
Submitted:
23 February 2024
Posted:
26 February 2024
You are already at the latest version
Abstract
Predominantly antibody deficiencies (PAD) represent the most common type of primary immu-nodeficiencies in humans, characterized by a wide variation in disease onset, clinical manifesta-tions, complications and outcome. Considering that the prevalence of PAD in Greece is unknown and there is limited knowledge on clinical and laboratory characteristics of affected patients, we conducted a nationwide study, including 153 patients (male/female: 66/87; median age: 43.0 years; range: 7.0-77.0) diagnosed and/or followed-up between April 2007 to September 2023. Furthermore, we classified our cohort into five Groups according to their medical history, im-munoglobulin levels, and CTLA4-mutational status: 123 had common variable immunodeficien-cy (CVID), 12 with CVID-like disease (with a history of a previous B-cell depletion immunother-apy for autoimmune or malignant disease), seven with combined IgA and IgG subclass deficien-cies, five with immune dysregulation syndrome and CTLA4 defects, and six with unclassified hypogammaglobulinemia. We demonstrated a remarkable delay of PAD diagnosis, several years after the onset of related symptoms (median: 9.0 years, range: 0-43.0). Family history of PAD was only 11.8%, with the majority of patients considered sporadic cases. Most patients were diag-nosed in the context of a diagnostic work-up for recurrent infections, or recurrent/resistant auto-immune cytopenias. Interestingly, ten patients (5.6%) had no history of infection, diagnosed due to either recurrent/resistant autoimmunity, or during a work-up of their medical/family history. Remarkable findings included an increased prevalence of lymphoproliferation (60.1%) and atopy (24.2%), while 39 patients (25.5%) developed bronchiectasis and 16 (10.5%) granulomatous dis-ease. Patients with CTLA4 defects displayed a variable clinical phenotype at diagnosis, with four patients diagnosed during a work-up of re-current infections, while one also displayed a massive splenomegaly. Cancer was a common complication in our cohort (25 patients, 16.3%), with B-cell malignancies representing the most common neoplasms (56.7%). Our findings indicate the necessity of awareness about PAD and their complications, aiming for early diagnosis and appropriate management of affected patients
Keywords:
predominantly antibody deficiencies
; common variable immunodeficiency
; IgA deficiency
; IgG subclass deficiency
; CTLA4
; diagnosis
1. Introduction
Inborn errors of Immunity (IEI) are a diverse Group of rare genetic disorders, affecting almost every aspect of the immune response. Currently, more than 480 different types of IEI have been described. However, several patients remain undiagnosed, with timely diagnosis representing a challenge for their management [1]. Among IEI, Predominantly Antibody Deficiencies (PAD) are the most common type, affecting both children and adults [2,3]. PAD predispose individuals to recurrent infections, with a high prevalence of autoimmune manifestations, lymphoproliferation and atopy, along with a high incidence of cancer, especially lymphomas of B cell origin [1,2,3]. Notably, approximately half of patients with PAD do not exhibit the initial disease manifestations during childhood, but rather during puberty or adolescence [3,4]. With the wide variety of symptoms, this in turn obscures their differential diagnosis leading to a long diagnostic delay, in some cases of up to 10 years or more [3,4]. As a result, PAD patients often present with severe and irreversible complications, including bronchiectasis and/or respiratory insufficiency, resulting in significant socioeconomic consequences [2,4,5].
Among PAD, common variable immunodeficiency (CVID) is the most prevalent disorder worldwide (approximately 1:25,000 to 1:75,000), although its incidence varies among countries [6,7,8]. The most common finding among CVID patients is hypogammaglobulinemia leading to recurrent and/or persistent bacterial infections [6,7], along with a wide range of other clinical manifestations (lymphoproliferation, autoimmunity, granulomas formation, etc.) indicative of an extensive immune dysregulation [5,6,9]. The primary criteria for CVID diagnosis include: (a) low serum levels of IgG, IgA and/or IgM, greater than two standard deviations below the normal mean for the age; (b) absent isohemagglutinins and poor responses to vaccines (especially the polysaccharide ones); and (c) an exclusion of other defined causes of hypogammaglobulinemia and/or other types of IEI [7,10]. However, some patients do not meet all the aforementioned CVID criteria, making their diagnosis rather challenging [9]. For instance, this is applicable in the case of combined IgA and IgG subclasses deficiencies, since affected patients may display recurrent infections mainly of the upper respiratory tract, along with non-infectious manifestations, including benign lymphoproliferation and/or autoimmunity [9,11].
Notably, the genetic defects leading to overt immunodeficiency are unknown for the majority of PAD patients, including patients with CVID and combined IgA and IgG subclass deficiencies [2]. Interestingly, in some patients with an initial diagnosis of CVID, CTLA4 mutations may be identified as the causative defects, leading to the reclassification of their condition as immune dysregulating syndrome [12,13]. These patients display low immunoglobulin levels along with severe autoimmunity, lymphadenopathy and/or inflammatory bowel disease. Similar clinical and laboratory findings, as presented in patients with CTLA4 mutations, are present in CVID patients with LRBA mutations (OMIM CVID8; https://www.omim.org/entry/614700); these patients exhibit defects in B cell activation and autophagy, with susceptibility to apoptosis, resulting in both hypogammaglobulinemia and autoimmunity [14]. Therefore, considering that PAD is a heterogeneous Group of disorders, their accurate clinical characterization and further genetic analysis may provide appropriate information to better understand, categorize, and manage the affected patients.
In an effort to unravel part of this complexity, many research Groups worldwide conduct national or sub-national registries reporting the clinical phenotypes, laboratory findings, and their correlations with outcomes of PAD patients [4,6,15,16]. In this context the prevalence of PAD in Greece is unknown, and limited knowledge exists about the clinical and laboratory characteristics of affected patients. This nationwide study reports for the first time in the literature, data on the clinical presentation, diagnosis delay and management of Greek PAD patients from 2016 to 2023, and subsequently provides the basis for the first nationwide registry of PAD patients in Greece.
2. Materials and Methods
A total of 153 patients (male/female: 66/87; median age: 43.0 years; range: 7.0-77.0) with a PAD diagnosis and follow-up between April 2007 to September 2023 were retrospectively enrolled in the study; 143 patients (93.46%) were adults (male/female: 60/83), nine (5,88 %; male/female: 6/3) were adolescents and only one patient was a child (female, seven years of age). The majority of participating centers were hospital-level care centers for adult patients. Most patients (140; 91.5%) fulfilled the diagnostic criteria of CVID (as mentioned above) [7,10], while 13 patients (8.5%) displayed combined IgA and IgG subclass deficiencies with a CVID-like clinical phenotype. Furthermore, we classified our cohort into Groups according to their medical history (previous immune/chemo-therapy or not), immunoglobulin levels, and the CTLA4 mutational status, as presented in detail below (see Results section).
Recorded parameters included demographics, disease symptoms onset (clinical manifestations, infections or not) that led to medical awareness, age at diagnosis, and duration of the diagnostic delay. Specific attention was given to recorded complications due to infections (nasal polyps, bronchiectasis, chronic obstructive and/or restrictive respiratory disease), development and location of granulomatous disease, presence of benign lymphoproliferation (splenomegaly, reactive lymphadenopathy, intestinal lymphoid infiltrates), as well as associated interventions related to the latter (adenoidectomy, tonsillectomy, splenectomy). We further recorded gastrointestinal manifestations including sprue-like disease, Crohn-like disease, non-specific colitis, and chronic diarrhea due to infections or other causes. The type of autoimmune manifestations and development of neoplasia were also recorded, with an emphasis on the type of autoimmunity and cancer, along with the time of emergence.
