
Submitted:
23 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
Systemic lupus erythematosus (SLE) is a prevalent autoimmune condition, yet its alignment with a specific host immunological pathway remains unclear. The THαβ host immunological pathway, recognized for its defense against viruses and prions, has recently emerged as a response to DNA, RNA, and protein pathogens. SLE patients often produce anti-double strand DNA antibodies and anti-nuclear antibodies, suggesting a potential association with the THαβ host immune reaction. Throughout the course of SLE, elevated levels of type 1 interferons, type 3 interferons, interleukin-10, IgG1, and IgA1 are commonly observed. These cytokines and antibody isotypes are indicative of the THαβ host immunological pathway. Similarly, Myasthenia gravis, Grave’s disease, Graft versus host disease, autoimmune hemolytic anemia, immune thrombocytopenia, dermatomyositis, and Sjogren’s syndrome are also linked to THαβ-related type 2 hypersensitivities. Considering the potential association of these diseases with dysregulated THαβ immune responses, therapeutic strategies such as anti-interleukin-10 or anti-interferons α/β could be explored to manage these disorders effectively.
Keywords:
autoimmune diseases
; type 2 hypersensitivity
; Tr1
; SLE
; Myasthenia gravis
; Grave’s disease
; GvHD
; Sjogren’s syndrome
; AIHA
; ITP
; dermatomyositis
Introduction
Systemic lupus erythematosus (SLE) is a relatively common autoimmune disorder, and there is ongoing discussion about its connection to the TH17 host immunological pathway. TH17 is known for its role in combating extracellular micro-organisms like bacteria, fungi, or protozoa. Despite this, there remains uncertainty regarding whether SLE is truly associated with the TH17 host immune response. The confusion arises because SLE autoantibodies typically target anti-double strand DNA and anti-nuclear antibodies, suggesting a potential link to viral infections rather than an alignment with the TH17 pathway designed for extracellular micro-organisms [1]. The development of autoimmunity in SLE is suggested to involve molecular mimicry, particularly through virus infections, such as Epstein-Barr virus (EBV) [2]. We suggest that the THαβ immune response, known for its defense against viruses and prions, is connected to the pathophysiology of SLE and it appears to align more closely with a type 2 hypersensitivity. In this type of hypersensitivity, an excessive type 1 interferon response plays a pivotal role in the pathophysiology. However, the hallmark of type 2 hypersensitivity is antibody-mediated cellular cytotoxicity.
Several type 2 hypersensitivity disorders, including Sjogren’s syndrome, Grave’s disease, autoimmune hemolytic anemia, immune thrombocytopenia, graft versus host disease, dermatomyositis, and Myasthenia gravis, do not align with TH1, TH2, or TH17 inflammatory disorders, creating uncertainty about their immunopathogenesis. In this context, we present evidence suggesting that these disorders are also linked to anti-viral THαβ dominant autoimmune reactions, possibly triggered by viral infections through molecular mimicry. The presence of anti-nuclear antigens in above mention diseases further supports the notion that these type 2 autoimmune conditions can be classified as THαβ dominant immune disorders. This perspective adds depth to our understanding of the immunological mechanisms underlying these diseases and their potential connection to viral infections.
The Framework of Host Immunological Pathways
The host’s immunological pathways exhibit a classification into two primary categories: IgG-dominant eradicable immune reactions and IgA-dominant tolerable immune reactions [3,4,5]. The host’s immunological pathways exhibit a classification into two primary categories: IgG-dominant eradicable immune reactions and IgA-dominant tolerable immune reactions [3,4,5]. Facilitating the development of eradicable immunity, follicular helper T cells play a crucial role by encouraging the antibody class switch from IgM to IgG. Within eradicable immune responses, four distinct types correspond to different pathogenic entities. TH1 immunity, for instance, serves as the host’s immunological pathway specifically designed to combat intracellular micro-organisms, encompassing intracellular bacteria, protozoa, and fungi. This categorization enhances our understanding of the intricate and specialized roles played by different immune pathways in responding to diverse types of pathogens [6]. TH1 immunity includes M1 macrophages, IFNγ CD4 T cells, iNKT1 cells, CD8 T cells (Tc1, EM4), and IgG3 B cells [7,8]. TH1 immunity is linked to type 4 delayed-type hypersensitivity, showcasing a delayed immune response. In contrast, TH2 immunity functions as the host’s immunological pathway specifically tailored to combat parasites. Notably, TH2 immunity comprises two distinct subtypes, emphasizing the nuanced and specialized nature of the immune response against parasitic invaders [9]. TH2a immunity is the host immune reaction against endoparasites (helminths). TH2b immunity is the host immune reaction against ectoparasites (insects). TH2a immunity includes inflammatory eosinophils (iEOS), interleukin-4/interleukin-5 CD4 T cells, mast cells-tryptase (MCt), iNKT2 cells, and IgG4 B cells [10,11,12,13]. TH2b immunity includes basophils, interleukin-13/interleukin-4 CD4 T cells, mast cells-tryptase/chymase (MCtc), iNKT2 cells, and IgE B cells [14,15,16]. The TH2 immunological pathway is intricately associated with type 1 allergic hypersensitivity, representing a specific immune response to allergens. On the other hand, TH22 immunity serves as the host’s specialized immune reaction targeted at extracellular micro-organisms, encompassing extracellular bacteria, protozoa, and fungi. The components of TH22 immunity include neutrophils (N1), CD4 T cells producing interleukin-22, iNKT17 cells, and IgG2 B cells, highlighting the diverse cellular players involved in orchestrating this immune response against extracellular threats [17,18]. TH22 immunity is associated with type 3 immune complex mediated hypersensitivity. THαβ immunity is the host immunological pathway against infectious particles (viruses and prions) [19,20,21]. THαβ immunity includes NK cells (NK1), interleukin-10 producing CD4 T cells, iNKT10 cells, CD8 T cells(Tc2,EM1), and IgG1 B cells [22]. THαβ immunity is associated with type 2 antibody dependent cytotoxic hypersensitivity.
The immunological pathways classified as tolerable are characterized by their dominance of IgA-mediated immune reactions, further subdivided into four distinct groups tailored to address different pathogens. Regulatory T cells play a pivotal role in facilitating the development of tolerable immune reactions by aiding in the antibody class switch to IgA [3]. TH1-like immunity is the host tolerable immune reaction against intracellular micro-organisms (intracellular bacteria, protozoa, and fungi). TH1-like immunity includes M2 macrophages, TGFβ/IFNγ CD4 T cells, iNKT1 cells, CD8 T cells (EM3), and IgA1 B cells [23]. TH1-like immunity is associated with type 4 delayed type hypersensitivity. TH9 immunity is the host tolerable immune reaction against parasites (insects and helminths). TH9 immunity includes regulatory eosinophils (rEOS), basophils, interleukin-9 CD4 T cells, iNKT2 cells, IL-9 related mast cells (MMC9), and IgA2 B cells [24,25]. TH9 immunity is associated with type 1 allergic hypersensitivity. TH17 immunity is the host tolerable immune reaction against extracellular micro-organisms (extracellular bacteria, protozoa, and fungi). TH17 immune reaction includes neutrophils (N2), interleukin-17 producing CD4 T cells, iNKT17 cells, and IgA2 B cells. TH17 immunity is associated with type 3 immune complex mediated hypersensitivity [26,27]. TH3 immunity is the host immune reaction against infectious particles (viruses and prions). TH3 immunity includes NK cells (NK2), interleukin-10/TGFβ CD4 T cells, iNKT10 cells, CD8 T cells (EM2), and IgA1 B cells [28,29]. TH3 immunity is associated with type 2 antibody dependent cytotoxic hypersensitivity.
