
Submitted:
22 February 2024
Posted:
23 February 2024
You are already at the latest version
Abstract
Now day; the interest for the synthesis of heterocyclic compounds containing hydroquinoline fragments has increased tremendously due to their wide variety of pharmaceutical and industrial applications. This work details the synthesis of linear and fused heterocyclic systems containing hydroquinoline fragments and the pharmacological activity spectra of the synthesized compounds was predicted by in silico method using Prediction of Activity Spectra of Substances (PASS) program. 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5a was reacted for three hours with a system of hydroxylammonium chloride/pyridinium chloride/toluene to produce 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-nitrile 7. N-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbaldehyde 6b and iodine reacted at room temperature in aqueous ammonia water, followed by aq. treatment with Na2S2O3 and produced N-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-nitrile 8. 30-60% 2-phenyl-1,3-oxazol-5(4H)-ones 9a,b; 10a,b were synthesized via the condensation reaction of N-alkylhydroquinoline-6-carbaldehydes 5a,b; 6a,b with hippuric acid in acetic acid. When activated 7-methylazolopyrimidines 11a,b were reacted with N-alkyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbaldehydes 6a,b, the reaction produced 60–70% yield of 7-[(E)-2,2,4-trimethylhydroquinolin-6-ylidenemethyl]azolo[1,5-a]pyrimidin-6-yl carboxylic acids 12a,b. The condensation reaction of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 3h with dimethylacetylenedicarboxylate (DMAD) and ethylacetoacetate afforded methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16 and 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17, respectively. The condensation reaction of 6-formyl-7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 5e with methylene active compounds ethylcyanoacetate/dimethyl-3-oxopentanedioate/ ethylacetoacetate/diethylmalonate/Meldrum’s acid afforded 3-substituted dihydroquinoline containg coumarins 19 & 21. A pentacyclic coumarin 22 was obtained via the random condensation reaction of malononitrile with 5e in the presence of catalytic amount of piperidine in ethanol. The potential biological activities of the synthesized compounds were evaluated using PASS computer program. According to the prognosis, (E)-4-(N-alkylhydroquinolin-6-yl)-3-buten-2-ones, 13a, b & 14 showed high probability of being active (Pa) as the Gluconate 2-dehydrogenase inhibitor, Anti-allergic, Anti-asthmatic, and Anti-arthritic with the Pa value of 0.849-0.870. It was also found out that hydroquinolinecarbonitrile compounds 7 & 8 have a propensity to act as a good progesterone antagonists, anti-allergic, anti-asthmatic, and anti-arthritic (Pa = 0.276-0.827). Among the coumarin moieties containing hydroquinolines, compounds 17, 19a, and 19c were predicted to be good progesterone antagonists with the Pa values of 0.710, 0.630 and 0.615; respectively.
Keywords:
Heterocyclic Compound
; Hydroquinoline
; Vilsmeier-Haack Formylation
; Quinoline
1. Introduction
Nowadays, heterocyclic compounds have gained a great deal of research interest because of their numerous significant medical and biological applications [1,2,3,4]. Quinoline derivatives belong to the N-containing heterocyclic compounds which are known for their applications in medicinal chemistry such as antimalarial, anti-inflammatory agents, anti-asthmatic, anti- bacterial, antihypertensive, and tyrosine kinase inhibiting agents [5,6,7,8,9,10,11]. Hence, the interest for the synthesis of heterocyclic compounds containing a quinoline scaffold has increased tremendously [12,13,14,15].
Alkyl derivatives of 1,2-H-dihydroquinoline belong to a large group of quinoline derivatives of great practical importance [16,17,18]. 2,2,4-trimethyl-1,2-H-dihydroquinoline is one of the most important representative, which is considered as a essential and effective anti-oxidant in rubber technologies [19]. The methods of the synthesis of 1,2-quinoline derivatives are based on the cyclization reaction, well known as Skraup reaction and various Skraup-based modification reactions such as Combes, Knorr, Doebner and Friedlander [12,17,19,20,21]. Tetrahydroquinoline is one of the most important simple nitrogen heterocycles, being widespread in nature and present in a broad variety of pharmacologically active compounds [22,23].
It is found out that heterocyclic compounds containing hydroquinoline derivatives are the basis for a wide range of biological activities [24,25,26] and also have industrial applications such as photo sensitizers in solar cells [27,28,29] and additives to polymers [30,31]. Hence, the construction of new heterocyclic assemblies and functionalization of hydroquinoline compounds have attracted the attention of synthetic organic chemists to further realize their versatile potential. Even though the methods of synthesis of annulated heterocyclic systems containing s1,2-dihydro- and 1,2,3,4-tetrahydroquinoline moieties are stated in the literature, the problem of synthesizing new classes of linear and fused heterocyclic systems with hydroquinoline fragment including those containing a chiral center in a certain configuration and various substituent’s, has not been completely solved.
In-silico studies are becoming more important in drug development from its initial stage to the approval of the drug. Using in silico tools to predict the biological activity and/or ADMET target for novel compounds has been a wide practice as it helps in decreasing the cost/time related to drug discovery [32,33].
Hence, in this study, the synthesis of new heterocyclic compounds containing hydroquinoline moieties such as carbonitriles, oxazolones, pyrazolo/triazolo[1,5-a]pyrimidines, and coumarins based on formylhydroquinolines was described. Moreover, the pharmacological activity spectrum of the synthesized compounds were predicted by in-silico method using the prediction of activity spectrum of substances (PASS) computer program [32,33,34].
2. Results and Discussion
2.1. Chemistry
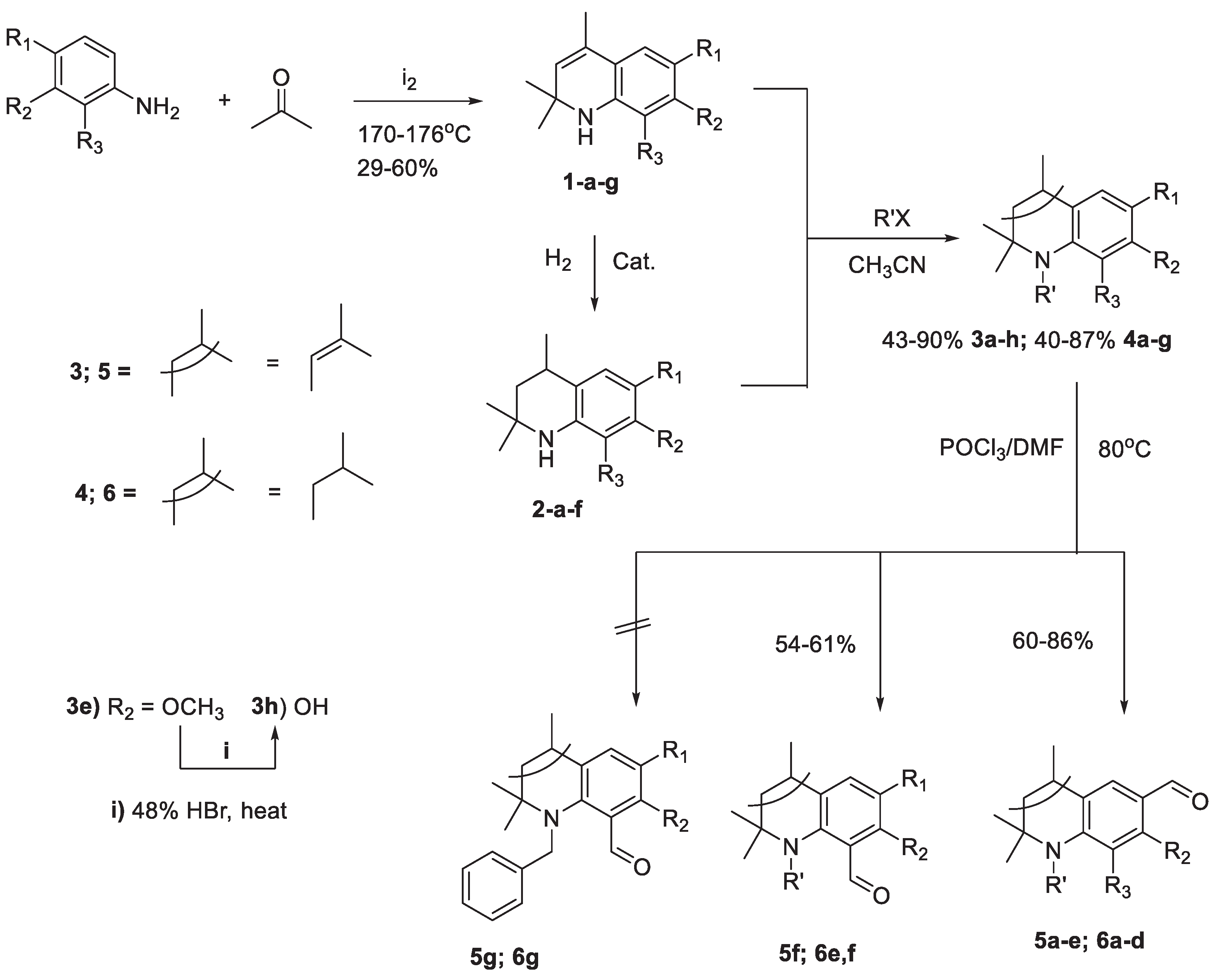
K. Tianet et. al. previously reported that the Vilsmeier—Haack (VH) formylation reaction of 1,2,2,4-tetramethyl-1,2-dihydroquinoline bearing unsubstituted benzene ring proceeded with the formation of 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde as the only product [35]. In our previous publication [36] and PhD dissertation [37], with the objective of broadening the scope of the synthesis of formyl hydroquinolines containing electron-donating substituents on the aromatic ring; we reported that the VH formylation reaction of 7(8)-substituted-N-alkyl-1,2-dihydro- and 1,2,3,4-tetrahydroquinolines leads to the exclusive formation of 6-formyl-7(8)-substituted- N-alkyl-1,2-dihydro- and 1,2,3,4-tetrahydroquinolines.
Whereas the Vilsmeier-Haack formylation reaction of 6-substituted-N-methyl-2,2,4-trimethyl-1,2-di(1,2,3,4-tetra)hydroquinolones afforded 8-formyl-6-substituted--1,2,2,4-tetramethyl-1,2-di(1,2,3,4-tetra)hydroquinolones, our attempt that was made to formylate 6-substituted-N-benzyl-2,2,4-trimethyl-1,2-dihydro(1,2,3,4-tetrahydro)quinolines to obtain 8-formyl-6-substituted-N-benzyl-2,2,4-trimethyl-1,2-di(1,2,3,4-tetra)hydroquinolines was not succeeded (Scheme 1 and Table 1). This can be explained mainly due to the steric impedance at the 8 position of the hydroquinoline ring which is introduced by the more bulky N-benzyl substituent compared to the methyl group.
2.1.1. Synthesis of N-alkyl-2,2,4-trimethylhydroquinolin-6-yl-carbonitriles 7, 8
One of the most important functional group transformations in organic synthesis is the conversion of aldehyde functional groups into nitriles. Nitriles are very versatile functional group that can be used as a starting material for the synthesis of a variety of biologically active compounds such as thiazole derivatives, oxazolines, imidazoles, triazolopyrimidines, benzamidines, etc [38]. In the majority of cases, nitriles are obtained via the initial formation of aldoxime which is obtained by the condensation reaction of hydroxylamine hydrochloride with aldehyde which is then subjected to dehydration effected by a variety of reagents to give nitriles. There are also a few methods known for the one-step conversion of aldehydes in to nitriles without separation of the aldoxime formed [38,39,40,41,42].
