
Submitted:
27 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
This study introduces an efficient strategy for synthesizing polyhydroxyurethane-based multicomponent hydrogels with enhanced rheological properties. In a single-step process, 3D materials composed of Polymer 1 (PHU) and Polymer 2 (PVA or gelatin) were produced. Polymer 1, a crosslinked polyhydroxyurethane (PHU) grew within a colloidal suspension of Polymer 2, forming an interconnected network. The synthesis of Polymer 1 utilized a Non-Isocyanate Polyurethane (NIPU) methodology based on the aminolysis of bis(cyclic carbonate) (bisCC) monomers derived from 1-thioglycerol and 1,2-dithioglycerol (monomers MA and ME, respectively). This method, applied for the first time in Semi Interpenetrating Network (SIPN) formation, demonstrated exceptional orthogonality, since the functional groups in Polymer 2 do not interfere with Polymer 1 formation. Optimizing PHU formation involved a twenty-trial methodology, identifying influential variables such as polymer concentration, temperature, solvent (an aprotic and a protic solvent), and the organo-catalyst used [a thiourea derivative (TU) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)]. The highest molecular weights were achieved under near-bulk polymerization conditions using TU-protic and DBU-aprotic as catalyst-solvent combinations. ME-based PHU exhibited higher (M_w ) ̅ than MA-based PHU (34,100 Da and 16,400 Da, respectively). Applying the enhanced methodology to prepare ten multicomponent hydrogels using PVA or gelatin as the polymer scaffold revealed superior rheological properties in PVA-based hydrogels, exhibiting solid-like gel behavior. Incorporating ME enhanced mechanical properties and elasticity (with loss tangent values of 0.09 and 0.14). SEM images unveiled distinct microstructures, including a sponge-like pattern in certain PVA-based hydrogels when MA was chosen, indicating the formation of highly superporous interpenetrated materials. In summary, this innovative approach presents a versatile methodology for obtaining advanced hydrogel-based systems with potential applications in various biomedical fields.
Keywords:
NIPU
; cyclic carbonates
; PHU
; functional polymers
; interpenetrated networks
; IPN
; SIPN
; porous materials
; rheological properties
1. Introduction
Multicomponent hydrogels, formed by combining two or more distinct polymers or other components, have garnered considerable attention for their unique properties and versatile applications in biomedicine [1,2,3,4]. Hydrogels provide a hydrated and mechanically supportive environment [1], making them cytocompatible and suitable for precise molecule release. Diverse chemical and physical approaches have been utilized to construct these hydrogel networks, incorporating both synthetic and biologically derived molecules to confer specific properties. These advancements have resulted in materials with heightened mechanical attributes and diverse biological functions, showcasing promise in tissue engineering, cancer treatment, and gene therapies [1,3]. Furthermore, the integration of modular and heterogeneous building blocks into well-defined hydrogel composites has given rise to artificial extracellular matrices (ECMs) that closely emulate the hybrid nature of natural ECMs [2]. Additionally, the progress in developing multicomponent hydrogels with stimuli-responsive properties may open avenues for applications in smart drug delivery systems (DDS) in which these hydrogels can respond to specific biomolecules through gel-sol transition behavior [4]. Nevertheless, the existing limitations in achieving these hydrogels via conventional one-step mixing protocols [4] underscore the imperative need for novel synthetic tools and methodologies. Although polyurethanes (PU) are commonly used biocompatible and biodegradable materials [5,6] employed from medical devices [7] (pump, valves, stems, etc.) to smart materials useful in the preparation of responsive drug delivery systems [8,9], multicomponent systems involving them have not been prepared yet.
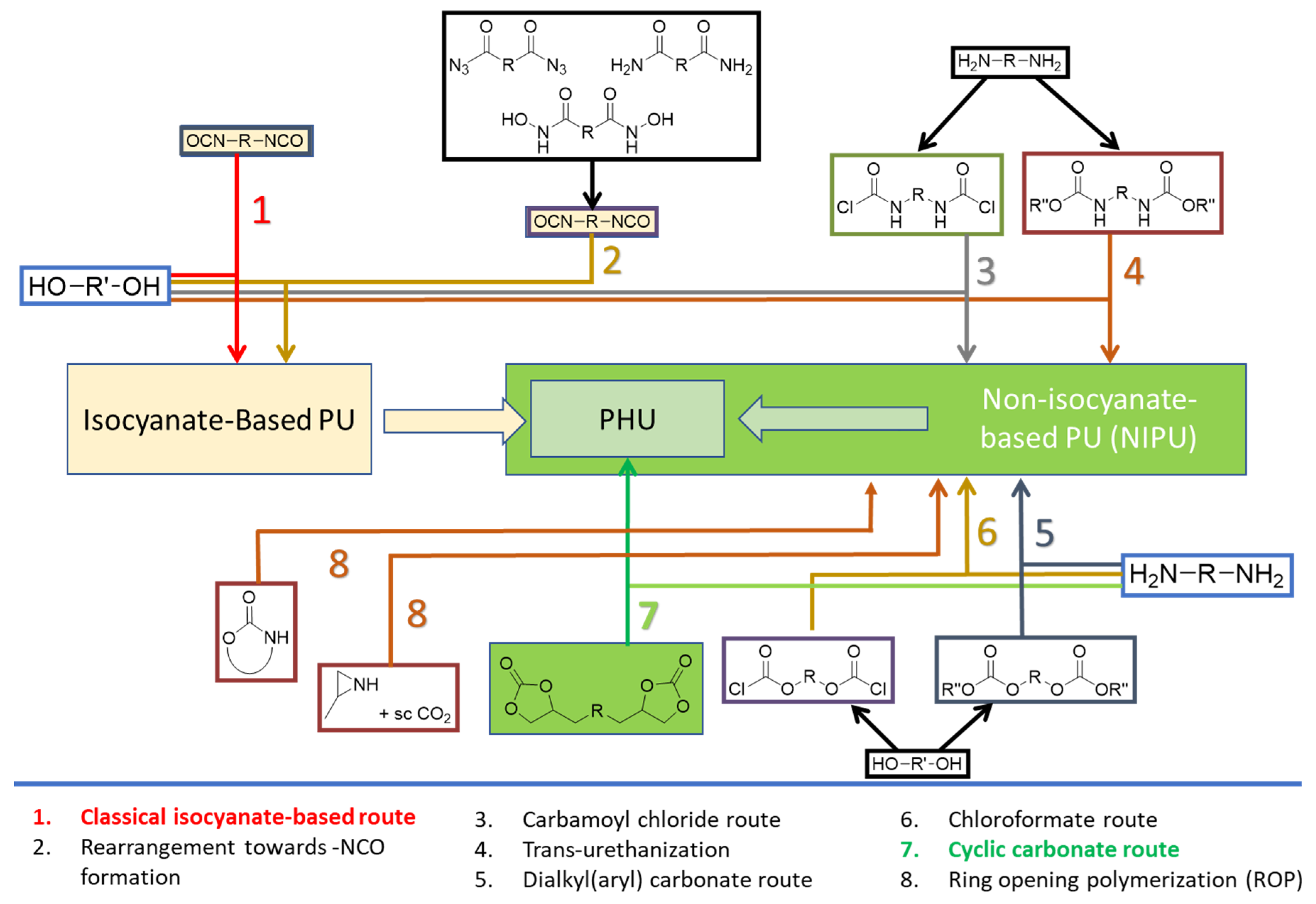
PU can be synthesized by several methods [5,6], being the most relevant those recorded in Figure 1. Traditional PU synthesis involves reacting di(multi)isocyanates and di(multi)alcohols under moisture-free, inert atmosphere conditions using metal catalysts like stannous octoate [10]. However, these catalysts pose environmental [11] and health risks [12]. Isocyanates, used in PU synthesis, are toxic and face stringent regulations. The need for eco-friendly alternatives has led to the development of non-isocyanate polyurethanes (NIPU) [13] with less demanding conditions, ideally at room or moderate temperatures.
NIPU are best obtained via carbamoyl chloride, dialkyl(aryl) carbonate, chloroformate, or cyclic carbonate (CC) routes or by ring opening polymerizations and trans-urethanizations, as shown in Figure 1 {preferred paths: routes 4 [13,14,15] and 7 [16,17,18]}.
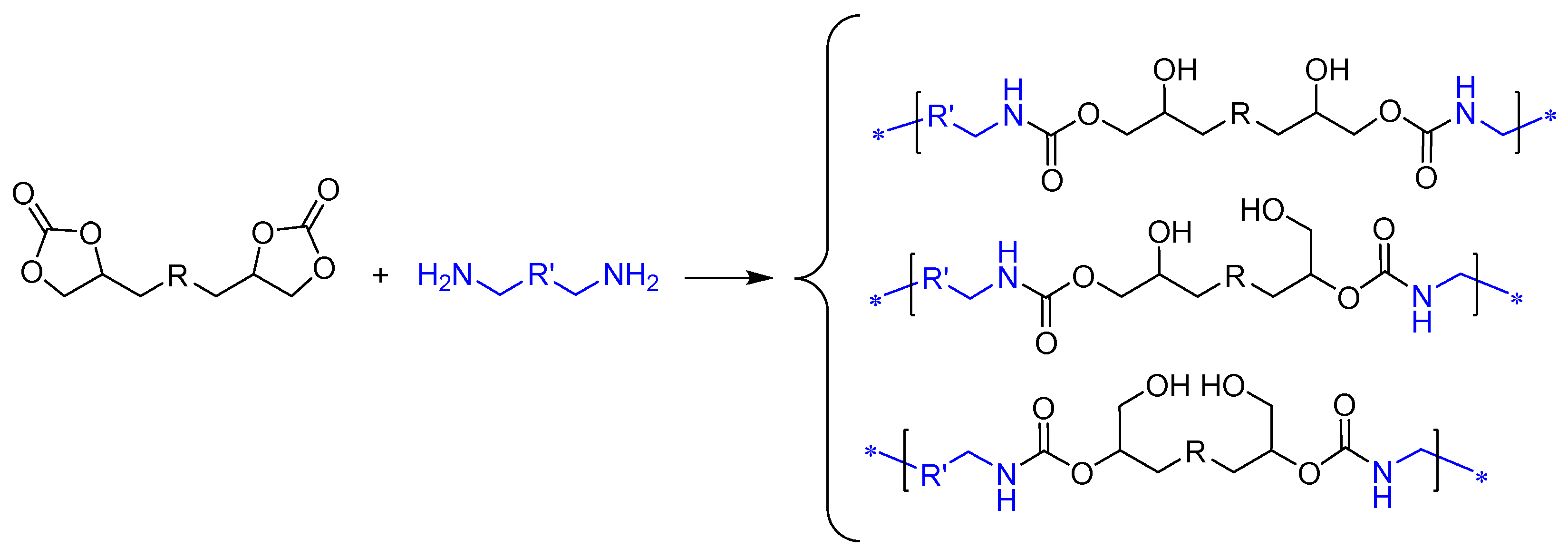
The CC route (Route 7, Figure 1) involves reaction of bis(multi)CC with di(multi)amines, resulting in the opening of cyclic carbonate rings (Scheme 1). The non-regioselective ring opening process yields polyhydroxyurethanes (PHU) characterized by enhanced hydrophilicity compared to conventional PU. This improved hydrophilicity offers an additional advantage for diverse biomedical applications [19]. This polymerization reaction exhibits orthogonality with a broad spectrum of biocompatible polymers and is unaltered by the presence of water or protic solvents, enabling its implementation under non-stringent mild conditions.
Multicomponent hydrogels, such as full- and semi-interpenetrating polymer networks (FIPN and SIPN, respectively), have emerged as innovative biomaterials for drug delivery [20] and as scaffolds for tissue engineering [21]. Typically, third generation IPN hydrogels are constituted by two polymers whose chains are entangled with each other giving rise to a stable and porous three-dimensional scaffold. Polymer 1 is formed within a colloidal suspension of biocompatible Polymer 2 by an orthogonal polymerization method [22]. Hydrophilic polymers (natural or synthetic), such as poly(vinyl alcohol) (PVA) [23], collagen [24] and gelatin [25] are among the most suitable materials for the manufacture of such hydrogels, since the presence of polar functional groups in their structure makes them soluble or swellable by water.