THαβ Immunological Pathway Related with SLE
While several previous studies have suggested an association between the TH17 immune response and SLE, the validity of this theory remains uncertain. The TH17 immunological pathway has been proposed to be linked to nearly all autoimmune disorders, but this concept is flawed. Conditions such as asthma, atopic dermatitis, and allergic rhinitis fall into the category of type 1 autoimmune disorders and are associated with the TH2 or TH9 immunological pathway. On the other hand, multiple sclerosis, contact dermatitis, and type 1 diabetes mellitus are classified as type 4 autoimmune diseases and are related to the TH1 immunological pathway. Consequently, the TH17 immunological pathway cannot comprehensively explain the mechanisms underlying all autoimmune disorders. It is crucial to recognize that the TH17 immunological pathway primarily serves as the host’s defense against extracellular micro-organisms, including extracellular bacteria, protozoa, or fungi. The existence of disease-specific antinuclear antibodies in SLE indicates a correlation with the host’s immune response against viruses. The THαβ immunological pathway, responsible for host eradicable immunity against viral infections, plays a role in this context. The relationship between SLE and the THαβ immune reaction will be explored further in this article.
THαβ immunity constitutes an anti-viral immune response. The initiation of the THαβ immune response involves innate lymphoid cells, specifically ILC10 cells. Additionally, plasmacytoid dendritic cells serve as antigen-presenting cells to initiate the THαβ immunological pathway. A prior study has established the significance of plasmacytoid dendritic cells in the initiation of systemic lupus erythematosus [30]. Type 1 interferons (interferons alpha and beta) are mainly produced by plasmacytoid dendritic cells. Type 1 interferons are the cytokines for the first line to fight against virus pathogens invading host. Type 1 interferons play key roles in the pathogenesis of systemic lupus erythematosus [31,32,33]. Monoclonal antibodies against type 1 interferons have been tested effective in SLE patients in several previous clinical trials [34,35]. Type 3 interferons such as interferon lambda have similar cellular functions against virus infection like type 1 interferons. Previous study also pointed out the relationship of type 3 interferons and the pathogenesis of SLE [36,37].
Besides, we can examine the Toll-like receptor patterns in initiating anti-viral immune reaction. TLR3, TLR7, and TLR9 are the major Toll-like receptor subtypes to trigger anti-viral immune response. TLR3 is activated by double strand RNA. TLR7 and TLR9 are activated by single strand RNA. TLR3, TLR7, and TLR9 all locate in the endosomal compartments in the cells. TLR3, TLR7, and TLR9 activation can lead dendritic cells, especially plasmacytoid dendritic cells, to produce type 1 interferons (interferon alpha and beta). TLR3, TLR7, and TLR9 are all found to be related to the pathophysiology of systemic lupus erythematosus [38,39,40,41,42,43,44]. Thus, toll-like receptors can be therapeutic targets for controlling SLE [38,45].
The central THαβ immunity cytokine is interleukin-10. Interleukin-10 can activate the immune activities of NK cell and CTL and cause B cell antibody isotype switch to IgG1. The serum level of interleukin-10 is actually elevated in patients of systemic lupus erythematosus [46,47]. And, anti-interleukin-10 monoclonal antibody can alleviate the symptoms and disease progression in systemic lupus erythematosus patients [48,49,50,51]. Thus, these evidences all point out that THαβ immune reaction plays a key role in the pathogenesis of SLE. IgG1 is the antibody subtype mainly against viral infection. In SLE patients, IgG1 level is elevated. These all imply the hypothesis that SLE is a THαβ immune disorder.
Besides THαβ eradicable host immunological pathway, TH3 tolerable immunity is also an immune response against virus infection [28]. This is usually seen in the chronic stage of systemic lupus erythematosus. In TH3 immunological pathway, serum antibody IgA1 will be elevated. This is actually seen in chronic SLE patients [52]. Besides. TH3 immunological pathway with Treg cells and TGFβ production can cause kidney fibrosis and finally lead renal failure. This is a common complication of chronic SLE. Thus, TH3 immunity also plays a vital role in the pathophysiology of SLE.
THαβ Immunological Pathway Related with Sjogren’s Syndrome
Sjogren’s syndrome manifests with symptoms such as dry eye, dry mouth, and dry mucosa, indicating an autoimmune condition. These clinical features, resembling those of Sjogren’s syndrome, can also be associated with various viral infections. Therefore, in the evaluation of clinical symptoms and signs resembling Sjogren’s syndrome, it is crucial to consider virus infections in the differential diagnosis. Furthermore, virus infections, notably Epstein-Barr virus (EBV) infection, have the potential to activate the molecular mimicry mechanism, leading to the subsequent development of autoimmune Sjogren’s syndrome. This highlights the importance of recognizing and investigating the role of viral infections in the etiology and pathogenesis of Sjogren’s syndrome [53]. Other viral infections including HIV, HCV, and HTLV can also cause dry eye, dry mouth, and dry mucosa to mimic the clinical symptoms and signs of Sjogren’s syndrome [53]. In the differential diagnosis of Sjogren’s syndrome, these viral infections must be ruled out before giving the diagnosis of Sjogren’s syndrome. Therefore, it is probable that Sjogren’s syndrome is associated with the anti-viral THαβ immunological pathway. Furthermore, the pathogenesis of Sjogren’s syndrome involves the destruction of cells in the salivary gland, lacrimal gland, and mucosal tissue. This destruction is characterized as a type 2 antibody-dependent cytotoxic hypersensitivity, which is linked to the anti-viral THαβ immune reaction. All these factors collectively indicate that Sjogren’s syndrome can be classified as a THαβ immune disorder.
Sjogren’s syndrome is categorized as a type 2 hypersensitivity disorder. This syndrome is related to the presence of ant-Ro and anti-La autoantibodies in patients’ serum [54]. Anti-Ro and anti-La autoantibodies are the antibodies against RNA molecules (Y RNA). Thus, the anti-nucleic acid antibodies associated with Sjogren’s syndrome also suggest that this syndrome is related to anti-viral THαβ immunity. Virus particles can be DNA or RNA viruses. Thus, RNA is also an inherited molecule of infectious viruses. SLE is also related to THαβ autoimmunity, and it is not uncommon that patients both suffer from SLE and Sjogren’s syndrome. These two autoimmune diseases can be overlapped. Besides, TLR3, TLR7, and TLR9 activations are related to the immunopathogenesis of Sjogren’s syndrome [55,56]. And, TLR3, TLR7, and TLR9 are toll-like receptors responding to DNA and RNA molecules during viral infections. Plasmacytoid dendritic cells, the antigen presenting cells for initiating THαβ immunity, are activated in Sjogren’s syndrome. Type 1 and type 3 interferons, which are responsible for THαβ anti-viral immunity, are also up-regulated in Sjogren’s syndrome [57]. Type 1 and type 3 interferons are driver cytokines to trigger THαβ immune reaction. There is a case report saying that interferon treatment for HCV Follicular helper T cells can help to trigger eradicable host immune reactions including THαβ immune response. Follicular helper T cells are also found to play a role in the pathogenesis of Sjogren’s syndrome [58]. Follicular dendritic cells and plasmacytoid dendritic cells are associated with Sjogren’s syndrome [59]. Follicular dendritic cells are related to up-regulation of follicular helper T cells. Plasmacytoid dendritic cells are related to TLR3, TLR7, and TLR9 up-regulation to trigger type 1 interferon responses to initiate THαβ anti-virus host immune reactions. The anti-virus antibody subtype, IgG1, are the major autoantibody found in Sjogren’s syndrome [54]. The central THαβ cytokine, interleukin-10, is also up-regulated in Sjogren’s syndrome, and it suggested that IL-10 plays a key role in the pathogenesis of Sjogren’s syndrome [60]. Another THαβ cytokine, interleukin-27, which can induce the production of IL-10, is also activated in Sjogren’s syndrome [61]. TCR receptor repertoire is related to the pathogenesis of Sjogren’s syndrome [62]. Cytotoxic T cells, which are the key effector cells in anti-viral THαβ immunity, are also stimulated in Sjogren’s syndrome [63]. Thus, T cell related cellular immune response including THαβ immune reaction is correlated to the pathophysiology of Sjogren’s syndrome. STAT1 and STAT3 are major transcription factors to mediate anti-viral THαβ immunological pathway. STAT1 and STAT3 are both found to be up-regulated in Sjogren’s syndrome [64,65]. CXCR3 are the major chemokine receptor presented in THαβ related T lymphocytes. There is also evidence that CXCR3 presenting lymphocytes are correlated to Sjogren’s syndrome [66]. Up-regulation of CXCR3 ligands including CXCL9, CXCL10, and CXCL11 are also found in the involved tissues of Sjogren’s syndrome. Another study found out that NKT cells, CD56+ NK cells are also play vital roles in the pathogenesis of Sjogren’s syndrome [67]. These evidences all point out the importance of THαβ immunity in the pathogenesis of Sjogren’s syndrome.
