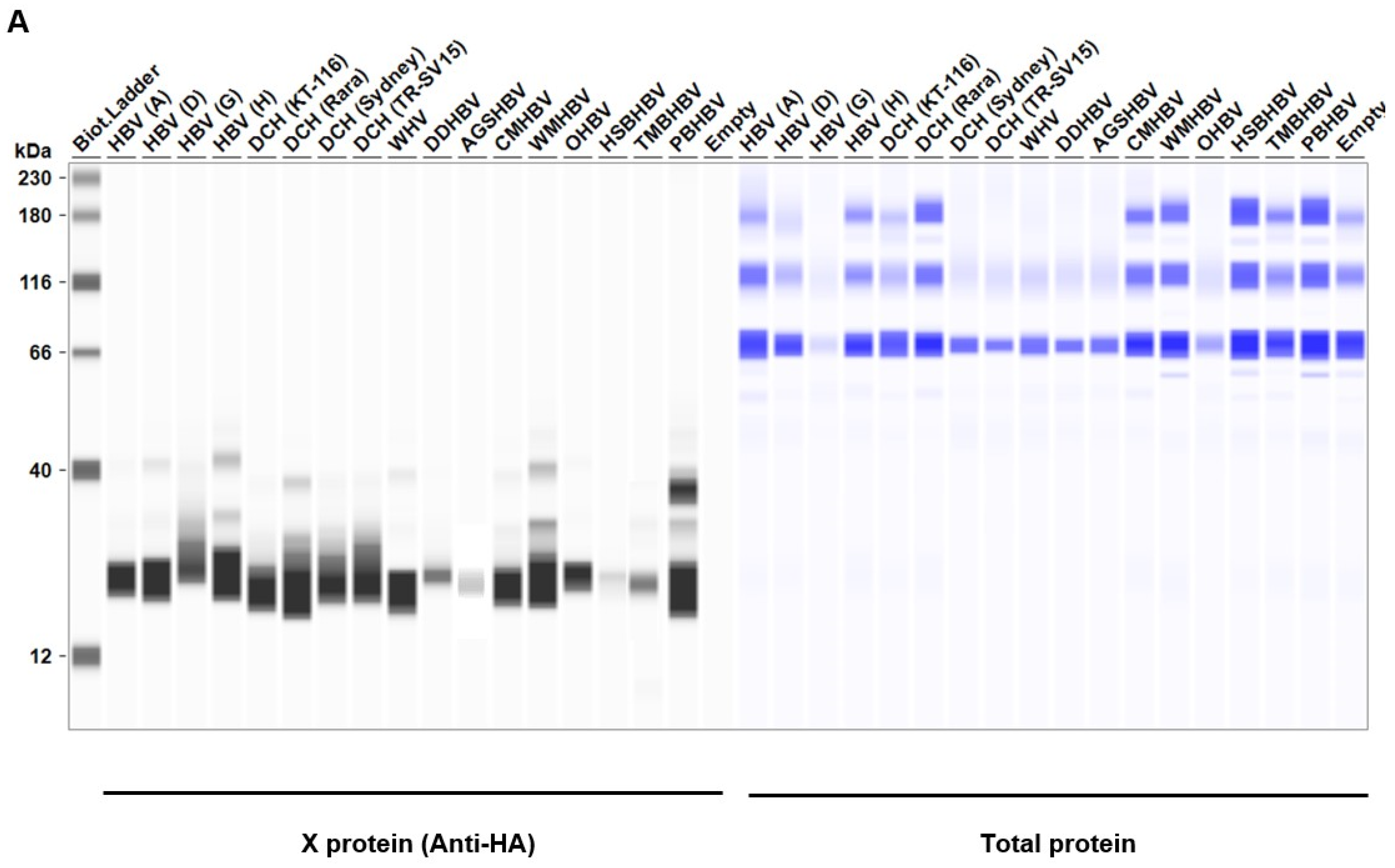
Figure 3.
Inhibitory effect of Orthohepadnavirus X proteins on IFN-β signaling. (A) Inhibitory effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with IFN-β Luc and TRIF plasmids. (B) Inhibitory effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with IgK-IFN Luc and MAVS plasmids. (C) Effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with IgK-IFN Luc and IKKe plasmids. (D) Inhibitory effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with ISRE Luc and IRF3 plasmids. Differences between cells transfected with HBV, DCH X protein plasmids, or an empty plasmid were examined by one-way ANOVA followed by Tukey’s multiple comparison test. ****p < 0.0001, **p < 0.01, and *p < 0.05.
Figure 3.
Inhibitory effect of Orthohepadnavirus X proteins on IFN-β signaling. (A) Inhibitory effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with IFN-β Luc and TRIF plasmids. (B) Inhibitory effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with IgK-IFN Luc and MAVS plasmids. (C) Effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with IgK-IFN Luc and IKKe plasmids. (D) Inhibitory effect of HBV (genotype A) and DCH (KT-116) upon co-transfection with ISRE Luc and IRF3 plasmids. Differences between cells transfected with HBV, DCH X protein plasmids, or an empty plasmid were examined by one-way ANOVA followed by Tukey’s multiple comparison test. ****p < 0.0001, **p < 0.01, and *p < 0.05.