The primary aim of this research is to explore the potential of the cyclic carbonate-based NIPU methodology as an orthogonal tool for the efficient development of multicomponent hydrogels with enhanced rheological properties. Prior to the formation of multicomponent hydrogels, the optimal polymerization conditions, including catalyst, solvent, monomer concentration, and temperature, will be investigated for the preparation of PHU using two new bisCC with two diamines [diethylenetriamine (DETA) and/or 1,6 hexamethylenediamine (HMDA)] as monomers. Emphasis will be placed on conducting polymerization tests at lower temperatures to produce the desired materials. The identified optimal polymerization conditions will then be applied to form multicomponent hydrogels using biocompatible hydrophilic polymers, specifically PVA and gelatin derived from porcine skin. The rheological properties of the resulting semi interpenetrating polymer networks (SIPN) will be thoroughly compared to investigate the influence of composition and experimental parameters.
2. Experimental Section
2.1. Materials and General Methods
All chemicals used were from Aldrich Chemical Co (Spain). 1,2-Dithioglycerol, triethylamine (TEA), diethylenetriamine (DETA) and tris(2-aminoethyl)amine (TAEA), were stored at r.t. whilst 1-thioglycerol was kept in the fridge at 4 °C under argon atmosphere until needed. Gelatin from porcine skin (type A) and PVA [hydrolysis degree: 89%; weight average molecular weight (, g/mol): 32,000] were also purchased from Aldrich Chemical Co (Spain) and used as received.
IR spectra were recorded on a Jasco FT/IR 4200 spectrometer equipped with ATR. NMR spectra were acquired at 300 K on either a Bruker Advance AVIII-300 MHz, or a Bruker AV NEO500 MHz (CITIUS, Universidad de Sevilla). Chemical shifts (δ) are reported as parts per million (ppm) downfield from Me4Si. Mass spectra were obtained using either an IRMS Thermo Scientific Delta V Plus instrument or a Thermo Scientific Orbitrap Elite instrument (CITIUS, Universidad de Sevilla).
Gel permeation chromatography (GPC) of the samples were conducted using Waters equipment (Milford, MA, USA) provided with a refractive-index detector 2414 (thermostated at 40 °C). N,N-dimethylformamide (DMF) containing LiBr (5.8 mM solution) was the mobile phase. Sample preparation: DMF solution (HPLC grade) approx. 4 mg in 2.0 mL, filtration (0.22 μm) and duplicate injection of the stock solution. Samples (100 μL of 0.2% (w/v) solution) were injected and chromatographed with a flow of 1 mL·min−1. HR3 and HR4 Waters Styragel columns (7.8 × 300 mm) were used, linked in series, and protected with a guard column, thermostated at 60 °C. Molar mass averages and their distributions were estimated against polystyrene standards.
The thermal behavior of the polymers was examined using differential scanning calorimetry (DSC) on a TA Instruments DSC Q200, which was calibrated with indium. DSC data were obtained from samples weighing 4-6 mg, with heating rates set at 10 °C per minute under a nitrogen flow. The glass transition temperatures (Tg) were determined using rapidly melt-quenched polymer samples, with a heating rate of 20 °C min-1. The testing procedure involved initially ramping up the temperature to 170 °C at a heating rate of 10 °C per minute, followed by a 2-minute hold at 170 °C to eliminate the thermal history of the sample. Subsequently, they were cooled from the melt at a rate of 10 °C per minute to -30 °C and held at -30 °C for 2 minutes. Finally, the temperature was increased up to 170 °C at a rate of 20 °C per minute. The resulting heating curve was recorded for further analysis.
In the analysis of the multicomponent hydrogels prepared, both rheological measurements and images from field emission scanning electron microscope were conducted/recorded at the CITIUS facilities of the Universidad de Sevilla. The rheological measurements of the hydrogels were performed in a Discovery HR-3 (TA Instruments, New Castle, DE, USA) rheometer, equipped with a Peltier temperature controller, at 25 °C, using a plate-plate geometry (diameter: 40 mm and 1 mm gap). Small-amplitude oscillatory shear (SAOS) tests from 0.12 to 62 rad/s, in the linear viscoelastic regime, were carried out. The linear viscoelasticity range was performed at a frequency of 1 Hz through strain amplitude sweeps. At least two replicates were done on fresh samples. Images were recorded specifically at the Microscopy Laboratories, by means of a field emission scanning electron microscope, Zeiss EVO, at an accelerating voltage of 10 kV using secondary electrons. The samples were dried by the critical point drying method to prevent the alteration of their surface topography. This protocol is widely applied for the analysis of biological tissues and was followed according to a previous study [26].
The synthesis and characterization of small molecules and monomers are described in the Supplementary Information document. All spectra of synthesized monomers and small molecules, as well as those of polymers (proton and carbon NMR, two-dimensional homo- and heteronuclear NMR spectra, ATR-FTIR spectra, and mass spectra), and the GPC chromatograms from the 30 polymerization experiments conducted under various conditions, are accessible for consultation in idUS (Depósito de Investigación de la Universidad de Sevilla) [27].
2.2. Preparation of PHU by Means of Non-Isocyanate Polyurethane (NIPU) Methodology
The synthesis of PHU have been conducted from five-membered bis (cyclic carbonate) (bisCC) monomers (MA and ME) following the schemes recorded next (Scheme 2 and Scheme 3) under a batch of polymerization conditions summarized in Tables S1 and S2.
2.2.1. Preparation of PHU-ADETA from MA and DETA
The preparation of PHU-ADETA were conducted by the aminolysis of MA with DETA (Scheme 2) under a variety of polymerization conditions recorded (Table S1).
As an example, the general in-solution polymerization procedure for the manufacture of PHU-ADETA was as follows: to a solution of MA (90 mg, 0.164 mmol, [monomer] = 165 mmol/L) and DETA (12 μL, 0.114 mmol) in the chosen solvent [whether DMSO or 2,2,2-trifluoroethanol (TFE)] the organo-catalyst (TU, DBU, or none) was added (8.26 μmol, 10% in mol regarding MA) and the reaction stirred at the chosen temperature (25 °C or 50 °C) for 24 hours (Table S1, entries from 1 to 12).
Similarly, quasi-in-bulk polymerizations ([monomer] = 1.8 mol/L) with the same solvents, catalysts, and selected temperatures were conducted (Table S1, entries from 13 to 20). In every case, once the polymerization had taken place, the polymer was precipitated over cold tert-butyl methyl ether, (tBME), washed with additional tBME and dried under vacuum for 48 hours (from 91% to quantitative yields). The mass average molecular weights () ranged from 3,400 Da to 16,400 Da (Table S1, entries 13-20).
Spectroscopic data for PHU-ADETA: IR (ν cm-1) 3301 (O-H), 2933, 2863 (C-H), 1698 (C=O urethane), 1537 (N-H, N-C=O urethane). 1H-NMR (500 MHz, CD3OD) δ (ppm) 7.94 (s, 2H, H-6), 7.34-7,11 (m, 2H, N-H urethane), 4.92 (bs, 1H, H-2), 4.68 (t, 4H, J7,8 = 6.5 Hz, H-7), 4.07, 4.02 (2 bs, 4H, H-10), 3.86 (bs, 4H, H-4), 3.77-3.31 (m, 2H, H-9, H-1), 3.24 (bs, 8H, H-8, H-11), 2.85-2.50 (m, 8H, H-3, H-12). 13C-NMR (125 MHz, CD3OD) δ (ppm) 159.1 (C=O), 143.7 (C-5), 125.0 (C-6), 75.6 (C-2), 70.5 (C-9), 68.5 (C-10), 63.6 (C-1), 50.1 (C-7), 41.5 (C-8), 39.0 (C-11), 36.1 (C-12) 33.1 (C-3), 27.4 (C-4).
2.2.2. Preparation of PHU-EDETA and PHU-EHMDA
The preparation of PHU-EDETA and PHU-EHMDA were conducted by the aminolysis of ME with DETA and HMDA, respectively, under a variety of polymerization conditions (Scheme 3, Table S2). In all cases the concentration of ME utilized was the highest tested for MA (1.8 mol/L), and the polymerization variables were temperature (25 °C or 50 °C), solvent (DMSO and EtOH as an aprotic and a protic solvent, respectively) and the organo-catalyst used (DBU or TU). The polymerization procedure was identical to that described for PHU-ADETA, rendering the polymeric materials with very high yields (> 90% to quantitatively). for PHU-EDETA ranged from 10,300 Da to 29,600 Da (Table S2, entries 1-5) and from 13,600 Da to 34,000 Da for PHU-EHMDA (Table S2, entries 6-10).
Spectroscopic data for PHU-EDETA: IR (ν cm-1) 3299 (O-H, N-H), 2930, 2857 (C-H), 1689 (C=O urethane), 1539 (N-H, N-C=O urethane). 1H-NMR (500 MHz, DMSO-d6) δ (ppm) 7.07, 7.03 (2 bs, 2H, N-H), 4.99-4.81 (m, 3H, OH), 4.70 (bs, 1H, H-2), 3.99-3.83 (m, 2H, H-9), 3.76 (bs, 1H, H-8), 3.71-3.64 (m, 2H, H-7), 3.62-3.50 (m, H-1), 3.14-3.04 (m, 4H, H-10), 2.94-2.81 (m, 1H, H-5), 2.80-2.68 (m, 2H, H-6), 2.67-2.58 (m, 8H, H-4, H-4’, H-11), 2.01-1.54 (m, 4H, H-3, H-3’). 13C-NMR (125 MHz, DMSO-d6) δ (ppm) 156.8 (C=O), 73.7 (C-2), 68.2 (C-9), 67.7 (C-8), 63.2 (C-7), 62.7 (C-1), 48.9 (C-11), 48.1 (C-5), 40.9 (C-10), 34.4, 34.2, 31.9, 31.5 (C-3, C-3’, C-6), 28.6, 27.1, 26.9 (C-4, C-4’).
Spectroscopic data for PHU-EHMDA: IR (ν cm-1) 3301 (O-H, N-H), 2929, 2860 (C-H), 1695 (C=O urethane), 1538 (N-H, N-C=O urethane). 1H-NMR (500 MHz, DMSO-d6) δ (ppm) 7.11, 7.06 (2 bs, 2H, N-H), 4.97-4.79 (m, 2H, OH), 4.68 (bs, 1H, H-2), 3.97-3.81 (m, 2H, H-9), 3.73 (bs, 1H, H-8), 3.70-3.61 (m, 2H, H-7), 3.60-3.51 (m, 2H, H-1), 3.07-2.96 (m, 4H, H-10), 2.95-2.80 (m, 1H, H-5), 2.79-2.66 (m, 2H, H-6), 2.46-2.58 (m, 4H, H-4, H-4’), 1.95-1.52 (m, 4H, H-3, H-3’), 1.43 (bs, 4H, H-11), 1.29 (bs, 4H, H-12). 13C-NMR (125 MHz, DMSO-d6) δ (ppm) 156.7 (C=O), 73.6 (C-2), 68.1 (C-9), 67.7 (C-8), 63.2 (C-7), 62.7 (C-1), 48.2 (C-5), 40.8 (C-10), 34.5, 31.9 (C-3, C-3’), 34.3 (C-6), 29,7 (C-11), 28.5, 26.7 (C-4, C-4’), 26.3 (C-12).
2.3. One-Step Procedure for the Manufacture of SIPN-Based Hydrogels. Characterization of the New Multicomponent Hydrogels.
All multicomponent hydrogels were prepared via a single step procedure: Polymer 1 (crosslinked PHU) was in-situ synthesized within a colloidal suspension of either PVA or gelatin (Polymer 2) leading to SIPN.
In a typical procedure (preparation of IPN 4, Table 1) ME (700 mg, 1.98 mmol), DETA (192 μL, 1.78 mmol), the catalyst DBU (29 μL, 0.198 mmol), and the appropriate amount of the covalent cross-linker tris(2-aminoethyl)amine [TAEA, 19 μL, 0.13 mmol (degree of crosslinking aimed: 10%] were added to a colloidal suspension of gelatin (900 mg) in 1:1 DMSO-H2O (7 mL) and the mixture was gently stirred. Additional volume of 1:1 DMSO-H2O solution was added (enough to a final volume equal to 9 mL). The system was gently stirred at 25 °C for 8 h and later heated up to 50 °C and agitation was maintained for other additional 16 h. Polymer 1/Polymer 2 ratio was 1:1 in weight.
All the SIPN systems were prepared in a manner analogous to the aforementioned process, following the reaction conditions outlined in Table 1.