THαβ Immunological Pathway Related with Myasthenia Gravis
Myasthenia gravis is also a common type 2 hypersensitivity disorder. Previous studies found that this illness is not TH1, TH2, nor TH17 disorder. Because type 2 hypersensitivity belongs to THαβ immunological pathway, we will provide evidences that Myasthenia gravis is closely related to THαβ immune response. The auto-antibody for Myasthenia gravis is mainly anti-acetylcholine receptor antibody. This type of auto-antibody is mainly a IgG1 antibody which is an anti-viral antibody in THαβ immunity [68]. Because acetylcholine receptors locate in cell membrane of neurons in the neuromuscular junction, anti-acetylcholine receptor IgG1 antibody can cause neuron cell antibody dependent cellular cytotoxicity via the help of NK cells. NK cell activities are also found to be up-regulated in Myasthenia gravis patients. After plasmapheresis for Myasthenia gravis treatment, natural killer cells activities will decrease [69]. Interleukin-10 polymorphism is associated in Myasthenia gravis, saying that THαβ immune response is related to this autoimmune disorder [70]. This whole mark of cellular immunity is the pathogenesis of Myasthenia gravis. Another study reported that anti-acetylcholine receptor antibody, interleukin-10, and Tr1 cells (THαβ CD4 T cells) are all correlated and up-regulated in Myasthenia gravis [71]. TLR3, TLR7, and TLR9 are toll-like receptors related to THαβ immune activation, and these toll-like receptors are over-expressed in thymus of Myasthenia gravis [72,73].
The central anti-viral THαβ immune cytokine is interleukin-10. And, interleukin-10 levels are elevated in patients with Myasthenia gravis. This also suggests that Myasthenia gravis is associated with THαβ immune reaction [74]. Another key THαβ cytokine, interleukin-27, which can induct the production of interleukin-10, is also up-regulated in Myasthenia gravis [75]. Besides, the initiators of THαβ immunity, type 1 interferons, are also up-regulated in Myasthenia gravis [76]. The up-regulated interferons in Myasthenia gravis include interferon-alpha and interferon-beta. These evidences all suggest that THαβ immunological pathway plays a vital role in the pathophysiology of Myasthenia gravis. Thymoma is often found in patients with Myasthenia gravis. Thymoma is usually associated with over-production of CD4 T cells and CD8 T cells. These two lymphocytes, especially cytotoxic CD8 T cells, are also important components of THαβ immune reaction. And, anti-double strand RNA, another autoantibody related to viral infection, is also related to the etiology of Myasthenia gravis [77]. Virus infection can usually cause the exacerbation of Myasthenia gravis, and this also pointed out the vital role of THαβ immunological pathway in the disease pathogenesis. EBV, parvovirus, and HSV infections have been reported to be associated with the pathogenesis of Myasthenia gravis [78,79]. Chemokine receptor and its ligand, CXCR3 and IP10, which are related to THαβ immunity, are overexpressed in T lymphocytes in Myasthenia gravis [80].
THαβ Immunological Pathway Related with Grave’s Disease
Autoimmune thyroiditis encompasses Hashimoto thyroiditis and Graves’ disease. Graves’ disease is classified as a type 2 hypersensitivity disorder, while Hashimoto thyroiditis falls under type 4 hypersensitivity. It is important to note that Graves’ disease is associated with hyperthyroidism, whereas Hashimoto thyroiditis is linked to hypothyroidism. In Graves’ disease, the presence of autoantibodies leads to thyroid hyperplasia, while in Hashimoto thyroiditis, these autoantibodies result in the destruction of thyroid tissue [81]. That is the main reason why the two autoimmune thyroiditis have different clinical pictures. It is decided by the characteristics of autoantibodies [82]. Like SLE, anti-nuclear antibodies can be detected in autoimmune thyroiditis, especially in Grave’s disease. Anti-double strand DNA autoantibodies are also found significantly in populations of Grave’s disease [83]. Chemokine receptor, CXCR3, and its ligand, CXCL10, are over-represented on THαβ immune cells, and they are both over-expressed in Grave’s disease [84]. Type 1 and type 3 interferons including interferon alpha/beta/lambda are the initiator of THαβ immune response, and they are also over-expressed in Grave’s disease [85]. The transcription factor, IRF7, which is related to the expression of type 1 interferon, is also over-represented in Grave’s disease. IgG1 is the ant-viral THαβ immune reaction antibody, and it is the major autoantibody IgG subtype found in Grave’s disease [86]. The central cytokine of THαβ immunological pathway, interleukin-10, is also over-expressed in the Grave’s disease [87]. Another important THαβ immunity cytokine, interleukin-27, is also related to the pathophysiology of Grave’s disease. Toll-like receptors: TLR3, TLR7, and TLR9 have vital roles in sensing virus antigens to trigger THαβ immune reaction, and these toll-like receptor subtypes are also over-expressed in Grave’s disease [88]. Natural killer cells are important immune effector cells against virus infection. However, NK cells activity can be suppressed by thyroid hormone. Thus, decreased NK cell activity is noted in Grave’s disease with hyperthyroidism [89].
THαβ Immunological Pathway Related with Graft Versus Host Disease
Graft versus host disease (GVHD) is classified as a type 2 hypersensitivity reaction and is identified as a THαβ dominant autoimmune disorder. In the context of transplantation, the activated natural killer cells and cytotoxic CD8 T cells originating from the graft target the recipient’s tissues that possess mismatched MHC molecules, leading to the development of GVHD [90]. The antibody dependent cellular cytotoxicity is over-activated during graft versus host disease. The activity of CD8 T cell perforins is over-expressed in graft versus host disease. Interleukin-10, the central cytokine of THαβ immunity, is related to the severity of graft versus host disease [91]. The THαβ immunity related chemokine receptor, CXCR3, is over-expressed in graft versus host disease [92]. The anti-viral THαβ immunity cytokine, interleukin-15, is also up-regulated in graft versus host disease [93]. Type 1 interferons, the vital THαβ immunity initiators, are also up-regulated in graft versus host disease [94]. The transcription factors related to type 1 interferons, IRF3 and IRF7, are also up-regulated in graft versus host disease [95,96]. The key THαβ immunity transcription factors, STAT1 and STAT3, are activated in graft versus host disease [97,98]. Another important THαβ immunity cytokine, interleukin-27, is up-regulated in graft versus host disease, too [99]. TLR7 and TLR9, the toll-like receptors sensing virus nucleic acid antigen, are also up-regulated in graft versus host disease [100,101]. The downstream signaling of these toll-like receptors including MyD88 and TRIF are also vital to the pathogenesis of graft versus host disease [102]. The antigen presenting cells for anti-viral THαβ immunity, plasmacytoid dendritic cells, are activated in graft versus host disease [103].