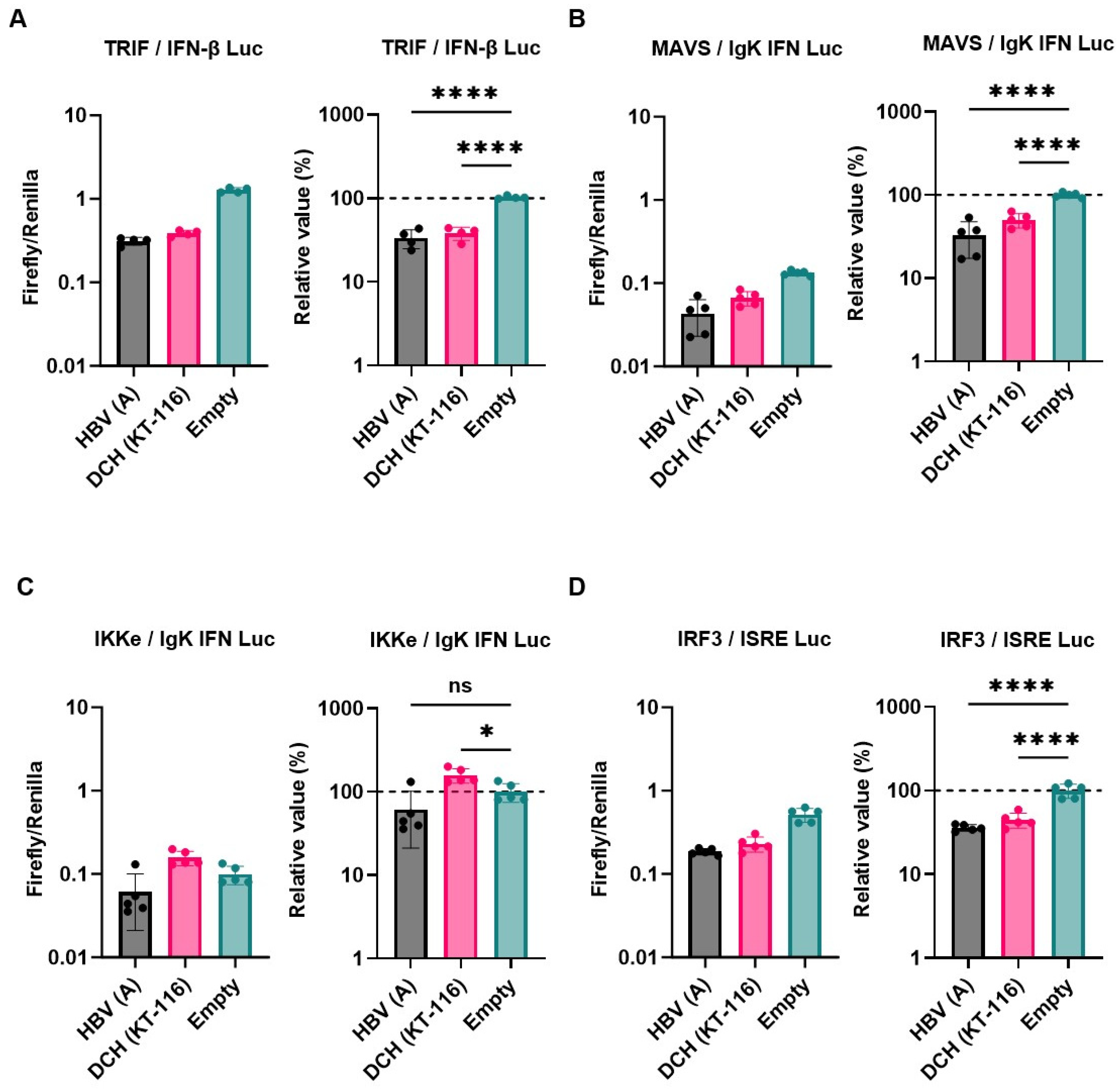
Figure 4.
Inhibitory effect of Orthohepadnavirus X proteins on TIR-domain-containing adaptor protein inducing IFN-β (TRIF)-mediated IFN-β signaling. (A) Raw data of the luciferase reporter assay. The RLU of Firefly luciferase was divided by the RLU of Renilla luciferase. (B) Relative value of IFN-β luciferase reporter assay. Differences between cells transfected with plasmids expressing Orthohepadnavirus X protein or an empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test.
Figure 4.
Inhibitory effect of Orthohepadnavirus X proteins on TIR-domain-containing adaptor protein inducing IFN-β (TRIF)-mediated IFN-β signaling. (A) Raw data of the luciferase reporter assay. The RLU of Firefly luciferase was divided by the RLU of Renilla luciferase. (B) Relative value of IFN-β luciferase reporter assay. Differences between cells transfected with plasmids expressing Orthohepadnavirus X protein or an empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test.
