
Submitted:
26 February 2024
Posted:
27 February 2024
You are already at the latest version
Abstract
DNA polymerases replicate cell genomes and/or participate in the maintenance of genome integrity. DNA polymerases sharing high sequence homology with E. coli DNA polymerase I (pol I), have been grouped in Family A. Pol I participates in Okazaki fragment maturation and in bacterial genome repair. Since its discovery in 1956, pol I has been extensively studied, primarily to get deeper insights into the mechanism of DNA replication. As research on DNA polymerases advances, many novel functions of this group of polymerases are uncovered. For example, human DNA polymerase θ (a Family A DNA pol) has been shown to synthesize DNA using RNA as a template, a function typically attributed to retroviral reverse transcriptase. A heightened interest in drug discovery against pol θ has emerged due to its roles in cancer. Likewise, Pol I family enzymes also appear attractive drug-development targets against microbial infections. Development of antimalarial compounds targeting apicoplast apPOL, an ortholog of Pol I, further extends the druggability of this family of enzymes. Here, we summarize reported drug-development efforts against Family A polymerases, and future perspective regarding these enzymes as antibiotic targets. Current techniques such artificial intelligence can be used to facilitate the goal of new drugs.
Keywords:
DNA polymerase I
; Polymerase θ
; reverse transcriptase
; homologous recombination
; antibiotics
; apicoplast
1. Introduction
DNA replication and repair are essential for the propagation of all forms of life. DNA polymerases (DNA pols) replicate genomic DNA or maintain the integrity of the host cell genome. These enzymes catalyze phosphoryl transfer reactions and incorporate deoxynucleotide monophosphate (dNMP) at the 3′-end of the growing chain by hydrolyzing deoxynucleotide triphosphates (dNTPs). Broadly, DNA pols can be divided into two groups: (i) replication DNA pols, and (ii) repair DNA pols. The replication DNA pols are required only once in the lifetime of a cell, whereas the repair DNA pols are needed throughout the lifespan of a cell as a mammalian cell is subject to ~ 70,000 lesions per day [1]. Generally, DNA pols have high fidelity and processivity and carry out template-dependent DNA synthesis. However, some DNA pols conduct error-prone DNA synthesis (low fidelity) and have low processivity, i.e. they conduct distributive DNA synthesis. A few DNA pols such as terminal deoxynucleotidyl transferase (TdT) synthesize DNA in a template-independent manner [2].
Using sequence homology, DNA pols have been divided into 8 major families: A, B, C, D, X, Y, RT (reverse transcriptase), and AEP (archaeo-eukaryotic primase) [3,4,5,6,7,8,9]. DNA pols sharing sequence homology with E. coli DNA polymerase I, the first discovered DNA pol [3,9]. DNA pols sharing sequence homology with E. coli DNA polymerases I, II, and III were grouped into A, B, and C Families, respectively [3,9]. E. coli DNA pol I (pol I) is one of the most studied Family A DNA polymerases. Additionally, a rigorous strategy was employed to reclassify Family A pols into 19 subfamilies [10]. Pol I has three functions: (i) 5′ – 3′ DNA synthesis, (ii) 3′-5′ exonuclease (proofreading) activity, and (iii) 5′ – nuclease (also known as flap-endonuclease) activity. All these functions reside on the same polypeptide but on three structurally distinct domains [11]. Limited proteolysis of pol I results in two active fragments [11,12]: A large fragment of ~600 C-terminal residues known as Klenow fragment (KF), which possesses both DNA synthesis and 3′-5′ exonuclease activities, and a smaller fragment (~300 amino acids) that contains 5′ – nuclease activity [12]. All known bacterial pol I homologs have high structural similarity and contain three distinct structural domains. However, some members do not have 3′ – 5′ exonuclease activity despite the presence of the structural domain [13,14]. The pol I orthologs in eukaryotes (except yeast) only have KF-equivalent proteins. For example, the catalytic subunit of mammalian polymerase γ (pol γ) has only the polymerase and proofreading domains [15]. Similarly, mammalian DNA polymerases θ and ν (pol θ and pol ν) have polymerase and proofreading domains and no 5′ – nuclease domain [16,17,18,19,20,21]. While mammalian pol γ possesses proofreading activity, both pol θ and pol ν lack a conserved 3′–5′ exonuclease motif DxE therefore, they do not perform 3′-5′ exonuclease function [10]. Nonetheless, owing to structural homology with pol I, pol γ, pol θ, and pol ν have been conveniently referred to as Family A DNA pols [10].
The Family A pols have been identified in almost all forms of biological entities including viruses, plants, and parasitic organisms [10,22,23]. However, a pol I homologue in yeast is yet to be discovered. In recent years, there has been a heightened interest among researchers in these enzymes due their role in diseases such as cancer and malaria. Thus, inhibitors targeting DNA synthesis function of human DNA pol θ [24,25,26,27] and P. falciparum apicoplast apPOL [28] have been reported. Development of competitive inhibitors with respect to dNTP substrate, and allosteric inhibitors have been reported [24]. However, only one allosteric inhibitor has recently been cleared for Phase I/II clinical trials. Discovery of allosteric inhibitors targeting pol θ paves the way for developing the compounds against bacterial Family A DNA polymerase as these enzymes share a high sequence and structural homology with allosteric inhibitor binding pockets of pol θ and apPOL (discussed in following sections), thereby providing opportunities for the development of a novel class of antibiotics. Due to low structural and sequence homology of DNA pol γ with pol θ, it appears that it is unlikely that the same approach can be used to develop inhibitors against pol γ even though pol γ has been associated with variety of disorders [29].
2. Pol θ as Drug-Development Target in HR-Deficient Cancers
2.1. Human DNA pol θ
Human DNA pol θ, a multifunctional protein of 2590 amino acids (~290 kDa) is encoded by POLQ gene. Pol θ has three distinct domains: (i) a 899 amino acid long N-terminal SF2 helicase domain, (ii) a ~771 amino acid long C-terminal DNA polymerase domain, and (iii) a 920 amino acid long central domain [30]. The structure of N-terminal domain resembles to SF2 helicase and conducts NTPase activity, but the nucleic acid unwinding function is yet to be demonstrated [31]. The structure, and the function of middle domain is not known. The C-terminal domain shares structural homology with the KF of E. coli DNA pol I [13,32]. The role of pol θ in maintaining genome integrity and DNA repair has been extensively studied [30]. Some of the documented functions of pol θ include translesion DNA synthesis (TLS) past variety of DNA lesions (bulky adducts, abasic sites, and interstrand crosslinks) [33], template-independent DNA synthesis [34], RNA-dependent DNA synthesis (reverse transcriptase activity) and RNA-dependent DNA repair [35] as well as pol θ mediated end joining (TMEJ) [36,37]. Reports have shown that pol θ is overexpressed in breast, prostate, and lung cancer, and its inhibition can sensitize cancer cells to chemotherapy and radiotherapy [38]. A recent review by Wood and Doublie [30] details all the functions of pol θ. Therefore, we will only focus on recent drug-discovery efforts targeting this enzyme. Additionally, we have restricted this report only to the polymerase domain of pol θ despite the fact that the helicase domain is as significant as the polymerase domain.
2.2. Human DNA pol θ as Double Strand Break (DSB) Repair Enzyme
In healthy cells BRCA1 and BRCA2 serve as “tumor suppressor” genes. The proteins encoded by these genes (BRCA1 and BRCA2) repair double stranded breaks (DSBs) [39]. Deficiency in DSB repair mediated by BRCA1 and BRCA2 can lead to the proliferation of cancer cells due to the accumulation of driver mutations. Alternate DNA repair pathways attempt to take over [40] in the event of compromised BRCA-mediated DSB repair [41]. All components of alternate DSB repair pathways are attractive anti-cancer drug-development targets as cancer cells are prone to mutations and DSB generation forms the backbone of cancer therapies such as radiation-based therapies. One of the well-studied, and successful proteins of alternate DBS repair pathway is Poly (ADP-ribose) polymerase 1 (PARP1) [42]. Thus, a handful anticancer drugs have been developed and approved against PARP [43,44] (Figure 1). Unfortunately, resistance to PARP inhibitors is an extremely common phenomenon in clinics, and more than 40% BRCA1/BRCA2-deficient patients fail to respond to PARP inhibitors [45] (Figure 1). Pol θ has been shown to conducts microhomology mediated end joining (MMEJ) (also known as theta mediate end-joining or TMEJ) in the absence of functional BRCA1 and BRCA2 in cancer cells. Therefore, pol θ is considered an attractive anticancer drug development target for drug resistant HR cancers. It should be noted that recent advancements in the field indicate that BRCA mutants which exhibit DNA end section have a heightened sensitivity to pol θ inhibitors [46]. Genomic expression data published in The Cancer Genome Atlas (TGCA) show that POLQ is significantly overexpressed in cancer when BRCA1 or BRCA2 are altered or mutated (Figure 2). Therefore, the inhibition of pol θ is anticipated to be less toxic than the conventional chemotherapy. Reports showed that inhibition of pol θ using siRNA resulted in cancer cell death in vitro [41]. These results have triggered identifying new therapeutics such as small molecule inhibitors against pol θ.