THαβ Immunological Pathway Related with Immune Thrombocytop Enia
The disease, immune thrombocytopenia, is formerly called idiopathic thrombocytopenia. It is a disease of immune mediated destruction of platelets. Virus infections could cause the consequence of immune thrombocytopenia. Thus, it is reasonable that immune thrombocytopenia is a THαβ related immune disorder. Anti-nuclear autoantibody is found in immune thrombocytopenia, especially in chronic immune thrombocytopenia. B cells and T cells, adaptive immunity, play important roles in the pathogenesis of immune thrombocytopenia. Cytotoxic T lymphocytes and plasmacytoid dendritic cells, both of which are important effector cells in THαβ immunological pathway, also play key roles in the pathophysiology of immune thrombocytopenia [104].
The key cytokines of THαβ immunological pathway also play key roles in the pathophysiology of immune thrombocytopenia. The central THαβ cytokine, interleukin-10, is closely associated with the disease process of immune thrombocytopenia. Genetic polymorphism of interleukin-10 is related to immune thrombocytopenia [105]. Interleukin-10 can also induce immune thrombocytopenia [106]. Another important THαβ immunity cytokine, interleukin-27, is also elevated in patients of immune thrombocytopenia [107]. Besides, interleukin-27 polymorphism is also associated with the risk of immune thrombocytopenia [108]. Type 1 interferons, the initiators of THαβ immunity, are also important in the pathogenesis of immune thrombocytopenia [109]. Interferonα can induce immune thrombocytopenia. In immune thrombocytopenia, a lot of interferon regulated genes will be up-regulated. IgG elevation is associated with the disease of immune thrombocytopenia. IgG1, the anti-viral THαβ immunity antibody subtype, is over-represented in the immune thrombocytopenia [110,111].
Signaling pathways involved in THαβ immune reaction are also up-regulated in immune thrombocytopenia. The toll-like receptor relating to anti-viral immunity, TLR7, is up-regulated in immune thrombocytopenia [110]. Type 1 interferon signaling related transcription factor, IRF3, and adaptive immunity related transcription factor, NFkB, are both over-represented in immune thrombocytopenia [112]. The master THαβ immune transcription factors, STAT1 and STAT3, are also over-represented in immune thrombocytopenia [113,114]. These above evidences all showed that THαβ immune reaction is important to the pathophysiology of immune thrombocytopenia.
THαβ Immunological Pathway Related with Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia falls under the category of type 2 autoimmune disorders, wherein red blood cells bound with IgG antibodies are targeted and destroyed by the immune cells, leading to hemolysis. This condition is identified as a THαβ immunological disorder as well. Notably, our findings provide evidence that type 1 regulatory T cells, specifically THαβ CD4 T cells, play a crucial role in the development and progression of autoimmune hemolytic anemia [115]. Anti-nuclear autoantibody is also seen in autoimmune hemolytic anemia [116,117]. It implies that certain viral related antigens can trigger autoimmune hemolytic anemia via molecular mimicry. For example: enterovirus 71 can induce autoimmune hemolytic anemia. CD4 T cells and CD8 T cells are also important in the disease process of autoimmune hemolytic anemia [118]. Follicular helper T cells (Tfh), which is the initiator of eradicable immunity, can promote autoimmune hemolytic anemia[119]. On the other hand, regulatory T cells (Treg), which drive to tolerable immune response, can control autoimmune hemolytic anemia [119]. Plasmacytoid dendritic cells with IRF8 up-regulation play important roles in triggering autoimmune hemolytic anemia [30].
The key THαβ immunological pathway related cytokines play important roles in triggering autoimmune hemolytic anemia. Interleukin-10 levels are associated with autoimmune hemolytic anemia [120]. There are a positive correlation between interleukin-10 levels and reticulocyte counts, and a negative correlation between interleukin-10 and haptoglobin in hemolytic anemia [121]. Type 1 interferons including interferon alpha and interferon beta can trigger the disorder of autoimmune hemolytic anemia [122]. IgG1, the anti-viral THαβ immune related antibody, is related to the pathogenesis of autoimmune hemolytic anemia [116]. STAT1 and STAT3, key transcription factors of THαβ immune response, are also important in the pathogenesis of autoimmune hemolytic anemia [123,124]. The chemokine receptor CXCR3, which is important in the THαβ immunological pathway, is also involved in the pathophysiology of autoimmune hemolytic anemia [125].
THαβ Immunological Pathway Related with Dermatomyositis
Dermatomyositis is classified as a type 2 autoimmune disorder, specifically falling under the category of inflammatory myopathy. This condition is characterized by an association with interferon over-representation, particularly the over-expression of type 1 interferon, within the spectrum of inflammatory myopathies [126]. Therefore, dermatomyositis is characterized as an autoimmune disorder with a dominance of THαβ cells. The majority of individuals with dermatomyositis exhibit the presence of the Anti-Jo1 autoantibody, which targets histidyl-tRNA synthetase [127]. Because THαβ immune reaction is to fight against viruses derived DNA and RNA molecules, dermatomyositis is reasonable to associated with anti-viral THαβ immunity [128]. B cells, CD4 T cells, CD8 T cells, and NK cells all are important components of THαβ immune reaction, and are all important in the pathogenesis of dermatomyositis [128,129,130,131,132]. The central THαβ immunity related cytokine, interleukin-10, is up-regulated in dermatomyositis and is related to its pathophysiology [133,134,135]. Another important THαβ immune reaction associated cytokine, interleukin-27, is also over-represented in dermatomyositis [136]. These all point out the importance of THαβ immunity in the pathogenesis of dermatomyositis.
As for the signaling pathway of THαβ immunity, these signaling molecules are also over-represented in dermatomyositis. MHC1 molecule, the antigen presenting molecule for CD8 T cells, is over-represented in dermatomyositis [137]. Type 1 interferon related transcription factors, IRF3 and IRF7, are also over-expressed in dermatomyositis [138]. JAK signaling for activating STAT immune master transcription factors is also important in the pathogenesis of dermatomyositis [139]. TLR9 and TLR7, the toll-like receptors related to activating anti-viral THαβ immunity, are also up-regulated in dermatomyositis [132,140]. CXCR3+ lymphocytes and its ligands, CXCL9 and CXCL10, correlating with the disease process of dermatomyositis [141]. These chemokine ligands and chemokine receptor are important in mediating THαβ immunity.
Conclusions
Understanding that the etiologies of type 2 hypersensitivities, encompassing disorders like SLE, Myasthenia gravis, Grave’s disease, graft versus host disease, immune thrombocytopenia, autoimmune hemolytic anemia, dermatomyositis, and Sjogren’s syndrome, are linked to the anti-viral THαβ immunological pathway opens the door to improved diagnosis and treatment approaches for these debilitating conditions. Current management often involves the use of steroid agents, which, while effective, pose significant risks by impacting adaptive immunity and increasing susceptibility to virus or bacterial infections. If THαβ immune response is identified as the primary pathogenic mechanism underlying these diseases, a more targeted approach could involve blocking central THαβ cytokines such as type 1 interferons, interleukin-10, and interleukin-27 using specific inhibitors. This strategic intervention aims to halt the progression of the diseases while circumventing the infection-related drawbacks associated with steroid treatments. This innovative management strategy holds considerable promise in providing more effective and tailored therapeutic options for these autoimmune disorders.