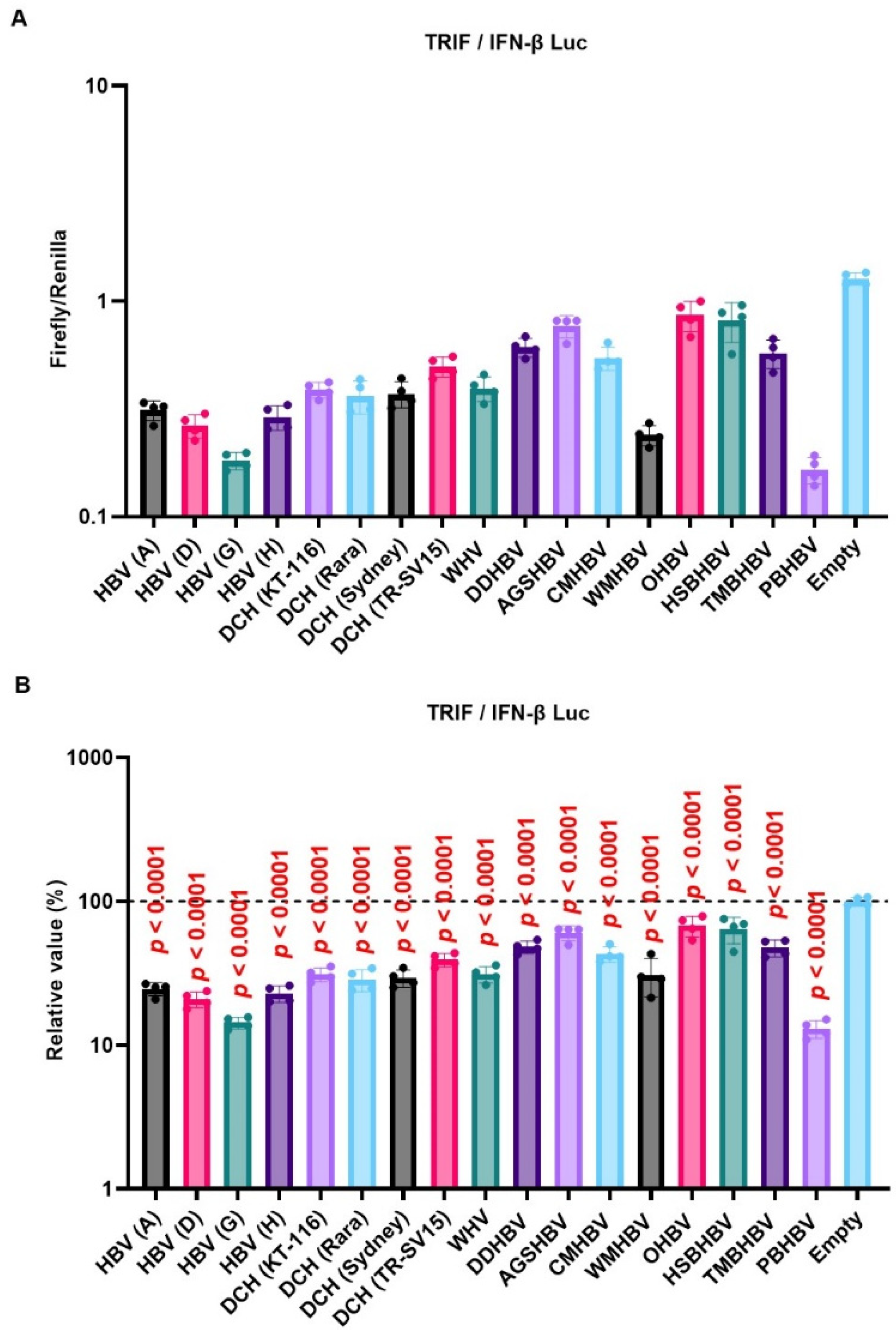
Figure 5.
The C-terminus transactivation domain of X protein plays an important role in protein stability and function. (A) Amino acid alignment of HBV (genotype A) and DCH (KT-116) X proteins. (B) Deletion mutants of X proteins. (C) Expression levels of mutant X proteins derived from HBV (genotype A) and DCH (KT-116) in Lenti-X 293T cells. (D) Raw data from the luciferase reporter assay. The RLU of Firefly luciferase was divided by the RLU of Renilla luciferase. (E) Relative value of the IFN-β luciferase reporter assay. Differences between cells transfected with Orthohepadnavirus X protein plasmids or an empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test.
Figure 5.
The C-terminus transactivation domain of X protein plays an important role in protein stability and function. (A) Amino acid alignment of HBV (genotype A) and DCH (KT-116) X proteins. (B) Deletion mutants of X proteins. (C) Expression levels of mutant X proteins derived from HBV (genotype A) and DCH (KT-116) in Lenti-X 293T cells. (D) Raw data from the luciferase reporter assay. The RLU of Firefly luciferase was divided by the RLU of Renilla luciferase. (E) Relative value of the IFN-β luciferase reporter assay. Differences between cells transfected with Orthohepadnavirus X protein plasmids or an empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test.