Our study furthermore focused on the possible consanguinity among ancestors, as well as family medical history of IEI, autoimmune disorders and/or cancer. Finally, we recorded the type and duration of immunoglobulin replacement treatment (IgRT), along with administration of other type of therapy that patients received (i.e., for autoimmune manifestations, cancer etc.). In terms of laboratory testing, we recorded the quantitative immunoglobulin serum levels (IgG with isotypes, IgA, and IgM) at the time of diagnosis. Completed forms were imported in a Microsoft Excel database that is maintained at the Department of Immunology and Histocompatibility, University of Thessaly, Greece.
Written informed consent was obtained from each individual or an accompanying relative, for a few patients whose consent was not legally applicable (e.g., children). The study was designed according to Helsinki II declaration ethics and approved by the ethical committee of the Faculty of Medicine, University of Thessaly, Greece (6/18.3.2015). At the end, descriptive statistics for quantitative variables and frequencies for qualitative variables were calculated through GraphPad Prism Software (San Diego, California USA; version 10.1.1).
3. Results
3.1. Diagnosis, family history and further subclassification of PAD study patients
As presented in Table 1, the major problem that emerged from recording patients’ data was the remarkable delay of PAD diagnosis, several years after the onset of relevant symptoms (median: 9.0 years, range: 0-43). Most patients were diagnosed with hypogammaglobulinemia in the context of a diagnostic work-up for recurrent infections, or recurrent/resistant autoimmune cytopenias. Moreover, 18 patients were relatives of probands who were initially diagnosed with PAD; thus, family history of PAD in our cohort was only 11.8%, with the majority of PAD patients considered as sporadic cases. Conversely, medical history records revealed that a great majority of patients (63, 43.1%) had a family history of autoimmune diseases, while 52 patients (33.9%) also had a family history of cancer.
As mentioned in the Material and Methods section, we further classified our patients into five (5) subGroups according to their medical history, immunoglobulin levels, and CTLA4 mutational status. In particular, Group A consisted of 123 patients (male/female: 50/73; median age at analysis: 46.0 years; range 7.0-77.0; median age at diagnosis: 37.0 years; range: 4-69) who fulfilled the classical diagnostic criteria of CVID, as mentioned above [7,10] (Table 1).
In Group B, we included 12 patients (male/female: 9/3; median age at analysis: 39.0 years; range: 26-65; median age at diagnosis: 32.0 years; range: 11-60) (Table 1), who also fulfilled all the aforementioned CVID diagnostic criteria, but displayed a medical history of immunotherapy and/or chemotherapy due to either a hematological malignancy or autoimmune cytopenias, several years before PAD diagnosis. In particular, seven patients with a history of a B cell-malignancy (five with non-Hodgkin lymphomas, and two with acute lymphoblastic leukemia) were treated with regimens containing rituximab, remaining in complete hematological remission for many years before PAD diagnosis (median: 10 years; range: 7-19). The latter was performed in the context of a diagnostic approach of recurrent infections. Similarly, five patients had a history of autoimmune cytopenias (three with Evans syndrome, and two with autoimmune thrombocytopenia) who had received immunosuppressant treatment, including rituximab several years before PAD diagnosis (median: 8 years; range: 6-11). Among them, two patients (40.0%) were diagnosed at the relapse of autoimmunity, while the remaining patients were diagnosed during a diagnostic work-up of recurrent infections.
In Group C, we included 7 patients who displayed combined IgA and IgG4 subclass deficiencies with a CVID-like clinical phenotype (recurrent infections, and/or lymphoproliferation, and/or autoimmune manifestations, and/or enteropathy; and/or granulomas formation; Table 1). Among them, three patients also displayed lower levels of IgG2, three had lower levels of IgG1, and one patient had lower levels of IgG3; however, no patients had IgG1/IgG2/IgG3 deficiency. Their median age at diagnosis was 33.0 years (range: 13-44), and the median age at time of analysis was 45.0 years (range: 19-47) (Table 1).
In Group D, we included 5 patients (male/female: 1/4; median age at diagnosis: 19.0; range: 7-44) who had an initial diagnosis of CVID, but genetic analysis revealed the presence of pathogenic CTLA4 mutations. The molecular and clinical characteristics of these patients are presented in detail in Table 2, with more clinical data provided in the next subsection.
Finally, in Group E we included 6 patients (male/female: 1/5; median age at diagnosis: 59.0; range: 51-69) who displayed mild to moderate hypogammaglobulinemia with recurrent infections and a negative work-up for secondary immunodeficiencies, but did not fulfill diagnostic criteria of CVID, displaying for example appropriate immune responses after vaccination. Their median age at diagnosis was 59.0 years (range: 51-69), and their median age at time of analysis was 73.0 years (range: 54-71; Table 1).
3.2. Clinical and laboratory characteristics of the study patients
As presented in Table 1, the prevalent clinical problem of PAD patients were recurrent and/or severe infections, with an incidence according to their location detailed in Table 1 and Figure 1. Thus, the most common locations were those of upper and lower respiratory tracks. Central nervous system (CNS) infections (meningitis and/or encephalitis) were less common (8 patients; 5.2%), including a case of brain cysticercosis and another one with severe systemic nocardiosis with brain involvement. Parasitic infections were recorded in seven patients (4.6%), involving colon, lungs and brain, while 12 patients (7.8%) exhibited at least one attack of herpes zoster. It is noteworthy that patients with combined IgA and IgG subclass deficiencies (Group C) did not display gastrointestinal infections, despite the undetectable IgA levels in their blood (Table 1).
Interestingly, 55 patients (35.9%) were diagnosed after a work-up for other clinical manifestations, especially of recurrent/resistant autoimmune hematologic manifestations (autoimmune thrombocytopenia, autoimmune hemolytic anemia, Evans syndrome), or unexplained lymphoproliferation (splenomegaly and/or lymphadenopathy). However, most revealed an additional history of recurrent infections (that was considered by the patients as a “normal” condition due to its chronicity), and only 10 patients (6.5%) had no history of infections. The latter Group displayed recurrent hematologic or hepatic autoimmunity (6 and two patients, respectively), while two further patients were diagnosed during a conventional work-up. In particular, the first patient displayed a history of acute lymphoblastic leukemia after immunotherapy (being in remission, but without any recovery of immunoglobulin levels more than 7 years after treatment), and the second patient was the brother of a proband with CVID carrying the same pathogenic IKZF1 (Ikaros) mutation (CVID13, OMIM #616873).
In addition, remarkable findings were the increased prevalence of lymphoproliferation and atopy in our cohort of patients, regardless of their subclassification (Table 1, Figure 2). Moreover, 16 patients (10.5%) developed granulomatous disease, 13 in Group A and one patient in each of the other Groups, with the exception of Group E (Table 1). Lungs were the most frequent location (10 patients; 62.5%), but granulomas were also found in the liver (5 patients; 31.2%), skin (two patients; 12.5%), spleen (one patient; 6.3%), and lymph nodes (one patient; 6.3%). Only three CVID patients exhibited multisystemic granulomatosis, with granulomas being formed in more than one location apart from liver (lymph nodes, spleen and lungs, respectively). It should be noted that two of these 16 patients (12.5%) were initially misdiagnosed with sarcoidosis, receiving corticosteroid treatment for one and three years respectively, resulting in severe lower respiratory infections in one case and severe lung parasitosis in the other, since they were not receiving IgRT.
Interestingly, patients with CTLA4 defects (all located into exon 2 of CTLA4 gene) displayed a variable clinical phenotype at diagnosis, as presented in detail in Table 2. Thus, four out of 5 patients were diagnosed during a work-up of recurrent infections, while one of these patients also displayed a massive splenomegaly. The fifth patient was diagnosed after a traffic accident, when a diffuse lymphoid hyperplasia, splenomegaly and hypogammaglobulinemia - found incidentally - initially aroused suspicion of a lymphoid malignancy. After a comprehensive work-up and worsening hypogammaglobulinemia over time, the patient was diagnosed with PAD, while the final diagnosis of a CTLA4 defect and the immune dysregulating syndrome were made postmortem. It is worth noting that two patients with CTLA4 defects displayed no autoimmune manifestations, while the remaining three suffered from autoimmune thyroiditis, and one also suffered from pernicious anemia (Table 2). Clinical and laboratory data for three of 5 patients with CTLA4 defects, along with detailed functional studies for one of them, were also presented by us in previous manuscripts [17,18].
