Submitted:
27 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
Forensic hospitals throughout the country house individuals with severe mental illness and history of criminal violations. Insomnia affects 67.4% of hospitalized patients with chronic neuropsychiatric disorders, indicating that these conditions may hijack human somnogenic pathways. Conversely, somnolence is a common adverse effect of many antipsychotic drugs, further highlighting a common etiopathogenesis.
The role of dysfunctional mitochondria in psychopathology is well-established, however, the association of these organelles with sleep physiology is novel. Indeed, reducing neuronal oxidative stress by importing mitochondria from astrocytes, may be the purpose of human slumber. This model may explain mitochondrial dysfunction during anesthesia as well as in the rare genetic disease, fatal familial insomnia.
In this narrative review, we focus on the salience network of the brain, a common denominator for insomnia, neuropsychiatric and neurodegenerative disorders. We also discuss mitochondria-protecting strategies, including membrane lipid replacement, natural and synthetic phenazine and phenothiazine derivatives.
Keywords:
Von Economo Neuron
; interoceptive awareness
; frontotemporal dementia behavioral variant
; phenazines
Introduction
One of the most common sleep disorders in the United States, primary insomnia, is usually defined as long sleep latency, difficulty staying asleep, prolonged nighttime wakefulness, and/or early morning awakening [1]. In prison, approximately 60% of inmates experience insomnia, a prevalence 6-10 times higher than in the population at large [2]. Moreover, insomnia is present in 67.4% of hospitalized patients with severe mental illness, suggesting that the pathways of sleep and neuropathology are highly intertwined [3].
Forensic psychiatric hospitals admit patients with schizophrenia (SCZ) or schizophrenia-like disorders (SLDs) and criminal violations. Insomnia is common in this population and failure to address this condition may increase healthcare expenditure due to medical complications, including metabolic, cardiovascular, and neurodegenerative disorders. The salience network (SN), comprised of insular cortex (IC), anterior cingulate cortex (ACC) and several subcortical nodes, has recently been implicated in the etiopathogenesis of insomnia, SCZ, and neurodegenerative disorders [4,5,6,7,8,9]. Von Economo neurons (VENS), a special class of large, spindle-shaped cells found only in humans and superior mammals, are believed to drive empathy, social awareness, fairness, and alertness, connecting sleep with the higher brain functions [10,11]. VENS reside in the SN and play a key role in switching the attentional focus from interoception to exteroception as required by each situation.
At the molecular level, incarceration, insomnia, and severe mental illness have been associated with premature cellular senescence, a phenotype marked by increased intracellular iron and mitochondrial depletion [11,12,13,14,15,16,17,18]. Premature cellular senescence may be triggered by activating the master regulator of cellular aging, aryl hydrocarbon receptor (AhR), residing in both the cytosol and mitochondria [19,20,21]. Senescent cells upregulate intracellular iron which in the proximity of cytosolic fats, increases the risk of lipid peroxidation and neuronal demise by ferroptosis [22,23,24]. Ferroptosis is a programmed, cell death induced by iron in the context of antioxidant failure marked by depletion of glutathione peroxidase-4 (GPX-4) [25,26]. GPX-4 is a mitochondrial enzyme which averts ferroptosis by repairing the oxidized phospholipids and cholesterol in mitochondrial and neuronal membranes [27].
Antipsychotic drugs are known for causing somnolence, indicating a likely interference with the human sleep pathways. For example, phenothiazines, induce sleep by antagonizing histamine H1 and alpha1 adrenergic receptors [28]. Clozapine, an AhR-activating ligand, may induce somnolence by altering the expression of circadian clock genes, some of which are controlled by the AhR [29,30]. Aside from clozapine, oxidized cell membrane lipids also bind AhR, possibly interfering with sleep physiology.
The phenothiazine class of antipsychotic drugs are potent inhibitors of cholesterol metabolism as they lower 7-dehydrocholesterol reductase (7DHC), upregulating 7-dehydrocholesterol (7DHC), a lipid which gets incorporated into the plasma and mitochondrial membranes, strengthening the lipid bilayer [31]. For example, trifluoperazine was shown to protect mitochondria by inhibiting membrane permeability and pore formation [32]. Moreover, phenothiazines intercalate themselves into the lipid bilayer of plasma and mitochondrial membranes, inhibiting peroxidation, thus, protecting the neurons from ferroptosis [33,34,35]. Interestingly, chlorpromazine was found effective against prion diseases, emphasizing a likely beneficial role in fatal familial insomnia (FFI) [36].
Dysfunctional mitochondria and impaired oxidative phosphorylation (OXPHOS), increases glycolysis and lactic acid levels, a metabolic pattern characteristic of SCZ or SLDs [37]. Indeed, increased lactate, considered a marker of sleep deprivation, likely activates mitochondrial AhR (mitoAhR), disrupting the organelle [38,39,40]. This is significant as lactate and neuro-metabolism likely comprise another sleep pathway hijacked by mental illness.
To compensate for dysfunctional mitochondria, neurons import these organelles from glial cells, especially the astrocyte [41,42]. In large cells, such as VENS, mitochondria are more vulnerable to damage and autophagic elimination as they undergo more wear and tear during their journey through the long axons of these neurons [43]. Due to their small number (around 193, 000) and their large sizes, VENS are more susceptible to plasma membrane oxidative stress, which may trigger significant pathology even after a limited neuronal loss, a pathology encountered in frontotemporal dementia behavioral variant (bvFTD).
Since mitochondria are crucial for neuronal function, preserving the integrity of these organelles via membrane lipid replacement (MLR) and other natural strategies, is of utmost importance. Microbial phenazines and the novel antioxidant phenothiazine derivatives, offer new opportunities to combat insomnia, psychosis, and neurodegeneration at the level of cell and mitochondrial membranes.
SN in sleep and neuropathology
The SN is comprised of anterior cingulate cortex (ACC) and anterior insular cortex (AIC) which along with subcortical nodes in the hypothalamus, thalamus, striatum, and midbrain process salient stimuli [44,45]. SN functions as a switch between exteroception and interoception or central executive network (CEN) and default mode network (DMN), depending on stimulus relevance [46]. Switching from CEN to DMN and vice versa is impaired in severe mental illness, insomnia, and neurodegenerative disorders [47]. Several antipsychotic drugs are known to lower the assignment of salience to objects and events, restoring the SN function, likely ameliorating both the psychotic symptoms and insomnia [48].
The SN harbors VENs, which are large, corkscrew neurons located in layer V of the IC and ACC. These non-telencephalic cells are believed to drive the prosocial cognition, empathy, and emotional intelligence. As parts of the SN, VENS respond to endogenous or exogenous stimuli in the order of priority. VENS are selectively eliminated in bvFTD, a disorder marked by criminal violations, lack of empathy, poor insight, and sleep impairment [49,50,51,52,53].
Under physiological circumstances, sleep is driven by the ventrolateral preoptic nucleus (VLPO) of the anterior hypothalamus which releases inhibitory neurotransmitters, including, γ-aminobutyric acid (GABA), and galanin [54]. The opposing system, orexin (hypocretin) neurons in the lateral hypothalamus, inhibit VLPO [55,56,57]. In addition, orexin/hypocretin neurons induce wakefulness by blocking the melanin concentrating hormone (MCH), a somnogen released by the hypothalamus and zona incerta [58,59]. Orexin and DA, the key players of saliency, have been implicated in the neuropsychiatric disorders associated with sleep disturbances, including narcolepsy, attention-deficit/hyperactivity disorder (ADHD), and Parkinson’s disease (PD) [60]. Histamine is another wakefulness-promoting neurotransmitter implicated in SCZ and SLDs and a novel target for treating the negative and cognitive symptoms [61].
To better comprehend the pathogenesis of insomnia, it is necessary to study the pathways of wakefulness, a brain state driving self-awareness and probably consciousness [62]. Early studies on this subject have focused on the locus coeruleus, midbrain tegmentum, pons, and parabrachial nucleus, as neurons in these regions are active during wakefulness [63,64]. In the early 1900s, while studying encephalitis lethargica, Constantin von Economo found that lesions in the posterior hypothalamus were associated with sleep, hypothesizing that this area contained the “center of wakefulness” [65,66,67].
FFI, a rare autosomal dominant disease, is marked by hypometabolism and neuronal loss in the thalamus and cingulate cortex, linking this condition to the SN [68]. Indeed, dysfunctional salience perception in FFI is reflected in sleep disturbances, psychiatric disorders, and autonomic dysregulation, pathologies previously linked to AIC and ACC [69,70,71,72]. The role of SN in sleep physiology and pathology is further highlighted by the fact that anesthetics, especially propofol, lower salience processing, inducing sleep [68,69,70,71,72,73,74,75,76,77,78]. Moreover, recent studies on sleep deprived human volunteers and patients with primary insomnia demonstrated altered connectivity in AIC, further linking SN to sleep and wakefulness [79,80]. Furthermore, several preclinical studies are in line with the findings in humans, implicating the SN in slumber homeostasis [74,81].
Aside from insomnia and neuropsychiatric pathology, the SN connectivity is disrupted in neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and bvFTD, suggesting that insomnia and neuropathology are highly intertwined [82,83,84,85,86]. Indeed, dysfunctional AIC and ACC connectivity may account for the criminal violations in patients with bvFTD in which breaking the law may often be the initial dementia symptom [87,88].
bvFTD as a secondary psychopathy
The second most common neurodegenerative disorder after AD, bvFTD, is marked by inappropriate emotional responses and disinhibited behaviors, often leading to criminal violations [52,89]. In forensic institutions, individuals with first incarceration after the age 55 may suffer from bvFTD, an entity difficult to diagnose as memory may remain intact for longer periods of time. As a result, bvFTD is often missed or misdiagnosed as antisocial personality disorder (APD), SCZ, or SLDs [90].
Over the past two decades, the number of senior first offenders has grown in parallel with the prevalence of young-onset dementia (YOD)(emergence of symptoms before age 65), a subgroup of neurodegenerative disorders, which may include bvFTD [91,92]. Indeed, recent studies have revealed that the prevalence of bvFTD has increased from 15/100, 000 in 2013 to 119 per 100, 000 in 2021, mirroring the growing number of forensic detainees with this diagnosis [92,93].
Compared to AD in which 12% of patients exhibit criminal behavior, bvFTD is associated with a crime rate of 54%, suggesting an acquired psychopathy [94]. Frontotemporal lobar degeneration (FTLD), the pathology driving bvFTD, is believed to selectively eliminate the “honesty cells”, VENS, predisposing to impulsivity and criminal violations [50,51]. Indeed, due to their large size, VENs may be particularly vulnerable to oxidative stress and mitochondrial depletion [95]. The latter is likely due to autophagy of damaged organelles traveling through the long VENS axons. Indeed, lysosomal aggregates, hallmarks of hyperactive autophagy, were demonstrated in VENS derived from patients with bvFTD and SCZ, suggesting excessive mitophagy [95,96,97]. Depletion of VENS has been associated with lack of empathy, aggressive behavior, and criminal violations documented in bvFTD and severe mental illness [51,52]. For example, homicide or attempted homicide have been documented in bvFTD, indicating that criminal behavior and murder can sometimes be the earliest manifestation of this disorder [98,99]. Since VENS are only present in large mammals, including humans, great apes, macaques, cetaceans, and elephants, but not in rodents, these cells are difficult to study in vivo [10]. VENS are larger than pyramidal neurons and drive interoceptive awareness, the ability to detect and process internal cues, such as heartbeat, respiration and the overall visceral state [100,101]. VENS are components of the SN, a large neuronal assembly which responds to intrinsic or extrinsic stimuli, shifting attention from CEN to DMN and vice versa [102,103].
Recent transcriptomic studies found that VENS express monoaminergic proteins, including vesicular monoamine transporter 2 (VMAT2) and adrenergic receptor α-1A (ADRA1A), suggesting involvement in autonomic functions, including the circadian rhythm [104,105,106]. Indeed, impaired monoaminergic signaling has been documented in insomnia, bvFTD, SCZ, and SLDs, implicating VENS in these pathologies [107,108,109,110,111].
Sleep and glial cells
Astrocytes, the most numerous brain cells communicate with each other via calcium waves, attaining synchronization with neurons which supports the slow-wave sleep [112,113]. Moreover, astrocytes release somnogenic molecules, including adenosine, lactate, glutamate, GABA, and interleukin-1 (IL-1), which influence the status of neuronal cells, predisposing to sleep [114].