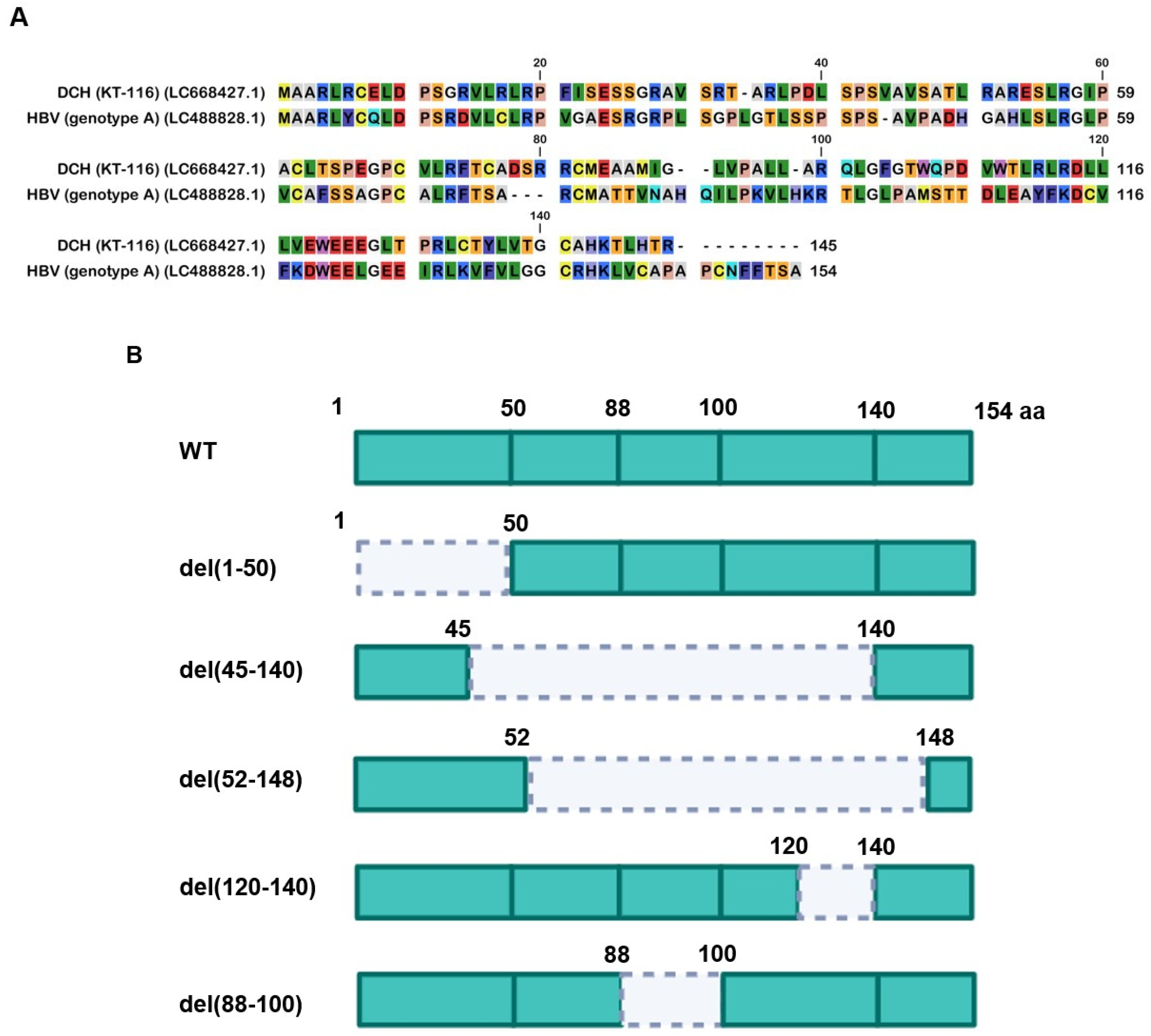
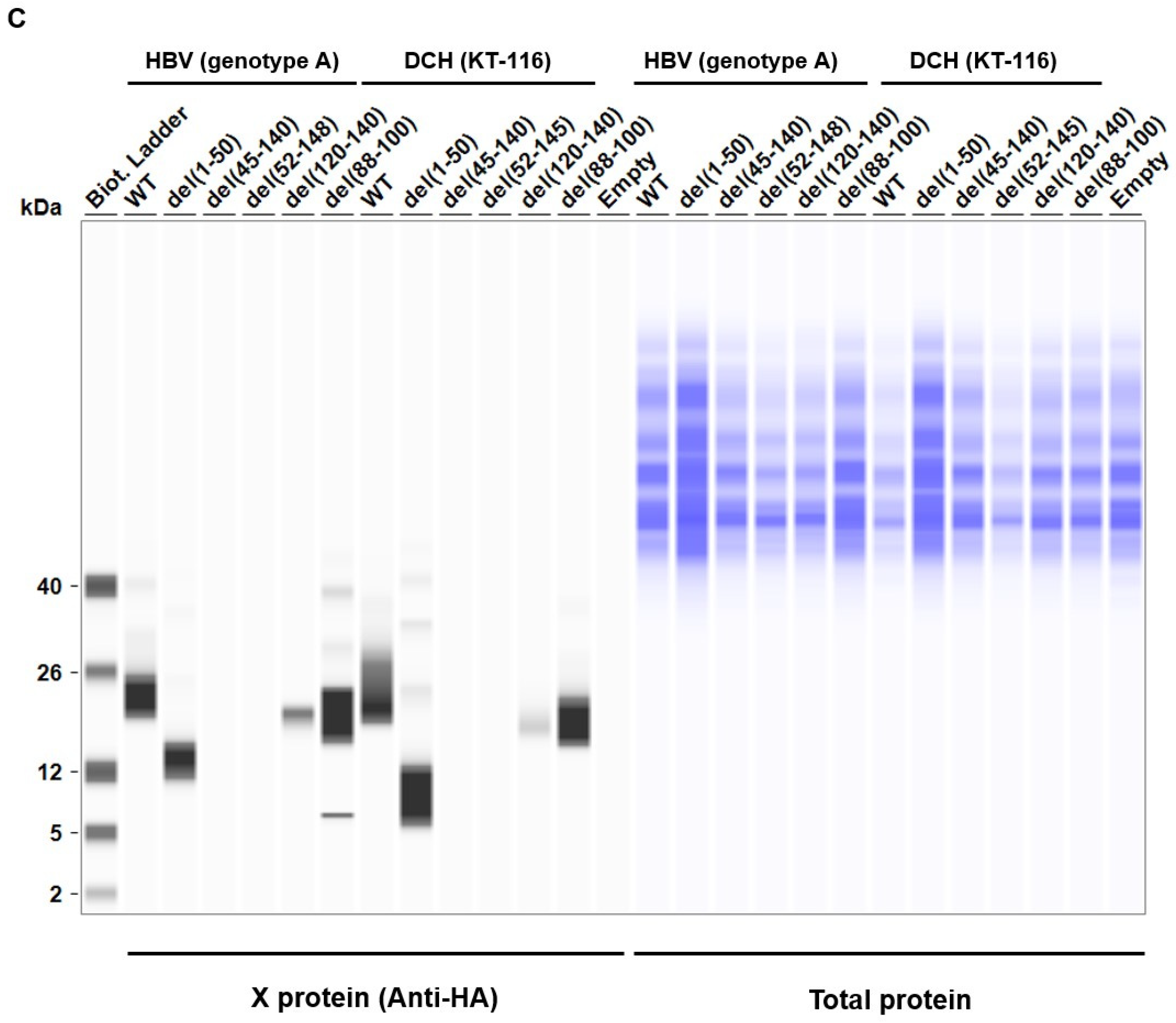