Only 21 patients (13.7%, male/female: 11/12; median age of disease onset: 20 years, range: 1-48; median age at diagnosis: 37.0 years, range: 4-53) displayed only infections, without any other manifestation of PAD (including lymphoproliferation, autoimmunity, atopy, enteropathy, or granulomatous disease) (Figure 2); this included 16 patients from Group A, two from Group B and two from Group E (Figure 2). All these patients were alive in September 2023, with a median follow-up period of 5 years (range: 5-21).
Considering the laboratory findings of our patients, a notable result was the almost complete absence of immunoglobulins (IgG levels undetectable or below 12 mg/dL, along with undetectable IgM and IgA levels, namely a presence of agammaglobulinemia) in peripheral blood of 9 PAD patients (5.9%); among them 8 belonged to Group A and one to Group B.
In addition, four male patients (2.6% of the total) with an onset of disease in adulthood (mean age of onset: 31 years, range: 18-44) exhibited undetectable levels of IgG and IgA at diagnosis, along with IgM levels into the normal range. Molecular analyses for the known causatives of hyper-IgM syndromes were negative, while a patient carried the pathogenic CVID mutation TNFRSF13B-p.C104R (rs34557412) in heterozygous state.
The majority of our PAD patients (142 of 153, 92.8% in total; 118 patients, 95.3% of Group A; 10 patients, 83.3% of Group B; four patients, 57.1% of Group C; 5 patients, 100.0% of Group D; 5 patients, 83.3% of Group E) systematically received IgRT, with most of them subcutaneous IgRT (108 patients; 76.1%). As mentioned above, 11 patients did not receive IgRT due to an absence of infections, especially in cases with moderate IgG hypogammaglobulinemia (levels above 400 mg/dL); however, two patients denied IgRT (despite recommendations), receiving only antibiotic treatment during infections.
3.3. Complications in our cohort of PAD patients
The most common complication was the development of severe chronic respiratory disease, either chronic bronchiectasis or chronic obstructive and/or chronic restrictive pulmonary disease, or chronic sinusitis with nasal polyps (Table 1). Bronchiectasis was present in 39 patients (25.5%) being one of the prevalent complications in Group A patients (37; 30.0%), including an individual that underwent lobectomy due to extensive bronchiectasis, leading eventually to defective lung function. Conversely, only one patient (14.3%) in Group C and one in Group E (16.7%) developed bronchiectasis, while no patient from Groups B and D developed this complication (Table 1).
Moreover, two patients with CVID (Group A) developed nodular regenerative hyperplasia (NRH) of the liver, which was confirmed by histology (Table 1); one of them died due to its complications.
Eighteen patients (11.8%) underwent splenectomy either at diagnosis in the context of differential diagnosis of a potential lymphoproliferative neoplasm (5 patients, 27.8%), or during the follow-up period for therapeutic reasons (13 patients, 72.2%), including refractory autoimmune hematologic manifestations (9 patients with refractory autoimmune thrombocytopenia or Evans syndrome), or severe hypersplenism (4 patients) leading to spontaneous spleen rupture in one patient.
Furthermore, neoplasia was a common complication in our cohort (25 patients, 16.3%), considering however that several patients had developed cancer many years before PAD diagnosis (Table 1). It is worth noting that five patients displayed more than one type of neoplasia; in particular, two patients with an initial diagnosis of Hodgkin lymphoma relapsed with Non-Hodgkin lymphomas (Burkitt and marginal zone, respectively), a patient with an initial diagnosis of Burkitt lymphoma relapsed with Hodgkin lymphoma, a patient with marginal zone lymphoma relapsed with a diffuse large B cell lymphoma (DLBCL), and another patient developed both, cervical and colon cancern. Nevertheless, in the majority of cases there was a past history of recurrent infections, but either no test for immunoglobulin levels had been performed appropriately, or an established hypogammaglobulinemia was not considered (as in a patient diagnosed with severe hypogammaglobulinemia and splenomegaly/splenectomy with granulomas formation 7 years before developing Hodgkin disease and a concomitant PAD diagnosis in a tertiary University Hospital). As presented in Figure 3, hematological malignancies of B-cell origin represent the most common neoplasms developed in our cohort (17 out of 30 cases, 56.7%). Complications of malignancies were the cause of death in three patients (12.0%).
In the end, 14 patients (9.2%) died during the follow-up period (Table 1); with the exception of one patient who passed away due to an automobile accident, in all other cases the cause of death was a PAD complication (severe infections in four patients – in two with CNS involvement; COVID-19 in two patients; autoimmune complications in two patients; pulmonary hypertension in one patient; NRH complications in one patient; neoplasia in three patients – two due to Non-Hodgkin lymphomas and one due to colon cancer).
4. Discussion
Our study represents the first nationwide study recording the clinical and laboratory manifestations of PAD patients in Greece. It is clear that the major problem in our cohort was a remarkable delay of the precise diagnosis. Thus, many patients experienced incorrect recognition of their condition for several years, resulting in a worsening of their medical status when the final diagnosis was performed. This is a very common phenomenon worldwide, since PAD patients remain largely mis- and/or under-diagnosed, a fact substantially affecting patients’ prognosis and disease outcome. In this context, several Groups have conducted national, sub-national or multinational studies collecting data in an attempt to record clinical and laboratory characteristics of PAD, while also raising awareness about the importance of timely diagnosis [4,8,11,16].
In our study most PAD patients suffer from CVID (Group A, Table 1). As mentioned above, the delay of disease diagnosis was higher in our cohort compared to other European countries (median: 9.0 vs 2.1 and 1.8 years in the Netherlands and Poland, respectively), or to the entire European continent based on ESID data (median 4.0 years) [4]. However, in other countries the diagnostic delay was higher as reported in studies from Mexico and Turkey (median: 12.5 and 14.0 years, respectively) [19,20,21]. Clearly, these differences are due to patients’ selection criteria and data. The majority of our patients were adults (Table 1) without early disease onset in childhood; it should be considered that when the disease manifests in early life, there is an increased suspicion of genetic disorders by pediatricians, with attention for adequate disease diagnosis and patients’ management. Obviously, an awareness of early disease recognition and diagnosis in adulthood is also necessary, as delayed diagnosis is usually complicated by severe and irreversible manifestations (for example bronchiectasis, chronic obstructive pulmonary disease, respiratory insufficiency etc.) [2,3,4,5].
One of the most interesting study findings was the demonstration of CVID clinical and laboratory phenotypes in patients of Group B, namely in patients initially diagnosed with hematological or autoimmune diseases, receiving immunosuppression therapy against B cells (rituximab in all cases) and developing a sustained immunodeficiency that never recovered several years after initial diagnosis (Table 1). It is known and expected that in many patients, the administration of rituximab results in a reduction of B cells and immunoglobulin levels, but these effects are transient and recovered over time. However, as already described by other groups, rituximab may represent a triggering factor for the emergence of an overt disease in individuals with a genetic predisposition for immunodeficiency [22,23,24]. Therefore, it is necessary to consider this uncommon “complication” in patients receiving rituximab or similar agents for appropriate monitoring, early diagnosis of PAD and proper management. Clearly, further genetic studies may clarify the predisposition of genetic factors for this uncommon “complication”.