The rheological properties of the hydrogels were studied and compared with the colloidal suspensions of Polymer 2 on its own (Blanks: entries 1-4, Table 1). In these tests, the elastic and viscous moduli ( and , respectively) were obtained, together with the loss tangent [. Furthermore, the values for the plateau modulus (), and tan (δ) at 1 rad/s were selected and tabulated to improve the comparison of the different systems (Table 3). The plateau modulus, , was estimated as the value at a frequency for which the loss tangent is minimum [28]. It may be considered as a measure of the aggregation of the dispersed structural units or the density of physical entanglements, and, consequently, it is related to the strength of the microstructural network [29].
The microstructure of selected multicomponent hydrogels was investigated through images produced by a field emission scanning electron microscope on samples dried by the critical point drying method to avoid alteration of their surface topography.
3. Results and Discussion
In the present work, 10 multicomponent hydrogels with improved rheological properties have been prepared by means of a one-step process in which a crosslinked functional PHU (Polymer 1) grows within the colloidal suspension of a selected biocompatible polymer (Polymer 2: gelatin or PVA, Table 1). Polymer 1 was formed by the aminolysis reaction of firstly synthesized 5CC (MA and/or ME) with DETA or HMDA, using TAEA as a crosslinker (PHU-ADETA, PHU-EDETA and PHU-EHMDA, Scheme 2 and 3). Optimization of such PHU production was carried out to discover relevant findings on polymerization conditions that can help in the further development of the multicomponent hydrogels aimed.
3.1. Synthesis of Monomer MA and Preliminary Studies on PHU-ADETA Formation
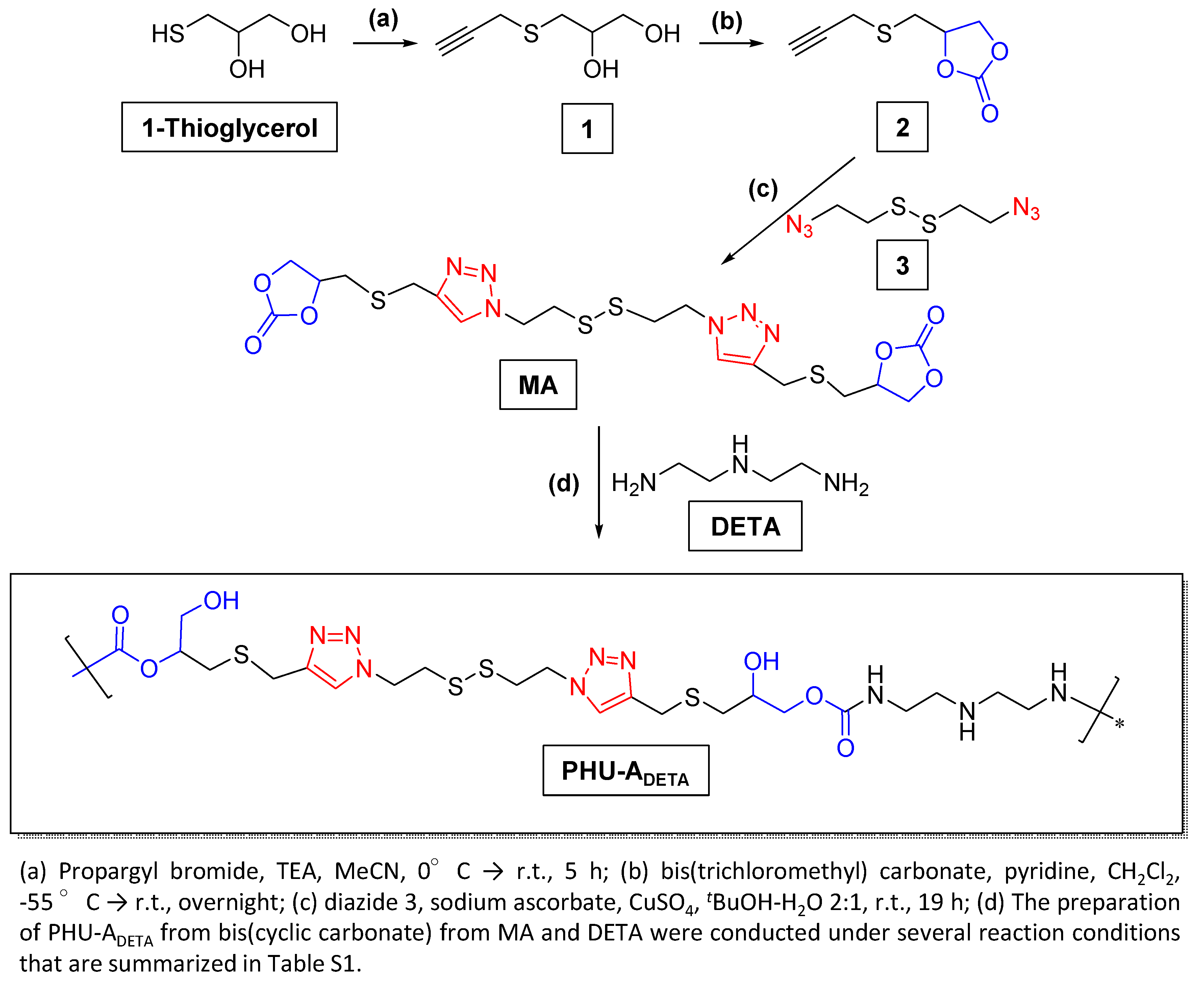
Monomer A (MA), selected for its disulfide bonds and interest in developing new materials for biomedical applications [8,9], was synthesized through a three step synthetic process from 1-thioglycerol, as outlined in Scheme 2. The nucleophilic thiol group in the starting material facilitated functionalization with a propargyl moiety (compound 1) via an SN2 reaction. The formation of 5- and 6-membered cyclic carbonates from 1,2- and 1,-3-diols, respectively, can be carried out successfully by reaction with electrophilic carbonates [30]. In our study, the synthesis of compound 2 was successfully achieved by reacting diol 1 with bis(trichloromethyl) carbonate resulting in the formation of propargyl CC 2 with excellent yields (96%). The versatile nature of the building block propargyl cyclic carbonate 2, obtained with an overall yield of 86.4% from 1-thioglycerol, expands the design possibilities for new CC monomers and cross-linkers. This facilitates the implementation of click azide-alkyne cycloaddition reactions, involving compound 2 and diazides or multiazides, whether commercially available or synthesized de novo, to produce a diverse range of compounds suitable for PHU preparation.
In this investigation, the chosen diazide (compound 3, [23]) features a disulfide linkage, making it responsive to the natural tripeptide glutathione [9,32]. The final step for the synthesis of MA followed a procedure similar to that used for preparing other click azide–alkyne adducts [26], yielding moderate yields.
To elucidate the impact of polymerization conditions — specifically temperature, solvent, monomer concentration and catalyst— on PHU formation, we conducted a comparative study on the synthesis of PHU-ADETA. This involved the aminolysis of monomer A with DETA, as outlined in Scheme 2, under various experimental conditions (Table S1). The objective was to explore lower temperatures and eliminate the need for metal catalysts in the production of these novel materials.
PHU-ADETA was obtained through organo-catalyzed experiments and characterized by NMR and FTIR spectroscopies. Figure 2 presents the 1H NMR spectrum of the synthesized PHU, with its structure conclusively confirmed through mono and bi-dimensional NMR experiments (1H NMR, 13C NMR, COSY and HSQC experiments). Detailed peak assignments are documented in the Experimental Section. Notably, the spectrum not only reveals the presence of urethane bonds, formed by the reaction between the cyclic carbonate and amine groups from DETA (the carbon of the urethane bonds appears at δ 159.1 ppm in the 13C NMR spectrum), but also the disappearance of peaks attributable to the CH protons from the cyclic carbonate groups (at δ 5.00 ppm). Additionally, peaks corresponding to protons from the DETA monomer in the polymer backbone can be identified at δ 3.24 and 2.85-2.50 ppm.
To verify the formation of PHU-ADETA, attenuated total reflection (ATR)—Fourier Transform (FT) IR spectroscopy was employed, providing complementary evidence to NMR experiments. The ATR-FTIR Spectra of MA and PHU-ADETA are recorded in Figure 3.
The polymer spectrum confirms the opening of the cyclic carbonate ring through several indicators: firstly, the broad band centered at νmax 3301 cm−1, primarily attributed to the formation of primary and secondary hydroxyl groups, overlaps with the stretch bands corresponding to N-H bonds from DETA secondary amines and the newly generated urethane groups. Secondly, the presence of urethane linkages is substantiated by characteristic bands at νmax 1698 cm−1 (associated with the stretching vibration of the C=O) and 1537 cm−1 [related to the stretching vibration of the N-C(=O) bond and the N-H flexion band]. Thirdly, the near disappearance of the strong band at νmax 1790 cm−1, indicative of the vibration of C=O bonds from the cyclic carbonate monomer, conclusively validates the procedure. Notably, the band corresponding to the stretching vibration of C-H bonds from the triazole rings (≈ 3135 cm-1) is evident in both spectra.
It has been documented the formation of urea by products when methanol serves as the solvent, as evidenced by the stretching of the urea C=O group in the range of νmax 1665-1630 cm-1 [18]. The authors attributed this phenomenon to the protic nature of the solvent. Markedly, no such occurrence was observed in any of the tested polymerization conditions, whether employing the protic solvent TFE or the aprotic solvent DMSO (refer to Figure 3, PHU ADETA-13; polymerization conducted in TFE, Table S1). In summary, FTIR-based findings align with conclusions drawn from NMR spectra, confirming the efficient production of linear PHU-ADETA under the specified conditions.
3.2. Optimization of Polymerization Conditions for PHU-ADETA Formation
The formation of CC-based NIPU often results in low molecular weight polymers due to the limited reactivity of CC with amino groups during PHU formation [19]. To improve CC aminolysis, organo-catalysts such as N’-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexylthiourea (TU), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) have been employed, with TU [34] and DBU [22] exhibiting optimal results. Consequently, DBU and TU were chosen as catalysts based on their favorable performance in this polymerization reaction. Alternatively, the polymerization process typically occurs in bulk or in the presence of aprotic polar solvents such as dimethylsulfoxide (DMSO) [35], N,N-dimethylformamide (DMF) [17], or N,N’-dimethylacetamide (DMAc) [36]. DMSO has demonstrated excellence as a solvent for NIPU formation [35,37]; therefore, its inclusion in the experimental batches is warranted. Moreover, considering that strong hydrogen bonds between PHU chains are known to impede an increase in polymer molar mass [38], we advocate for the utilization of a protic solvent in this study, to potentially improve PHU formation by interfering with hydrogen bonding among polymer chains. While there are limited precedents for the use of volatile protic solvents in this type of polymerization [18], exploring their potential as an alternative is justified. TFE was chosen as the protic solvent for several trials due to its high solubility of MA and DETA, which may positively correlate with higher NIPU molecular weights [19]. While high temperatures are often employed [16], we aimed for milder conditions and ensured that polymerization temperatures did not exceed 50 °C. Interesting precedents in polymerization procedures performed at room temperature have been published [39,40].
To enhance polymerization efficiency while maintaining mild reaction conditions, we systematically assessed twenty different conditions (Table S1) Samples were designated based on their composition as ADETA (monomer A and monomer DETA), with a numerical identifier distinguishing between tested conditions (from ADETA-1 to ADETA-20). The parameters explored during the polymerization encompassed: (a) monomer concentration, spanning from in-solution polymerizations (entries 1-12, Table S1) to near-bulk polymerizations (entries 13-20, Table S1); (b) temperature, offering choices of 25 °C or 50 °C; (c) solvent selection, featuring a protic polar solvent (TFE) or an aprotic polar solvent (DMSO); and (d) incorporation of an organo-catalyst, such as TU, DBU, or no catalyst. These variations were systematically investigated to pinpoint the optimal set of conditions for the polymerization process.
The principal aim was to evaluate the viability of the polymerization process without the involvement of a catalyst. FTIR-based kinetic studies demonstrated the non-occurrence of aminolysis of CC at room temperature, as indicated by the absence of the urethane bond-associated band at νmax 1700 cm-1 (entries 1 and 3, Table S1). In contrast, at 50 °C (entries 2 and 4, Table S1), the consumption of CC functional groups and the formation of carbamate bonds were observed even in the absence of a catalyst, albeit resulting in low molecular weights. This observation is consistent with prior research indicating that aminolysis of CC occurs at higher temperatures, either in-bulk or in-solution, even when no catalyst is employed [17,18,41]. However, achieving higher molecular weights under mild conditions necessitates the inclusion of an organo-catalyst in the formulation.