Astrocytes are central to the neurovascular unit (NVU) and bridge the gap between the neuron and brain microvessels, regulating the flow of interstitial fluid through the aquaporin 4 (AQP-4) receptors [115] (Figure 1). The volume of the brain interstitial fluid (ISF) fluctuates in a circadian manner as it flows through the glymphatic system, a mechanism for clearing misfolded proteins during sleep [116]. The glymphatic system can also carry extracellular vesicles containing mitochondria from astrocytes to neurons [117]. Astrocytes support the neurons by generating GPX-4 to avert neuronal death by ferroptosis. GPX-4 functions to repair oxidized lipids and oxysterols, including 7-ketocholesterol (7KCl), toxins that disrupt plasma and mitochondrial membranes, triggering neuronal death [118]. As mitochondria play a key role in sleep homeostasis, insomnia may be the result of plasma or mitochondrial membrane oxidation [119]. Indeed, it has been suggested that sleep is necessary for abrogating neuronal oxidative stress [120].
Intracellular iron is stored in ferritin and released for intracellular needs via ferritinophagy (ferritin autophagy) in lysosomes. Several antipsychotic drugs, including haloperidol, accumulate in lysosomes disrupting ferritinophagy, which in return lowers intracellular iron, averting ferroptosis [121,122] (Figure 2). This may highlight a DA-independent, antipsychotic action of haloperidol, suggesting that dopaminergic blockade is not the only psychosis-deterring mechanism of this drug. Indeed, ferroptosis of hippocampal neurons, documented in AD and severe mental illness, is the likely cause of cognitive impairment and negative symptoms in these conditions [123,124]. Prolonged insomnia was demonstrated to damage the astrocyte which in return may trigger neuronal demise [125]. Moreover, chronic sleep loss was demonstrated to activate both astrocytes and microglia, turning these cells into neurotoxic phenotypes capable of eliminating healthy neurons and synapses [126,127,128].
Mitochondria and aryl hydrocarbon receptor
Recent studies have implicated mitochondria in the pathophysiology of sleep and neurodegenerative disorders, while the role of these organelles in severe mental illness, including SCZ and SLDs, has been previously established [129,130]. Lipid peroxidation of mitochondrial membrane and iron upregulation can trigger ferroptosis and organelle demise [131,132,133,134]. Indeed, lipid peroxides and oxysterols, such as 7KCl, are mitoAhR ligands, contributing to mitochondrial dysfunction and autophagic elimination [135]. AhR is a xenobiotic sensor which regulates cytochrome p450 and binds the environmental toxin, dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin). Other AhR ligands, include somnogens, such as phenazines, melatonin, and tryptophan derivatives, which participate in the physiology of sleep, wakefulness, and the circadian rhythm [136,137,138]. In addition, reactive oxygen species (ROS), known to induce sleep via a redox-sensitive potassium channel, are AhR ligands, bringing this transcription factor in the arena of slumber, mental illness, and neurodegeneration [131,139]. Indeed, microbial phenazines, including pyocyanin and 1-hydroxyphenazine, activate AhR, influencing the transcription of many genes, including those involved in sleep regulation [140,141].
The importance of mitochondria in sleep physiology is further substantiated by the organelle involvement in FFI as well as in general anesthesia [142,143]. Indeed, general anesthetics are known to inhibit N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors, while stimulating GABA. NMDA and AMPA upregulate intracellular and mitochondrial calcium, inducing cell and organelle demise [144]. Interestingly, elevated mitochondrial calcium, a characteristic of prion diseases, may link these organelles to FFI [145,146]. Indeed, the prion peptide causes calcium inflow via L-type calcium channels, triggering neuronal damage and apoptosis [147]. In contrast, the typical antipsychotic, chlorpromazine, not only induces sleep, but also exerts anti-prion properties, probably by promoting autophagy of the misfolded protein [148,149,150].
Mitochondrial trafficking from astrocytes to neurons, supports neuronal bioenergetic needs, especially in large pyramidal cells or VENs. Mitochondria can be imported via cell-cell fusion, tunneling nanotubes (cytoskeletal protrusions reaching to other cells) as well as transported by extracellular vesicles [151,152] (Figure 2). Moreover, astrocytes generate GPX-4 from cysteine obtained via the cystine/glutamate antiporter system (Xc−) or by transmethylation of methionine. Glutathione is generated from cysteine and glutathione disulfide (GSSC) [153] (Figure 2).
Mitochondrial trafficking as well as autophagy (mitophagy) occur during sleep, probably explaining the reason most living beings require rest [154]. Interestingly, serotonin (5-HT) promotes mitochondrial transport in hippocampal neurons, suggesting that antidepressant drugs, serotonin reuptake inhibitors (SSRIs), may “exert their action by supplying healthy mitochondria to stressed neurons [155]. This may imply that ROS accumulation during wakefulness may induce slumber to repair oxidized lipids and import mitochondria from glial cells [120,131,139]. In addition, accumulation of intracellular microtubule-associated protein tau (MAPT) in VENS likely impairs mitochondrial transport, contributing to bvFTD pathogenesis [156].
Four cases of bvFTD from our hospital and the community
Case #1 The teacher who shot her neighbor
Ms. KS (initials changed), a Caucasian female, age 68, divorced, retired elementary school teacher, lived alone prior to her admission to Patton State Hospital. Ms. KS did not have a psychiatric history until the age of 56 when she purchased a gun and shot her neighbor in the shoulder. She stated that she attacked the man because he was spying on her and intruded into her house during the night. She was convicted of attempted murder and sent to prison, where her condition deteriorated, prompting transfer to our forensic institution. KS was diagnosed with SCZ and admitted as a forensic detainee.
During her hospital stay, KS was treated with various antipsychotic drugs with minimal symptomatic relief. She was unaware that she did anything wrong and her poor insight and impulsivity were documented during her six years of hospital stay. Because of poor insight, KS never met criteria for the conditional release program (CONREP).
In 2014, KS became more forgetful, required assistance with most activities of daily living (ADLs), and exhibited a change in her dietary preferences. For example, she asked for ice cream daily, although earlier in her life she detested ice cream. In time, KS became more apathetic and often refused to get out of bed. The internal medicine consultant performed a dementia workup, but the laboratory studies came back normal, except for mild anemia, and a vitamin D level of 29.3 nmol/L. KS scored 25/30 on Mini Mental Status Exam (MMSE) and when a Montreal Cognitive Assessment (MoCA) was administered, the score was 23/30, consistent with executive dysfunction. Neuropsychology consult was called, and after a battery of tests, bvFTD was diagnosed.
With this information, the treatment team petitioned the Court, arguing that KS did not benefit from hospitalization in a forensic institution as she was not expected to recover. The judge agreed with the treating clinicians and ordered placement in a facility specialized in dementia.
Due to the numerous clinical and legal ramifications (discussed below), this case was featured in the mass media at the time:
Case #2 The attorney with a sweet tooth
An outpatient we treated in 2013, was a 72 years old, retired attorney, arrested because he stole chocolate from a grocery store while casually conversing with the owner. When confronted, he replied: “what’s the problem, I have a sweet tooth”. According to the family, the patient came across as careless and indifferent of his children and the spouse, being either apathetic or angry and irritable. For example, when he learned that his son-in-law died unexpectedly, he responded by saying “let’s go out to eat”. His eating habits had changed dramatically, according to his wife, consuming mostly sweets which previously he had avoided. When told to eat more nutritious food, he often became angry.
Case #3 The psychiatrist turned a drug dealer
Dr. Joel Stanley Dreyer was a well-respected psychiatrist who practiced in Riverside, California. In 1990s, Dr. Dreyer was diagnosed with bvFTD but continued to practice psychiatry, and in 2010 was convicted for prescribing, selling, and distributing large amounts of addictive painkillers. As a result of careless prescribing, one person died of an overdose and Dr. Dreyer was convicted and served ten years in prison despite having been diagnosed with bvFTD prior to his crime. This case emphasizes that some jurisdictions do not recognize bvFTD as an attenuating circumstance. The court ruling was based on the testimony of the prison psychiatrist who did not challenge the diagnosis of bvFTD but stated that since not all individuals with this disorder engage in criminal behavior, “direct causality” between Dr. Dreyer’s crime and bvFTD could not be established. A detailed history of this case can be found at the link below:
Case #4 The Buick murderer
On July 16, 2003, Mr. GRW, an 83 years old man crashed his Buick LeSabre in an open-air market in Santa Monica, California, killing 10 and injuring 63 individuals. Despite the catastrophic event he caused, GRW did not express remorse, showed indifference, callousness, and lack of empathy. In the court, he appeared apathetic, angry, and unapologetic, stating that he was sorry the dead and injured could not “enjoy the value of their purchases”. No psychiatric evaluation was ordered because there was no previous history, however, criminal behavior may often represent the first symptom of bvFTD. Despite never being diagnosed with a neurodegenerative disorder, people who knew GRW noticed a drastic personality change in the years prior to this event, indicative of bvFTD. His neighbors, friends, and his pastor, described GRW as caring, pleasant, and friendly individual. He had been married for over 60 years, was compassionate, involved in peoples’ lives, and after retirement, volunteered with various civic organizations. Although GRW was never officially diagnosed with bvFTD, this case illustrates the difficulty clinicians encounter because this neurodegenerative disorder affects the executive function, leaving memory intact for many years. Indeed, shortly before his crime, GRW was able to pass his DMV license renewal test, suggesting that his memory was unaffected. Since in California drivers who are 70 or older must renew their driver’s license in person, GRW did not raise a dementia red flag with the DMV worker.
Discussion
Since bvFTD comprises 2.7% of all dementias and in early stages, patients retain their cognitive abilities, this condition is often misdiagnosed as SCZ, depression, or bipolar disorder, and frequently admitted to psychiatric institutions. Patients with bvFTD respond poorly to antipsychotic drugs, are often labeled “treatment resistant”, and prescribed additional psychotropics [157]. Moreover, as criminal behavior is frequently the initial manifestation of bvFTD, clinicians rarely suspect this condition when examining an incarcerated individual. However, there are several characteristics of this disorder which should prompt the clinician to think of a neurodegenerative condition. These include absence of psychiatric history at a younger age, first legal violation after the age of 55, poor insight despite a previously successful life, sudden change in eating habits, altered sleep pattern, lack of empathy, and engaging in criminal acts despite the presence of witnesses.
Mitochondria-protective treatments
The key role of mitochondria in sleep disorders, SCZ, SLDs, and neurodegeneration, highlights the importance of mitoprotective approaches to resuscitate, replace, or increase the import of mitochondria from glial cells. For example, treatment with SSRIs during the early stages of dementias, may delay the onset of cognitive decline. Along this line, a recent study found that treatment with SSRIs slowed the conversion of mild cognitive impairment to frank dementia, suggesting that prophylactic treatment with these agents may be beneficial [158]. In addition, natural anti-ferroptosis drugs and iron chelators, such as halogenated phenazines, may improve the course of neurodegenerative disorders, suggesting novel therapeutic strategies [159,160].
Membrane Lipid Replacement (MLR)
MLR refers to the oral supplementation with natural, cell membrane glycerophospholipids and kaempferol (3,4′,5,7-tetrahydroxyflavone), a natural flavonoid found in tea, broccoli, cabbage, kale, beans, endive, leek, tomato, strawberries, and grapes [161]. Kaempferol is a glycogen synthase kinase-3β (GSK-3β) inhibitor which prevents sleep deprivation-induced cognitive decline [162,163]. Like lithium and several antipsychotic drugs, kaempferol blocks GSK-3β, an enzyme previously implicated in SCZ and circadian rhythm disorders, suggesting that this natural compound may exert antipsychotic properties without the adverse effects of conventional therapeutics [164,165,166,167].
The aim of MLR + kaempferol is gradual replacement of damaged phospholipids and oxysterols from neuronal and/or mitochondrial membranes with natural glycerophospholipids and a polyphenol. Indeed, oxidized membrane lipids have been implicated in SCZ, SLDs, insomnia, and neurodegeneration, while MLR and kaempferol offer a dual mechanism of action: 1) elimination of lipid peroxides and 2) GSK-3β inhibition [168]. Replacing oxidized plasma and/or mitochondrial membrane fats with healthy natural lipids, averts deformation of neuronal membrane and misalignment of neuroreceptors. Conversely, oxidized membrane lipids and ferroptosis alter the biophysical properties of membranes, disrupting neuronal functions [169].