Consistent with other studies in the literature [19,21], recurrent respiratory infections were the most frequent infection type in our cohort, and the most common symptom at disease onset (Table 1). It has been established that chronic inflammatory stimulation due to recurrent respiratory infections leads to structural damages in the respiratory system. This presents as nasal polyps, bronchiectasis, chronic obstructive and/or restrictive respiratory disease, resulting in the reduction of respiratory capacity. Therefore, similar to previous studies [8,25,26], we observed a rather high prevalence of bronchiectasis in our cohort that was more prevalent in CVID patients (Group A; 30.0%), while it was absent in patients with CLTA4 mutations (Group D). Unexpectedly, bronchiectasis formation was also absent in Group B patients (CVID-like phenotype after immunosuppression treatment); we suggest this finding (in Groups B and D) may be due to a rather shorter time between disease onset and patient enrollment, compared to other groups (Table 1). However, these findings and their potential explanation would be confirmed in further studies.
Similarly to other studies [4,15,16,19,27], we demonstrated that autoimmune thyroid disease, autoimmune thrombocytopenia and autoimmune hemolytic anemia were the most prevalent autoimmune conditions in PAD patients. As mentioned in the Results section, it should be noted that recurrent and/or resistant autoimmune manifestations were the reason behind disease diagnosis in several patients (55, 35.9%), despite the presence of a past history of infections. Moreover, 10 of 153 patients in our cohort (6.5%) had no history of infections; eight displayed only autoimmune manifestations (two with recurrent/resistant autoimmune thrombocytopenia, one with recurrent autoimmune hemolytic anemia, three with Evans syndrome and two with hepatic autoimmunity - Table 1). Obviously, the presence of recurrent and/or resistant autoimmune manifestations - especially in the context of hematologic autoimmunity - should increasingly raise both suspicion of an underlying immunodeficiency and the need for appropriate diagnostic work-up.
As detailed in the Results section and Table 1, lymphoproliferation (splenomegaly, lymphadenopathy, and intestinal lymphoid infiltrates) was quite common in our cohort (60.8%). Specifically, splenomegaly rates were higher in our study (52.4%) compared to the studies conducted by Ho and Cunningham (20.9%)[28], Chapel et al. [26], (30%) and Wehr et al. (40.5%) [29], but lower compared to a study by Graziano et al. (65%) [30]. Moreover, the prevalence of lymphadenopathy in our cohort (44.1%) was also higher compared to studies by Wehr et al. (26.2%) [29] and Chapel et al. (30%) [26], but closer to prevalence reported by Musabak et al. (48.4%) [20]. Notable differences in the incidence of lymphoproliferation rates between the aforementioned studies likely correspond to the disease heterogeneity worldwide. Further studies incorporating molecular data through genetic analysis of enrolled patients, may clarify the association between clinical presentation of PAD and the genetic background of affected patients.
In this study, we also reported on patients with an initial diagnosis of CVID, who were eventually categorized as suffering from immune dysregulation syndrome due to their CTLA4 mutational status (Table 1 and Table 2). Heterozygous CTLA4 mutations have been implicated in the emergence of a complex immune dysregulation syndrome with features of autoinflammation, autoimmunity and immunodeficiency [12,13,31,32], as well as increased risk of cancer development [32]. Schwab et al. characterized a worldwide cohort of 133 CTLA4 mutation carriers concerning their clinical and laboratory features; similar to our study, they observed that most mutations were located in exon 2 of the CTLA4 gene, encoding the ligand binding and dimerization domains [31]. However, a rather incomplete clinical penetrance has been reported by us and others for the CTLA4 haploinsufficiency [13,17,18,31], raising the question of additional genetic lesions that may contribute to the emergence and clinical phenotype of the disease. In this context, it is also interesting that we describe a new patient with a CTLA4 defect (#116, Table 2), without any sign of autoimmunity or autoinflammation, displaying only recurrent infections and splenomegaly. This suggests that CTLA4 molecular analysis is necessary in all patients with PAD.
One of our study limitations is the fact that we did not have molecular data for the entire cohort of patients (with the exception of the molecular analysis of CTLA4 gene for all and TNFRSF13B/TACI for the great majority of the enrolled patients [33]. Consequently, we have not performed association studies of the genetic background of affected individuals with the phenotype of their disease. However, the principal purpose of our nationwide study was to initially select the demographic, clinical and laboratory data from Greek patients with PAD, in order to proceed with the organization of a National registry which could facilitate collaboration between centers, and promoting the molecular analysis of patients and their families.
5. Conclusions
In conclusion, this is the first nationwide study of PAD patients in Greece. We demonstrated a remarkable delay of disease diagnosis, while we also observed a variable disease phenotype, with a proportion of patients developing PAD several years after an initial diagnosis of autoimmunity or malignancy, and their appropriate therapy. Clearly, these findings indicate the necessity of awareness about PAD and their complications, aiming for early diagnosis and appropriate management of affected patients.
Author Contributions
Conceptualization, M.S.; methodology, A.K.,F.K.,S.S., E.F., N.A., I.K., A.K., V.L., C.K., G.T., M.D., T.V., M.V., I.O., E.P., S.P., E.G-B., A.S., C.H., A.E.G., M.S.; validation, F.K., C.H. and. M.S; formal analysis, M.S.; investigation, A.K., F.K.; resources, F.K., E.F., N.A., I.K., A.K., V.L., C.K., G.T., M.D., T.V., M.V., I.O., E.P., S.P., E.G-B., A.S., M.S.; data curation, M.S..; writing—original draft preparation, A,K., M.S.; writing—review and editing, M.S., F.K., S.S.; visualization, M.S.; supervision, M.S.; project administration, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was designed according to Helsinki II declaration ethics and approved by the ethical committee of the Faculty of Medicine, University of Thessaly, Greece (6/18.3.2015).
Informed Consent Statement
Written informed consent was obtained from each individual or an accompanying relative, for a few patients whose consent was not legally applicable (e.g., children).
Data Availability Statement
Data available on request on request from the corresponding author.
Acknowledgments
The authors express their sincere gratitude to Ms Lemonia Anagnostopoulos for her invaluable assistance in providing important comments on the quality of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bousfiha, A.; Moundir, A.; Tangye, S. G.; Picard, C.; Jeddane, L.; Al-Herz, W.; Rundles, C. C.; Franco, J. L.; Holland, S. M.; Klein, C.; et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol 2022, 42, 1508–520. [Google Scholar] [CrossRef] [PubMed]
- Durandy, A.; Kracker, S.; Fischer, A. Primary Antibody Deficiencies. Nat Rev Immunol 2013, 13, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Yazdani, R. Predominantly Antibody Deficiencies. Immunology and Genetics Journal 2018, 52–80. [Google Scholar] [CrossRef]
- Gathmann, B.; Mahlaoui, N.; CEREDIH; Gérard, L.; Oksenhendler, E.; Warnatz, K.; Schulze, I.; Kindle, G.; Kuijpers, T. W.; Dutch, W.I.D.; et al. European Society for Immunodeficiencies Registry Working Party. Clinical Picture and Treatment of 2212 Patients with Common Variable Immunodeficiency. J Allergy Clin Immunol 2014, 134, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Rundles, C. The Many Faces of Common Variable Immunodeficiency. Hematology Am Soc Hematol Educ Program 2012, 2012, 301–305. [Google Scholar] [CrossRef]
- Hammarström, L.; Vorechovsky, I.; Webster, D. Selective IgA Deficiency (SIgAD) and Common Variable Immunodeficiency (CVID). Clin Exp Immunol 2000, 120, 225–231. [Google Scholar] [CrossRef]
- Ameratunga, R. Assessing Disease Severity in Common Variable Immunodeficiency Disorders (CVID) and CVID-Like Disorders. Front Immunol 2018, 9, 2130. [Google Scholar] [CrossRef] [PubMed]
- Selenius, J. S.; Martelius, T.; Pikkarainen, S.; Siitonen, S.; Mattila, E.; Pietikäinen, R.; Suomalainen, P.; Aalto, A. H.; Saarela, J.; Einarsdottir, E.; et al. Unexpectedly High Prevalence of Common Variable Immunodeficiency in Finland. Front Immunol 2017, 8, 1190. [Google Scholar] [CrossRef]
- Filion, C. A.; Taylor-Black, S.; Maglione, P. J.; Radigan, L.; Cunningham-Rundles, C. Differentiation of Common Variable Immunodeficiency From IgG Deficiency. J Allergy Clin Immunol Pract 2019, 7, 1277–1284. [Google Scholar] [CrossRef]
- ESID—European Society for Immunodeficiencies. Available online: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (accessed on 15 February 2024).