Moderate molecular weights for PHU synthesis were attainable at 25 °C through quasi-bulk or in-solution polymerizations, contingent upon the inclusion of an organo-catalyst [() of 16,400 Da and 14,300 Da for quasi-bulk (Table S1, entry 13) and in solution (Table S1, entry 11), respectively]. Notably, two out of the twenty trials conducted yielding the highest molecular weights were carried out under near-bulk polymerizations conditions [entries 13 and 20, Table S1; () of 16,400 Da and 14,700 Da]. Under such conditions, the optimal combinations were TU-TFE and DBU-DMSO, respectively. In the case of in-solution trials, DBU emerged as the most advantageous catalyst for any solvent-catalyst combination tested.
3.3. Synthesis of Bis(Cyclic Carbonate) Monomer ME and PHU-EDETA and PHU-EHMDA
The more polar and flexible monomer, ME, was prepared through a click thiol-ene reaction (Scheme 3) in high yields (89 %) employing a procedure previously established for other thiol-ene combinations [20]. For the formation of ME-based PHU, two amine-based monomers, DETA and HMDA, were employed. Similarly to ADETA polymers, samples were designated based on their composition (EDETA and EHMDA, respectively) with a numerical identifier distinguishing between tested conditions (EDETA-1 to EDETA-5; EHMDA-1 to EHMDA-5).
Building upon the prior findings in MA-based PHU synthesis, ME polymerization was conducted under quasi-in-bulk conditions, chosen over in-solution conditions, resulting in elevated molecular weights compared to those obtained for MA-based PHU [() of 34,100 Da and 29,600 Da for PHU-EHMDA (Table S2, entry 7) and PHU-EDETA (Table S2, entry 4), respectively]. This outcome can be ascribed not only to ME’s improved solubility in various solvents but also to its flexibility, characteristics that can potentially enhance the interaction and subsequent reaction between the CC functional groups and the primary amino groups located at the ends of the growing polymer chains. Similar to the trials conducted with MA, the most favorable co catalyst-solvent combinations were TU-protic (Table S2, entry 4) and DBU-aprotic (Table S2, entry 7). However, the highest values were achieved when the reaction mixture was maintained at 50 °C although satisfactory results were also obtained at room temperature (Table S2, entry 6, 20,300 Da).
The polymers’ thermal behavior was assessed through differential scanning calorimetry (DSC). All examined polymers were found to be amorphous (Table 2). Typically, aliphatic NIPU polymers exhibit a Tg below zero, attributed to their relatively low molecular weight [15] and the low rigidity introduced by a hydrocarbon chain (length of 4, 6, 8 carbon atoms) between the urethane groups (Tg from -18 °C to -26 °C) [14]. However, the Tg values observed in this study for the PHU synthesized were relatively high compared to reported values for similar PHU, likely due to the comparatively higher of the obtained materials. Specifically, the Tg of PHU-ADETA (ADETA-13, Table 2) was higher than its counterpart (EDETA-4, Table 2), both remaining below zero degrees Celsius. This distinction may be attributed to the presence of triazole groups, contributing to the stiffness of the final materials. Notably, the Tg of EHMDA-2 PHU was significantly above zero degrees Celsius (Tg 14.2 °C).
3.4. One-Step Procedure for the Manufacture of PHU-Based Multicomponent Hydrogels
To assess the efficacy of the described NIPU methodology for producing hybrid materials, we fabricated ten multicomponent hydrogels based on interpenetrated networks (SIPN) formation [Figure 4 and Table 1 (entries 5-14; Table 1 displayed in Experimental Section)]. Each procedure involved the in-situ generation of a cross-linked PHU within a colloidal suspension of a biocompatible polymer (either PVA or gelatin type A) to reinforce its rheological properties (Figure 4). Additionally, we aimed to demonstrate the orthogonal nature of the NIPU formation process by reacting di(multi)CC with di(multi)amines within various polymer suspensions. To prepare crosslinked Polymer 1 (PHU) in PVA or gelatin suspensions, the two bisCC monomers (ME or MA) and two diamines (DETA or HMDA) were employed, along with the crosslinking agent tris(2-aminoethyl)amine (TAEA). As monomers MA and ME are not soluble in water, a miscible cosolvent was employed during hydrogel formation [DMSO-H2O (1:1) or EtOH-H2O (1:1)].
The manufacture of the multicomponent hydrogels in a single step was confirmed through FTIR spectroscopy, as depicted in Figure 5. The presence of Polymer 1 (PHU) in the colloidal suspension of Polymer 2 (PVA or gelatin) was clearly observed. Interestingly, the absence of the band at νmax 1778 cm-1, which corresponds to the stretching band of C=O groups from CC (ME, Figure 5a), in the built SIPN indicates the aminolysis of these functional groups during the PHU formation process (Figure 5c). Furthermore, the IPN1 FTIR spectrum displayed two distinct bands at νmax 1645 cm-1 and 1535 cm-1 (Figure 5c). They can be attributed to the production of crosslinked PHU network intertwined with PVA chains. Specifically, the band at 1645 cm-1 appears to be broadened due to its overlap with the band associated with C=O from the acetate groups present in PVA (νmax 1732 cm-1, Figures 5b and 5c). These findings provide compelling evidence of successful fabrication of SIPN. The distinctive spectral features observed in the FTIR analysis confirm the generation of new chemical bonds and underscore the effectiveness of the SIPN formation process.
3.5. Characterization of Multicomponent Hydrogels
The success of this single-step procedure was further validated by studying the rheological properties of the multicomponent hydrogels and comparing them with the colloidal suspensions of Polymer 2 (Table 1). In these tests, the elastic and viscous moduli ( and , respectively) were obtained, together with the loss tangent [. Furthermore, the values for the plateau modulus (), and tan (δ) at 1 rad/s were selected and tabulated to improve the comparison of the different systems (Table 1). The plateau modulus, , was estimated as the value at a frequency for which the loss tangent is minimum [28]. It may be considered as a measure of the aggregation of the dispersed structural units or the density of physical entanglements, and, consequently, it is related to the strength of the microstructural network [29].
The rheological properties of these hydrogels, as described in detail below (Table 1), were highly dependent on factors such as the choice of monomers, the polymer scaffold, and the cosolvent employed. Interestingly, substituting DMSO with ethanol in the solvent mixture had a dual effect. Firstly, when comparing the blanks in both solvents (entries 1 and 2 for gelatin; entries 3 and 4 for PVA), it was observed that the plateau modulus increased for both gelatin and PVA suspensions, with a more significant impact on the former. However, when SIPN were prepared in ethanol-water mixtures, the rheological properties were slightly enhanced for gelatin-based hydrogels (IPN3), but significantly deteriorated for PVA-based hydrogels (IPN6). Therefore, the DMSO-water mixture was preferentially used in the preparation of the remaining SIPN.
Regarding the PVA-based SIPN (in DMSO-H2O), all the systems, including the blank (PVA, Polymer 2, entry 3), demonstrated significantly higher values of the storage modulus () compared to the loss modulus () across the entire frequency range studied (Table 1, Figure 6). This behavior is consistent with that of solid-like gels [42] where a distinct “plateau region” in the mechanical spectrum was clearly observed in all cases.
It was also verified that the diamine monomer (DETA or HMDA) did not significantly affect the evolution of the SAOS functions with frequency. However, when bisCC MA was used a tendency to a crossover between and was noticed at high frequencies (Figure 6).
Moreover, ME exhibited remarkable potential as the preferred bisCC in producing optimized sol-like hydrogels, as evidenced by the successful formation of IPN4 and IPN5. Additionally, a comparative analysis of both SIPN led to the conclusion that DETA (IPN4) was the most suitable diamine for enhancing the SAOS functions of the blank. Consequently, DETA was employed in the formation of IPN8 in conjunction with MA. However, it should be noted that the utilization of MA in this particular case resulted in a significant reduction of the viscoelastic functions and plateau modulus.
The hydrogels IPN4 and IPN5 demonstrated remarkably low loss tangent (0.09 and 0.14, respectively), indicating a higher degree of relative elasticity. This finding further supports the notion that these hydrogels achieved an intertwined micro-structured system, resulting in improved mechanical properties compared to the blank (colloidal suspension of PVA). It was hypothesized that this enhancement occurred due not only to the crosslinking of the PHU chains through chemical crosslinking via TAEA but also to the formation of stabilizing hydrogen bonds between Polymer 1 (PHU) and Polymer 2 (PVA).
The incorporation of monomer MA (IPN8) resulted in slight improvements in the rheological properties of the PVA-based blank. However, these improvements were not as significant as when ME was chosen as the bisCC monomer in the formation of Polymer 1. This difference can be attributed to the higher stiffness of MA, which may have an impact on entanglements with PVA chains. Additionally, it is postulated that the lower density of free hydroxyl groups in MA-based PHU, compared to ME-PHU, substantially reduced the number of stabilizing hydrogen bonds.
To explore the correlation between monomer composition and rheological properties, two additional SIPN were prepared with bisCC ME/MA ratios of 80:20 and 20:80 (co-IPN1 and co-IPN2, respectively; Figure 7). In both cases, the elastic component of the systems exhibited a significant increase of over two orders of magnitude compared to the blank and showed a slight frequency-dependent increase. Furthermore, displayed a minimum and the values were even higher than those observed for IPN4 (Table 1). Notably, a tendency to crossover at low frequencies was observed. Moreover, co-IPN1 and co-IPN2 demonstrated an increase of tan(δ) at a frequency of 1 rad/s (from 0.21 for the blank to 0.32 and 0.35 for co-IPN1 and co-IPN2, respectively), indicating a lower relative elasticity. These unexpected findings will be compared with ongoing studies involving various systems, further contributing to the understanding of these phenomena.
Figure 8 depicts the trends observed in studies where Polymer 2 consisted of gelatin (in DMSO-H2O, 1:1). In these cases, IPN7 (utilizing bisCC monomer MA) exhibited higher values compared to the blank (Table 1), making it the most promising in terms of rheological performance when compared to Polymer 2 alone (gelatin). This improvement can be attributed to the hydrophobic interactions between the heterocycles present in MA-based PHU and the hydrophobic amino acid residues present in gelatin. In gelatin-based SIPN, the formation of hydrogen bonds between Polymer 1 and Polymer 2 was not as significant as observed in cases where the biocompatible polymer was PVA.
On the other hand, a typical sol–gel transition response, with a crossover occurring at medium frequencies, was displayed by IPN1. As widely acknowledged [43], the gel strength of dispersed biopolymer systems —from dilute solutions to fully crosslinked gels— can be quantified from SAOS measurements. Firstly, the slopes of and versus frequency plots provide insights into the dependence and on frequency. Secondly, the relative values of the viscoelastic functions, particularly the loss tangent, enable the comparison of relative elasticity. Thus, and regarding the loss tangent at 1 rad/s for IPN1, a lower relative elasticity was observed which was conclusive of a low level of crosslinking and physical entanglements.
The morphology of the IPN was studied by scanning electron microscopy (SEM). Before SEM observations, the samples underwent critical point drying (CPD) to prevent alteration of their surface topography [26]. The CPD technique is suitable for maintaining the microstructure of hydrogels due to its gentle and controlled drying process. By avoiding the abrupt phase change associated with other drying methods, such as air drying or freeze drying, CPD minimizes the formation of artifacts or structural distortions that could alter the hydrogel’s microstructure.
Regarding multicomponent hydrogels formed with monomer MA, PVA-based IPN displayed sponge-like microstructure as evident from the SEM images of IPN8 shown in Figure 9a. Conversely, when SEM images of gelatin-based IPN were examined, a non-porous structure was observed as seen in the SEM images of IPN7 shown in Figure 9b. Similar findings were observed for IPN2 (Figure 10c). It should be noted that these SEM photos of bulk samples may not fully capture the true internal microstructure of the hydrogel as pointed out by other researchers in the case of gelatin-based matrices [44]. Notably, the 3D networks of ME-based multicomponent hydrogels were clearly visible when Polymer 2 was PVA, as shown in Figures 10a and 10b.