Phenazines and phenothiazine derivatives
Phenazines are nitrogen-containing heterocyclic compounds produced by various marine and terrestrial microorganisms which participate in microbial clearance, iron signaling, and biofilm formation [170]. Phenazines can be natural (bacteria-derived) or synthetic.
Natural phenazines, such as iodinin (1.6-dihydroxy-N5, N10-dioxide phenazine) and myxin, are antibiotics which have been known for several decades [171]. The newer, terpenoid, glycosylated and fused phenazines, are derived from various Streptomyces species and exert antibiotic and anticancer effects. For example, geranylphenazinediol is an inhibitor of human acetylcholinesterase with potential benefit in neurodegenerative disorders without the adverse effects of the manufactured drugs [172]. Other natural phenazines, including baraphenazines, leucanicidin and endophenasides, exert antimicrobial, anticancer activity, and very likely possess antipsychotic properties [173,174,175].
Synthetic phenazine derivatives consist of over 6,000 compounds, exerting antimicrobial, antiparasitic, neuroprotective, anti-inflammatory, and anticancer activities [176,177,178]. To the best of our knowledge, natural or synthetic phenazines have not been tested for SCZ, insomnia, or neurodegeneration. Pontemazines A and B are neuroprotective phenazine derivatives which in animal studies have rescued hippocampal neurons from glutamate cytotoxicity, highlighting their pro-cognitive properties which could benefit patients with negative symptoms of SCZ or neurodegenerative disorders [176].
Synthetic phenazines exert antioxidant and radical-scavenging properties, inhibit lipid peroxidation, suggesting beneficial effects in severe insomnia, mental illness and neurodegeneration [179,180] (Figure 3). Moreover, halogenated phenazines act as iron chelators, likely preventing neuronal ferroptosis [181]. We believe that Pontemazines and halogenated phenazines should be assessed for antipsychotic/anti-neurodegenerative properties.
From the biochemical standpoint, phenazines are almost identical to phenothiazine antipsychotics and likely possess similar properties (Figure 4). Phenothiazines are typical antipsychotic drugs utilized primarily for SCZ and SLDs which block dopaminergic transmission at the level of postsynaptic neuron. Several phenothiazines influence other receptors, including adrenergic, histaminergic, and cholinergic, exerting various clinical effects as well as adverse reactions. Aside from psychotic disorders, phenothiazines are also used for the treatment of migraine headaches, hiccups, nausea, vomiting, and cancer [182]. Like phenazines, phenothiazines intercalate themselves into the lipid bilayer of plasma and mitochondrial membranes, disrupting the curvature and receptor alignment on neuronal/mitochondrial surfaces [183] (Figure 3). In contrast, oxidized lipids, including 7-ketocholesterol (7KCl), form looped structures, generating membrane curvatures and pores, that may trigger cell death [184].
Antioxidant phenothiazine and their derivatives have recently been developed for cancer, cardiovascular disease (CVD), Mycobacterium leprae and other antibiotic-resistant microbes [185,186].
Phenothiazine derivatives exert anti-peroxidation properties and protect against lipid pathology and ferroptosis, suggesting efficacy as antipsychotic drugs [187]. In addition, antioxidant phenothiazines are likely beneficial for insomnia and neurodegenerative disorders, suggesting that these compounds should be tested for neuropsychiatric pathology [186].
Propenylphenothiazine is a potent antioxidant with electron-donor capability that could prevent gray matter loss, a hallmark of SCZ and SLDs [188,189]. Electron-donating psychotropic drugs have been known to preserve the brain volume, suggesting that propenylphenothiazine may treat psychosis, without reducing the gray matter volume. The majority of conventional antipsychotic drugs are electron-acceptors which often lower the brain volume as documented by many neuroimaging studies [190,191,192,193]. An even newer category of tetracyclic and pentacyclic phenothiazines with antioxidant properties have recently been developed, suggesting likely efficacy for cognitive impairment and negative SCZ symptoms [194]. Moreover, the N10-carbonyl-substituted phenothiazines were demonstrated to inhibit lipid peroxidation, suggesting superior antipsychotic efficacy [187].
Mitochondrial transfer and transplantation
The early studies on mitochondrial transplantation, from the 1980s, utilized co-incubation of various cell types with naked mitochondria, hoping that cells would internalize the organelles from the extracellular environment [195]. Later, HeLa cells and mesenchymal stem cells were used as mitochondrial sources and found that successful organelle uptake occurred in a short time interval of 1-2 hours [196,197,198]. At present, mitochondrial transplantation into cardiomyocytes has been accomplished successfully and confirmed by mitochondrial DNA (mtDNA) detected in host cells [199,200].
Mitochondrial transplantation and neuronal rescue from ferroptosis have been performed successfully in both animals and humans, suggesting a novel strategy for neurometabolic disorders [201]. To our knowledge, mitochondrial transplantation has not been attempted in sleep disorders, while in mental illness, it has been tried in animal models only [132]. Trafficking mitochondria from astrocytes and microglia to neurons can take place spontaneously after brain injuries, reflecting a likely compensatory mechanism to preserve neuronal viability [202]. In addition, it has been established that SSRIs, GJA1-20K, and CD38 signaling can facilitate mitochondrial transfer, emphasizing potential strategies for insomnia, severe mental illness, and neurodegeneration [203,204].
Conclusions
Forensic institutions throughout the country house individuals with severe mental illness and often comorbid insomnia, suggesting overlapping pathogeneses. Loss of neurons due to impaired sleep along with SCZ or SLDs-related gray matter depletion, may trigger the premature development of dementias and other medical complications. These comorbidities increase healthcare expenditures and shorten patients’ lifespan, therefore, identifying and treating these conditions early is essential.
YOD, a category of neurodegenerative disorders which include bvFTD, has been on the rise over the past few decades as evidenced by the increased number of first offenders younger than 65. Selective loss of VENS in bvFTD is likely due to the large size of these cells, predisposing to peroxidation of plasma membrane lipids and mitochondrial loss by autophagy.
At the molecular level, AhR is the equivalent of cerebral VENS, as this protein responds to both endogenous and exogenous ligands, including the lipid peroxides and other insomnia and psychosis-related molecules.
Novel AhR ligands, phenazine and phenothiazine derivatives, as well as mitochondrial transfer or transplantation are potential new strategies for treating psychosis, insomnia, and neurodegeneration without additional loss of brain volume.
References
- Sateia, M.J.; Doghramji, K.; Hauri, P.J.; Morin, C.M. Evaluation of chronic insomnia. An American Academy of Sleep Medicine review. Sleep 2000, 23, 243–308. [Google Scholar] [CrossRef] [PubMed]
- Dewa, L.H.; Thibaut, B.; Pattison, N.; Campbell, S.J.; Woodcock, T.; Aylin, P.; Archer, S. Treating insomnia in people who are incarcerated: a feasibility study of a multi-component treatment pathway. SLEEP Adv. 2024, 5, zpae003. [Google Scholar] [CrossRef] [PubMed]
- Talih, F.; Ajaltouni, J.; Ghandour, H.; Abu-Mohammad, A.S.; Kobeissy, F. Insomnia in hospitalized psychiatric patients: prevalence and associated factors. Neuropsychiatr. Dis. Treat. 2018, ume 14, 969–975. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Li, W.; Xiao, L.; Huo, X.; Ding, J.; Sun, T. Is the insula linked to sleep? A systematic review and narrative synthesis. Heliyon 2022, 8, e11406. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dong, M.; Yin, Y.; Hua, K.; Fu, S.; Jiang, G. Aberrant Effective Connectivity of the Right Anterior Insula in Primary Insomnia. Front. Neurol. 2018, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.P.; Tregellas, J.R. The role of the insula in schizophrenia. Schizophr. Res. 2010, 123, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Fathy, Y.Y.; Hoogers, S.E.; Berendse, H.W.; van der Werf, Y.D.; Visser, P.J.; de Jong, F.J.; van de Berg, W.D. Differential insular cortex sub-regional atrophy in neurodegenerative diseases: a systematic review and meta-analysis. Brain Imaging Behav. 2019, 14, 2799–2816. [Google Scholar] [CrossRef]
- Koutsouleris, N.; Pantelis, C.; Velakoulis, D.; McGuire, P.; Dwyer, D.B.; Urquijo-Castro, M.-F.; Paul, R.; Dong, S.; Popovic, D.; Oeztuerk, O.; et al. Exploring Links Between Psychosis and Frontotemporal Dementia Using Multimodal Machine Learning. JAMA Psychiatry 2022, 79, 907–919. [Google Scholar] [CrossRef]
- Triarhou, L.C. The percipient observations of Constantin von Economo on encephalitis lethargica and sleep disruption and their lasting impact on contemporary sleep research. Brain Res. Bull. 2006, 69, 244–258. [Google Scholar] [CrossRef]
- Allman, J.M.; Tetreault, N.A.; Hakeem, A.Y.; Manaye, K.F.; Semendeferi, K.; Erwin, J.M.; Park, S.; Goubert, V.; Hof, P.R. The von Economo neurons in frontoinsular and anterior cingulate cortex in great apes and humans. Anat. Embryol. 2010, 214, 495–517. [Google Scholar] [CrossRef]
- Berg, M.T.; Rogers, E.M.; Lei, M.-K.; Simons, R.L. Losing Years Doing Time: Incarceration Exposure and Accelerated Biological Aging among African American Adults. J. Heal. Soc. Behav. 2021, 62, 460–476. [Google Scholar] [CrossRef]
- Kaiksow, F.A.; Brown, L.; Merss, K.B. Caring for the Rapidly Aging Incarcerated Population: The Role of Policy. J. Gerontol. Nurs. 2023, 49, 7–11. [Google Scholar] [CrossRef]