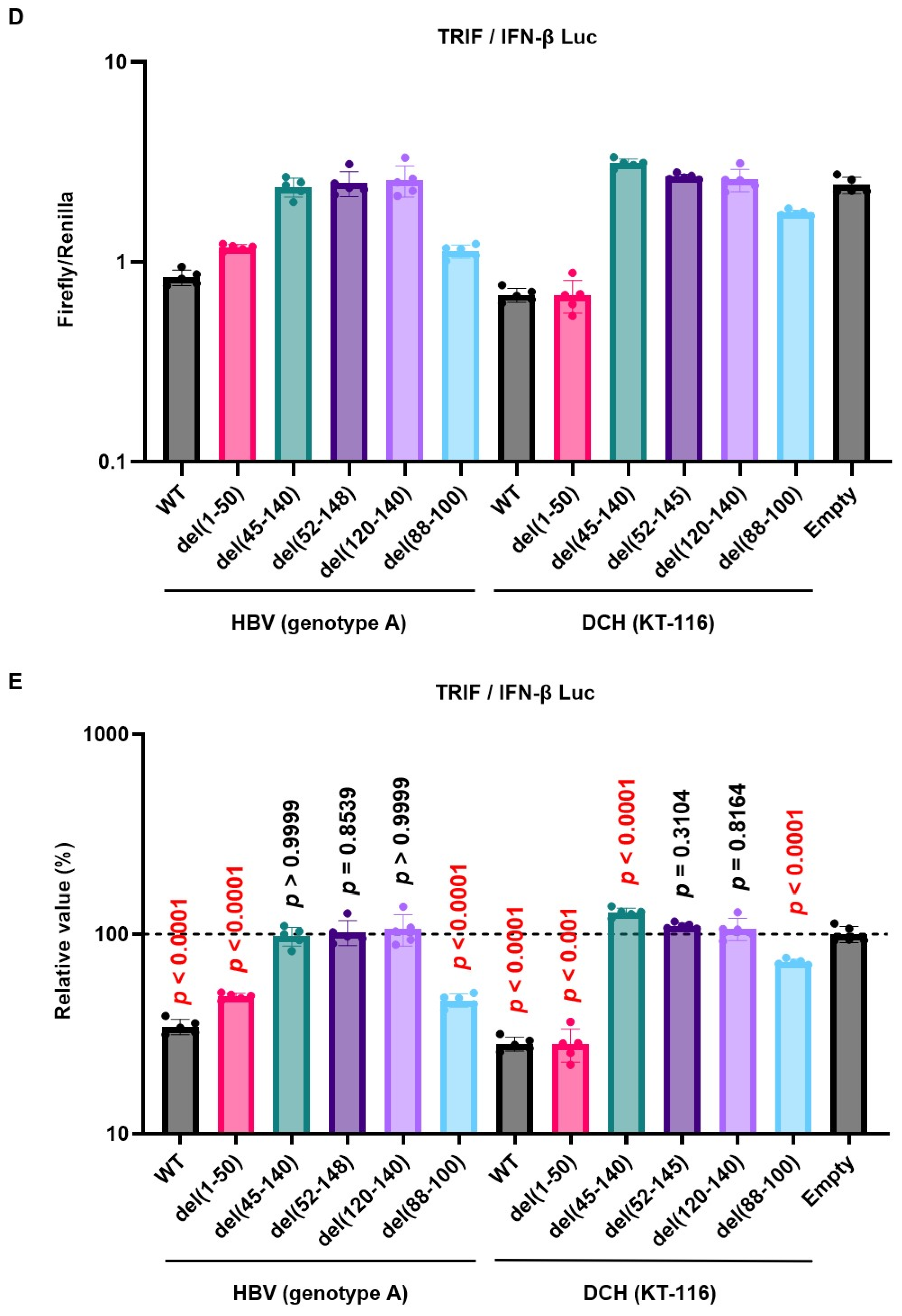
Figure 6.