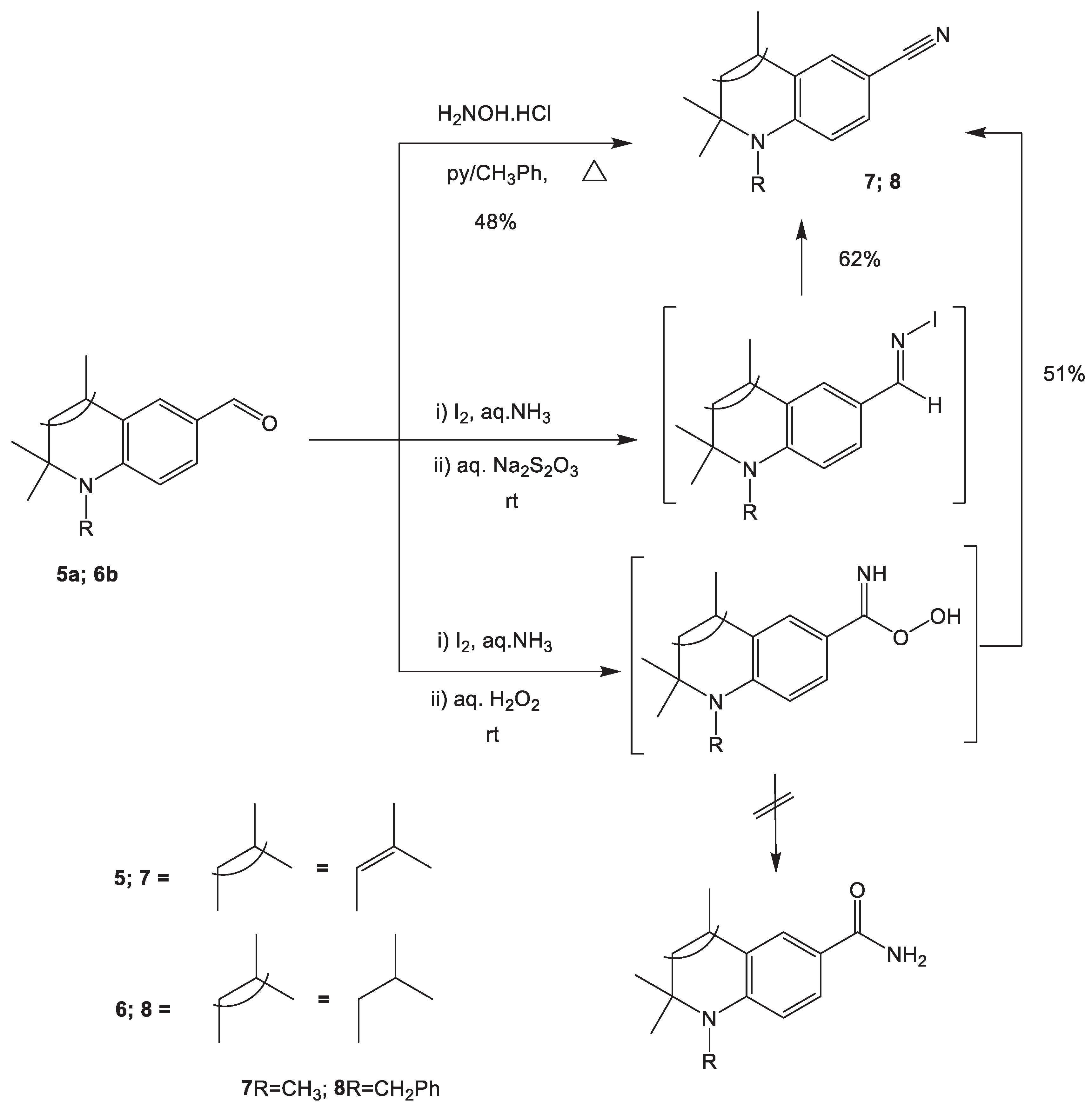
Hence, with the objective of further functionalizing the hydroquinoline carbaldehydes, we have attempted to synthesis carbonitrile via the two adopted methodologies. First, the hydroxylammonium chloride/pyridium/toluene system with the azeotropic separation of water was employed. The pyridinium chloride formed participates in the aldoxime-nitrile conversion; thus, the presence of a dehydrating agent is not necessary. The reaction of 6-formyl-N-methyl-2,2,4-trimethyl-1,2-dihydroquinoline 5a with H2NOH.HCl/C5H5N/C7H8 were completed within 3 hours with the reaction yield of 48% to afford 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-nitrile 7. The disappearance of the aldehydic proton signal that typically resonates in the range of 9.88 – 10.22 ppm on the1H-NMR spectrum of the starting material is evidence that the reaction has gone to completion (supporting information- Figure 1).
The second methodology we employed was via the treatment of the respective aldehyde with iodine in aqueous ammonia water at room temperature [42]. We speculated that the reaction proceeded via the oxidation of aldimine with iodine to give an N-iodoaldimine intermediate, which eliminated an HI molecule in ammonia solution to afford the nitrile product. It was found out that this procedure was applicable in our case using the 6-formyl-N-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline 6b that afforded N-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-nitrile 8 (Scheme 2 & supporting information- Figure 2).
We have also made an attempt to synthesis an amide from 6-formyl-N- benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline 6b with the treatment of aqueous hydrogen peroxide at room temperature, reportedly with the intermediate formation of peroxocarboxamic acid [43]. However, a 4 hour exposure to hydrogen peroxide at room temperature did not lead to the formation of the expected corresponding amide, yet the isolated compound was identified as the previously obtained nitrile 8. This might be due to the antioxidant properties of the tetrahydroquinoline fragment.
2.1.2. Synthesis of 2-Phenyl-1,3-oxazol-5(4Н)-ones 9, 10
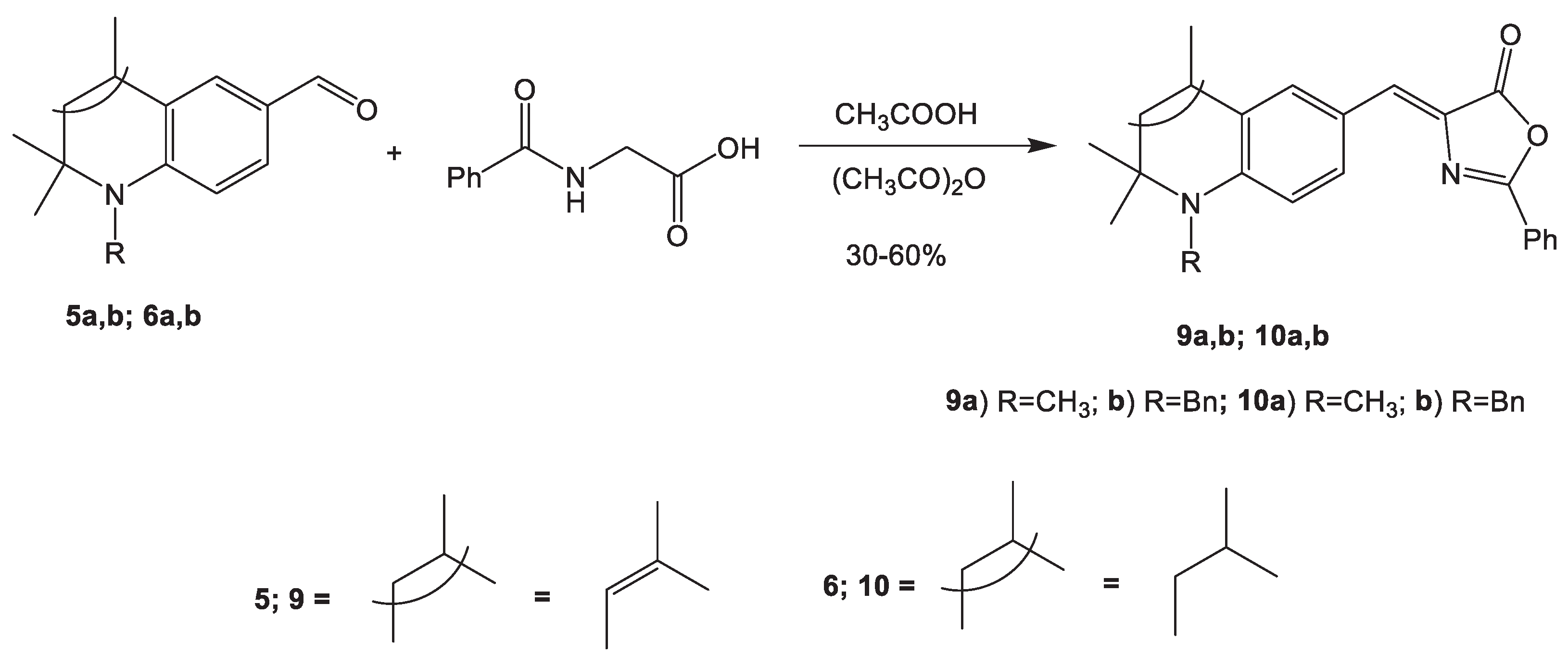
Benzoxazole derivatives have been reported in the literature that they possess anti-inflammatory, analgesic, anti-bacterial, trichomonacidal, anthelmintic, fungicidal, antiviral and kinase inhibition activities [44]. The method for the synthesis of 4-arylidene-2-aryloxazol-5-ones has been known for a long time and is widely used in synthetic organic chemistry, which consists in the condensation of aromatic/ heterocyclic/ aldehydes with hippuric acid or its heteroanalogues in glacial acetic acid in the presence of anhydrous sodium acetate [45]. In our case, we used a solution of acetic anhydride in acetic acid for the condensation of hydroquinolinecarbaldehydes 5, 6 with hippuric acid and we obtained 2-aryl-4-arylidene-5(4H)-oxazolones 9a,b; 10a,b with moderate yield of 30-60% (Scheme 3). Sidhu and coworkers confirmed that the 1H-NMR spectra of oxazolones show the deshielding influence of the cis-N=C-C6H5 moiety on the olefinic hydrogen atom which shifts more downfield than when the olefinic hydrogen is cis to the carbonyl group [45]. In our case, the olefinic protons were found to resonate in the chemical shift range of δ = 7.18-7.22 ppm, which is relatively up field than expected (nearly δ = 7.60-7.85 ppm). Hence, based on the 1H-NMR spectral data these compounds have been assigned the (Z) configuration at the 4 position (supporting information- Figures 3–6).
2.1.3. Condensation of Hydroquinolinecarbaldehydes with Reagents Containing an Activated Methyl Group 12, 13 14
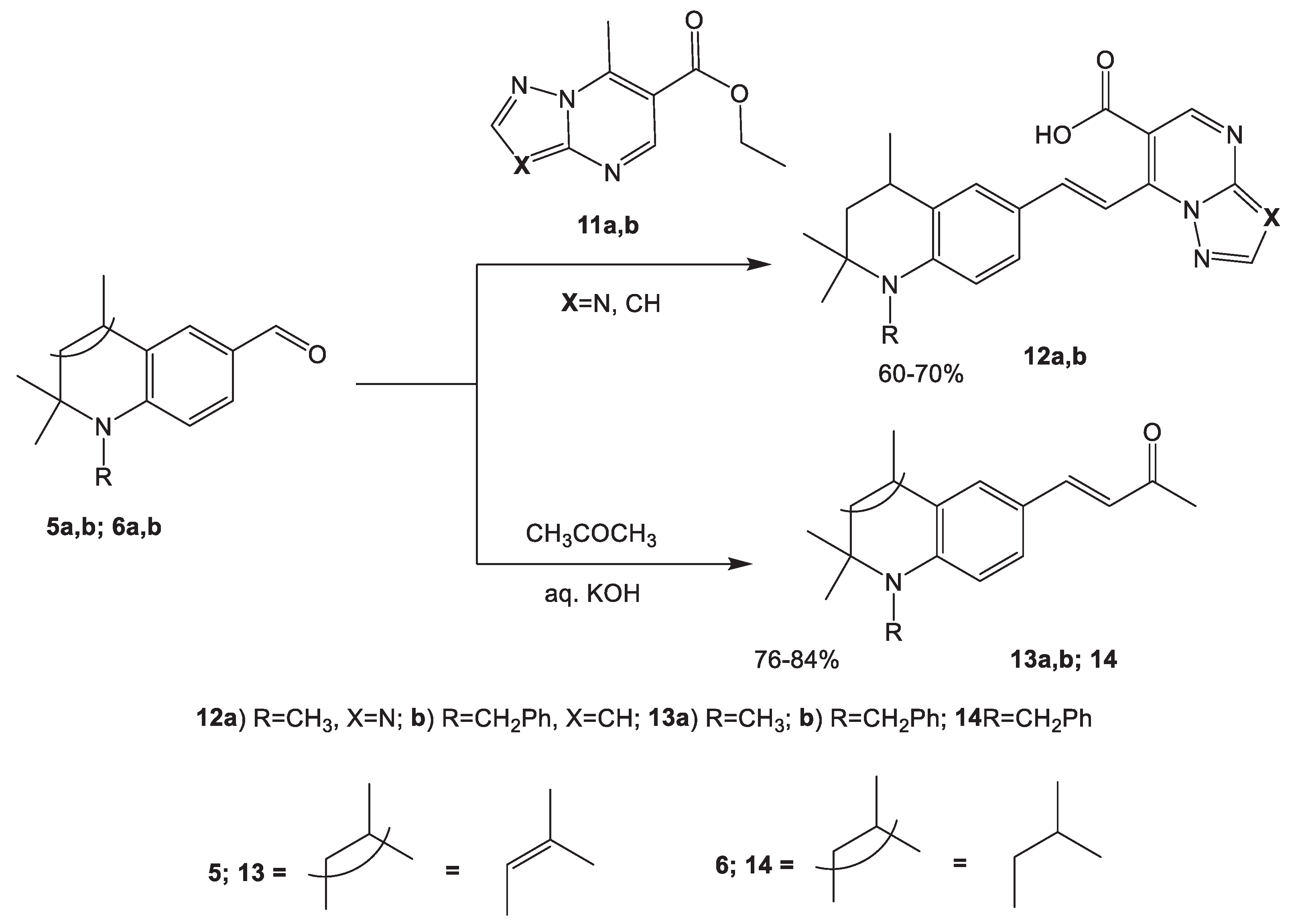
In order to search for potential biologically active compounds, we extended our work with the reaction of 6-formyl-N-alkyl-2,2,4-trimethyldihydroquinolines 5a,b; 6a,b with ethyl-7-methylpyrazolo/triazolo/[1,5-a]pyrimidine-6-carboxylates 11a,b (Scheme 4).