2.3. Status of Drug-Development against Human DNA pol θ
To date, four studies have reported inhibitors of pol θ that target its polymerase function [24,25,26,27]. Additionally, one study reported a class of small molecules that inhibit helicase function of pol θ [47]. A summary of pol θ inhibitors has been presented by Pismataro et al. [48]. Pol θ inhibitors targeting polymerase domain can be divided into two classes: (i) competitive inhibitors (termed as dxNTPs) with respect to dNTPs [24], and (ii) allosteric inhibitors [25,26,27]. Structurally related allosteric inhibitors ART558, ART812, and ART899 [25,27,49] bind in the ‘fingers’ subdomain of the polymerase domain and inhibit polymerase function of pol θ (Figure 3), most likely via as mechanism similar to that of non-nucleoside RT inhibitors (NNRTIs). A small molecule inhibitor GSK101 (IDE705) inhibits ATPase function of pol θ. An IND (Investigational New Drug) application has been cleared by the US FDA (August 2023). Other notable pol θ inhibitors are novobiocin (NVB), and RP-6685 [20,22,23]. NVB inhibits ATPase function [47], whereas RP-6685 is an allosteric polymerase inhibitor that binds in the ‘fingers’ sub domain of pol θ [26]. NVB is an antibiotic, which was discovered as the inhibitor of DNA gyrase [50]). It has been demonstrated that NVB and ART558 have high synergy with PARP inhibitors and decreases the IC50 values significantly in HR-deficient cells [49]. RP-6685 was identified in 2022 and mouse models bearing BRCA-deficient tumors showed treated with RP-6685 a decreased tumor volume when treated with RP-6685, although a slight increase was observed after the 21st day of treatment [20,24].
3. Apicoplast DNA Polymerase (apPOL) as Antimalaria Target
3.1. Status of Drug-Development against apPOL
According to the World Malaria Report 2022 (https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022), malaria kills more than half a million people every year. Malaria is caused by the parasites of the Plasmodium genus of Apicomplexa phylum [51]. Majority of organisms belonging to Apicomplexa phylum contain an apicoplast that is evolutionarily related to the chloroplast [51,52,53,54]. Apicoplast participates several metabolic pathways, and it is essential for survival of the parasite [55]. A Family A DNA polymerase known as apPOL is an integral part of replisome that copies 35 kb genome of apicoplast [54,56]. The phylogenetic analyses showed that apPOL has low sequence homology (23%) with human DNA pol θ. However, the crystal structure of showed that apPOL has a canonical KF fold (PDB files 7SXL and 7SXQ) [28] and the polymerase domain of apPOL superposes on the polymerase domain of pol θ (PDB file 8E23) with ~1.5 Å root mean square deviation (RMSD), and with ~1.9 Å with the Klenow fragment equivalent of Bacillus stearothermophilus despite low (25%) sequence homology [57].
3.2. Apicoplast as Antimalaria Drug Target
The majority of drug-development against apPOL has been conducted by Nelson and colleagues. Screening of compounds from the Open Access Malaria Box drug collection resulted in the identification of compound MMV666123 [52]. Further attempts at structure-activity relationship (SAR) studies of MMV66123 (termed as compound 5a) did not provide a compound with better potency [28]. Attempts to solve the crystal structure of apPOL in complex with 5a did not succeed. However, mutational data showed that compound 5a binds close to W512. The equivalent residues in KF and pol θ are Y801 and F2426, respectively. In the crystal structure of pol θ in complex with RP-6685 (PDB file 8E24) [26], residue F2426 is not within interacting distance with the compound. Therefore, F2426 may serve as the ‘gate’ as suggested by Chheda et al for W512 [28]. It is possible that RP6685 and related compounds bind in the ‘fingers’ subdomain of apPOL at the RP-6685 equivalent site involving the residues of O, O1, and O2 helices as the pol θ residues interacting with RP-6685 are well-conserved across Family A DNA pols (Section 4.4). The other antimalarial compounds under development are pyrrole-hydroxybutenolide hybrids [58]. It is also possible that W512 and I422 of apPOL act as E138 of p51 subunit K101 of p66 subunit of HIV-1 RT, which serve as ‘gate’ for the binding of 2nd-generation non-nucleoside inhibitors (NNRTI) (PDB files 3MEE and 2ZD1) [59,60,61].
4. Pol I as Target for Combating Antimicrobial Resistance
4.1. Antimicrobial Resistance: A Global Health Concern
Antimicrobial resistance (AMR) is a serious public health concern. According to the Antibiotic Resistance Threats Report 2019 from the CDC, ~2.8 million AMR infections occur each year causing ~36,000 deaths [62]. Globally, these numbers are significantly higher. In 2019, ~4.3 million deaths were associated with bacterial antibiotic resistance, of which 1.27 million deaths were directly attributed to bacterial AMR [63]. Closely connected humans, animals, and environmental habitats contribute to the emergence, evolution, and spread of bacterial AMR [64]. Thus, major multidrug-resistant (MDR) organisms have been recovered from humans, animals, and the environmental habitat [64]. The drivers of bacterial AMR are manifold, but excessive antibiotic use and misuse in humans and animals are two major drivers of AMR [65,66,67]. Other socioeconomic factors such as the paucity of clean water have led to the development of resistance to almost all currently used antibiotics [62]. All of the above-mentioned factors have sparked renewed interest of researchers to investigate alternatives to currently prescribed antibiotics as treatment options against drug-resistant bacterial infections [68,69].
4.2. Family A Polymerase as a Novel Antibacterial Target
Pol I group of enzymes participate in the Okazaki fragment maturation during bacterial genome replication [70]. This group of enzymes is also crucial for cell survival following DNA damage. Due to their ability of nick-translation, pol I enzymes function as the effectors for the ligase-mediated sealing of single-stranded nicks. A nick-sealing property does not appear to be present in other DNA [71]. Mutations in E. coli pol I has been shown to confer high sensitivity to UV radiation and methyl methanesulphonate [72]. Pathogenically, Pol I has been shown to be essential for the viability of, otherwise impaired, dnaN159 E. coli [73], and the survival of H. pylori [74]. In S. Typhimurium LT2, pol I is required for the uses of ethanolamine, 1,2-propanediol, or propionate as the carbon and energy source [75]. These examples underscore the importance of bacterial DNA pol I as drug targets.
4.3. Pol I Inhibitors Acting through a Mechanism Analogous to NNRTI Inhibition of HIV-1 RT
The structure of KF (the first DNA polymerase structure) [13] showed that the polymerase domain of these enzymes consists of subdomains that resemble a half-open right hand, leading to the nomenclature of these domains as the ‘thumb’, fingers’ and the ‘palm’. Subsequent structures of pol I enzymes showed that the ‘thumb’ and ‘fingers’ subdomains undergo substantial conformational changes to form a catalytically competent ternary complex consisting of template-primer and dNTP substrate [76]. A comparison of HIV-1 RT in complex with NNRTI nevirapine [77], and HIV-1 RT in complex with DNA and dNTP [78] showed that the binding of NNRTI restricts the movement of the ‘thumb’ subdomain among other conformational changes called ‘molecular arthritis’ [77,79]. Reported compounds that bind in the ‘fingers’ subdomain of pol θ appear to inhibit it via a mechanism that is analogous to NNIRT inhibition of HIV-1 RT [80].
