Submitted:
25 February 2024
Posted:
26 February 2024
You are already at the latest version
Abstract
Despite advances in our understanding of molecular aspects of oncogenesis, cancer remains a leading cause of death. The malignant behavior of a cancer cell is driven by the inappropriate activation of transcription factors. In particular, signal transducers and activators of transcription (STATs), which regulate many critical cellular processes such as proliferation, apoptosis, and differentiation, are frequently activated inappropriately in a wide spectrum of human cancers. Multiple signaling pathways converge on the STATs, highlighting their importance in the development and progression of oncogenic diseases. STAT3 and STAT5 are two members of the STAT protein family that are the most frequently activated in cancers and can drive cancer pathogenesis directly. The development of inhibitors targeting STAT3 and STAT5 has been the subject of intense investigations in the last decade, although effective treatment options remain limited. In this review, we investigate the specific roles of STAT3 and STAT5 in normal physiology and cancer biology, discuss the opportunities and challenges in pharmacologically targeting STAT proteins and their upstream activators, and offer insights into novel therapeutic strategies to identify STAT inhibitors as cancer therapeutics.
Keywords:
STAT3
; STAT5
; cancer
; immunity
; cancer therapy
1. Introduction: STAT transcription factors
Cancer is the second leading cause of death after cardiovascular diseases worldwide, accounting for nearly 10 million deaths in 2020, or nearly one in six deaths [1]. Despite advances in our understanding of mechanisms of cancer pathogenesis and the development of novel modes of therapy, most advanced cancers remain incurable. To develop novel therapies that have greater efficacy and less toxicity, there is an interest in identifying cellular pathways on which malignant cells, but not normal cells, are dependent. Since the phenotype of a cancer cell, including properties such as invasion and metastasis, are driven by the pattern of gene expression, there is a particular interest in identifying transcription factors that control critical cellular processes, and that become activated inappropriately in cancer. One such family of transcription factors is the STAT family.
The STAT family was discovered as key mediators of cytokine signaling and interferon (IFN)-related anti-viral activity in the late 1980s and early 1990s [2,3,4]. STATs, an acronym for signal transducers and activator of transcription, comprise seven members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. These proteins share a conserved structure and a common mechanism of action, but have different functions in normal cells and in tumor biology [5]. Their structure is characterized in sequence by an N-terminal domain, a coiled-coil domain, a DNA-binding domain, a linker region, a phosphotyrosine-binding Src homology 2 (SH2) domain, and a C-terminal transactivation domain. Under basal conditions, STATs are found in the cytoplasm of cells as inactive dimers. When they become phosphorylated on a single tyrosine residue towards the carboxy terminus, they undergo a conformational change leading to reciprocal phosphotyrosine-SH2 interactions. This reveals a nuclear localization signal that allows the STAT dimers to translocate from the cytoplasm to the nucleus, and to bind specific nine or ten base pair motifs in the regulatory regions of target genes. Although their name indicates that they activate the transcription of target genes, like most transcription factors, STATs can also repress expression of genes in a context-dependent fashion.
STATs are commonly activated in response to cytokines, many of whose receptors are associated with one or more of the four JAK family kinases (JAK1, JAK2, JAK3, and TYK2). Hence, signaling through these transcription factors is often categorized by the shorthand “JAK-STAT” signaling. However, given the broad range of genes regulated by these seven STAT family members in different cell types, this designation over-simplifies the diversity and complexity of the role of these proteins. The designation “JAK-STAT” also does not take into consideration the fact that many tyrosine kinases other than JAK family members can phosphorylate and activate STAT proteins. This includes receptor tyrosine kinases (including the receptors for epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), and others) as well as non-receptor tyrosine kinases of the SRC family and others. Not only can STATs be activated in response to cytokines and growth factors, but STATs can also be activated downstream of cell-cell and cell-matrix interactions.
STAT tyrosine phosphorylation and nuclear translocation occurs within seconds of cytokine stimulation. Reflecting the critical functions of target genes regulated by STATs, they are also inactivated very rapidly by a number of mechanisms, including protein tyrosine phosphatases (PTPs), protein inhibitors of activated STAT (PIAS), and suppressors of cytokine signaling proteins (SOCS) [6]. Nuclear PTPs can dephosphorylate STAT proteins, which leads to their inactivation and subsequent transport out of the nucleus [7]. SOCS proteins function in the cytoplasm and can bind various phosphotyrosines on intracellular receptors, blocking STATs from their native docking sites [8]. PIAS proteins can bind to phosphorylated STAT dimers, thus preventing DNA recognition and STAT-mediated signaling cascades [9,10].
2. Inappropriate activation of STATs in cancer
As noted, the seven STAT family members are activated in a wide array of cell types and regulate expression of many different genes. However, reflecting their role in directing key biological programs in response to cytokines and other extracellular cues, the target genes of STATs have certain commonalities [11]. Many STAT target genes regulate core cellular processes such as survival, proliferation, self-renewal, and differentiation. Functionally, STAT activation underlies processes such as cell motility, invasion, angiogenesis, and immune function and recognition [12,13]. Notably, STATs also regulate the expression of other transcription factors (including themselves). Thus, STAT activation leads to the initiation of multiple transcriptional programs in a coordinated way and amplifies its own response. Consequently, the tight regulation of STAT function is important for physiologic homeostasis. On the other hand, inappropriate activation of STATs, either through increased activity of upstream kinases, decreased expression or function of negative regulators, or both, can lead to significant perturbations in cellular function [12]. In fact, shortly after the discovery of these proteins, it was observed that inappropriate or constitutive phosphorylation and activation of these proteins occurred commonly in human cancer and model systems of malignancy, and this can occur through a variety of mechanisms [14,15] (Figure 1).
2.2. STAT3
STAT3, was originally described as “acute phase response factor (APRF),” in that it mediates the effects of interleukin-6 (IL-6) and related cytokines in the response to tissue injury, inflammation, and infection. Its target genes regulate cell proliferation, differentiation, apoptosis, angiogenesis, inflammation, and immune responses [16,17] (Table 1). Constitutive or inappropriate activation of STAT3 has been reported in many types of cancers [18,19,20]. Generally, this is due to increased phosphorylation or decreased inactivation of STAT3. For example, heat shock protein 110 can directly bind to STAT3 and facilitate its phosphorylation, contributing to tumor growth in colon cancers [21]. Activated Notch1 receptor increases the level of phosphorylated STAT3, promoting cancer progression in gastric cancer [22]. Silencing of SOCS3 causes decreased inactivation of STAT3, which also contributes to its constitutive activation and thus the progression of cancers such as hepatocellular carcinoma and cholangiocarcinoma [23,24,25]. Persistent STAT3 activation plays a central role in tumorigenesis [26,27]. It can promote the transcription of target genes such as BCL-6, MCL1, MYC to promote proliferation and survival [28]. it can also activate genes such as HGF, VEGF, and HIF-1α while decreasing the expression of target genes such as IL-12 and p53 to promote angiogenesis [29]. In addition, STAT3 is widely involved in metastasis by increasing the expression of genes such as MMP2/9, Twist, and Vimentin [30].
In some forms of cancer, such as large granular lymphocytic T-cell leukemia, mutations occurring in STAT3 itself leads to increased magnitude or duration of STAT3 phosphorylation [5,42,43,44]. Large granular lymphocyte (LGL) leukemia is an indolent lymphoproliferative disorder that belongs to mature T and natural killer (NK) cell neoplasms in the 2016 World Health Organization classification [45,46]. There are two types of LGL leukemia: T-cell (T-LGL) and natural killer cell (NK-LGL). Somatic gain-of-function STAT3 mutations are demonstrated in 28-75% of T-LGL leukemia and 30-48% of NK-LGL leukemia [47]. Most STAT3 mutations are detected in the SH2 domain, which drives the dimerization and activation of the STAT protein [48]. The amino acid changes result in a more hydrophobic protein surface and are associated with phosphorylation of STAT3 and its localization in the nucleus [49]. Mutations outside the SH2 domain are rare but have been found in the DNA-binding and coiled-coil domains [50]. It has been suggested that STAT3 mutations may not be the initial trigger of the leukemic process in LGL leukemia [51]. This is supported by pre-clinical evidence that expression of a STAT3 mutant alone is not sufficient to induce LGLL in animal models and that inhibition of STAT3 restores apoptosis of LGL cells regardless of the STAT3 mutation status [47,52]. This is also supported by clinical evidence that STAT3-unmutated patients show hyperactive STAT3 but STAT3-mutated cases may remain in subclinical states for a very long time, sometimes indefinitely [51,53]. Rather, STAT3 mutations have been found to cause a higher level of transcription of survival components, thus conferring a competitive growth advantage on clonal accumulation and autoimmunity [53,54,55].
Aberrant STAT3 activation can promote oncogenesis by its cell-autonomous effects in a cancer cell [16]. Other non-malignant cells in the surrounding area can also have increased STAT3 activation since STAT3 can become activated in the tumor microenvironment in part due to the presence of cytokines that activate this protein, like IL-6 [56]. This can be of particular significance in immune cells, in which enhanced STAT3 activation can lead to decreased antigen presentation and immune effector function [57]. In innate immunity, STAT3 regulates critical steps during emergency granulopoiesis to help contain infection, restrains neutrophil production to limit inflammatory responses, and suppresses the maturation and activation of dendritic cells to induce immunosuppressive effects [58,59,60,61]. In adaptive immunity, STAT3 positively regulates an early step in B-cell development and promotes the differentiation and maturation of plasma cells [62,63]. It can also promote the proliferation and diversity of CD4+ T cells as well as generate stable, long-lived CD8+ memory T cells [64,65]. However, in aggregate, STAT3 activation in immune cells helps create an immunosuppressive microenvironment that is permissive to the maintenance and spread of cancer cells [66].
2.3. STAT5
STAT5 refers to two highly related proteins, STAT5A and STAT5B, that are encoded by adjacent highly homologous genes which likely arose through gene duplication. STAT5 transduces signals from a number of cytokines that regulate hematopoiesis at the level of hematopoietic stem cells, hematopoietic progenitor cells, and mature cell populations [67] (Table 2). In hematopoietic stem and progenitor cells, STAT5 is required for cellular “fitness”, with STAT5 deficiency resulting in greatly impaired long-term multilineage competitive repopulation capacity [68,69,70]. In natural killer cells, STAT5 helps mediate cell development, maturation, and homoeostasis [71]. In adaptive immune cells, STAT5 plays a critical role in differentiation and development [72,73] and promotes the differentiation of B cells [74]. It also promotes cell proliferation and active cellular uptake of carnitine in CD4+ T cells, and enhances BCL2 expression and cell survival of CD8+ T cells [75,76,77,78]. It becomes aberrantly activated mostly due to increased phosphorylation or due to the loss of negative regulators [79]. Constitutive activation of STAT5 has been shown to be a direct leukemia driver [80]. As with STAT3, STAT5 can rarely become activated through mutation in a STAT5 isoform itself. STAT5 mutations are commonly found in human hematologic cancers, such as T-cell prolymphocytic leukemia, B-cell and T-cell acute lymphoblastic leukemia, and γδ T-cell-derived lymphomas [5,81,82,83].
STAT5 is expressed in a wide array of mammalian tissues [85]. It was first identified as “mammary gland factor,” a transcription factor that mediates the effects of prolactin on mammary epithelial cells. It is critical for the growth and differentiation of alveolar progenitors as well as the survival of secretory mammary epithelial cells during normal mammary gland development [86]. Aberrant activation of STAT5 is commonly found in breast cancer, though it is generally associated with more differentiated hormone-responsive tumors, reflecting its physiologic role [87]. It promotes cell survival and instigates breast tumor formation, drug resistance, and metastatic capabilities of breast cancers [88].
In some tumor types, such as acute leukemias and breast cancer, either STAT3, STAT5, or both can be found to be activated inappropriately [89,90]. Although the canonical binding sites for STAT3 and STAT5 appear identical in isolated DNA, their transcriptional and biological effects are not identical [89,90]. For example, STAT3 and STAT5 have antagonistic effects in the regulation of the transcriptional modulator BCL6, which is a master transcription factor in the regulation and proliferation of B cells and T follicular helper cells [91]. While STAT5 and STAT3 can compete for binding sites to regulate BCL6 expression and lead to opposite effects, STAT5-mediated repression of BCL6 is usually dominant over STAT3-mediated induction because it can displace STAT3 from regulatory regions to which it binds [90,92]. Reflecting these transcriptional effects, co-activation of STAT3 and STAT5 in breast cancer is associated with the more differentiated phenotype of breast cancers than with STAT3 activation alone [89]. This finding may have implications for the effects of inhibitors of specific STATs in cancers in which more than one STAT is activated inappropriately.
2.4. Other STATs
Although STAT3 and STAT5 are the STAT family members most widely associated with cancer pathogenesis, other STATs can become activated inappropriately in cancer cells and play important biological roles. For example, STAT1 can be activated by various ligands including IFN-α, IFN-γ, EGF, platelet-derived growth factor, and IL-6. It has a key role in regulating genes that modulate cell survival, viability, and pathogen response. Reflecting its key role as a mediator of interferon signaling, germline mutations in STAT1 are associated with immunodeficiency [93]. STAT1 mainly acts as an inhibitor of cancer as its expression is associated with a better prognosis [94]. However, reflecting the context-dependency of all STATs, STAT1 was also found to act as a cancer promoter in a mouse model of leukemia [95].
STAT2 is principally activated by type I IFNs [96], and it can form a complex with STAT1 to mediate innate antiviral activity. Mutations in this gene result in immunodeficiency [97]. It can either inhibit or promote tumorigenesis depending on the unique environment presented by each type of cancer [98]. Overexpression of STAT2 is associated with either better or worse outcome in human cancers of the skin, head and neck, kidney, lung, ovary, and endometrium [99,100,101,102].
STAT4 can be activated by cytokines like IFN-α, IFN-β, IL-12, and IL-23 [103,104]. It is required for the maturation of T cells and for IFN-γ production [105]. Overexpression of STAT4 can be associated with either better or worse outcome in cancers depending on the types of cancer, and can induce apoptosis in cervical cancer cells [106,107,108].
Similarly, as other STAT family proteins, STAT6 can be activated by growth factors and cytokines such as IL-4 and IL-13 [109]. STAT6 is required for the differentiation of T helper 2 cells, the maturation of B cells, as well as the M2 subtype activation of macrophages [110,111]. STAT6 has been reported to be highly expressed in several types of cancer, including breast, pancreatic, prostate and colorectal cancer [112] It promotes cancer growth, development, invasion and metastasis by mechanisms such as decreasing the cytotoxic activity of CD8 T cells and favoring the expression of anti-apoptotic proteins and epithelial cell growth [113,114].
