
Submitted:
22 February 2024
Posted:
23 February 2024
You are already at the latest version
Abstract
The initiation reactions of DNA synthesis are central processes during human chromosomal DNA replication. They are separated into two main processes: the initiation events at replication origins, the start of the leading strand synthesis for each replicon, and the numerous initiation events taking place during lagging strand DNA synthesis. In addition, a third mechanism is the re-initiation of DNA synthesis after replication fork stalling, which takes place when DNA lesions hinder the progression of DNA synthesis. The initiation of leading strand synthesis at replication origin is regulated at multiple levels, from the origin recognition to the assembly and activation of replicative helicase, the Cdc45-MCM2-7-GINS (CMG) complex. In addition, the multiple interactions of the CMG complex with the eukaryotic replicative DNA polymerases such as DNA polymerase α-primase (Pol α), DNA polymerase δ and ε at replication forks play pivotal roles in the mechanism of initiation reactions including the initiation of signalling unperturbed and stalled replication forks, ‘replication stress’ events, via ATR (ATM-Rad 3-related protein kinase). All these processes are essential for the accurate transfer of the cells’ genetic information to their daughters. Failures and dysfunctions in these processes gives rise to genome instability followed by genetic diseases including cancer. Thus, in their review ‘Hallmarks of Cancer: New Dimensions‘ Hanahan and Weinberg (2022) called genome instability a fundamental function in the development process of cancer cells. In recent years, the understanding of the initiation processes and mechanisms of human DNA replication has made substantial progress at all levels which will be discussed in the review.
Keywords:
DNA replication
; origin of replication
; initiation
; replication fork
; Okazaki fragment
; DNA damage signalling
; ATR kinase
; telomeres
1. Introduction
DNA replication is a fundamental process in all living organisms to accurately and timely duplicate the genetic information of their cells before dividing into two genetically identical daughter cells [1,2,3,4,5,6,7,8,9]. In eukaryotes, DNA replication takes place in a specific time window of a cell division cycle called S phase or synthesis phase [10,11]. Replication of eukaryotic chromosomal DNA requires tight regulations to ensure that the genome of each cell is duplicated once and only once per cell cycle cells [5,6,8,12,13]. Failures to do so can have detrimental results and lead to diseases such as cancer. DNA replication is a highly conserved process at molecular levels in all eukaryotes [1,2,3,4,5,6,8,9]. However, major differences between human, yeast, and other eukaryotes exist and have been studied [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. For example, origins of replication have specific sequence requirements in some yeast such as Saccharomyces cerevisiae, whereas in most other eukaryotes replication origins are defined by protein-DNA complexes with little or no dependence on DNA sequence [15]. Moreover, the regulation of the origin recognition by the conserved protein complex called origin recognition complex (ORC) differs between eukaryotes. Yeast ORC binds to replication origins constitutively whereas in other organisms one subunit of ORC is destructed in a cell cycle-dependent manner [16,17,18,19,20,21,29,30]. Additionally, although the replicative helicase, the CDC45-MCM2-7-GINS (CMG) complex, is conserved from yeast to man its assembly and activation at replication origin requires several factors. In yeast, DNA polymerase ε (Pol ε) and MCM10 support its assembly and activation whereas in multicellular organisms the protein Donson, a protein not yet found in S. cerevisiae, assists the assembly of the CMG complex [28]. Interestingly, mutations in Donson, MCM2-7, CDC45, and in other replication initiation factors have been found to be the cause of genetic diseases such as Meier-Gorlin syndrome highlighting the importance to understand the mechanism of initiation in humans [9,22,24,25,26,27,28].
2. Establishing DNA Replication Forks—Origins of DNA Replication and Chromatin-Loading of Replication Proteins
The DNA replication process is divided into three separate steps: initiation, elongation, and termination [1,2,3,8]. The initiation step yields the unwinding of the double-stranded DNA and the formation of fundamental structures, called replication forks (RFs), which are Y-shaped DNA structures representing the two templates on which primase synthesises the initiator RNAs (ribonucleic acids) and the replicative DNA polymerases synthesise new DNAs (deoxyribonucleic acid) during the elongation step. When two opposing RFs from different origins meet the DNA synthesis ends (termination) [1,2,3]. In the current review, we will focus on the initiation reactions, which are highly regulated processes in all living organisms.
The process of replication origin activation during the initiation phase of DNA replication and the RF establishment have been best studied in yeast and Xenopus laevis, where biochemical replication system either with purified proteins or using Xenopus oocyte extracts have been established and are supported by genetic studies, especially in yeast. The yeast biochemical DNA replication system uses double-stranded DNA (dsDNA) containing an origin of DNA replication whereas the Xenopus laevis system, which is based on embryonic proteins, works with long stretches of DNA devoid of specific origin sequences and the established origins are spaced ~15 kB away [14,15,31]. Recent studies using synthesised, pre-assembled RFs and purified proteins have advanced the field in the elongation phase and can be combined with cell biological studies and advanced modern technologies including next generation sequencing as well as structural biology techniques [32,33,34,35,36,37,38,39]. Although origin-dependent replication initiation processes in higher eukaryotes are not as well understood as in yeast, combining results of multiple model systems have yielded a picture of highly conserved processes in combination with subtle changes as discussed below (summarised in Figure 1, [14,15,16,17,18,19,20,21,22,24,25,26,27,28]).
All DNA replication processes start at specific sequences in eukaryotic genomes named origins of DNA replication. These replication origins are well defined in the yeast S. cerevisiae and closely related yeasts. They are AT-rich and are show sequence-specific requirements ([15] Figure 1). In contrast, origins in other yeasts such as Schizosaccharomyces pombe (S. pombe) and higher multicellular organisms are less well defined and show less defined requirements. Origins of S. pombe and related yeasts preferentially contain AT-rich sequences and the origin-binding complex (ORC) with the AT-hook in ORC4 binds to the AT-rich sequence but otherwise there are no sequence specificities known which define those origins. In Metazoan, on the other hand, origins are GC-rich, they are epigenetically defined, and often associated with promoter regions [15]. However, the conserved six-subunit protein complex ORC, first described in yeast, binds to all eukaryotic origins, serves as a landing platform for replication initiation factors, and allows the origin-dependent initiation process to take place [15,40]. In somatic cells of Metazoan these origins are spaced far apart, ~50-100 kilobases (kba), whereas, in embryonic cells, origins are spaced as close as 15 kba from each other [14]. In summary, replication origins are chromosomal DNA sequences, which ORC recognises in an ATP-dependent manner, and which form the starting point of DNA replication or for Metazoan the DNA replication-starting region [15].
Figure 1.
The initiation process at eukaryotic origins of DNA replication. At the end of mitosis and in early G1 phase of the cell cycle, ORC, the origin-recognition complex, binds to eukaryotic origins of replication together with CDC6. In G1, CTD1 together with the MCM2-7 complex binds then the CDC6-ORC complex and the MCM2-7 proteins are loaded as helicase-inactive double hexamers (MCM-DHs) onto the chromatin forming the pre-replicative complex (Pre-RC) and license the origin. Modification of CDC6 and binding of CTD1 to Geminin inactivates the loading activity of these proteins with CDC6 binding degraded similarly as free CTD1. In the next step, Treslin-MTBP (SLD3-SLD7 in yeast) interacts with MCM-DHs at the chromatin, and the DBF4/DRF1 CDC7 kinase (DDK) phosphorylates the MCM2-7 proteins. The DDK-dependent phosphorylation can be reversed by RIF1-PP1 making this step reversible. Next, Cyclin-CDKs phosphorylate Treslin and stimulate the formation of Donson-TOPBP1 complexes, which in turn bind to MCM-DHs. Donson-TOPBP1 supports the loading of the GINS complex and its association with MCM-DHs. Binding of CDC45 leads to the formation of the CMG complex and its activation, whereas TOPBP1 and Treslin-MTBP are released from the chromatin. CryoEM data suggest that Donson associates as dimer with CMG, but only one Donson subunit binds to GINS and MCM2-7 proteins stabilising the CMG complex [28]. In the following, two replication forks (RFs) are formed and replication protein A (RPA) with the help of the CDC45 binds and stabilises the resulting ssDNA. The association of AND-1/CTF4/WDHD1 (shown as AND-1 in the diagram) with CMG allows the loading of an inactive Pol α (dark green) including its primase subunits to RFs. The activation of Pol α (light green) permits the primase subunit PRIM1/PRI1 with help of PRIM2/PRI2 and additional replication factors to synthesise the first RNA primer in origin sequences resulting in the completion of the initiation process at origins and the start of the elongation phase. Additional proteins associated with RFs such as the fork stabilising proteins Timeless, Tipin and Claspin plus Pol ε [3,41,42] were omitted in the diagram for simplification and clarity reasons providing a better overview. Adapted from [3,26,27,28,43,44,45] and created with BioRender.com.
Figure 1.
The initiation process at eukaryotic origins of DNA replication. At the end of mitosis and in early G1 phase of the cell cycle, ORC, the origin-recognition complex, binds to eukaryotic origins of replication together with CDC6. In G1, CTD1 together with the MCM2-7 complex binds then the CDC6-ORC complex and the MCM2-7 proteins are loaded as helicase-inactive double hexamers (MCM-DHs) onto the chromatin forming the pre-replicative complex (Pre-RC) and license the origin. Modification of CDC6 and binding of CTD1 to Geminin inactivates the loading activity of these proteins with CDC6 binding degraded similarly as free CTD1. In the next step, Treslin-MTBP (SLD3-SLD7 in yeast) interacts with MCM-DHs at the chromatin, and the DBF4/DRF1 CDC7 kinase (DDK) phosphorylates the MCM2-7 proteins. The DDK-dependent phosphorylation can be reversed by RIF1-PP1 making this step reversible. Next, Cyclin-CDKs phosphorylate Treslin and stimulate the formation of Donson-TOPBP1 complexes, which in turn bind to MCM-DHs. Donson-TOPBP1 supports the loading of the GINS complex and its association with MCM-DHs. Binding of CDC45 leads to the formation of the CMG complex and its activation, whereas TOPBP1 and Treslin-MTBP are released from the chromatin. CryoEM data suggest that Donson associates as dimer with CMG, but only one Donson subunit binds to GINS and MCM2-7 proteins stabilising the CMG complex [28]. In the following, two replication forks (RFs) are formed and replication protein A (RPA) with the help of the CDC45 binds and stabilises the resulting ssDNA. The association of AND-1/CTF4/WDHD1 (shown as AND-1 in the diagram) with CMG allows the loading of an inactive Pol α (dark green) including its primase subunits to RFs. The activation of Pol α (light green) permits the primase subunit PRIM1/PRI1 with help of PRIM2/PRI2 and additional replication factors to synthesise the first RNA primer in origin sequences resulting in the completion of the initiation process at origins and the start of the elongation phase. Additional proteins associated with RFs such as the fork stabilising proteins Timeless, Tipin and Claspin plus Pol ε [3,41,42] were omitted in the diagram for simplification and clarity reasons providing a better overview. Adapted from [3,26,27,28,43,44,45] and created with BioRender.com.