4. Conclusions
This study introduces an efficient strategy for synthesizing single-step process to produce intertwined 3D multicomponent hydrogels (SIPN) with enhanced rheological properties, showing promise for biomedical applications. The method demonstrates exceptional orthogonality, with the functional groups in Polymer 2 not interfering with Polymer 1 formation. Optimizing PHU formation identifies key variables, with the highest molecular weights being achieved under near-bulk polymerization conditions using TU-protic and DBU-aprotic as catalyst-solvent combinations. ME-based PHU displays higher molecular weights than MA-based PHU (34,100 Da and 16,400 Da, respectively). Applying the optimized methodology to prepare ten multicomponent hydrogels using PVA or gelatin as the polymer scaffold shows superior rheological properties in PVA-based hydrogels, exhibiting solid-like gel behavior. The incorporation of bisCC ME enhances mechanical properties and elasticity (with loss tangent values of 0.09 and 0.14). SIPN hydrogels with bisCC ME/MA ratios of 80:20 and 20:80 demonstrate significantly increased elastic components and minimal loss components. SEM images unveil distinct microstructures, including a sponge-like pattern in certain PVA-based hydrogels when MA was chosen, indicating the formation of highly superporous interpenetrated materials. In summary, this innovative approach presents a versatile methodology for obtaining advanced hydrogel-based systems with potential applications in various biomedical fields.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Ana I. Carbajo-Gordillo: Methodology, Validation, Investigation, Data Curation, Writing - Original Draft, visualization; Elena Benito: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Resources, Writing - Original Draft, Writing - Review & Editing, Visualization, Supervision; Elsa Galbis: Validation, Investigation; Roberto Grosso: Validation, Investigation; Nieves Iglesias: Validation, Investigation; M.-Gracia García-Martín: Conceptualization, Methodology, Formal analysis, Resources, Writing - Review & Editing, Supervision, Funding acquisition; Concepción Valencia: Validation, Formal analysis, Writing - Original Draft, visualization; Ricardo Lucas: Investigation; M.-Violante de-Paz: Conceptualization, Methodology, Validation, Formal analysis, Writing - Original Draft, Writing - Review & Editing, visualization, Supervision, Project administration, Funding acquisition.
Funding
The authors received with gratitude financial support from Ministerio de Ciencia e Innovación - Agencia Estatal de Investigación (MCIN/AEI) Grant Number PID2020-115916GB-I00, and Fondo Europeo de Desarrollo Regional (FEDER), and La Consejería de Economía y Conocimiento (Junta de Andalucía), Grant Number US-1380587.
Data Availability Statement
Data will be made available on request.
Acknowledgments
Authors want to acknowledge CITIUS for granting access to and their assistance with microscopy, NMR, functional characterization, biology and mass spectrometry services.
References
- Lau, H.K.; Kiick, K.L. Opportunities for multicomponent hybrid hydrogels in biomedical applications. Biomacromolecules 2015, 16, 28–42. [CrossRef]
- Jia, X.; Kiick, K.L. Hybrid multicomponent hydrogels for tissue engineering. Macromolecular Bioscience 2009, 9, 140–156. [CrossRef]
- Derkus, B.; Okesola, B.O.; Barrett, D.W.; D’Este, M.; Chowdhury, T.T.; Eglin, D.; Mata, A. Multicomponent hydrogels for the formation of vascularized bone-like constructs in vitro. Acta Biomaterialia 2020, 109, 82–94. [CrossRef]
- Tanaka, W.; Shigemitsu, H.; Fujisaku, T.; Kubota, R.; Minami, S.; Urayama, K.; Hamachi, I. Post-assembly Fabrication of a Functional Multicomponent Supramolecular Hydrogel Based on a Self-Sorting Double Network. Journal of the American Chemical Society 2019, 141, 4997–5004. [CrossRef]
- Galbis, J.A.; García-Martín, M. de G.; de Paz, M.V.; Galbis, E. Synthetic Polymers from Sugar-Based Monomers. Chemical Reviews 2016, 116, 1600–1636. [CrossRef]
- Galbis, J.A.; García-Martín, M.G.; De-Paz, M.-V.; Galbis, E. Bio-based Polyurethanes from Carbohydrate Monomers. In Aspects of Polyurethanes; Yilmaz, F., Ed.; Intech: Rijeka, Croatia, 2017; pp. 155–192 ISBN 978-953-51-3545-6.
- Santerre, J.P.; Woodhouse, K.; Laroche, G.; Labow, R.S. Understanding the biodegradation of polyurethanes: from classical implants to tissue engineering materials. Biomaterials 2005, 26, 7457–70. [CrossRef]
- Ferris, C.; de Paz, M.V.; Aguilar-de-Leyva, A.; Caraballo, I.; Galbis, J.A. Reduction-sensitive functionalized copolyurethanes for biomedical applications. Polymer Chemistry 2014, 5, 2370–2381. [CrossRef]
- Benito, E.; Romero-Azogil, L.; Galbis, E.; De-Paz, M.V.; García-Martín, M.G. Structurally simple redox polymersomes for doxorubicin delivery. European Polymer Journal 2020, 137, 109952 (1–11). [CrossRef]
- De Paz, M.V.; Marin, R.; Zamora, F.; Hakkou, K.; Alla, A.; Galbis, J.A.; Munoz-Guerra, S. Linear polyurethanes derived from alditols and diisocyanates. Journal of Polymer Science, Part A: Polymer Chemistry 2007, 45, 4109–4117. [CrossRef]
- Mehta, R.; Kumar, V.; Bhunia, H.; Upadhyay, S.N. Synthesis of Poly(Lactic Acid): A Review. Journal of Macromolecular Science, Part C 2005, 45, 325–349. [CrossRef]
- Robert, J.L.; Aubrecht, K.B. Ring-Opening Polymerization of Lactide To Form a Biodegradable Polymer. Journal of Chemical Education 2008, 85, 258. [CrossRef]
- Valette, V.; Kébir, N.; Tiavarison, F.B.; Burel, F.; Lecamp, L. Preparation of flexible biobased non-isocyanate polyurethane (NIPU) foams using the transurethanization approach. Reactive and Functional Polymers 2022, 181. [CrossRef]
- Wołosz, D.; Parzuchowski, P.G.; Świderska, A. Synthesis and characterization of the non-isocyanate poly(carbonate-urethane)s obtained via polycondensation route. European Polymer Journal 2021, 155. [CrossRef]
- He, X.; Xu, X.; Wan, Q.; Bo, G.; Yan, Y. Solvent- and catalyst-free synthesis, hybridization and characterization of biobased nonisocyanate polyurethane (NIPU). Polymers 2019, 11. [CrossRef]
- Monie, F.; Grignard, B.; Thomassin, J.M.; Mereau, R.; Tassaing, T.; Jerome, C.; Detrembleur, C. Chemo- and Regioselective Additions of Nucleophiles to Cyclic Carbonates for the Preparation of Self-Blowing Non-Isocyanate Polyurethane Foams. Angewandte Chemie - International Edition 2020, 59, 17033–17041. [CrossRef]
- Mao, H.I.; Chen, C.W.; Yan, H.C.; Rwei, S.P. Synthesis and characteristics of nonisocyanate polyurethane composed of bio-based dimer diamine for supercritical CO2 foaming applications. Journal of Applied Polymer Science 2022, 139. [CrossRef]
- Ke, J.; Li, X.; Jiang, S.; Liang, C.; Wang, J.; Kang, M.; Li, Q.; Zhao, Y. Promising approaches to improve the performances of hybrid non-isocyanate polyurethane. Polymer International 2019, 68, 651–660. [CrossRef]
- Cornille, A.; Auvergne, R.; Figovsky, O.; Boutevin, B.; Caillol, S. A perspective approach to sustainable routes for non-isocyanate polyurethanes. European Polymer Journal 2017, 87, 535–552. [CrossRef]
- Grosso, R.; Benito, E.; Carbajo-Gordillo, A.I.; García-Martín, M.G.; Perez-Puyana, V.; Sánchez-Cid, P.; De-Paz, M.-V. Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept. International Journal of Molecular Sciences 2023, 24, 2281 (1–23). [CrossRef]
- Matricardi, P.; Di Meo, C.; Coviello, T.; Hennink, W.E.; Alhaique, F. Interpenetrating polymer networks polysaccharide hydrogels for drug delivery and tissue engineering. Advanced Drug Delivery Reviews 2013, 65, 1172–1187. [CrossRef]
- Sánchez-Cid, P.; Romero, A.; Díaz, M.J.; De-Paz, M. V.; Perez-Puyana, V. Chitosan-based hydrogels obtained via photoinitiated click polymer IPN reaction. Journal of Molecular Liquids 2023, 379, 121735 (1–11). [CrossRef]
- Heo, J.Y.; Noh, J.H.; Park, S.H.; Ji, Y.B.; Ju, H.J.; Kim, D.Y.; Lee, B.; Kim, M.S. An injectable click-crosslinked hydrogel that prolongs dexamethasone release from dexamethasone-loaded microspheres. Pharmaceutics 2019, 11, 438. [CrossRef]
- León-Campos, M.I.; Claudio-Rizo, J.A.; Rodriguez-Fuentes, N.; Cabrera-Munguía, D.A.; Becerra-Rodriguez, J.J.; Herrera-Guerrero, A.; Soriano-Corral, F. Biocompatible interpenetrating polymeric networks in hydrogel state comprised from jellyfish collagen and polyurethane. Journal of Polymer Research 2021, 28, 291. [CrossRef]
- Mushtaq, F.; Raza, Z.A.; Batool, S.R.; Zahid, M.; Onder, O.C.; Rafique, A.; Nazeer, M.A. Preparation, properties, and applications of gelatin-based hydrogels (GHs) in the environmental, technological, and biomedical sectors. International Journal of Biological Macromolecules 2022, 218, 601–633. [CrossRef]
- Bray D. Critical Point Drying of Biological Specimens for Scanning Electron Microscopy. In: Williams JR, Clifford AA, editors. Supercritical Fluid Methods and Protocols. Methods In Biotechnology. Totowa, New Jersey: Humana Press; 2000. p. 235-43. [CrossRef]
- Carbajo-Gordillo, A.I.; Benito, E.; Galbis, E.; Grosso, R.; Iglesias, N.; García-Martín, M.-G.; De-Paz, M.-V. Spectra and GPC Data for Polyhydroxyurethanes Formation from Bis(cyclic carbonate) Monomers in Multicomponent Semi-IPN Hydrogels Fabrication [Dataset] 2024, 1–90. [CrossRef]
- Liu, C.; He, J.; Ruymbeke, E. van; Keunings, R.; Bailly, C. Evaluation of different methods for the determination of the plateau modulus and the entanglement molecular weight. Polymer 2006, 47, 4461–4479. [CrossRef]
- Baumgaertel, M.; Winter, H.H. Interrelation between continious and discrete time spectra. Journal of Non-Newtonian Fluid Mechanics 1992, 44, 15–36. [CrossRef]
- Besse, V.; Foyer, G.; Auvergne, R.; Caillol, S.; Boutevin, B.; Burk, R.M.; Roof, M.B. A safe and efficient method for conversion of 1,2- and 1,3-diols to cyclic carbonates utilizing triphosgene. Tetrahedron Letters 1993, 51, 3284–3296. [CrossRef]
- Wang, Y.; Zhang, R.; Xu, N.; Du, F.S.; Wang, Y.L.; Tan, Y.X.; Ji, S.P.; Liang, D.H.; Li, Z.C. Reduction-degradable linear cationic polymers as gene carriers prepared by Cu(I)-catalyzed azide-alkyne cycloaddition. Biomacromolecules 2011, 12, 66–74. [CrossRef]
- de Paz, M.V.; Zamora, F.; Begines, B.; Ferris, C.; Galbis, J.A. Glutathione-Mediated Biodegradable Polyurethanes Derived from L -Arabinitol. Biomacromolecules 2010, 11, 269–276. [CrossRef]
- Martínez-Bailén, M.; Galbis, E.; Carmona, A.T.; De-Paz, M.-V.; Robina, I. Preparation of water-soluble glycopolymers derived from five-membered iminosugars. European Polymer Journal 2019, 119, 213–221. [CrossRef]
- Blain, M.; Yau, H.; Jean-Gérard, L.; Auvergne, R.; Benazet, D.; Schreiner, P.R.; Caillol, S.; Andrioletti, B. Urea-and thiourea-catalyzed aminolysis of carbonates. ChemSusChem 2016, 9, 2269–2272. [CrossRef]
- Mhd. Haniffa, M.A.C.; Munawar, K.; Ching, Y.C.; Illias, H.A.; Chuah, C.H. Bio-based Poly(hydroxy urethane)s: Synthesis and Pre/Post-Functionalization. Chemistry – An Asian Journal 2021, 16, 1281–1297. [CrossRef]
- Blattmann, H.; Fleischer, M.; Bähr, M.; Mülhaupt, R. Isocyanate- and phosgene-free routes to polyfunctional cyclic carbonates and green polyurethanes by fixation of carbon dioxide. Macromolecular Rapid Communications 2014, 35, 1238–1254. [CrossRef]
- Schmidt, S.; Gatti, F.J.; Luitz, M.; Ritter, B.S.; Bruchmann, B.; Mülhaupt, R. Erythritol dicarbonate as intermediate for solvent-and isocyanate-free tailoring of bio-based polyhydroxyurethane thermoplastics and thermoplastic elastomers. Macromolecules 2017, 50, 2296–2303. [CrossRef]
- Blain, M.; Cornille, A.; Boutevin, B.; Auvergne, R.; Benazet, D.; Andrioletti, B.; Caillol, S. Hydrogen bonds prevent obtaining high molar mass PHUs. Journal of Applied Polymer Science 2017, 134, 44958. [CrossRef]
- Cornille, A.; Blain, M.; Auvergne, R.; Andrioletti, B.; Boutevin, B.; Caillol, S. A study of cyclic carbonate aminolysis at room temperature: effect of cyclic carbonate structures and solvents on polyhydroxyurethane synthesis. Polymer Chemistry 2017, 8, 592–604. [CrossRef]
- Cornille, A.; Guillet, C.; Benyahya, S.; Negrell, C.; Boutevin, B.; Caillol, S. Room temperature flexible isocyanate-free polyurethane foams. European Polymer Journal 2016, 84, 873–888. [CrossRef]
- Guzmán Agudelo, A.F.; Pérez-Sena, W.Y.; Kebir, N.; Salmi, T.; Ríos, L.A.; Leveneur, S. Influence of steric effects on the kinetics of cyclic-carbonate vegetable oils aminolysis. Chemical Engineering Science 2020, 228. [CrossRef]
- Almdal, K.; Dyre, J.; Hvidt, S.; Kramer, O. Towards a phenomenological definition of the term “gel.” Polymer Gels and Networks 1993, 1, 5–17. [CrossRef]
- Lu, L.; Liu, X.; Tong, Z. Critical exponents for sol-gel transition in aqueous alginate solutions induced by cupric cations. Carbohydrate Polymers 2006, 65, 544–551. [CrossRef]
- Sachlos, E.; Wahl, D.A.; Triffitt, J.T.; Czernuszka, J.T. The impact of critical point drying with liquid carbon dioxide on collagen-hydroxyapatite composite scaffolds. Acta Biomaterialia 2008, 4, 1322–1331. [CrossRef]
Figure 1.