3. Targeting STATs for cancer therapy
It is clear that inappropriately activated STATs directly drive the malignant phenotype
of cancer cells. However, the key issue in developing cancer therapeutics is to have an acceptable therapeutic index, the ability to kill cancer cells without harming normal cells [115]. Given the central role that STATs play in so many central physiologic processes, there was a concern that STAT inhibitors might have unacceptable toxicities [12]. However, evidence form a number of areas has suggested that STAT inhibition can be well-tolerated, especially for relatively brief intervals needed for cancer therapy [116,117,118].
One piece of evidence of the tolerability of STAT3 inhibition came from the discovery that the inherited hyper-IgE syndrome (sometimes referred to as Job’s syndrome) is caused by one of several mutations in STAT3 [119]. Not only do these mutations inactivate the transcriptional function of the affected allele but given the requirement that STAT3 be in a dimer to mediate transcriptional regulation, these mutant forms of STAT3 act in a dominant inhibitory manner [120]. Consequently, individual with these inherited mutations have greatly reduced STAT3 transcriptional activity from the time of conception [121]. While the hyper-IgE syndrome is characterized by a variety of inflammatory and immune-deficient effects, these individuals otherwise develop normally [122]. Thus, severe attenuation of STAT3 function in every cell in the body throughout development and beyond can be tolerated. This finding, coupled with evidence from animal models and the use of pharmacologic inhibitors of STAT function have reinforced the conjecture that inhibition of STAT transcriptional function could be a targeted form of cancer therapy which would be well tolerated [123,124]. Furthermore, since STATs sit at a convergence point of multiple upstream kinases, targeting STATs hold the potential to have efficacy in a broad array of cancers.
One of the most significant recent advances in cancer therapy has come through the development of kinase inhibitors, which can target mutated, over-expressed, or even normally expressed wildtype kinases to which the cancer cell has become dependent or “addicted” [125,126]. While these agents can be enormously effective in treating patients even with advanced disease, a near-universal issue with these drugs, however, is the emergence of resistance [127,128]. Resistance to targeted anti-cancer therapies like kinase inhibitors often arises from the activation of another tyrosine kinase or a parallel signaling pathway. For example, one mechanism for the emergence of resistance to inhibitors of the EGF receptor tyrosine kinase in patients with non-small cell lung cancer is the activation of the MET receptor tyrosine kinase [129]. These parallel signaling pathways often still converge on the same small number of oncogenic transcription factors, like STATs [130,131]. Therefore, in addition to the direct therapeutic benefits from targeting STAT proteins, their position as convergence points for multiple signaling pathways holds the promise that it will be more difficult for resistance to develop to these agents, and that they may be particularly useful in combination with other targeted therapies [132].
In considering how best to therapeutically target STATs, three main approaches have been pursued, which will be discussed in more detail. The first is to target STAT proteins directly. This has often been challenging, as transcription factors have generally been viewed as structurally difficult to inhibit with small molecules [133]. This reflects the fact that, as opposed to kinases, which have discrete ATP-binding domains in which a small organic molecule can be designed to bind, transcription factors have large flat surfaces that allow them to interact with DNA or other proteins [134]. Nonetheless, STATs possess structural elements like SH2 domains that have been used for direct targeting [135,136]. The second approach is to use screening strategies, such as with chemical biology or computational methods, to identify compounds that specifically inhibit STAT-dependent transcription [137,138]. The third approach is to target steps in STAT activation that are directly upstream of STAT phosphorylation [139]. Most therapeutic development in this area has been directed at STAT3, though, as noted, other STATs may also play pathogenic roles in a variety of cancers (Figure 2).
4. Strategies to directly target STATs
4.1. Direct STAT binding molecules
Almost all compounds designed to target STAT3 in clinical development currently do so by targeting the SH2 domain of the protein [135] (Table 3). Since the STAT3 SH2 domain participates in dimerization with another STAT3 protein to form a STAT3 homodimer, or with a STAT1 protein to form a STAT1-STAT3 heterodimer, peptides that can bind to the STAT3 SH2 domain are relatively easy to design [140]. However, small peptides, particularly ones that need to be phosphorylated or otherwise have a strong negative charge like one that would bind to the STAT3 SH2 domain, generally have unfavorable pharmacologic characteristics including rapid degradation and limited intracellular penetration [141]. Nonetheless, non-peptide molecules derived from this approach have been made, and have shown some efficacy in model systems [136,142]. One potential shortcoming of this approach reflects the fact that SH2 domains have some latitude in the sequences that they can bind and different SH2 domains can bind to the same tyrosine-phosphorylated sequence [143]. Consequently, it is difficult to develop molecules that can bind to the STAT3 SH2 domain with a high degree of specificity. Nonetheless, a molecule that bound with high specificity to the STAT3 SH2 domain would be appealing, as it would both inhibit the recruitment of STAT3 to activated receptor-kinase complexes and would also block the activating dimerization of STAT3.
Among the small molecules, BP-1-102 and its two analogs were designed as direct STAT3 inhibitors with reasonable in vivo tumor-inhibiting activity by binding specifically to the STAT3 SH2 domain [158]. N4 is another small molecule that has potent antitumor bioactivity. It directly binds to the STAT3 SH2 domain, and thereby inhibits the STAT3 dimerization and STAT3-NF-κB cross-talk [136]. Betulinic acid is a plant-derived compound that has been found to induce cells apoptosis in cervical cancer. It has a very high affinity to STAT3 SH2, and thus also inhibits STAT3 phosphorylation, as would be predicted by this mechanism of action [159,160].
4.2. STAT degraders
Although direct STAT binding molecules such as small-molecule STAT3 inhibitors are promising therapeutic strategies, they often have selectivity problems and show very limited clinical activity [130]. In recent years, targeted protein degradation (TPD) strategies that leverage the endogenous protein destruction machinery of a cell to remove specific disease-associated proteins have emerged as promising approaches. Proteolysis-targeting chimeras (PROTAC) and molecular glues are two examples of TPD strategies [161].
PROTACs, also known as bivalent chemical protein degraders, are heterobifunctional molecules that degrade specific endogenous proteins through the E3 ubiquitin ligase pathway [162,163,164]. PROTACs have the potential to be better tolerated than traditional small molecule inhibitors as PROTACs exert their effects through a repeated and iterative mode of action to induce target protein degradation rather than competing with active sites like traditional small molecule inhibitors [165]. Several PROTAC degraders have been developed to target STATs [164]. Many of these molecules employ a STAT3-binding component that binds to the STAT3 SH2 domain. For example, SD-36 consists of an analogue of the CRBN ligand lenalidomide, a linker, and the SH2-targeting STAT3 inhibitor SI-109 [166]. It achieves tumor regression in multiple xenograft models, including acute myeloid leukemia, anaplastic large-cell lymphoma, and glioma [118,167]. SD-91 is another STAT3 degrader that is capable of achieving complete and long-lasting tumor regression in xenograft models of megakaryoblastic leukemia [168].
Recently, a phase 1 clinical trial was initiated using the STAT3 degrader KT-333 in a variety of hematologic cancers and solid tumors [169]. In addition to evidence of some clinical activity, robust pharmacodynamic data are being obtained. These show significant down regulation of STAT3 protein expression, reflecting an on-target effect [169]. Furthermore, the canonical STAT3 target gene SOCS3 was downregulated, as were the STAT3-dependent acute-phase inflammatory markers C-reactive protein (CRP) and serum amyloid A (SAA) protein [169].
Molecular glues are small molecules that can induce protein–protein interactions between a ubiquitin ligase and a target protein, which leads to protein ubiquitination and subsequent proteasome-based degradation [170]. Molecular glues may have advantages over heterobifunctional PROTACs, including favorable physicochemical properties [171]. Although no molecular glues targeting STAT3 have been designed, this technology holds great potential to be exploited [172].
5. Targeting upstream kinases: STAT phosphorylation as a biomarker for on-target effects
Since STAT activation in the tumor microenvironment is often driven, at least in part, by the presence of cytokines that can activate STATs, targeting these effects has been an appealing strategy. However, for a number of reasons, targeting upstream steps in these pathways has been therapeutically useful only in a relatively small number of circumstances.
IL-6 can activate STAT3, and IL-6 is often elevated systemically and in the tumor microenvironment in patients with cancer [173]. Furthermore, IL-6 levels can be a negative prognostic indicator in cancer patients. Since antibody-based therapeutics that either bind directly to IL-6 or block the effect of IL-6 by binding to its receptor are already in use for inflammatory and rheumatologic disorders [174,175], several clinical trials have tested this approach (NCT04333706, NCT04940299, NCT02644967). However, by and large, these strategies have shown little clinical benefit. There are several reasons for this. The first is that while IL-6 can lead to the activation of STAT3 in the tumor microenvironment, many other cytokines and growth factors can do so as well. Multiple STAT3-activating cytokines can also be detected in the conditioned medium of primary cancer cells or cancer cell lines grown in vitro [130]. Thus, blocking a single cytokine has limited therapeutic efficacy. Even where successful, the rapid emergence of resistance is likely to occur. While it is true that elevated levels of IL-6 may be associated with a worse prognosis in cancer patients [176], this can also reflect the production of IL-6 in response to the physiologic effects of having advanced cancer, whereby the acute phase response is induced [177]. In that setting, merely inhibiting IL-6 is unlikely to have significant therapeutic benefit.
A second approach is to target kinases downstream of cytokines, such as JAK inhibitors [178]. While JAK inhibitors can decrease STAT3 tyrosine phosphorylation in tumor cells, this approach suffers from two major shortcomings. The first reflects the fact that cytokine receptors are generally associated with two different JAK family members. Receptors that mediate signaling via the gp130 receptor chain (which transduces signals from IL-6, LIF, oncostatin M, IL-13, CNTF, and others) associates with three different JAK family members (JAK1, JAK2, and TYK2) [179]. Therefore, to effectively suppress signaling through these receptors, effective kinase inhibitors would need to suppress almost all cytokine signaling in a patient [180]. Such an approach would lead to unacceptable toxicity [181].
The second limitation of JAK inhibitors, even more selective ones, reflects the fact that the immune response to cancer is often a critical component of the therapeutic response. A key mediator of this effect, both in making tumor cells more visible to immune cells and activating immune cell function, are IFNs [182,183]. JAK inhibitors will generally suppress IFN signaling (which also uses JAK1, JAK2, and TYK2), and thus suppress critical immune effects [6]. This has also limited the clinical applicability of JAK inhibitors in cancer therapy.
Despite these limitations, as outlined below, there are some clinical situations in which inhibition of upstream kinases is an effective approach to suppressing pathogenic STAT signaling. Furthermore, measuring STAT phosphorylation and target gene expression can also be an effective pharmacodynamic marker for the therapeutic activity of kinase inhibitors.
5.1. Inhibiting STAT5 in chronic myeloid leukemia
It has been known since the 1970s that chronic myeloid leukemia (CML) is almost universally associated with a translocation of chromosomes 9 and 22, which leads to a fusion oncoprotein, BCR-ABL1 [184]. This fusion tyrosine kinase can phosphorylate STAT5 directly and also lead to the activation of JAK2 in both CML and some forms of acute lymphoid leukemia (ALL) [185,186,187,188]. As noted, cellular and enzymatic analyses suggest that STAT5 is phosphorylated by BCR-ABL1 directly, and that STAT5 is indispensable for the initial transformation of the leukemia [189]. In contrast, initial myeloid transformation and leukemia maintenance were independent of JAK2. These results suggests that it is more effective to target STAT5 rather than JAK2 to treat BCR-ABL+ diseases [189]. Inhibiting BCR-ABL1 with tyrosine kinase inhibitors like imatinib has revolutionized the treatment of CML, and suppresses STAT5 phosphorylation [190]. Resistance to first generation BCR-ABL1 kinase inhibitors like imatinib can occur. However, compounds that inhibit STAT5 activation through kinase independent mechanisms, like pimozide, still have activity in this setting [191] . Of note, combined inhibition of STAT5 with pimozide may synergize with JAK2 inhibition in models of other myeloproliferative neoplasms [192].
5.2. Targeting kinases upstream of STATs in AML
Given the importance of STATs in mediating the oncogenic effects of upstream signaling pathways, monitoring the phosphorylation of STATs in clinical samples may prove to be an important method to ensure on-target effects by kinase inhibitors. This approach may also be useful as an early means to detect resistance to these therapies. This type of strategy was exemplified in a recent clinical trial for patients with relapsed or refractory acute myeloid leukemia (AML). Mesenchymal Epithelial Transition (MET) is an oncogenic receptor tyrosine kinase that is upregulated or overly activated in many cancers, including hematopoietic cancers like AML [193]. Autocrine production of HGF can activate MET by binding to its ligand FGF receptor (FGFR), and eventually lead to myeloblast growth and survival in acute myeloid leukemia [194,195]. MET inhibitors such as merestinib exhibit activity in AML preclinical studies, but HGF upregulation by the FGFR pathway is a common mechanism of resistance [196]. Based on this background and in vitro studies that suggested efficacy from targeting both the MET and FGF receptor pathways simultaneously in AML, a phase 1 clinical trial using the rational combination of the MET inhibitor merestinib and the FGFR inhibitor LY2874455 was conducted in patients with relapsed or refractory acute myeloid leukemia [197]. Although the study was restricted by limitations in the supply of the drugs, one key finding was that, at least in some subjects, phosphorylation of STAT3 and/or STAT5 correlated closely with clinical response. This suggests that readily available techniques such as flow cytometry could be useful in monitoring and regulating the dose of kinase inhibitors in certain settings. This study also found that in the setting of progressive disease, genes that were upregulated could activate STAT3 and/or STAT5 through alternate mechanisms. This finding again emphasizes the point that STATs, which sit at a convergence point for multiple upstream kinase pathways including MET [198], may be a more effective target than upstream kinases alone [199].
6. Novel ways to identify STAT transcriptional inhibitors
Searching for structural elements in a protein through which therapeutic inhibitors can be developed is a standard approach to drug development [200]. However, transcription factors are not optimal proteins for this strategy. This reflects their relative lack of surfaces to which small organic compounds can bind to lead to functional inhibition [201]. As noted, the potentially targetable motifs that they do possess, such as the SH2 domain, may not confer optimal specificity for drug development [141]. Therefore, alternate approaches to developing compounds that can block STAT-dependent transcriptional activity is appealing. In addition to leading to novel therapeutic compounds, this strategy can help to uncover unappreciated aspects of STAT-dependent gene regulation. In addition, these types of approaches may also be able to identify compounds that can enhance STAT-dependent transcriptional activity, which may be useful in certain applications. Finally, these broad-based and unbiased strategies can also be applied to other transcription factors.