It was found out that the condensation of N-alkyl-1,2,3,4-tetrahydroquinoline-6-carbaldehydes 56a,b with 7-methylazolopyrimidines 11a,b occurs in the presence of the strong base potassium tert-butoxide and the reaction product is accompanied by the hydrolysis of the ester group under the action of the water released during condensation reaction. The downfield broad signals observed on the 1H-NMR spectra at 13.31 and 13.50 ppm are assignable to carboxylic acid protons of 12a, b, respectively (supporting information- Figure 7 & 8). The resulting 7-[(E)-2,2,4-trimethylhydroquinolin-6-ylidenemethyl]triazolo/pyrazolo[1,5-a]pyrimidin-6-yl carboxylic acids 12 a,b are red crystalline compounds which are sparingly soluble in water.
Analysis of the 1H NMR spectra of carboxylic acids 12, b showed that they are trans isomers. This is evidenced by the fact that the coupling constant of the alkene protons which resonate as doublets at δ 8.23 and 9.05 ppm is 16 Hz; which is the typical characteristic of trans- isomer substituted alkenes. The13C NMR spectrum of 12b shows a signal at 166.78 ppm which is assignable to the carbonyl carbon atom of the carboxylic acid functional group (supporting information- Figure 9).
The condensation of N-alkylhydroquinoline-6-carbaldehydes 5a, b; 6a, b with acetone proceeds under milder conditions and leads to the previously unknown 4-(2,2,4-trimethylhydroquinoline- 6-yl)-3-buten-2-ones 13 a, b and 14, which are promising synthons for synthesizing biologically active heterocyclic systems.
In the 1H NMR spectra of compounds 13 a,b, and 14, the olefinic protons signals are observed in the form of two doublets in the regions of 6.50-6.54 and 7.44-7.49 ppm, and the value of the coupling constant is 16.4 Hz (supporting information- Figure 10-12). There is the typical characteristics for 1,2-disubstituted alkenes with the trans configuration. On this basis, the compounds obtained were assigned the structure (E)-4-(1,2,2-trimethylhydroquinolin-6-yl)but-3-en-2-ones (Scheme 4).
2.1.4. Cyclizations of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 3h and 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde, 5e with Methylene Active Compounds 16, 17, 19, 21, 22
Dimethylacetylenedicarboxylate (DMAD) is an electron-deficient acetylenic compound which is widely used in cyclization reactions. With phenol, DMAD usually gives adducts, which are the result of addition of the hydroxyl group under both basic and acid catalysis. However, with pyridine in the presence of sufficiently strong CH-acids, DMAD forms stable 1,4-diionic compounds of the betaine type [46]. The formation of such compounds leads to the fact that the product of the reaction between DMAD and various phenols is annelated α-methylene-γ-butyrolactones [47].
In 7-hydroxy-1,2,3,4-tetramethyl-1,2-dihydroquinoline, 3h, positions 6 and 8 exhibit pronounced CH-acidity associated with the coordinated orientation of the –OH and disubstituted amine groups, which made it possible to hope for the preparation of the corresponding cyclic derivatives under pyridine catalysis conditions. Since position 8 is relatively sterically hindered as compared with position 6, only one cycling center remains, which is position 6.
Scheme 5.
Synthesis of methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16 and 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17.
Scheme 5.
Synthesis of methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16 and 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17.
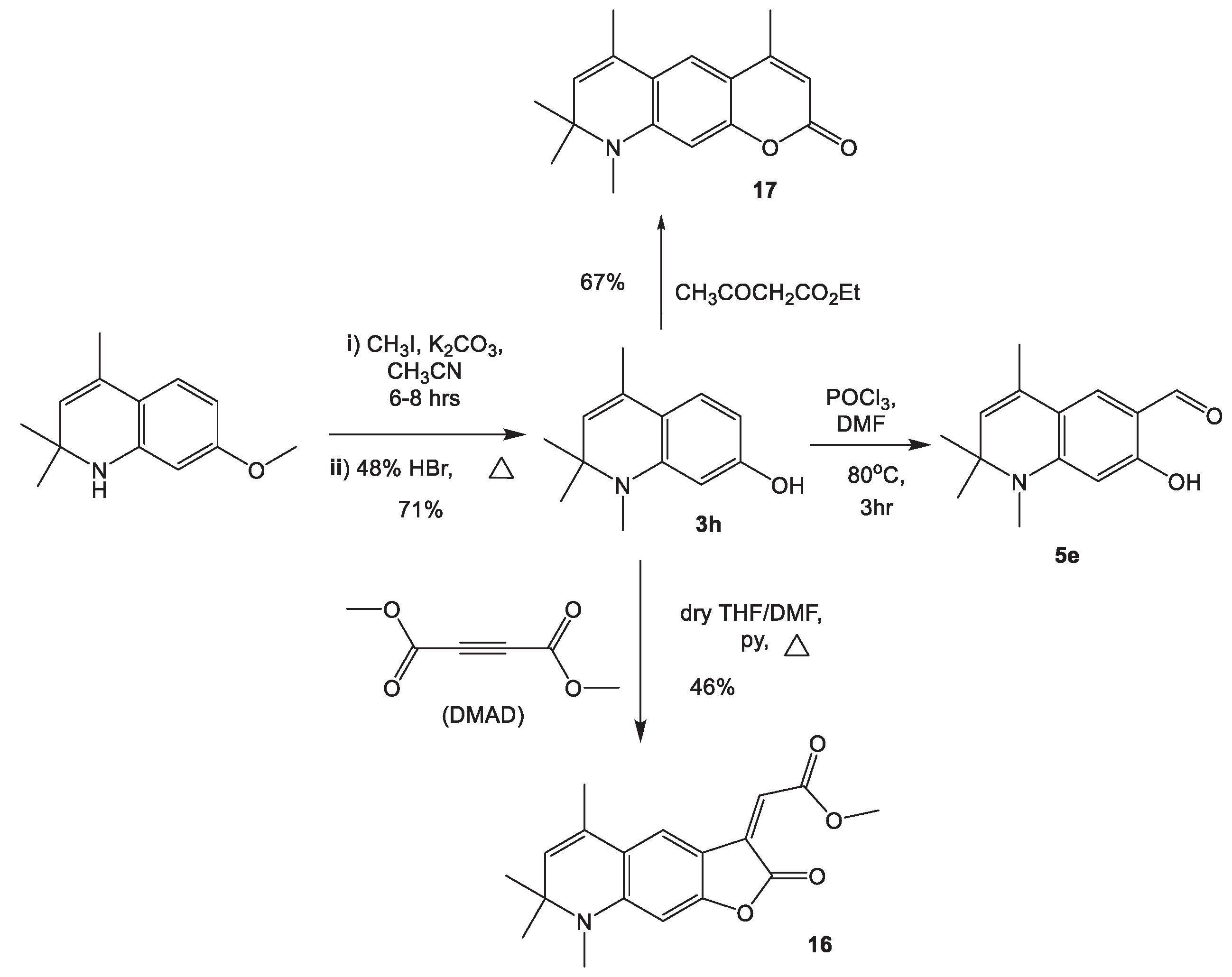
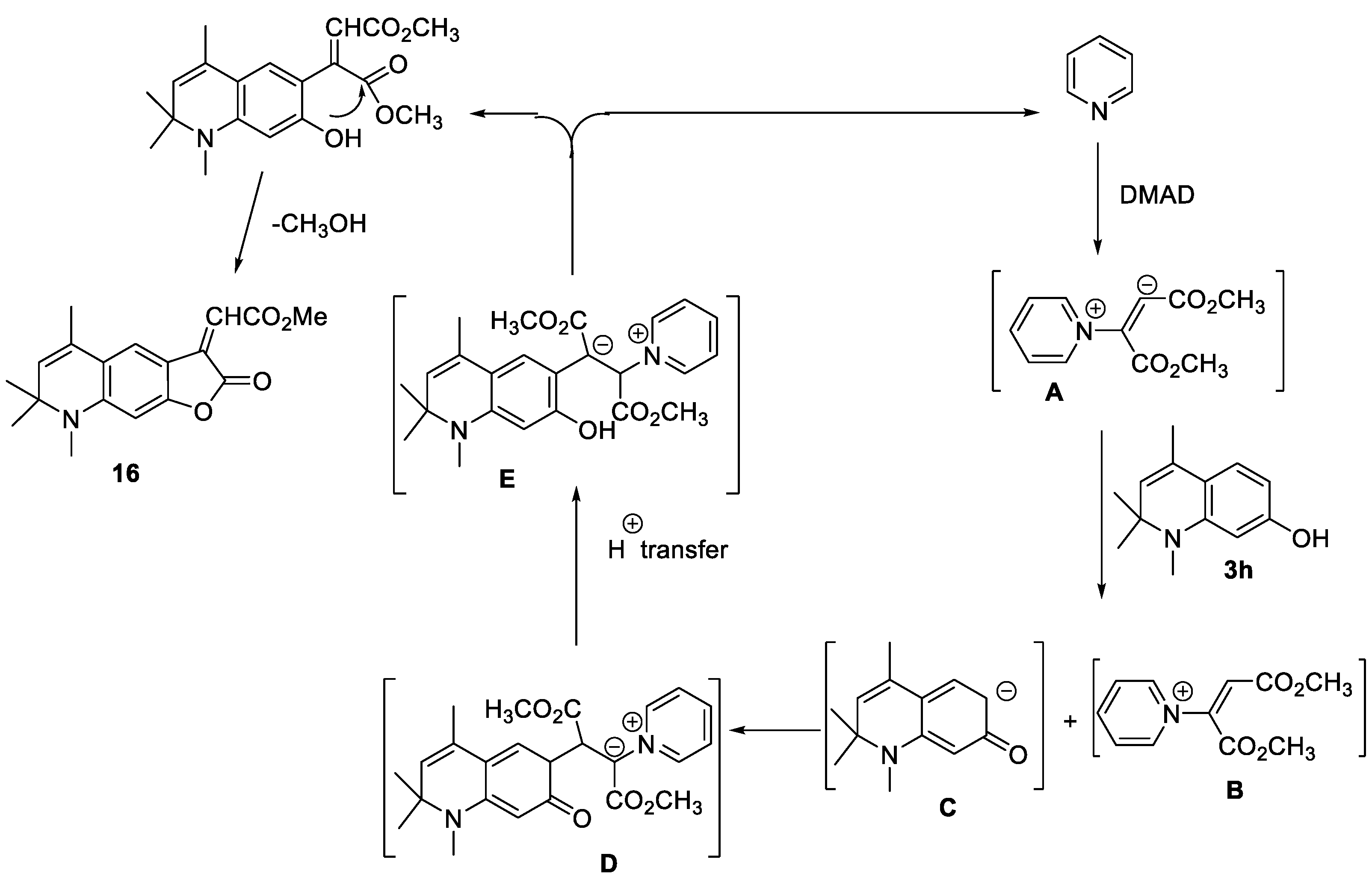
The reaction of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 3h with DMAD in the presence of catalytic amount of pyridine afforded 16 with 46% reaction yield The 1H NMR spectrum of 16 indicates a set of signals corresponding to the tricyclic structure of annulated α-methylene-γ-butyrolactone (supporting information- Figure 13). The plausable reaction mechanism was proposed as shown in Scheme 6. In the presence of pyridine, DMAD gives a betaine derivative of dimethyl maleate A, which is stabilized by interaction with the phenolic derivative 3h (B and C, here in D).
Subsequent intramolecular proton transfer leads to an intermediate E, which decomposes with the regeneration of pyridine and the formation of a compound in which the phenolic hydroxyl group and one of the ester groups of the dimethylmaleate fragment are spatially closed. The subsequent intramolecular transesterification leads to the elimination of methanol and the formation of lactone 16.
The Pechmann reaction introduces one of the most significant and simple methods for the synthesis of a variety of coumarin derivatives [48]. Pechmann condensation of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 3h with ethylacetoacetate in the presence of catalytic amount of sulphuric acid afforded 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17 in 67% yield (Scheme 5).The 1H-NMR spectrum of 17 indicates that there are two singlet signals at δ 6.36 & 7.16 ppm each signal integrable to one proton which are assignable to H-10 and H-5 aromatic protons, respectively. This clearly indicated that the condensation reaction occurred via position 6, not through position 8 of the aromatic ring of 3h. If the condensation had been occurred via position 8 of the ring, there would have been two ortho- substituted aromatic protons which would result in two doublet aromatic signals with each signal integrable to one proton with the J-value of nearly7(8) Hz. The preference of position 6 over position 8 can be rationalized due to the relative steric hindrance of position 8 over 6 of the aromatic ring of the starting material 3h (supporting information- Figure 14).
Coumarins are natural plant species with diverse biological activities including anti-inflammatory, anti-tumor, anti-oxidant, and antidiabetic. Coumarin derivatives have been also considered as neuroprotective agents against oxidative stress and free radical generation [49]. Several methods have been employed for the synthesis of coumarins such as Von Pechmann, Perkin, Knoevenagel, Reformatsky, Wittig, and Claisen Rearrangement reactions [50].