4.4. Comparison of Allosteric Inhibitor Binding Site in pol I Enzymes from Diverse Species
As reported for bacterial pol I [76], the ‘fingers’ subdomain of pol θ undergoes substantial conformational change to a closed conformation (Figure 4). A comparison of pol θ in complex with template-primer (t/p) and dideoxyguanosine 5′-triphosphate (ddGTP) (PDB file 8E24) [26] and pol θ bound to RNA/DNA (PDB file 6XBU) [35] shows that the O-helix significantly rotates towards the ‘palm’ subdomain (Figure 4). The binding of allosteric inhibitor RP-6685 to the ternary complex pol θ consisting of t/p and ddGTP (PDB file 8E24) locks O-helix in its closed conformation (Figure 4) [26], and restricts the conformational change of O-helix since as a part of the inhibitor is at the position where O-helix is expected to be during pyrophosphate release and the translocation steps of DNA polymerase catalysis (in the open fingers conformation) [81,82] (Figure 4).
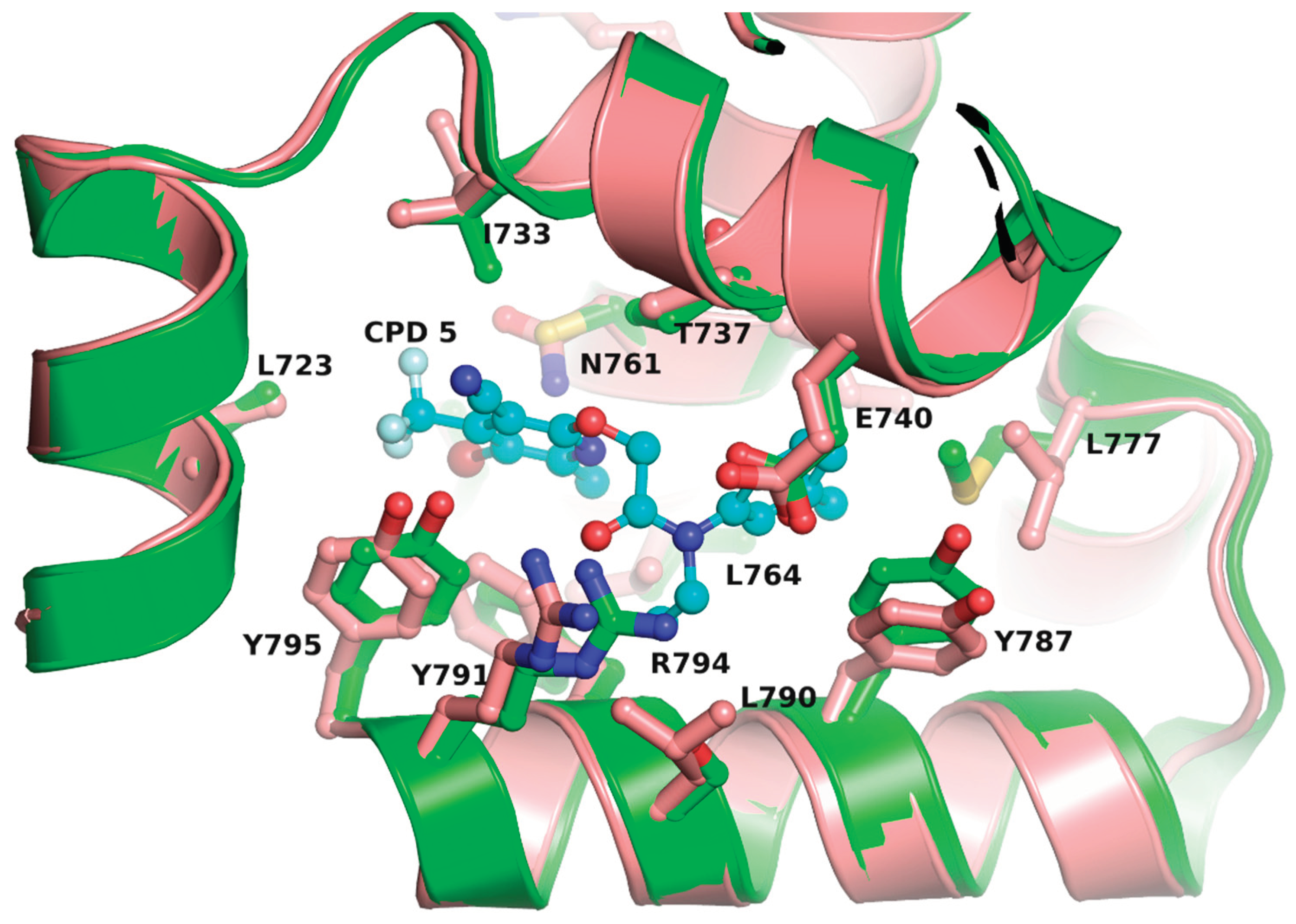
To assess if the pol θ allosteric inhibitor binding pocket is also present in other pol I enzymes at the topological position, we generated structures of pol I enzymes using pol θ in complex with p/t, ddGTP and compound 5 (PDB file 7ZX0) [27] as a template. We then docked compound 5 in a pocket formed by N, O, O1, and O2-helices of pol I enzymes from 12 bacterial species, apPOL and human pol ν. Compound 5 was docked with a high docking score in the ‘fingers’ subdomain of all bacterial pol I enzymes, and the crystal structures of apPOL and pol ν. The best docking pose of 5 in E. coli pol I is shown in Figure 5. a superposition of ‘fingers’ subdomain of pol It is clear from this figure that the majority of compound 5 interacting amino acid sidechains are conserved between pol θ and E. coli pol I (8 out of 11) (Figure 5). Additionally, compound 5 interacting residues are conserved across pol I family suggesting that structure-activity relationship approaches can be used to design species-specific allosteric inhibitors of pol I family of enzymes.
5. Conclusions
In response to the challenge of mutations under pressure of drugs to treat cancers, malaria and microbes, novel solutions are continuously explored. These solutions include novel targets, and application of novel drugs to different targets. For example, cancers with HR deficiencies can be targeted by developing inhibitors against pol θ, the inhibitors of pol θ can also act against opportunistic infections such bacterial and parasitic infections due to the structural homology seen in Family A polymerases. The structure-activity relationship strategies can be applied to develop disease-specific inhibitors. Furthermore, compounds against unexplored targets such as bacterial DNA pol I can be expected to inhibit multi-drug resistant bacteria and thereby help in combating AMR.
Author Contributions
Conceptualization, K. S., S. R. K., and S. W. G.; methodology, K. S., S. R. K., and S. W. G.; formal analysis, K. S., S. R. K., and S. W. G.; data curation, K. S., S. R. K., and S. W. G.; writing—original draft preparation, K.S. and S. R. K.; writing—review and editing, K. S., C.L.L., S. W. G., and W. P., supervision, K.S.; funding acquisition, K.S. All authors have read and agreed to the published version of the manuscript.
Funding
K Singh was partially funded the University of Missouri startup support.
Data Availability Statement
Not applicable.
Acknowledgments
K. Singh acknowledges the computation facilities of the Molecular Interactions Core at the University of Missouri, Columbia, MO 65212. We also thank numerous laboratories that have enormously contributed to pol I research, but we missed to cite their work.
Conflicts of Interest
C.L. Lorson is cofounder and chief scientific officer of Shift Pharmaceuticals and shares patents on compounds licensed by Shift Pharmaceuticals and planned patents. K Singh is chief scientific officer for Sanctum Therapeutics Corporation K Singh and MU share patents on compounds licensed by Sanctum Therapeutics Corporation and planned patents
References
- Lindahl, T.; Barnes, D.E.2000 Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol, 65, 127-133. [CrossRef]
- Modak, M.J.1978 Biochemistry of terminal deoxynucleotidyltransferase: Mechanism of inhibition by adenosine 5’-triphosphate. Biochemistry, 17, 3116-3120. [CrossRef]
- Delarue, M.; Poch, O.; Tordo, N.; Moras, D.; Argos, P.1990 An attempt to unify the structure of polymerases. Protein Eng, 3, 461-467. [CrossRef]
- Ishino, Y.; Komori, K.; Cann, I.K.; Koga, Y.1998 A novel DNA polymerase family found in archaea. J Bacteriol, 180, 2232-2236. [CrossRef]
- Braithwaite, D.K.; Ito, J.1993 Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res, 21, 787-802. [CrossRef]
- Cann, I.K.; Ishino, Y.1999 Archaeal DNA replication: Identifying the pieces to solve a puzzle. Genetics, 152, 1249-1267. [CrossRef]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; Prakash, L.; Prakash, S.; Todo, T.; Walker, G.C.; Wang, Z.; Woodgate, R.2001 The y-family of DNA polymerases. Mol Cell, 8, 7-8.