Given that much is known about molecular aspects of STAT transcriptional regulation, two screening strategies that have leveraged this information have been particularly fruitful, which will be described below. One is dependent on screening compounds that specifically alter the transcriptional activity of a specific STAT family member [202]. The second uses a strategy that leverages large databases of gene expression changes induced by drugs to infer compounds that can specifically modulate STATs [203]. Both of these approaches have led to the identification of STAT inhibitors that have been introduced into therapeutic clinical trials for cancer therapy.
6.1. Chemical Biology approaches
The general motif for the binding of STATs to gene regulatory regions have been well-defined. For most STATs, the consensus genomic cognate binding site conforms to the general nine base pair sequence TTCNNNGAA [7]. One can therefore generate heterologous reporter genes in which one or more STAT consensus sequences are placed upstream of a reporter gene such as luciferase [204]. These constructs can then be introduced into cells with undetectable basal STAT activation, in which a specific STAT can be activated with an appropriate cytokine [204]. Such a system can then be tested on large chemical libraries to identify molecules that can inhibit (or enhance) the STAT-dependent luciferase activity (Figure 3). To ensure the specificity of any molecules identified, the chemical library would also be screened against parallel systems in which expression of the reporter gene was driven by other transcription factors.
This approach has identified a number of compounds that act through a variety of mechanisms. One drug identified in this manner as an inhibitor of STAT3 transcriptional function is nifuroxazide [202]. Nifuroxazide appears to act mainly through inhibition of JAK kinase activity and showed activity against STAT3-dependent multiple myeloma cell lines and primary cells. As expected from this mechanism of action, it was also able to overcome the pro-survival effects afforded by co-culture with bone marrow stromal cells [202].
While nifuroxazide acts through a fairly conventional mechanism, other compounds identified through this approach reflected novel mechanisms. For example, the diphenylbutylpiperidine anti-psychotic drug pimozide, which is used to treat the tics associated with Tourette syndrome, was identified to inhibit both STAT5 and STAT3 transcriptional activity [191]. In contrast to nifuroxazide, pimozide is not a direct inhibitor of any JAK family member or BCR-ABL1 [192]. Although the exact mechanism of action is still being elucidated, pimozide appears to act by enhancing the activity of negative regulators of STAT signaling [205]. By virtue of having this kinase-independent effect, pimozide has shown efficacy in leukemic models driven by BCR-ABL1, mutated JAK2, and mutated FLT3 [206].
Both nifuroxazide and pimozide decrease the activating tyrosine phosphorylation of STATs. Another compound identified through this chemical biology approach, pyrimethamine, decreases STAT3-dependent transcription, but does not appear to significantly decreases STAT3 phosphorylation, nuclear localization, or DNA binding [207,208,209,210]. Pyrimethamine likely inhibits the interaction between STAT3 and other proteins necessary for transcriptional activation. This discovery through an unbiased approach may reveal other approaches to targeting STAT3-dependent gene expression [203].
Since pyrimethamine is already used as an anti-parasitic agent, treating diseases such as toxoplasmosis and malaria, there are abundant human pharmacokinetic and safety data available about this drug [211]. Pyrimethamine inhibits STAT3-dependent transcription at low micromolar concentrations, which are known to be safely achieved in humans for months at a time [211]. This allowed the planning of a clinical trial of pyrimethamine in patients with chronic lymphocytic leukemia (CLL), the most common form of leukemia in much of the world [147]. CLL is characterized by the transcriptional activation of STAT3 in almost every patient, though it is through a non-canonical phosphorylation event [212,213]. In this clinical trial, patients with CLL that had progressed despite multiple lines of standard therapy were treated with single agent pyrimethamine [147]. Although the mean plasma levels of pyrimethamine (6.17 mM) were at the lower end of the concentration needed to inhibit STAT3 transcriptional activity, half of the patients achieved stable disease [147]. To understand whether this drug was inhibiting STAT3 transcriptional activity in the leukemia cells of these patients, cells were isolated from their blood, and the expression of a panel of STAT3-dependent genes was analyzed by RT-PCR. Suppression of STAT3-dependent gene expression was seen in 50% of the patients. Notably, at the time of disease progression, increased expression of the STAT3 signature genes was generally observed, suggesting that this drug was working through an on-target mechanism [147]. This type of integrated analysis of clinical efficacy with pharmacodynamic measurements will be important in the further therapeutic development of STAT inhibitors.
While it is clearly therapeutically useful to identify inhibitors of oncogenic STAT family members that drive cancer pathogenesis, these types of chemical biology approaches can also be useful in identifying putative activators of transcription factors as well. As noted earlier, STAT1 mediates the effects of IFNs, and is a key component of the innate immune system response to viral infections and other pathogens [214]. Activated STAT1 can promote cell cycle arrest, apoptosis, differentiation, and enhanced immune recognition [6]. It can also mediate anti-angiogenic effects [215]. In fact, inhibition of STAT1 may mediate the immunosuppressive effects of drugs like fludarabine [216]. A similar chemical biology approach has been used to identify activators of STAT1-dependent transcription [217]. This strategy identified 2-(1,8-naphthyridin-2-yl)phenol (2-NP) as a compound that could enhance STAT1-dependent gene expression. It extends the duration of IFN-g-induced STAT1 tyrosine phosphorylation, and in this way may amplify signals through this transcription factor. 2-NP enhanced the ability of IFN-g to decrease the proliferation of human breast cancer and sarcoma cell lines. Indicating that this effect was mechanism-specific, cells that lacked STAT1 were unaffected by either IFN-g or 2-NP [217]. It remains to be seen whether enhancing STAT1 activity will be a worthwhile addition to anti-cancer therapy.
6.2. Computational approaches leveraging transcriptional signatures
In addition to chemical biology approaches, other open-ended and unbiased strategies have also been useful in identifying inhibitors of STAT-dependent gene expression. The target genes directly modulated by STAT transcription factors have been increasingly defined, particularly those regulated by STAT3 in cancer cells [218]. At the same time, data sets, such as the Connectivity Map, have become publicly available, providing large amounts of data on how a wide variety of chemical compounds alter gene expression in a variety of cell types [219]. STAT3 target genes can then be ordered from the ones most highly induced by STAT3 to those most highly repressed by STAT3. Data sets such as the Connectivity Map can then be employed to identify compounds that are associated with the exact opposite effect—decreasing expression of STAT3 induced genes and increasing expression of STAT3-repressed genes [220]. The hypothesis is that the compounds identified by this strategy would have a high likelihood of being inhibitors of STAT3 transcriptional function (Figure 4).
Using this approach, it was found that atovaquone induced gene expression changes that were highly anti-correlated from a STAT3 gene expression signature [219]. From this purely computational or in silico method, subsequent experiments revealed that atovaquone did in fact decrease STAT3 tyrosine phosphorylation. Atovaquone is not a kinase inhibitor, but rather decreased signaling through the gp130 (CD130) receptor chain used by IL-6 and many other family members to cause the phosphorylation of STAT3. Atovaquone decreased survival of STAT3-dependent cell lines and primary leukemic cells [150]. Atovaquone is widely used in oncology to prevent the development of Pneumocystis pneumonia in immunosuppressed patients [221]. From the pharmacokinetic data available, it is clear that levels of atovaquone sufficient to suppress STAT3 phosphorylation were readily and safely achieved in patients [203,222]. In fact, serum from patients taking atovaquone can be shown to have anti-leukemic effects compared to serum from patients taking other drugs for prophylaxis of Pneumocystis pneumonia [223]. Consequently, clinical trials testing the anti-cancer effects of atovaquone are currently in progress for STAT3-driven cancers including ovarian cancer (ClinicalTrials.gov NCT05998135) and AML (ClinicalTrials.gov NCT03568994).
7. Conclusions and future directions
Oncogenic transcription factors such as STAT3 and STAT5, which sit at convergence points of multiple upstream pathways, are appealing targets for cancer therapy. Although they are not commonly mutated themselves, they mediate the oncogenic effects of many diverse upstream oncogenic events. Since so many pathways converge on a relatively small number of proteins that regulate the genes mediating malignant cellular behavior, inhibition of STATs holds the promise of low rates of resistance arising from the activation of alternate or parallel pathways [12]. Finally, the fact that loss of STAT activity is tolerated well in healthy cells suggests that targeting STAT transcription factors will have a high therapeutic index.
While targeting transcription factors can be challenging, the increasing success in achieving this goal makes the oft-used term “undruggable” inappropriate. Through a combination of direct targeting approach and chemical biology and computational strategies, novel ways to target this pathway are becoming apparent. The agents uncovered from these approaches may be useful therapeutic agents themselves. Perhaps more importantly, they may lead to unappreciated mechanisms that can be exploited further with sophisticated medicinal chemistry approaches.
One other aspect of targeting STATs for cancer therapy should be noted. As with most cancer treatments, single agent therapy may not be fully successful [224]. While an oncogenic transcription factor such as STAT3 or STAT5 may regulate genes that underlie a cancer phenotype, interrupting these pathways may not be sufficient to kill established tumors. However, inhibiting oncogenic STATs in cancer may set the stage for synthetic lethal combination strategies [124]. For example, many target genes of STATs encode pro-survival proteins such as BCL2, BCL-XL, MCL1, and survivin [225]. By virtue of inhibiting the expression of these proteins, STAT inhibitors may allow synergy with cytotoxic drugs or radiation.
In addition, STAT target genes can affect DNA repair mechanisms, suggesting that combinations of STAT inhibitors with PARP inhibitors or telomerase inhibitors may show enhanced efficacy [226].
Furthermore, STAT3 mediates the physiologic acute phase response. One component of this response involves protecting healthy cells from being killed by infiltrating immune cells in the setting of tissue injury, inflammation, and infection [227,228]. Increased STAT3 activity in a tumor likewise protects a tumor from immune-based destruction. Therefore, combinations of STAT inhibitors and immune activating therapies, both immune checkpoint inhibitors and engineered cellular therapies, may be a promising approach [229].
Given these ongoing discoveries, there is a high likelihood that in the coming years further advances in targeting STATs and other oncogenic transcription factors will be a major component of a new generation of cancer therapies that display increased efficacy and decreased toxicity.
Author Contributions
Conceptualization, D.A.F., writing—original draft preparation, D.A.F., W.W.; writing—review and editing, D.A.F., W.W.; visualization, D.A.F, M.C.L.M, R.H., T.H., W.W.; supervision, D.A.F.; funding acquisition, D.A.F. All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Acknowledgments
This work was supported by research funding from Michael Deutsch, Esq. to DAF..
Conflicts of Interest
DAF has received research support from Kymera Therapeutics.
References
- “Global Cancer Facts & Figures.” Accessed: Aug. 24, 2023. [Online]. Available: https://www.cancer.org/research/cancer-facts-statistics/global.html.
- H. B. Sadowski, K. Shuai, J. E. Darnell, and M. Z. Gilman, “A common nuclear signal transduction pathway activated by growth factor and cytokine receptors,” Science, vol. 261, no. 5129, pp. 1739–1744, Sep. 1993. [CrossRef]
- U. M. Wegenka et al., “The interleukin-6-activated acute-phase response factor is antigenically and functionally related to members of the signal transducer and activator of transcription (STAT) family,” Mol Cell Biol, vol. 14, no. 5, pp. 3186–3196, May 1994. [CrossRef]
- J. E. Darnell, I. M. Kerr, and G. R. Stark, “Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins,” Science, vol. 264, no. 5164, pp. 1415–1421, 1994. [CrossRef]
- A. Orlova et al., “Direct Targeting Options for STAT3 and STAT5 in Cancer,” Cancers (Basel), vol. 11, no. 12, p. 1930, Dec. 2019. [CrossRef]
- W. Wang et al., “The complementary roles of STAT3 and STAT1 in cancer biology: insights into tumor pathogenesis and therapeutic strategies,” Front Immunol, vol. 14, p. 1265818, 2023. [CrossRef]
- T. J. Mitchell and S. John, “Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas,” Immunology, vol. 114, no. 3, pp. 301–312, Mar. 2005. [CrossRef]
- A. Yoshimura, T. Naka, and M. Kubo, “SOCS proteins, cytokine signalling and immune regulation,” Nat Rev Immunol, vol. 7, no. 6, pp. 454–465, Jun. 2007. [CrossRef]
- C. D. Chung et al., “Specific Inhibition of Stat3 Signal Transduction by PIAS3,” Science, vol. 278, no. 5344, pp. 1803–1805, Dec. 1997. [CrossRef]
- L. N. Heppler and D. A. Frank, “Rare mutations provide unique insight into oncogenic potential of STAT transcription factors,” J Clin Invest, vol. 128, no. 1, pp. 113–115, Jan. 2018. [CrossRef]
- M. Bar-Natan, E. A. Nelson, M. Xiang, and D. A. Frank, “STAT signaling in the pathogenesis and treatment of myeloid malignancies,” JAKSTAT, vol. 1, no. 2, pp. 55–64, Apr. 2012. [CrossRef]
- D. A. Frank, “STAT signaling in the pathogenesis and treatment of cancer,” Mol Med, vol. 5, no. 7, pp. 432–456, Jul. 1999. [CrossRef]
- V. Boudny and J. Kovarik, “JAK/STAT signaling pathways and cancer. Janus kinases/signal transducers and activators of transcription,” Neoplasma, vol. 49, no. 6, pp. 349–355, 2002.
- R. Catlett-Falcone et al., “Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells,” Immunity, vol. 10, no. 1, pp. 105–115, Jan. 1999. [CrossRef]
- J. F. Bromberg et al., “Stat3 as an Oncogene,” Cell, vol. 98, no. 3, pp. 295–303, Aug. 1999. [CrossRef]
- H. Lee, A. J. Jeong, and S.-K. Ye, “Highlighted STAT3 as a potential drug target for cancer therapy,” BMB Rep, vol. 52, no. 7, pp. 415–423, Jul. 2019. [CrossRef]
- T. Xia et al., “Advances in the role of STAT3 in macrophage polarization,” Frontiers in Immunology, vol. 14, 2023, Accessed: Nov. 29, 2023. [Online]. Available: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1160719.