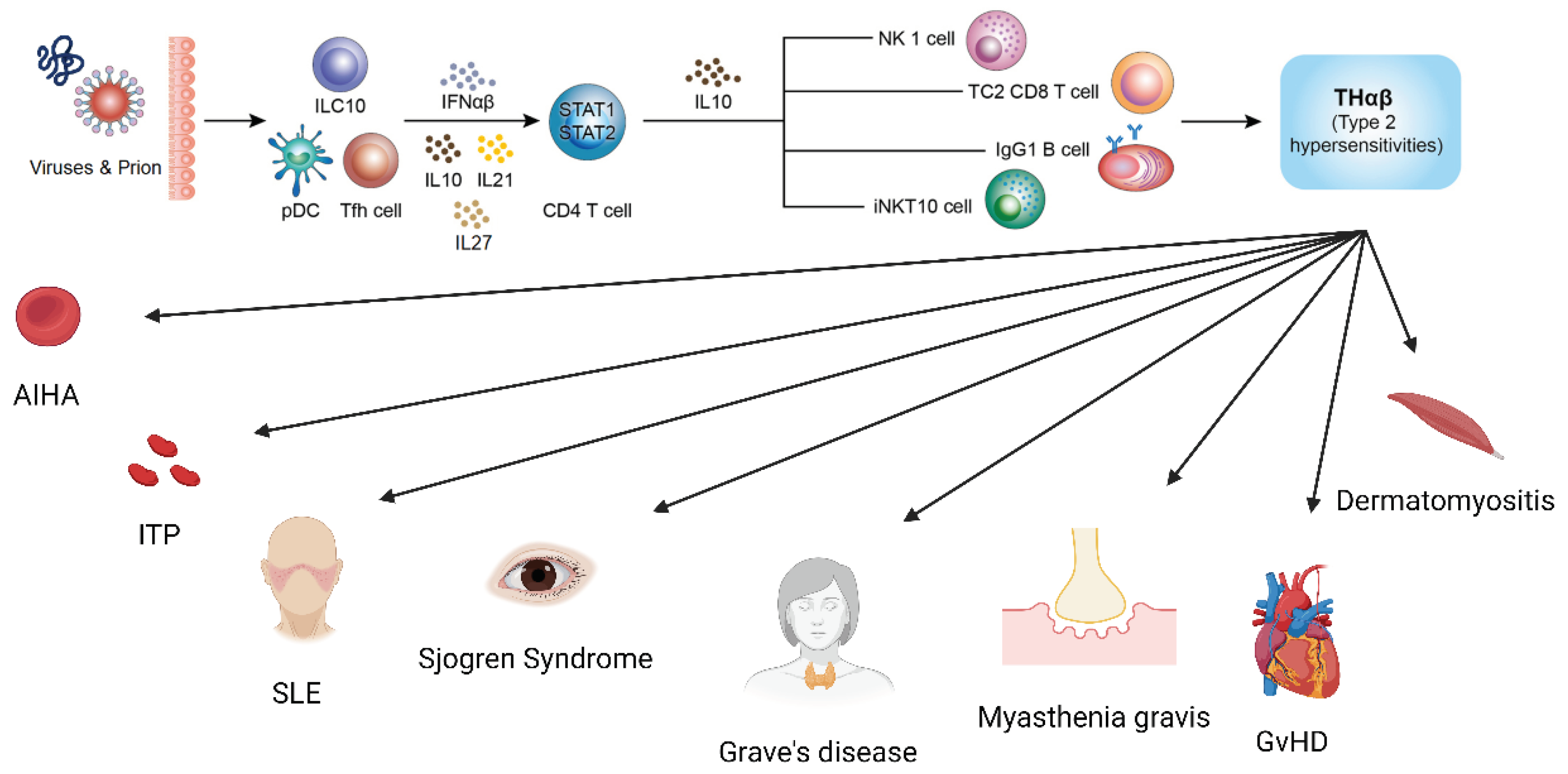
Figure 1.
The anti-viral THαβ immunological pathway and its relations to type 2 hypersensitivity disorders including autoimmune hemolytic anemia, immune thrombocytopenia, systemic lupus erythematosus, Sjogren’s syndrome, Grave’s disease, Myasthenia gravis, graft versus host disease, and dermatomyositis.
Figure 1.
The anti-viral THαβ immunological pathway and its relations to type 2 hypersensitivity disorders including autoimmune hemolytic anemia, immune thrombocytopenia, systemic lupus erythematosus, Sjogren’s syndrome, Grave’s disease, Myasthenia gravis, graft versus host disease, and dermatomyositis.
Author Contributions
YMH, LJS, KWT, TWH, and WCH conducted the study and wrote the manuscript. LJS, KCL, and WCH aided in collecting reference literature and assisted in drafting the manuscript. TWH is helpful for the manuscript draft writing. YMH, LJS and WCH handled the data processing and created tables and figures. YMH, KWT, KCL and WCH oversaw the study and edited the manuscript.
Funding
This study was supported by grants from Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (TCRD-TPE-110-02(2/3) and TCRD-TPE-111-01(3/3)).
Ethical Statement and Consent
This review article examined literature on Type 2 hypersensitivity disorders, encompassing THαβ dominant autoimmune diseases such as Systemic lupus erythematosus, Sjogren’s syndrome, Grave’s disease, Myasthenia Gravis, immune thrombocytopenia, autoimmune hemolytic anemia, dermatomyositis, and graft versus host disease. References were sourced from PubMed and Medline, obviating the need for ethical statements and informed consent in the article’s preparation and completion.
Data Availibility Statement
Data sharing not applicable – no new data generated.
Acknowledgments
The authors would also like to thank the Core Laboratory at the Department of Research, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, for their technical support and use of their facilities.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
References
- Villalta, D.; Bizzaro, N.; Bassi, N.; Zen, M.; Gatto, M.; Ghirardello, A.; Iaccarino, L.; Punzi, L.; Doria, A. Anti-dsDNA antibody isotypes in systemic lupus erythematosus: IgA in addition to IgG anti-dsDNA help to identify glomerulonephritis and active disease. PLoS One 2013, 8, e71458. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Zan, H.; Cerutti, A.; Dramitinos, P.; Schaffer, A.; Casali, P. CD40 engagement triggers switching to IgA1 and IgA2 in human B cells through induction of endogenous TGF-beta: evidence for TGF-beta but not IL-10-dependent direct S mu-->S alpha and sequential S mu-->S gamma, S gamma-->S alpha DNA recombination. J Immunol 1998, 161, 5217–5225. [Google Scholar] [CrossRef] [PubMed]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med 2000, 192, 1545–1552. [Google Scholar] [CrossRef]
- Hu, W.C. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front Immunol 2020, 11, 1992. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 1986, 136, 2348–2357. [Google Scholar] [CrossRef]
- Kobayashi, N.; Kondo, T.; Takata, H.; Yokota, S.; Takiguchi, M. Functional and phenotypic analysis of human memory CD8+ T cells expressing CXCR3. J Leukoc Biol 2006, 80, 320–329. [Google Scholar] [CrossRef]
- Tomiyama, H.; Takata, H.; Matsuda, T.; Takiguchi, M. Phenotypic classification of human CD8+ T cells reflecting their function: inverse correlation between quantitative expression of CD27 and cytotoxic effector function. Eur J Immunol 2004, 34, 999–1010. [Google Scholar] [CrossRef]
- Wen, T.H.; Tsai, K.W.; Wu, Y.J.; Liao, M.T.; Lu, K.C.; Hu, W.C. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef]
- Farne, H.A.; Wilson, A.; Powell, C.; Bax, L.; Milan, S.J. Anti-IL5 therapies for asthma. Cochrane Database Syst Rev 2017, 9, Cd010834. [Google Scholar] [CrossRef]
- Masure, D.; Vlaminck, J.; Wang, T.; Chiers, K.; Van den Broeck, W.; Vercruysse, J.; Geldhof, P. A role for eosinophils in the intestinal immunity against infective Ascaris suum larvae. PLoS Negl Trop Dis 2013, 7, e2138. [Google Scholar] [CrossRef]
- Vliagoftis, H.; Lacy, P.; Luy, B.; Adamko, D.; Hollenberg, M.; Befus, D.; Moqbel, R. Mast cell tryptase activates peripheral blood eosinophils to release granule-associated enzymes. Int Arch Allergy Immunol 2004, 135, 196–204. [Google Scholar] [CrossRef]
- Komai-Koma, M.; Brombacher, F.; Pushparaj, P.N.; Arendse, B.; McSharry, C.; Alexander, J.; Chaudhuri, R.; Thomson, N.C.; McKenzie, A.N.; McInnes, I.; et al. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naive mice. Allergy 2012, 67, 1118–1126. [Google Scholar] [CrossRef]
- Obata, K.; Mukai, K.; Tsujimura, Y.; Ishiwata, K.; Kawano, Y.; Minegishi, Y.; Watanabe, N.; Karasuyama, H. Basophils are essential initiators of a novel type of chronic allergic inflammation. Blood 2007, 110, 913–920. [Google Scholar] [CrossRef]
- Romagnani, P.; De Paulis, A.; Beltrame, C.; Annunziato, F.; Dente, V.; Maggi, E.; Romagnani, S.; Marone, G. Tryptase-Chymase Double-Positive Human Mast Cells Express the Eotaxin Receptor CCR3 and Are Attracted by CCR3-Binding Chemokines. The American Journal of Pathology 1999, 155, 1195–1204. [Google Scholar] [CrossRef]
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075. [Google Scholar] [CrossRef]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.C. Human immune responses to Plasmodium falciparum infection: molecular evidence for a suboptimal THalphabeta and TH17 bias over ideal and effective traditional TH1 immune response. Malaria journal 2013, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.C. The Central THalphabeta Immunity Associated Cytokine: IL-10 Has a Strong Anti-Tumor Ability Toward Established Cancer Models In Vivo and Toward Cancer Cells In Vitro. Front Oncol 2021, 11, 655554. [Google Scholar] [CrossRef] [PubMed]