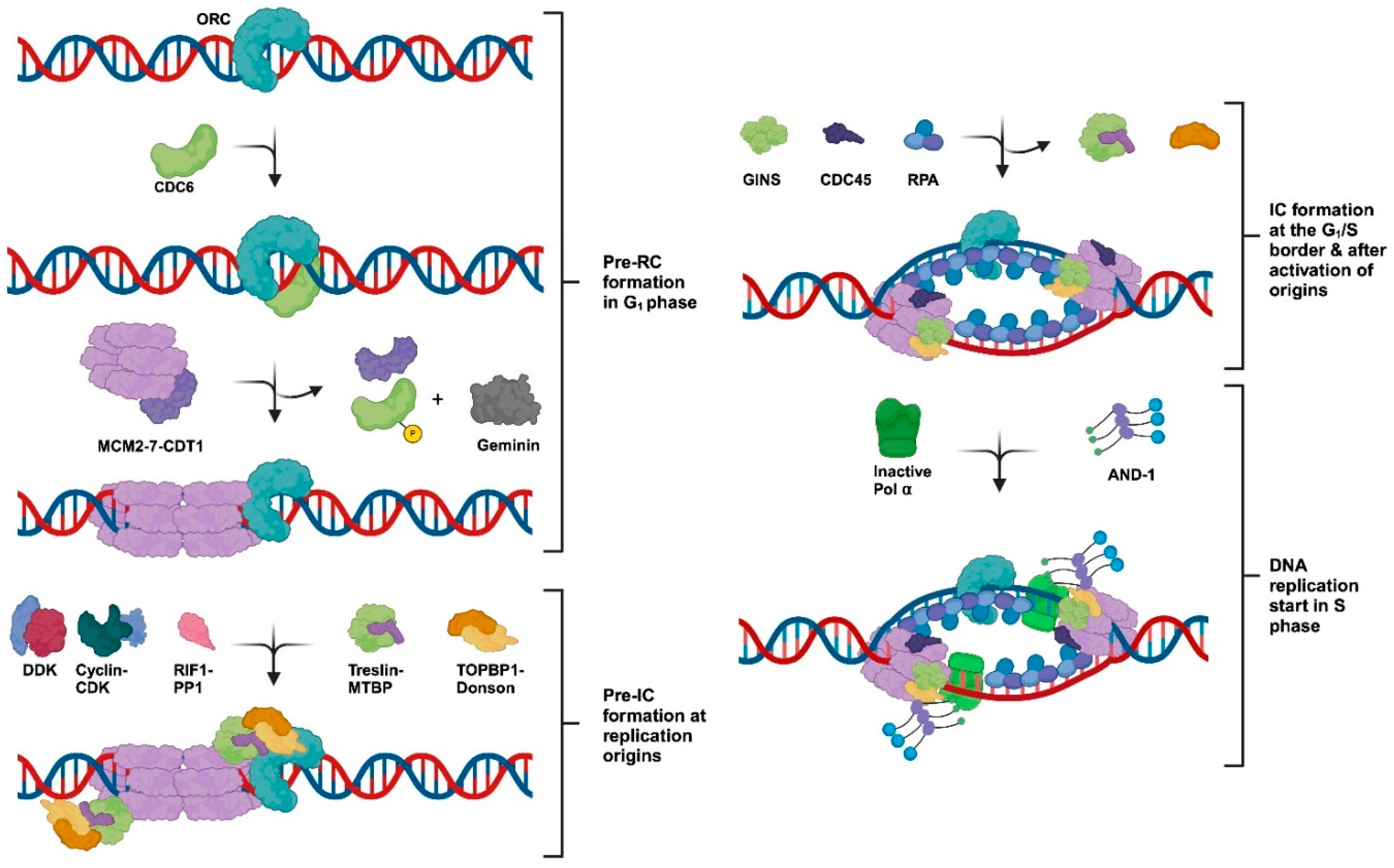
During the initiation process of eukaryotes, these ORC-DNA complexes serve as landing platforms for additional proteins called CDC6 and CTD1-MCM2-7 in G1 phase of the cell cycle when the activity of cyclin-dependent kinases (CDKs) is low [3,9,30,46,47,48,49]. Here, MCM2-7 hexamers bind to the origin chromatin forming a head-to-head dimer, MCM2-7 double hexamer (MCM-DH), on the DNA [50,51]. These proteins form the so-called Pre-RC complex, which is first step of the formation and activation of the replicative DNA helicase containing MCM2-7 as a core, and results in the licensing of replication origins [3,12,52]. In the next step, CDC6 and CTD1 leave the complex and are inactivated e.g., depending on the organism, CDC6 is phosphorylated and degraded. In contrast, CTD1 binds Geminin and forms a stable, inactive CTD1-Geminin complex, whereas free CTD1 is proteolytically degraded [53,54]. In some organisms including human, CTD1 is completely proteolytically degraded in a ubiquitin-dependent manner in late G1/early S phase [55]. In the following, the proteins Treslin and MTBP, named SLD3 and SLD5 in yeast, bind to MCM-DHs [2,3,55]. Then DBF4/DRF1-CDC7 kinase (DDK) phosphorylates MCM-DHs preparing the latter for the next step. Cyclin-CDK complexes e.g., CycE-CDK2, phosphorylate Treslin in complex with MCM-DHs and stimulate Donson-TOPBP1 complex formation. TOPBP1 has Dpb11 as its equivalent in budding yeast, whereas the Donson-equivalent protein has not yet been found in yeast [28]. In yeast, MCM10 and/or Pol ε may take over some functions of Donson during CMG formation [37,50,56,57]. Then TOPBP1 binds to the GINS complex and P-Treslin bringing the GINS proteins to the MCM-DHs located at origin sequences. This complex formation allows CDC45 to associate with GINS and MCM2-7 forming the CMG complex, the eukaryotic replicative helicase [22,24,25,26,27,28]. Next, TOPBP1 and the Treslin/MTBP complex leave the CMG complexes but two Donson molecules remain associated with each active CMG complex [22,28]. These activated helicase complexes start to untwist and unwind dsDNA establishing replication bubbles in origins and move in opposite directions forming two RFs at the active origins [3,50]. To stabilise the ssDNA and protect the ssDNA against nucleases, CMG complexes load the eukaryotic ssDNA-binding protein, replication protein A (RPA), a heterotrimer consisting of RPA70, RPA32 and RPA14, onto the unwound ssDNA strands [58,59,60,61]. In the following, Pol α associates with the CMG complex via the AND-1/CTF4/WDGD homotrimer and in turn the primase function of Pol α forms RNA primers on ssDNA templates at origins, the start of eukaryotic DNA replication [1,9,45,60,62,63].
3. The Initiation of DNA Synthesis at Origins
The binding of the MCM2-7 complexes and their activation are well understood as summarised above but the transition from the dsDNA binding modus of MCM2-7 proteins and the CMG to the unwinding of the dsDNA origin to form stretches of ssDNA bound by RPA is less well understood. However, recently cryoEM studies have revealed some insights into this process in human and yeast [50,64]. After the loading of MCM-DHs on origin dsDNA in an ATP hydrolysis-dependent manner, a stable ADP-MCM2-7 complex is formed, and the ‘MCM6 wedge’ (N-terminal pore loop in Mcm6) interacts with dsDNA at MCM-DH interface and an initial open structure at the hexamer junction is generated [50,64]. In the following, the formation of Pre-IC (pre-initiation complex) occurs which includes DDK and CDK phosphorylation as well as the association of CDC45 and GINS resulting in CMG complex formation (Figure 1, [50]). The ATP-hydrolysis-depending on opening and untwisting of dsDNA follows a mechanism of pushing of one strand and pulling of the other strand by MCM2 components plus interactions of the MCM6 wedge with the partially single-stranded bases allowing the first base pairs in the dsDNA to be broken [50,64]. Next, a putative further rotation of the DNA-bound CMG and the reconfiguration of the CMG-DNA complex with the help of the MCM10 protein plus further ATP hydrolysis, two RFs with Pol ε-CMG complexes as the replicative helicase complex on the leading strands are formed in budding yeast [50]. Then, like SV40 T antigen in the viral DNA replication, CDC45 of the CMG complex loads RPA on the emerging ssDNA, and these ssDNA sequences are further extended with the help of RPA and the two CMG molecules move on ssDNA in 3′-5′ direction yielding two RFs [58,60,65,66].
In the next step, primase consisting of PRIM1/PRI1, the catalytic subunit [67], and PRIM2/PRI2, the regulatory subunit [68,69], is recruited to the RF as part of the four-subunit Pol α complex via a AND-1/CTF4/WDHD1 trimer. The latter associates with the CMG complex at RFs (Figure 1 and Figure 2, [1,45,63,70]). On the ssDNA template, primase synthesises short RNA primers on the CMG-RPA-bound ssDNA templates initiating the leading strand DNA synthesis (Figure 2, [43,60,71,72,73,74]). Next, a protein complex involving RPA, PRIM2/PRI2-CT, and other partners of the initiation complex hand over the RNA annealed to the ssDNA template to the catalytic centre of PolA1 of Pol α, which then elongates the primer with a short stretch of DNA (additional ~20 nts, Figure 2, [1,43,60,75]). After synthesising this short DNA sequence, Pol α leaves the template, RFC-PCNA binds this RNA-DNA primer, and associates with DNA polymerase δ (Pol δ). Pol δ is a four-subunit protein complex with the largest subunit p125/Pol3 having DNA polymerase activity, which elongates the RNA-DNA. The p125/Pol3 subunit, different to Pol α-p180, also contains an active 3′-5′ proofreading exonuclease function [1,76,77,78]. Then, in the model called ‘division of labour’, during leading strand DNA synthesis, Pol δ finally hands the DNA molecule over to DNA polymerase ε (Pol ε), which has a high intrinsic processivity and can synthesise the leading strand DNA as long as a full replicon in one go, associates with the PCNA-bound DNA and elongates it. Pol ε also consists of four subunits, of which the largest subunit PolE1/Pol2 has DNA polymerase and a proofreading 3′-5′ exonuclease to correct falsely incorporated nucleotides in the leading strand [1,76]. Although Pol ε can synthesise a full replicon size worth of DNA in one processivity cycle, under exceptional circumstances the DNA synthesis will stall e.g., when Pol ε encounters a DNA lesion, and replication fork stabilisation mechanisms will be activated as discussed below (Section 5, [42,72,73,79]).
4. Initiation Processes at DNA replication forks—Okazaki Fragment Synthesis
During the initiation of DNA replication at origins of DNA replication, the CMG helicase produces two RFs with each having two ssDNA templates [1,43,81]. The antiparallel characteristics of the two strands yield that at each RF, one ssDNA template has a 3′-5′ orientation and serves as template of the leading strand synthesis, which is continuous as described above. In contrast, the second strand has a 5′-3′ direction and does not allow continuous DNA synthesis by DNA polymerases. To overcome this problem, the second strand, the lagging strand, is synthesised discontinuously in short fragments with length of 200-300 nucleotides in eukaryotes, called Okazaki fragments. Thus, to replicate the human genome completely, for each cell duplication the cellular replication machinery must initiate, elongate, and maturate ~30 million Okazaki fragments on the lagging strand for complete replication of a human genome [1,32,43].
Recent cryo-EM structure studies of budding yeast and human replisomes as well as of the CST-Pol α complexes have given a profound insight into the mechanism of the initiation of Okazaki fragment synthesis at RFs and telomere sequences [39,62,82,83]. The RF structures by Jones et al. show that Pol α has multiple interactions with the CMG complex and the AND-1/CTF4/WDHD1 complex (summarised in Figure 2, [62]). Here, especially MCM3/P1, and the GINS subunits have direct contacts with the primase subunit PRIM2/PRI2, and to the second largest Pol α subunit PolA2/Pol12 [62,84,85,86,87]. Interestingly, most of these interaction sites of the CMG helicase and AND-1 with Pol α are localised within intrinsic disordered regions (IDRs), which have some intrinsic structural flexibility. The sites of these interactions may allow the movement of Pol α on the lagging strand template during primer synthesis due to the flexibility of the IDR structure, whereas CMG advances on the leading strand template unwinding the dsDNA [43,45,62]. Importantly, these protein-protein and CMG-DNA interactions position the catalytic primase subunit of Pol α, PRIM1/PRI1, close to MCM5 and the opening of the exit channel for the lagging-strand template ssDNA [62]. In the yeast cryo-EM structure, the MCM5 Zn finger has physical contacts with yPRI1 but this interaction is not conserved in the human Pol α-AND-1-CMG structure [62].
During primer synthesis, PRIM1/PRI1 and PRIM2/PRI2 bind to the ssDNA template to catalyse the formation of the initial dinucleotide, the rate-limiting step of primer synthesis, using two incoming ribonucleoside 5′-triphosphates and divalent metal cations [67,68,70,88,89,90,91,92,93,94]. The primase regulatory subunit PRIM2/PRI2 consists of two domains, N- and C-terminal domain, PRIM2N/PRI2N and PRIM2C/PRI2C, respectively [43,60,70,75,82,88,94,95,96]. Here, PRIM2N/PRI2N serves as a protein interaction platform and links the catalytic PRIM1/PRI1 subunit to the C-terminal domain of PolA1. The latter serves as a scaffold for Pol α complex formation including binding the second largest subunit PolA2/p70/68/B-subunit, an essential regulatory subunit of Pol α without known enzyme activity, and the primase dimer. After the initial dinucleotide formation, PRIM1/PRI1 elongates the molecule to a size of 7-10 nucleotides in a distributive manner since PRIM1/PRI1 and its active site frequently detache e.g., after three nucleotides, from the newly formed oligoribonucleotides due to weak interactions of PRIM1/PRI1 with di- and tri-nucleotide substrates [94,97]. PRIM2C supports the catalytic cycle by functioning as a processivity factor and staying bound to the 5′-end of the primer, allowing the re-association of PRIM1/PRI1 to the 3′-end of the RNA primer to elongate the primer [68,70,88,91,94,95]. However, when the primer reaches a size of ~10 nucleotides further elongation PRIM2C can no longer support the enzyme activity of PRIM1/PRI1 due to a steric clash between PRIM2C and PRIM2N [70,88,95]. Instead, PRIM2C hands over the RNA primer with help of auxiliary mediator proteins such as RPA and CST to PolA1 for further extension [43,60,70,75,82,83,94,95,96]. For this to happen, the primer must be at least 7 nucleotides in length as PolA1 cannot associate with shorter primers. When the primer is at least 7 nucleotides long, a competition for primer binding between PRIM1/PRI1 and PolA1 occurs [94]. PolA1 has a higher affinity for primers of such a size making its binding more favourable, enabling the hand-over. PolA1 adds then ~20 deoxyribonucleoside monophosphate (dNMP) molecules to the primer creating an RNA-DNA hybrid primer with a size of ~30 nucleotides [92,96,97,98].