- Iyer, L.M.; Koonin, E.V.; Leipe, D.D.; Aravind, L.2005 Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: Structural insights and new members. Nucleic Acids Res, 33, 3875-3896. [CrossRef]
- Ito, J.; Braithwaite, D.K.1991 Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res, 19, 4045-4057. [CrossRef]
- Czernecki, D.; Nourisson, A.; Legrand, P.; Delarue, M.2023 Reclassification of family a DNA polymerases reveals novel functional subfamilies and distinctive structural features. Nucleic Acids Res, 51, 4488-4507. [CrossRef]
- Kornberg, A.; Baker, T.A. DNA replication. W.H. Freeman: New York, 1992.
- Klenow, H.; Henningsen, I.1970 Selective elimination of the exonuclease activity of the deoxyribonucleic acid polymerase from escherichia coli b by limited proteolysis. Proc Natl Acad Sci U S A, 65, 168-175. [CrossRef]
- Ollis, D.L.; Brick, P.; Hamlin, R.; Xuong, N.G.; Steitz, T.A.1985 Structure of large fragment of escherichia coli DNA polymerase i complexed with dtmp. Nature, 313, 762-766. [CrossRef]
- Kim, Y.; Eom, S.H.; Wang, J.; Lee, D.S.; Suh, S.W.; Steitz, T.A.1995 Crystal structure of thermus aquaticus DNA polymerase. Nature, 376, 612-616. [CrossRef]
- Carrodeguas, J.A.; Kobayashi, R.; Lim, S.E.; Copeland, W.C.; Bogenhagen, D.F.1999 The accessory subunit of xenopus laevis mitochondrial DNA polymerase gamma increases processivity of the catalytic subunit of human DNA polymerase gamma and is related to class ii aminoacyl-tRNA synthetases. Mol Cell Biol, 19, 4039-4046. [CrossRef]
- Malaby, A.W.; Martin, S.K.; Wood, R.D.; Doublie, S.2017 Expression and structural analyses of human DNA polymerase theta (polq). Methods Enzymol, 592, 103-121. [CrossRef]
- Leonhardt, E.A.; Henderson, D.S.; Rinehart, J.E.; Boyd, J.B.1993 Characterization of the mus308 gene in drosophila melanogaster. Genetics, 133, 87-96. [CrossRef]
- Kent, T.; Mateos-Gomez, P.A.; Sfeir, A.; Pomerantz, R.T.2016 Polymerase theta is a robust terminal transferase that oscillates between three different mechanisms during end-joining. Elife, 5. [CrossRef]
- Marini, F.; Kim, N.; Schuffert, A.; Wood, R.D.2003 Poln, a nuclear pola family DNA polymerase homologous to the DNA cross-link sensitivity protein mus308. J Biol Chem, 278, 32014-32019. [CrossRef]
- Takata, K.; Shimizu, T.; Iwai, S.; Wood, R.D.2006 Human DNA polymerase n (poln) is a low fidelity enzyme capable of error-free bypass of 5s-thymine glycol. J Biol Chem, 281, 23445-23455. [CrossRef]
- Sharief, F.S.; Vojta, P.J.; Ropp, P.A.; Copeland, W.C.1999 Cloning and chromosomal mapping of the human DNA polymerase theta (polq), the eighth human DNA polymerase. Genomics, 59, 90-96. [CrossRef]
- Inagaki, S.; Suzuki, T.; Ohto, M.A.; Urawa, H.; Horiuchi, T.; Nakamura, K.; Morikami, A.2006 Arabidopsis tebichi, with helicase and DNA polymerase domains, is required for regulated cell division and differentiation in meristems. Plant Cell, 18, 879-892. [CrossRef]
- Schoenfeld, T.W.; Murugapiran, S.K.; Dodsworth, J.A.; Floyd, S.; Lodes, M.; Mead, D.A.; Hedlund, B.P.2013 Lateral gene transfer of family a DNA polymerases between thermophilic viruses, aquificae, and apicomplexa. Mol Biol Evol, 30, 1653-1664. [CrossRef]
- Kent, T.; Rusanov, T.D.; Hoang, T.M.; Velema, W.A.; Krueger, A.T.; Copeland, W.C.; Kool, E.T.; Pomerantz, R.T.2016 DNA polymerase theta specializes in incorporating synthetic expanded-size (xdna) nucleotides. Nucleic Acids Res, 44, 9381-9392. [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; Majithiya, J.; McWhirter, C.; Martin, N.M.B.; Moore, S.; Neves, J.; Rajendra, E.; Ranzani, M.; Schaedler, T.; Stockley, M.; Wiggins, K.; Brough, R.; Sridhar, S.; Gulati, A.; Shao, N.; Badder, L.M.; Novo, D.; Knight, E.G.; Marlow, R.; Haider, S.; Callen, E.; Hewitt, G.; Schimmel, J.; Prevo, R.; Alli, C.; Ferdinand, A.; Bell, C.; Blencowe, P.; Bot, C.; Calder, M.; Charles, M.; Curry, J.; Ekwuru, T.; Ewings, K.; Krajewski, W.; MacDonald, E.; McCarron, H.; Pang, L.; Pedder, C.; Rigoreau, L.; Swarbrick, M.; Wheatley, E.; Willis, S.; Wong, A.C.; Nussenzweig, A.; Tijsterman, M.; Tutt, A.; Boulton, S.J.; Higgins, G.S.; Pettitt, S.J.; Smith, G.C.M.; Lord, C.J.2021 Poltheta inhibitors elicit brca-gene synthetic lethality and target parp inhibitor resistance. Nat Commun, 12, 3636. [CrossRef]
- Bubenik, M.; Mader, P.; Mochirian, P.; Vallee, F.; Clark, J.; Truchon, J.F.; Perryman, A.L.; Pau, V.; Kurinov, I.; Zahn, K.E.; Leclaire, M.E.; Papp, R.; Mathieu, M.C.; Hamel, M.; Duffy, N.M.; Godbout, C.; Casas-Selves, M.; Falgueyret, J.P.; Baruah, P.S.; Nicolas, O.; Stocco, R.; Poirier, H.; Martino, G.; Fortin, A.B.; Roulston, A.; Chefson, A.; Dorich, S.; St-Onge, M.; Patel, P.; Pellerin, C.; Ciblat, S.; Pinter, T.; Barabe, F.; El Bakkouri, M.; Parikh, P.; Gervais, C.; Sfeir, A.; Mamane, Y.; Morris, S.J.; Black, W.C.; Sicheri, F.; Gallant, M.2022 Identification of rp-6685, an orally bioavailable compound that inhibits the DNA polymerase activity of poltheta. J Med Chem, 65, 13198-13215. [CrossRef]
- Stockley, M.L.; Ferdinand, A.; Benedetti, G.; Blencowe, P.; Boyd, S.M.; Calder, M.; Charles, M.D.; Edwardes, L.V.; Ekwuru, T.; Finch, H.; Galbiati, A.; Geo, L.; Grande, D.; Grinkevich, V.; Holliday, N.D.; Krajewski, W.W.; MacDonald, E.; Majithiya, J.B.; McCarron, H.; McWhirter, C.L.; Patel, V.; Pedder, C.; Rajendra, E.; Ranzani, M.; Rigoreau, L.J.M.; Robinson, H.M.R.; Schaedler, T.; Sirina, J.; Smith, G.C.M.; Swarbrick, M.E.; Turnbull, A.P.; Willis, S.; Heald, R.A.2022 Discovery, characterization, and structure-based optimization of small-molecule in vitro and in vivo probes for human DNA polymerase theta. J Med Chem, 65, 13879-13891. [CrossRef]
- Chheda, P.R.; Nieto, N.; Kaur, S.; Beck, J.M.; Beck, J.R.; Honzatko, R.; Kerns, R.J.; Nelson, S.W.2022 Promising antimalarials targeting apicoplast DNA polymerase from plasmodium falciparum. Eur J Med Chem, 243, 114751. [CrossRef]
- Rahman, S.; Copeland, W.C.2019 Polg-related disorders and their neurological manifestations. Nat Rev Neurol, 15, 40-52. [CrossRef]
- Wood, R.D.; Doublie, S.2022 Genome protection by DNA polymerase theta. Annu Rev Genet, 56, 207-228. [CrossRef]
- Newman, J.A.; Cooper, C.D.O.; Aitkenhead, H.; Gileadi, O.2015 Structure of the helicase domain of DNA polymerase theta reveals a possible role in the microhomology-mediated end-joining pathway. Structure, 23, 2319-2330. [CrossRef]
- Zahn, K.E.; Averill, A.M.; Aller, P.; Wood, R.D.; Doublie, S.2015 Human DNA polymerase theta grasps the primer terminus to mediate DNA repair. Nat Struct Mol Biol, 22, 304-311. [CrossRef]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.A.; McBride, K.M.; Wood, R.D.2014 Mechanism of suppression of chromosomal instability by DNA polymerase polq. PLoS Genet, 10, e1004654. [CrossRef]