- Tsou, A.; Chen, P.J.; Tsai, K.W.; Hu, W.C.; Lu, K.C. THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Krovi, S.H.; Gapin, L. Invariant Natural Killer T Cell Subsets-More Than Just Developmental Intermediates. Front Immunol 2018, 9, 1393. [Google Scholar] [CrossRef]
- Prochazkova, J.; Pokorna, K.; Holan, V. IL-12 inhibits the TGF-beta-dependent T cell developmental programs and skews the TGF-beta-induced differentiation into a Th1-like direction. Immunobiology 2012, 217, 74–82. [Google Scholar] [CrossRef]
- Anuradha, R.; George, P.J.; Hanna, L.E.; Chandrasekaran, V.; Kumaran, P.; Nutman, T.B.; Babu, S. IL-4-, TGF-beta-, and IL-1-dependent expansion of parasite antigen-specific Th9 cells is associated with clinical pathology in human lymphatic filariasis. J Immunol 2013, 191, 2466–2473. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nature immunology 2014, 15, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Backert, I.; Koralov, S.B.; Wirtz, S.; Kitowski, V.; Billmeier, U.; Martini, E.; Hofmann, K.; Hildner, K.; Wittkopf, N.; Brecht, K.; et al. STAT3 activation in Th17 and Th22 cells controls IL-22-mediated epithelial host defense during infectious colitis. J Immunol 2014, 193, 3779–3791. [Google Scholar] [CrossRef]
- Kumar, S.; Naqvi, R.A.; Khanna, N.; Pathak, P.; Rao, D.N. Th3 immune responses in the progression of leprosy via molecular cross-talks of TGF-beta, CTLA-4 and Cbl-b. Clin Immunol 2011, 141, 133–142. [Google Scholar] [CrossRef]
- Jiang, R.; Feng, X.; Guo, Y.; Lu, Q.; Hou, J.; Luo, K.; Fu, N.; Venuprasad, K.; Banchereau, J.; Ueno, H. T helper cells in patients with chronic hepatitis B virus infection. Chinese medical journal 2002, 115, 422–424. [Google Scholar] [CrossRef]
- Huang, X.; Dorta-Estremera, S.; Yao, Y.; Shen, N.; Cao, W. Predominant Role of Plasmacytoid Dendritic Cells in Stimulating Systemic Autoimmunity. Front Immunol 2015, 6, 526. [Google Scholar] [CrossRef]
- Elkon, K.B.; Stone, V.V. Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res 2011, 31, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Elkon, K.B.; Wiedeman, A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol 2012, 24, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Eloranta, M.L.; Ronnblom, L. Cause and consequences of the activated type I interferon system in SLE. J Mol Med (Berl) 2016, 94, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W.; investigators, C.D.s. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Amezcua-Guerra, L.M.; Marquez-Velasco, R.; Chavez-Rueda, A.K.; Castillo-Martinez, D.; Masso, F.; Paez, A.; Colin-Fuentes, J.; Bojalil, R. Type III Interferons in Systemic Lupus Erythematosus: Association Between Interferon lambda3, Disease Activity, and Anti-Ro/SSA Antibodies. J Clin Rheumatol 2017, 23, 368–375. [Google Scholar] [CrossRef]
- Goel, R.R.; Wang, X.; O’Neil, L.J.; Nakabo, S.; Hasneen, K.; Gupta, S.; Wigerblad, G.; Blanco, L.P.; Kopp, J.B.; Morasso, M.I.; et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc Natl Acad Sci U S A 2020, 117, 5409–5419. [Google Scholar] [CrossRef]
- Celhar, T.; Fairhurst, A.M. Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Front Pharmacol 2014, 5, 265. [Google Scholar] [CrossRef]
- Christensen, S.R.; Kashgarian, M.; Alexopoulou, L.; Flavell, R.A.; Akira, S.; Shlomchik, M.J. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med 2005, 202, 321–331. [Google Scholar] [CrossRef]
- Ehlers, M.; Fukuyama, H.; McGaha, T.L.; Aderem, A.; Ravetch, J.V. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 2006, 203, 553–561. [Google Scholar] [CrossRef]
- Elloumi, N.; Fakhfakh, R.; Abida, O.; Hachicha, H.; Marzouk, S.; Fourati, M.; Bahloul, Z.; Masmoudi, H. RNA receptors, TLR3 and TLR7, are potentially associated with SLE clinical features. Int J Immunogenet 2021, 48, 250–259. [Google Scholar] [CrossRef]
- Enevold, C.; Kjaer, L.; Nielsen, C.H.; Voss, A.; Jacobsen, R.S.; Hermansen, M.L.; Redder, L.; Oturai, A.B.; Jensen, P.E.; Bendtzen, K.; et al. Genetic polymorphisms of dsRNA ligating pattern recognition receptors TLR3, MDA5, and RIG-I. Association with systemic lupus erythematosus and clinical phenotypes. Rheumatol Int 2014, 34, 1401–1408. [Google Scholar] [CrossRef]
- Fillatreau, S.; Manfroi, B.; Dorner, T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol 2021, 17, 98–108. [Google Scholar] [CrossRef]
- Shen, N.; Fu, Q.; Deng, Y.; Qian, X.; Zhao, J.; Kaufman, K.M.; Wu, Y.L.; Yu, C.Y.; Tang, Y.; Chen, J.Y.; et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A 2010, 107, 15838–15843. [Google Scholar] [CrossRef]
- Horton, C.G.; Pan, Z.J.; Farris, A.D. Targeting Toll-like receptors for treatment of SLE. Mediators Inflamm 2010, 2010. [Google Scholar] [CrossRef]
- Baglaenko, Y.; Manion, K.P.; Chang, N.H.; Gracey, E.; Loh, C.; Wither, J.E. IL-10 Production Is Critical for Sustaining the Expansion of CD5+ B and NKT Cells and Restraining Autoantibody Production in Congenic Lupus-Prone Mice. PLoS One 2016, 11, e0150515. [Google Scholar] [CrossRef]
- Beebe, A.M.; Cua, D.J.; de Waal Malefyt, R. The role of interleukin-10 in autoimmune disease: systemic lupus erythematosus (SLE) and multiple sclerosis (MS). Cytokine Growth Factor Rev 2002, 13, 403–412. [Google Scholar] [CrossRef]
- Sung, Y.K.; Park, B.L.; Shin, H.D.; Kim, L.H.; Kim, S.Y.; Bae, S.C. Interleukin-10 gene polymorphisms are associated with the SLICC/ACR Damage Index in systemic lupus erythematosus. Rheumatology (Oxford) 2006, 45, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Facciotti, F.; Larghi, P.; Bosotti, R.; Vasco, C.; Gagliani, N.; Cordiglieri, C.; Mazzara, S.; Ranzani, V.; Rottoli, E.; Curti, S.; et al. Evidence for a pathogenic role of extrafollicular, IL-10-producing CCR6(+)B helper T cells in systemic lupus erythematosus. Proc Natl Acad Sci U S A 2020, 117, 7305–7316. [Google Scholar] [CrossRef] [PubMed]