In the next step, the RNA-DNA primer is handed over to Pol δ with the help of RPA and RFC. The latter loads the homotrimer PCNA, also called the replication clamp, onto the primed DNA and supports the Pol δ-primer interaction to elongate the Okazaki fragment in processive manner [1,81,99,100,101,102,103]. When Pol δ reaches the 5′-end of the previous Okazaki fragment it starts to displace the RNA and parts of the Pol α-synthesised DNA as a first step of Okazaki fragment maturation (Figure 2, inserted panels, [1,35,102,103,104,105,106,107,108,109]). The resulting flap is then cleaved by FEN1 (flap endonuclease 1) which associates with PCNA in parallel with Pol δ and LIG1 (Ligase 1, [102,103,104,105,108]). The FEN1 cleavage yields two adjacent DNA strands with a nick, the perfect substrate for LIG1, which ligates them together in an ATP-dependent manner yielding a continuous stretch of lagging strand DNA (Figure 2, [102,103,110]). Alternatively, RPA binds to the Pol δ-displaced flap DNA, which FEN1 then can no longer access, and recruits endonuclease/helicase DNA2 to the sequence. The latter cleaves the flap just one nucleotide adjacent to the dsDNA part of the sequence and LIG1 cannot ligate the DNA2 product. Instead, FEN1 cuts off the remaining single nucleotide and yields a nicked dsDNA sequence as described above, which LIG1 ligates [102,103,110]. Interestingly, the maturation enzyme DNA2 serves as one of the sensors for ongoing unperturbed DNA replication (see Figure 3 and Section 5, [111,112]. As the removal of the RNA part during the maturation process in the above described is slow, pre-removal of the RNA primer by the RNAse H accelerates the maturation rate by about one order of magnitude [102].
5. Challenges at Replication Forks—Replication Fork Stalling
RFs are tightly controlled, and stable structures formed during eukaryotic chromosomal DNA replication in S phase [42,74,81,116,118,121,122,123,124,125,126]. Nevertheless, various problems and challenges including DNA lesions, difficult-to-replicate DNA sequences, and collisions with transcription machineries may happen and interfere with the progression of RFs, which are summarised as ‘replication stress’ (Figure 3 and reviewed [8,41,42,72,116,118,125,126,127]. Such stress can yield the stalling of DNA polymerases or slow-down of the RF movement, which in turn threatens the timely proceeding and fidelity of human genome duplication. Stalled RFs can lead to challenges of genome stability such as DNA double strand breaks (DSBs), or incomplete sister chromatid separation during mitosis. If arrested RFs are not appropriately processed and restarted, they become unstable, and the collapse of forks may occur [8,41,42,72,116,118,125,126,127]. Numerous different issues such as shortages of nucleotides or replication factors, the misalignment of nucleotides, lesions of DNA template, or malfunctions during the unwinding process can cause replication stress. Although these challenges normally arise sporadically, oncogenic activation of proteins such as E2F, and mutations of tumour suppressor proteins such as p53 and pRB also frequently cause replication stress [41,42,72,116,118,125,127].
However, eukaryotic cells have adapted to such challenges and several pathways, the so called the DNA damage response (DDR) pathways, have evolved to stabilize, repair, and restart the stalled forks to prevent additional DNA lesions and faulty structures from progressing further through the cycle [41,42,72,116,118,125,127,128]. These DDR pathways, which consist of numerous proteins including protein kinases such as the ATM (Ataxia telangiectasia-mutated) and especially ATR (ATM-Rad 3-related protein), have been well described in recent reviews [118,125,126,128,129,130,131]. In the present review, we will focus on the roles of DNA synthesis initiation reactions in the establishment of the replication stress signal and their connections to DDR pathways (Figure 3) as well as how they support the restart of DNA replication.
It is important to note that ATR is not only active during replication stress but also during normal replication processes when ATR monitors RF progression and slows down RFs to avoid e. g., nucleotide shortages (Figure 3 top panel, [111,112,126,132]). These ATR-dependent pathways may depend on the loading of the 9-1-1 complex to the 5′-ends of Okazaki fragments, the interaction of TOPBP1 with the 9-1-1 checkpoint clamp (called Rad9-Rad1-Hus1 in S. pombe and humans, Ddc1-Rad17-Mec3 in S. cerevisiae), DNA2, and Pol α (Figure 3, [111,112,119,120]). A second function of ATR (via the so-called ‘canonical’ ATR pathway [119,126]) is to monitor and signal replication stress when RF progress through DNA sequences such as repetitive sequences and sequences containing G-quadruplexes (G4s), or encounter DNA lesions yielding difficulties to progress. Lesions or problematic DNA sequences on the lagging strand are easier to handle due to its discontinuous nature. They can be easily bypassed by initiating new Okazaki fragments to allow later repair of the lesion or its bypass via translesion DNA synthesis of specialised DNA polymerases [117,119,120]. Nevertheless, these non-maturated Okazaki fragments may cause or contribute to replication stress signals (Figure 3B).
In contrast, on leading strand templates, Pol ε encountering a lesion stops and disengages with the CMG helicase or modifies its activity. As a result, the CMG helicase continues to unwind DNA and load RPA on the leading strand template ssDNA, whereas the lagging strand synthesis continues and Pol α produces RNA-DNA primers. RFC-PCNA-Pol δ can extend these primers to form Okazaki fragment, but if the latter are not maturated they produce ssDNA-dsDNA/RNA products with free 5′-ends, which are recognised by Rad17/24-RFC, the 9-1-1 clamp loader, loading the 9-1-1 clamp on these structures [114,116,129,133,134,135,136,137]. Here, it is important to note that RNA primers produced by primase are sufficient for the checkpoint initiation [114,138]. The 9-1-1 complex and Pol α on the lagging strand template then recruit TOPBP1 to the stalled RF whereas RPA, an ATR substrate, on the leading binds to the ATR-binding protein ATRIP (Figure 3B, [61,116,119,139]). In addition, the stalled Pol ε may interact with TOPBP1 and stabilise its interaction with the stalled RF. In summary, this protein complex with TOPBP1 at its centres forms a protein bridge linking the leading and lagging strand templates. This arrangement of the binding and activation of ATR, the so-called canonical ATR activation pathway, now initiates DDR pathways and helps to stabilise the RF under replication stress conditions as well as by activating checkpoint kinases such as RAD53 in yeast and CHK1 in Metazoan [111,116,118,119]. The activation of ATR by ETAA1 (Ewing’s Tumor-Associated Antigen 1) with the help of RPA-ATRIP complexes on the leading strand could serve as an additional, TOPBP1-independent branch to activate or amplify ATR signals at stalled RFs [116,126]. This model describes the cooperation of multiple key factors to activate DDR pathways resulting in slowing down RFs and preventing origin firing, which are not yet active.
The stalled RF can be release by a partial exchange of RPA to RAD51 with the help of BRCA2 and RF reversal or, as a bypass mechanism. For the latter, RPA recruits prim-pol (primase-polymerase), a second eukaryotic DNA polymerase with associated primase activity different to Pol α, to the leading strand template and induces prim-pol to synthesise a primer to restart leading strand DNA synthesis [127,140]. Importantly, Pol α associated with CMG at the RF cannot initiate on the leading strand outside the replication origin but its activity is important for the checkpoint initiation, a clear separation of tasks between Pol α and prim-pol [71,72,127,140]. However, to restart leading strand synthesis in G4-rich sequence the CTC1-STN1-TEN1 (CST complex), which normally acts at telomere sequences (see Section 6), can recruit Pol α to G4-rich templates and activate the enzyme complex to synthesise RNA primer [141,142]. As an additional alternative to start DNA replication, dormant origins can be activated to restart leading and lagging strand DNA synthesis [143,144,145]. These processes would then in turn solve the replication problem and allow continuation of replication of genomic DNA. However, in case that the replication problem(s) cannot be resolved, the situation may lead to cell death pathways e.g., via ATR-dependent CHK2 activation [146], or continuous hyperactivation of ATR yielding senescence of these cells [147].
6. Okazaki Fragment Synthesis and the End-Replication Problem at Telomeres
The end replication problem hypothesis refers to the challenge that eukaryotic cells faced by the lagging strand replication machinery to fully replicate DNA at the end of their linear chromosomes first hypothesised by Olovnikov [148]. At telomeric ends of chromosomes on the lagging strand, primase synthesises the last oligoribonucleotide at or close to the end of the ssDNA template [149]. Thus, the removal of the RNA primers and putatively parts of the Pol α-synthesised DNA results in a shortened lagging strand, which cannot be fully synthesised by the DNA replication machinery [150]. This yields the shortening of one of the newly synthesised chromosomes and the loss of genetic material in the next DNA replication round [151]. Multiple replication rounds will finally result in the loss of genetic information. Telomeres, nucleoprotein complexes consisting of repeated ‘TTAGGG’ sequences and proteins at the end of chromosomes, are responsible for the protection of eukaryotic genomes [152,153,154]. Telomeres have a special DNA structure including a region of dsDNA ending with a 3′ tail containing ssDNA known as the G overhang. Additionally, telomeres consist of proteins including the shelterin complex, which plays a crucial role in the regulation and protection of telomeres [151,155]. At telomere ends, the shelterin complex also controls the activity of telomerase, a reverse transcriptase, which has its own RNA template and synthesis telomeric DNA repeats to the G overhang. On one hand, the RPA-like ssDNA-binding protein complex consisting of CTC1, STN1 and TEN1, the CST complex, diminishes DNA synthesis by telomerase. On the other hand, CST binds to Pol α, recruits the enzyme complex to telomere sequences, and remodels Pol α from an inactive to initiation-active enzyme complex, which, in turn, starts to synthesise the RNA primer close to the 5′ of the template strand starting C strand synthesis [43,75,96,151]. Next, CST and PRIM2/PRI2-CT hand over the RNA primer to the DNA polymerase domain of PolA1 in the Pol α complex, which adds dNMPs to the strand. PCNA-Pol δ then elongates the RNA-DNA primer until the C strand is fully synthesised [82,83,96]. The importance of the process described above is underline by human disease such as Coats plus syndrome having mutations in CST subunits [156,157]. Recent findings suggest that the CST-Pol α complex solves an until recently little recognised second telomere end-replication problem since a lack of CST-Pol α yields not only shortening of the lagging strand at telomeres but also a resection of the leading strand DNA in the next round of DNA replication [150].
7. Outlook
The faithful replication of cellular DNA including replication initiation reactions are central processes for the maintenance of human cell functions and avoiding genetic diseases including cancer [158,159,160]. Cancer cells proliferate and replicate their DNA in an uncontrolled manner, which leads to abnormal cell numbers. The lack of control of replication initiation mechanisms is often a cause of genome instability, which in turn is a property that enables cancer development [159,160]. However, beyond cancer genetic diseases such as Meier-Gorlin syndrome, a form of microcephalic primordial dwarfism, and Coats plus syndrome (also called Cerebro retinal Microangiopathy with Calcifications and Cysts (CRMCC)), a multi-organ symptom including brain, eye, and gastrointestinal tract, have mutations in central players of the initiation of DNA synthesis such as MCM2-7, CDC45 and the CST complex [28,83,156,161].