Orthohepadnavirus X protein degrades TIR-domain-containing adaptor protein inducing IFN-β (TRIF). (A and C) Expression levels of HA-tagged Orthohepadnavirus X proteins in transfected Lenti-X 293T cells. (B and D) Expression levels of Myc-tagged human TRIF in transfected Lenti-X 293T cells. (E) Relative values of TRIF expression. Relative value was calculated from the corrected area of the TRIF band with Compass for Simple Western software.
Figure 6.
Orthohepadnavirus X protein degrades TIR-domain-containing adaptor protein inducing IFN-β (TRIF). (A and C) Expression levels of HA-tagged Orthohepadnavirus X proteins in transfected Lenti-X 293T cells. (B and D) Expression levels of Myc-tagged human TRIF in transfected Lenti-X 293T cells. (E) Relative values of TRIF expression. Relative value was calculated from the corrected area of the TRIF band with Compass for Simple Western software.
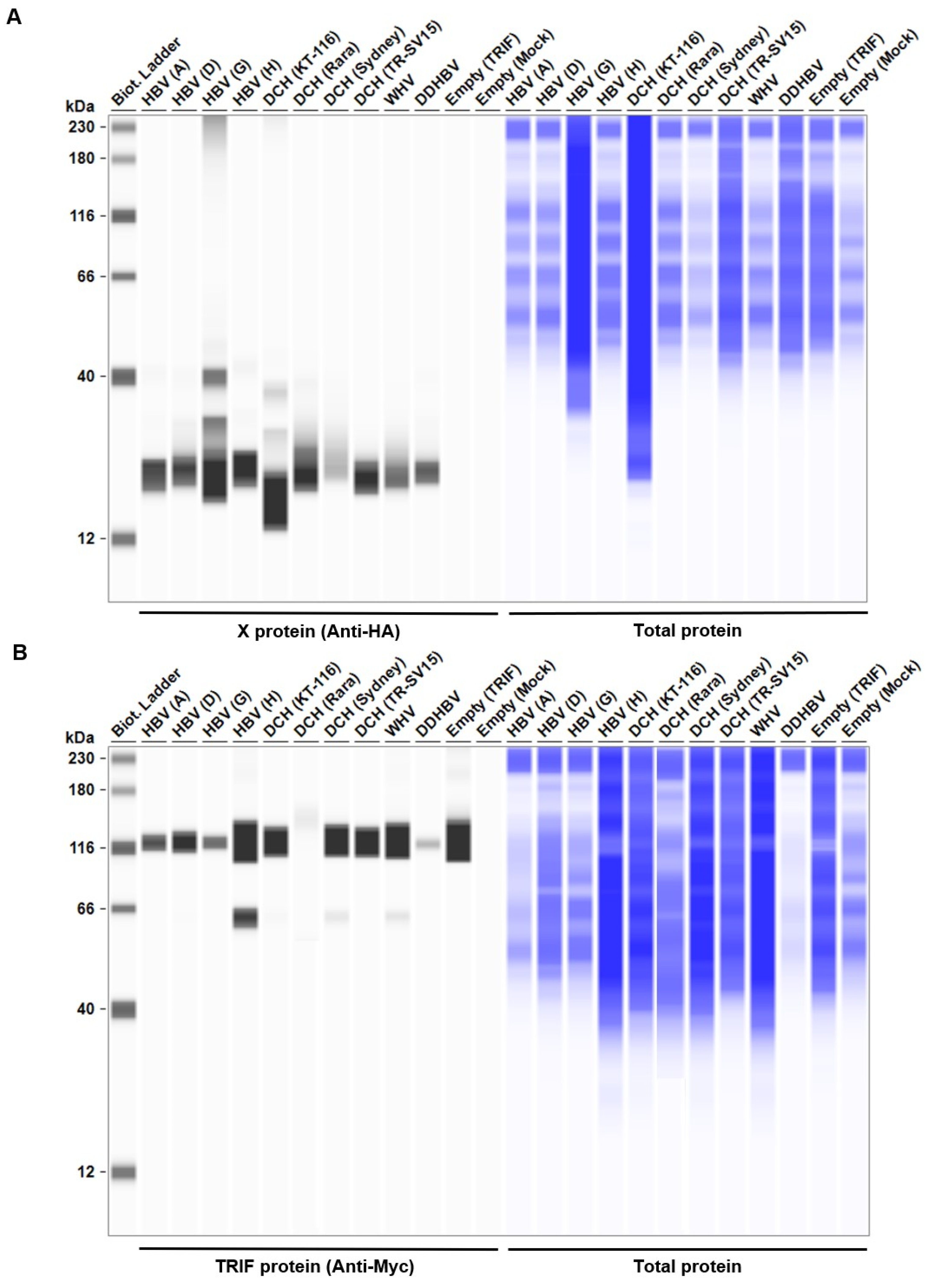
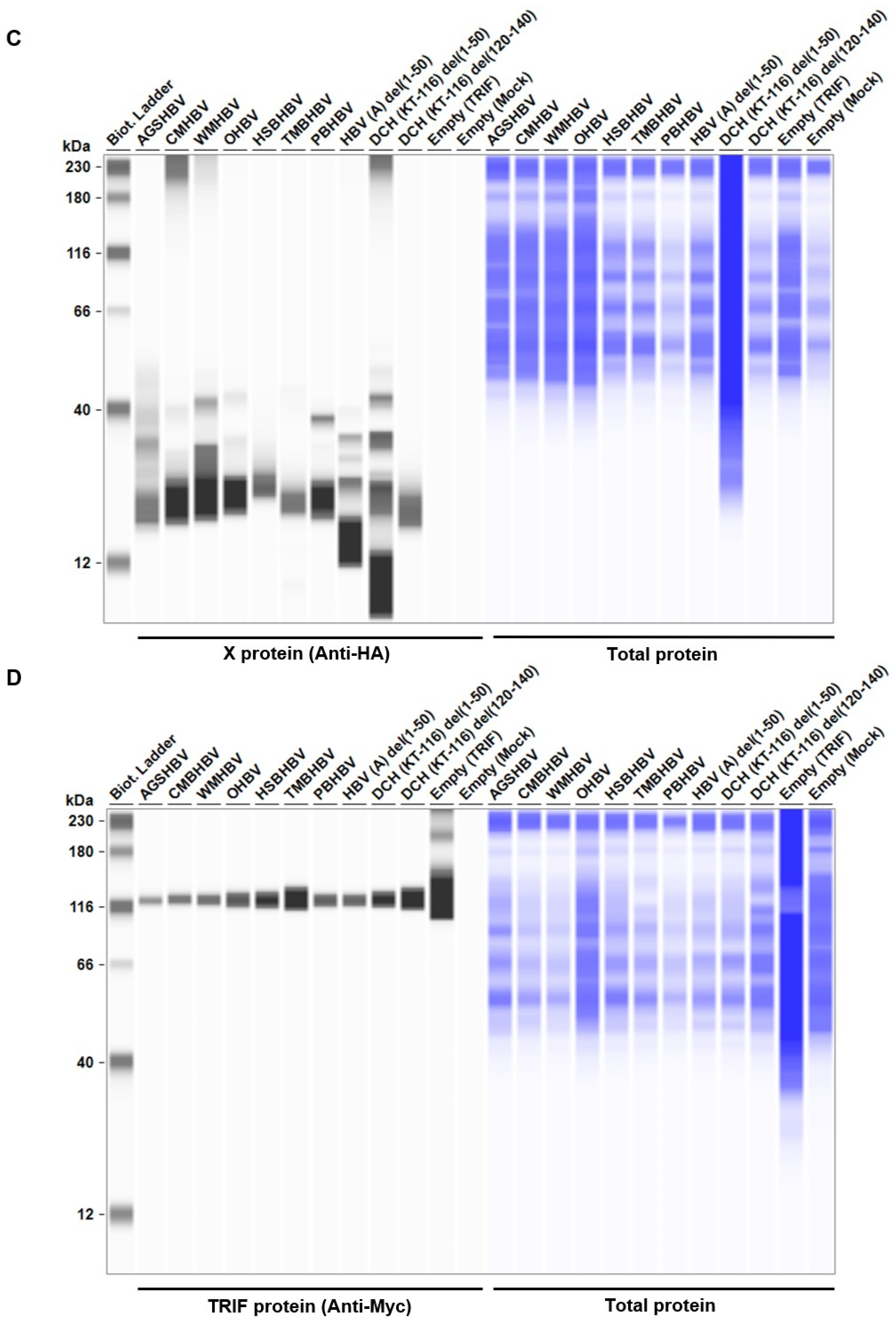
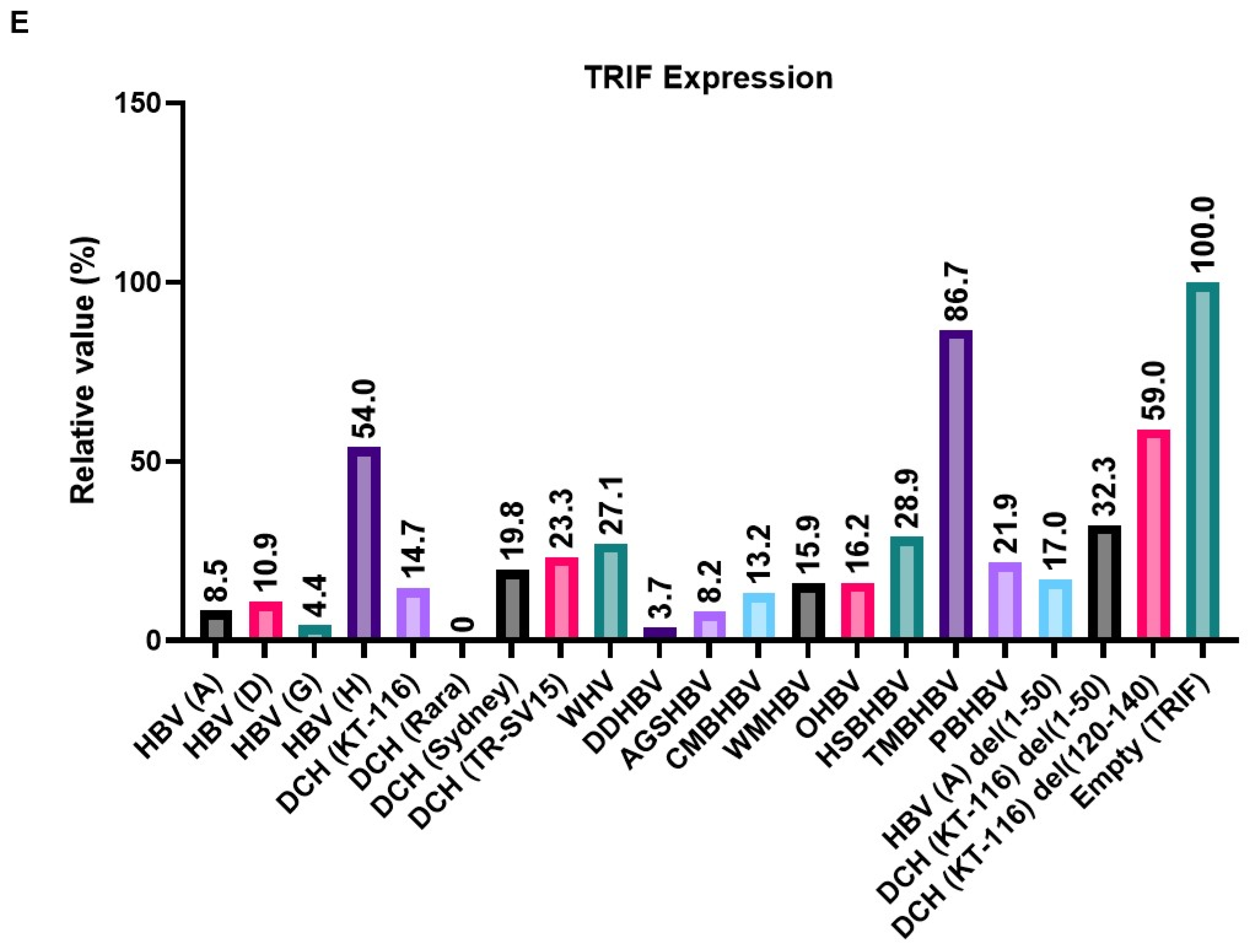
Figure 7.
Variations in the C-terminus transactivation domains of DCH X proteins determine inhibitory effects on interferon-stimulated response element (ISRE)-mediated IFN-β signaling. (A and E) Raw data of the luciferase reporter assay. The RLU of Firefly luciferase was divided by the RLU of Renilla luciferase. (B and F) Relative value of IFN-β luciferase reporter assay. Differences between cells transfected with Orthohepadnavirus X protein plasmids or an empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test. (C) Amino acid alignment of DCH (KT-116), DCH (Rara), DCH (Sydney), and DCH (TR-SV15) X proteins. (D) Schematic representation of chimeric X proteins between DCH (Sydney) and DCH (TR-SV15) X proteins.
Figure 7.
Variations in the C-terminus transactivation domains of DCH X proteins determine inhibitory effects on interferon-stimulated response element (ISRE)-mediated IFN-β signaling. (A and E) Raw data of the luciferase reporter assay. The RLU of Firefly luciferase was divided by the RLU of Renilla luciferase. (B and F) Relative value of IFN-β luciferase reporter assay. Differences between cells transfected with Orthohepadnavirus X protein plasmids or an empty plasmid were examined by one-way ANOVA followed by Dunnett’s multiple comparison test. (C) Amino acid alignment of DCH (KT-116), DCH (Rara), DCH (Sydney), and DCH (TR-SV15) X proteins. (D) Schematic representation of chimeric X proteins between DCH (Sydney) and DCH (TR-SV15) X proteins.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