- J. R. Grandis et al., “Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo,” Proceedings of the National Academy of Sciences, vol. 97, no. 8, pp. 4227–4232, Apr. 2000. [CrossRef]
- J. Bromberg, “Stat proteins and oncogenesis,” J Clin Invest, vol. 109, no. 9, pp. 1139–1142, May 2002. [CrossRef]
- H. Lee et al., “Persistently Activated Stat3 Maintains Constitutive NF-κB Activity in Tumors,” Cancer Cell, vol. 15, no. 4, pp. 283–293, Apr. 2009. [CrossRef]
- K. Berthenet et al., “HSP110 promotes colorectal cancer growth through STAT3 activation,” Oncogene, vol. 36, no. 16, pp. 2328–2336, Apr. 2017. [CrossRef]
- K.-W. Hsu et al., “Activation of the Notch1/STAT3/Twist signaling axis promotes gastric cancer progression,” Carcinogenesis, vol. 33, no. 8, pp. 1459–1467, Aug. 2012. [CrossRef]
- Y. Niwa et al., “Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma,” Oncogene, vol. 24, no. 42, pp. 6406–6417, Sep. 2005. [CrossRef]
- H. Isomoto et al., “Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing,” Gastroenterology, vol. 132, no. 1, pp. 384–396, Jan. 2007. [CrossRef]
- H. Ogata et al., “Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production,” Oncogene, vol. 25, no. 17, pp. 2520–2530, Apr. 2006. [CrossRef]
- M. Z. Kamran, P. Patil, and R. P. Gude, “Role of STAT3 in Cancer Metastasis and Translational Advances,” Biomed Res Int, vol. 2013, p. 421821, 2013. [CrossRef]
- M. Tolomeo and A. Cascio, “The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy,” Int J Mol Sci, vol. 22, no. 2, p. 603, Jan. 2021. [CrossRef]
- L. Lin et al., “STAT3 is necessary for proliferation and survival in colon cancer-initiating cells,” Cancer Res, vol. 71, no. 23, pp. 7226–7237, Dec. 2011. [CrossRef]
- G. Niu et al., “Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis,” Oncogene, vol. 21, no. 13, pp. 2000–2008, Mar. 2002. [CrossRef]
- N. Priego et al., “STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis,” Nat Med, vol. 24, no. 7, pp. 1024–1035, Jul. 2018. [CrossRef]
- Q. Xu et al., “Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways,” Oncogene, vol. 24, no. 36, pp. 5552–5560, Aug. 2005. [CrossRef]
- S. K. Tripathi et al., “Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation,” Cell Reports, vol. 19, no. 9, pp. 1888–1901, May 2017. [CrossRef]
- J. Odajima et al., “Full oncogenic activities of v-Src are mediated by multiple signaling pathways. Ras as an essential mediator for cell survival,” J Biol Chem, vol. 275, no. 31, pp. 24096–24105, Aug. 2000. [CrossRef]
- J. Luo, R. Yan, X. He, and J. He, “Constitutive activation of STAT3 and cyclin D1 overexpression contribute to proliferation, migration and invasion in gastric cancer cells,” Am J Transl Res, vol. 9, no. 12, pp. 5671–5677, Dec. 2017.
- C. Yue et al., “STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis,” Cancer Immunol Res, vol. 3, no. 8, pp. 864–870, Aug. 2015. [CrossRef]
- T. Wang et al., “Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells,” Nat Med, vol. 10, no. 1, pp. 48–54, Jan. 2004. [CrossRef]
- J.-P. Herbeuval, E. Lelievre, C. Lambert, M. Dy, and C. Genin, “Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6,” J Immunol, vol. 172, no. 7, pp. 4630–4636, Apr. 2004. [CrossRef]
- T.-X. Xie et al., “Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis,” Oncogene, vol. 23, no. 20, pp. 3550–3560, Apr. 2004. [CrossRef]
- H.-W. Lo et al., “Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression,” Cancer Res, vol. 67, no. 19, pp. 9066–9076, Oct. 2007. [CrossRef]
- T. N. Dechow et al., “Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C,” Proc Natl Acad Sci U S A, vol. 101, no. 29, pp. 10602–10607, Jul. 2004. [CrossRef]
- Y. Wu, I. Diab, X. Zhang, E. S. Izmailova, and Z. E. Zehner, “Stat3 enhances vimentin gene expression by binding to the antisilencer element and interacting with the repressor protein, ZBP-89,” Oncogene, vol. 23, no. 1, pp. 168–178, Jan. 2004. [CrossRef]
- R. Garcia et al., “Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells,” Oncogene, vol. 20, no. 20, pp. 2499–2513, May 2001. [CrossRef]
- J. V. Alvarez et al., “A STAT3 Gene Expression Signature in Gliomas is Associated with a Poor Prognosis,” Transl Oncogenomics, vol. 2, pp. 99–105, 2007. [CrossRef]
- R. L. Carpenter and H.-W. Lo, “STAT3 Target Genes Relevant to Human Cancers,” Cancers (Basel), vol. 6, no. 2, pp. 897–925, Apr. 2014. [CrossRef]
- S. H. Swerdlow et al., “The 2016 revision of the World Health Organization classification of lymphoid neoplasms,” Blood, vol. 127, no. 20, pp. 2375–2390, May 2016. [CrossRef]
- A. Moignet and T. Lamy, “Latest Advances in the Diagnosis and Treatment of Large Granular Lymphocytic Leukemia,” Am Soc Clin Oncol Educ Book, vol. 38, pp. 616–625, May 2018. [CrossRef]
- T. Lamy, A. Moignet, and T. P. Loughran, “LGL leukemia: from pathogenesis to treatment,” Blood, vol. 129, no. 9, pp. 1082–1094, Mar. 2017. [CrossRef]
- A. Fasan, W. Kern, V. Grossmann, C. Haferlach, T. Haferlach, and S. Schnittger, “STAT3 mutations are highly specific for large granular lymphocytic leukemia,” Leukemia, vol. 27, no. 7, pp. 1598–1600, Jul. 2013. [CrossRef]
- H. L. M. Koskela et al., “Somatic STAT3 Mutations in Large Granular Lymphocytic Leukemia,” N Engl J Med, vol. 366, no. 20, pp. 1905–1913, May 2012. [CrossRef]
- E. Andersson et al., “Activating somatic mutations outside the SH2-domain of STAT3 in LGL-Leukemia,” Leukemia, vol. 30, no. 5, pp. 1204–1208, May 2016. [CrossRef]
- G. Semenzato, A. Teramo, G. Calabretto, V. R. Gasparini, and R. Zambello, “All that glitters is not LGL Leukemia,” Leukemia, vol. 36, no. 11, pp. 2551–2557, Nov. 2022. [CrossRef]
- A. Dutta, D. Yan, R. E. Hutchison, and G. Mohi, “STAT3 mutations are not sufficient to induce large granular lymphocytic leukaemia in mice,” Br J Haematol, vol. 180, no. 6, pp. 911–915, Mar. 2018. [CrossRef]
- A. Teramo et al., “STAT3 mutation impacts biological and clinical features of T-LGL leukemia,” Oncotarget, vol. 8, no. 37, pp. 61876–61889, Jun. 2017. [CrossRef]
- T. P. Loughran et al., “Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998),” Leukemia, vol. 29, no. 4, pp. 886–894, Apr. 2015. [CrossRef]
- E. Masle-Farquhar et al., “STAT3 gain-of-function mutations connect leukemia with autoimmune disease by pathological NKG2Dhi CD8+ T cell dysregulation and accumulation,” Immunity, vol. 55, no. 12, pp. 2386-2404.e8, Dec. 2022. [CrossRef]
- S. Grivennikov et al., “IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer,” Cancer Cell, vol. 15, no. 2, pp. 103–113, Feb. 2009. [CrossRef]
- H. Kitamura et al., “Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy,” Cancer Science, vol. 108, no. 10, pp. 1947–1952, 2017. [CrossRef]
- F. Cheng et al., “A critical role for Stat3 signaling in immune tolerance,” Immunity, vol. 19, no. 3, pp. 425–436, Sep. 2003. [CrossRef]
- M. L. McLemore et al., “STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation,” Immunity, vol. 14, no. 2, pp. 193–204, Feb. 2001. [CrossRef]
- H. Zhang, H. Nguyen-Jackson, A. D. Panopoulos, H. S. Li, P. J. Murray, and S. S. Watowich, “STAT3 controls myeloid progenitor growth during emergency granulopoiesis,” Blood, vol. 116, no. 14, pp. 2462–2471, Oct. 2010. [CrossRef]
- C. Lee et al., “STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation,” Immunity, vol. 17, no. 1, pp. 63–72, Jul. 2002. [CrossRef]
- W.-C. Chou, D. E. Levy, and C.-K. Lee, “STAT3 positively regulates an early step in B-cell development,” Blood, vol. 108, no. 9, pp. 3005–3011, Nov. 2006. [CrossRef]
- S. A. Diehl, H. Schmidlin, M. Nagasawa, B. Blom, and H. Spits, “IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells,” Immunol Cell Biol, vol. 90, no. 8, pp. 802–811, Sep. 2012. [CrossRef]
- L. Durant et al., “Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis,” Immunity, vol. 32, no. 5, pp. 605–615, May 2010. [CrossRef]
- S. M. Kaech and W. Cui, “Transcriptional control of effector and memory CD8+ T cell differentiation,” Nat Rev Immunol, vol. 12, no. 11, pp. 749–761, Nov. 2012. [CrossRef]
- X. Y. Fu, “STAT3 in immune responses and inflammatory bowel diseases,” Cell Res, vol. 16, no. 2, Art. no. 2, Feb. 2006. [CrossRef]
- Z. Wang and K. D. Bunting, “STAT5 in hematopoietic stem cell biology and transplantation,” JAKSTAT, vol. 2, no. 4, p. e27159, Oct. 2013. [CrossRef]
- K. D. Bunting, H. L. Bradley, T. S. Hawley, R. Moriggl, B. P. Sorrentino, and J. N. Ihle, “Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5,” Blood, vol. 99, no. 2, pp. 479–487, Jan. 2002. [CrossRef]
- Z. Wang, G. Li, W. Tse, and K. D. Bunting, “Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement,” Blood, vol. 113, no. 20, pp. 4856–4865, May 2009. [CrossRef]
- G. Li, Z. Wang, K. L. Miskimen, Y. Zhang, W. Tse, and K. D. Bunting, “Gab2 Promotes Hematopoietic Stem Cell Maintenance and Self-Renewal Synergistically with STAT5,” PLoS One, vol. 5, no. 2, p. e9152, Feb. 2010. [CrossRef]
- J.-X. Lin et al., “Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells,” Nat Commun, vol. 8, no. 1, p. 1320, Nov. 2017. [CrossRef]
- L. M. Heltemes-Harris et al., “Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia,” J Exp Med, vol. 208, no. 6, pp. 1135–1149, Jun. 2011. [CrossRef]
- M. A. Burchill et al., “Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells,” J Immunol, vol. 171, no. 11, pp. 5853–5864, Dec. 2003. [CrossRef]
- S. L. Nutt and B. L. Kee, “The Transcriptional Regulation of B Cell Lineage Commitment,” Immunity, vol. 26, no. 6, pp. 715–725, Jun. 2007. [CrossRef]
- T. W. Hand et al., “Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival,” Proceedings of the National Academy of Sciences, vol. 107, no. 38, pp. 16601–16606, Sep. 2010. [CrossRef]
- P. Tripathi et al., “STAT5 IS CRITICAL TO MAINTAIN EFFECTOR CD8+ T CELL RESPONSES,” J Immunol, vol. 185, no. 4, pp. 2116–2124, Aug. 2010. [CrossRef]
- J.-H. Park et al., “Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells,” Nat Immunol, vol. 11, no. 3, pp. 257–264, Mar. 2010. [CrossRef]
- T. Kanai et al., “Identification of STAT5A and STAT5B Target Genes in Human T Cells,” PLoS One, vol. 9, no. 1, p. e86790, Jan. 2014. [CrossRef]
- C. E. Halim, S. Deng, M. S. Ong, and C. T. Yap, “Involvement of STAT5 in Oncogenesis,” Biomedicines, vol. 8, no. 9, p. 316, Aug. 2020. [CrossRef]
- D. A. Frank, “StAT signaling in cancer: insights into pathogenesis and treatment strategies,” Cancer Treat Res, vol. 115, pp. 267–291, 2003. [CrossRef]
- E. A. Nelson, S. R. Walker, J. V. Alvarez, and D. A. Frank, “Isolation of Unique STAT5 Targets by Chromatin Immunoprecipitation-based Gene Identification *,” Journal of Biological Chemistry, vol. 279, no. 52, pp. 54724–54730, Dec. 2004. [CrossRef]
- Y. Verhoeven et al., “The potential and controversy of targeting STAT family members in cancer,” Seminars in Cancer Biology, vol. 60, pp. 41–56, Feb. 2020. [CrossRef]
- W. Lin, J. W. Schmidt, B. A. Creamer, A. A. Triplett, and K.-U. Wagner, “Gain-of-Function of Stat5 Leads to Excessive Granulopoiesis and Lethal Extravasation of Granulocytes to the Lung,” PLOS ONE, vol. 8, no. 4, p. e60902, Apr. 2013. [CrossRef]
- J. C. G. van der Zwet et al., “STAT5 does not drive steroid resistance in T-cell acute lymphoblastic leukemia despite the activation of BCL and BCLXL following glucocorticoid treatment,” Haematologica, vol. 108, no. 3, pp. 732–746, Jun. 2022. [CrossRef]
- I. Barash, “Stat5 in breast cancer: potential oncogenic activity coincides with positive prognosis for the disease,” Carcinogenesis, vol. 33, no. 12, pp. 2320–2325, Dec. 2012. [CrossRef]
- K.-U. Wagner and H. Rui, “Jak2/Stat5 Signaling in Mammogenesis, Breast Cancer Initiation and Progression,” J Mammary Gland Biol Neoplasia, vol. 13, no. 1, pp. 93–103, Mar. 2008. [CrossRef]
- “Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers - Cotarla - 2004 - International Journal of Cancer - Wiley Online Library.” Accessed: Jan. 07, 2024. [Online]. Available: https://onlinelibrary.wiley.com/doi/10.1002/ijc.11619.