Here, the understanding of replication processes such as initiation of DNA replication at origins and Okazaki fragment synthesis may help to develop treatments of these rare diseases. Progress in the knowledge of the structure, protein-protein interactions, and PTMs (post translational modifications) of replication factors involved initiation processes and the replication fork using Cryo-EM and modern mass spectrometry including single-cell analysis approaches will bring about the knowledge to develop treatments [8,62,162,163,164,165]. Next generation sequencing techniques with the adaptation to DNA replication processes as seen for genomic Okazaki fragment distributions will help to map origins and their dynamic during normal and perturbed DNA replication. These methods will not only be used in yeast but also in human cells and other organisms, which in part have started, will bring a better understanding of the regulation of DNA replication during normal undisturbed and perturbed cell cycles as well as dysfunctional replication control in cancer cells [35,166]. The latter may help to identify new Achilles heels of cancer cells, which can be used as targets for treatment development, or for cancer cell diagnosis and cancer prognosis. Examples of recent developments are the cancer diagnostics using the overexpression of MCM2-7 proteins shown in a variety of cancers such as renal cell carcinomas, breast, prostate, and lung cancer. Increased levels of MCM2-7 have usually associated with a poor prognosis of these cancers and an aggressive tumour behaviour [167,168,169]. Additionally, the fundamental research of the replication initiation pathways has yielded the development of new inhibitors for cancer therapy using such as CDC7 and the CMG helicase as targets [170,171,172,173,174].
Abbreviations
| 9-1-1 | Rad 9-Hus 1-Rad 1 complex (fission yeast and human) equivalent to the |
| complex | Rad 17-Mec3-Ddc1 complex in budding yeast |
| aa | amino acid |
| AND-1/CTF4/ | acidic and nucleoplasmic DNA- binding protein/Chromosome - |
| WDHD1 | Transmission Fidelity 4/WD repeat and HMG-box DNA-binding protein 1 |
| A-T | Ataxia telangiectasia |
| ATM | Ataxia telangiectasia-mutated |
| ATR | ATM-Rad 3-related protein kinase |
| ATRIP | ATR interacting protein |
| ba | bases |
| BLM | Bloom’s helicase |
| CDC | cell division cycle |
| CDK | cyclin-dependent kinase |
| CMG | CDC45-MCM2-7-GINS |
| CST | CTC1-STN1-TEN1 |
| DBD | DNA-binding domain |
| DDR | DNA damage response |
| DH | double hexamer |
| DSB | double-strand break |
| dsDNA | double-stranded DNA |
| ETAA1 | Ewing’s Tumor-Associated Antigen 1 |
| G4 | G-quadruplexes |
| IC | initiation complex |
| mt | mitochondrial |
| MCM | minichromosome maintenance |
| hPolprim1 | human primase-polymerase 1 |
| MCM | Minichromosome maintainance |
| MTBP | Mouse double minute 2-binding protein |
| nt | nucleotide |
| PCNA | proliferating cell nuclear antigen |
| PIKK | phosphotnositol-3 kinase-like protein kinase |
| Pol α | DNA polymerase α–primase |
| Pol δ | DNA polymerase δ |
| Pol ε | DNA polymerase ε |
| POT1 | protector of telomeres 1 |
| Pre-RC | pre-replicative complex |
| Pre-IC | pre-initiation complex |
| RAD | radiation-induced mutation |
| RFC | replication factor C |
| RPA | replication protein A |
| SSB | single-stranded DNA-binding protein |
| ssDNA | single-stranded DNA |
| S. cerevisiae | Saccharomyces cerevisiae |
| S. pombe | Schizosaccharomyces pombe |
| SV40 | simian virus 40 |
| Tp53 | tumour suppressor protein p53 |
| TOPBP1 | topoisomerase II-binding protein 1 |
References
- Burgers, P.M.J. and T.A. Kunkel. Eukaryotic DNA Replication Fork. Annu Rev Biochem, 2017. 86: p. 417-438. [CrossRef]
- Bleichert, F., M.R. Botchan, and J.M. Berger. Mechanisms for initiating cellular DNA replication. Science, 2017. 355: p. eaah6317. [CrossRef]
- Costa, A. and J.F.X. Diffley. The Initiation of Eukaryotic DNA Replication. Annu Rev Biochem, 2022. 91: p. 107-131. [CrossRef]
- Costa, A., I.V. Hood, and J.M. Berger. Mechanisms for initiating cellular DNA replication. Annu Rev Biochem, 2013. 82: p. 25-54. [CrossRef]
- Masai, H., S. Matsumoto, Z. You, N. Yoshizawa-Sugata, and M. Oda. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem, 2010. 79: p. 89-130. [CrossRef]
- Nasheuer, H.P.; Pospiech, H.; Syväoja, J. Progress towards the anatomy of the eukaryotic DNA replication fork. In Genome Integrity: Facets and Perspectives; Lankenau, D.H., Ed.; Springer: Berlin-Heidelberg-NewYork, 2007; Genome Dynamics & Stability; Volume 1, pp. 27–68. [Google Scholar] [CrossRef]
- O’Donnell, M., L. Langston, and B. Stillman. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol, 2013. 5. [CrossRef]
- González-Acosta, D. and M. Lopes. DNA replication and replication stress response in the context of nuclear architecture. Chromosoma, 2023. [CrossRef]
- Attali, I., M.R. Botchan, and J.M. Berger. Structural Mechanisms for Replicating DNA in Eukaryotes. Annu Rev Biochem, 2021. 90: p. 77-106. [CrossRef]
- Howard, A.; Pelc, S.R. Synthesis of deoxyribonucleic acid in normal and irradiated cells and its relation to chromosome breakage. Heredity 1953, 6, 261–273. [Google Scholar]
- Voorhees, J.J., E.A. Duell, D.A. Chambers, and C.L. Marcelo. Regulation of Cell Cycles. Journal of Investigative Dermatology, 1976. 67: p. 15-19. [CrossRef]
- Blow, J.J.; Dutta, A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005, 6, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Laskey, R.A. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature 1988, 332, 546–548. [Google Scholar] [CrossRef] [PubMed]
- Mechali, M.; Kearsey, S. Lack of specific sequence requirement for DNA replication in Xenopus eggs compared with high sequence specificity in yeast. Cell 1984, 38, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Stillman, B. Origins of DNA replication in eukaryotes. Mol. Cell 2023, 83, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F.X. Early events in eukaryotic DNA replication. Trends Cell Biol. 1992, 2, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F.X.; Cocker, J.H. Protein-DNA interactions at a yeast replication origin. Nature 1992, 357, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Natale, D.A., C.J. Li, W.H. Sun, and M.L. DePamphilis. Selective instability of Orc1 protein accounts for the absence of functional origin recognition complexes during the M-G(1) transition in mammals. Embo J, 2000. 19: p. 2728-2738. [CrossRef]
- Sun, W.H., T.R. Coleman, and M.L. DePamphilis. Cell cycle-dependent regulation of the association between origin recognition proteins and somatic cell chromatin. Embo J, 2002. 21: p. 1437-46. [CrossRef]
- DePamphilis, M.L. The ‘ORC cycle’: a novel pathway for regulating eukaryotic DNA replication. Gene 2003, 310, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Saha, T., S. Ghosh, A. Vassilev, and M.L. Depamphilis. Ubiquitylation, phosphorylation and Orc2 modulate the subcellular location of Orc1 and prevent it from inducing apoptosis. J Cell Sci, 2006. [CrossRef]
- Cvetkovic, M.A., P. Passaretti, A. Butryn, A. Reynolds-Winczura, G. Kingsley, A. Skagia, C. Fernandez-Cuesta, D. Poovathumkadavil, R. George, A.S. Chauhan, S.S. Jhujh, G.S. Stewart, A. Gambus, and A. Costa. The structural mechanism of dimeric DONSON in replicative helicase activation. Mol Cell, 2023. 83: p. 4017-4031.e9. [CrossRef]
- Dhar, S.K., L. Delmolino, and A. Dutta. Architecture of the human origin recognition complex. J Biol Chem, 2001. 276: p. 29067-29071. [CrossRef]
- Evrin, C., V. Alvarez, J. Ainsworth, R. Fujisawa, C. Alabert, and K.P. Labib. DONSON is required for CMG helicase assembly in the mammalian cell cycle. EMBO Rep, 2023. 24: p. e57677. [CrossRef]
- Hashimoto, Y., K. Sadano, N. Miyata, H. Ito, and H. Tanaka. Novel role of DONSON in CMG helicase assembly during vertebrate DNA replication initiation. Embo j, 2023. 42: p. e114131. [CrossRef]
- Kingsley, G., A. Skagia, P. Passaretti, C. Fernandez-Cuesta, A. Reynolds-Winczura, K. Koscielniak, and A. Gambus. DONSON facilitates Cdc45 and GINS chromatin association and is essential for DNA replication initiation. Nucleic Acids Res, 2023. 51: p. 9748-9763. [CrossRef]
- Lim, Y., L. Tamayo-Orrego, E. Schmid, Z. Tarnauskaite, O.V. Kochenova, R. Gruar, S. Muramatsu, L. Lynch, A.V. Schlie, P.L. Carroll, G. Chistol, M.A.M. Reijns, M.T. Kanemaki, A.P. Jackson, and J.C. Walter. In silico protein interaction screening uncovers DONSON’s role in replication initiation. Science, 2023. 381: p. eadi3448. [CrossRef]
- Xia, Y., R. Sonneville, M. Jenkyn-Bedford, L. Ji, C. Alabert, Y. Hong, J.T.P. Yeeles, and K.P.M. Labib. DNSN-1 recruits GINS for CMG helicase assembly during DNA replication initiation in Caenorhabditis elegans. Science, 2023. 381: p. eadi4932. [CrossRef]
- Vashee, S., P. Simancek, M.D. Challberg, and T.J. Kelly. Assembly of the human origin recognition complex. J Biol Chem, 2001. 276: p. 26666-26673. [CrossRef]
- Pellegrini, L. The CMG DNA helicase and the core replisome. Curr Opin Struct Biol, 2023. 81: p. 102612. [CrossRef]
- Blow, J.J., P.J. Gillespie, D. Francis, and D.A. Jackson. Replication origins in Xenopus egg extract Are 5-15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol, 2001. 152: p. 15-25. [CrossRef]
- Smith, D.J. and I. Whitehouse. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature, 2012. 483: p. 434-8. [CrossRef]
- Jones, M.L., Y. Baris, M.R. Taylor, and J.T.P. Yeeles. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. Embo j, 2023. 42: p. e115685. [CrossRef]
- Baretić, D., M. Jenkyn-Bedford, V. Aria, G. Cannone, M. Skehel, and J.T.P. Yeeles. Cryo-EM Structure of the Fork Protection Complex Bound to CMG at a Replication Fork. Mol Cell, 2020. 78: p. 926-940.e13. [CrossRef]
- Sriramachandran, A.M., G. Petrosino, M. Méndez-Lago, A.J. Schäfer, L.S. Batista-Nascimento, N. Zilio, and H.D. Ulrich. Genome-wide Nucleotide-Resolution Mapping of DNA Replication Patterns, Single-Strand Breaks, and Lesions by GLOE-Seq. Mol Cell, 2020. 78: p. 975-985.e7. [CrossRef]
- Blin, M., L. Lacroix, N. Petryk, Y. Jaszczyszyn, C.L. Chen, O. Hyrien, and B. Le Tallec. DNA molecular combing-based replication fork directionality profiling. Nucleic Acids Res, 2021. 49: p. e69. [CrossRef]