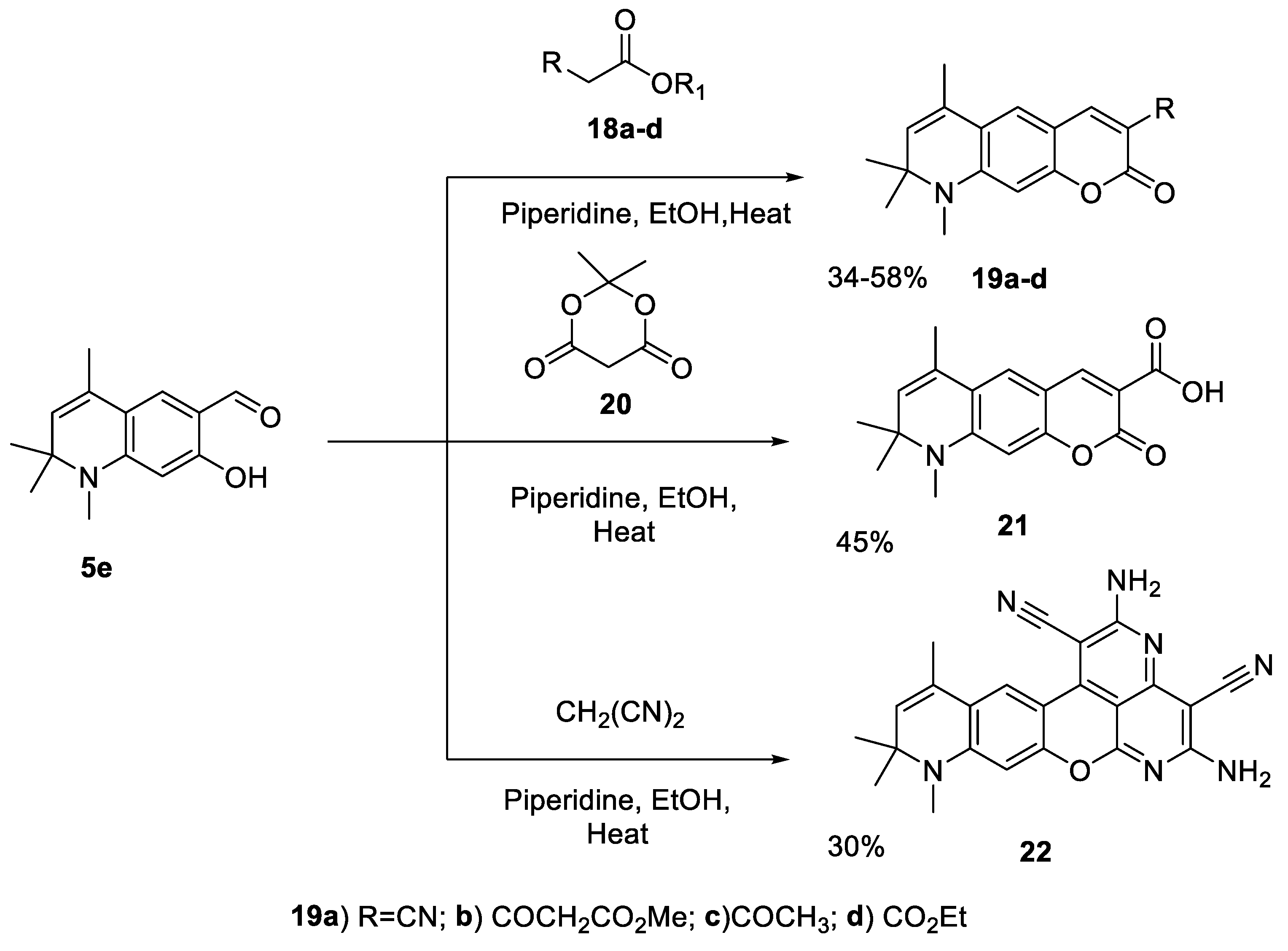
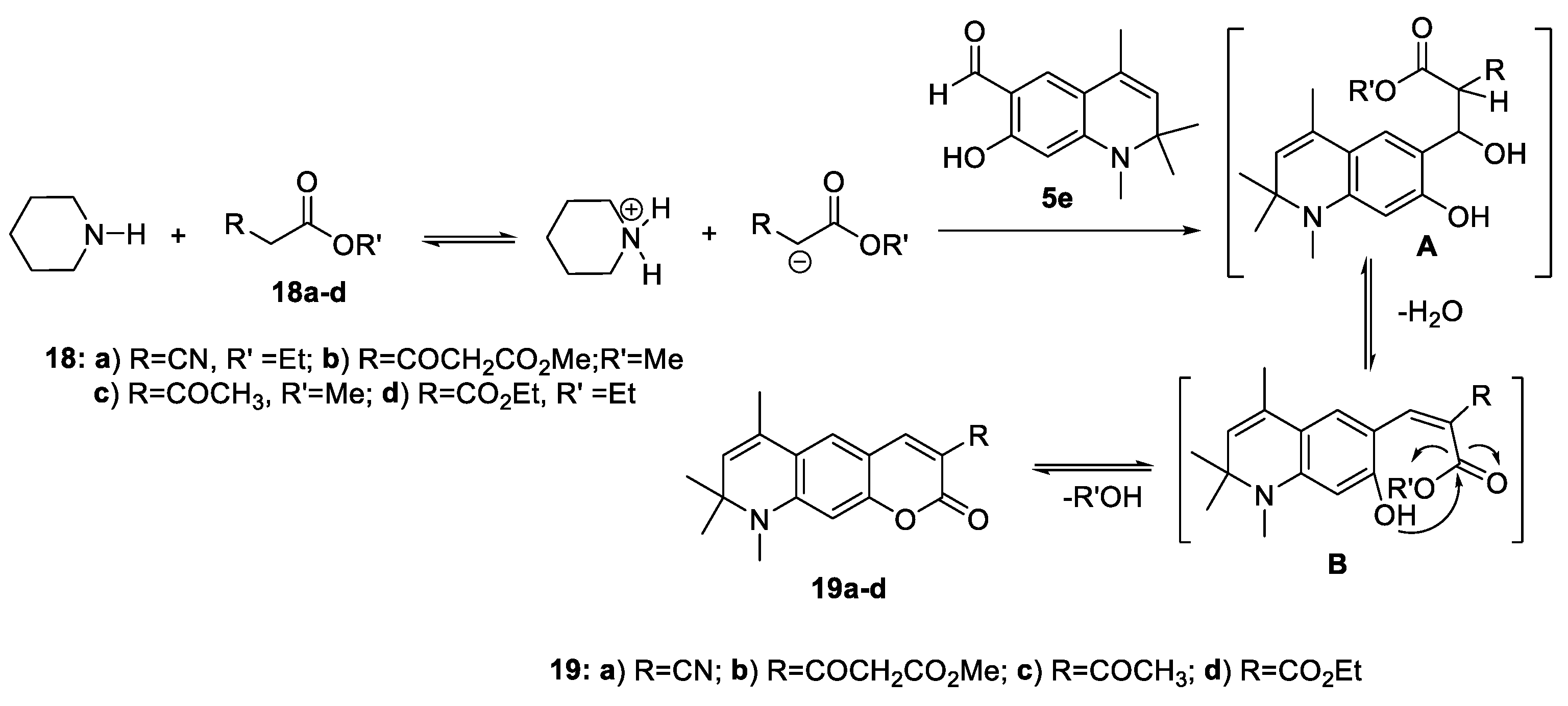
In the condensation reaction with 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5e in the presence of catalytic amount of secondary amine piperidine in ethanol, we have used methylene active compounds such as ethylcyanoacetate (18а), dimethyl-3-oxopentanedioate (18b), ethylacetoacetate (18с), and diethylmalonate (18d) (Scheme 7); and 34-58% yield of 3-substituted dihydroquinoline containing coumarins 19a-d were obtained.
The plausible reaction mechanism of formation of 3-R-6,8,8,9-tetramethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 19a-d is shown in Scheme 8. Common to all methylene active compounds, 18a-d is the presence of an ester functional group, which enters into the intramolecular transesterification reaction of the arylidene derivative B, which is formed after dehydration of the Knoevenagel adduct A.
The 1H-NMR spectrum of 19a shows that there are two singlet signals, each integrable to one proton at the chemical shifts δ = 6.44 and 8.48 ppm which are assignable to the aromatic protons H-10 and H-5, respectively. The downfield signal at δ = 7.25 ppm belongs to the olefinic proton H-4 which is beta to the electron-withdrawing cyano group. The mass spectrum of compound 19a indicates that it has a [M]+ value of 280.12 (supporting information- Figure 15).
The 1H-NMR spectrum of compound 19b indicates that the singlet signal at δ = 8.53 and 6.43 ppm belong to the aromatic protons H-5 and H-10, respectively. The singlet signal at δ = 3.96 ppm, which is integrable to two protons, belongs to the methylene protons, whereas the singlet signals at δ = 3.62 ppm and 7.46 ppm are assignable to the methoxy and olefinic (H-4) protons. The 13C-NMR spectrum of 19b shows the chemical shift values at 189.14, 168.48 and 160.07 ppm which are assignable to the carbonyl carbon of ketone, lactone and acyclic ester, respectively (Supporting information- Figure 16 & 17). The 1H-NMR spectrum of compound 19c designates that the singlet signal at δ = 8.47 and 6.40 ppm belongs to the aromatic protons H-5 and H-10, respectively. The singlet signal at δ = 2.51, which is integrable to three protons, belongs to the acetyl protons (supporting information- Figure 18).
The 1H-NMR spectrum of compound 19d designates that there is a triplet signal at δ = 1.28 ppm with the spin-spin coupling constant value of 7.10 Hz;,which is integrable to three protons belongs to the methyl protons of ethoxy group. The quartet signal at δ = 4.22 ppm J = 7.10 Hz assignable to the methylene protons. The singlet signal at δ = 8.54 and 6.37 ppm belong to the aromatic protons H-5 and H-10, respectively (supporting information- Figure 19).
Another highly reactive methylene compound is Meldrum’s acid 20, which we also used in the Knoevenagel condensation reaction with 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5e. In this case, the arylidene derivative B (Scheme 9) undergoes the following transformation: the intramolecular nucleophilic attack of the phenol hydroxyl group on the carbonyl group of Meldrum’s acid leads to the elimination of the acetone molecule and, after protonation, to the formation of acid 21. The formation of analogous compounds is described by the interaction of salicylaldehyde with acid [51].
The 1H-NMR spectrum of compound 21 indicates that there is downfield singlet signal at δ = 7.39 ppm which can be assignable to the olefinic proton H-4 which is beta to the electron-withdrawing carboxylic group (supporting information- Figure 20).
Unlike other methylene active compounds, malononitrile (CH2(CN)2) is capable of undergoing a cascade condensation reactions with salicylaldehyde and its analogues. At the first stage of the cascade, 2-imino-2H-chromene-3-carbonitrile is formed; who’s analog is the intermediate C (Scheme 10). At an equimolar ratio of reactants and under conditions of catalysis with metal halides [52] or ion-exchange resin [53], this primary condensation product can be isolated; however, with an excess of malononitrile, it enters into further condensation.
2-Imino-2H-chromene-3-carbonitrile or its analogue С contains an electrophilic carbon atom in position 4, which reacts with N- and S-nucleophiles under mild conditions [54]. With an excess of malononitrile under the conditions of basic catalysis, this reaction center enters into a Knoevenagel condensation with a C-nucleophile, which is the bridging carbon atom of malononitrile. As a result, an intermediate D is formed, the further transformation of which requires the transformation of the nitrile group, for example, into an amidine group, which occurs when ammonium acetate is the catalyst of the transformation.
However, the third equivalent of malononitrile [55] can act as such a nucleophile, under the action of which F is formed, the next participant in the domino reaction, which is oxidized under the action of intermediate C or atmospheric oxygen to the tetracyclic intermediate G, which is closed into the final pentacyclic product 22. Previously, a similar scheme was proposed for the interaction of salicylaldehyde with an excess of malononitrile [56].
The molecular ion peak [M]+ at m/z 408, which was expected on the mass spectrum of compound 22, was not observed. However, there are notable peaks at m/z (rel. intensity, %) 31(60), 44(100), 197(30), and 394(53). The value of the heaviest peak at m/z 394 corresponds to [M-14]+. The broad singlet signal at δ = 7.28 ppm in the 1H-NMR spectrum of compound 22, which can be integrated to a total of four protons, is attributed to the two amine groups (supporting information- Figure 21).