- Papanastasiou, E.; Gaughran, F.; Smith, S. Schizophrenia as segmental progeria. J. R. Soc. Med. 2011, 104, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Killilea, D.W.; Wong, S.L.; Cahaya, H.S.; Atamna, H.; Ames, B.N. Iron Accumulation during Cellular Senescence. Ann. N. Y. Acad. Sci. 2004, 1019, 365–367. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Nãºã±Ez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Carvalhas-Almeida, C.; Cavadas, C.; Álvaro, A.R. The impact of insomnia on frailty and the hallmarks of aging. Aging Clin. Exp. Res. 2022, 35, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.E.; Prather, A.A. Sleep and biological aging: A short review. Curr. Opin. Endocr. Metab. Res. 2021, 18, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna-Żydecka, K.; Jamioł-Milc, D.; Borecki, K.; Stachowska, E.; Zabielska, P.; Kamińska, M.; Karakiewicz, B. The Prevalence of Insomnia and the Link between Iron Metabolism Genes Polymorphisms, TF rs1049296 C>T, TF rs3811647 G>A, TFR rs7385804 A>C, HAMP rs10421768 A>G and Sleep Disorders in Polish Individuals with ASD. Int. J. Environ. Res. Public Heal. 2020, 17, 400. [Google Scholar] [CrossRef]
- Nacarino-Palma, A.; Rico-Leo, E.M.; Campisi, J.; Ramanathan, A.; González-Rico, F.J.; Rejano-Gordillo, C.M.; Ordiales-Talavero, A.; Merino, J.M.; Fernández-Salguero, P.M. Aryl hydrocarbon receptor blocks aging-induced senescence in the liver and fibroblast cells. Aging 2022, 14, 4281–4304. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Peng, V.; Sudan, R.; Antonova, A.U.; Di Luccia, B.; Ohara, T.E.; Fachi, J.L.; Grajales-Reyes, G.E.; Jaeger, N.; Trsan, T.; et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity 2023, 56, 797–812. [Google Scholar] [CrossRef]
- Hwang, H.J.; Dornbos, P.; Steidemann, M.; Dunivin, T.K.; Rizzo, M.; LaPres, J.J. Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome. Toxicol. Appl. Pharmacol. 2016, 304, 121–132. [Google Scholar] [CrossRef]
- Dietrich-Muszalska, A.; Kontek, B. Lipid peroxidation in patients with schizophrenia. Psychiatry Clin. Neurosci. 2010, 64, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Chen, J.; Qu, C.; Yang, L.; Wu, X.; Wang, S.; Yang, T.; Liu, H.; Fang, Y.; Sun, P. Identification of Ferroptosis-Related Genes in Schizophrenia Based on Bioinformatic Analysis. Genes 2022, 13, 2168. [Google Scholar] [CrossRef] [PubMed]
- Gulec, M.; Ozkol, H.; Selvi, Y.; Tuluce, Y.; Aydin, A.; Besiroglu, L.; Ozdemir, P.G. Oxidative stress in patients with primary insomnia. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2012, 37, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Van Remmen, H.; Frohlich, V.; Lechleiter, J.; Richardson, A.; Ran, Q. Gpx4 protects mitochondrial ATP generation against oxidative damage. Biochem. Biophys. Res. Commun. 2007, 356, 893–898. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2018, 133, 144–152. [Google Scholar] [CrossRef]
- Cole-Ezea, P.; Swan, D.; Shanley, D.; Hesketh, J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelial cells. Free. Radic. Biol. Med. 2012, 53, 488–497. [Google Scholar] [CrossRef]
- Fujii, R.; Hasuo, H.; Sakuma, H.; Okada, M.; Uchitani, K. The efficacy and safety of intravenous chlorpromazine treatment for sleep disturbance in patients with incurable cancer, with oral administration difficulty: a 1-week, prospective observational study. Ann. Palliat. Med. 2021, 10, 8547–8556. [Google Scholar] [CrossRef]
- Tischkau, S.A. Mechanisms of circadian clock interactions with aryl hydrocarbon receptor signalling. Eur. J. Neurosci. 2019, 51, 379–395. [Google Scholar] [CrossRef]
- Fehsel, K.; Schwanke, K.; Kappel, B.; Fahimi, E.; Meisenzahl-Lechner, E.; Esser, C.; Hemmrich, K.; Haarmann-Stemmann, T.; Kojda, G.; Lange-Asschenfeldt, C. Activation of the aryl hydrocarbon receptor by clozapine induces preadipocyte differentiation and contributes to endothelial dysfunction. J. Psychopharmacol. 2022, 36, 191–201. [Google Scholar] [CrossRef]
- Korade; Liu, W.; Warren, E.B.; Armstrong, K.; Porter, N.A.; Konradi, C. Effect of psychotropic drug treatment on sterol metabolism. Schizophr. Res. 2017, 187, 74–81. [Google Scholar] [CrossRef]
- Kiani, A.; Nik, S.H.; Khodadoost, A.; Salimi, A.; Pourahmad, J. Trifluoperazine an Antipsychotic Drug and Inhibitor of Mitochondrial Permeability Transition Protects Cytarabine and Ifosfamide-Induced Neurotoxicity. Drug Res. 2020, 70, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Pathak, D.N.; Singh, R. Effects of Chlorpromazine on the Activities of Antioxidant Enzymes and Lipid Peroxidation in the Various Regions of Aging Rat Brain. J. Neurochem. 1984, 42, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Ficarra, S.; Russo, A.; Barreca, D.; Giunta, E.; Galtieri, A.; Tellone, E. Short-Term Effects of Chlorpromazine on Oxidative Stress in Erythrocyte Functionality: Activation of Metabolism and Membrane Perturbation. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Brunauer, L.S.; Chu, F.C.; Helsel, C.M.; Gedde, M.M.; Huestis, W.H. Selective amphipathic nature of chlorpromazine binding to plasma membrane bilayers. Biochim. et Biophys. Acta (BBA) - Biomembr. 2003, 1616, 95–105. [Google Scholar] [CrossRef]
- Korth, C.; May, B.C.H.; Cohen, F.E.; Prusiner, S.B. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. 2001, 98, 9836–9841. [Google Scholar] [CrossRef]
- Henkel, N.D.; Wu, X.; O’donovan, S.M.; Devine, E.A.; Jiron, J.M.; Rowland, L.M.; Sarnyai, Z.; Ramsey, A.J.; Wen, Z.; Hahn, M.K.; et al. Schizophrenia: a disorder of broken brain bioenergetics. Mol. Psychiatry 2022, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Kempf, A. Mitochondrial control of sleep. Curr. Opin. Neurobiol. 2023, 81, 102733. [Google Scholar] [CrossRef]
- Richardson, R.B.; Mailloux, R.J. Mitochondria Need Their Sleep: Redox, Bioenergetics, and Temperature Regulation of Circadian Rhythms and the Role of Cysteine-Mediated Redox Signaling, Uncoupling Proteins, and Substrate Cycles. Antioxidants 2023, 12, 674. [Google Scholar] [CrossRef]
- Denis, A.A.; Toledo, D.; Hakim, Q.A.; Quintana, A.A.; Escobar, C.R.; Oluwole, S.A.; Costa, A.; Gil Garcia, E.; Hill, A.R.; Agatemor, C. Ligand-Independent Activation of Aryl Hydrocarbon Receptor and Attenuation of Glutamine Levels by Natural Deep Eutectic Solvent. ChemBioChem 2023, 24, e202300540. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Gollihue, J.; Norris, C. Astrocyte mitochondria: Central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res. Rev. 2020, 59, 101039–101039. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Downar, J.; Crawley, A.P.; Mikulis, D.J.; Davis, K.D. A multimodal cortical network for the detection of changes in the sensory environment. Nat. Neurosci. 2000, 3, 277–283. [Google Scholar] [CrossRef]
- Wolff, M.; Vann, S.D. The Cognitive Thalamus as a Gateway to Mental Representations. J. Neurosci. 2018, 39, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, D.; Levitin, D.J.; Menon, V. A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc. Natl. Acad. Sci. 2008, 105, 12569–12574. [Google Scholar] [CrossRef] [PubMed]
- Ueno, D.; Matsuoka, T.; Kato, Y.; Ayani, N.; Maeda, S.; Takeda, M.; Narumoto, J. Individual Differences in Interoceptive Accuracy Are Correlated With Salience Network Connectivity in Older Adults. Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef]
- Blessing, W.W.; Blessing, E.M.; Mohammed, M.; Ootsuka, Y. Clozapine, chlorpromazine and risperidone dose-dependently reduce emotional hyperthermia, a biological marker of salience. Psychopharmacol. 2017, 234, 3259–3269. [Google Scholar] [CrossRef]
- Seeley, W.W. The Salience Network: A Neural System for Perceiving and Responding to Homeostatic Demands. J. Neurosci. 2019, 39, 9878–9882. [Google Scholar] [CrossRef]
- Pasquini, L.; Nana, A.L.; Toller, G.; A Brown, J.; Deng, J.; Staffaroni, A.; Kim, E.-J.; Hwang, J.-H.L.; Li, L.; Park, Y.; et al. Salience Network Atrophy Links Neuron Type-Specific Pathobiology to Loss of Empathy in Frontotemporal Dementia. Cereb. Cortex 2020, 30, 5387–5399. [Google Scholar] [CrossRef]
- Mendez, M.F.; Anderson, E.; Shapira, J.S. An Investigation of Moral Judgement in Frontotemporal Dementia. Cogn. Behav. Neurol. 2005, 18, 193–197. [Google Scholar] [CrossRef]
- Mendez, M.F. The Neurobiology of Moral Behavior:Review and Neuropsychiatric Implications. CNS Spectrums 2009, 14, 608–620. [Google Scholar] [CrossRef]
- Boeve, B.F. Behavioral Variant Frontotemporal Dementia. Contin. Lifelong Learn. Neurol. 2022, 28, 702–725. [Google Scholar] [CrossRef]
- Arrigoni, E.; Fuller, P.M. The Sleep-Promoting Ventrolateral Preoptic Nucleus: What Have We Learned over the Past 25 Years? Int. J. Mol. Sci. 2022, 23, 2905. [Google Scholar] [CrossRef]
- De Luca, R.; Nardone, S.; Grace, K.P.; Venner, A.; Cristofolini, M.; Bandaru, S.S.; Sohn, L.T.; Kong, D.; Mochizuki, T.; Viberti, B.; et al. Orexin neurons inhibit sleep to promote arousal. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Inutsuka, A.; Yamanaka, A. The regulation of sleep and wakefulness by the hypothalamic neuropeptide orexin/hypocretin. . 2013, 75, 29–36. [Google Scholar]
- Yin, D.; Dong, H.; Wang, T.-X.; Hu, Z.-Z.; Cheng, N.-N.; Qu, W.-M.; Huang, Z.-L. Glutamate Activates the Histaminergic Tuberomammillary Nucleus and Increases Wakefulness in Rats. Neuroscience 2019, 413, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Konadhode, R.R.; Pelluru, D.; Shiromani, P.J. Neurons containing orexin or melanin concentrating hormone reciprocally regulate wake and sleep. Front. Syst. Neurosci. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Weber, F.; Zhong, P.; Tan, C.L.; Nguyen, T.N.; Beier, K.T.; Hörmann, N.; Chang, W.-C.; Zhang, Z.; Do, J.P.; et al. Identification of preoptic sleep neurons using retrograde labelling and gene profiling. Nature 2017, 545, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Bandarabadi, M.; Li, S.; Aeschlimann, L.; Colombo, G.; Tzanoulinou, S.; Tafti, M.; Becchetti, A.; Boutrel, B.; Vassalli, A. Inactivation of hypocretin receptor-2 signaling in dopaminergic neurons induces hyperarousal and enhanced cognition but impaired inhibitory control. Mol. Psychiatry 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Gao, C.; Han, F.; Cheng, H. Histamine H1 receptor in basal forebrain cholinergic circuit: A novel target for the negative symptoms of schizophrenia? Neurosci. Bull. 2022, 38, 558–560. [Google Scholar] [CrossRef]
- Grady, F.S.; Boes, A.D.; Geerling, J.C. A Century Searching for the Neurons Necessary for Wakefulness. Front. Neurosci. 2022, 16, 930514. [Google Scholar] [CrossRef]
- Kerkhofs, M.; Lavie, P. HISTORICAL NOTE: Frédéric Bremer 1892–1982: a pioneer in sleep research. Sleep Med. Rev. 2000, 4, 505–514. [Google Scholar] [CrossRef]