- Langston, L.D., R.E. Georgescu, and M.E. O’Donnell. Mechanism of eukaryotic origin unwinding is a dual helicase DNA shearing process. Proc Natl Acad Sci U S A, 2023. 120: p. e2316466120. [CrossRef]
- Yuan, Z., L. Bai, J. Sun, R. Georgescu, J. Liu, M.E. O’Donnell, and H. Li. Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat Struct Mol Biol, 2016. 23: p. 217-24. [CrossRef]
- Georgescu, R.E., G.D. Schauer, N.Y. Yao, L.D. Langston, O. Yurieva, D. Zhang, J. Finkelstein, and M.E. O’Donnell. Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. Elife, 2015. 4: p. e04988. [CrossRef]
- Bell, S.P. and B. Stillman. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature, 1992. 357: p. 128-134. [CrossRef]
- Qiu, S., G. Jiang, L. Cao, and J. Huang. Replication Fork Reversal and Protection. Front Cell Dev Biol, 2021. 9: p. 670392. [CrossRef]
- Vipat, S. and T.N. Moiseeva. The TIMELESS Roles in Genome Stability and Beyond. J Mol Biol, 2024. 436: p. 168206. [CrossRef]
- Nasheuer, H.P. and N.O. Onwubiko. Lagging Strand Initiation Processes in DNA Replication of Eukaryotes-Strings of Highly Coordinated Reactions Governed by Multiprotein Complexes. Genes (Basel), 2023. 14. [CrossRef]
- Nasheuer, H.P., R. Smith, C. Bauerschmidt, F. Grosse, and K. Weisshart. Initiation of eukaryotic DNA replication: regulation and mechanisms. Prog Nucleic Acid Res Mol Biol, 2002. 72: p. 41-94. [CrossRef]
- Kilkenny, M.L., A.C. Simon, J. Mainwaring, D. Wirthensohn, S. Holzer, and L. Pellegrini. The human CTF4-orthologue AND-1 interacts with DNA polymerase α/primase via its unique C-terminal HMG box. Open Biol, 2017. 7. [CrossRef]
- Yeeles, J.T., T.D. Deegan, A. Janska, A. Early, and J.F. Diffley. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature, 2015. 519: p. 431-5. [CrossRef]
- On, K.F., F. Beuron, D. Frith, A.P. Snijders, E.P. Morris, and J.F. Diffley. Prereplicative complexes assembled in vitro support origin-dependent and independent DNA replication. Embo j, 2014. 33: p. 605-20. [CrossRef]
- Labib, K., J.F. Diffley, and S.E. Kearsey. G1-phase and B-type cyclins exclude the DNA-replication factor Mcm4 from the nucleus. Nat Cell Biol, 1999. 1: p. 415-422. [CrossRef]
- Deegan, T.D. and J.F. Diffley. MCM: one ring to rule them all. Curr Opin Struct Biol, 2016. 37: p. 145-51. [CrossRef]
- Lewis, J.S., M.H. Gross, J. Sousa, S.S. Henrikus, J.F. Greiwe, A. Nans, J.F.X. Diffley, and A. Costa. Mechanism of replication origin melting nucleated by CMG helicase assembly. Nature, 2022. 606: p. 1007-1014. [CrossRef]
- Frigola, J., D. Remus, A. Mehanna, and J.F. Diffley. ATPase-dependent quality control of DNA replication origin licensing. Nature, 2013. 495: p. 339-43. [CrossRef]
- Romanowski, P., M.A. Madine, A. Rowles, J.J. Blow, and R.A. Laskey. The Xenopus origin recognition complex is essential for DNA replication and MCM binding to chromatin. Curr Biol, 1996. 6: p. 1416-25. [CrossRef]
- McGarry, T.J. and M.W. Kirschner. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell, 1998. 93: p. 1043-1053. [CrossRef]
- Ferenbach, A., A. Li, M. Brito-Martins, and J.J. Blow. Functional domains of the Xenopus replication licensing factor Cdt1. Nucleic Acids Res, 2005. 33: p. 316-24. [CrossRef]
- Pozo, P.N. and J.G. Cook. Regulation and Function of Cdt1; A Key Factor in Cell Proliferation and Genome Stability. Genes (Basel), 2016. 8. [CrossRef]
- Yuan, Z., R. Georgescu, L. Bai, D. Zhang, H. Li, and M.E. O’Donnell. DNA unwinding mechanism of a eukaryotic replicative CMG helicase. Nat Commun, 2020. 11: p. 688. [CrossRef]
- Xu, Z., J. Feng, D. Yu, Y. Huo, X. Ma, W.H. Lam, Z. Liu, X.D. Li, T. Ishibashi, S. Dang, and Y. Zhai. Synergism between CMG helicase and leading strand DNA polymerase at replication fork. Nat Commun, 2023. 14: p. 5849. [CrossRef]
- Szambowska, A., I. Tessmer, P. Prus, B. Schlott, H. Pospiech, and F. Grosse. Cdc45-induced loading of human RPA onto single-stranded DNA. Nucleic Acids Res, 2017. [CrossRef]
- Chen, R. and M.S. Wold. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays, 2014. 36: p. 1156-61. [CrossRef]
- Nasheuer, H.P., A.M. Meaney, T. Hulshoff, I. Thiele, and N.O. Onwubiko. Replication Protein A, the Main Eukaryotic Single-Stranded DNA Binding Protein, a Focal Point in Cellular DNA Metabolism. Int J Mol Sci, 2024. 25: p. 588. [CrossRef]
- Fousek-Schuller, V.J. and G.E.O. Borgstahl. The Intriguing Mystery of RPA Phosphorylation in DNA Double-Strand Break Repair. Genes, 2024. 15: p. 167. [CrossRef]
- Jones, M.L., V. Aria, Y. Baris, and J.T.P. Yeeles. How Pol α-primase is targeted to replisomes to prime eukaryotic DNA replication. Mol Cell, 2023. 83: p. 2911-2924.e16. [CrossRef]
- Villa, F., A.C. Simon, M.A. Ortiz Bazan, M.L. Kilkenny, D. Wirthensohn, M. Wightman, D. Matak-Vinkovíc, L. Pellegrini, and K. Labib. Ctf4 Is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol Cell, 2016. 63: p. 385-96. [CrossRef]
- Li, J., J. Dong, W. Wang, D. Yu, X. Fan, Y.C. Hui, C.S.K. Lee, W.H. Lam, N. Alary, Y. Yang, Y. Zhang, Q. Zhao, C.L. Chen, B.K. Tye, S. Dang, and Y. Zhai. The human pre-replication complex is an open complex. Cell, 2023. 186: p. 98-111.e21. [CrossRef]
- Jiang, X., V. Klimovich, A.I. Arunkumar, E.B. Hysinger, Y. Wang, R.D. Ott, G.D. Guler, B. Weiner, W.J. Chazin, and E. Fanning. Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. Embo J, 2006. 25: p. 5516-26. [CrossRef]
- Broderick, R., M.D. Rainey, C. Santocanale, and H.P. Nasheuer. Cell cycle-dependent formation of Cdc45-Claspin complexes in human cells are compromized by UV-mediated DNA damage. FEBS J, 2013. 280: p. 4888-4902. [CrossRef]
- Nasheuer, H.P. and F. Grosse. DNA polymerase α-primase from calf thymus. Determination of the polypeptide responsible for primase activity. J. Biol. Chem., 1988. 263: p. 8981-8988. [CrossRef]
- Zerbe, L.K. and R.D. Kuchta. The p58 subunit of human DNA primase is important for primer initiation, elongation, and counting. Biochemistry, 2002. 41: p. 4891-4900. [CrossRef]
- Baranovskiy, A.G., N.D. Babayeva, Y. Zhang, J. Gu, Y. Suwa, Y.I. Pavlov, and T.H. Tahirov. Mechanism of Concerted RNA-DNA Primer Synthesis by the Human Primosome. J Biol Chem, 2016. 291: p. 10006-20. [CrossRef]
- Baranovskiy, A.G. and T.H. Tahirov. Elaborated Action of the Human Primosome. Genes (Basel), 2017. 8: p. 62. [CrossRef]
- Taylor, M.R.G. and J.T.P. Yeeles. Dynamics of Replication Fork Progression Following Helicase-Polymerase Uncoupling in Eukaryotes. J Mol Biol, 2019. 431: p. 2040-2049. [CrossRef]
- Taylor, M.R.G. and J.T.P. Yeeles. The Initial Response of a Eukaryotic Replisome to DNA Damage. Mol Cell, 2018. 70: p. 1067-1080.e12. [CrossRef]
- Jones, M.L., Y. Baris, M.R.G. Taylor, and J.T.P. Yeeles. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. Embo j, 2021. 40: p. e108819. [CrossRef]
- Guilliam, T.A. and J.T.P. Yeeles. An updated perspective on the polymerase division of labor during eukaryotic DNA replication. Crit Rev Biochem Mol Biol, 2020. 55: p. 469-481. [CrossRef]
- Barbour, A.T. and D.S. Wuttke. RPA-like single-stranded DNA-binding protein complexes including CST serve as specialized processivity factors for polymerases. Curr Opin Struct Biol, 2023. 81: p. 102611. [CrossRef]
- Jain, R., A.K. Aggarwal, and O. Rechkoblit. Eukaryotic DNA polymerases. Curr Opin Struct Biol, 2018. 53: p. 77-87. [CrossRef]
- Liu, L., J. Mo, E.M. Rodriguez-Belmonte, and M.Y. Lee. Identification of a fourth subunit of mammalian DNA polymerase delta. J Biol Chem, 2000. 275: p. 18739-18744. [CrossRef]
- Zhang, S., H.H. Chao, X. Wang, Z. Zhang, E.Y.C. Lee, and M. Lee. Loss of the p12 subunit of DNA polymerase delta leads to a defect in HR and sensitization to PARP inhibitors. DNA Repair (Amst), 2019. 73: p. 64-70. [CrossRef]
- Cali, F., S.K. Bharti, R. Di Perna, R.M. Brosh, Jr., and F.M. Pisani. Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res, 2016. 44: p. 705-17. [CrossRef]
- Prindle, M.J. and L.A. Loeb. DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen, 2012. 53: p. 666-82. [CrossRef]
- Zhang, D. and M. O’Donnell. The Eukaryotic Replication Machine. Enzymes, 2016. 39: p. 191-229. [CrossRef]
- He, Q., X. Lin, B.L. Chavez, S. Agrawal, B.L. Lusk, and C.J. Lim. Structures of the human CST-Polα–primase complex bound to telomere templates. Nature, 2022. 608: p. 826-832. [CrossRef]
- Cai, S.W., J.C. Zinder, V. Svetlov, M.W. Bush, E. Nudler, T. Walz, and T. de Lange. Cryo-EM structure of the human CST–Polα/primase complex in a recruitment state. Nature Structural & Molecular Biology, 2022. 29: p. 813-819. [CrossRef]
- Thömmes, P., R. Fett, B. Schray, R. Burkhart, M. Barnes, C. Kennedy, N.C. Brown, and R. Knippers. Properties of the nuclear P1 protein, a mammalian homologue of the yeast Mcm3 replication protein. Nucleic Acids Res., 1992. 20: p. 1069-1074. [CrossRef]
- Maine, G.T., P. Sinha, and B.K. Tye. Mutants of S. cerevisiae defective in the maintenance of minichromosomes. Genetics, 1984. 106: p. 365-85. [CrossRef]
- Tye, B.K. The MCM2-3-5 proteins: are they replication licensing factors? Trends Cell Biol, 1994. 4: p. 160-6. [CrossRef]
- Takayama, Y., Y. Kamimura, M. Okawa, S. Muramatsu, A. Sugino, and H. Araki. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev, 2003. 17: p. 1153-1165. [CrossRef]
- Baranovskiy, A.G., Y. Zhang, Y. Suwa, N.D. Babayeva, J. Gu, Y.I. Pavlov, and T.H. Tahirov. Crystal structure of the human primase. J Biol Chem, 2015. 290: p. 5635-46. [CrossRef]
- Arezi, B. and R.D. Kuchta. Eukaryotic DNA primase. Trends Biochem Sci, 2000. 25: p. 572-576. [CrossRef]
- Kirk, B.W. and R.D. Kuchta. Human DNA primase: anion inhibition, manganese stimulation, and their effects on in vitro start-site selection. Biochemistry, 1999. 38: p. 10126-10134. [CrossRef]
- Kirk, B.W. and R.D. Kuchta. Arg304 of human DNA primase is a key contributor to catalysis and NTP binding: primase and the family X polymerases share significant sequence homology. Biochemistry, 1999. 38: p. 7727-7736. [CrossRef]
- Grosse, F. and G. Krauss. The primase activity of DNA polymerase α from calf thymus. J Biol Chem, 1985. 260: p. 1881-1888.