- Hogg, M.; Sauer-Eriksson, A.E.; Johansson, E.2012 Promiscuous DNA synthesis by human DNA polymerase theta. Nucleic Acids Res, 40, 2611-2622. [CrossRef]
- Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick, T.; Lopezcolorado, F.W.; Kent, T.; Siddique, L.A.; Mallon, J.; Huhn, J.; Shoda, Z.; Kashkina, E.; Brambati, A.; Stark, J.M.; Chen, X.S.; Pomerantz, R.T.2021 Poltheta reverse transcribes RNA and promotes RNA-templated DNA repair. Sci Adv, 7. [CrossRef]
- Chen, C.C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M.2018 Homology-directed repair and the role of brca1, brca2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol, 2, 313-336. [CrossRef]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P.2022 Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat Rev Mol Cell Biol, 23, 125-140. [CrossRef]
- Goff, J.P.; Shields, D.S.; Seki, M.; Choi, S.; Epperly, M.W.; Dixon, T.; Wang, H.; Bakkenist, C.J.; Dertinger, S.D.; Torous, D.K.; Wittschieben, J.; Wood, R.D.; Greenberger, J.S.2009 Lack of DNA polymerase theta (polq) radiosensitizes bone marrow stromal cells in vitro and increases reticulocyte micronuclei after total-body irradiation. Radiat Res, 172, 165-174. [CrossRef]
- Welcsh, P.L.; King, M.C.2001 Brca1 and brca2 and the genetics of breast and ovarian cancer. Hum Mol Genet, 10, 705-713. [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; Ramsden, D.A.; Gupta, G.P.2019 Genetic determinants of cellular addiction to DNA polymerase theta. Nat Commun, 10, 4286. [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; Yusufzai, T.; D’Andrea, A.D.2015 Homologous-recombination-deficient tumours are dependent on poltheta-mediated repair. Nature, 518, 258-262. [CrossRef]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P.2021 Alternative non-homologous end-joining: Error-prone DNA repair as cancer’s achilles’ heel. Cancers (Basel), 13. [CrossRef]
- Du, Y.; Yamaguchi, H.; Hsu, J.L.; Huang, M.C.2017 Parp inhibitors as precision medicine for cancer treatment. Natl. Sci. Rev., 4, 576–592. [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E.2020 Parp inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol, 8, 564601. [CrossRef]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J.2020 Parp inhibitor resistance: The underlying mechanisms and clinical implications. Mol Cancer, 19, 107. [CrossRef]
- Krais, J.J.; Glass, D.J.; Chudoba, I.; Wang, Y.; Feng, W.; Simpson, D.; Patel, P.; Liu, Z.; Neumann-Domer, R.; Betsch, R.G.; Bernhardy, A.J.; Bradbury, A.M.; Conger, J.; Yueh, W.T.; Nacson, J.; Pomerantz, R.T.; Gupta, G.P.; Testa, J.R.; Johnson, N.2023 Genetic separation of brca1 functions reveal mutation-dependent poltheta vulnerabilities. Nat Commun, 14, 7714. [CrossRef]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yucel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; Tainer, J.A.; Blagg, B.S.J.; Ceccaldi, R.; D’Andrea, A.D.2021 A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat Cancer, 2, 598-610. [CrossRef]
- Pismataro, M.C.; Astolfi, A.; Barreca, M.L.; Pacetti, M.; Schenone, S.; Bandiera, T.; Carbone, A.; Massari, S.2023 Small molecules targeting DNA polymerase theta (poltheta) as promising synthetic lethal agents for precision cancer therapy. J Med Chem, 66, 6498-6522. [CrossRef]
- Rodriguez-Berriguete, G.; Ranzani, M.; Prevo, R.; Puliyadi, R.; Machado, N.; Bolland, H.R.; Millar, V.; Ebner, D.; Boursier, M.; Cerutti, A.; Cicconi, A.; Galbiati, A.; Grande, D.; Grinkevich, V.; Majithiya, J.B.; Piscitello, D.; Rajendra, E.; Stockley, M.L.; Boulton, S.J.; Hammond, E.M.; Heald, R.A.; Smith, G.C.M.; Robinson, H.M.R.; Higgins, G.S.2023 Small-molecule poltheta inhibitors provide safe and effective tumor radiosensitization in preclinical models. Clin Cancer Res, 29, 1631-1642. [CrossRef]
- Gellert, M.; O’Dea, M.H.; Itoh, T.; Tomizawa, J.1976 Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc Natl Acad Sci U S A, 73, 4474-4478. [CrossRef]
- Kalanon, M.; McFadden, G.I.2010 Malaria, plasmodium falciparum and its apicoplast. Biochem Soc Trans, 38, 775-782. [CrossRef]
- Kaur, S.; Nieto, N.S.; McDonald, P.; Beck, J.R.; Honzatko, R.B.; Roy, A.; Nelson, S.W.2022 Discovery of small molecule inhibitors of plasmodium falciparum apicoplast DNA polymerase. J Enzyme Inhib Med Chem, 37, 1320-1326. [CrossRef]
- Sato, S.2011 The apicomplexan plastid and its evolution. Cell Mol Life Sci, 68, 1285-1296. [CrossRef]
- Wilson, R.J.; Denny, P.W.; Preiser, P.R.; Rangachari, K.; Roberts, K.; Roy, A.; Whyte, A.; Strath, M.; Moore, D.J.; Moore, P.W.; Williamson, D.H.1996 Complete gene map of the plastid-like DNA of the malaria parasite plasmodium falciparum. J Mol Biol, 261, 155-172. [CrossRef]
- Seeber, F.; Soldati-Favre, D.2010 Metabolic pathways in the apicoplast of apicomplexa. Int Rev Cell Mol Biol, 281, 161-228. [CrossRef]
- Seow, F.; Sato, S.; Janssen, C.S.; Riehle, M.O.; Mukhopadhyay, A.; Phillips, R.S.; Wilson, R.J.; Barrett, M.P.2005 The plastidic DNA replication enzyme complex of plasmodium falciparum. Mol Biochem Parasitol, 141, 145-153. [CrossRef]
- Milton, M.E.; Choe, J.Y.; Honzatko, R.B.; Nelson, S.W.2016 Crystal structure of the apicoplast DNA polymerase from plasmodium falciparum: The first look at a plastidic a-family DNA polymerase. J Mol Biol, 428, 3920-3934. [CrossRef]
- Pandey, A.R.; Singh, S.P.; Joshi, P.; Srivastav, K.S.; Srivastava, S.; Yadav, K.; Chandra, R.; Bisen, A.C.; Agrawal, S.; Sanap, S.N.; Bhatta, R.S.; Tripathi, R.; Barthwal, M.K.; Sashidhara, K.V.2023 Design, synthesis and evaluation of novel pyrrole-hydroxybutenolide hybrids as promising antiplasmodial and anti-inflammatory agents. Eur J Med Chem, 254, 115340. [CrossRef]
- Lansdon, E.B.; Brendza, K.M.; Hung, M.; Wang, R.; Mukund, S.; Jin, D.; Birkus, G.; Kutty, N.; Liu, X.2010 Crystal structures of HIV-1 reverse transcriptase with etravirine (tmc125) and rilpivirine (TMC278): Implications for drug design. J Med Chem, 53, 4295-4299. [CrossRef]
- Das, K.; Bauman, J.D.; Clark, A.D., Jr.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E.2008 High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci U S A, 105, 1466-1471. [CrossRef]
- Singh, K.; Marchand, B.; Rai, D.K.; Sharma, B.; Michailidis, E.; Ryan, E.M.; Matzek, K.B.; Leslie, M.D.; Hagedorn, A.N.; Li, Z.; Norden, P.R.; Hachiya, A.; Parniak, M.A.; Xu, H.T.; Wainberg, M.A.; Sarafianos, S.G.2012 Biochemical mechanism of HIV-1 resistance to rilpivirine. J Biol Chem, 287, 38110-38123. [CrossRef]
- Centers for disease control and prevention. Antibiotic resistance threats in the united states. U.S. Department of health and human services 2019. Available at https://www.Cdc.Gov/drugresistance/biggest-threats.Html. Accessed on oct. 10, 2023. Available at https://www.cdc.gov/DrugResistance/Biggest-Threats.html.