- Fuller, P.; Sherman, D.; Pedersen, N.P.; Saper, C.B.; Lu, J. Reassessment of the structural basis of the ascending arousal system. J. Comp. Neurol. 2010, 519, 933–956. [Google Scholar] [CrossRef] [PubMed]
- Lavie, P. The sleep theory of Constantin von Economo. J. Sleep Res. 1993, 2, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; De Jesus, O. Von Economo Encephalitis. 2023 Aug 23. In StatPearls (Internet); StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Rosen, D. Asleep: the Forgotten Epidemic That Remains One of Medicine’s Greatest Mysteries. J Clin Sleep Med. 2010, 6, 299. [Google Scholar] [CrossRef]
- Cortelli, P.; Perani, D.; Parchi, P.; Grassi, F.; Montagna, P.; De Martin, M.; Castellani, R.; Tinuper, P.; Gambetti, P.; Lugaresi, E.; et al. Cerebral metabolism in fatal familial insomnia: Relation to duration, neuropathology, and distribution of protease-resistent prion protein. Neurology 1997, 49, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Gallassi, R.; Morreale, A.; Montagna, P.; Cortelli, P.; Avoni, P.; Castellani, R.; Gambetti, R.; Lugaresi, E. Fatal familial insomnia. Neurology 1996, 46, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Sturm, V.E.; Brown, J.A.; Hua, A.Y.; Lwi, S.J.; Zhou, J.; Kurth, F.; Eickhoff, S.B.; Rosen, H.J.; Kramer, J.H.; Miller, B.L.; et al. Network Architecture Underlying Basal Autonomic Outflow: Evidence from Frontotemporal Dementia. J. Neurosci. 2018, 38, 8943–8955. [Google Scholar] [CrossRef] [PubMed]
- Mallikarjun, P.K.; Lalousis, P.A.; Dunne, T.F.; Heinze, K.; Reniers, R.L.; Broome, M.R.; Farmah, B.; Oyebode, F.; Wood, S.J.; Upthegrove, R. Aberrant salience network functional connectivity in auditory verbal hallucinations: a first episode psychosis sample. Transl. Psychiatry 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Cracco, L.; Appleby, B.S.; Gambetti, P. Fatal familial insomnia and sporadic fatal insomnia. Handb Clin Neurol. 2018, 153, 271–299. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Li, W.; Xiao, L.; Huo, X.; Ding, J.; Sun, T. Is the insula linked to sleep? A systematic review and narrative synthesis. Heliyon 2022, 8, e11406. [Google Scholar] [CrossRef]
- Levichkina, E.V.; Busygina, I.I.; Pigareva, M.L.; Pigarev, I.N. The Mysterious Island: Insula and Its Dual Function in Sleep and Wakefulness. Front. Syst. Neurosci. 2021, 14. [Google Scholar] [CrossRef]
- Guo, Y.; Zou, G.; Shao, Y.; Chen, J.; Li, Y.; Liu, J.; Yao, P.; Zhou, S.; Xu, J.; Hu, S.; et al. Increased connectivity of the anterior cingulate cortex is associated with the tendency to awakening during N2 sleep in patients with insomnia disorder. Sleep 2022, 46. [Google Scholar] [CrossRef]
- Guldenmund, P.; Demertzi, A.; Boveroux, P.; Boly, M.; Vanhaudenhuyse, A.; Bruno, M.-A.; Gosseries, O.; Noirhomme, Q.; Brichant, J.-F.; Bonhomme, V.; et al. Thalamus, Brainstem and Salience Network Connectivity Changes During Propofol-Induced Sedation and Unconsciousness. Brain Connect. 2013, 3, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Luo, L.; Zhou, Z.; Xu, K.; Zhang, L.; Liu, X.; Tan, X.; Zhang, J.; Ye, X.; Gao, J.; et al. Functional Connectivity of Anterior Insula Predicts Recovery of Patients With Disorders of Consciousness. Front. Neurol. 2018, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Mashour, G.A. Anesthetizing the Self. Anesthesiology 2016, 124, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Li, B.-Z.; Zhang, Y.; Pan, B.; Gao, Y.-H.; Zhan, H.; Liu, Y.; Shao, Y.-C.; Zhang, X. Altered insula-prefrontal functional connectivity correlates to decreased vigilant attention after total sleep deprivation. Sleep Med. 2021, 84, 187–194. [Google Scholar] [CrossRef]
- Li, C.; Dong, M.; Yin, Y.; Hua, K.; Fu, S.; Jiang, G. Aberrant Effective Connectivity of the Right Anterior Insula in Primary Insomnia. Front. Neurol. 2018, 9, 317. [Google Scholar] [CrossRef]
- Chen, M.C.; Chiang, W.-Y.; Yugay, T.; Patxot, M.; Ozcivit, I.B.; Hu, K.; Lu, J. Anterior Insula Regulates Multiscale Temporal Organization of Sleep and Wake Activity. J. Biol. Rhythm. 2016, 31, 182–93. [Google Scholar] [CrossRef] [PubMed]
- Palaniyappan, L.; White, T.; Liddle, P. The Concept of Salience Network Dysfunction in Schizophrenia: From Neuroimaging Observations to Therapeutic Opportunities. Curr. Top. Med. Chem. 2012, 12, 2324–2338. [Google Scholar] [CrossRef]
- Huang, H.; Chen, C.; Rong, B.; Wan, Q.; Chen, J.; Liu, Z.; Zhou, Y.; Wang, G.; Wang, H. Resting-state functional connectivity of salience network in schizophrenia and depression. Sci. Rep. 2022, 12, 1–8. [Google Scholar] [CrossRef]
- He, X.; Qin, W.; Liu, Y.; Zhang, X.; Duan, Y.; Song, J.; Li, K.; Jiang, T.; Yu, C. Abnormal salience network in normal aging and in amnestic mild cognitive impairment and Alzheimer’s disease. Hum. Brain Mapp. 2013, 35, 3446–3464. [Google Scholar] [CrossRef] [PubMed]
- Putcha, D.; Ross, R.S.; Cronin-Golomb, A.; Janes, A.C.; Stern, C.E. Salience and Default Mode Network Coupling Predicts Cognition in Aging and Parkinson’s Disease. J. Int. Neuropsychol. Soc. 2016, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Day, G.S.; Farb, N.A.S.; Tang-Wai, D.F.; Masellis, M.; Black, S.E.; Freedman, M.; Pollock, B.G.; Chow, T.W. Salience Network Resting-State Activity. JAMA Neurol. 2013, 70, 1249–1253. [Google Scholar] [CrossRef]
- Sheffield, J.M.; Rogers, B.P.; Blackford, J.U.; Heckers, S.; Woodward, N.D. Insula functional connectivity in schizophrenia. Schizophr. Res. 2020, 220, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; David, A.S. Patterns of anterior cingulate activation in schizophrenia: a selective review. Neuropsychiatr. Dis. Treat. 2007, 3, 87–101. [Google Scholar] [CrossRef]
- Finger, E.C. Frontotemporal Dementias. Contin. Lifelong Learn. Neurol. 2016, 22, 464–489. [Google Scholar] [CrossRef] [PubMed]
- Zago, S.; Scarpazza, C.; Difonzo, T.; Arighi, A.; Hajhajate, D.; Torrente, Y.; Sartori, G. Behavioral Variant of Frontotemporal Dementia and Homicide in a Historical Case. 2021, 49, 219–227. [CrossRef]
- Nilsson, C.; Waldö, M.L.; Nilsson, K.; Santillo, A.; Vestberg, S. Age-Related Incidence and Family History in Frontotemporal Dementia: Data from the Swedish Dementia Registry. PLOS ONE 2014, 9, e94901. [Google Scholar] [CrossRef]
- Hendriks, S.; Peetoom, K.; Bakker, C.; van der Flier, W.M.; Papma, J.M.; Koopmans, R.; Verhey, F.R.J.; de Vugt, M.; Köhler, S.; Withall, A.; et al. Global Prevalence of Young-Onset Dementia. JAMA Neurol. 2021, 78, 1080–1090. [Google Scholar] [CrossRef]
- Onyike, C.U.; Diehl-Schmid, J. The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 2013, 25, 130–137. [Google Scholar] [CrossRef]
- Diehl-Schmid, J.; Perneczky, R.; Koch, J.; Nedopil, N.; Kurz, A. Guilty by Suspicion? Criminal Behavior in Frontotemporal Lobar Degeneration. Cogn. Behav. Neurol. 2013, 26, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Theiss, C.; Brüne, M. Ultrastructural Alterations of Von Economo Neurons in the Anterior Cingulate Cortex in Schizophrenia. Anat. Rec. 2017, 300, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Nana, A.L.; Sidhu, M.; Gaus, S.E.; Hwang, J.-H.L.; Li, L.; Park, Y.; Kim, E.-J.; Pasquini, L.; Allen, I.E.; Rankin, K.P.; et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2018, 137, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Vohryzek, J.; Cabral, J.; Vuust, P.; Deco, G.; Kringelbach, M.L. Understanding brain states across spacetime informed by whole-brain modelling. Philos. Trans. R. Soc. A: Math. Phys. Eng. Sci. 2022, 380, 20210247. [Google Scholar] [CrossRef] [PubMed]
- Nathani, M.; Jaleel, V.; Turner, A.; Dirvonas, C.; Suryadevara, U.; Tandon, R. When you hear hoofbeats, think horses and zebras: The importance of a wide differential when it comes to frontotemporal lobar degeneration. Asian J. Psychiatry 2019, 47, 101875. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.M.; Herbert, C.; Pollatos, O. On the Relationship Between Interoceptive Awareness and Alexithymia: Is Interoceptive Awareness Related to Emotional Awareness? J. Pers. 2011, 79, 1149–1175. [Google Scholar] [CrossRef] [PubMed]
- Quadt, L.; Critchley, H.D.; Garfinkel, S.N. The neurobiology of interoception in health and disease. Ann. New York Acad. Sci. 2018, 1428, 112–128. [Google Scholar] [CrossRef]
- Cauda, F.; Geminiani, G.C.; Vercelli, A. Evolutionary appearance of von Economo’s neurons in the mammalian cerebral cortex. Front. Hum. Neurosci. 2014, 8, 104. [Google Scholar] [CrossRef]
- Menon, V.; Uddin, L.Q. Saliency, switching, attention and control: a network model of insula function. Brain Struct. Funct. 2010, 214, 655–667. [Google Scholar] [CrossRef]
- López-Ojeda, W.; Hurley, R.A. Von Economo Neuron Involvement in Social Cognitive and Emotional Impairments in Neuropsychiatric Disorders. J. Neuropsychiatry 2022, 34, 302–306. [Google Scholar] [CrossRef]
- Hodge, R.D.; Miller, J.A.; Novotny, M.; Kalmbach, B.E.; Ting, J.T.; Bakken, T.E.; Aevermann, B.D.; Barkan, E.R.; Berkowitz-Cerasano, M.L.; Cobbs, C.; et al. Transcriptomic evidence that von Economo neurons are regionally specialized extratelencephalic-projecting excitatory neurons. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- A Dijkstra, A.; Lin, L.-C.; Nana, A.L.; E Gaus, S.; Seeley, W.W. Von Economo Neurons and Fork Cells: A Neurochemical Signature Linked to Monoaminergic Function. Cereb. Cortex 2016, 28, 131–144. [Google Scholar] [CrossRef]
- Azizi, S.A. Monoamines: Dopamine, Norepinephrine, and Serotonin, Beyond Modulation, “Switches” That Alter the State of Target Networks. Neurosci. 2020, 28, 121–143. [Google Scholar] [CrossRef]
- Valli, M.; Cho, S.S.; Uribe, C.; Masellis, M.; Chen, R.; Mihaescu, A.; Strafella, A.P. VMAT2 availability in Parkinson’s disease with probable REM sleep behaviour disorder. Mol. Brain 2021, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Broese, M.; Riemann, D.; Hein, L.; Nissen, C. α-Adrenergic Receptor Function, Arousal and Sleep: Mechanisms and Therapeutic Implications. Pharmacopsychiatry 2012, 45, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Mancama, D.; Kerwin, R.W.; Arranz, M.J. Expression of the α1A-adrenergic receptor in schizophrenia. Neurosci. Lett. 2006, 401, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, M.; Minthon, L.; Passant, U.; Blennow, K.; Wallin, A. Decreased monoamine metabolites in frontotemporal dementia and Alzheimer’s disease. Neurobiol. Aging 1998, 19, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.C.; Kay, D.B.; Buysse, D.J. The Pathophysiology of Insomnia. Chest 2015, 147, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Kiyoshi, C.M. Astrocyte syncytium: a functional reticular system in the brain. Neural Regen. Res. 2019, 14, 595–596. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, S.; Picard, K.; Limatola, C.; Nadjar, A.; Pascual, O.; Tremblay, M.E. Role of Glia in the Regulation of Sleep in Health and Disease. 10, 101.245. Compr Physiol 2020, 10, 101.245. [Google Scholar] [CrossRef]