- Vaithiyalingam, S., D.R. Arnett, A. Aggarwal, B.F. Eichman, E. Fanning, and W.J. Chazin. Insights into eukaryotic primer synthesis from structures of the p48 subunit of human DNA primase. J Mol Biol, 2014. 426: p. 558-69. [CrossRef]
- Cordoba, J.J., E.A. Mullins, L.E. Salay, B.F. Eichman, and W.J. Chazin. Flexibility and Distributive Synthesis Regulate RNA Priming and Handoff in Human DNA Polymerase α-Primase. J Mol Biol, 2023. 435: p. 168330. [CrossRef]
- He, Q., A.G. Baranovskiy, L.M. Morstadt, A.E. Lisova, N.D. Babayeva, B.L. Lusk, C.J. Lim, and T.H. Tahirov. Structures of human primosome elongation complexes. Nat Struct Mol Biol, 2023. 30: p. 579-583. [CrossRef]
- Zaug, A.J., K.J. Goodrich, J.J. Song, A.E. Sullivan, and T.R. Cech. Reconstitution of a telomeric replicon organized by CST. Nature, 2022. 608: p. 819-825. [CrossRef]
- Kuchta, R.D., B. Reid, and L.M. Chang. DNA primase. Processivity and the primase to polymerase alpha activity switch. J Biol Chem, 1990. 265: p. 16158-65.
- Kaguni, L.S., J.M. Rossignol, R.C. Conaway, and I.R. Lehman. Isolation of an intact DNA polymerase-primase from embryos of Drosophila melanogaster. Proc Natl Acad Sci U S A, 1983. 80: p. 2221-5. [CrossRef]
- Hübscher, U., G. Maga, and S. Spadari. Eukaryotic DNA polymerases. Annu Rev Biochem, 2002. 71: p. 133-63. [CrossRef]
- Donnianni, R.A., Z.X. Zhou, S.A. Lujan, A. Al-Zain, V. Garcia, E. Glancy, A.B. Burkholder, T.A. Kunkel, and L.S. Symington. DNA Polymerase Delta Synthesizes Both Strands during Break-Induced Replication. Mol Cell, 2019. 76: p. 371-381.e4. [CrossRef]
- Yeeles, J.T.P., A. Janska, A. Early, and J.F.X. Diffley. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol Cell, 2017. 65: p. 105-116. [CrossRef]
- Raducanu, V.S., M. Tehseen, A. Al-Amodi, L.I. Joudeh, A. De Biasio, and S.M. Hamdan. Mechanistic investigation of human maturation of Okazaki fragments reveals slow kinetics. Nat Commun, 2022. 13: p. 6973. [CrossRef]
- Stodola, J.L. and P.M. Burgers. Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat Struct Mol Biol, 2016. 23: p. 402-8. [CrossRef]
- Kao, H.I., J.L. Campbell, and R.A. Bambara. Dna2p helicase/nuclease is a tracking protein, like FEN1, for flap cleavage during Okazaki fragment maturation. J Biol Chem, 2004. 279: p. 50840-9. [CrossRef]
- Kao, H.I. and R.A. Bambara. The protein components and mechanism of eukaryotic Okazaki fragment maturation. Crit Rev Biochem Mol Biol, 2003. 38: p. 433-52. [CrossRef]
- Williams, J.S., P.P. Tumbale, M.E. Arana, J.A. Rana, R.S. Williams, and T.A. Kunkel. High-fidelity DNA ligation enforces accurate Okazaki fragment maturation during DNA replication. Nature Communications, 2021. 12: p. 482. [CrossRef]
- Beattie, T.R. and S.D. Bell. The role of the DNA sliding clamp in Okazaki fragment maturation in archaea and eukaryotes. Biochem Soc Trans, 2011. 39: p. 70-6. [CrossRef]
- Maga, G., G. Villani, V. Tillement, M. Stucki, G.A. Locatelli, I. Frouin, S. Spadari, and U. Hübscher. Okazaki fragment processing: modulation of the strand displacement activity of DNA polymerase delta by the concerted action of replication protein A, proliferating cell nuclear antigen, and flap endonuclease-1. Proc Natl Acad Sci USA, 2001. 98: p. 14298-14303. [CrossRef]
- Lin, S.H., X. Wang, S. Zhang, Z. Zhang, E.Y. Lee, and M.Y.W.T. Lee. Dynamics of enzymatic interactions during short flap human Okazaki fragment processing by two forms of human DNA polymerase δ. DNA Repair (Amst), 2013. 12: p. 922-35. [CrossRef]
- Ayyagari, R., X.V. Gomes, D.A. Gordenin, and P.M. Burgers. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J Biol Chem, 2003. 278: p. 1618-25. [CrossRef]
- Bastos de Oliveira, F.M., D. Kim, J.R. Cussiol, J. Das, M.C. Jeong, L. Doerfler, K.H. Schmidt, H. Yu, and M.B. Smolka. Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell, 2015. 57: p. 1124-1132. [CrossRef]
- Mutreja, K., J. Krietsch, J. Hess, S. Ursich, M. Berti, F.K. Roessler, R. Zellweger, M. Patra, G. Gasser, and M. Lopes. ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep, 2018. 24: p. 2629-2642.e5. [CrossRef]
- Lanz, M.C., S. Oberly, E.J. Sanford, S. Sharma, A. Chabes, and M.B. Smolka. Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev, 2018. 32: p. 822-835. [CrossRef]
- Michael, W.M., R. Ott, E. Fanning, and J. Newport. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science, 2000. 289: p. 2133-2137. [CrossRef]
- Yan, S. Yan, S. and W.M. Michael. TopBP1 and DNA polymerase alpha-mediated recruitment of the 9-1-1 complex to stalled replication forks: implications for a replication restart-based mechanism for ATR checkpoint activation. Cell Cycle, 2009. 8: p. 2877-84. [CrossRef]
- Saldivar, J.C., D. Cortez, and K.A. Cimprich. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol, 2017. 18: p. 622-636. [CrossRef]
- Hadjicharalambous, A., A.J. Whale, G. Can, J.M. Skehel, J.M. Houseley, and P. Zegerman. Checkpoint kinase interaction with DNA polymerase alpha regulates replication progression during stress. Wellcome Open Res, 2023. 8: p. 327. [CrossRef]
- McClure, A.W., B. Canal, and J.F.X. Diffley. A DNA replication fork-centric view of the budding yeast DNA damage response. DNA Repair (Amst), 2022. 119: p. 103393. [CrossRef]
- Nam, E.A. and D. Cortez. ATR signalling: more than meeting at the fork. Biochem J, 2011. 436: p. 527-36. [CrossRef]
- Yan, S. and W.M. Michael. TopBP1 and DNA polymerase-alpha directly recruit the 9-1-1 complex to stalled DNA replication forks. J Cell Biol, 2009. 184: p. 793-804. [CrossRef]
- Labib, K. and A. Gambus. A key role for the GINS complex at DNA replication forks. Trends Cell Biol, 2007. 17: p. 271-8. [CrossRef]
- O’Donnell, M. and H. Li. The Eukaryotic Replisome Goes Under the Microscope. Curr Biol, 2016. 26: p. R247-56. [CrossRef]
- Grabarczyk, D.B. The Fork Protection Complex: A Regulatory Hub at the Head of the Replisome. Subcell Biochem, 2022. 99: p. 83-107. [CrossRef]
- Bell, S.P. and K. Labib. Chromosome Duplication in Saccharomyces cerevisiae. Genetics, 2016. 203: p. 1027-67. [CrossRef]
- Liao, H., F. Ji, T. Helleday, and S. Ying. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep, 2018. 19. [CrossRef]
- Simoneau, A. and L. Zou. An extending ATR-CHK1 circuitry: the replication stress response and beyond. Curr Opin Genet Dev, 2021. 71: p. 92-98. [CrossRef]
- Bainbridge, L.J., R. Teague, and A.J. Doherty. Repriming DNA synthesis: an intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Res, 2021. 49: p. 4831-4847. [CrossRef]
- Kieffer, S.R. and N.F. Lowndes. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front Genet, 2022. 13: p. 793884. [CrossRef]
- Hustedt, N., S.M. Gasser, and K. Shimada. Replication checkpoint: tuning and coordination of replication forks in s phase. Genes (Basel), 2013. 4: p. 388-434. [CrossRef]
- Tye, S., G.E. Ronson, and J.R. Morris. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin Cell Dev Biol, 2021. 113: p. 14-26. [CrossRef]
- Kondratick, C.M., M.T. Washington, and M. Spies. Making Choices: DNA Replication Fork Recovery Mechanisms. Semin Cell Dev Biol, 2021. 113: p. 27-37. [CrossRef]
- Petermann, E. and K.W. Caldecott. Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle, 2006. 5: p. 2203-9. [CrossRef]
- Castaneda, J.C., M. Schrecker, D. Remus, and R.K. Hite. Mechanisms of loading and release of the 9-1-1 checkpoint clamp. Nat Struct Mol Biol, 2022. 29: p. 369-375. [CrossRef]
- Majka, J., S.K. Binz, M.S. Wold, and P.M. Burgers. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J Biol Chem, 2006. 281: p. 27855-61. [CrossRef]
- Majka, J. and P.M. Burgers. Function of Rad17/Mec3/Ddc1 and its partial complexes in the DNA damage checkpoint. DNA Repair (Amst), 2005. 4: p. 1189-94. [CrossRef]
- Majka, J. and P.M. Burgers. The PCNA-RFC families of DNA clamps and clamp loaders. Prog Nucleic Acid Res Mol Biol, 2004. 78: p. 227-60. [CrossRef]
- Green, C.M., H. Erdjument-Bromage, P. Tempst, and N.F. Lowndes. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr Biol, 2000. 10: p. 39-42. [CrossRef]
- Van, C., S. Yan, W.M. Michael, S. Waga, and K.A. Cimprich. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J Cell Biol, 2010. 189: p. 233-46. [CrossRef]
- Mordes, D.A., G.G. Glick, R. Zhao, and D. Cortez. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev, 2008. 22: p. 1478-89. [CrossRef]
- Zabrady, K., A.W.H. Li, and A.J. Doherty. Mechanism of primer synthesis by Primase-Polymerases. Curr Opin Struct Biol, 2023. 82: p. 102652. [CrossRef]
- Wang, F., J.A. Stewart, C. Kasbek, Y. Zhao, W.E. Wright, and C.M. Price. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep, 2012. 2: p. 1096-103. [CrossRef]
- Stewart, J.A., F. Wang, M.F. Chaiken, C. Kasbek, P.D. Chastain, 2nd, W.E. Wright, and C.M. Price. Human CST promotes telomere duplex replication and general replication restart after fork stalling. Embo j, 2012. 31: p. 3537-49. [CrossRef]
- Blow, J.J., X.Q. Ge, and D.A. Jackson. How dormant origins promote complete genome replication. Trends Biochem Sci, 2011. 36: p. 405-14. [CrossRef]
- Blow, J.J. and X.Q. Ge. Replication forks, chromatin loops and dormant replication origins. Genome Biol, 2008. 9: p. 244. [CrossRef]
- Woodward, A.M., T. Göhler, M.G. Luciani, M. Oehlmann, X. Ge, A. Gartner, D.A. Jackson, and J.J. Blow. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol, 2006. 173: p. 673-83. [CrossRef]
- Pabla, N., S. Huang, Q.S. Mi, R. Daniel, and Z. Dong. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem, 2008. 283: p. 6572-83. [CrossRef]
- Toledo, L.I., M. Murga, P. Gutierrez-Martinez, R. Soria, and O. Fernandez-Capetillo. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev, 2008. 22: p. 297-302. [CrossRef]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol, 1973. 41: p. 181-90. [CrossRef]
- de Lange, T. Protection of mammalian telomeres. Oncogene, 2002. 21: p. 532-540. [CrossRef]
- Takai, H., V. Aria, P. Borges, J.T.P. Yeeles, and T. de Lange. CST-Polymeraseα-primase solves a second telomere end-replication problem. bioRxiv, 2023. [CrossRef]
- Lim, C.J. and T.R. Cech. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat Rev Mol Cell Biol, 2021. 22: p. 283-298. [CrossRef]
- Greider, C.W. and E.H. Blackburn. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell, 1987. 51: p. 887-98. [CrossRef]
- Szostak, J.W. and E.H. Blackburn. Cloning yeast telomeres on linear plasmid vectors. Cell, 1982. 29: p. 245-55. [CrossRef]
- Ohki, R., T. Tsurimoto, and F. Ishikawa. In vitro reconstitution of the end replication problem. Mol Cell Biol, 2001. 21: p. 5753-66. [CrossRef]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu Rev Genet, 2018. 52: p. 223-247. [CrossRef]
- Ganduri, S. and N.F. Lue. STN1-POLA2 interaction provides a basis for primase-pol α stimulation by human STN1. Nucleic Acids Research, 2017. 45: p. 9455-9466. [CrossRef]