- Ozkan, H.; Atlihan, F.; Genel, F.; Targan, S.; Gunvar, T. IgA and/or IgG Subclass Deficiency in Children with Recurrent Respiratory Infections and Its Relationship with Chronic Pulmonary Damage. J Investig Allergol Clin Immunol 2005, 15, 69–74. [Google Scholar]
- Kuehn, H. S.; Ouyang, W.; Lo, B.; Deenick, E. K.; Niemela, J. E.; Avery, D. T.; Schickel, J.-N.; Tran, D. Q.; Stoddard, J.; Zhang, Y.; et al. Immune Dysregulation in Human Subjects with Heterozygous Germline Mutations in CTLA4. Science 2014, 345, 1623–1627. [Google Scholar] [CrossRef]
- Schubert, D.; Bode, C.; Kenefeck, R.; Hou, T. Z.; Wing, J. B.; Kennedy, A.; Bulashevska, A.; Petersen, B.-S.; Schäffer, A. A.; Grüning, B. A.; et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans with CTLA4 Mutations. Nat Med 2014, 20, 1410–1416. [Google Scholar] [CrossRef]
- Lopez-Herrera, G.; Tampella, G.; Pan-Hammarström, Q.; Herholz, P.; Trujillo-Vargas, C. M.; Phadwal, K.; Simon, A. K.; Moutschen, M.; Etzioni, A.; Mory, A.; et al. Deleterious Mutations in LRBA Are Associated with a Syndrome of Immune Deficiency and Autoimmunity. Am J Hum Genet 2012, 90, 986–1001. [Google Scholar] [CrossRef]
- Resnick, E. S.; Moshier, E. L.; Godbold, J. H.; Cunningham-Rundles, C. Morbidity and Mortality in Common Variable Immune Deficiency over 4 Decades. Blood 2012, 119, 1650–1657. [Google Scholar] [CrossRef]
- Quinti, I.; Soresina, A.; Spadaro, G.; Martino, S.; Donnanno, S.; Agostini, C.; Claudio, P.; Franco, D.; Maria Pesce, A.; Borghese, F.; et al. Italian Primary Immunodeficiency Network. Long-Term Follow-up and Outcome of a Large Cohort of Patients with Common Variable Immunodeficiency. J Clin Immunol 2007, 27, 308–316. [Google Scholar] [CrossRef]
- Sic, H.; Speletas, M.; Cornacchione, V.; Seidl, M.; Beibel, M.; Linghu, B.; Yang, F.; Sevdali, E.; Germenis, A. E.; Oakeley, E. J.; et al. An Activating Janus Kinase-3 Mutation Is Associated with Cytotoxic T Lymphocyte Antigen-4-Dependent Immune Dysregulation Syndrome. Front Immunol 2017, 8, 1824. [Google Scholar] [CrossRef]
- Kelaidi, C.; Tzotzola, V.; Polychronopoulou, S. The Paradigm of Hematological Malignant versus Non-Malignant Manifestations, Driven by Primary Immunodeficiencies: A Complex Interplay. Fam Cancer 2021, 20, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Vargas, N.; Arablin-Oropeza, S. E.; Mojica-Martínez, D.; Yamazaki-Nakashimada, M. A.; de la Luz García-Cruz, M.; Terán-Juárez, L. M.; Cortés-Grimaldo, R. M.; Torres-Lozano, C.; Madrigal-Beas, I.; Ortega-Cisneros, M.; et al. Clinical and Immunological Features of Common Variable Immunodeficiency in Mexican Patients. Allergol Immunopathol (Madr) 2014, 42, 235–240. [Google Scholar] [CrossRef]
- Muşabak, U. H.; Demirel, F.; Yeşillik, S.; Baysan, A.; Selçuk, A.; Kartal, Ö.; Güleç, M.; Öktenli, Ç.; Şener, O. Adults with Common Variable Immunodeficiency: A Single-Center Experience. Turk J Med Sci 2017, 47, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nepesov, S.; Aygun, F. D.; Firtina, S.; Cokugras, H.; Camcioglu, Y. Clinical and Immunological Features of 44 Common Variable Immunodeficiency Patients: The Experience of a Single Center in Turkey. Allergol Immunopathol (Madr) 2020, 48, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Diwakar, L.; Gorrie, S.; Richter, A.; Chapman, O.; Dhillon, P.; Al-Ghanmi, F.; Noorani, S.; Krishna, M. T.; Huissoon, A. Does Rituximab Aggravate Pre-Existing Hypogammaglobulinaemia? J Clin Pathol 2010, 63, 275–277. [Google Scholar] [CrossRef]
- Kaplan, B.; Kopyltsova, Y.; Khokhar, A.; Lam, F.; Bonagura, V. Rituximab and Immune Deficiency: Case Series and Review of the Literature. J Allergy Clin Immunol Pract 2014, 2, 594–600. [Google Scholar] [CrossRef]
- Mogensen, T. H.; Bernth-Jensen, J. M.; Petersen, C. C.; Petersen, M. S.; Nyvold, C.; Gadegaard, K. H.; Hokland, M.; Hokland, P.; Larsen, C. S. Common Variable Immunodeficiency Unmasked by Treatment of Immune Thrombocytopenic Purpura with Rituximab. BMC Hematol 2013, 13, 4. [Google Scholar] [CrossRef]
- Odnoletkova, I.; Kindle, G.; Quinti, I.; Grimbacher, B.; Knerr, V.; Gathmann, B.; Ehl, S.; Mahlaoui, N.; Van Wilder, P.; Bogaerts, K.; et al. Plasma Protein Therapeutics Association (PPTA) Taskforce. The Burden of Common Variable Immunodeficiency Disorders: A Retrospective Analysis of the European Society for Immunodeficiency (ESID) Registry Data. Orphanet J Rare Dis 2018, 13, 201. [Google Scholar] [CrossRef]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B.; Fieschi, C.; Thon, V.; Abedi, M. R.; Hammarstrom, L. Common Variable Immunodeficiency Disorders: Division into Distinct Clinical Phenotypes. Blood 2008, 112, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Cunningham-Rundles, C. Autoimmunity in Common Variable Immunodeficiency. Curr Allergy Asthma Rep 2009, 9, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.-E.; Cunningham-Rundles, C. Non-Infectious Complications of Common Variable Immunodeficiency: Updated Clinical Spectrum, Sequelae, and Insights to Pathogenesis. Front Immunol 2020, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Wehr, C.; Kivioja, T.; Schmitt, C.; Ferry, B.; Witte, T.; Eren, E.; Vlkova, M.; Hernandez, M.; Detkova, D.; Bos, P. R.; Poerksen, G.; et al. The EUROclass Trial: Defining Subgroups in Common Variable Immunodeficiency. Blood 2008, 111, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Graziano, V.; Pecoraro, A.; Mormile, I.; Quaremba, G.; Genovese, A.; Buccelli, C.; Paternoster, M.; Spadaro, G. Delay in Diagnosis Affects the Clinical Outcome in a Cohort of Cvid Patients with Marked Reduction of Iga Serum Levels. Clin Immunol 2017, 180, 1–4. [Google Scholar] [CrossRef]
- Schwab, C.; Gabrysch, A.; Olbrich, P.; Patiño, V.; Warnatz, K.; Wolff, D.; Hoshino, A.; Kobayashi, M.; Imai, K.; Takagi, M.; et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4-Insufficient Subjects. J Allergy Clin Immunol 2018, 142, 1932–1946. [Google Scholar] [CrossRef]
- Verma, N.; Burns, S. O.; Walker, L. S. K.; Sansom, D. M. Immune Deficiency and Autoimmunity in Patients with CTLA-4 (CD152) Mutations. Clin Exp Immunol 2017, 190, 1–7. [Google Scholar] [CrossRef]
- Kakkas, I.; Tsinti, G.; Kalala, F.; Farmaki, E.; Kourakli, A.; Kapousouzi, A.; Dimou, M.; Kalaitzidou, V.; Sevdali, E.; Peristeri, et. al. TACI Mutations in Primary Antibody Deficiencies: A Nationwide Study in Greece. Medicina (Kaunas) 2021, 57, 827. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Location and incidence of infections in the patients of the study.