2.2. In-silico studies
2.2.1. PASS the predicted activity spectrum of test synthesized compounds
PASS predicted spectrum of activity of the test compounds (Table 2) showed that 4-aryl substituted-3-buten-2-one compounds 13a,13b, and 14 possess a good probability of exhibiting gluconate 2-dehydrogenase (acceptor) inhibitor activity with the Pa values of 0.849–0.870. Whereas hydroquinolinecarbonitrile containing compounds 7 & 8 have a propensity to act as a good antiallergic, antiasthmatic, antiarthritic, and progesterone antagonists (Pa values 0.276–0.827), phenyloxazolones 9, 10(a,b) indicated low probability of being pharmaceutically active. Coumarins containing compounds 17, 19a, and 19c were predicted to be a good progesterone antagonists with the Pa values of 0.710, 0.63, and 0.615, respectively.
3. Experimental Section
3.1. General
The starting materials hydroquinolinecarbaldehydes are synthesized via the Vilsmeier-Haack formylation reaction of the respective N-alkylhydroquinolines. All other reacting materials were commercially accessible from Sigma-Aldrich suppliers. In this study, analytical grade reagents and solvents were employed except for the solvents THF and DMF where further purifications were done. The reaction progress and purity of the individual synthesized compounds were monitored using precoated silica gel TLC plates (Merck TLC Silica gel 60 F254 plates, eluent chloroform, methanol, hexane, ethyl acetate in various proportions) and visualization of the spots was carried out using a UV lamp. The 1Н and 13С NMR spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) at 500.13 and 125.03 MHz, respectively, on a Bruker DRX instrument. Chemical shifts (δH) are recorded relative to the internal standard, tetramethyl silane (TMS). Mass spectra were recorded using Agilent Technologies LCMS6230B (chemical ionization, 200 eV) and GCMS7890B instruments (carrier gas, EI, energy of ionization 70 eV). Melting points (mp) were measured uncorrected using a Stuart SMP30 melting point instrument.
3.2. Synthesis of 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbonitrile 7.
1,2,2,4-Tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5а (2.0 g, 9.28 mmol) was added to a flask containing a solution of hydroxylamine hydrochloride (0.65 g, 9.28 mmol) in 2 mL of pyridine with continuous stirring.After 5 minutes of stirring on a magnetic stirrer, 10 ml of toluene was added and the mixture was refluxed with a Dean-Stark trap.Upon completion of the process (TLC, hexane/ethyl acetate 8:2), the reaction mass was cooled, filtered off from the precipitate of pyridine hydrochloride, the filtrate was washed with distilled water, dried over anhydrous sodium sulfate, and toluene was removed on a rotary evaporator.The product was recrystallized from aqueous acetonitrile.
Yield: 48%, Mp. 80-82 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.32 (s, 6H, (CH3)2); 1.91 (s, 3H, CH3-C4); 2.81 (s, 3H, N-CH3); 5.43 (s, 1H, CH-C3); 6.54 (d, 1H, J=8.67, H-8); 7.23 (d, 1H, J=2.03, H-5); 7.41 (dd, 1H, J=8.62, J=2.04, H-7).
3.3. Synthesis of 1-Benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbonitrile, 8
A. Hydroquinolinecarbaldehyde 6b (5 mmol) was treated with iodine (5.5 mmol) in a mixture of aqueous ammonia (25% 30 ml) and THF (5 ml) at room temperature for 8 hours.During the reaction, the solution gradually became colorless as the iodine was consumed.An aqueous solution of Na2S2O3 (1M, 10 ml) was added dropwise to the reaction mixture, stirred for 3 hours, and extracted with methylene chloride.The organic phase was washed with water, dried and evaporated.The residue was recrystallized from aqueous acetonitrile.
B. Hydroquinolinecarbaldehyde 6b (5 mmol) was treated with iodine (5.5 mmol) in a mixture of aqueous ammonia (25% 30 ml) and THF (5 ml) at room temperature for 8 hours.During the reaction, the solution gradually became colorless as the iodine was consumed.An aqueous solution of H2O2 (35%, 3 ml) was added dropwise to the reaction mixture, stirred for 4 hours, and extracted with methylene chloride.The organic phase was washed with water, dried and evaporated.The residue was recrystallized from aqueous acetonitrile.
Yields: 62% (Method A) and 51% (Method B), Mp. 115-117 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.26 (s, 6H, (CH3)2); 1.34 (d, 3H, J=6.60, CH3-C4); 1.63 (t, 1H, J=13.01, CH2A-C3); 1.92 (dd, 1H, J=13.14, J=4.61, CH2B-C3); 2.95 (m, 1H, CH-C4); 4.36 (d, 1H, J=18.12, Bn-Ha); 4.83 (d, 1H, J=17.99, Bn-Hb); 6.24 (d, 1H, J=8.78, arom.); 7.15-7.45 (m, 7H, arom.).
3.4. Synthesis of 2-phenyl-1,3-oxazol-5(4Н)-ones 9, 10 (General procedure)
A solution of the corresponding hydroquinolinecarbaldehyde 5 or 6 (3.4 mmol) in 3 ml of acetic acid was added dropwise to a stirred solution of hippuric acid (3.4 mmol), acetic anhydride (20 ml) and acetic acid.The reaction mixture was stirred at 70°C for 3 hours, poured into crushed ice, and stirred for 30 minutes.The product was extracted with chloroform, washed, and the chloroform was evaporated using a rotary evaporator, and the residue was recrystallized from isopropanol.
3.4.1. (Z)-2-Phenyl-4-((1,2,2,4-tetramethyl-1,2-dihydroquinolin-6-yl)methylene)oxazol-5(4H)-one (9a)
Yield: 60%, Mp. 180-182 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.37 (s, 6H, C(CH3)2); 2.02 (s, 3H, CH3-4); 2.91 (s, 3H, N-CH3); 5.48 (s, 1H, CH-3); 6.64 (d, 1H, J=8.8, arom.); 7.21 (s, 1H, CH-methylene); 7.60-8.20 (m, 7H, arom.)
3.4.2. (Z)-4-((1-Benzyl-2,2,4-trimethyl-1,2-dihydroquinolin-6-yl)methylene)-2-phenyloxazol-5(4H)-one (9b)
Yield: 30%, Mp. 130-132 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.41 (s, 6H, C(CH3)2); 2.08 (s, 3H, CH3-4); 4.73 (s, 2H, CH2-Bn); 5.57 (s, 1H, CH-3); 6.33 (d, J=8.86, 1H, arom.); 7.18 (s, 1H, CH-methylene); 7.21-8.20 (m, 12H, arom.)
3.4.3. (Z)-2-Phenyl-4-((1,2,2,4-tetramethyl-1,2,3,4-tetrahydroquinolin-6-yl)methylene)oxazol-5(4H)-one (10a)
Yield: 41%, Mp. 140-142 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.25 (s, 3H, C(CH3)2A), 1.32 (s, 3H, C(CH3)2B); 1.39 (d, J=6.56, 3H, CH3-4); 1.45 (t, J=13.12, 1H, CH2A-3); 1.89 (dd, J=13.12, J=4.20, 1H, CH2B-3); 2.85 (m, 1H, CH-4); 2.92 (s, 3H, N-CH3); 6.67 (d, J=8.86, 1H, arom.); 7.22 (s, 1H, CH-methylene); 7.55-8.20 (m, 7H, arom.).
3.4.4. (Z)-4-((1-Benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)methylene)-2-phenyloxazol-5(4H)-one (10b)
Yield: 47%, Mp. 195-197oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.29 (s, 3H, C(CH3)2A); 1.32 (s, 3H, C(CH3)2B); 1.44 (d, J=6.54, 3H, CH3-4); 1.69 (t, J=13.05, 1H, CH2A-3); 1.96 (dd, J=13.18, J=4.47, 1H, CH2B-3); 3.03 (m, 1H, CH-3); 4.48 (d, J=18.19, 1H, CH2A-Bn); 4.88 (d, J=18.21, 1H, CH2B-Bn); 6.33 (d, J=8.91, 1H, arom.); 7.18 (s, 1H, CH-methylene); 7.20-8.30 (m, 12H, arom.).
3.5. Synthesis of (E)-7-[2,2,4-trimethylhydroquinolin-6-ylidenemethyl]triazolo(pyrazolo)[1,5-a]pyrimidin-6-ylcarboxylic acid 12 (General procedure)
A mixture of 2 mmol of 6-formyltetrahydroquinoline (6a,b), 2 mmol of 7-methylazolopyrimidine 11a,b, 2.2 mmol of potassium tert-butoxide, and 20 ml of methanol was refluxed for 6 hours, cooled, and poured into 100 ml of 10% acetic acid solution.The precipitate formed was filtered off, washed with water, and after drying, was recrystallized from ethanol.
3.5.1. (E)-7-(2-(1,2,2,4-Tetramethyl-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylic acid (12a)
Yield: 67%, Mp. 255-257 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.04 (d, 3H, J=6.7, CH3); 1.23 (s., 3H, CH3); 1.31 (s, 3H, CH3); 1.39-1,48 (m, 1H, СН2а); 1,88 (dd, 1H, J=13.0, J=4.2 CH2b); 2.80-2.86 (m, 1H, CH); 2,88 (s, 3Н, N-СН3); 6.45 (d, 1H, J=8.5, CH arom.); 7.42-7.46 (m, 2H, СН arom.); 8.23 (d, 1H, J=16.0, СН vinyl); 8,81 (s, 1Н, СНtriazol.); 9,05 (d, 1Н, J=16.0, СН vinyl); 9,14 (s, 1Н, СН pyrim.); 13,31 (s, 1Н, СООН).
3.5.2. (E)-7-(2-(1-Benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)pyrazolo[1,5-a]pyrimidine-6-carboxylic acid (12b)
Yield: 60%, Mp. 291-293 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.29 (s, 6H, (CH3)2); 1.41 (d, 3H, J=6.7, CH3); 1.68 (t, 1H, J=12.8, CH2A); 1.94 (dd, 1H, J=12.9, J=4.7, CH2B); 3.02 (p, 1H, J=5.8, CH); 4.41 (d, 1H, J=18.0, Bn-СH2а); 4.81 (d, 1H, J=18.0, Bn-СH2b); 6.32 (d, 1H, J=8.6, СН arom.); 6.80 (d, 1H, J=2.4, СHpyraz.); 7.20-7.28 (m, 7H аrom.); 7.50 (s, 1H, СH arom.); 8,23 (d, 1Н, J=16.0, СН vinyl); 8,43 (d, 1Н, J=2,4, СН pyriaz.);8,87 (s, 1Н, СН pyrim.); 9,05(d, 1Н, J=16.0, СН vinyl); 13,50 (s, 1Н, СООН).
13C NMR (DMSO-d6, 125.03 MHz, δ( ppm): 19,69 (1С, СН3); 24,90 (1С, СН3); 26,65 (1С, СН3); 28,75 (1С, СН); 45,58 (1С, СН2); 48,38 (1С, СН2); 45,58 (1С, СН2); 55,14 (1С, С(СН3)2); 96,86 (1С, СНpyraz.); 109,20 (1С, С-6pyrim.); 110,27 (1С, СНbenz.); 112,63 (1С, СНvinyl); 123,42 (1С, СНbenz.); 125,96 (2С, o-СН phenyl); 126,46 (1С, p-СН phenyl); 126,57 (1С, Сpyraz-pyrim.); 126,96 (1С, Сbenz); 126,96 (1С, Сbenz); 127,72 (1С, С-СООН); 128,65 (2С, m-СНphenyl); 128,99 (1С, СНbenz); 139,60 (1С, СНpyraz.); 144,55 (1С, Сphenyl); 146,96 (1С, Сbenz); 147,57 (1С, Сbenz-pyr.); 149,64 (1С, СНvinyl; 150,12 (1С, Сpyrim.); 166,78 (1С, СООН).
3.6. Synthesis of (E)-4-(2,2,4-trimethylhydroquinolin-6-yl)-3-buten-2-ones 13, 14.
To a solution of 2 mol of the corresponding N-alkylhydroquinoline-6-carbaldehyde, 5,6 in 15 ml of acetone was slowly added to a stirring solution of 1 g of potassium hydroxide in 20 ml of water.The solution was stirred at room temperature for 5 h. The precipitate formed was filtered off, washed with water, dried, and recrystallized from a mixture of acetone water (1:1).
3.6.1. (E)-4-(1,2,2,4-Tetramethyl-1,2-dihydroquinolin-6-yl)but-3-en-2-one (13a)
Yield: 76%, Mp. 68-70 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.30 (s, 6H, (CH3)2); 1.96 (s, 3H, CH3-C’4); 2.26 (s, 3H, CH3-CO-); 2.81 (s, 3H, N-CH3); 5.40 (s, 1H, CH-DHQ); 6.52 (d, 1H, J=8.67, H’-8); 6.55 (d, 1H, J=16.14, CH-C3); 7.28 (d, 1H, J=2.14, H’5); 7.39 (dd, 1H, J=8.56, J=2.14, H’7); 7.49 (d, 1H, J=16.17, CH-C4).
3.6.2. (E)-4-(1-Benzyl-2,2,4-trimethyl-1,2-dihydroquinolin-6-yl)but-3-en-2-one (13b)
Yield: 80%, Mp. 70-72 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.35 (s, 6H, (CH3)2); 2.01 (s, 3H, CH3-C’4); 2.23 (s, 3H, Bn); 5.49 (s, 1H, CH-C’3); 6.22 (d, 1H, J=8.67, H’8); 6.50 (d, 1H, J=16.15, H-3); 7.15-7.35 (m, 7H, аrom.); 7.44 (d, 1H, J=16.17, H-4).