- Anderson, B.H., P.R. Kasher, J. Mayer, M. Szynkiewicz, E.M. Jenkinson, S.S. Bhaskar, J.E. Urquhart, S.B. Daly, J.E. Dickerson, J. O’Sullivan, E.O. Leibundgut, J. Muter, G.M. Abdel-Salem, R. Babul-Hirji, P. Baxter, A. Berger, L. Bonafé, J.E. Brunstom-Hernandez, J.A. Buckard, D. Chitayat, W.K. Chong, D.M. Cordelli, P. Ferreira, J. Fluss, E.H. Forrest, E. Franzoni, C. Garone, S.R. Hammans, G. Houge, I. Hughes, S. Jacquemont, P.Y. Jeannet, R.J. Jefferson, R. Kumar, G. Kutschke, S. Lundberg, C.M. Lourenço, R. Mehta, S. Naidu, K.K. Nischal, L. Nunes, K. Ounap, M. Philippart, P. Prabhakar, S.R. Risen, R. Schiffmann, C. Soh, J.B. Stephenson, H. Stewart, J. Stone, J.L. Tolmie, M.S. van der Knaap, J.P. Vieira, C.N. Vilain, E.L. Wakeling, V. Wermenbol, A. Whitney, S.C. Lovell, S. Meyer, J.H. Livingston, G.M. Baerlocher, G.C. Black, G.I. Rice, and Y.J. Crow. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet, 2012. 44: p. 338-42. [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov, 2022. 12: p. 31-46. [CrossRef]
- Hanahan, D. and R.A. Weinberg. Hallmarks of cancer: the next generation. Cell, 2011. 144: p. 646-74. [CrossRef]
- Hanahan, D. and R.A. Weinberg. The hallmarks of cancer. Cell, 2000. 100: p. 57-70. [CrossRef]
- Fenwick, A.L., M. Kliszczak, F. Cooper, J. Murray, L. Sanchez-Pulido, S.R. Twigg, A. Goriely, S.J. McGowan, K.A. Miller, I.B. Taylor, C. Logan, S. Bozdogan, S. Danda, J. Dixon, S.M. Elsayed, E. Elsobky, A. Gardham, M.J. Hoffer, M. Koopmans, D.M. McDonald-McGinn, G.W. Santen, R. Savarirayan, D. de Silva, O. Vanakker, S.A. Wall, L.C. Wilson, O.O. Yuregir, E.H. Zackai, C.P. Ponting, A.P. Jackson, A.O. Wilkie, W. Niedzwiedz, and L.S. Bicknell. Mutations in CDC45, Encoding an Essential Component of the Pre-initiation Complex, Cause Meier-Gorlin Syndrome and Craniosynostosis. Am J Hum Genet, 2016. 99: p. 125-38. [CrossRef]
- Piersimoni, L., P.L. Kastritis, C. Arlt, and A. Sinz. Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein–Protein Interactions─A Method for All Seasons. Chemical Reviews, 2022. 122: p. 7500-7531. [CrossRef]
- Munden, A., M.T. Wright, D. Han, R. Tirgar, L. Plate, and J.T. Nordman. Identification of replication fork-associated proteins in Drosophila embryos and cultured cells using iPOND coupled to quantitative mass spectrometry. Sci Rep, 2022. 12: p. 6903. [CrossRef]
- Lenz, S., L.R. Sinn, F.J. O’Reilly, L. Fischer, F. Wegner, and J. Rappsilber. Reliable identification of protein-protein interactions by crosslinking mass spectrometry. Nature Communications, 2021. 12: p. 3564. [CrossRef]
- Ho, B., A. Baryshnikova, and G.W. Brown. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst, 2018. 6: p. 192-205.e3. [CrossRef]
- Petryk, N., M. Kahli, Y. d’Aubenton-Carafa, Y. Jaszczyszyn, Y. Shen, M. Silvain, C. Thermes, C.L. Chen, and O. Hyrien. Replication landscape of the human genome. Nat Commun, 2016. 7: p. 10208. [CrossRef]
- Tanigawa, K., Y. Tomioka, S. Misono, S. Asai, N. Kikkawa, Y. Hagihara, T. Suetsugu, H. Inoue, K. Mizuno, and N. Seki. Minichromosome maintenance proteins in lung adenocarcinoma: Clinical significance and therapeutic targets. FEBS Open Bio, 2023. 13: p. 1737-1755. [CrossRef]
- Mukherjee, G., B. Muralidhar, U.D. Bafna, R.A. Laskey, and N. Coleman. MCM immunocytochemistry as a first line cervical screening test in developing countries: a prospective cohort study in a regional cancer centre in India. Br J Cancer, 2007. 96: p. 1107-11. [CrossRef]
- Scarpini, C., V. White, B. Muralidhar, A. Patterson, N. Hickey, N. Singh, J. Mullerat, M. Winslet, R.J. Davies, M.L. Phillips, P. Stacey, R.A. Laskey, R. Miller, M. Nathan, and N. Coleman. Improved screening for anal neoplasia by immunocytochemical detection of minichromosome maintenance proteins. Cancer Epidemiol Biomarkers Prev, 2008. 17: p. 2855-64. [CrossRef]
- Simon, N.E. and A. Schwacha. The Mcm2-7 replicative helicase: a promising chemotherapeutic target. Biomed Res Int, 2014. 2014: p. 549719. [CrossRef]
- Montagnoli, A., P. Tenca, F. Sola, D. Carpani, D. Brotherton, C. Albanese, and C. Santocanale. Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res, 2004. 64: p. 7110-7116. [CrossRef]
- Swords, R., D. Mahalingam, M. O’Dwyer, C. Santocanale, K. Kelly, J. Carew, and F. Giles. Cdc7 kinase—a new target for drug development. Eur J Cancer, 2010. 46: p. 33-40. [CrossRef]
- Menichincheri, M., A. Bargiotti, J. Berthelsen, J.A. Bertrand, R. Bossi, A. Ciavolella, A. Cirla, C. Cristiani, V. Croci, R. D’Alessio, M. Fasolini, F. Fiorentini, B. Forte, A. Isacchi, K. Martina, A. Molinari, A. Montagnoli, P. Orsini, F. Orzi, E. Pesenti, D. Pezzetta, A. Pillan, I. Poggesi, F. Roletto, A. Scolaro, M. Tatò, M. Tibolla, B. Valsasina, M. Varasi, D. Volpi, C. Santocanale, and E. Vanotti. First Cdc7 kinase inhibitors: pyrrolopyridinones as potent and orally active antitumor agents. 2. Lead discovery. J Med Chem, 2009. 52: p. 293-307. [CrossRef]
- Iwai, K., T. Nambu, R. Dairiki, M. Ohori, J. Yu, K. Burke, M. Gotou, Y. Yamamoto, S. Ebara, S. Shibata, R. Hibino, S. Nishizawa, T. Miyazaki, M. Homma, Y. Oguro, T. Imada, N. Cho, N. Uchiyama, A. Kogame, T. Takeuchi, O. Kurasawa, K. Yamanaka, H. Niu, and A. Ohashi. Molecular mechanism and potential target indication of TAK-931, a novel CDC7-selective inhibitor. Sci Adv, 2019. 5: p. eaav3660. [CrossRef]
Figure 2.
Leading and lagging strand synthesis at a eukaryotic replication fork. In this RF model, CMG helicase (CDC45-MCM2-7-GINS with CDC45 in dark blue, the MCM2-7 hexamer in purple, and GINS in light green) unwinds the parental dsDNA into the leading and lagging strand templates (dark-red and dark blue, respectively). The protein Donson associates with the CMG complex during its formation and remains attached to it during the unwinding reaction. Additionally, the diagram shows the replication proteins that are involved in DNA synthesis and maturation of Okazaki fragments. As seen in the model, RPA heterotrimers (three shades of blue) bind to the unwound ssDNAs preventing hairpin formation and nuclease-dependent ssDNA degradation. RFC (blue) loads the PCNA ring (red brown) onto primed template DNA. The latter stabilises Pol ε (light blue) on the template DNA when synthesising the leading strand (light blue DNA). Pol ε also associates with the CMG complex to support its unwinding activity but this interaction might also be important during replication fork stalling (see Section 5). For lagging strand DNA synthesis, the AND-1/CTF4/WDHD1 homotrimer (named AND-1 in the diagram with one subunit consisting of an HMG (green), SepB (dark blue), and WD (blue) domain) links CMG to the Pol α complex (green). The primase function of Pol α synthesises the RNA primer (light grey) starting Okazaki fragment synthesis during lagging strand synthesis. After the initiation step, primase hands over the RNA primer to the DNA polymerase domain of Pol ⍺ on PolA1 (first polymerase transition). The latter extends the RNA primer and synthesises a short RNA-DNA fragment before leaving the template. RFC (blue) replaces Pol ⍺ with the help of RPA and loads PCNA, the DNA clamp, on the primed DNA. This RFC-PCNA complex allows Pol δ (pink) to associate with the RNA-DNA primer (2nd polymerase transition). The RFC-PCNA-Pol δ complex elongates this RNA-DNA in a processive manner until it reaches the next Okazaki fragment. Then Pol δ slows down but continues to elongate the newly synthesised DNA. The polymerase displaces the RNA and parts of the Pol α-synthesised DNA of the Okazaki fragment in front (strand displacement). Thus, Pol δ produces an RNA-DNA flap, which is recognised and cleaved by PCNA-associated FEN1 (blue), creating a perfect product, nicked DNA, for LIG1 (top panel on the left; the two inserted panels provide insights into the different pathways of Okazaki fragment maturation process). The DNA ligase LIG1, which is also bound to PCNA along with Pol δ and FEN1, then ligates the two DNA fragments yielding a continuous stretch of DNA. In an alternate pathway, RPA binds the flap structure produced by Pol δ competing with FEN1 (lower inserted panel). RPA recruits the DNA2 helicase/endonuclease to the flap structure. The latter in turn cleaves the ssDNA but leaving an extra nucleotide remaining, which results in a product that LIG1 does not ligate. After RPA and DNA2 left the DNA, FEN1 cuts off the remaining base and LIG1 ligates the two DNA fragments. Adapted from [2,60,62,80] and created with BioRender.com.
Figure 2.
Leading and lagging strand synthesis at a eukaryotic replication fork. In this RF model, CMG helicase (CDC45-MCM2-7-GINS with CDC45 in dark blue, the MCM2-7 hexamer in purple, and GINS in light green) unwinds the parental dsDNA into the leading and lagging strand templates (dark-red and dark blue, respectively). The protein Donson associates with the CMG complex during its formation and remains attached to it during the unwinding reaction. Additionally, the diagram shows the replication proteins that are involved in DNA synthesis and maturation of Okazaki fragments. As seen in the model, RPA heterotrimers (three shades of blue) bind to the unwound ssDNAs preventing hairpin formation and nuclease-dependent ssDNA degradation. RFC (blue) loads the PCNA ring (red brown) onto primed template DNA. The latter stabilises Pol ε (light blue) on the template DNA when synthesising the leading strand (light blue DNA). Pol ε also associates with the CMG complex to support its unwinding activity but this interaction might also be important during replication fork stalling (see Section 5). For lagging strand DNA synthesis, the AND-1/CTF4/WDHD1 homotrimer (named AND-1 in the diagram with one subunit consisting of an HMG (green), SepB (dark blue), and WD (blue) domain) links CMG to the Pol α complex (green). The primase function of Pol α synthesises the RNA primer (light grey) starting Okazaki fragment synthesis during lagging strand synthesis. After the initiation step, primase hands over the RNA primer to the DNA polymerase domain of Pol ⍺ on PolA1 (first polymerase transition). The latter extends the RNA primer and synthesises a short RNA-DNA fragment before leaving the template. RFC (blue) replaces Pol ⍺ with the help of RPA and loads PCNA, the DNA clamp, on the primed DNA. This RFC-PCNA complex allows Pol δ (pink) to associate with the RNA-DNA primer (2nd polymerase transition). The RFC-PCNA-Pol δ complex elongates this RNA-DNA in a processive manner until it reaches the next Okazaki fragment. Then Pol δ slows down but continues to elongate the newly synthesised DNA. The polymerase displaces the RNA and parts of the Pol α-synthesised DNA of the Okazaki fragment in front (strand displacement). Thus, Pol δ produces an RNA-DNA flap, which is recognised and cleaved by PCNA-associated FEN1 (blue), creating a perfect product, nicked DNA, for LIG1 (top panel on the left; the two inserted panels provide insights into the different pathways of Okazaki fragment maturation process). The DNA ligase LIG1, which is also bound to PCNA along with Pol δ and FEN1, then ligates the two DNA fragments yielding a continuous stretch of DNA. In an alternate pathway, RPA binds the flap structure produced by Pol δ competing with FEN1 (lower inserted panel). RPA recruits the DNA2 helicase/endonuclease to the flap structure. The latter in turn cleaves the ssDNA but leaving an extra nucleotide remaining, which results in a product that LIG1 does not ligate. After RPA and DNA2 left the DNA, FEN1 cuts off the remaining base and LIG1 ligates the two DNA fragments. Adapted from [2,60,62,80] and created with BioRender.com.