- M. Lin et al., “STAT5 confers lactogenic properties in breast tumorigenesis and restricts metastatic potential,” Oncogene, vol. 41, no. 48, pp. 5214–5222, Nov. 2022. [CrossRef]
- S. R. Walker et al., “Reciprocal Effects of STAT5 and STAT3 in Breast Cancer,” Molecular Cancer Research, vol. 7, no. 6, pp. 966–976, Jun. 2009. [CrossRef]
- B. Wingelhofer et al., “Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer,” Leukemia, vol. 32, no. 8, Art. no. 8, Aug. 2018. [CrossRef]
- K. Basso and R. Dalla-Favera, “Germinal centres and B cell lymphomagenesis,” Nat Rev Immunol, vol. 15, no. 3, pp. 172–184, Mar. 2015. [CrossRef]
- S. R. Walker, E. A. Nelson, J. E. Yeh, L. Pinello, G.-C. Yuan, and D. A. Frank, “STAT5 Outcompetes STAT3 To Regulate the Expression of the Oncogenic Transcriptional Modulator BCL6,” Mol Cell Biol, vol. 33, no. 15, pp. 2879–2890, Aug. 2013. [CrossRef]
- “STAT1 signal transducer and activator of transcription 1 [Homo sapiens (human)] - Gene - NCBI.” Accessed: Jan. 07, 2024. [Online]. Available: https://www.ncbi.nlm.nih.gov/gene/6772.
- S. E. Dovhey, N. S. Ghosh, and K. L. Wright, “Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line,” Cancer Res, vol. 60, no. 20, pp. 5789–5796, Oct. 2000.
- B. Kovacic et al., “STAT1 acts as a tumor promoter for leukemia development,” Cancer Cell, vol. 10, no. 1, pp. 77–87, Jul. 2006. [CrossRef]
- C. Park, S. Li, E. Cha, and C. Schindler, “Immune Response in Stat2 Knockout Mice,” Immunity, vol. 13, no. 6, pp. 795–804, Dec. 2000. [CrossRef]
- S. Hambleton et al., “STAT2 deficiency and susceptibility to viral illness in humans,” Proceedings of the National Academy of Sciences, vol. 110, no. 8, pp. 3053–3058, Feb. 2013. [CrossRef]
- J. Canar, kennedy Darling, R. Dadey, and A. M. Gamero, “The duality of STAT2 mediated type I interferon signaling in the tumor microenvironment and chemoresistance,” Cytokine, vol. 161, p. 156081, Jan. 2023. [CrossRef]
- C.-J. Lee et al., “FBXW7-mediated stability regulation of signal transducer and activator of transcription 2 in melanoma formation,” Proceedings of the National Academy of Sciences, vol. 117, no. 1, pp. 584–594, Jan. 2020. [CrossRef]
- L. Zhou, Y. Li, Z. Li, and Q. Huang, “Mining therapeutic and prognostic significance of STATs in renal cell carcinoma with bioinformatics analysis,” Genomics, vol. 112, no. 6, pp. 4100–4114, Nov. 2020. [CrossRef]
- X.-Y. Zhou, H.-Y. Dai, H. Zhang, J.-L. Zhu, and H. Hu, “Signal transducer and activator of transcription family is a prognostic marker associated with immune infiltration in endometrial cancer,” Journal of Clinical Laboratory Analysis, vol. 36, no. 4, p. e24315, 2022. [CrossRef]
- Y.-C. Huang, J.-L. Huang, L.-C. Tseng, P.-H. Yu, S.-Y. Chen, and C.-S. Lin, “High Expression of Interferon Pathway Genes CXCL10 and STAT2 Is Associated with Activated T-Cell Signature and Better Outcome of Oral Cancer Patients,” J Pers Med, vol. 12, no. 2, p. 140, Jan. 2022. [CrossRef]
- K. B. Nguyen et al., “Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection,” Science, vol. 297, no. 5589, pp. 2063–2066, Sep. 2002. [CrossRef]
- C. Parham et al., “A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R,” J Immunol, vol. 168, no. 11, pp. 5699–5708, Jun. 2002. [CrossRef]
- C. M. Bacon et al., “Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes.,” Proc Natl Acad Sci U S A, vol. 92, no. 16, pp. 7307–7311, Aug. 1995. [CrossRef]
- L. Zhao et al., “An integrated analysis identifies STAT4 as a key regulator of ovarian cancer metastasis,” Oncogene, vol. 36, no. 24, pp. 3384–3396, Jun. 2017. [CrossRef]
- M. Nishi et al., “High STAT4 Expression Indicates Better Disease-free Survival in Patients with Gastric Cancer,” Anticancer Research, vol. 37, no. 12, pp. 6723–6729, Dec. 2017. [CrossRef]
- K. Anderson, N. Ryan, G. Volpedo, S. Varikuti, A. R. Satoskar, and S. Oghumu, “Immune Suppression Mediated by STAT4 Deficiency Promotes Lymphatic Metastasis in HNSCC,” Frontiers in Immunology, vol. 10, 2020, Accessed: Jan. 07, 2024. [Online]. Available: https://www.frontiersin.org/articles/10.3389/fimmu.2019.03095.
- S. Goenka and M. H. Kaplan, “Transcriptional regulation by STAT6,” Immunol Res, vol. 50, no. 1, pp. 87–96, May 2011. [CrossRef]
- J. Zhu and W. E. Paul, “CD4 T cells: fates, functions, and faults,” Blood, vol. 112, no. 5, pp. 1557–1569, Sep. 2008. [CrossRef]
- K. Binnemars-Postma, R. Bansal, G. Storm, and J. Prakash, “Targeting the Stat6 pathway in tumor-associated macrophages reduces tumor growth and metastatic niche formation in breast cancer,” FASEB J, vol. 32, no. 2, pp. 969–978, Feb. 2018. [CrossRef]
- Y. Delgado-Ramirez, V. Colly, G. V. Gonzalez, and S. Leon-Cabrera, “Signal transducer and activator of transcription 6 as a target in colon cancer therapy,” Oncol Lett, vol. 20, no. 1, pp. 455–464, Jul. 2020. [CrossRef]
- A. B. Di Stefano et al., “Survivin is regulated by interleukin-4 in colon cancer stem cells,” J Cell Physiol, vol. 225, no. 2, pp. 555–561, Nov. 2010. [CrossRef]
- A. Jayakumar and A. L. M. Bothwell, “Stat6 Promotes Intestinal Tumorigenesis in a Mouse Model of Adenomatous Polyposis by Expansion of MDSCs and Inhibition of Cytotoxic CD8 Response,” Neoplasia, vol. 19, no. 8, pp. 595–605, Aug. 2017. [CrossRef]
- J. Iqbal et al., “Nanomedicines for developing cancer nanotherapeutics: from benchtop to bedside and beyond,” Appl Microbiol Biotechnol, vol. 102, no. 22, pp. 9449–9470, Nov. 2018. [CrossRef]
- D. Hong et al., “AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer,” Sci Transl Med, vol. 7, no. 314, p. 314ra185, Nov. 2015. [CrossRef]
- M. J. Reilley et al., “STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial,” J Immunother Cancer, vol. 6, no. 1, p. 119, Nov. 2018. [CrossRef]
- L. Bai et al., “A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo,” Cancer Cell, vol. 36, no. 5, pp. 498-511.e17, Nov. 2019. [CrossRef]
- S. M. Holland et al., “STAT3 mutations in the hyper-IgE syndrome,” N Engl J Med, vol. 357, no. 16, pp. 1608–1619, Oct. 2007. [CrossRef]
- Y. Minegishi et al., “Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome,” Nature, vol. 448, no. 7157, Art. no. 7157, Aug. 2007. [CrossRef]
- D. E. Levy and C. A. Loomis, “STAT3 Signaling and the Hyper-IgE Syndrome,” New England Journal of Medicine, vol. 357, no. 16, pp. 1655–1658, Oct. 2007. [CrossRef]
- R. T. Rhodes, “Understanding Hyper IgE Syndrome,” 2020.
- M. Furqan, A. Akinleye, N. Mukhi, V. Mittal, Y. Chen, and D. Liu, “STAT inhibitors for cancer therapy,” Journal of Hematology & Oncology, vol. 6, no. 1, p. 90, Dec. 2013. [CrossRef]
- U. Bharadwaj, M. M. Kasembeli, and D. J. Tweardy, “STAT3 Inhibitors in Cancer: A Comprehensive Update,” in STAT Inhibitors in Cancer, A. C. Ward, Ed., in Cancer Drug Discovery and Development., Cham: Springer International Publishing, 2016, pp. 95–161. [CrossRef]
- K. S. Bhullar et al., “Kinase-targeted cancer therapies: progress, challenges and future directions,” Molecular Cancer, vol. 17, no. 1, p. 48, Feb. 2018. [CrossRef]
- P. Cohen, D. Cross, and P. A. Jänne, “Kinase drug discovery 20 years after imatinib: progress and future directions,” Nat Rev Drug Discov, vol. 20, no. 7, Art. no. 7, Jul. 2021. [CrossRef]
- C. M. Lovly and A. T. Shaw, “Molecular Pathways: Resistance to Kinase Inhibitors and Implications for Therapeutic Strategies,” Clin Cancer Res, vol. 20, no. 9, pp. 2249–2256, May 2014. [CrossRef]
- S. A. Rosenzweig, “Acquired Resistance to Drugs Targeting Tyrosine Kinases,” Adv Cancer Res, vol. 138, pp. 71–98, 2018. [CrossRef]
- E. Benedettini et al., “Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis,” Am J Pathol, vol. 177, no. 1, pp. 415–423, Jul. 2010. [CrossRef]
- S. Zou, Q. Tong, B. Liu, W. Huang, Y. Tian, and X. Fu, “Targeting STAT3 in Cancer Immunotherapy,” Molecular Cancer, vol. 19, no. 1, p. 145, Sep. 2020. [CrossRef]
- P. Demela, N. Pirastu, and B. Soskic, “Cross-disorder genetic analysis of immune diseases reveals distinct gene associations that converge on common pathways,” Nat Commun, vol. 14, no. 1, Art. no. 1, May 2023. [CrossRef]
- A. Sarapultsev, E. Gusev, M. Komelkova, I. Utepova, S. Luo, and D. Hu, “JAK-STAT signaling in inflammation and stress-related diseases: implications for therapeutic interventions,” Mol Biomed, vol. 4, p. 40, Nov. 2023. [CrossRef]
- A. N. Koehler, “A complex task? Direct modulation of transcription factors with small molecules,” Curr Opin Chem Biol, vol. 14, no. 3, pp. 331–340, Jun. 2010. [CrossRef]
- X. Xie et al., “Recent advances in targeting the ‘undruggable’ proteins: from drug discovery to clinical trials,” Sig Transduct Target Ther, vol. 8, no. 1, Art. no. 1, Sep. 2023. [CrossRef]
- X. Xu, M. M. Kasembeli, X. Jiang, B. J. Tweardy, and D. J. Tweardy, “Chemical probes that competitively and selectively inhibit Stat3 activation,” PLoS One, vol. 4, no. 3, p. e4783, 2009. [CrossRef]
- H. Chen et al., “Targeting STAT3 by a small molecule suppresses pancreatic cancer progression,” Oncogene, vol. 40, no. 8, p. 1440, 2021. [CrossRef]
- E. A. Nelson, S. V. Sharma, J. Settleman, and D. A. Frank, “A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation,” Oncotarget, vol. 2, no. 6, pp. 518–524, Jun. 2011. [CrossRef]
- P. J. M. Jackson, S. Jamshidi, and D. B. Farag, “Computational Approaches in the Development of Small-molecule Transcription Factor Inhibitors,” Sep. 2018. [CrossRef]
- P. Yue and J. Turkson, “Targeting STAT3 in cancer: how successful are we?,” Expert Opin Investig Drugs, vol. 18, no. 1, pp. 45–56, Jan. 2009. [CrossRef]
- U. Hemmann et al., “Differential Activation of Acute Phase Response Factor/Stat3 and Stat1 via the Cytoplasmic Domain of the Interleukin 6 Signal Transducer gp130,” Journal of Biological Chemistry, vol. 271, no. 22, pp. 12999–13007, May 1996. [CrossRef]
- A. Diop et al., “SH2 Domains: Folding, Binding and Therapeutical Approaches,” Int J Mol Sci, vol. 23, no. 24, p. 15944, Dec. 2022. [CrossRef]
- U. Bharadwaj et al., “Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma,” Oncotarget, vol. 7, no. 18, pp. 26307–26330, Mar. 2016. [CrossRef]
- R. A. Cerulli and J. A. Kritzer, “Phosphotyrosine Isosteres: Past, Present and Future,” Org Biomol Chem, vol. 18, no. 4, pp. 583–605, Jan. 2020. [CrossRef]
- Z. Shi et al., “Silibinin inhibits endometrial carcinoma via blocking pathways of STAT3 activation and SREBP1-mediated lipid accumulation,” Life Sci, vol. 217, pp. 70–80, Jan. 2019. [CrossRef]
- “BP-1-102 exerts antitumor effects on T-cell acute lymphoblastic leukemia cells by suppressing the JAK2/STAT3/c-Myc signaling pathway - PubMed.” Accessed: Feb. 17, 2024. [Online]. Available: https://pubmed.ncbi.nlm.nih.gov/37020528/.
- R. Zhang, X. Chen, S. Fu, L. Xu, and J. Lin, “A small molecule STAT3 inhibitor, LLL12, enhances cisplatin- and paclitaxel-mediated inhibition of cell growth and migration in human ovarian cancer cells,” Oncol Rep, vol. 44, no. 3, pp. 1224–1232, Sep. 2020. [CrossRef]
- J. R. Brown et al., “Targeting constitutively active STAT3 in chronic lymphocytic leukemia: A clinical trial of the STAT3 inhibitor pyrimethamine with pharmacodynamic analyses,” Am J Hematol, vol. 96, no. 4, pp. E95–E98, Apr. 2021. [CrossRef]
- “STAT3 Inhibitor OPB-51602 Is Cytotoxic to Tumor Cells Through Inhibition of Complex I and ROS Induction - PubMed.” Accessed: Feb. 17, 2024. [Online]. Available: https://pubmed.ncbi.nlm.nih.gov/33305182/.