3.6.3. (E)-4-(1-Benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)but-3-en-2-one (14)
Yield: 84%, Mp. 100-102 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.25 (s, 3H, (CH3)2A); 1.26 (s, 3H, (CH3)2B); 1.37 (d, 3H, J=6.59, CH3-C’4); 1.62 (t, 1H, J=12.97, CH2A-C’3); 1.91 (dd, 1H, J=13.05, J=4.61, CH2B-C’3); 2.23 (s, 3H, CH3-CO-); 2.97 (m, 1H, CH-C’4); 4.33 (d, 1H, J=18.09, Bn-Ha); 4.80 (d, 1H, J=18.06, Bn-Hb); 6.21 (d, 1H, J=8.72, Arom.); 6.50 (d, 1H, J=16.12, H-3); 7.15-7.40 (m, 7H, аrom.); 7.45 (d, 1H, J=16.27, H-4).
3.8. Synthesis of methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinoline-3(2H)-ylidene)acetate 16
To a stirred solution of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5e (5 mmol) and dimethylacetylenedicarboxylate (5 mmol) in ml of anhydrous THF/DMF (4:1) was added dropwise pyridine (0.5 mmol) at room temperature. The reaction mixture was stirred at room temperature for 30 min and the mixture was refluxed until the reaction was complete. The solvent was removed on a rotary evaporator, the residue was separated by column chromatography using n-hexane/ethyl acetate (4:1) as an eluent.
Yield: 46%, Mp. 195-197 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.35 (s, 6H, C(CH3)2); 1.93 (s, 3H, CH3-5); 2.87 (s, 3H, N-CH3); 3.91 (s, 3H, CH3O-); 5.50 (s, 1H, CH-5); 6.38 (s, 1H, H-8); 6.43 (s, 1H, H-3); 7.63 (s, 1H, CH-2).
3.9. Synthesis of 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17
A mixture of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 3h (1 mmol) and ethylacetoacetate (1.2mmol) was heated to 80oC in the presence of sulfuric acid (2 mL) until reaction completion. After completion of the reaction (monitored by TLC), the mixture was cooled to room temperature and extracted with ethylacetate (2X). The solution was concentrated and the crude product was recrystallized from ethanol to afford pure 4-substituted hydroquinoline containing coumarin 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17.
Yield: 68%, Mp. 170-172 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.32 (s,6H, (CH3)2); 1.98 (s, 3H. C6-CH3); 2.35 (s, 3H, C4-CH3); 2.83 (s, 3H, N-CH3); 5.46 (s, 1H, H-8); 5.94 (s, 1H, H-3); 6.36 (s, 1H, H-10 (arom.); 7.16 (s, 1H, H-5 (arom.))
3.10. General procedure for the synthesis of 6,8,8,9-tetramethyl-8,9-dihydropyrano[3,2-g]quinolin-2-ones 19, 21 and, 22
A mixture of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5e (8.65 mmol), the corresponding active methylene compound 18, 20 (8.64 mmol, 25.95 mmol in the case of synthesis 21) and a few drops of piperidine in 10 ml of ethanol was boiled until the end of the reaction, after which the reaction mixture was cooled to room temperature and poured into 5 ml of chilled water. The resulting precipitate was filtered off and recrystallized from ethanol.
3.10.1. 6,8,8,9-Tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carbonitrile (19a)
Yield: 51%, Mp. 190-192 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.38 (s, 6H, C(CH3)2); 1.94 (s, 3H, CH3-6); 2.94 (s, 3H, N-CH3); 5.54 (s, 1H, CH-7); 6.44 (s, 1H, H-10); 7.25 (s, 1H, H-4); 8.48 (s, 1H, H-5).
3.10.2. Methyl 3-oxo-3-(6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinolin-3-yl)propanoate (19b)
Yield: 58%, Mp. 179-181 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.39 (s, 6H, C(CH3)2); 1.95 (s, 3H, CH3-6); 2.95 (s, 3H, N-CH3); 3.62 (s, 3H, CH3O-); 3.96 (s, 2H, CH2); 5.53 (s, 1H, CH-7); 6.43 (s, 1H, H-10); 7.46 (s, 1H, CH-4); 8.53 (s, 1H, H-5).
13C NMR (DMSO-d6, 125.03 MHz, δ( ppm): 18.19, 28.67, 28.74, 32.04, 48.11, 51.74, 58.41, 95.14, 107.83, 113.42, 119.97, 124.73, 125.09, 130.30, 148.15, 151.40, 158.81, 160.02, 168.48, 189.14
3.10.3. 3-Acetyl-6,8,8,9-tetramethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one (19c)
Yield: 39%, Mp. 200-202 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.38 (s, 6H, C(CH3)2); 1.94 (s, 3H, CH3-6); 2.51 (s, 3H, CH3-CO-); 2.93 (s, 3H, N-CH3); 5.51 (s, 1H, CH-7); 6.40 (s, 1H, H-10); 7.42 (s, 1H, H-4); 8.47 (s, 1H, H-5).
3.10.4. Ethyl 6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carboxylate (19d)
Yield: 34%, Mp. 130-132 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.28 (t, 3H, J=7.1, CH3CH2O-); 1.37 (s, 6H, C(CH3)2); 1.94 (s, 3H, CH3-6); 2.91 (s, 3H, N-CH3); 4.22 (q, 2H, J=7.1, CH3CH2O-); 5.58 (s, 1H, CH-7); 6.37 (s, 1H, H-10); 7.41 (s, 1H, CH-4); 8.54 (s, 1H, H-5).
3.10.5. 6,8,8,9-Tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carboxylic acid (21)
Yield: 45%, Mp. >250 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.36 (s, 6H, C(CH3)2); 1.94 (s, 3H, CH3-6); 2.90 (s, 3H, N-CH3); 5.49 (s, 1H, CH-7); 6.39 (s, 1H, H-10); 7.39 (s, 1H, CH-4); 8.51 (s, 1H, H-5).
3.10.6. 2,5-Diamino-9,10,10,12-tetramethyl-9,10-dihydropyrido[4′,3′,2′:8,1]isochromeno[4,3-g]quinoline-1,4-dicarbonitrile (22)
Yield: 30%, Mp. >250 oC. 1H NMR (DMSO-d6, 500.13 MHz, δ( ppm), J (Hz)): 1.38 (s, 6H, C(CH3)2); 1.95 (s, 3H, CH3-12); 2.89 (s, 3H, N-CH3); 5.51 (s, 1H, CH-11); 6.37 (s, 1H, H-8); 7.28 (bro. s, 4H, 2xNH2); 8.44 (s, 1H, H-13). MS (EI, 70 ev), m/z (Irel (%)): 394 [M-14]+ (53), 197 (30), 44 (100), 31 (60).
3.11. In Silico PASS Prediction
Prediction of Activity Spectra of Substances (PASS) was used to predict the pharmacological activities of the synthesized compounds. The synthesized compounds were first converted into the SMILES format using ChemBioDraw and then predicted using the PASS online web tool/ http://www.way2drug.com/PASSOnline/predict.php/. PASS indicates the probable activity (Pa) and probable inactivity (Pi) of ‘drug-like’ substances.
4. Conclusion
In this study, heterocyclic compounds containing hydroquinoline fragment such as carbonitriles, oxazolones, azolo[1,5-a]pyrimidines and coumarins were synthesized. A three hours reflux of 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5a with a system of hydroxylammonium chloride/pyridinium chloride/toluene afforded 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-nitrile 7 in moderate yield. N-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbaldehyde 6b and iodine reacted at room temperature in aqueous ammonia water, followed by aqueous treatment with Na2S2O3 and produced N-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-nitrile 8 in 62%. 30-60% 2-phenyl-1,3-oxazol-5(4H)-ones 9a,b; 10a,b were synthesized via the condensation reaction of N-alkylhydroquinoline-6-carbaldehydes 5a,b; 6a,b with hippuric acid in acetic acid. When activated 7-methylazolopyrimidines 11a,b were reacted with N-alkyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbaldehydes 6a,b, the reaction produced 60–70% yield of 7-[(E)-2,2,4-trimethylhydroquinolin-6-ylidenemethyl]azolo[1,5-a]pyrimidin-6-yl carboxylic acids 12a,b.
The condensation reaction of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 3h with dimethylacetylenedicarboxylate (DMAD) and ethylacetoacetate afforded methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16 and 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17, respectively. The condensation reaction of 6-formyl-7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline 5e with methylene active compounds ethylcyanoacetate/dimethyl-3-oxopentanedioate/ ethylacetoacetate/diethylmalonate/Meldrum’s acid afforded 3-substituted dihydroquinoline containg coumarins 19 - 21. The reaction of 5e with malononitrile afforded a pentacyclic compound 22.
The potential biological activities of the synthesized compounds were evaluated using the PASS computer program. According to the prognosis, the (E)-4-(N-methyl(benzyl)-2,2,4-trimethyl-1,2-dihydroquinolin-6-yl)but-3-en-2-ones 13 a, b and (E)-4-(1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)but-3-en-2-one 14 are most likely to have inhibitory activity against gluconate dehydrogenase, as well as anti-arthritic, anti-allergic, and anti-asthmatic properties. It was also found out that hydroquinolinecarbonitrile compounds 7 & 8 have a tendency to act as a good progesterone antagonists, anti-allergic, anti-asthmatic, and anti-arthritic with the Pa values ranging from 0.276 to 0.827. Among the coumarin moieties containing hydroquinolines, compounds 17, 19a, and 19c were predicted to be a good progesterone antagonists with the Pa values of 0.710, 0.630 and 0.615; respectively.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. A separate Supporting Information consisting of the spectroscopic data of the synthesized compounds is provided with this manuscript. Figure 1. 1H-NMR spectrum of 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbonitrile 7. Figure 2. 1H-NMR spectrum of 1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbonitrile 8. Figure 3. 1H-NMR spectrum of (Z)-2-phenyl-4-((1,2,2,4-tetramethyl-1,2-dihydroquinolin-6-yl)methylene)oxazol-5(4H)-one 9a. Figure 4. 1H-NMR spectrum of (Z)-4-((1-benzyl-2,2,4-trimethyl-1,2-dihydroquinolin-6-yl)methylene)-2-phenyloxazol-5(4H)-one 9b. Figure 5. 1H-NMR spectrum of (Z)-2-phenyl-4-((1,2,2,4-tetramethyl-1,2,3,4-tetrahydroquinolin-6-yl)methylene)oxazol-5(4H)-one 10a. Figure 6. 1H-NMR spectrum of (Z)-4-((1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)methylene)-2-phenyloxazol-5(4H)-one 10b. Figure 7. 1H-NMR spectrum of (E)-7-(2-(1,2,2,4-tetramethyl-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylic acid 12a. Figure 8. 1H-NMR spectrum of (E)-7-(2-(1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)pyrazolo[1,5-a]pyrimidine-6-carboxylic acid 12b. Figure 9. 13C-NMR spectrum of (E)-7-(2-(1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)pyrazolo[1,5-a]pyrimidine-6-carboxylic acid 12b. Figure 10. 1H-NMR spectrum of (E)-4-(1,2,2,4-tetramethyl-1,2-dihydroquinolin-6-yl)but-3-en-2-one 13a. Figure 11. 1H-NMR spectrum of (E)-4-(1-benzyl-2,2,4-trimethyl-1,2-dihydroquinolin-6-yl)but-3-en-2-one 13b. Figure 12. 1H-NMR spectrum of (E)-4-(1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinolin-6-yl)but-3-en-2-one 14. Figure 13. 1H-NMR spectrum of methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16. Figure 14. 1H-NMR spectrum of 4,6,8,8,9-pentamethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 17. Figure 15. 1H-NMR Spectrum of 6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carbonitrile 19a. Figure 16. 1H-NMR spectrum of methyl 3-oxo-3-(6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinolin-3-yl)propanoate 19b. Figure 17. 13C-NMR Spectrum of of methyl 3-oxo-3-(6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinolin-3-yl)propanoate 19b. Figure 18. 1H-NMR Spectrum of 3-acetyl-6,8,8,9-tetramethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 19c. Figure 19. 1H-NMR Spectrum of ethyl 6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carboxylate 19d. Figure 20. 1H-NMR Spectrum of 6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carboxylic acid 21. Figure 21. 1H-NMR Spectrum of 2,5-diamino-9,10,10,12-tetramethyl-9,10-dihydropyrido[4′,3′,2′:8,1]isochromeno[4,3-g]quinoline-1,4-dicarbonitrile 22.
Data Availability Statement
The data used to support the findings of this study are included within the supplementary information file.
Acknowledgments
The authors acknowledge the Ministry of Education and Science of the Russian Federation for financial support (Project No. 4.2100.2014/К).