- Que, M.; Li, Y.; Wang, X.; Zhan, G.; Luo, X.; Zhou, Z. Role of astrocytes in sleep deprivation: accomplices, resisters, or bystanders? Front. Cell. Neurosci. 2023, 17, 1188306. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef] [PubMed]
- Leow, D.M.-K.; Cheah, I.K.-M.; Fong, Z.W.-J.; Halliwell, B.; Ong, W.-Y. Protective Effect of Ergothioneine against 7-Ketocholesterol-Induced Mitochondrial Damage in hCMEC/D3 Human Brain Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 5498. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. Mitochondrial Dysfunction and Chronic Disease: Treatment With Natural Supplements. Integr Med (Encinitas). 2014, 13, 35–43. [Google Scholar] [PubMed]
- Hill, V.M.; O’connor, R.M.; Sissoko, G.B.; Irobunda, I.S.; Leong, S.; Canman, J.C.; Stavropoulos, N.; Shirasu-Hiza, M. A bidirectional relationship between sleep and oxidative stress in Drosophila. PLOS Biol. 2018, 16, e2005206. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Cai, R.; Volchuk, A.; Steinberg, B.E.; Saito, Y.; Matsuzawa, A.; Grinstein, S.; Freeman, S.A. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr. Biol. 2023, 33, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Heckers, S.; Konradi, C. Hippocampal neurons in schizophrenia. J. Neural Transm. 2002, 109, 891–905. [Google Scholar] [CrossRef]
- Padurariu., M.; Ciobica, A.; Mavroudis, I.; Fotiou, D.; Baloyannis, S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. 2012 Jun;24(2):152-8. Psychiatr Danub. 2012, 24, 152–158. [Google Scholar]
- Zhang, P.; Li, Y.-X.; Zhang, Z.-Z.; Yang, Y.; Rao, J.-X.; Xia, L.; Li, X.-Y.; Chen, G.-H.; Wang, F. Astroglial Mechanisms Underlying Chronic Insomnia Disorder: A Clinical Study. Nat. Sci. Sleep 2020, ume 12, 693–704. [Google Scholar] [CrossRef]
- Bellesi, M.; de Vivo, L.; Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2017, 285, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, T.; Howes, O. The role of mitochondria in the pathophysiology of schizophrenia: A critical review of the evidence focusing on mitochondrial complex one. Neurosci. Biobehav. Rev. 2021, 132, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Beaupre, L.M.M.; Brown, G.M.; Braganza, N.A.; Kennedy, J.L.; Gonçalves, V.F. Mitochondria’s role in sleep: Novel insights from sleep deprivation and restriction studies. World J. Biol. Psychiatry 2021, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.B.; Mailloux, R.J. Mitochondria Need Their Sleep: Redox, Bioenergetics, and Temperature Regulation of Circadian Rhythms and the Role of Cysteine-Mediated Redox Signaling, Uncoupling Proteins, and Substrate Cycles. Antioxidants 2023, 12, 674. [Google Scholar] [CrossRef] [PubMed]
- Ene, H.M.; Karry, R.; Farfara, D.; Ben-Shachar, D. Mitochondria play an essential role in the trajectory of adolescent neurodevelopment and behavior in adulthood: evidence from a schizophrenia rat model. Mol. Psychiatry 2022, 28, 1170–1181. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. (BBA)—Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.J.; Suh, J.H.; Lee, S.H.; Poulsen, K.L.; An, Y.A.; Moorthy, B.; Hartig, S.M.; Moore, D.D.; Kim, K.H. Aryl hydrocarbon receptor maintains hepatic mitochondrial homeostasis in mice. Mol. Metab. 2023, 72, 101717. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Dornbos, P.; Steidemann, M.; Dunivin, T.K.; Rizzo, M.; LaPres, J.J. Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome. Toxicol. Appl. Pharmacol. 2016, 304, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Cenijn, P.H.; Brouwer, A.; Denison, M.S. Activation of the Ah Receptor by Tryptophan and Tryptophan Metabolites. Biochemistry 1998, 37, 11508–11515. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Slominski, R.M.; Song, Y.; Qayyum, S.; Placha, W.; Janjetovic, Z.; Kleszczyński, K.; Atigadda, V.; Song, Y.; et al. Melatonin and Its Metabolites Can Serve as Agonists on the Aryl Hydrocarbon Receptor and Peroxisome Proliferator-Activated Receptor Gamma. Int. J. Mol. Sci. 2023, 24, 15496. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-X.; Wang, C.; Krager, S.L.; Bottum, K.M.; Tischkau, S.A. Aryl Hydrocarbon Receptor Activation Attenuates Per1 Gene Induction and Influences Circadian Clock Resetting. Toxicol. Sci. 2013, 132, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Kempf, A. Mitochondrial control of sleep. Curr. Opin. Neurobiol. 2023, 81, 102733. [Google Scholar] [CrossRef]
- Wei, Y.-D.; Helleberg, H.; Rannug, U.; Rannug, A. Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem. Interactions 1998, 110, 39–55. [Google Scholar] [CrossRef]
- Ziv-Gal, A.; Flaws, J.A.; Mahoney, M.M.; Miller, S.R.; Zacur, H.A.; Gallicchio, L. Genetic polymorphisms in the aryl hydrocarbon receptor-signaling pathway and sleep disturbances in middle-aged women. Sleep Med. 2013, 14, 883–887. [Google Scholar] [CrossRef]
- Frau-Méndez, M.A.; Fernández-Vega, I.; Ansoleaga, B.; Tech, R.B.; Tech, M.C.; del Rio, J.A.; Zerr, I.; Llorens, F.; Zarranz, J.J.; Ferrer, I. Fatal familial insomnia: mitochondrial and protein synthesis machinery decline in the mediodorsal thalamus. Brain Pathol. 2016, 27, 95–106. [Google Scholar] [CrossRef]
- Kishikawa, J.-I.; Inoue, Y.; Fujikawa, M.; Nishimura, K.; Nakanishi, A.; Tanabe, T.; Imamura, H.; Yokoyama, K. General anesthetics cause mitochondrial dysfunction and reduction of intracellular ATP levels. PLOS ONE 2018, 13, e0190213–e0190213. [Google Scholar] [CrossRef]
- Wei, H. The Role of Calcium Dysregulation in Anesthetic-Mediated Neurotoxicity. Anesth. Analg. 2011, 113, 972–974. [Google Scholar] [CrossRef]
- Lee, H.-G.; Choi, S.-I.; Park, S.-K.; Park, S.-J.; Kim, N.-H.; Choi, E.-K. Alteration of glutathione metabolism and abnormal calcium accumulation in the mitochondria of hamster brain infected with scrapie agent. Neurobiol. Aging 2000, 21, 151. [Google Scholar] [CrossRef]
- Glatzel, M.; Sepulveda-Falla, D. Losing sleep over mitochondria: a new player in the pathophysiology of fatal familial insomnia. Brain Pathol. 2016, 27, 107–108. [Google Scholar] [CrossRef]
- Moon, J.-H.; Park, S.-Y. Prion peptide-mediated calcium level alteration governs neuronal cell damage through AMPK-autophagy flux. Cell Commun. Signal. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Matteoni, S.; Matarrese, P.; Ascione, B.; Ricci-Vitiani, L.; Pallini, R.; Villani, V.; Pace, A.; Paggi, M.G.; Abbruzzese, C. Chlorpromazine induces cytotoxic autophagy in glioblastoma cells via endoplasmic reticulum stress and unfolded protein response. J. Exp. Clin. Cancer Res. 2021, 40, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.L.; Iraci, N.; Biggi, S.; Cecchetti, V.; Biasini, E. Pharmacological Agents Targeting the Cellular Prion Protein. Pathogens 2018, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Korth, C.; May, B.C.H.; Cohen, F.E.; Prusiner, S.B. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. 2001, 98, 9836–9841. [Google Scholar] [CrossRef] [PubMed]
- Khattar, K.E.; Safi, J.; Rodriguez, A.-M.; Vignais, M.-L. Intercellular Communication in the Brain through Tunneling Nanotubes. Cancers 2022, 14, 1207. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gerdes, H.-H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Savaskan, N.E.; Borchert, A.; Bräuer, A.U.; Kuhn, H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: Specific induction of enzyme expression in reactive astrocytes following brain injury. Free. Radic. Biol. Med. 2007, 43, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Mauri, S.; Favaro, M.; Bernardo, G.; Mazzotta, G.M.; Ziviani, E. Mitochondrial autophagy in the sleeping brain. Front. Cell Dev. Biol. 2022, 10, 956394. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Owens, G.C.; Crossin, K.L.; Edelman, D.B. Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol. Cell. Neurosci. 2007, 36, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Nana, A.L.; Hepker, M.; Hwang, J.-H.L.; Gaus, S.E.; Spina, S.; Cosme, C.G.; Gan, L.; Grinberg, L.T.; Geschwind, D.H.; et al. Preferential tau aggregation in von Economo neurons and fork cells in frontotemporal lobar degeneration with specific MAPT variants. Acta Neuropathol. Commun. 2019, 7, 1–10. [Google Scholar] [CrossRef]
- Hogan, D.B.; Jetté, N.; Fiest, K.M.; Roberts, J.I.; Pearson, D.; Smith, E.E.; Roach, P.; Kirk, A.; Pringsheim, T.; Maxwell, C.J. The Prevalence and Incidence of Frontotemporal Dementia: a Systematic Review. Can. J. Neurol. Sci. / J. Can. des Sci. Neurol. 2016, 43, S96–S109. [Google Scholar] [CrossRef]
- Course, M.M.; Wang, X. Transporting mitochondria in neurons. F1000Research 2016, 5, 1735. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, M.T.; Chana-Cuevas, P. New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 109. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Youdim, M.B.H. The Neuroprotective Activities of the Novel Multi-Target Iron-Chelators in Models of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis and Aging. Cells 2023, 12, 763. [Google Scholar] [CrossRef]
- Calderon-Montaño, J.M.; Burgos-Morón, E.; Perez-Guerrero, C.; Lopez-Lazaro, M. A Review on the Dietary Flavonoid Kaempferol. Mini-Reviews Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef]
- Du, Y.-Y.; Sun, T.; Yang, Q.; Liu, Q.-Q.; Li, J.-M.; Yang, L.; Luo, L.-X. Therapeutic Potential of Kaempferol against Sleep Deprivation-Induced Cognitive Impairment: Modulation of Neuroinflammation and Synaptic Plasticity Disruption in Mice. ACS Pharmacol. Transl. Sci. 2023, 6, 1934–1944. [Google Scholar] [CrossRef]
- Saleem, A.; Ain, Q.U.; Akhtar, M.F. Alternative Therapy of Psychosis: Potential Phytochemicals and Drug Targets in the Management of Schizophrenia. Front. Pharmacol. 2022, 13, 895668. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ren, H.; Han, J.; Wang, W.; Zheng, Q.; Wang, D. Protective Effects of Kaempferol against Myocardial Ischemia/Reperfusion Injury in Isolated Rat Heart via Antioxidant Activity and Inhibition of Glycogen Synthase Kinase-3β. Oxidative Med. Cell. Longev. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Roh, M.-S. Glycogen Synthase Kinase-3 (GSK3) in Psychiatric Diseases and Therapeutic Interventions. Curr. Drug Targets 2006, 7, 1421–1434. [Google Scholar] [CrossRef]