Main routes for the synthesis of polyurethanes.
Scheme 1.
Synthesis of polyhydroxyurethanes (PHU) from bis(cyclic carbonate)s and diamines.
Scheme 2.
Synthetic scheme for the preparation of MA and PHU-ADETA.
Scheme 3.
Synthetic scheme for the preparation of ME and PHU-EDETA and PHU-EHMDA.
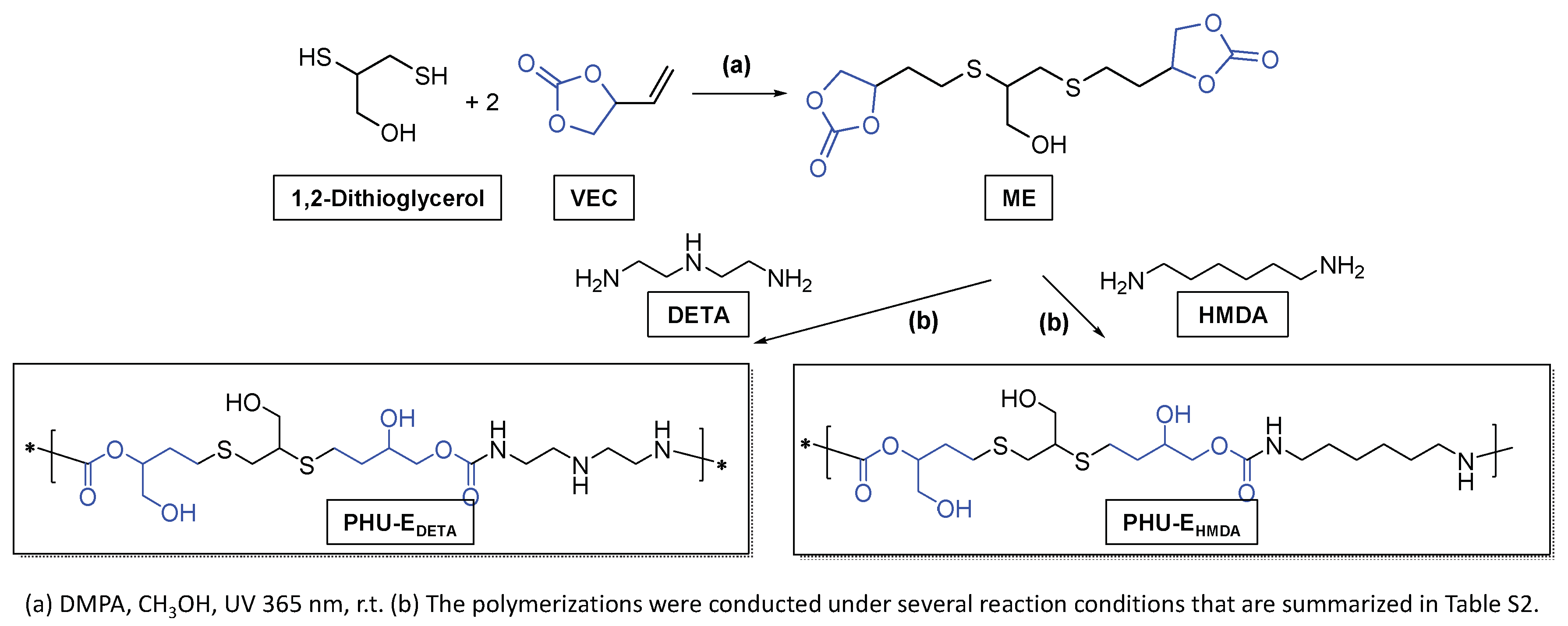
Figure 2.
1H NMR of PHU-ADETA [500 MHz, CD3OD, (entry 15, Table S1)].
Figure 2.
1H NMR of PHU-ADETA [500 MHz, CD3OD, (entry 15, Table S1)].
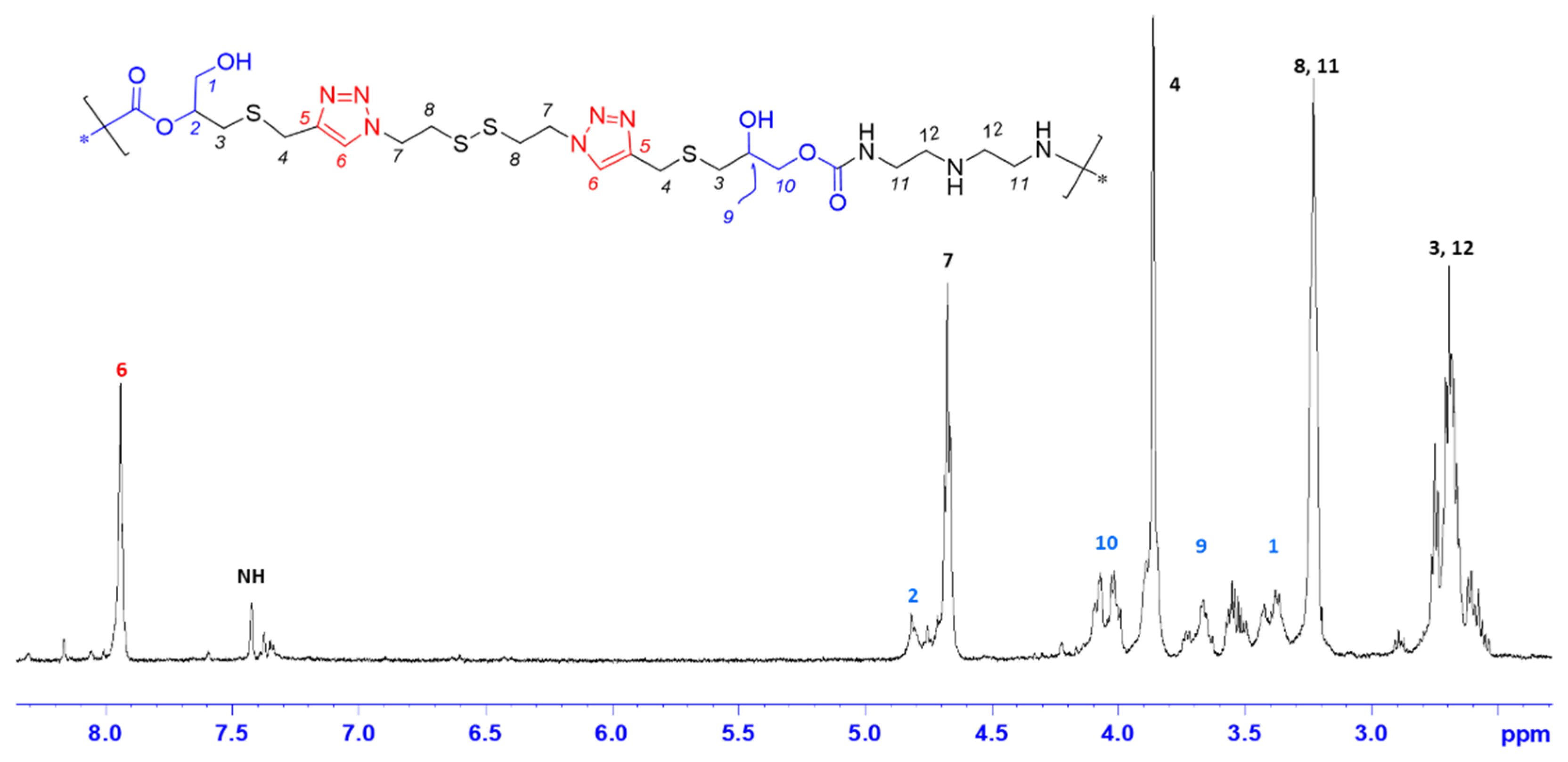
Figure 3.
FT-IR spectra of MA (green) and PHU-ADETA (black) (Table S1, entry 13).
Figure 3.
FT-IR spectra of MA (green) and PHU-ADETA (black) (Table S1, entry 13).