- M. Skwarski et al., “Mitochondrial Inhibitor Atovaquone Increases Tumor Oxygenation and Inhibits Hypoxic Gene Expression in Patients with Non-Small Cell Lung Cancer,” Clin Cancer Res, vol. 27, no. 9, pp. 2459–2469, May 2021. [CrossRef]
- A. M. Stevens et al., “Atovaquone is active against AML by upregulating the integrated stress pathway and suppressing oxidative phosphorylation,” Blood Adv, vol. 3, no. 24, pp. 4215–4227, Dec. 2019. [CrossRef]
- X. Qi et al., “Trichothecin Inhibits Cancer-Related Features in Colorectal Cancer Development by Targeting STAT3,” Molecules, vol. 25, no. 10, p. 2306, May 2020. [CrossRef]
- L. H et al., “Design, synthesis, and biological characterization of a potent STAT3 degrader for the treatment of gastric cancer,” Frontiers in pharmacology, vol. 13, Aug. 2022. [CrossRef]
- A. Kaneshige et al., “A selective small-molecule STAT5 PROTAC degrader capable of achieving tumor regression in vivo,” Nat Chem Biol, vol. 19, no. 6, pp. 703–711, Jun. 2023. [CrossRef]
- M. Roschewski et al., “Phase I Study of Acalabrutinib Plus Danvatirsen (AZD9150) in Relapsed/Refractory Diffuse Large B-Cell Lymphoma Including Circulating Tumor DNA Biomarker Assessment,” Clin Cancer Res, vol. 29, no. 17, pp. 3301–3312, Sep. 2023. [CrossRef]
- T. Nishina, T. Fujita, N. Yoshizuka, K. Sugibayashi, K. Murayama, and Y. Kuboki, “Safety, tolerability, pharmacokinetics and preliminary antitumour activity of an antisense oligonucleotide targeting STAT3 (danvatirsen) as monotherapy and in combination with durvalumab in Japanese patients with advanced solid malignancies: a phase 1 study,” BMJ Open, vol. 12, no. 10, p. e055718, Oct. 2022. [CrossRef]
- G. Casas, F. Perche, P. Midoux, C. Pichon, and J.-M. Malinge, “DNA minicircles as novel STAT3 decoy oligodeoxynucleotides endowed with anticancer activity in triple-negative breast cancer,” Mol Ther Nucleic Acids, vol. 29, pp. 162–175, Sep. 2022. [CrossRef]
- M. Aftabizadeh et al., “Potent antitumor effects of cell-penetrating peptides targeting STAT3 axis,” JCI Insight, vol. 6, no. 2, p. e136176. [CrossRef]
- P. Oleksak et al., “Design, synthesis, and in vitro evaluation of BP-1-102 analogs with modified hydrophobic fragments for STAT3 inhibition,” J Enzyme Inhib Med Chem, vol. 36, no. 1, pp. 410–424. [CrossRef]
- Y. Gu, I. S. Mohammad, and Z. Liu, “Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors (Review),” Oncology Letters, vol. 19, no. 4, pp. 2585–2594, Apr. 2020. [CrossRef]
- D. Wang, M. Xu, F. Li, Y. Gao, and H. Sun, “Target Identification-Based Analysis of Mechanism of Betulinic Acid-Induced Cells Apoptosis of Cervical Cancer SiHa,” Natural Product Communications, vol. 17, no. 7, p. 1934578X221115528, Jul. 2022. [CrossRef]
- L. Zhao, J. Zhao, K. Zhong, A. Tong, and D. Jia, “Targeted protein degradation: mechanisms, strategies and application,” Sig Transduct Target Ther, vol. 7, no. 1, Art. no. 1, Apr. 2022. [CrossRef]
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews, and R. J. Deshaies, “Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation,” Proceedings of the National Academy of Sciences, vol. 98, no. 15, pp. 8554–8559, Jul. 2001. [CrossRef]
- F. Potjewyd et al., “Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader,” Cell Chem Biol, vol. 27, no. 1, pp. 47-56.e15, Jan. 2020. [CrossRef]
- S.-M. Qi, J. Dong, Z.-Y. Xu, X.-D. Cheng, W.-D. Zhang, and J.-J. Qin, “PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy,” Front Pharmacol, vol. 12, p. 692574, May 2021. [CrossRef]
- M. He, W. Lv, and Y. Rao, “Opportunities and Challenges of Small Molecule Induced Targeted Protein Degradation,” Frontiers in Cell and Developmental Biology, vol. 9, 2021, Accessed: Nov. 29, 2023. [Online]. Available: https://www.frontiersin.org/articles/10.3389/fcell.2021.685106.
- H. Zhou et al., “Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein,” J. Med. Chem., vol. 62, no. 24, pp. 11280–11300, Dec. 2019. [CrossRef]
- S. Kong et al., “SD-36 promotes growth inhibition and induces apoptosis via suppression of Mcl-1 in glioma,” J Cell Mol Med, vol. 25, no. 17, pp. 8261–8270, Sep. 2021. [CrossRef]
- H. Zhou et al., “SD-91 as A Potent and Selective STAT3 Degrader Capable of Achieving Complete and Long-Lasting Tumor Regression,” ACS Med Chem Lett, vol. 12, no. 6, pp. 996–1004, Jun. 2021. [CrossRef]
- A. Shastri, “Preliminary Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of KT-333, a Targeted Protein Degrader of STAT3, in Patients with Relapsed or Refractory Lymphomas, Large Granular Lymphocytic Leukemia, and Solid Tumors,” presented at the 65th ASH Annual Meeting & Exposition, ASH, Dec. 2023. Accessed: Jan. 04, 2024. [Online]. Available: https://ash.confex.com/ash/2023/webprogram/Paper181130.html.
- J. M. Sasso, R. Tenchov, D. Wang, L. S. Johnson, X. Wang, and Q. A. Zhou, “Molecular Glues: The Adhesive Connecting Targeted Protein Degradation to the Clinic,” Biochemistry, vol. 62, no. 3, pp. 601–623, Jul. 2022. [CrossRef]
- G. Dong, Y. Ding, S. He, and C. Sheng, “Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery,” J. Med. Chem., vol. 64, no. 15, pp. 10606–10620, Aug. 2021. [CrossRef]
- W. den Besten and J. R. Lipford, “Prospecting for molecular glues,” Nat Chem Biol, vol. 16, no. 11, pp. 1157–1158, Nov. 2020. [CrossRef]
- D. E. Johnson, R. A. O’Keefe, and J. R. Grandis, “Targeting the IL-6/JAK/STAT3 signalling axis in cancer,” Nat Rev Clin Oncol, vol. 15, no. 4, pp. 234–248, Apr. 2018. [CrossRef]
- Y. Ohsugi and N. Tsuchimoto, “[Pharmacological and clinical profile of anti-human IL-6 receptor antibody (tocilizumab, ACTEMRA), a novel therapeutic drug for Castleman’s disease],” Nihon Yakurigaku Zasshi, vol. 126, no. 6, pp. 419–425, Dec. 2005. [CrossRef]
- M. Sheppard, F. Laskou, P. P. Stapleton, S. Hadavi, and B. Dasgupta, “Tocilizumab (Actemra),” Hum Vaccin Immunother, vol. 13, no. 9, pp. 1972–1988, Sep. 2017. [CrossRef]
- R. Salgado et al., “Circulating interleukin-6 predicts survival in patients with metastatic breast cancer,” Int J Cancer, vol. 103, no. 5, pp. 642–646, Feb. 2003. [CrossRef]
- B. E. Lippitz and R. A. Harris, “Cytokine patterns in cancer patients: A review of the correlation between interleukin 6 and prognosis,” Oncoimmunology, vol. 5, no. 5, p. e1093722, May 2016. [CrossRef]
- X. Hu, J. Li, M. Fu, X. Zhao, and W. Wang, “The JAK/STAT signaling pathway: from bench to clinic,” Sig Transduct Target Ther, vol. 6, no. 1, Art. no. 1, Nov. 2021. [CrossRef]
- R. Morris, N. J. Kershaw, and J. J. Babon, “The molecular details of cytokine signaling via the JAK/STAT pathway,” Protein Science, vol. 27, no. 12, pp. 1984–2009, 2018. [CrossRef]
- D. M. Schwartz, Y. Kanno, A. Villarino, M. Ward, M. Gadina, and J. J. O’Shea, “JAK inhibition as a therapeutic strategy for immune and inflammatory diseases,” Nat Rev Drug Discov, vol. 17, no. 1, p. 78, Dec. 2017. [CrossRef]
- K. L. Winthrop and S. B. Cohen, “Oral surveillance and JAK inhibitor safety: the theory of relativity,” Nat Rev Rheumatol, vol. 18, no. 5, Art. no. 5, May 2022. [CrossRef]
- M. von Locquenghien, C. Rozalén, and T. Celià-Terrassa, “Interferons in cancer immunoediting: sculpting metastasis and immunotherapy response,” J Clin Invest, vol. 131, no. 1, p. e143296. [CrossRef]
- A. Ribas, W. N. Haining, and T. N. M. Schumacher, “When Cancer Cells Become the Enablers of an Antitumor Immune Response,” Cancer Discovery, vol. 12, no. 10, pp. 2244–2248, Oct. 2022. [CrossRef]
- J. V. Melo, “The molecular biology of chronic myeloid leukaemia,” Leukemia, vol. 10, no. 5, pp. 751–756, May 1996.
- D. A. Frank and L. Varticovski, “BCR/abl leads to the constitutive activation of Stat proteins, and shares an epitope with tyrosine phosphorylated Stats,” Leukemia, vol. 10, no. 11, pp. 1724–1730, Nov. 1996.
- N. Carlesso, D. A. Frank, and J. D. Griffin, “Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl,” J Exp Med, vol. 183, no. 3, pp. 811–820, Mar. 1996. [CrossRef]
- K. Yanagisawa, H. Yamauchi, M. Kaneko, H. Kohno, H. Hasegawa, and S. Fujita, “Suppression of cell proliferation and the expression of a bcr-abl fusion gene and apoptotic cell death in a new human chronic myelogenous leukemia cell line, KT-1, by interferon-alpha,” Blood, vol. 91, no. 2, pp. 641–648, Jan. 1998. [CrossRef]
- A. Hoelbl et al., “Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia,” EMBO Mol Med, vol. 2, no. 3, pp. 98–110, Mar. 2010. [CrossRef]
- O. Hantschel et al., “BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia,” Nat Chem Biol, vol. 8, no. 3, Art. no. 3, Mar. 2012. [CrossRef]
- F. Rossari, F. Minutolo, and E. Orciuolo, “Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy,” Journal of Hematology & Oncology, vol. 11, no. 1, p. 84, Jun. 2018. [CrossRef]
- E. A. Nelson et al., “The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors,” Blood, vol. 117, no. 12, pp. 3421–3429, Mar. 2011. [CrossRef]
- M. Bar-Natan, E. A. Nelson, S. R. Walker, Y. Kuang, R. J. Distel, and D. A. Frank, “Dual inhibition of Jak2 and STAT5 enhances killing of myeloproliferative neoplasia cells,” Leukemia, vol. 26, no. 6, pp. 1407–1410, Jun. 2012. [CrossRef]
- A. Kentsis et al., “Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia,” Nat Med, vol. 18, no. 7, pp. 1118–1122, Jul. 2012. [CrossRef]
- L. Naldini et al., “Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor,” EMBO J, vol. 10, no. 10, pp. 2867–2878, Oct. 1991. [CrossRef]
- S. McGee et al., “Biological properties of ligand-dependent activation of the MET receptor kinase in acute myeloid leukemia,” Leukemia, vol. 29, no. 5, pp. 1218–1221, May 2015. [CrossRef]
- B. Kim et al., “Synthetic lethal screening reveals FGFR as one of the combinatorial targets to overcome resistance to Met-targeted therapy,” Oncogene, vol. 34, no. 9, pp. 1083–1093, Feb. 2015. [CrossRef]
- E. C. Chen et al., “Targeting MET and FGFR in Relapsed or Refractory Acute Myeloid Leukemia: Preclinical and Clinical Findings, and Signal Transduction Correlates,” Clinical Cancer Research, vol. 29, no. 5, pp. 878–887, Mar. 2023. [CrossRef]
- C. Zhao, H. Li, H.-J. Lin, S. Yang, J. Lin, and G. Liang, “Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism,” Trends Pharmacol Sci, vol. 37, no. 1, pp. 47–61, Jan. 2016. [CrossRef]
- L. N. Heppler and D. A. Frank, “Targeting Oncogenic Transcription Factors: Therapeutic Implications of Endogenous STAT Inhibitors,” Trends Cancer, vol. 3, no. 12, pp. 816–827, Dec. 2017. [CrossRef]
- C. L. Verlinde and W. G. Hol, “Structure-based drug design: progress, results and challenges,” Structure, vol. 2, no. 7, pp. 577–587, Jul. 1994. [CrossRef]
- A. Chen and A. N. Koehler, “Transcription Factor Inhibition: Lessons Learned and Emerging Targets,” Trends Mol Med, vol. 26, no. 5, pp. 508–518, May 2020. [CrossRef]
- E. A. Nelson et al., “Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3,” Blood, vol. 112, no. 13, pp. 5095–5102, Dec. 2008. [CrossRef]
- M. Xiang et al., “Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent,” Blood, vol. 128, no. 14, pp. 1845–1853, Oct. 2016. [CrossRef]
- M. Hirschenberger, M. Hayn, A. Laliberté, L. Koepke, F. Kirchhoff, and K. M. J. Sparrer, “Luciferase reporter assays to monitor interferon signaling modulation by SARS-CoV-2 proteins,” STAR Protoc, vol. 2, no. 4, p. 100781, Aug. 2021. [CrossRef]
- S. R. Walker and D. A. Frank, “Screening approaches to generating STAT inhibitors,” JAKSTAT, vol. 1, no. 4, pp. 292–299, Oct. 2012. [CrossRef]
- E. A. Nelson et al., “The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations,” Genes Cancer, vol. 3, no. 7–8, pp. 503–511, Jul. 2012. [CrossRef]
- A. Takakura et al., “Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways,” Hum Mol Genet, vol. 20, no. 21, pp. 4143–4154, Nov. 2011. [CrossRef]
- M. W. Khan et al., “The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer,” Cancer Immunol Immunother, vol. 67, no. 1, pp. 13–23, Jan. 2018. [CrossRef]
- L. N. Heppler et al., “The antimicrobial drug pyrimethamine inhibits STAT3 transcriptional activity by targeting the enzyme dihydrofolate reductase,” J Biol Chem, vol. 298, no. 2, p. 101531, Feb. 2022. [CrossRef]
- J. I. Brown et al., “Investigating the anti-cancer potential of pyrimethamine analogues through a modern chemical biology lens,” Eur J Med Chem, vol. 264, p. 115971, Jan. 2024. [CrossRef]
- C.-C. for D. C. and Prevention, “CDC - Toxoplasmosis - Resources for Health Professionals.” Accessed: Jan. 09, 2024. [Online]. Available: https://www.cdc.gov/parasites/toxoplasmosis/health_professionals/index.html.