- Mohammad, M.K.; Al-Masri, I.M.; Taha, M.O.; Al-Ghussein, M.A.; AlKhatib, H.S.; Najjar, S.; Bustanji, Y. Olanzapine inhibits glycogen synthase kinase-3β: An investigation by docking simulation and experimental validation. Eur. J. Pharmacol. 2008, 584, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Shilovsky, G.A.; Putyatina, T.S.; Morgunova, G.V.; Seliverstov, A.V.; Ashapkin, V.V.; Sorokina, E.V.; Markov, A.V.; Skulachev, V.P. A Crosstalk between the Biorhythms and Gatekeepers of Longevity: Dual Role of Glycogen Synthase Kinase-3. Biochem. (Moscow) 2021, 86, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Dozza, B.; Smith, M.A.; Perry, G.; Tabaton, M.; Strocchi, P. Regulation of glycogen synthase kinase-3β by products of lipid peroxidation in human neuroblastoma cells. J. Neurochem. 2004, 89, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Serafim, B.; Bernardino, A.R.; Freitas, F.; Torres, C.A.V. Recent Developments in the Biological Activities, Bioproduction, and Applications of Pseudomonas spp. Phenazines. Molecules 2023, 28, 1368. [Google Scholar] [CrossRef]
- Jiang, J.; Beltran, D.G.; Schacht, A.; Wright, S.; Zhang, L.; Du, L. Functional and Structural Analysis of Phenazine O-Methyltransferase LaPhzM from Lysobacter antibioticus OH13 and One-Pot Enzymatic Synthesis of the Antibiotic Myxin. ACS Chem. Biol. 2018, 13, 1003–1012. [Google Scholar] [CrossRef]
- Ohlendorf, B.; Schulz, D.; Erhard, A.; Nagel, K.; Imhoff, J.F. Geranylphenazinediol, an Acetylcholinesterase Inhibitor Produced by a Streptomyces Species. J. Nat. Prod. 2012, 75, 1400–1404. [Google Scholar] [CrossRef]
- Rusman, Y.; Oppegard, L.M.; Hiasa, H.; Gelbmann, C.; Salomon, C.E. Solphenazines A–F, Glycosylated Phenazines from Streptomyces sp. Strain DL-93. J. Nat. Prod. 2013, 76, 91–96. [Google Scholar] [CrossRef]
- Wang, X.; Abbas, M.; Zhang, Y.; Elshahawi, S.I.; Ponomareva, L.V.; Cui, Z.; Van Lanen, S.G.; Sajid, I.; Voss, S.R.; Shaaban, K.A.; et al. Baraphenazines A–G, Divergent Fused Phenazine-Based Metabolites from a Himalayan Streptomyces. J. Nat. Prod. 2019, 82, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- van Wezel, G. P.; McKenzie, N. L.; Nodwell, J. R. Chapter 5 Applying the genetics of secondary metabolism in model actinomycetes to the discovery of new antibiotics, 1st ed.; Elsevier Inc., 2009; Vol. 458. [Google Scholar]
- Cha, J.W.; Lee, S.I.; Kim, M.C.; Thida, M.; Lee, J.W.; Park, J.-S.; Kwon, H.C. Pontemazines A and B, phenazine derivatives containing a methylamine linkage from Streptomyces sp. UT1123 and their protective effect to HT-22 neuronal cells. Bioorganic Med. Chem. Lett. 2015, 25, 5083–5086. [Google Scholar] [CrossRef]
- Krishnaiah, M.; Rodriguez de Almeida, N.; Udumula, V.; Song, Z.; Chhonker, Y.S.; Abdelmoaty, M.M.; Aragao do Nascimento, V.; Murry, D.J.; Conda-Sheridan, M. Synthesis, biological evaluation, and metabolic stability of phenazine derivatives as antibacterial agents. Eur. J. Med. Chem. 2018, 143, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Lavaggi, M.L.; Aguirre, G.; Boiani, L.; Orelli, L.; García, B.; Cerecetto, H.; González, M. Pyrimido[1,2-a]quinoxaline 6-oxide and phenazine 5,10-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi. Eur. J. Med. Chem. 2008, 43, 1737–1741. [Google Scholar] [CrossRef]
- Kato, S.; Shindo, K.; Yamagishi, Y.; Matsuoka, M.; Kawai, H.; Mochizuki, J. Phenazoviridin, a novel free radical scavenger from Streptomyces sp. Taxonomy, fermentation, isolation, structure elucidation and biological properties. J. Antibiot. 1993, 46, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Laxmi, M.; Bhat, S.G. Characterization of pyocyanin with radical scavenging and antibiofilm properties isolated from Pseudomonas aeruginosa strain BTRY1. 3 Biotech 2016, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Alatawneh, N.; Meijler, M.M. Unraveling the Antibacterial and Iron Chelating Activity ofN-Oxide Hydroxy-Phenazine natural Products and Synthetic Analogs againstStaphylococcus aureus. Isr. J. Chem. 2023, 63. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Armistead, G.; A Rosa, C.; Anderson, A.; Patil, R.; Cornett, E.M.; Murnane, K.S.; Kaye, A.M.; Kaye, A.D. Phenothiazines and their Evolving Roles in Clinical Practice: A Narrative Review. Heal. Psychol. Res. 2022, 10, 38930. [Google Scholar] [CrossRef]
- Heitmann, A.S.B.; Zanjani, A.A.H.; Klenow, M.B.; Mularski, A.; Sønder, S.L.; Lund, F.W.; Boye, T.L.; Dias, C.; Bendix, P.M.; Simonsen, A.C.; et al. Phenothiazines alter plasma membrane properties and sensitize cancer cells to injury by inhibiting annexin-mediated repair. J. Biol. Chem. 2021, 297, 101012. [Google Scholar] [CrossRef]
- Boonnoy, P.; Jarerattanachat, V.; Karttunen, M.; Wong-Ekkabut, J. Bilayer Deformation, Pores, and Micellation Induced by Oxidized Lipids. J. Phys. Chem. Lett. 2015, 6, 4884–4888. [Google Scholar] [CrossRef]
- Wu, C.-H.; Bai, L.-Y.; Tsai, M.-H.; Chu, P.-C.; Chiu, C.-F.; Chen, M.Y.; Chiu, S.-J.; Chiang, J.-H.; Weng, J.-R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents–A drug repurposing strategy. Sci. Rep. 2016, 6, 27540. [Google Scholar] [CrossRef]
- Voronova, O.; Zhuravkov, S.; Korotkova, E.; Artamonov, A.; Plotnikov, E. Antioxidant Properties of New Phenothiazine Derivatives. Antioxidants 2022, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Keynes, R.G.; Karchevskaya, A.; Riddall, D.; Griffiths, C.H.; Bellamy, T.C.; Chan, A.W.E.; Selwood, D.L.; Garthwaite, J. N10-carbonyl-substituted phenothiazines inhibiting lipid peroxidation and associated nitric oxide consumption powerfully protect brain tissue against oxidative stress. Chem. Biol. Drug Des. 2019, 94, 1680–1693. [Google Scholar] [CrossRef] [PubMed]
- Iuga, C.; Campero, A.; Vivier-Bunge, A. Antioxidant vs. prooxidant action of phenothiazine in a biological environment in the presence of hydroxyl and hydroperoxyl radicals: a quantum chemistry study. RSC Adv. 2015, 5, 14678–14689. [Google Scholar] [CrossRef]
- Yue, Y.; Kong, L.; Wang, J.; Li, C.; Tan, L.; Su, H.; Xu, Y. Regional Abnormality of Grey Matter in Schizophrenia: Effect from the Illness or Treatment? PLOS ONE 2016, 11, e0147204–e0147204. [Google Scholar] [CrossRef]
- Martínez, A.; Ibarra, I.A.; Vargas, R. A quantum chemical approach representing a new perspective concerning agonist and antagonist drugs in the context of schizophrenia and Parkinson’s disease. PLOS ONE 2019, 14, e0224691. [Google Scholar] [CrossRef]
- Goode-Romero, G.; Dominguez, L.; Martínez, A. Electron Donor–Acceptor Properties of Different Muscarinic Ligands: On the Road to Control Schizophrenia. J. Chem. Inf. Model. 2021, 61, 5117–5124. [Google Scholar] [CrossRef]
- Ho BC, Andreasen NC, Ziebell S, et al. Long-term antipsychotic treatment and brain volumes: a longitudinal study of first-episode schizophrenia. Arch Gen Psychiatry 2011; 68: 128–137. [CrossRef]
- Veijola, J.; Guo, J.Y.; Moilanen, J.S.; Jääskeläinen, E.; Miettunen, J.; Kyllönen, M.; Haapea, M.; Huhtaniska, S.; Alaräisänen, A.; Mäki, P.; et al. Longitudinal Changes in Total Brain Volume in Schizophrenia: Relation to Symptom Severity, Cognition and Antipsychotic Medication. PLOS ONE 2014, 9, e101689. [Google Scholar] [CrossRef]
- Engwa, G.A.; Ayuk, E.L.; Igbojekwe, B.U.; Unaegbu, M. Potential Antioxidant Activity of New Tetracyclic and Pentacyclic Nonlinear Phenothiazine Derivatives. Biochem. Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Clark, M.A.; Shay, J.W. Mitochondrial transformation of mammalian cells. Nature 1982, 295, 605–607. [Google Scholar] [CrossRef]
- Katrangi, E.; D’Souza, G.; Boddapati, S.V.; Kulawiec, M.; Singh, K.K.; Bigger, B.; Weissig, V. Xenogenic Transfer of Isolated Murine Mitochondria into Human ρ0 Cells Can Improve Respiratory Function. Rejuvenation Res. 2007, 10, 561–570. [Google Scholar] [CrossRef]
- Pacak, C.A.; Preble, J.M.; Kondo, H.; Seibel, P.; Levitsky, S.; del Nido, P.J.; Cowan, D.B.; McCully, J.D. Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol. Open 2015, 4, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Pour, P.A.; Hosseinian, S.; Kheradvar, A. Mitochondrial transplantation in cardiomyocytes: foundation, methods, and outcomes. Am. J. Physiol. Physiol. 2021, 321, C489–C503. [Google Scholar] [CrossRef]
- Cowan, D.B.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; del Nido, P.J.; McCully, J.D. Transit and integration of extracellular mitochondria in human heart cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-G.; Miao, C.-Y. Mitochondrial transplantation as a promising therapy for mitochondrial diseases. Acta Pharm. Sin. B 2023, 13, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Ren, D.; Zheng, P.; Zou, S.; Gong, Y.; Wang, Y.; Duan, J.; Deng, J.; Chen, H.; Feng, J.; Zhong, C.; et al. GJA1-20K Enhances Mitochondria Transfer from Astrocytes to Neurons via Cx43-TnTs After Traumatic Brain Injury. Cell. Mol. Neurobiol. 2021, 42, 1887–1895. [Google Scholar] [CrossRef]
Figure 1.
Astrocytes contact cerebral microvessels with their end-feet processes, delineating a pathway for the flow of extracellular fluid, known as the glymphatic system. The volume of interstitial fluid (ISF) in the brain parenchyma varies with the brain work. During high intensity work, AQP-4 water receptors are upregulated in the end-feet, pumping the ISF into astrocytes. This results in low ISF (hypovolemia). During sleep (low intensity brain work), less ISF enters the astrocyte. The circulation of ISF clears the molecular debris (including beta amyloid) from the extracellular space.
Figure 1.
Astrocytes contact cerebral microvessels with their end-feet processes, delineating a pathway for the flow of extracellular fluid, known as the glymphatic system. The volume of interstitial fluid (ISF) in the brain parenchyma varies with the brain work. During high intensity work, AQP-4 water receptors are upregulated in the end-feet, pumping the ISF into astrocytes. This results in low ISF (hypovolemia). During sleep (low intensity brain work), less ISF enters the astrocyte. The circulation of ISF clears the molecular debris (including beta amyloid) from the extracellular space.
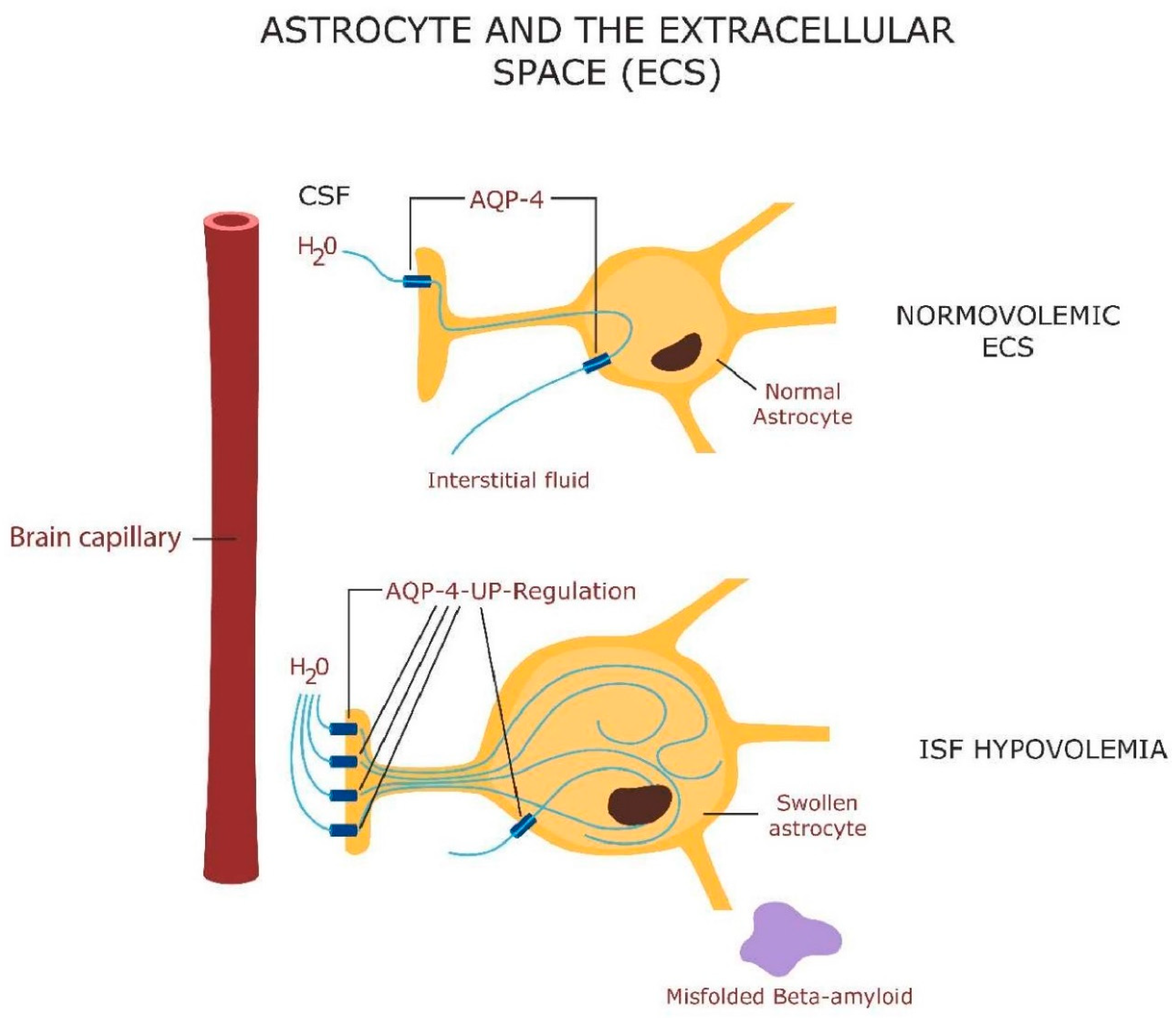
Figure 2.