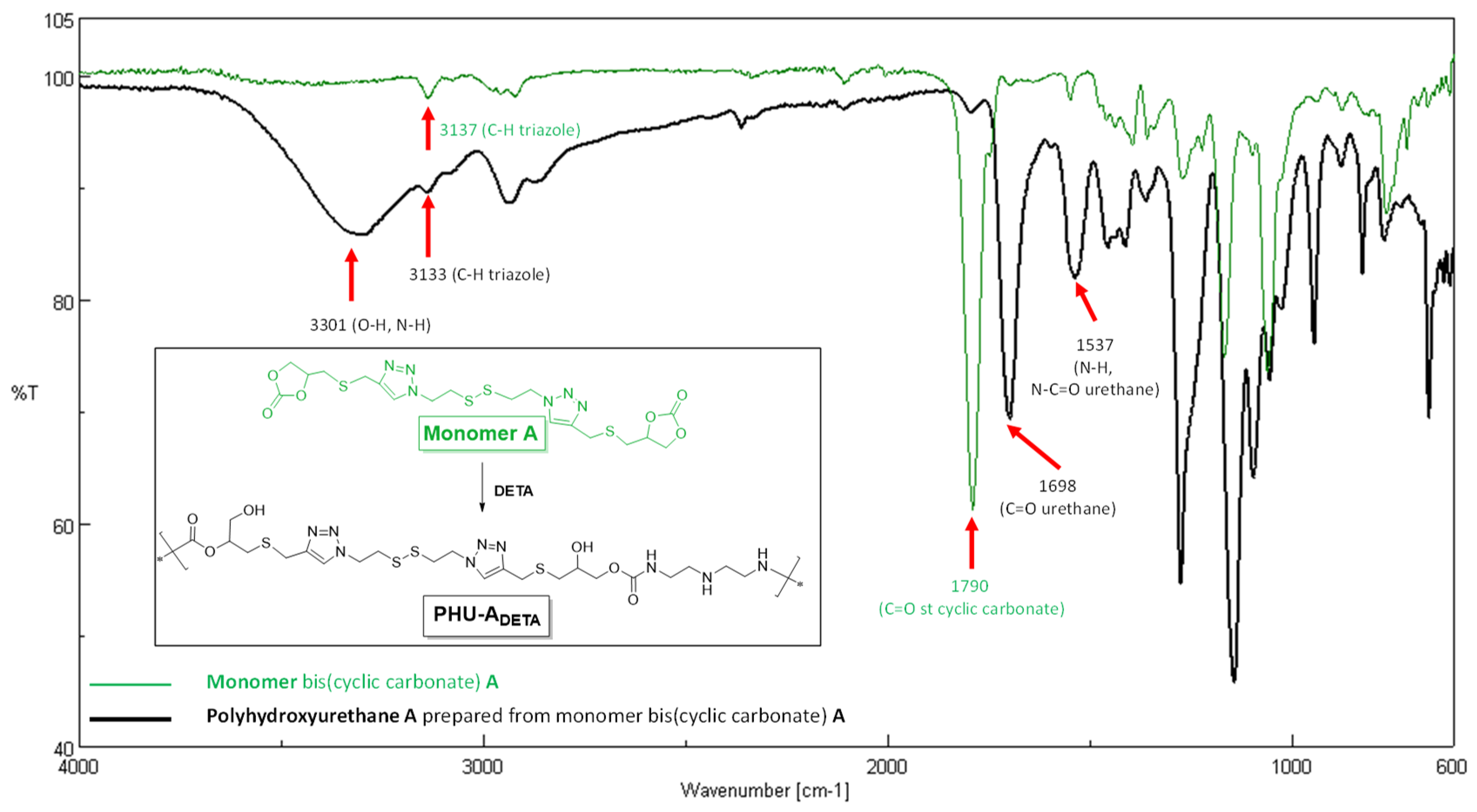
Figure 4.
A Unified Approach for Single-Step Multicomponent Hydrogel Synthesis.
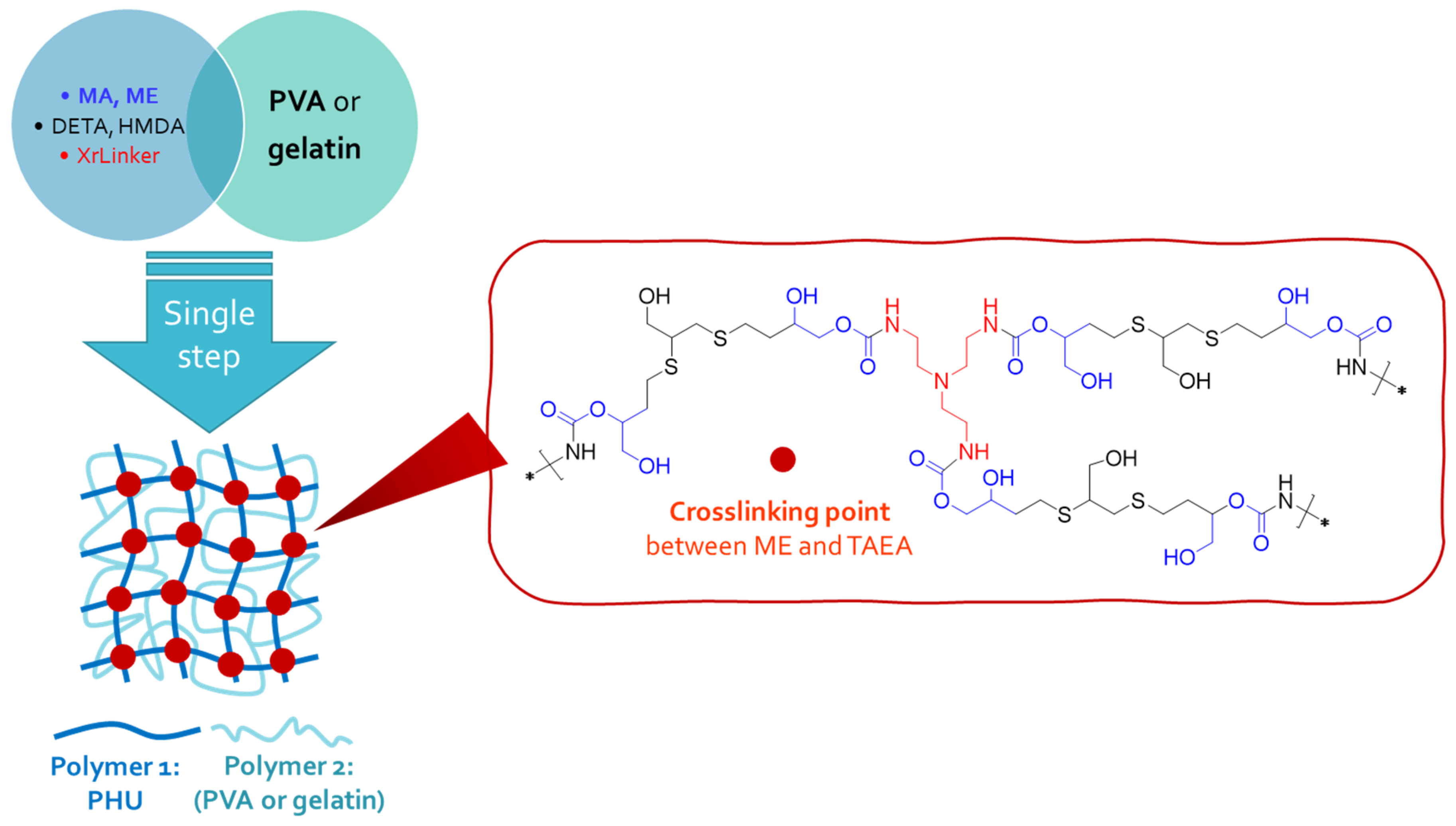
Figure 5.
ATR-FTIR spectra of (a) monomer E (ME); (b) PVA used in fabrication of PVA-based SIPN; (c) multicomponent hydrogel IPN1: EDETA-PVA.
Figure 5.
ATR-FTIR spectra of (a) monomer E (ME); (b) PVA used in fabrication of PVA-based SIPN; (c) multicomponent hydrogel IPN1: EDETA-PVA.
Figure 6.
Comparative studies of the rheological properties of PVA-based SIPN and its blank. Evolution of storage modulus, and loss modulus, with the frequency for SIPN hydrogels prepared by aminolysis of bisCC monomers MA or ME in colloidal suspensions of PVA.
Figure 6.
Comparative studies of the rheological properties of PVA-based SIPN and its blank. Evolution of storage modulus, and loss modulus, with the frequency for SIPN hydrogels prepared by aminolysis of bisCC monomers MA or ME in colloidal suspensions of PVA.
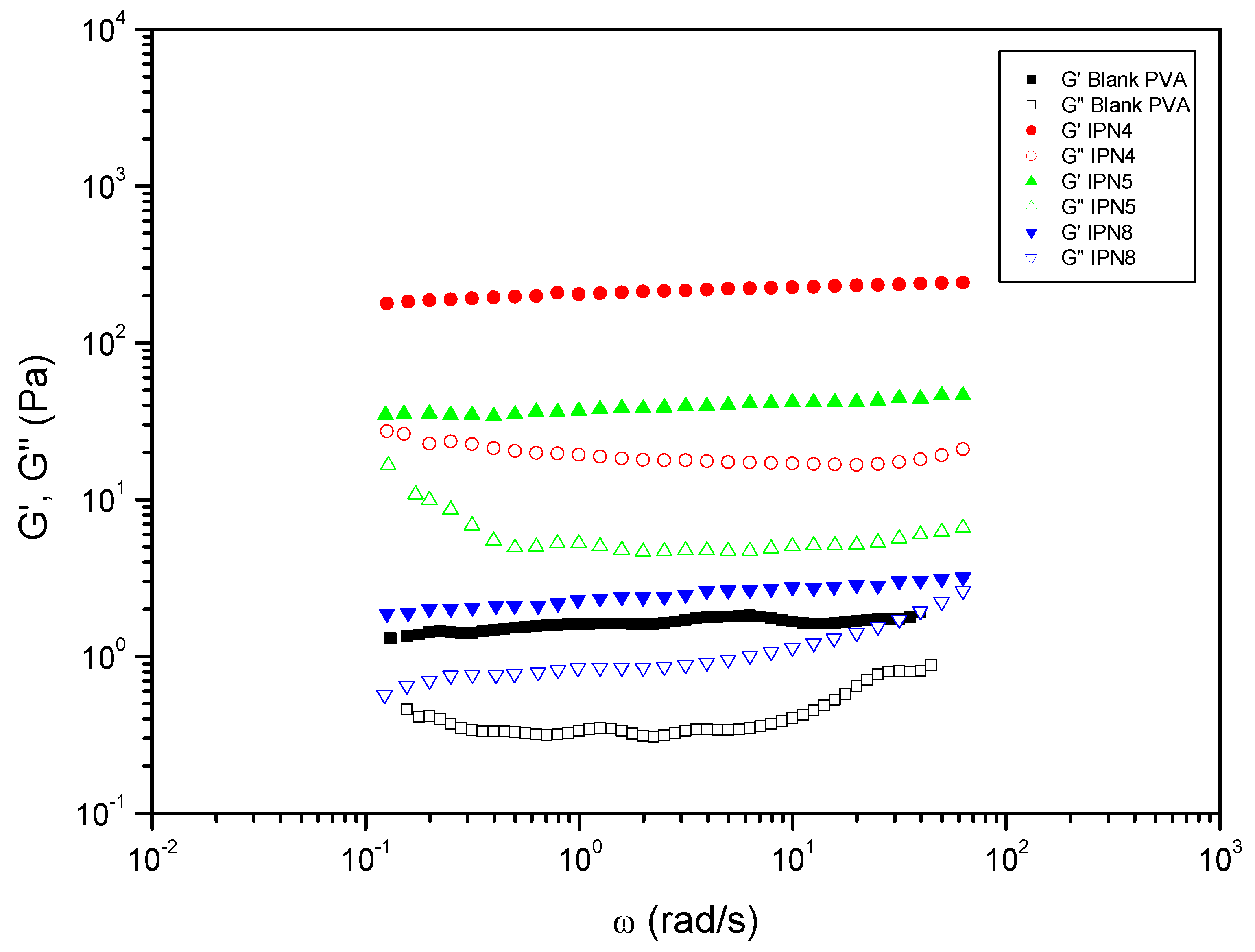
Figure 7.
Comparative studies of the rheological properties of PVA-based co-IPN and its blank. Evolution of storage modulus, and loss modulus, with the frequency for co-IPN hydrogels prepared by the aminolysis of bisCC monomers MA and ME in colloidal suspensions of PVA.
Figure 7.
Comparative studies of the rheological properties of PVA-based co-IPN and its blank. Evolution of storage modulus, and loss modulus, with the frequency for co-IPN hydrogels prepared by the aminolysis of bisCC monomers MA and ME in colloidal suspensions of PVA.