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- Kabir, E.; Uzzaman, M. A review on biological and medicinal impact of heterocyclic compounds. Results Chem. 2022, 4. [Google Scholar] [CrossRef]
- Al-Mulla, A. A Review: Biological Importance of Heterocyclic Compounds. Der Pharma Chem. 2017, 9. [Google Scholar]
- Qadir, T.; Amin, A.; Sharma, P.K.; Jeelani, I.; Abe, H. A Review on Medicinally Important Heterocyclic Compounds. Open Med. Chem. J. 2022, 16. [Google Scholar] [CrossRef]
- Mermer, A.; Keles, T.; Sirin, Y. Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: A review. Bioorganic Chem. 2021, 114, 105076. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Fang, K.C.; Sheu, J.Y.; Hsu, S.L.; Tzeng, C.C. Synthesis and antibacterial evaluation of certain quinolone derivatives. J. Med. Chem. 2001, 44, 2374–2377. [Google Scholar] [CrossRef]
- Yadav, P.; Shah, K. Quinolines, a perpetual, multipurpose scaffold in medicinal chemistry. Bioorganic Chem. 2021, 109, 104639. [Google Scholar] [CrossRef]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorganic Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef] [PubMed]
- Dib, K.; Ennibi, O.; Alaoui, K.; Cherrah, Y.; Filali-Maltouf, A. Antibacterial activity of plant extracts against periodontal pathogens: A systematic review. J. Herb. Med. 2021, 29, 100493. [Google Scholar] [CrossRef]
- Synthetic Approaches and Biological Activities of Quinoline Derivatives: A Review. Int. J. Pharm. Res. 2021, 13. [CrossRef]
- Updates on Synthesis and Biological Activities of Quinoline Derivatives: A Review. Int. J. Pharm. Res. 2021, 13. [CrossRef]
- Kaur, T.; Bhandari, D.D. Annotated Review on Various Biological Activities of Quinoline Molecule. Biointerface Res. Appl. Chem. 2023, 13. [Google Scholar] [CrossRef]
- Patel, A.; Patel, S.; Mehta, M.; Patel, Y.; Patel, R.; Shah, D.; Patel, D.; Shah, U.; Patel, M.; Patel, S.; et al. A review on synthetic investigation for quinoline- recent green approaches. Green Chem. Lett. Rev. 2022, 15, 337–372. [Google Scholar] [CrossRef]
- Marella, A.; Tanwar, O.P.; Saha, R.; Ali, M.R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 2013, 21. [Google Scholar] [CrossRef]
- Yang, J.M.; Wu, L.; Fang, D.; Cao, J.; Zhou, Y.J.; Wang, X.S. Iodine-catalyzed Povarov reaction for synthesis of cyclobuta[c]quinoline derivatives. Res. Chem. Intermed. 2014, 40, 1103–1113. [Google Scholar] [CrossRef]
- Ghoshal, A.; Yugandhar, D.; Nanubolu, J.B.; Srivastava, A.K. An Efficient one-pot synthesis of densely functionalized fused-quinolines via sequential Ugi4CC and acid-mediated povarov-type reaction. ACS Comb. Sci. 2017, 19, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, S.M.; Plaksina, M.E.; Shikhaliev, K.S. The synthesis of 6-R-2,2,4-trimethyl-1,2-dihydroquinoline- and 6-R-4-R’-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-8-carboxylic acids—The structural analogues of helquinoline. Žurnal organìčnoï ta Farm. hìmìï 2017, 13, 21–25. [Google Scholar] [CrossRef]
- Durgadas, S.; Chatare, V.K.; Mukkanti, K.; Pal, S. Ceric Ammonium Nitrate: An Efficient Catalyst for One-Pot Synthesis of 2,2,4-Trimethyl-1,2-dihydroquinolines. Lett. Org. Chem. 2010, 7, 306–310. [Google Scholar] [CrossRef]
- Patel, M.M.; Patel, L.J. Design, synthesis, molecular docking, and antibacterial evaluation of some novel flouroquinolone derivatives as potent antibacterial agent. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, J.; Jaroszewska, K.; Nowakowska-Bogdan, E.; Szmatoła, M.; Iłowska, J. Synthesis of 2,2,4-trimethyl-1,2- H -dihydroquinoline (TMQ) over selected organosulfonic acid silica catalysts: Selectivity aspects. Mol. Catal. 2018, 454, 94–103. [Google Scholar] [CrossRef]
- Dadhania, H.; Raval, D.; Dadhania, A. A Highly Efficient and Solvent-Free Approach for the Synthesis of Quinolines and Fused Polycyclic Quinolines Catalyzed by Magnetite Nanoparticle-Supported Acidic Ionic Liquid. Polycycl. Aromat. Compd. 2021, 41, 440–453. [Google Scholar] [CrossRef]
- Dabiri, M.; Azimi, S.C.; Bazgir, A. An efficient and rapid approach to quinolines via Friedländer synthesis catalyzed by silica gel supported sodium hydrogen sulfate under solvent-free conditions. Monatshefte Fuer Chemie/chemical Mon. 2007, 138, 659–661. [Google Scholar] [CrossRef]
- Muthukrishnan, I.; Sridharan, V.; Menéndez, J.C. Progress in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2019, 119, 5057–5191. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rachwal, S.; Rachwal, B. Recent progress in the synthesis of 1,2,3,4,-tetrahydroquinolines. Tetrahedron 1996, 52, 15031–15070. [Google Scholar] [CrossRef]
- Saadeh, H.A.; Sweidan, K.A.; Mubarak, M.S. Recent advances in the synthesis and biological activity of 8-hydroxyquinolines. Molecules 2020, 25, 4321. [Google Scholar] [CrossRef] [PubMed]
- Kljun, J.; León, I.E.; Peršič, Š.; Cadavid-Vargas, J.F.; Etcheverry, S.B.; He, W.; Bai, Y.; Turel, I. Synthesis and biological characterization of organoruthenium complexes with 8-hydroxyquinolines. J. Inorg. Biochem. 2018, 186, 187–196. [Google Scholar] [CrossRef]
- Heiskanen, J.P.; Omar, W.A.E.; Ylikunnari, M.K.; Haavisto, K.M.; Juan, M.J.; Hormi, O.E.O. Synthesis of 4-alkoxy-8-hydroxyquinolines. J. Org. Chem. 2007, 72, 920–922. [Google Scholar] [CrossRef]
- Zhang, L.; Wen, G.; Xiu, Q.; Guo, L.; Deng, J.; Zhong, C. Synthesis and photovoltaic properties of polymeric metal complexes containing 8-hydroxyquinoline as dye sensitizers for dye-sensitized solar cells. J. Co-ord. Chem. 2012, 65, 1632–1644. [Google Scholar] [CrossRef]
- Chen, R.; Yang, X.; Tian, H.; Wang, X.; Hagfeldt, A.; Sun, L. Effect of tetrahydroquinoline dyes structure on the performance of organic dye-sensitized solar cells. Chem. Mater. 2007, 19, 4007–4015. [Google Scholar] [CrossRef]
- Tian, H.; Yang, X.; Cong, J.; Chen, R.; Teng, C.; Liu, J.; Hao, Y.; Wang, L.; Sun, L. Effect of different electron donating groups on the performance of dye-sensitized solar cells. Dye. Pigment. 2010, 84, 62–68. [Google Scholar] [CrossRef]
- El-Ghaffar, M.A.A.; Shaffei, K.A.; Abdelwahab, N. Evaluation of some conducting polymers as novel antioxidants for rubber vulcanizates. Int. J. Polym. Sci. 2014, 2014. [Google Scholar] [CrossRef]
- Šmejkal, F.; Číhová, A.; Popl, M.; Novák, J. The analysis of polymeric 2,2,4-trimethyl-1,2-dihydroquinoline. Die Angew. Makromol. Chemie 1980, 88. [Google Scholar] [CrossRef]
- Saldanha, L.; Langel, Ü.; Vale, N. In Silico Studies to Support Vaccine Development. Pharmaceutics 2023, 15, 654. [Google Scholar] [CrossRef]
- Ratra, S.; Pant, B.; Roy, K.; Manohar, S.; Kumar, P.; Singh, S.; Tumba, K.; Kumari, K.; Singh, P. A review on synthesis of antiviral drugs, in silico studies and their toxicity. J. Indian Chem. Soc. 2023, 100. [Google Scholar] [CrossRef]
- Matin, M.M.; Bhattacharjee, S.C.; Chakraborty, P.; Alam, M.S. Synthesis, PASS predication, in vitro antimicrobial evaluation and pharmacokinetic study of novel n-octyl glucopyranoside esters. Carbohydr. Res. 2019, 485, 107812. [Google Scholar] [CrossRef]
- Tian, K.; Hu, D.; Hu, R.; Wang, S.; Li, S.; Li, Y.; Yang, G. Multiple fluorescence ΔCIE and ΔRGB codes for sensing volatile organic compounds with a wide range of responses. Chem. Commun. 2011, 47, 10052–10054. [Google Scholar] [CrossRef] [PubMed]
- Manahelohe, G.M.; Potapov, A.Y.; Shikhaliev, K.S. Synthesis of new hydroquinolinecarbaldehydes. Russ. Chem. Bull. 2016, 65, 1145–1147. [Google Scholar] [CrossRef]
- Манахелoхе, Г.М. СИНТЕЗ НОВЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СИСТЕМ НА ОСНОВЕ ФОРМИЛГИДРОХИНОЛИНОВ. ДИССЕРТАЦИЯ, ВОРОНЕЖСКИЙ ГОСУДАРСТВЕННЫЙУНИВЕРСИТЕТ, 2015.
- Wang, E.-C.; Lin, G.-J. ChemInform Abstract: A New One-Pot Method for the Conversion of Aldehydes into Nitriles Using Hydroxyamine and Phthalic Anhydride. ChemInform 2010, 29. [Google Scholar] [CrossRef]
- Wang, E.-C.; Lin, G.-J. A new one pot method for the conversion of aldehydes into nitriles using hydroxyamine and phthalic anhydride. Tetrahedron Lett. 1998, 39, 4047–4050. [Google Scholar] [CrossRef]
- Sampath Kumar, H.M.; Reddy, B.V.S.; Reddy, P.T.; Yadav, J.S. Efficient one-pot preparation of nitriles from aldehydes using N- methyl-pyrrolidone. Synthesis 1999, 1999, 586–587. [Google Scholar] [CrossRef]
- Sosnovsky, G.; Krogh, J.A.; Umhoefer, S.G. A one-flask conversion of aldehydes to nitriles using hydroxylamine hydrochloride and selenium dioxide. Synthesis (Stuttg) 1979, 1979, 722–724. [Google Scholar] [CrossRef]
- Talukdar, S.; Hsu, J.-L.; Chou, T.-C.; Fang, J.-M. Direct transformation of aldehydes to nitriles using iodine in ammonia water. Tetrahedron Lett. 2001, 42, 1103–1105. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M. Direct conversion of aldehydes to amides, tetrazoles, and triazines in aqueous media by one-pot tandem reactions. J. Org. Chem. 2003, 68, 1158–1160. [Google Scholar] [CrossRef]
- El-Hady, H.A.; Abubshait, S.A. Synthesis and anticancer evaluation of imidazolinone and benzoxazole derivatives. Arab. J. Chem. 2017, 10, S3725–S3731. [Google Scholar] [CrossRef]
- Rao, Y.S.; Filler, R. Geometric Isomers of 2-Aryl(Aralkyl)-4-arylidene(alkylidene)-5(4H)-oxazolones. Synthesis 1975, 1975, 749–764. [Google Scholar] [CrossRef]
- Yavari, I.; Anary-Abbasinejad, M.; Alizadeh, A. A Simple Synthesis of Stable 1,4-Diionic Pyridinium Betaines. ChemInform 2003, 34. [Google Scholar] [CrossRef]
- Yavari, I.; Hossaini, Z. Synthesis of fused α-methylene-γ-butyrolactone derivatives through pyridine-induced addition of phenols to dimethyl acetylenedicarboxylate. Tetrahedron Lett. 2006, 47, 4465–4468. [Google Scholar] [CrossRef]
- De, S.K.; Gibbs, R.A. An Efficient and Practical Procedure for the Synthesis of 4-Substituted Coumarins. Synthesis (Stuttg) 2005, 2005, 1231–1233. [Google Scholar] [CrossRef]
- Hirbod, K.; Jalili-Baleh, L.; Nadri, H.; Ebrahimi, S.E.S.; Moradi, A.; Pakseresht, B.; Foroumadi, A.; Shafiee, A.; Khoobi, M. Coumarin derivatives bearing benzoheterocycle moiety: synthesis, cholinesterase inhibitory, and docking simulation study. Iran. J. Basic Med. Sci. 2017, 20, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Alheety, K.A.; Jamel, N.M.; Ahmed, B.J. Synthesis of coumarin by Pechman reaction - A Review. J. Pharm. Sci. Res. 2019, 11. [Google Scholar]
- Undale, K.A.; Gaikwad, D.S.; Shaikh, T.S.; Desai, U.V.; Pore, D.M. Potassium phosphate catalyzed efficient synthesis of 3-carboxycoumarins. Indian J. Chem. - Sect. B Org. Med. Chem. 2012, 51. [Google Scholar]
- Prajapati, D.; Sandhu, J.S. Cadmium iodide as a new catalyst for Knoevenagel condensations. J. Chem. Soc. Perkin Trans. 1 1993, 739–740. [Google Scholar] [CrossRef]
- Mhiri, C.; El Gharbi, R.; Le Bigot, Y. Polymer supported reagents: Novel methodology for selective and general synthesis of iminocoumarins. Synth. Commun. 1999, 29, 3385–3399. [Google Scholar] [CrossRef]
- Brahmachari, G.; Das, S. Sodium Formate-Catalyzed One-Pot Synthesis of Benzopyranopyrimidines and 4-Thio-substituted 4H-Chromenes via Multicomponent Reaction at Room Temperature. J. Heterocycl. Chem. 2015, 52, 653–659. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Kireev, A.S.; Yakovenko, A.A.; Antipin, M.Y.; Magedov, I.V.; Kornienko, A. Convenient one-step synthesis of a medicinally relevant benzopyranopyridine system. Tetrahedron Lett. 2006, 47, 9309–9312. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, N.M.; Kireev, A.S.; Yakovenko, A.A.; Antipin, M.Y.; Magedov, I.V.; Kornienko, A. One-Step Synthesis of Heterocyclic Privileged Medicinal Scaffolds by a Multicomponent Reaction of Malononitrile with Aldehydes and Thiols. ChemInform 2007, 38. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of 6(8)-Formyl-N-alkyl-2,2,4-trimethyl-1,2-di(1,2,3,4-tetra)hydroquinolines 5a-f and 6a-f via VH formylation reaction.