- Antimicrobial Resistance, C.2022 Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet, 399, 629-655.
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L.J.N.m.2019 Defining and combating antibiotic resistance from one health and global health perspectives. 4, 1432-1442. [CrossRef]
- McEwen, S.A.; Collignon, P.J.J.M.s.2018 Antimicrobial resistance: A one health perspective. 6, 6.2. 10. [CrossRef]
- Chatterjee, A.; Modarai, M.; Naylor, N.R.; Boyd, S.E.; Atun, R.; Barlow, J.; Holmes, A.H.; Johnson, A.; Robotham, J.V.2018 Quantifying drivers of antibiotic resistance in humans: A systematic review. Lancet Infect Dis, 18, e368-e378. [CrossRef]
- Tamma, P.D.; Cosgrove, S.E.2020 Unlikely bedfellows: The partnering of antibiotic stewardship programs and the pharmaceutical industry. Clin Infect Dis, 71, 682-684. [CrossRef]
- Blaskovich, M.A.J.A.I.D. Antibiotic alternatives special issue. ACS Publications: 2021; Vol. 7, pp 2025-2026. [CrossRef]
- O’Neill, J.2016 Tackling drug-resistant infections globally: Final report and recommendations.
- Kornberg, A.; Baker, T.A. DNA replication. W. H. Greeman: 1992; Vol. 3.
- Hejna, J.A.; Moses, R.E. DNA replication. Elsevier Inc.: 2009.
- De Lucia, P.; Cairns, J.1969 Isolation of an e. Coli strain with a mutation affecting DNA polymerase. Nature, 224, 1164-1166. [CrossRef]
- Maul, R.W.; Sanders, L.H.; Lim, J.B.; Benitez, R.; Sutton, M.D.2007 Role of escherichia coli DNA polymerase i in conferring viability upon the dnan159 mutant strain. J Bacteriol, 189, 4688-4695. [CrossRef]
- Garcia-Ortiz, M.V.; Marsin, S.; Arana, M.E.; Gasparutto, D.; Guerois, R.; Kunkel, T.A.; Radicella, J.P.2011 Unexpected role for helicobacter pylori DNA polymerase i as a source of genetic variability. PLoS Genet, 7, e1002152. [CrossRef]
- Rondon, M.R.; Horswill, A.R.; Escalante-Semerena, J.C.1995 DNA polymerase i function is required for the utilization of ethanolamine, 1,2-propanediol, and propionate by salmonella typhimurium lt2. J Bacteriol, 177, 7119-7124. [CrossRef]
- Li, Y.; Korolev, S.; Waksman, G.1998 Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of thermus aquaticus DNA polymerase i: Structural basis for nucleotide incorporation. EMBO J, 17, 7514-7525. [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A.1992 Crystal structure at 3.5 a resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science, 256, 1783-1790. [CrossRef]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C.1998 Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science, 282, 1669-1675. [CrossRef]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E.2012 HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat Struct Mol Biol, 19, 253-259. [CrossRef]
- Singh, K.; Flores, J.A.; Kirby, K.A.; Neogi, U.; Sonnerborg, A.; Hachiya, A.; Das, K.; Arnold, E.; McArthur, C.; Parniak, M.; Sarafianos, S.G.2014 Drug resistance in non-B subtype HIV-1: Impact of HIV-1 reverse transcriptase inhibitors. Viruses, 6, 3535-3562. [CrossRef]
- Franklin, M.C.; Wang, J.; Steitz, T.A.2001 Structure of the replicating complex of a pol alpha family DNA polymerase. Cell, 105, 657-667. [CrossRef]
- Johnson, K.A.1993 Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem, 62, 685-713. [CrossRef]
Figure 1.
Role of BRCA1/2 proteins, PARP and pol θ in cancers. Mutations or alterations in BRCA1/2 genes results in compromised homologous recombination (HR) in cancer cells. As a result of the genome instability, cancel cells start proliferating. These cells rely on the PARP1 gene for HR, which is overexpressed in cancer cells. Therefore, inhibitors targeting PARP have been designed and approved to suppress the growth of cancer. However, ~40% patients develop resistance to PARP inhibitors. BRCA-deficient cells also rely on pol θ for their continued growth. Thus, pol θ is an important target for inhibitors to prevent the growth of cancer cells.
Figure 1.
Role of BRCA1/2 proteins, PARP and pol θ in cancers. Mutations or alterations in BRCA1/2 genes results in compromised homologous recombination (HR) in cancer cells. As a result of the genome instability, cancel cells start proliferating. These cells rely on the PARP1 gene for HR, which is overexpressed in cancer cells. Therefore, inhibitors targeting PARP have been designed and approved to suppress the growth of cancer. However, ~40% patients develop resistance to PARP inhibitors. BRCA-deficient cells also rely on pol θ for their continued growth. Thus, pol θ is an important target for inhibitors to prevent the growth of cancer cells.
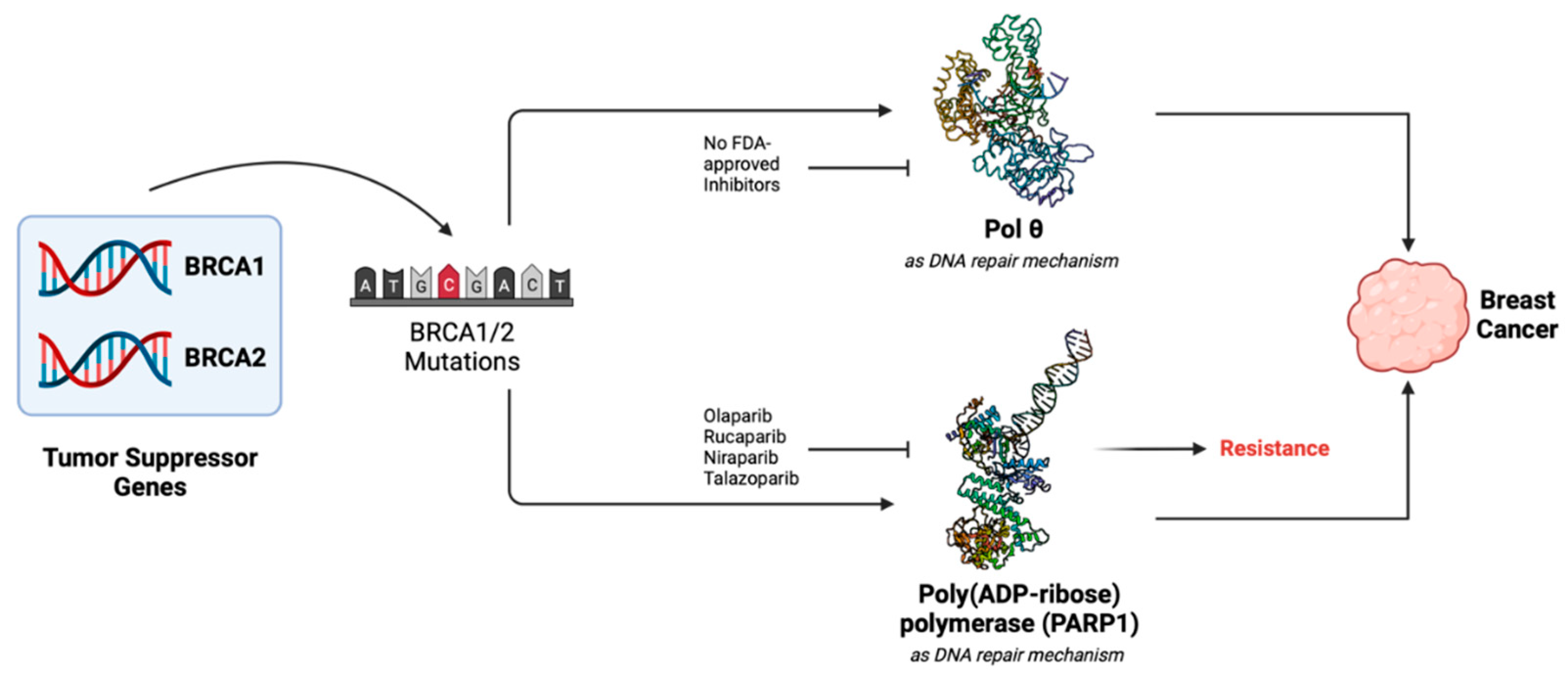
Figure 2.