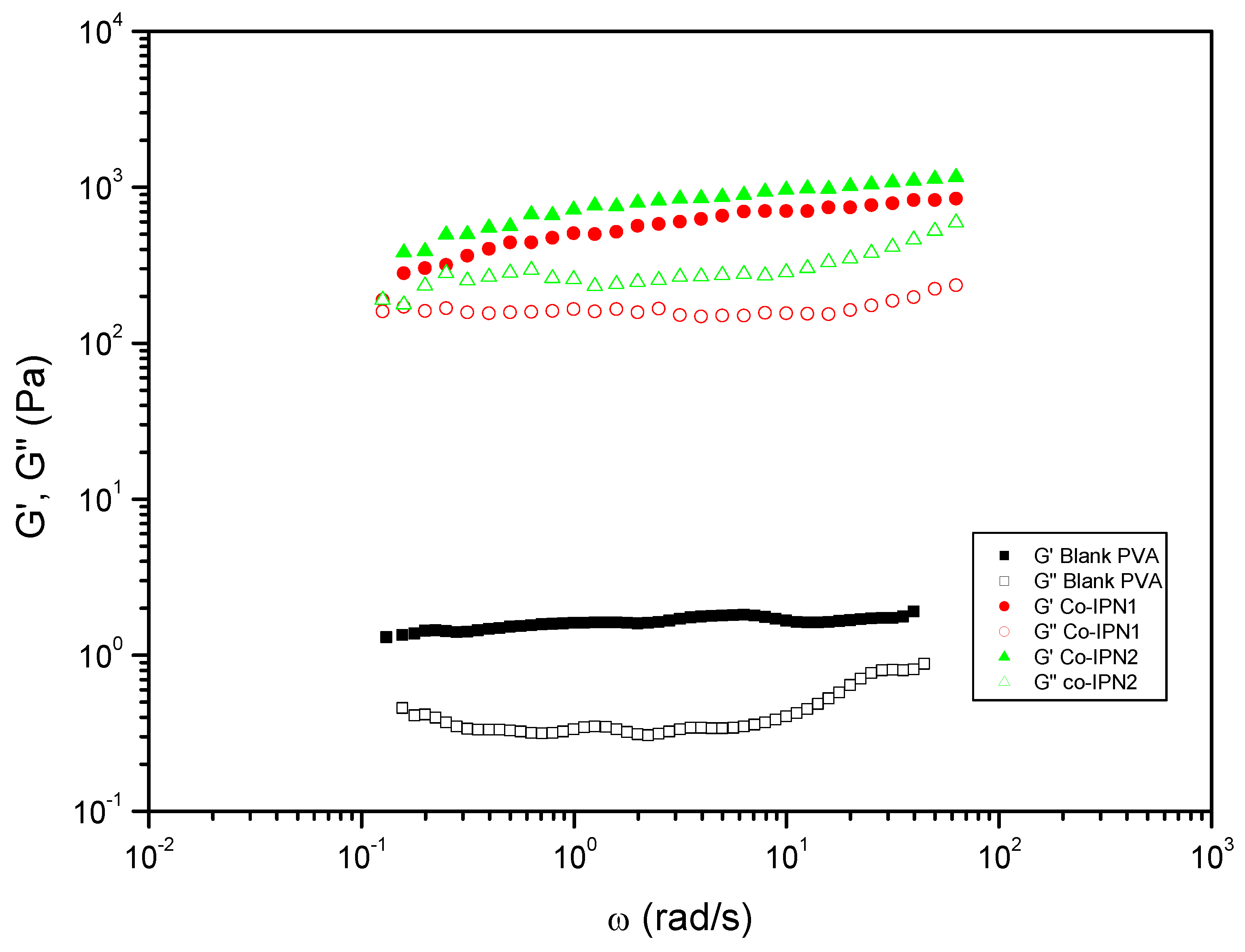
Figure 8.
Comparative studies of the rheological properties of gelatin-based IPN and its blank. Evolution of storage modulus, , and loss modulus, , with the frequency for IPN hydrogels prepared by aminolysis of bisCC monomers MA (IPN7) or ME (IPN1) in colloidal suspensions of gelatin.
Figure 8.
Comparative studies of the rheological properties of gelatin-based IPN and its blank. Evolution of storage modulus, , and loss modulus, , with the frequency for IPN hydrogels prepared by aminolysis of bisCC monomers MA (IPN7) or ME (IPN1) in colloidal suspensions of gelatin.
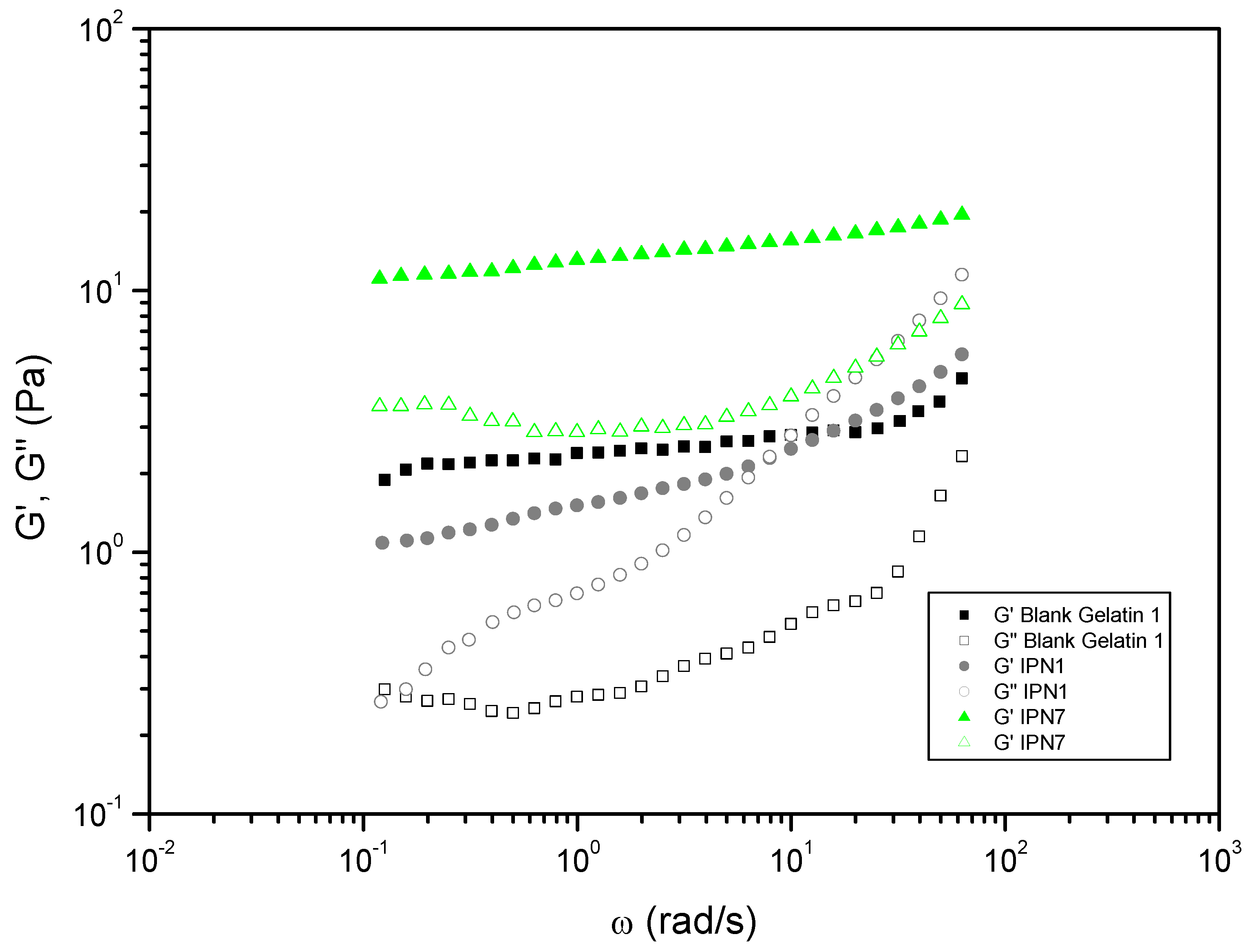
Figure 9.
SEM images of bulk samples from MA-based multicomponent hydrogels. (a) IPN8; (b): IPN7; Magnification 1.63 K & 5.00 K and 1.50 K, respectively.
Figure 9.
SEM images of bulk samples from MA-based multicomponent hydrogels. (a) IPN8; (b): IPN7; Magnification 1.63 K & 5.00 K and 1.50 K, respectively.

Figure 10.
SEM images of bulk samples fom ME-based SIPN (freeze-dried samples). (a) IPN4; (b): IPN5; (c) IPN2. Magnification 264, 293 and 641, respectively.
Figure 10.
SEM images of bulk samples fom ME-based SIPN (freeze-dried samples). (a) IPN4; (b): IPN5; (c) IPN2. Magnification 264, 293 and 641, respectively.


Table 1.
Semi-IPN (SIPN) compositions of the hydrogels formed through the aminolysis of bisCC monomers MA and ME in the colloidal suspension of either PVA or gelatin. Plateau modulus, loss tangent at 1 rad/s of the obtained PVA and gelatin-based hydrogels.
Table 1.
Semi-IPN (SIPN) compositions of the hydrogels formed through the aminolysis of bisCC monomers MA and ME in the colloidal suspension of either PVA or gelatin. Plateau modulus, loss tangent at 1 rad/s of the obtained PVA and gelatin-based hydrogels.
| Entry | Hydrogels prepared and blanks | Polymer 1 (PHU) Conc.: 10% w/v |
Polymer 2 (Polymer scaffold) Conc.: 10% w/v |
Solvents | Catalyst |
(Pa) |
||
|---|---|---|---|---|---|---|---|---|
| Monomer 1 (bisCC) |
Monomer 2 (diamine) |
|||||||
| 1 | Blank | --- | --- | Gelatin 1 | DMSO-H2O 1:1 | 2.24 | 0.10 | |
| 2 | Blank | --- | --- | Gelatin 1 | EtOH-H2O 1:1 | 1240.46 | 0.12 | |
| 3 | Blank | --- | --- | PVA | DMSO-H2O 1:1 | 1.86 | 0.21 | |
| 4 | Blank | --- | --- | PVA | EtOH-H2O 1:1 | 11.9 | 0.35 | |
| 5 | IPN1 | ME | DETA | Gelatin 1 | DMSO-H2O 1:1 | DBU | 1.45 | 0.44 |
| 6 | IPN2 | ME | HMDA | Gelatin 1 | DMSO-H2O 1:1 | DBU | 4.29 | 0.20 |
| 7 | IPN3 | ME | DETA | Gelatin 1 | EtOH-H2O 1:1 | TU | 5.19 | 0.81 |
| 8 | IPN4 | ME | DETA | PVA | DMSO-H2O 1:1 | DBU | 225.44 | 0.09 |
| 9 | IPN5 | ME | HMDA | PVA | DMSO-H2O 1:1 | DBU | 40.97 | 0.14 |
| 10 | IPN6 | ME | DETA | PVA | EtOH-H2O 1:1 | TU | 8.28 | 0.19 |
| 11 | IPN7 | MA | DETA | Gelatin 1 | DMSO-H2O 1:1 | DBU | 14.34 | 0.22 |
| 12 | IPN8 | MA | DETA | PVA | DMSO-H2O 1:1 | DBU | 2.39 | 0.38 |
| 13 | co-IPN1 | ME80/MA20 | DETA | PVA | DMSO-H2O 1:1 | DBU | 742.12 | 0.32 |
| 14 | co-IPN2 | ME20/MA80 | DETA | PVA | DMSO-H2O 1:1 | DBU | 958.39 | 0.35 |
| (a) (): Plateau modulus; (b) tan (δ) at 1 rad/s DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene; DETA: diethylenetriamine; DMSO: dimethylsulfoxide; EtOH: ethanol; HMDA: 1,6-hexamethylenediamine; MA: monomer bisCC A; ME: monomer bisCC E; TU: N′-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexylthiourea. | ||||||||
Table 2.
Comparison of values, as determined by DSC, for the three types of PHU synthesized in this study.
Table 2.
Comparison of values, as determined by DSC, for the three types of PHU synthesized in this study.
| Sample | BisCC | Diamine | [Monomer] | Solvent- | Temp. (°C) |
Catalyst |
g/mol |
(°C) |
(°C) |
|---|---|---|---|---|---|---|---|---|---|
| ADETA-13(d) | MA | DETA | 1.8 mol/L | TFE | 25 | TU | 16,400 | -3.0 | -- |
| EDETA-4(e) | ME | DETA | 1.8 mol/L | EtOH | 50 | TU | 29,600 | -10.3 | -- |
| EHMDA-2(e) | ME | HMDA | 1.8 mol/L | DMSO | 50 | DBU | 34,100 | 14.2 | -- |
| (a) Weight average molecular weight () calculated by gel permeation chromatography (GPC); (b) Glass transition temperature () determined by differential scanning calorimetry (DSC); (c) Melting temperature () determined by differential scanning calorimetry (DSC); (d) Table S1; (e) Table S2; DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene; DETA: diethylenetriamine; DMSO: dimethylsulfoxide; EtOH: ethanol; HMDA: 1,6-hexamethylenediamine; MA: monomer A; ME: monomer B; TFE: 2,2,2-trifluoroethanol; TU: N′-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexylthiourea. | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