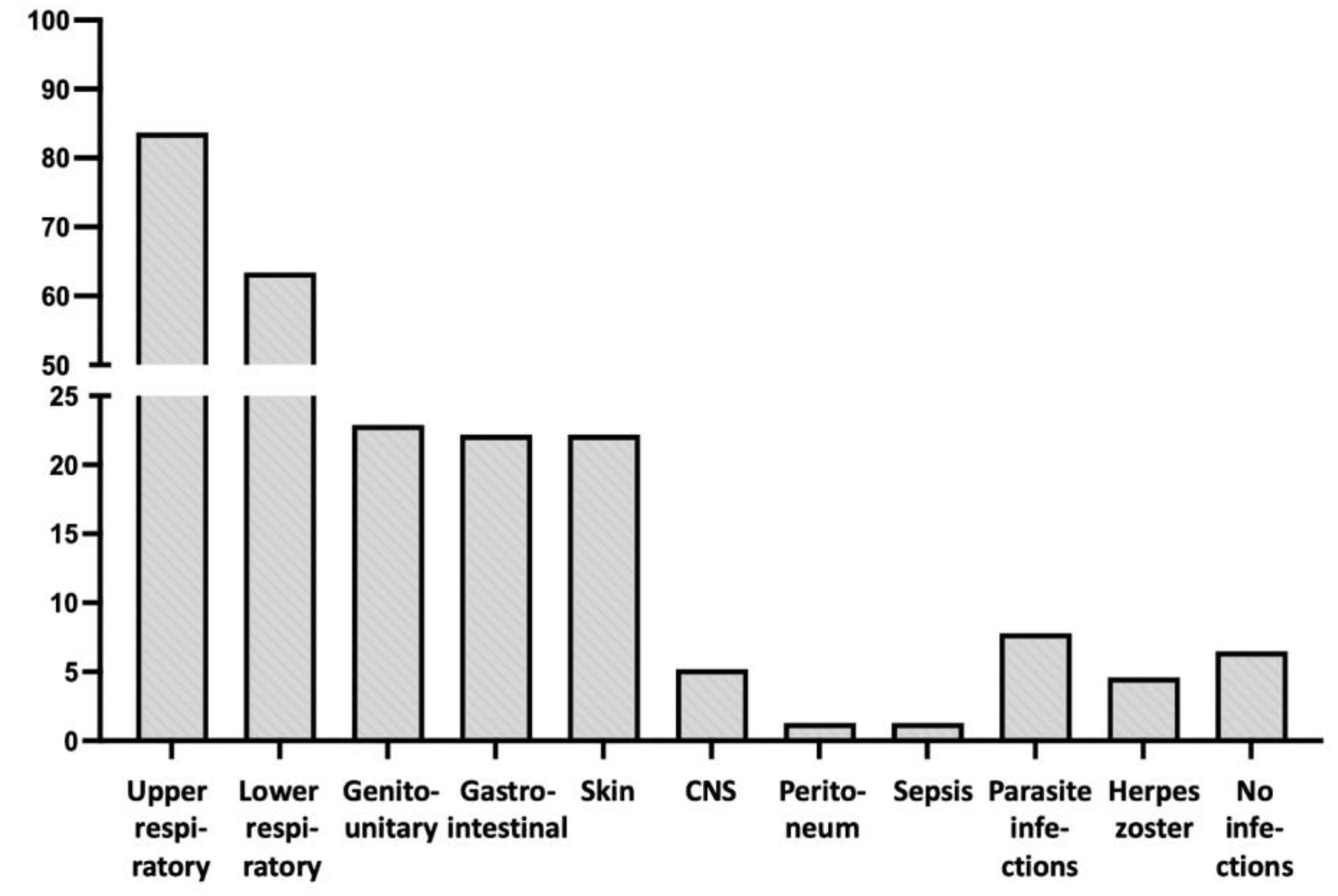
Figure 2.
An overview of the clinical manifestations and complications of the patients of the study.
Figure 2.
An overview of the clinical manifestations and complications of the patients of the study.
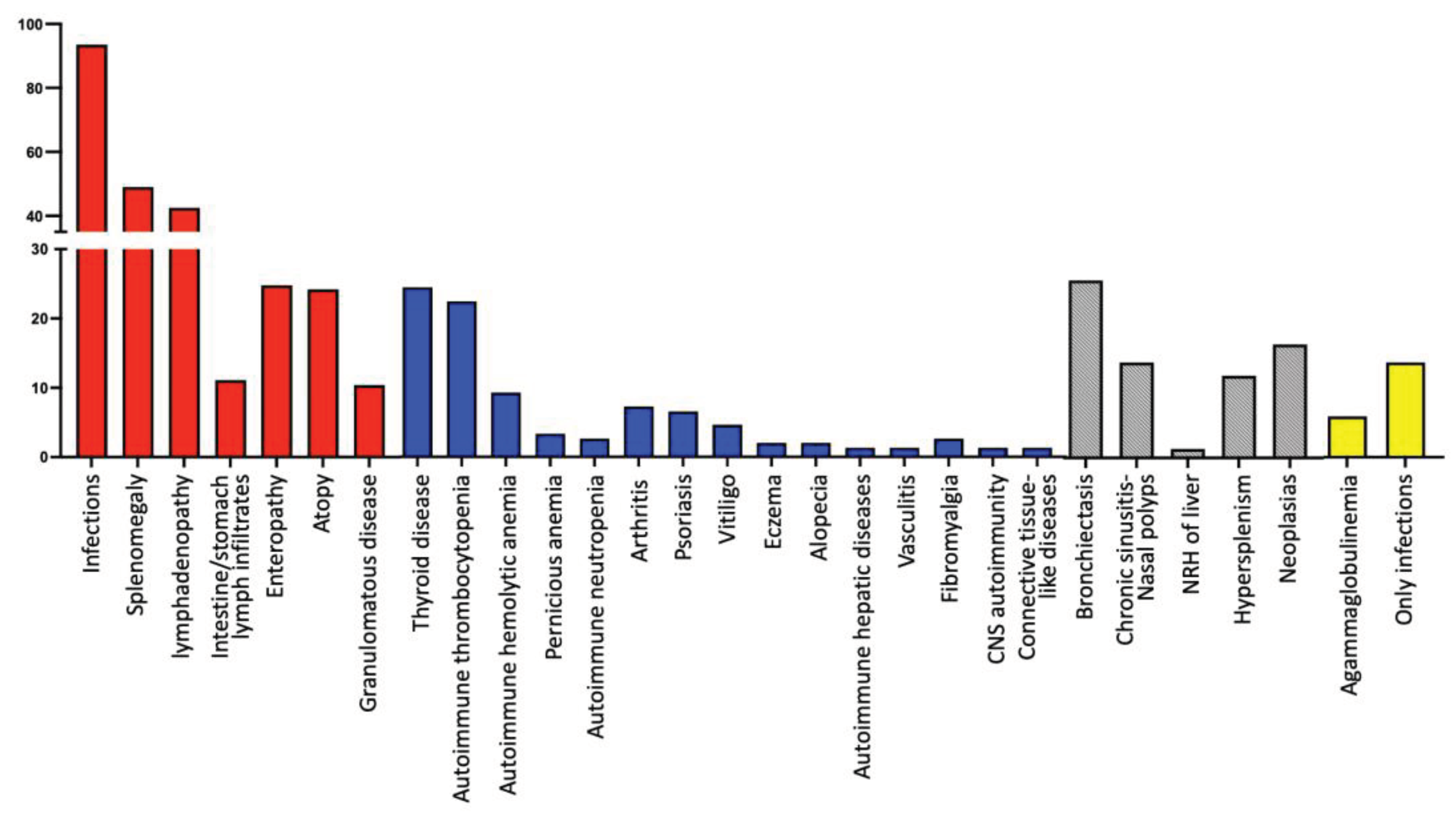
Figure 3.