- D. A. Frank, S. Mahajan, and J. Ritz, “B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues,” J Clin Invest, vol. 100, no. 12, pp. 3140–3148, Dec. 1997. [CrossRef]
- I. Hazan-Halevy et al., “STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells,” Blood, vol. 115, no. 14, pp. 2852–2863, Apr. 2010. [CrossRef]
- M. Tolomeo, A. Cavalli, and A. Cascio, “STAT1 and Its Crucial Role in the Control of Viral Infections,” Int J Mol Sci, vol. 23, no. 8, p. 4095, Apr. 2022. [CrossRef]
- T. E. Battle, R. A. Lynch, and D. A. Frank, “Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis,” Cancer Res, vol. 66, no. 7, pp. 3649–3657, Apr. 2006. [CrossRef]
- D. A. Frank, S. Mahajan, and J. Ritz, “Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling,” Nat Med, vol. 5, no. 4, pp. 444–447, Apr. 1999. [CrossRef]
- R. A. Lynch, J. Etchin, T. E. Battle, and D. A. Frank, “A small-molecule enhancer of signal transducer and activator of transcription 1 transcriptional activity accentuates the antiproliferative effects of IFN-gamma in human cancer cells,” Cancer Res, vol. 67, no. 3, pp. 1254–1261, Feb. 2007. [CrossRef]
- J. V. Alvarez, P. G. Febbo, S. Ramaswamy, M. Loda, A. Richardson, and D. A. Frank, “Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors,” Cancer Res, vol. 65, no. 12, pp. 5054–5062, Jun. 2005. [CrossRef]
- J. Lamb, “The Connectivity Map: a new tool for biomedical research,” Nat Rev Cancer, vol. 7, no. 1, Art. no. 1, Jan. 2007. [CrossRef]
- J. Lamb et al., “The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease,” Science, vol. 313, no. 5795, pp. 1929–1935, Sep. 2006. [CrossRef]
- W. T. Hughes, “The Role of Atovaquone Tablets in Treating Pneumocystis carinii Pneumonia,” JAIDS Journal of Acquired Immune Deficiency Syndromes, vol. 8, no. 3, p. 247, Mar. 1995. [CrossRef]
- Z. Lv, X. Yan, L. Lu, C. Su, and Y. He, “Atovaquone enhances doxorubicin’s efficacy via inhibiting mitochondrial respiration and STAT3 in aggressive thyroid cancer,” J Bioenerg Biomembr, vol. 50, no. 4, pp. 263–270, Aug. 2018. [CrossRef]
- A. M. Stevens et al., “Repurposing Atovaquone as a Therapeutic against Acute Myeloid Leukemia (AML): Combination with Conventional Chemotherapy Is Feasible and Well Tolerated,” Cancers, vol. 15, no. 4, Art. no. 4, Jan. 2023. [CrossRef]
- S. S. Bashraheel, A. Domling, and S. K. Goda, “Update on targeted cancer therapies, single or in combination, and their fine tuning for precision medicine,” Biomed Pharmacother, vol. 125, p. 110009, May 2020. [CrossRef]
- K. A. Z. Siddiquee and J. Turkson, “STAT3 as a target for inducing apoptosis in solid and hematological tumors,” Cell Res, vol. 18, no. 2, pp. 254–267, Feb. 2008. [CrossRef]
- N. Jin, Y. Xia, and Q. Gao, “Combined PARP inhibitors and small molecular inhibitors in solid tumor treatment (Review),” Int J Oncol, vol. 62, no. 2, p. 28, Jan. 2023. [CrossRef]
- L. Chen et al., “Inflammatory responses and inflammation-associated diseases in organs,” Oncotarget, vol. 9, no. 6, pp. 7204–7218, Dec. 2017. [CrossRef]
- KB. Megha, X. Joseph, V. Akhil, and PV. Mohanan, “Cascade of immune mechanism and consequences of inflammatory disorders,” Phytomedicine, vol. 91, p. 153712, Oct. 2021. [CrossRef]
- W. Wang, F. Burton, S. Herter, L. Codarri Deak, C. Klein, and D. Frank, “Pharmacologic Inhibitors of STAT3 or BCL6 Transcriptional Function Sensitize Lymphoma Cells to the Novel PD-1 Cis-Targeted PD1-IL2v Immunocytokine in a Murine Model,” Blood, vol. 140, no. Supplement 1, pp. 8835–8836, Nov. 2022. [CrossRef]
Figure 1.
Mechanisms of inappropriate STAT activation in cancer cells. STATs can be activated constitutively in cancer cells through a variety of mechanisms. This can include autocrine or paracrine production of cytokines that can activate these pathways; activation of upstream kinases through mutation (such as in JAKs), overexpression (such as EGFR), inappropriate activation (such as SRC), or activating translocations (such as in BCR-ABL); and loss of negative regulators (such as SOCS3). Rarely, the STATs can be activated by mutation within the STAT itself.
Figure 1.
Mechanisms of inappropriate STAT activation in cancer cells. STATs can be activated constitutively in cancer cells through a variety of mechanisms. This can include autocrine or paracrine production of cytokines that can activate these pathways; activation of upstream kinases through mutation (such as in JAKs), overexpression (such as EGFR), inappropriate activation (such as SRC), or activating translocations (such as in BCR-ABL); and loss of negative regulators (such as SOCS3). Rarely, the STATs can be activated by mutation within the STAT itself.
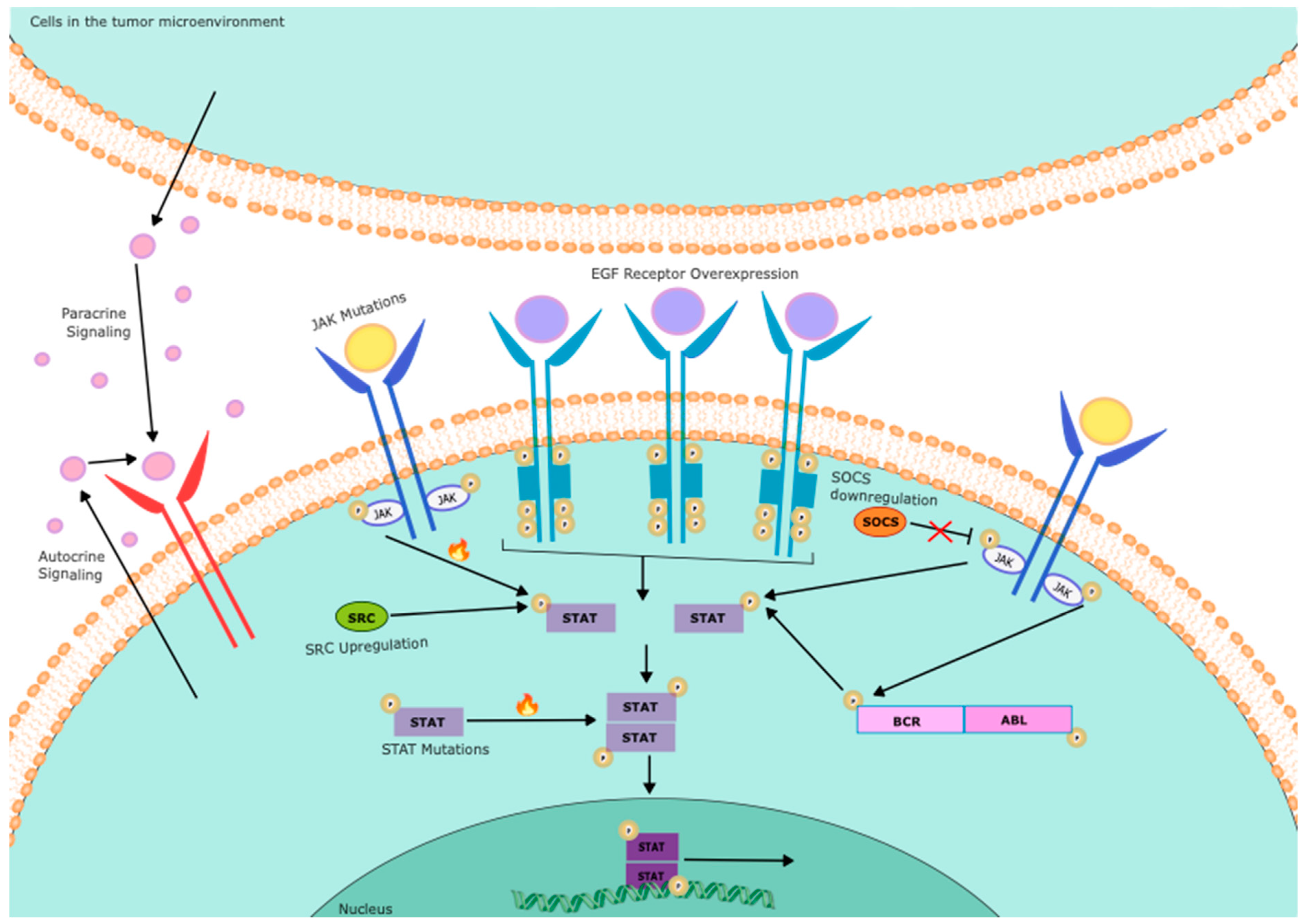
Figure 2.
Strategies to inhibit STATs for cancer therapy. Several targets have been established to mediate anti-cancer effects within the STAT signaling pathway. These include inhibitors of upstream kinases, such as JAKs, SRC, and receptor tyrosine kinases; inhibitors of receptor chains like GP130 (such as atovaquone); activators of negative regulators such as SOCS family members; degraders of STATs; and, agents that interfere with recruitment of transcriptional complexes, such as pyrimethamine.
Figure 2.
Strategies to inhibit STATs for cancer therapy. Several targets have been established to mediate anti-cancer effects within the STAT signaling pathway. These include inhibitors of upstream kinases, such as JAKs, SRC, and receptor tyrosine kinases; inhibitors of receptor chains like GP130 (such as atovaquone); activators of negative regulators such as SOCS family members; degraders of STATs; and, agents that interfere with recruitment of transcriptional complexes, such as pyrimethamine.
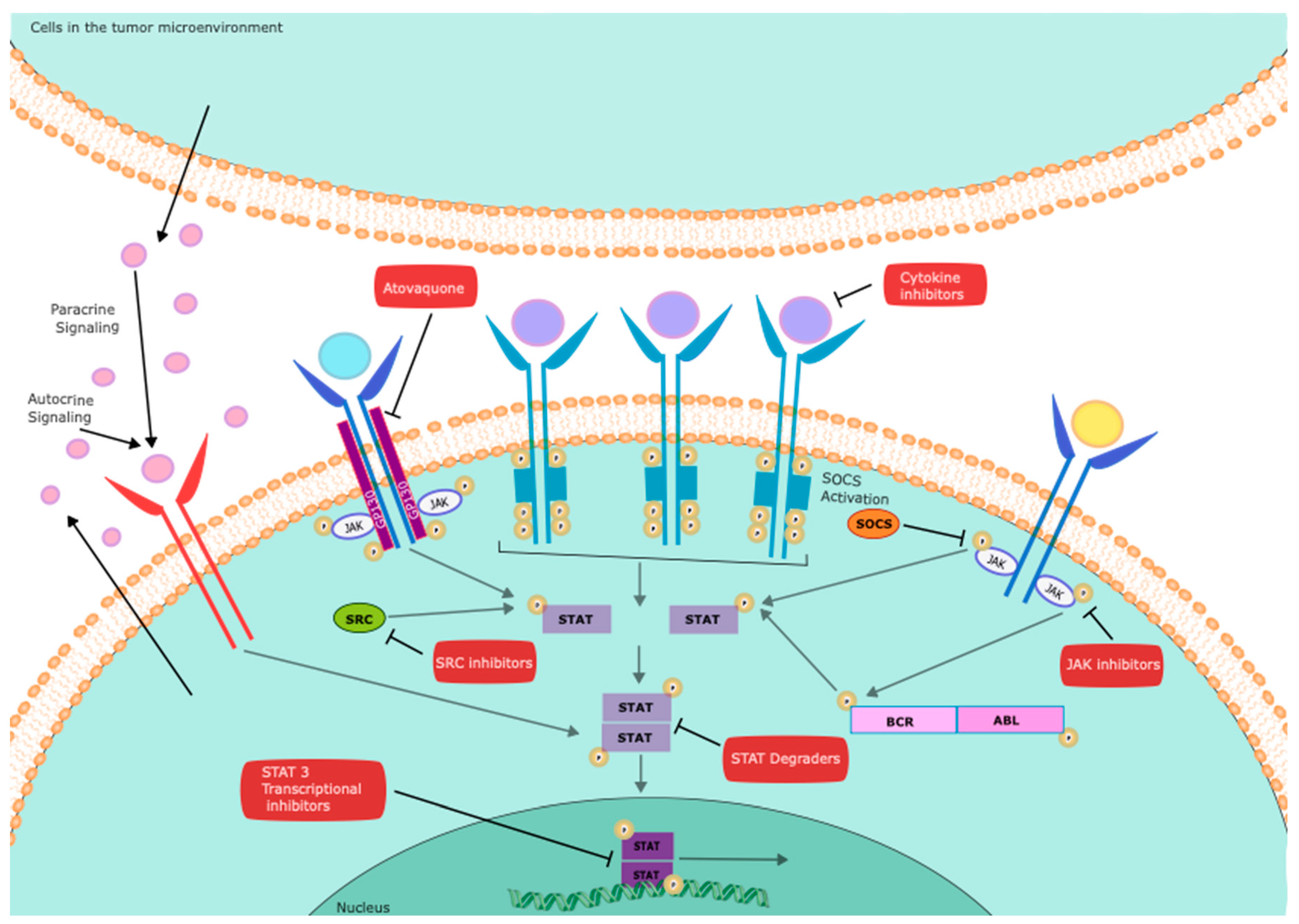
Figure 3.
Development of a cell-based system to screen compounds for the ability to inhibit STAT-dependent transcriptional activity. The stable introduction of a luciferase reporter gene under the control of a STAT-responsive promoter can enable a system to rapidly screen large numbers of compounds for the ability to inhibit STAT-dependent gene transcription (as measured by luminometry). Cellular systems can be designed that only respond to a specific STAT family member. It is also essential to counter-screen against similar systems under the control of unrelated transcription factors to exclude compounds that appear active in this assay but act through non-specific effects like general cytotoxicity.