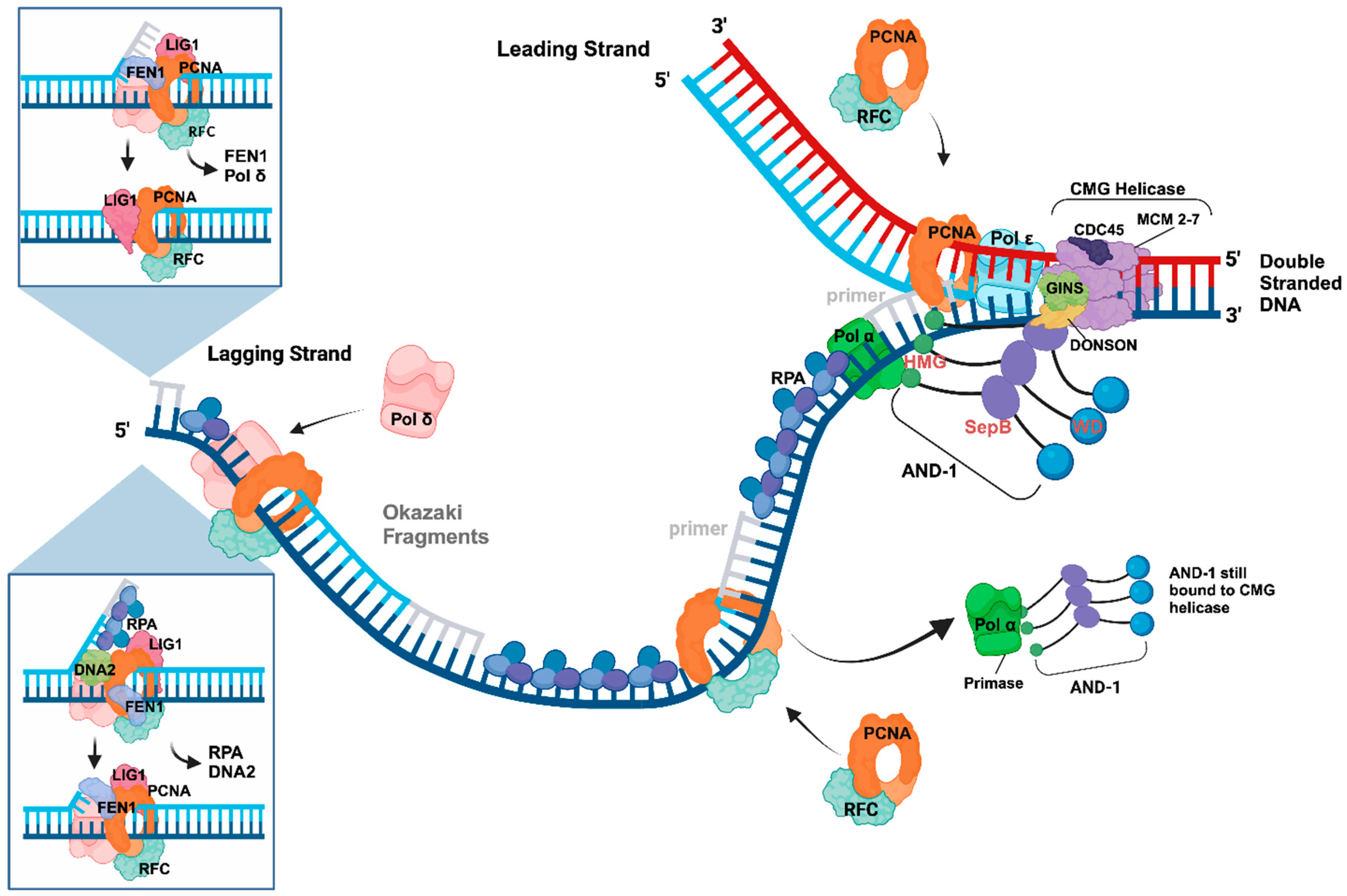
Figure 3.
Signalling at unperturbed and perturbed replication forks. In these simplified RF models, ATR/MEC1 and CHK1/RAD53 kinases initiate signalling pathways at RFs prior to and after passing damaged DNA resulting in a stalled leading strand synthesis, are summarised in panel A,B, respectively. During normal, unperturbed replication processes on leading and lagging strand, the Okazaki fragment synthesis on the lagging strand produces a signal via ATR/MEC1 (light red) and CHK1/RAD53 to slow down the RF via modulating e.g., the CMG helicase, to synchronise DNA synthesis and nucleotide synthesis (panel A). This ATR/MEC1 activity requires Pol α and DNA2 (Okazaki fragment initiation and maturation) and signals unperturbed RFs [111,113]. When Pol ε on the leading strand encounters a DNA lesion or is exposed to nucleotide depletion, the enzyme stops DNA synthesis and disengages with or modulates the CMG complex. Then the latter continues to unwind DNA yielding stretches of ssDNA bound to RPA. These RPA-ssDNA structures recruit ATRIP and ATR (dark red) to the leading strand in addition to ATR’s binding via TOPBP1, its main activator, to the lagging strand with Pol α and 9-1-1 as partners (panel B). Additionally, binding of Pol ε to TOPBP1, which may occur via a binding site of Pol ε to TOPBP1 hidden when associated with the CMG complex, may enhance this DDR signalling. It is important to note that Pol α and the RNA primer synthesis are key for the initiation of replication stress signals via ATR/MEC1 and that at RFs Pol α does not synthesise RNA primers on the leading strand [62,71,72,114,115]. The brown and red arrows indicate ATR/MEC1 and CHK1/RAD53 signalling. This model suggests that ATR signalling requires multiple key partners located on both template strands of a stalled RF. Importantly, on the sequence level, human and yeast CHK1 are homologues whereas human CHK2 is the orthologue of yeast RAD53, but regarding the ATR pathway during replication stress, human CHK1 and RAD53 are functionally equivalent [116]. The figure is created by Bio Render using published results [62,71,72,111,113,114,116,117,118,119,120].
Figure 3.
Signalling at unperturbed and perturbed replication forks. In these simplified RF models, ATR/MEC1 and CHK1/RAD53 kinases initiate signalling pathways at RFs prior to and after passing damaged DNA resulting in a stalled leading strand synthesis, are summarised in panel A,B, respectively. During normal, unperturbed replication processes on leading and lagging strand, the Okazaki fragment synthesis on the lagging strand produces a signal via ATR/MEC1 (light red) and CHK1/RAD53 to slow down the RF via modulating e.g., the CMG helicase, to synchronise DNA synthesis and nucleotide synthesis (panel A). This ATR/MEC1 activity requires Pol α and DNA2 (Okazaki fragment initiation and maturation) and signals unperturbed RFs [111,113]. When Pol ε on the leading strand encounters a DNA lesion or is exposed to nucleotide depletion, the enzyme stops DNA synthesis and disengages with or modulates the CMG complex. Then the latter continues to unwind DNA yielding stretches of ssDNA bound to RPA. These RPA-ssDNA structures recruit ATRIP and ATR (dark red) to the leading strand in addition to ATR’s binding via TOPBP1, its main activator, to the lagging strand with Pol α and 9-1-1 as partners (panel B). Additionally, binding of Pol ε to TOPBP1, which may occur via a binding site of Pol ε to TOPBP1 hidden when associated with the CMG complex, may enhance this DDR signalling. It is important to note that Pol α and the RNA primer synthesis are key for the initiation of replication stress signals via ATR/MEC1 and that at RFs Pol α does not synthesise RNA primers on the leading strand [62,71,72,114,115]. The brown and red arrows indicate ATR/MEC1 and CHK1/RAD53 signalling. This model suggests that ATR signalling requires multiple key partners located on both template strands of a stalled RF. Importantly, on the sequence level, human and yeast CHK1 are homologues whereas human CHK2 is the orthologue of yeast RAD53, but regarding the ATR pathway during replication stress, human CHK1 and RAD53 are functionally equivalent [116]. The figure is created by Bio Render using published results [62,71,72,111,113,114,116,117,118,119,120].
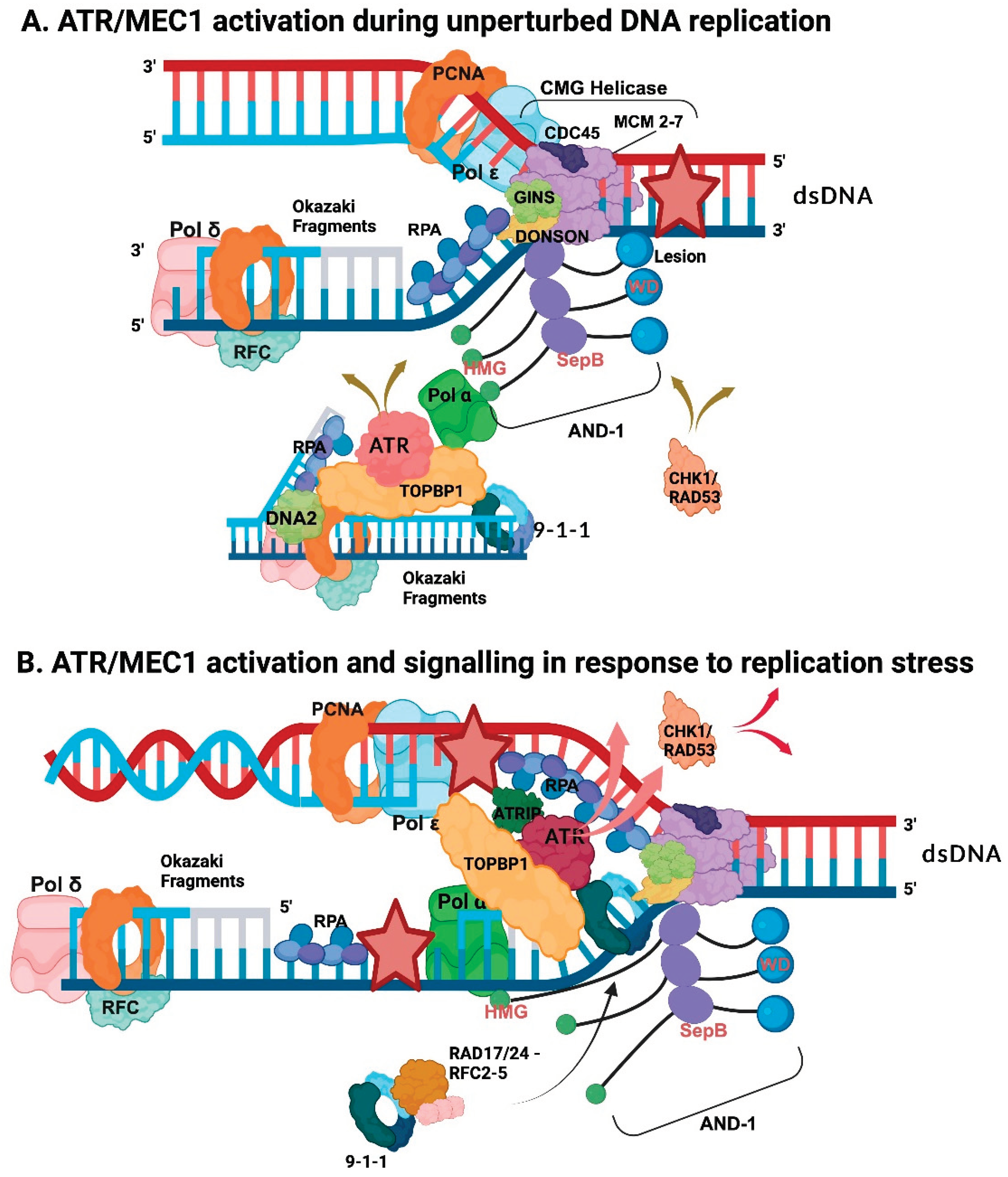
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