Scheme 1.
Synthesis of 6(8)-Formyl-N-alkyl-2,2,4-trimethyl-1,2-di(1,2,3,4-tetra)hydroquinolines 5a-f and 6a-f via VH formylation reaction.
Scheme 2.
Synthesis of 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbonitrile 7 and 1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbonitrile 8.
Scheme 2.
Synthesis of 1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbonitrile 7 and 1-benzyl-2,2,4-trimethyl-1,2,3,4-tetrahydroquinoline-6-carbonitrile 8.
Scheme 3.
Synthesis of 2-phenyl-1,3-oxazol-5(4H)-ones 9, 10.
Scheme 4.
Synthesis of triazolo/pyrazolo[1,5-a]pyrimidin-6-yl carboxylic acids 12a,b.
Scheme 6.
Plausible reaction mechanism of methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16.
Scheme 6.
Plausible reaction mechanism of methyl (Z)-2-(5,7,7,8-tetramethyl-2-oxo-7,8-dihydrofuro[3,2-g]quinolin-3(2H)-ylidene)acetate 16.
Scheme 7.
Condensation reaction of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5e with methylene active compounds.
Scheme 7.
Condensation reaction of 7-hydroxy-1,2,2,4-tetramethyl-1,2-dihydroquinoline-6-carbaldehyde 5e with methylene active compounds.
Scheme 8.
Plausible reaction mechanism of formation of 3-R-6,8,8,9-tetramethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 19a-d.
Scheme 8.
Plausible reaction mechanism of formation of 3-R-6,8,8,9-tetramethyl-8,9-dihydro-2H-pyrano[3,2-g]quinolin-2-one 19a-d.
Scheme 9.
Plausible reaction mechanism of formation of 6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carboxylic acid 21.
Scheme 9.
Plausible reaction mechanism of formation of 6,8,8,9-tetramethyl-2-oxo-8,9-dihydro-2H-pyrano[3,2-g]quinoline-3-carboxylic acid 21.
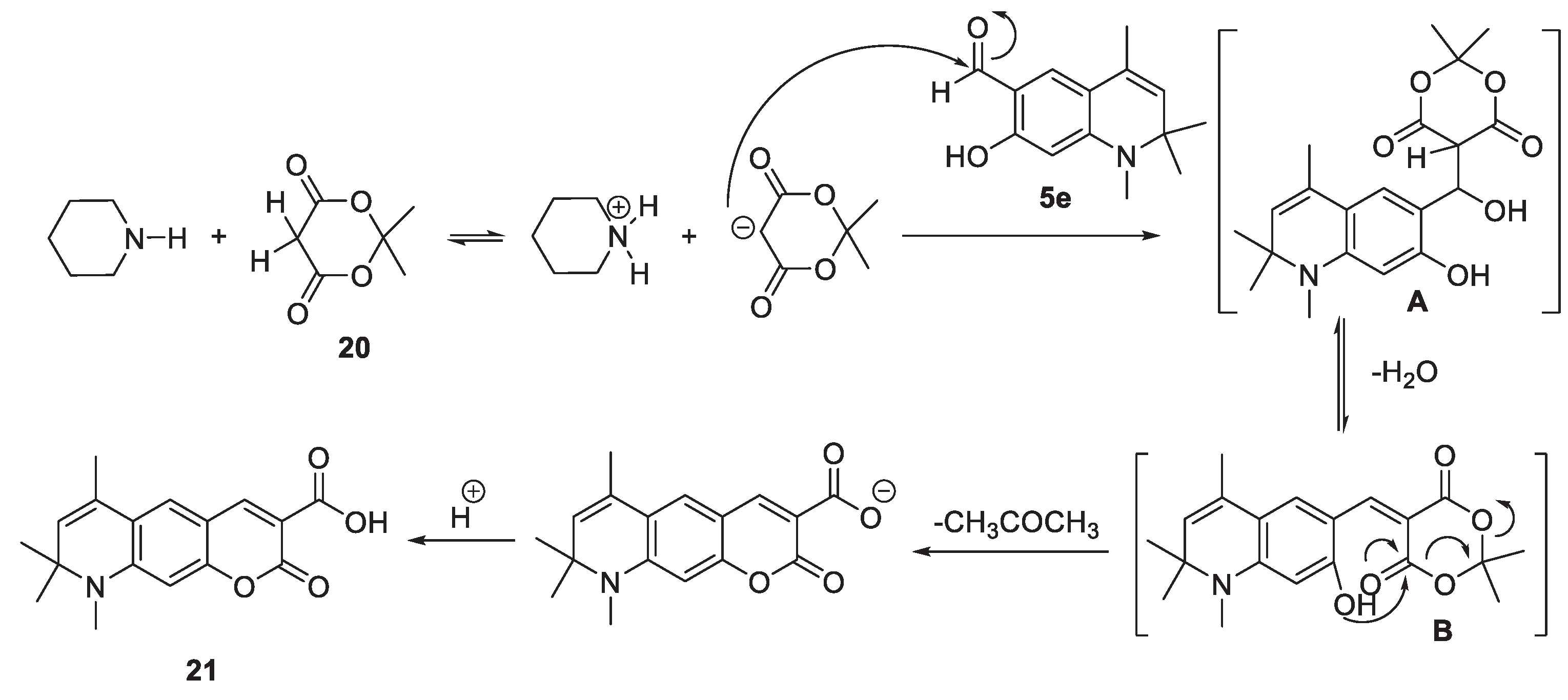
Scheme 10.
Plausible reaction mechanism of formation of 2,5-diamino-9,10,10,12-tetramethyl-9,10-dihydropyrido[4′,3′,2′:8,1]isochromeno[4,3-g]quinoline-1,4-dicarbonitrile 22.
Scheme 10.
Plausible reaction mechanism of formation of 2,5-diamino-9,10,10,12-tetramethyl-9,10-dihydropyrido[4′,3′,2′:8,1]isochromeno[4,3-g]quinoline-1,4-dicarbonitrile 22.
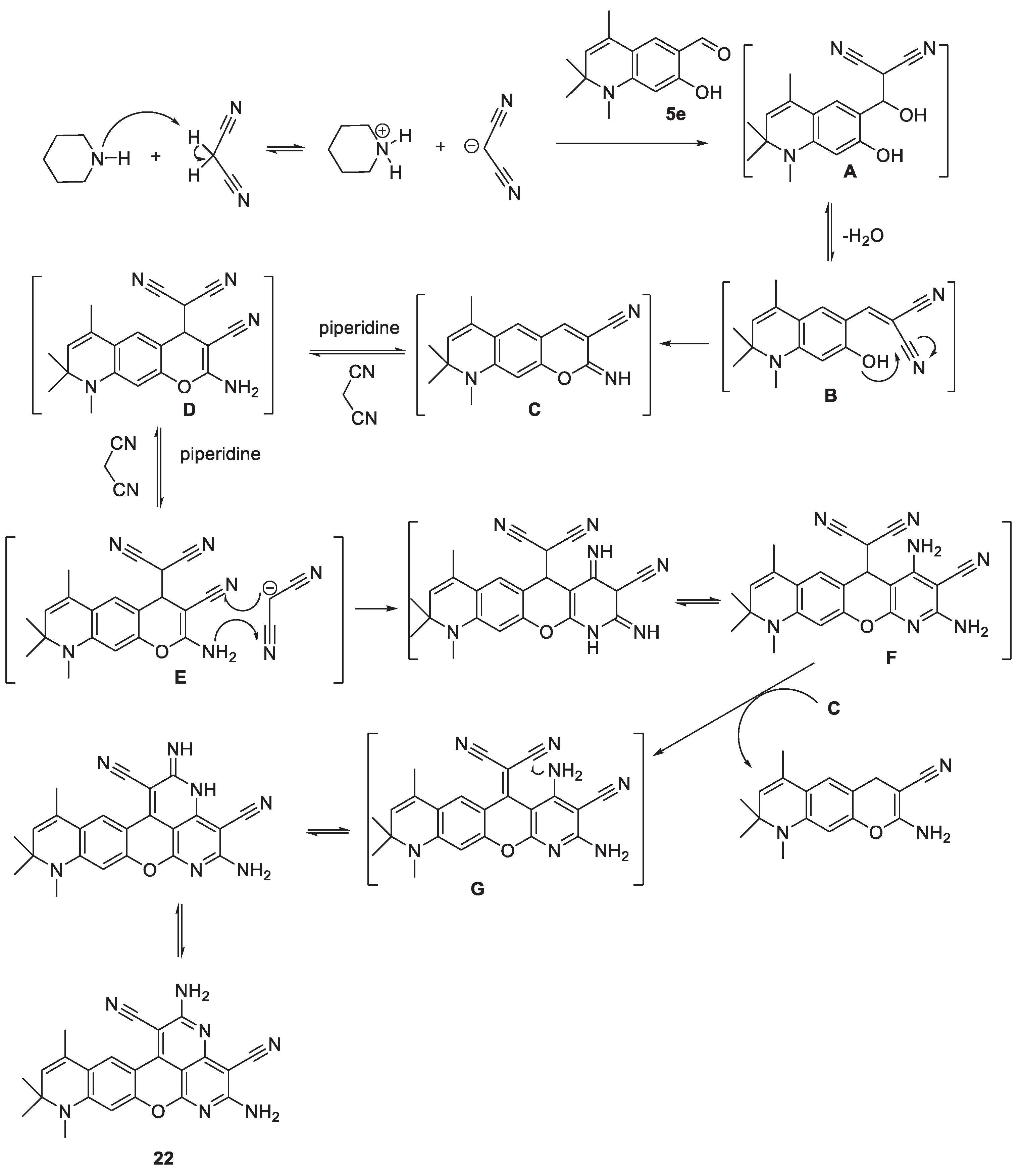

Table 1.
Substituents of the Synthesized 6(8)-Formyl-N-alkyl-2,2,4-trimethyl-1,2-di(1,2,3,4-tetra)hydroquinolines 5 and 6.
Table 1.
Substituents of the Synthesized 6(8)-Formyl-N-alkyl-2,2,4-trimethyl-1,2-di(1,2,3,4-tetra)hydroquinolines 5 and 6.
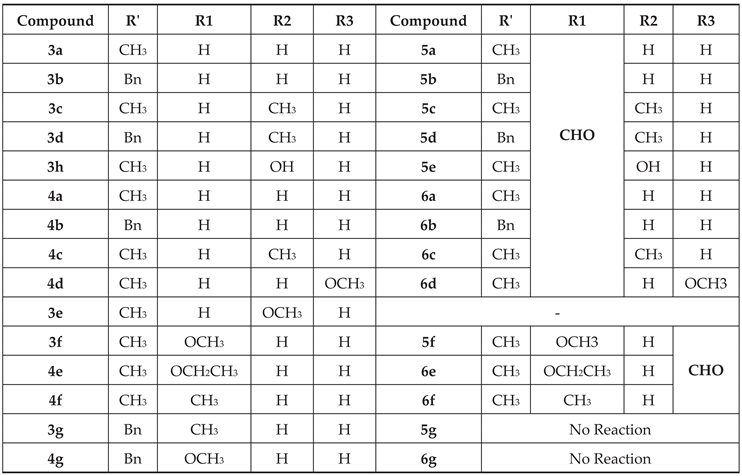
Table 2.
PASS the predicted activity of test compounds.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