Figure 3.
Development of a cell-based system to screen compounds for the ability to inhibit STAT-dependent transcriptional activity. The stable introduction of a luciferase reporter gene under the control of a STAT-responsive promoter can enable a system to rapidly screen large numbers of compounds for the ability to inhibit STAT-dependent gene transcription (as measured by luminometry). Cellular systems can be designed that only respond to a specific STAT family member. It is also essential to counter-screen against similar systems under the control of unrelated transcription factors to exclude compounds that appear active in this assay but act through non-specific effects like general cytotoxicity.
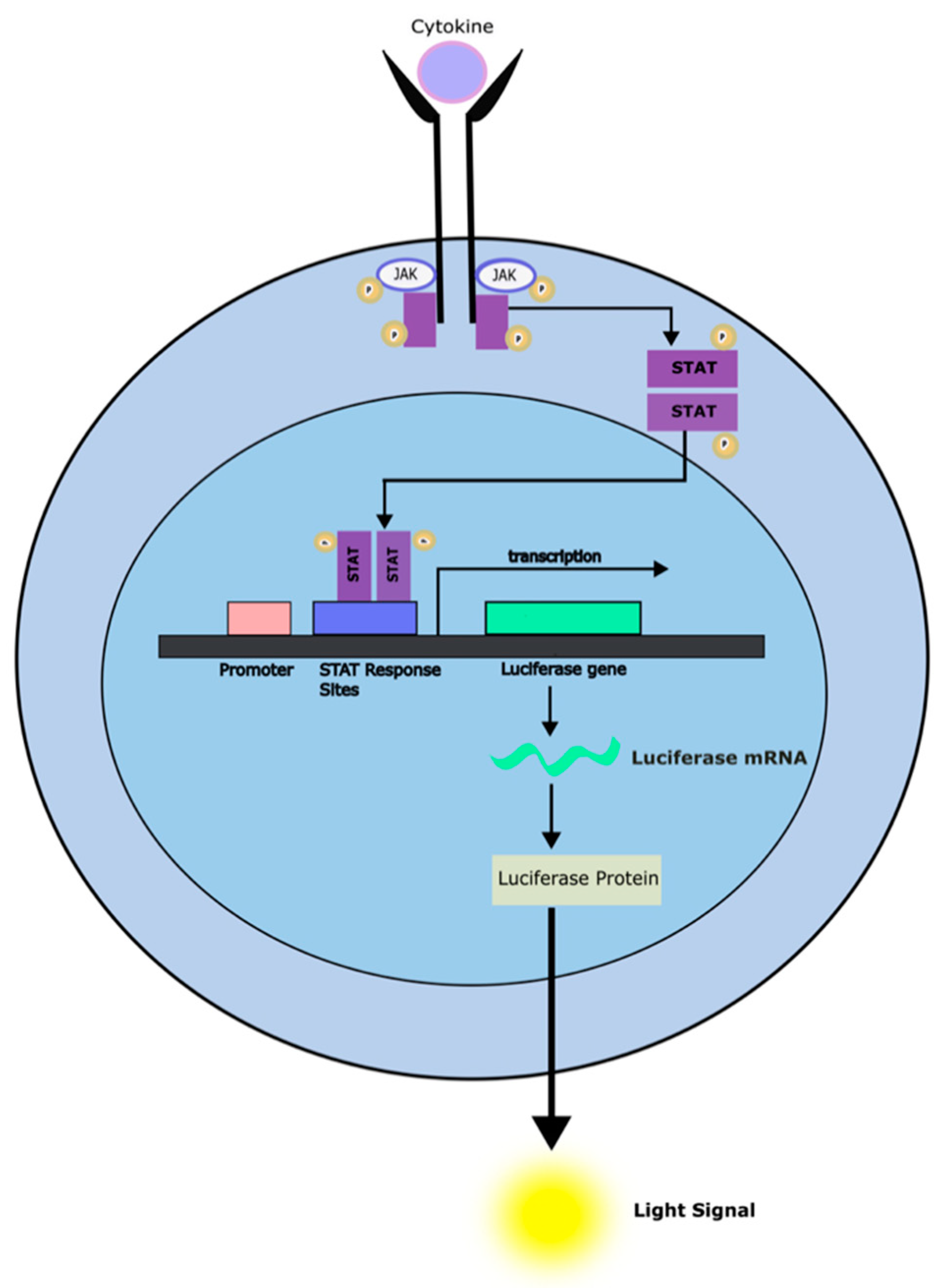
Figure 4.
Gene expression signatures coupled with databases of drug-induced changes in gene expression can be used to identify potential transcriptional inhibitors. General signatures of gene expression changes mediated by specific transcription factors have been defined. The availability of large databases, such as the Connectivity Map, provides an opportunity to query large number of drugs to identify those that induce gene expression changes most negatively correlated with the gene signature being queried. “Hits” from this computational approach hold the potential to be inhibitors of the specific pathway. Through this approach, the anti-parasitic drug atovaquone was identified as a therapeutically accessible inhibitor of STAT3-dependent transcription.
Figure 4.
Gene expression signatures coupled with databases of drug-induced changes in gene expression can be used to identify potential transcriptional inhibitors. General signatures of gene expression changes mediated by specific transcription factors have been defined. The availability of large databases, such as the Connectivity Map, provides an opportunity to query large number of drugs to identify those that induce gene expression changes most negatively correlated with the gene signature being queried. “Hits” from this computational approach hold the potential to be inhibitors of the specific pathway. Through this approach, the anti-parasitic drug atovaquone was identified as a therapeutically accessible inhibitor of STAT3-dependent transcription.
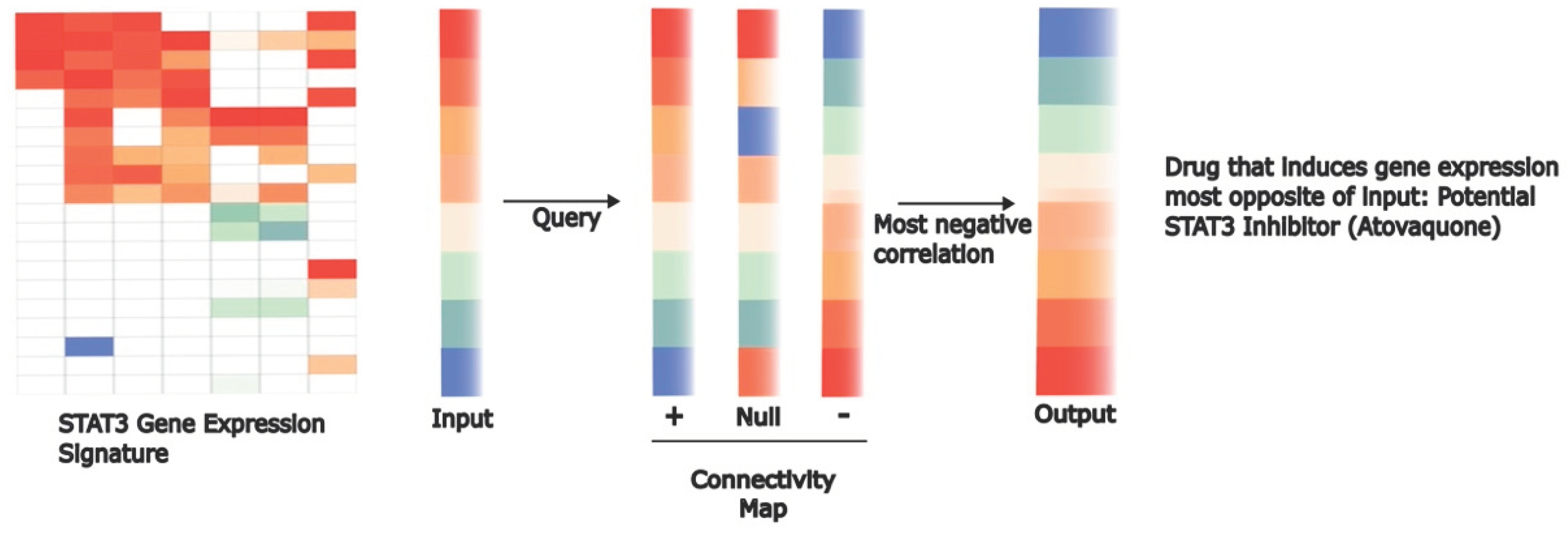

Table 1.
List of STAT3 target genes.
| Gene | Function | Status | Cell source | References |
|---|---|---|---|---|
| AKT1 | Proliferation | Upregulated | Various human cancer cells | [31] |
| BATF | Differentiation | Upregulated | Human Th17 cells | [32] |
| Bcl-xL | Anti-apoptosis | Upregulated | Human U266 cells | [14] |
| BCL6 | Proliferation | Upregulated | Human Th17 cells | [32] |
| MYC | Proliferation | Upregulated | Murine Ba/F3 cells | [33] |
| CCND1 | Proliferation | Upregulated | Human gastric cancer cells | [34] |
| CDKN2C | Cell cycle inhibition | Downregulated | Human Th17 cells | [32] |
| CREM | Spermatogenesis | Downregulated | Human Th17 cells | [32] |
| CXCL10 | Angiogenesis, Immune escape | Downregulated | Human CD8+ T cells | [35] |
| FOSL2 | Differentiation | Upregulated | Human Th17 cells | [32] |
| IKZF2 | Lymphocyte development | Downregulated | Human Th17 cells | [32] |
| IL6 | Immune escape | Upregulated | Murine melanoma cells | [36] |
| IL10 | Immune escape | Upregulated | Human colon Carcinoma | [37] |
| MMP2 | Immune escape | Upregulated | Murine melanoma cells | [38] |
| MMP9 | Immune escape | Upregulated | Murine fibroblasts | [39] |
| Ccl5 | Immune escape | Downregulated | Murine melanoma cells | [36] |
| RBPJ | Differentiation | Upregulated | Human Th17 cells | [32] |
| SMAD7 | Differentiation | Downregulated | Human Th17 cells | [32] |
| STAT1 | Differentiation | Downregulated | Human Th17 cells | [32] |
| STAT2 | Antiviral activity | Downregulated | Human Th17 cells | [32] |
| STAT3 | Differentiation | Upregulated | Human Th17 cells | [32] |
| TWIST | Immune escape | Upregulated | Human breast carcinomas | [40] |
| VEGF | Angiogenesis, immune escape | Upregulated | Murine fibroblasts | [39] |
| VIM | Immune escape | Upregulated | Monkey kidney cells | [41] |
Table 2.
List of STAT5 target genes.
| Gene | Function | Status | Cell source | References |
|---|---|---|---|---|
| Arnt | Protein sumoylation | Downregulated | Mouse proB cells | [81] |
| BCL2 | Anti-apoptosis | Upregulated | Human T cells | [84] |
| Bcl2l1 | Apoptosis | Upregulated | Mouse proB cells | [81] |
| BCLXL | Anti-apoptosis | Upregulated | Human T cells | [84] |
| C3ar1 | Chemotaxis | Upregulated | Murine proB cells | [78] |
| CISH | STAT inhibitor | Upregulated | Human T cells | [84] |
| Dusp1 | Anti-inflammation | Upregulated | Murine proB cells | [78] |
| DUSP5 | Anti-proliferation | No change | Human T cells | [84] |
| GTF2H5 | DNA repair | Downregulated | Human T cells | [84] |
| MBP | Inflammation | No change | Human T cells | [84] |
| Myc | Proliferation | Upregulated | Murine proB cells | [78] |
| OSM1 | Metabolic process | Upregulated | Human T cells | [84] |
| PIM1 | Proliferation, survival | Upregulated | Human T cells | [84] |
| Pim2 | Cell survival | Upregulated | Murine proB cells | [78] |
| Ro60 | Sperm antigen | Downregulated | Mouse proB cells | [81] |
| Ryk | Proliferation | Upregulated | Murine proB cells | [78] |
| Serpina3g | Proliferation | Upregulated | Murine proB cells | [78] |
| SGK1 | Proliferation | Downregulated | Human T cells | [84] |
| SLC22A5 | Carnitine uptake | Downregulated | Human T cells | [84] |
| Socs1 | Apoptosis | Upregulated | Murine proB cells | [78] |
| SOCS2 | inflammation | Upregulated | Human T cells | [84] |
| Srp9 | RNA binding | Upregulated | Mouse proB cells | [81] |
| Tnfrsf13b | B cell homeostasis | Upregulated | Murine proB cells | [78] |
Table 3.
List of STAT3/5 inhibitors.
| Type | Agent | Target | Cancer type | ClinicalTrial.gov identifier | Phase | References |
|---|---|---|---|---|---|---|
| Small molecules | Silibinin | STAT3 | Endometrial carcinoma | Preclinical | [144] | |
| SD-36 | STAT3 | Acute myeloid leukemia and anaplastic large cell lymphoma | Preclinical | [118] | ||
| BP-1-102 | STAT3 | Acute lymphoblastic leukemia | Preclinical | [145] | ||
| LLL12 | STAT3 | Ovarian cancer | Preclinical | [146] | ||
| Pyrimethamine | STAT3 | Chronic lymphocytic leukemia | NCT01066663 | Phase 1/2 | [147] | |
| OPB-51602 | STAT3 | Nasopharyngeal carcinoma | NCT01184807 | Phase 1 | [148] | |
| N4 | STAT3 | Pancreatic cancer | Preclinical | [136] | ||
| Atovaquone | STAT3 | Non-small cell lung cancer | NCT02628080 | Phase 1 | [149] | |
| STAT3 | Acute myeloid leukemia | Preclinical | [150] | |||
| Trichothecin | STAT3 | Colorectal Cancer | Preclinical | [151] | ||
| SDL-1 | STAT3 | Gastric cancer | Preclinical | [152] | ||
| AK-2292 | STAT5 | Chronic myeloid leukemia | Preclinical | [153] | ||
| Oligonucleotides | Danvatirsen | STAT3 | Diffuse large B-cell lymphoma | NCT03527147 | Phase 1 | [154] |
| STAT3 | Myelodysplastic syndromes, acute myeloid leukemia | NCT05986240 | Phase 1 | [155] | ||
| Double-stranded minicircles | STAT3 | Triple-negative breast cancer | Preclinical | [156] | ||
| Peptides | OPB-31121 | STAT3 | Hepatocellular carcinoma | NCT01406574 | Phase 1/2 | [154] |
| PS-acet.-STAT3 peptide | STAT3 | Melanoma | Preclinical | [157] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