The type of malignancies developed in the patients of the study, before and after PAD diagnosis.
Figure 3.
The type of malignancies developed in the patients of the study, before and after PAD diagnosis.
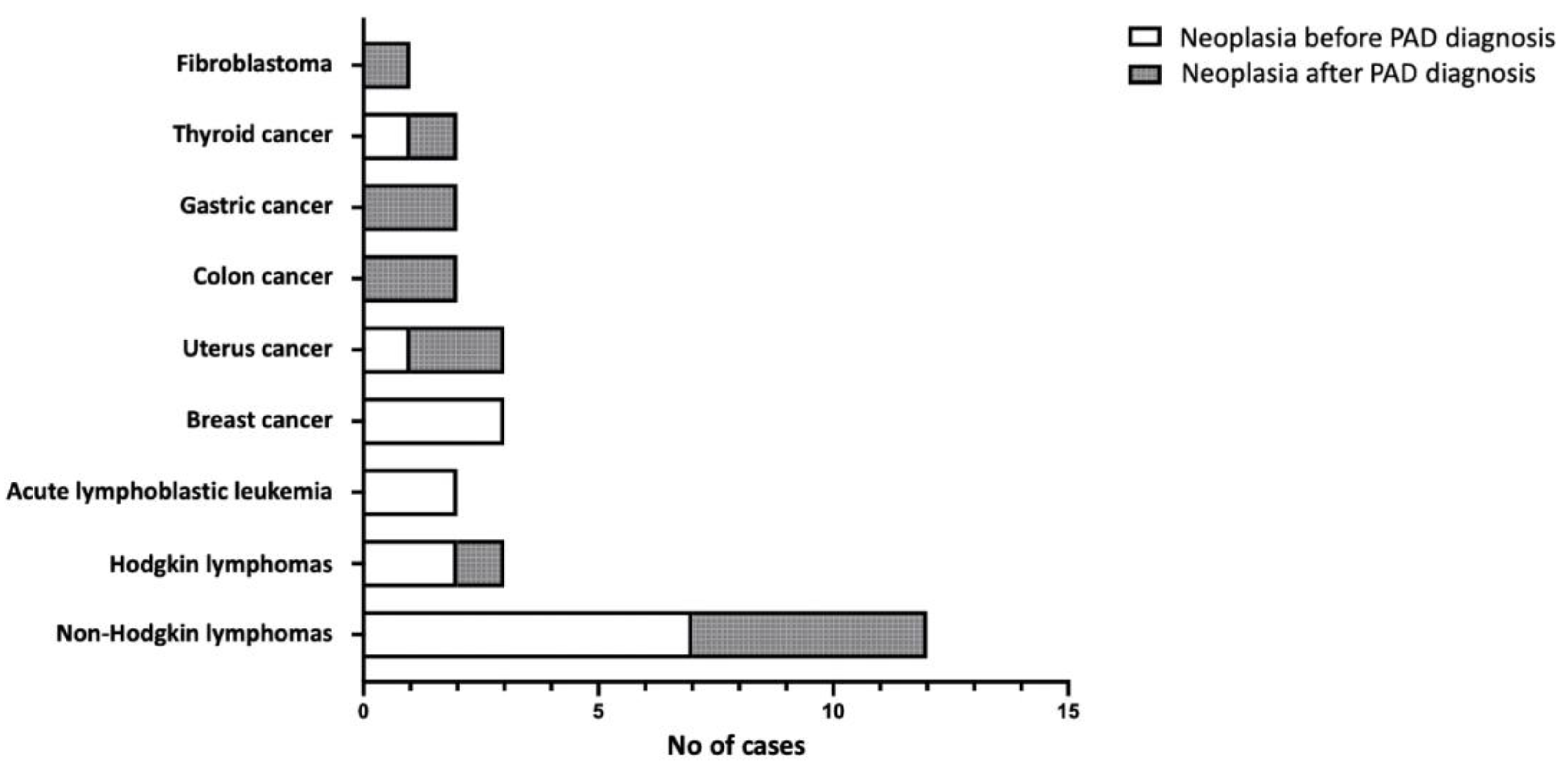

Table 1.
Overview of demographic, clinical and laboratory characteristics of the patients of the study.
Table 1.
Overview of demographic, clinical and laboratory characteristics of the patients of the study.
| Total | Group A (CVID) |
Group B (CVID-like) |
Group C (c.IgAD & IgGsD) |
Group D (CLT4 deficiency) |
Group E (unclassified hypogamma) |
|
| No | 153 | 123 | 12 | 7 | 5 | 6 |
| Sex (male/female) | 66/87 | 50/73 | 9/3 | 5/2 | 1/4 | 1/5 |
| Age of analysis (years; median, range) | 43.0 (7.0-77.0) |
44.0 (7.0-77.0) |
35 .0 (22.0-61.0) |
42.0 (15.0-45.0) |
28.0 (21.0-48.0) |
66.0 (54.0-71.0) |
| Age of diagnosis (years; median, range) | 37.0 (4.0-69.0) |
37.0 (4.0-69.0) |
32.0 (11.0-60.0) |
33.0 (13.0-44.0) |
19.0 (7.0-44.0) |
58.0 (46.0-69.0) |
| Delay of diagnosis (years; median, range) | 9.0 (0-43.0) |
9.0 (0.0-43.0) |
6.5 (1.0-17.0) |
12.0 (0.0-36.0) |
4.0 (0.0-25.0) |
7.5 (2.0-33.0) |
| Clinical manifestations | ||||||
| Infections (no, %) | 143 (93.5) | 116 (94.3) | 9 (75.0) | 7 (100.0) | 5 (100.0) | 6 (100.0) |
| Upper respiratory (no, %) | 128 (83.7) | 106 (86.1) | 8 (66.7) | 6 (85.7) | 4 (80.0) | 4 (66.7) |
| Lower respiratory (no, %) | 97 (63.4) | 83 (67.5) | 5 (41.7) | 3 (42.9) | 3 (60.0) | 3 (50.0) |
| Genitourinary (no, %) | 35 (22.9) | 31 (25.2) | 1 (8.3) | 1 (14.3) | 0 (0) | 2 (33.3) |
| Gastrointestinal (no, %) | 34 (22.2) | 28 (22.7) | 1 (8.3) | 0 (0) | 2 (40.0) | 3 (50.0) |
| Skin (no, %) | 34 (22.2) | 30 (24.4) | 2 (16.7) | 0 (0) | 1 (20.0) | 1 (16.7) |
| CNS (no, %) | 8 (5.2) | 7 (5.7) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) |
| Peritonitis (no, %) | 2 (1.3) | 1 (8.1) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) |
| Sepsis (no, %) | 2 (1.3) | 2 (1.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Parasite infections (no, %) | 7 (4.6) | 7 (5.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Herpes zoster (no, %) | 12 (7.8) | 11 (8.9) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) |
| Lymphoproliferation (no, %) | 92 (60.1) | 79 (64.2) | 5 (41.7) | 3 (42.9) | 3 (60.0) | 2 (33.3) |
| Splenomegaly (no, %) | 75 (49.0) | 62 (50.4) | 5 (41.7) | 3 (42.9) | 3 (60.0) | 2 (33.3) |
| Lymphadenopathy (no, %) | 65 (42.5) | 56 (45.5) | 5 (41.7) | 2 (28.6) | 2 (40.0) | 0 (0) |
| Intestine infiltrates (no, %) | 17 (11.1) | 15 (12.2) | 0 (0) | 1 (14.3) | 1 (20.0) | 0 (0) |
| Autoimmunity (no, %) | 87 (56.9) | 69 (56.1) | 7 (58.3) | 6 (85.7) | 3 (60.0) | 2 (33.3) |
| ITP or/and AHA (no, %) | 38 (24.8) | 29 (23.6) | 5 (41.7) | 3 (42.9) | 0 (0) | 1 (16.7) |
| Thyroid disease (no, %) ^ | 37 (24.2) | 28 (22.8) | 1 (8.3) | 3 (42.9) | 3 (60.0) | 2 (33.3) |
| Arthritis (no, %) | 11 (7.2) | 11 (8.9) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Psoriasis (no, %) | 10 (6.5) | 8 (6.5) | 2 (16.7) | 0 (0) | 0 (0) | 0 (0) |
| Others (no, %) # | 35 (22.9) | 28 (22.8) | 2 (16.7) | 2 (28.6) | 2 (40.0) | 1 (16.7) |
| Granulomatosis (no, %) | 16 (10.4) | 13 (10.6) | 1 (8.3) | 1 (14.3) | 1 (20.0) | 0 (0) |
| Enteropathy (no, %) | 38 (24.8) | 29 (23.6) | 1 (8.3) | 2 (28.6) | 3 (60.0) | 3 (50.0) |
| Atopy (no, %) | 37 (24.2) | 32 (26.0) | 1 (8.3) | 1 (14.3) | 2 (40.0) | 1 (16.7) |
| Complications of the disease | ||||||
| Bronchiectasis (no, %) | 39 (25.5) | 37 (30.0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (16.7) |
| COPD (no, %) | 19 (12.4) | 15 (12.2) | 0 (0) | 2 (28.6) | 0 (0) | 2 (33.3) |
| CRPD (no, %) | 25 (16.3) | 24 (19.5) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) |
| Chronic sinusitis (no, %)* | 21 (13.7) | 17 (13.8) | 2 (16.7) | 1 (14.3) | 0 (0) | 1 (16.7) |