Expression of POLQ in normal cells and the cells containing altered (or mutant) BRCA1/2. This figure shows the difference in POLQ expression in normal and in cells with BRCA mutations. Statistically significant higher POLQ expression is noted between normal and BRCA mutant cells.
Figure 2.
Expression of POLQ in normal cells and the cells containing altered (or mutant) BRCA1/2. This figure shows the difference in POLQ expression in normal and in cells with BRCA mutations. Statistically significant higher POLQ expression is noted between normal and BRCA mutant cells.
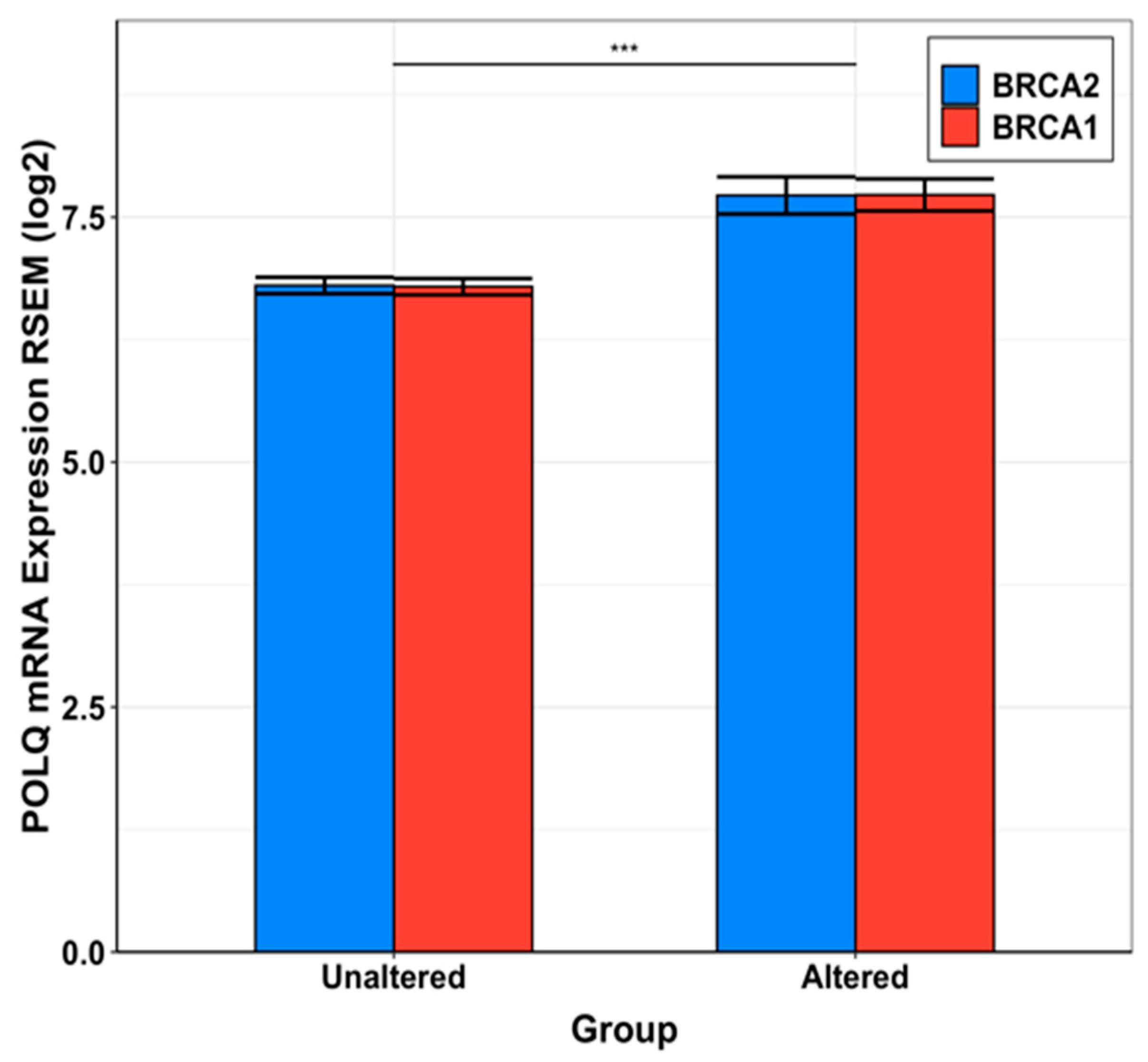
Figure 3.
Structure of human DNA pol θ in complex with allosteric inhibitor. This figure shows that compound 5 shown in surface representation [26] (PDB file 8E23) binds in the ‘fingers’ subdomain of human DNA pol θ. The ‘thumb’ ‘palm’, ‘fingers’ and 3′-5′ exonuclease domain of pol θ are shown as green, magenta, blue and gray ribbons, respectively.
Figure 3.
Structure of human DNA pol θ in complex with allosteric inhibitor. This figure shows that compound 5 shown in surface representation [26] (PDB file 8E23) binds in the ‘fingers’ subdomain of human DNA pol θ. The ‘thumb’ ‘palm’, ‘fingers’ and 3′-5′ exonuclease domain of pol θ are shown as green, magenta, blue and gray ribbons, respectively.
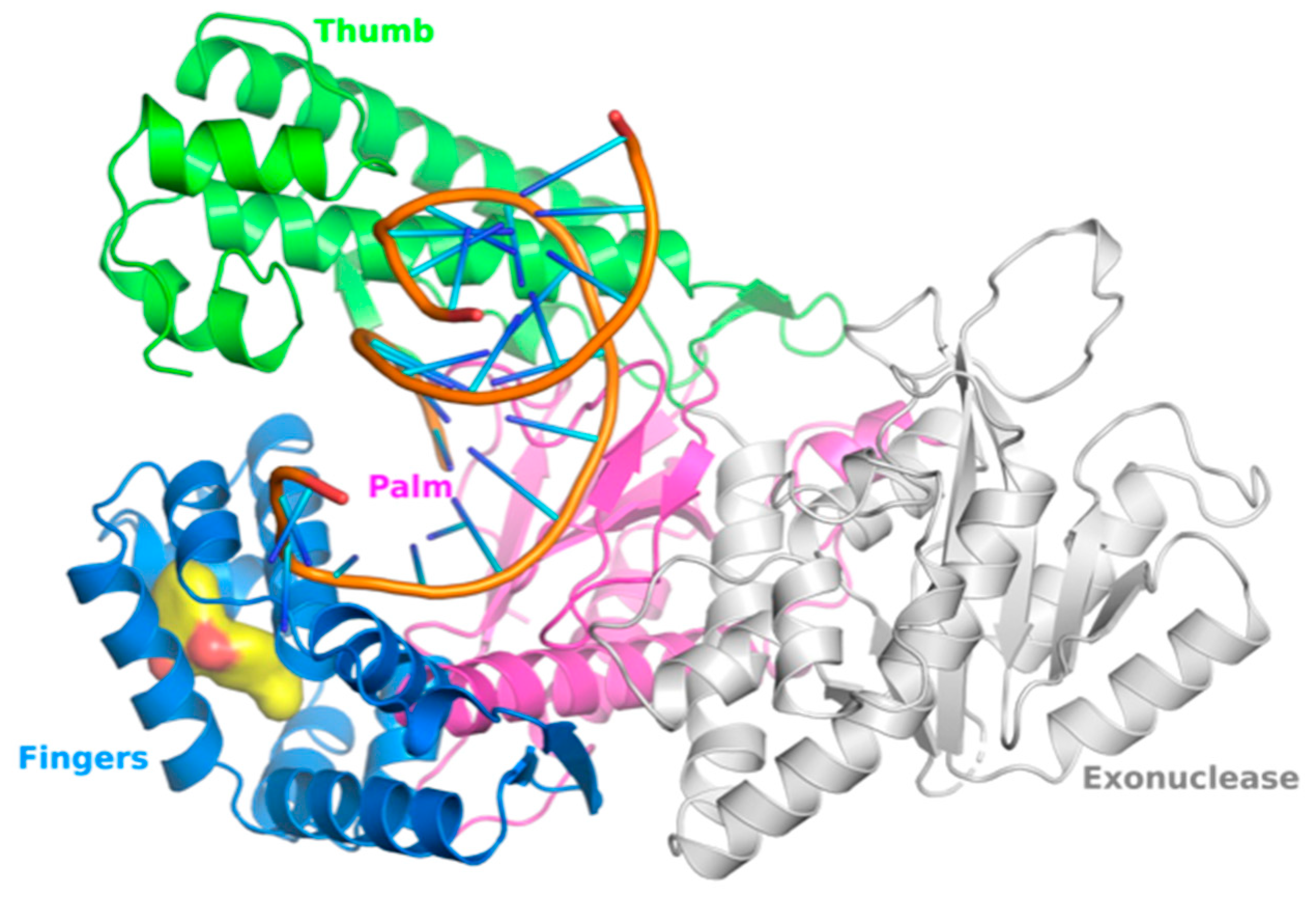
Figure 4.