- Geginat, J.; Vasco, M.; Gerosa, M.; Tas, S.W.; Pagani, M.; Grassi, F.; Flavell, R.A.; Meroni, P.; Abrignani, S. IL-10 producing regulatory and helper T-cells in systemic lupus erythematosus. Semin Immunol 2019, 44, 101330. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Muchamuel, T.; Sakaguchi, S.; Andrade, S.; Menon, S.; Howard, M. Continuous administration of anti-interleukin 10 antibodies delays onset of autoimmunity in NZB/W F1 mice. J Exp Med 1994, 179, 305–310. [Google Scholar] [CrossRef]
- Conley, M.E.; Koopman, W.J. Serum IgA1 and IgA2 in normal adults and patients with systemic lupus erythematosus and hepatic disease. Clin Immunol Immunopathol 1983, 26, 390–397. [Google Scholar] [CrossRef]
- Otsuka, K.; Sato, M.; Tsunematsu, T.; Ishimaru, N. Virus Infections Play Crucial Roles in the Pathogenesis of Sjogren’s Syndrome. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J. Preferentially immunoglobulin (IgG) subclasses production in primary Sjogren’s syndrome patients. Clin Chem Lab Med 2011, 50, 345–349. [Google Scholar] [CrossRef]
- Karlsen, M.; Hansen, T.; Nordal, H.H.; Brun, J.G.; Jonsson, R.; Appel, S. Expression of Toll-like receptor -7 and -9 in B cell subsets from patients with primary Sjogren’s syndrome. PLoS One 2015, 10, e0120383. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Horai, Y.; Suzuki, T.; Okada, A.; Ichinose, K.; Yamasaki, S.; Koji, T.; Kawakami, A. TLR3-mediated apoptosis and activation of phosphorylated Akt in the salivary gland epithelial cells of primary Sjogren’s syndrome patients. Rheumatol Int 2013, 33, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, I.L.A.; Versnel, M.A. Interferon activation in primary Sjogren’s syndrome: recent insights and future perspective as novel treatment target. Expert Rev Clin Immunol 2018, 14, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Kroese, F.G.M.; Bootsma, H. T cells in primary Sjogren’s syndrome: targets for early intervention. Rheumatology (Oxford) 2021, 60, 3088–3098. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kubo, S.; Nakayamada, S.; Shimajiri, S.; Zhang, X.; Yamaoka, K.; Tanaka, Y. Association of plasmacytoid dendritic cells with B cell infiltration in minor salivary glands in patients with Sjogren’s syndrome. Mod Rheumatol 2016, 26, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Bertorello, R.; Cordone, M.P.; Contini, P.; Rossi, P.; Indiveri, F.; Puppo, F.; Cordone, G. Increased levels of interleukin-10 in saliva of Sjogren’s syndrome patients. Correlation with disease activity. Clin Exp Med 2004, 4, 148–151. [Google Scholar] [CrossRef]
- Ciecko, A.E.; Foda, B.; Barr, J.Y.; Ramanathan, S.; Atkinson, M.A.; Serreze, D.V.; Geurts, A.M.; Lieberman, S.M.; Chen, Y.G. Interleukin-27 Is Essential for Type 1 Diabetes Development and Sjogren Syndrome-like Inflammation. Cell Rep 2019, 29, 3073–3086 e3075. [Google Scholar] [CrossRef]
- Lu, C.; Pi, X.; Xu, W.; Qing, P.; Tang, H.; Li, Y.; Zhao, Y.; Liu, X.; Tang, H.; Liu, Y. Clinical significance of T cell receptor repertoire in primary Sjogren’s syndrome. EBioMedicine 2022, 84, 104252. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjogren syndrome. Immunol Cell Biol 2017, 95, 684–694. [Google Scholar] [CrossRef]
- Pertovaara, M.; Silvennoinen, O.; Isomaki, P. Cytokine-induced STAT1 activation is increased in patients with primary Sjogren’s syndrome. Clin Immunol 2016, 165, 60–67. [Google Scholar] [CrossRef]
- Ramos, H.L.; Valencia-Pacheco, G.; Alcocer-Varela, J. Constitutive STAT3 activation in peripheral CD3(+) cells from patients with primary Sjogren’s syndrome. Scand J Rheumatol 2008, 37, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.C.; Park, C.S.; You, I.C.; Choi, H.J.; Lee, K.H.; Im, S.K.; Park, H.Y.; Pflugfelder, S.C. Expression of CXCL9, -10, -11, and CXCR3 in the tear film and ocular surface of patients with dry eye syndrome. Invest Ophthalmol Vis Sci 2010, 51, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Baber, A.; Nocturne, G.; Krzysiek, R.; Henry, J.; Belkhir, R.; Mariette, X.; Seror, R. Large granular lymphocyte expansions in primary Sjogren’s syndrome: characteristics and outcomes. RMD Open 2019, 5, e001044. [Google Scholar] [CrossRef]
- Leite, M.I.; Jacob, S.; Viegas, S.; Cossins, J.; Clover, L.; Morgan, B.P.; Beeson, D.; Willcox, N.; Vincent, A. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008, 131, 1940–1952. [Google Scholar] [CrossRef]
- Chien, P.J.; Yeh, J.H.; Chiu, H.C.; Hsueh, Y.M.; Chen, C.T.; Chen, M.C.; Shih, C.M. Inhibition of peripheral blood natural killer cell cytotoxicity in patients with myasthenia gravis treated with plasmapheresis. Eur J Neurol 2011, 18, 1350–1357. [Google Scholar] [CrossRef]
- Alseth, E.H.; Nakkestad, H.L.; Aarseth, J.; Gilhus, N.E.; Skeie, G.O. Interleukin-10 promoter polymorphisms in myasthenia gravis. J Neuroimmunol 2009, 210, 63–66. [Google Scholar] [CrossRef]
- Meng, H.; Zheng, S.; Zhou, Q.; Gao, Y.; Ni, Y.; Liang, H.; Chen, S. FoxP3(-) Tr1 Cell in Generalized Myasthenia Gravis and Its Relationship With the Anti-AChR Antibody and Immunomodulatory Cytokines. Front Neurol 2021, 12, 755356. [Google Scholar] [CrossRef]
- Cavalcante, P.; Barzago, C.; Baggi, F.; Antozzi, C.; Maggi, L.; Mantegazza, R.; Bernasconi, P. Toll-like receptors 7 and 9 in myasthenia gravis thymus: amplifiers of autoimmunity? Ann N Y Acad Sci 2018, 1413, 11–24. [Google Scholar] [CrossRef]
- Robinet, M.; Maillard, S.; Cron, M.A.; Berrih-Aknin, S.; Le Panse, R. Review on Toll-Like Receptor Activation in Myasthenia Gravis: Application to the Development of New Experimental Models. Clin Rev Allergy Immunol 2017, 52, 133–147. [Google Scholar] [CrossRef]