| NRH (no, %) | 2 (1.3) | 2 (1.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Hypersplenism (no, %) | 18 (11.8) | 15 (12.2) | 2 (16.7) | 0 (0) | 0 (0) | 0 (0) |
| Neoplasia-total (no, %) | 25 (16.3) | 16 (13.0) | 7 (58.3) | 0 (0) | 1 (20.0) | 1 (16.7) |
| Neoplasia after PAD diagnosis (no, %) | 11 (7.2) | 10 (8.1) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) |
| Death (no, %) | 14 (9.2) | 11 (8.9) | 0 (0) | 0 (0) | 2 (40.0) | 1 (16.7) |
| Immunoglobulin levels (mg/dl) | ||||||
| Total (mean±SDEV) | 356.7 ± 265.1 | 306.0 ± 222.2 | 410.9 ± 281.2 | 894.7 ± 307.6 | 582.5 ± 88.0 | 472.5 ± 238.6 |
| IgG (mean ± SDEV) | 299.6 ± 231.0 | 255.5 ± 188.1 | 331.6 ± 206.5 | 833.6 ± 292.9 | 504.7 ± 112.0 | 362.2 ± 173.1 |
| IgA (mean ± SDEV) | 21.2 ± 42.6 | 16.2 ± 31.5 | 46.9 ± 91.6 | 9.4 ± 16.0 | 46.8 ± 13.2 | 70.0 ± 69.6 |
| IgM (mean ± SDEV) | 41.4 ± 61.6 | 36.9 ± 47.9 | 82.2 ± 150.8 | 51.8 ± 25.2 | 31.0 ± 29.2 | 40.3 ± 13.1 |
| IgG1 (mean ± SDEV) | 230.0 ± 177.2 | 185.6 ± 138.2 | 239.5 ± 172.6 | 561.4 ± 201.4 | 372.5 ± 84.1 | 254.3 ± 141.2 |
| IgG2 (mean ± SDEV) | 73.8 ± 71.2 | 62.2 ± 61.9 | 72.5 ± 56.3 | 164.0 ± 119.1 | 124.5 ± 26.2 | 75.3 ± 61.4 |
| IgG3 (mean ± SDEV) | 18.7 ± 19.9 | 15.7 ± 16.0 | 12.8 ± 9.7 | 49.6 ± 34.7 | 48.6 ± 7.9 | 14.1 ± 15.7 |
| IgG4 (mean ± SDEV) | 6.0 ± 14.6 | 6.8 ± 16.4 | 6.4 ± 12.8 | 0.9 ± 1.0 | 1.9 ± 1.1 | 4.1 ± 5.4 |
Abbreviations: AHA, autoimmune hemolytic anemia; COPD, chronic obstructive pulmonary disease; CNS, central nervous system; CRPD, chronic restrictive pulmonary disease; CVID, common variable immunodeficiency; ITP, idiopathic thrombocytopenic purpura; NRH, nodular regenerative hyperplasia (of liver) ^ thyroid disease: hyper- or hypothyroidism # others included: vitiligo (7), pernicious anemia (5), fibromyalgia (4), eczema (3), alopecia (3), autoimmune neutropenia (4), autoimmune hepatitis/PBC (2), vasculitis (2), CNS myelitis (2), celiac-like disease (2), Sjogren-like syndrome (1), Raynaud syndrome (1) * including the development of nasal polyps (not only recurrent sinus infections).
Table 2.
Clinical and molecular data of patients with CTLA4 mutations*.
| Patient | Sex | Age onset | Age at diagnosis | Family history and clinical presentation | Molecular defect | Further complications and outcome |
| #68 | 2 | 4 | 18 | Proband. Recurrent upper respiratory infections, Crohn-like disease, autoimmune thyroiditis, pernicious anemia, tonsillar hypertrophy, sun-sensitive skin rash | c.267C>A, p.Y89X | No (alive in good condition under IgRT) |
| #69 | 2 | 20 | 44 | Mother of #68. Recurrent sinusitis and erycipelas, autoimmune thyroiditis, sun-sensitive skin rash | c.267C>A, p.Y89X | No (alive in good condition under IgRT) |
| #116 | 2 | 25 | 28 | Proband. Unknown family history. Recurrent CNS, upper & lower respiratory infections, nasal polyps, splenomegaly | c.224G>A, p.R75Q | No (alive in good condition under IgRT) |
| #133 | 1 | 15 | 19 | Proband. His father was a carrier of the same defect without any disease. Recurrent respiratory infections and autoimmune thyroiditis. Massive splenomegaly at diagnosis (diagnostic splenectomy). | c.267C>A, p.Y139C | Non-Hodgkin lymphoma 8 years after diagnosis (remission after treatment). Died 12 years after diagnosis due to a relapse of lymphoma |
| #134 | 2 | 7 | 7 | Proband. Her father was a carrier of the same defect without any disease. Splenomegaly (diagnostic splenectomy), lymphadenopathy, followed several years later by recurrent lower respiratory infections, spontaneous peritonitis, severe granulomatous disease (liver, spleen, lungs) | c.208C > T, p.R70W | Cirrhosis, renal insufficiency, pulmonary hypertension. Died 14 years after diagnosis due an attack of spontaneous bacterial peritonitis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