Mechanism of allosteric inhibition of pol θ. Superposition of complexes of d pol θ-RNA/DNA (PDB file 6XBU) (violet ribbons), and pol θ-t/p-ddGTP-RP6685 (PDB file 8E24) (green ribbons) providing a mechanism of inhibition of allosteric inhibitor bound at the ‘fingers’ subdomain. RP6685 is shown as ball-and-sticks (cyan carbons). O-helix residues known to participate in dNTP binding and pyrophosphate release, ddGTP, and one of the three catalytic site carboxylates (green carbons) are shown rendered in balls-and-sticks as references to the inhibitor binding site. Atoms nitrogen, oxygen, phosphorus, and fluorine are colored blue, red, orange and light cyan, respectively. This figure illustrates the movement of O-helix creates inhibitor binding pocket and bound inhibitor does restricts conformational changes of the O-helix.
Figure 4.
Mechanism of allosteric inhibition of pol θ. Superposition of complexes of d pol θ-RNA/DNA (PDB file 6XBU) (violet ribbons), and pol θ-t/p-ddGTP-RP6685 (PDB file 8E24) (green ribbons) providing a mechanism of inhibition of allosteric inhibitor bound at the ‘fingers’ subdomain. RP6685 is shown as ball-and-sticks (cyan carbons). O-helix residues known to participate in dNTP binding and pyrophosphate release, ddGTP, and one of the three catalytic site carboxylates (green carbons) are shown rendered in balls-and-sticks as references to the inhibitor binding site. Atoms nitrogen, oxygen, phosphorus, and fluorine are colored blue, red, orange and light cyan, respectively. This figure illustrates the movement of O-helix creates inhibitor binding pocket and bound inhibitor does restricts conformational changes of the O-helix.
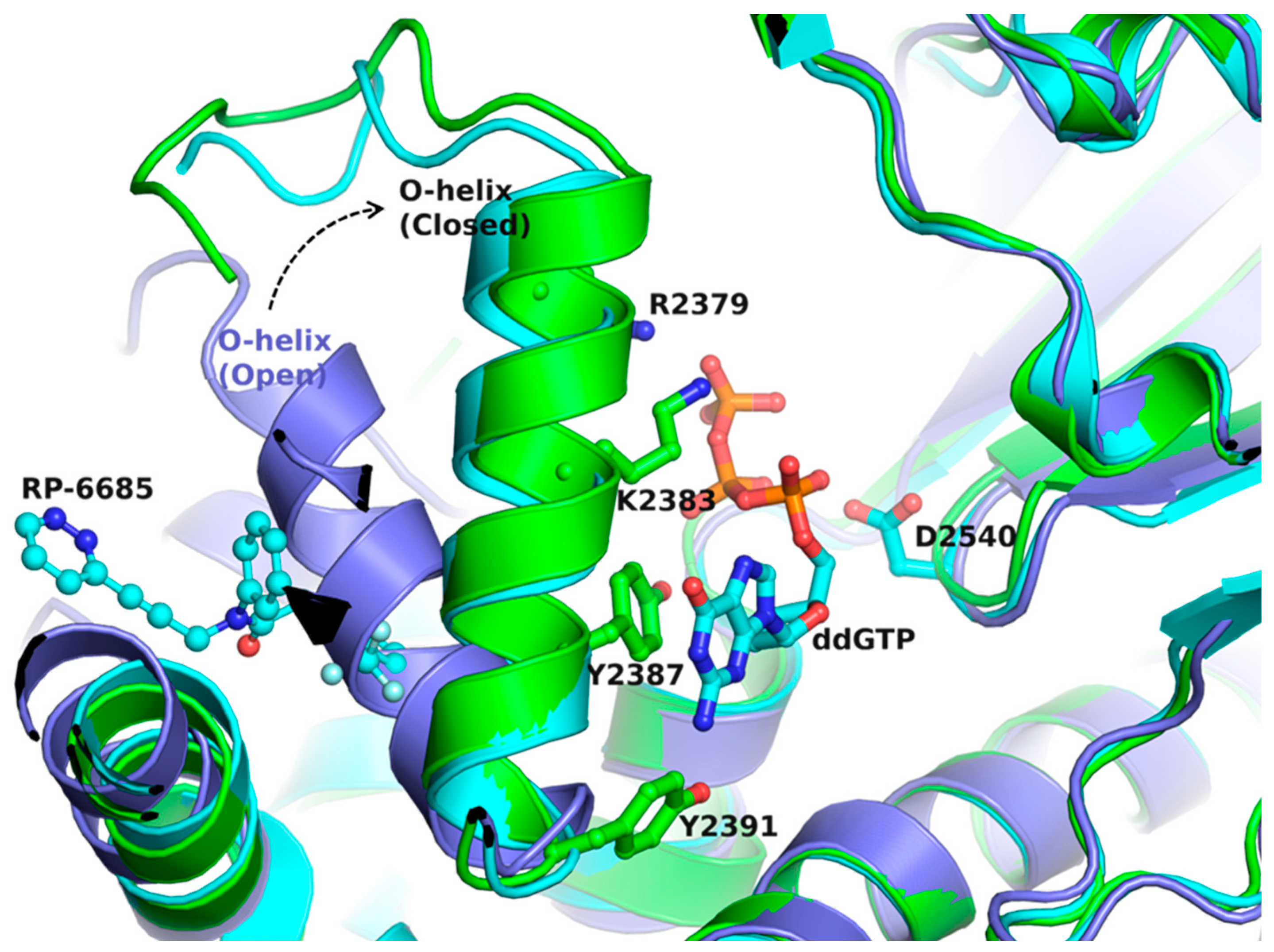
Figure 5.
Representative figure showing the conservation of inhibitor binding site residues. This figure shows the superposition pol θ (green ribbons) and KF (pink ribbons) bound to compound 5 (CPD 5), and the amino acid residues that interact with the compound are in ball-and-stick representation (pink carbons for E. coli pol I and green carbons for pol θ). RP6685 is also shown as ball-and-sticks (cyan carbons) but in thinner sticks compared to amino acid residues. Atoms nitrogen, oxygen, sulfur, and fluorine are colored blue, red, yellow, and light cyan, respectively. The numbering of amino acid residues corresponds to E. coli DNA pol I.
Figure 5.
Representative figure showing the conservation of inhibitor binding site residues. This figure shows the superposition pol θ (green ribbons) and KF (pink ribbons) bound to compound 5 (CPD 5), and the amino acid residues that interact with the compound are in ball-and-stick representation (pink carbons for E. coli pol I and green carbons for pol θ). RP6685 is also shown as ball-and-sticks (cyan carbons) but in thinner sticks compared to amino acid residues. Atoms nitrogen, oxygen, sulfur, and fluorine are colored blue, red, yellow, and light cyan, respectively. The numbering of amino acid residues corresponds to E. coli DNA pol I.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