Astrocytes support the postmitotic, long-lived, neurons by helping them avert death by ferroptosis and loss of mitochondria. The former is accomplished by exporting GPX-4 to neurons (to repair oxidized lipids), while the latter by exporting healthy mitochondria to neuronal cells (via tunneling nanotubules, extracellular vesicles, or cell-cell fusion). Astrocytes import cystine via cystine/glutamate antiporter (Xc-). Cystine is reduced to cysteine and generates glutathione and GPX-4 (which is transferred to neurons). Cysteine can also be derived from methionine, while glutathione can be generated from cysteine and glutathione disulfide (GSSC). In neurons, iron is stored in ferritin and when needed, ferritin undergoes ferritinophagy (autophagy) in lysosomes, releasing free iron. Iron ingresses the neuron via transferrin receptor 1 (TRF-1), while the excess intracellular iron is eliminated via ferroportin.
Figure 2.
Astrocytes support the postmitotic, long-lived, neurons by helping them avert death by ferroptosis and loss of mitochondria. The former is accomplished by exporting GPX-4 to neurons (to repair oxidized lipids), while the latter by exporting healthy mitochondria to neuronal cells (via tunneling nanotubules, extracellular vesicles, or cell-cell fusion). Astrocytes import cystine via cystine/glutamate antiporter (Xc-). Cystine is reduced to cysteine and generates glutathione and GPX-4 (which is transferred to neurons). Cysteine can also be derived from methionine, while glutathione can be generated from cysteine and glutathione disulfide (GSSC). In neurons, iron is stored in ferritin and when needed, ferritin undergoes ferritinophagy (autophagy) in lysosomes, releasing free iron. Iron ingresses the neuron via transferrin receptor 1 (TRF-1), while the excess intracellular iron is eliminated via ferroportin.
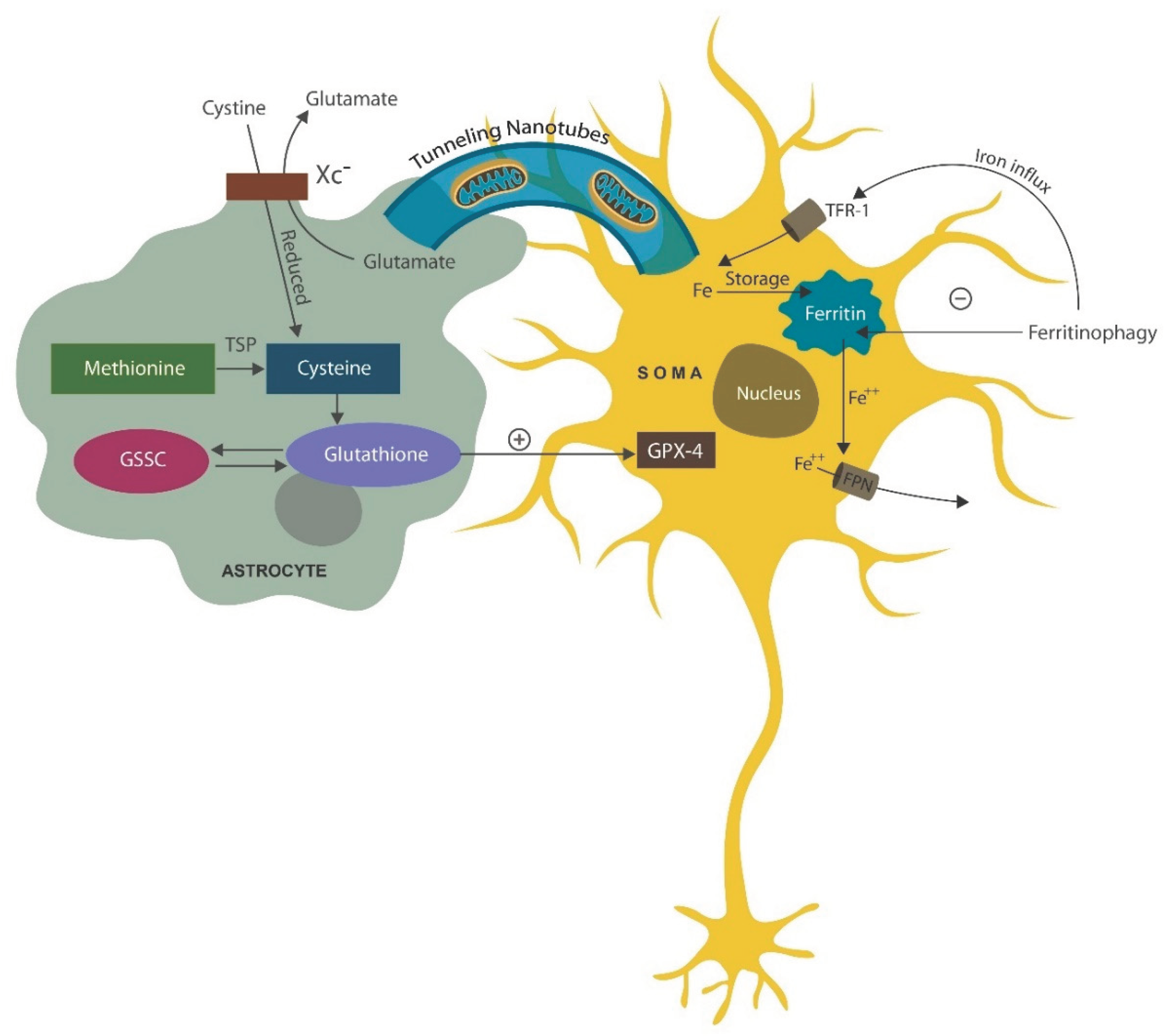
Figure 3.
The lipid bilayer of neuronal membrane is easily oxidated when intracellular iron is upregulated. Oxysterols, including 7-Ketocholesterol (a toxic oxide), and oxidated phospholipids alter the biophysical properties of cell membranes, disrupting neurotransmission. In addition, oxidized lipids activate AhR, triggering premature neuronal senescence. Phenazines, Phenothiazines, and their derivatives, intercalate themselves into the lipid bilayer, repairing the lipids in cellular and/or mitochondrial membranes.
Figure 3.
The lipid bilayer of neuronal membrane is easily oxidated when intracellular iron is upregulated. Oxysterols, including 7-Ketocholesterol (a toxic oxide), and oxidated phospholipids alter the biophysical properties of cell membranes, disrupting neurotransmission. In addition, oxidized lipids activate AhR, triggering premature neuronal senescence. Phenazines, Phenothiazines, and their derivatives, intercalate themselves into the lipid bilayer, repairing the lipids in cellular and/or mitochondrial membranes.
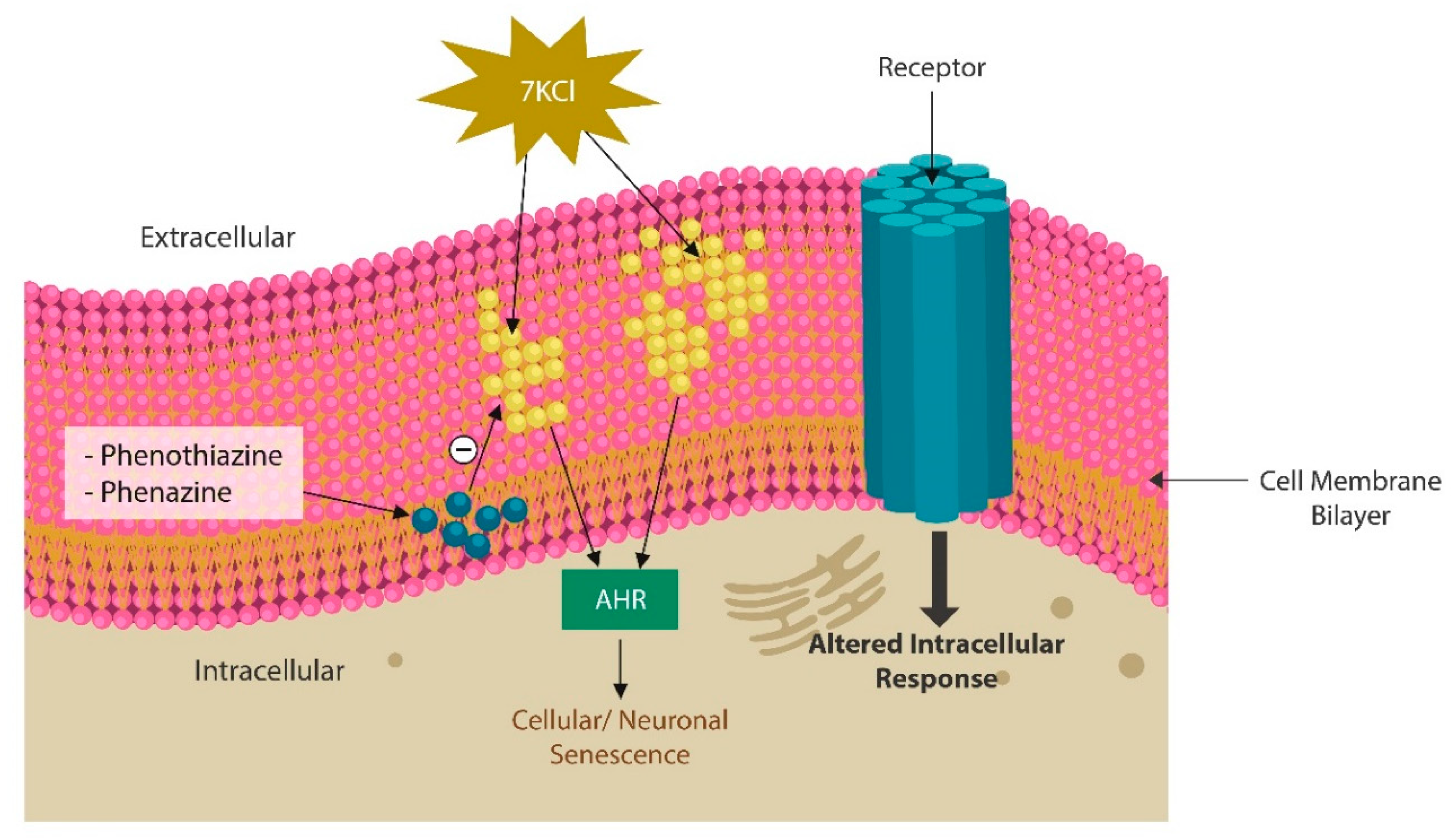
Figure 4.
Phenazine vs. Phenothiazine: similarities and differences.
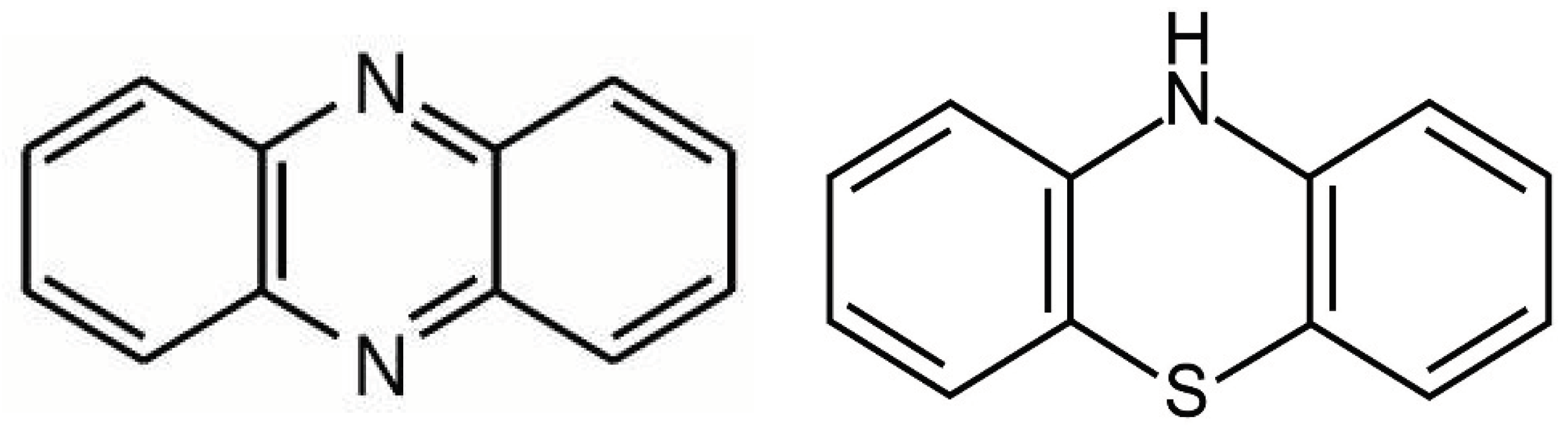
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