- Yapici, Z.; Tuzun, E.; Altunayoglu, V.; Erdogan, A.; Eraksoy, M. High interleukin-10 production is associated with anti-acetylcholine receptor antibody production and treatment response in juvenile myasthenia gravis. Int J Neurosci 2007, 117, 1505–1512. [Google Scholar] [CrossRef]
- Jeong, H.N.; Lee, J.H.; Suh, B.C.; Choi, Y.C. Serum interleukin-27 expression in patients with myasthenia gravis. J Neuroimmunol 2015, 288, 120–122. [Google Scholar] [CrossRef]
- Cufi, P.; Dragin, N.; Ruhlmann, N.; Weiss, J.M.; Fadel, E.; Serraf, A.; Berrih-Aknin, S.; Le Panse, R. Central role of interferon-beta in thymic events leading to myasthenia gravis. J Autoimmun 2014, 52, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Cufi, P.; Dragin, N.; Weiss, J.M.; Martinez-Martinez, P.; De Baets, M.H.; Roussin, R.; Fadel, E.; Berrih-Aknin, S.; Le Panse, R. Implication of double-stranded RNA signaling in the etiology of autoimmune myasthenia gravis. Ann Neurol 2013, 73, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, P.; Maggi, L.; Colleoni, L.; Caldara, R.; Motta, T.; Giardina, C.; Antozzi, C.; Berrih-Aknin, S.; Bernasconi, P.; Mantegazza, R. Inflammation and epstein-barr virus infection are common features of myasthenia gravis thymus: possible roles in pathogenesis. Autoimmune Dis 2011, 2011, 213092. [Google Scholar] [CrossRef]
- Leopardi, V.; Chang, Y.M.; Pham, A.; Luo, J.; Garden, O.A. A Systematic Review of the Potential Implication of Infectious Agents in Myasthenia Gravis. Front Neurol 2021, 12, 618021. [Google Scholar] [CrossRef] [PubMed]
- Feferman, T.; Maiti, P.K.; Berrih-Aknin, S.; Bismuth, J.; Bidault, J.; Fuchs, S.; Souroujon, M.C. Overexpression of IFN-induced protein 10 and its receptor CXCR3 in myasthenia gravis. J Immunol 2005, 174, 5324–5331. [Google Scholar] [CrossRef]
- Nisihara, R.; Pigosso, Y.G.; Prado, N.; Utiyama, S.R.R.; De Carvalho, G.A.; Skare, T.L. Rheumatic Disease Autoantibodies in Patients with Autoimmune Thyroid Diseases. Med Princ Pract 2018, 27, 332–336. [Google Scholar] [CrossRef]
- Ueki, I.; Abiru, N.; Kawagoe, K.; Nagayama, Y. Interleukin 10 deficiency attenuates induction of anti-TSH receptor antibodies and hyperthyroidism in a mouse Graves’ model. J Endocrinol 2011, 209, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Pedro, A.B.; Romaldini, J.H.; Americo, C.; Takei, K. Association of circulating antibodies against double-stranded and single-stranded DNA with thyroid autoantibodies in Graves’ disease and Hashimoto’s thyroiditis patients. Exp Clin Endocrinol Diabetes 2006, 114, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Giuggioli, D.; Ferrannini, E.; Ferri, C.; Fallahi, P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev 2014, 13, 272–280. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, K.W.; Choi, J.H.; An, J.H. Interferon-Alpha Induced Severe Hypothyroidism Followed by Graves’ Disease in a Patient Infected with Hepatitis C Virus. International Journal of Thyroidology 2015, 8. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Zhao, K.; Yu, N.; Li, Y.; Yu, Y.; Zhang, Y.; Song, Z.; Huang, Y.; Lu, G.; et al. Glycosylation of Anti-Thyroglobulin IgG1 and IgG4 Subclasses in Thyroid Diseases. Eur Thyroid J 2021, 10, 114–124. [Google Scholar] [CrossRef]
- Liu, N.; Lu, H.; Tao, F.; Guo, T.; Liu, C.; Cui, B.; Ning, G. An association of interleukin-10 gene polymorphisms with Graves’ disease in two Chinese populations. Endocrine 2011, 40, 90–94. [Google Scholar] [CrossRef]
- Peng, S.; Li, C.; Wang, X.; Liu, X.; Han, C.; Jin, T.; Liu, S.; Zhang, X.; Zhang, H.; He, X.; et al. Increased Toll-Like Receptors Activity and TLR Ligands in Patients with Autoimmune Thyroid Diseases. Front Immunol 2016, 7, 578. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, L.; Guo, C.; Kou, C.; Long, Y.; Li, J.; Zhang, H.-Q. Reduced proportion and activity of natural killer cells in patients with Graves’ disease. European Journal of Inflammation 2020, 18. [Google Scholar] [CrossRef]
- Villarroel, V.A.; Okiyama, N.; Tsuji, G.; Linton, J.T.; Katz, S.I. CXCR3-mediated skin homing of autoreactive CD8 T cells is a key determinant in murine graft-versus-host disease. J Invest Dermatol 2014, 134, 1552–1560. [Google Scholar] [CrossRef]
- Abraham, S.; Guo, H.; Choi, J.G.; Ye, C.; Thomas, M.B.; Ortega, N.; Dwivedi, A.; Manjunath, N.; Yi, G.; Shankar, P. Combination of IL-10 and IL-2 induces oligoclonal human CD4 T cell expansion during xenogeneic and allogeneic GVHD in humanized mice. Heliyon 2017, 3, e00276. [Google Scholar] [CrossRef] [PubMed]
- Duffner, U.; Lu, B.; Hildebrandt, G.C.; Teshima, T.; Williams, D.L.; Reddy, P.; Ordemann, R.; Clouthier, S.G.; Lowler, K.; Liu, C.; et al. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp Hematol 2003, 31, 897–902. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Blaser, B.W.; Freud, A.G.; Katz, K.; Bhatt, D.; Ferketich, A.K.; Bergdall, V.; Kusewitt, D.; Baiocchi, R.A.; Caligiuri, M.A. IL-15 but not IL-2 rapidly induces lethal xenogeneic graft-versus-host disease. Blood 2005, 106, 2433–2435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Y.; Zheng, H. The Effects of Interferons on Allogeneic T Cell Response in GVHD: The Multifaced Biology and Epigenetic Regulations. Front Immunol 2021, 12, 717540. [Google Scholar] [CrossRef]
- Martin-Antonio, B.; Suarez-Lledo, M.; Arroyes, M.; Fernandez-Avilés, F.; Martinez, C.; Rovira, M.; Espigado, I.; Gallardo, D.; Bosch, A.; Buno, I.; et al. A Gene Variant in IRF3 Impacts On the Clinical Outcome of Acute Myeloid Leukemia (AML) Patients Submitted to Allogeneic Stem Cell Transplantation (allo-SCT). Blood 2012, 120, 468–468. [Google Scholar] [CrossRef]
- Hakim, F.T.; Memon, S.; Jin, P.; Imanguli, M.M.; Wang, H.; Rehman, N.; Yan, X.Y.; Rose, J.; Mays, J.W.; Dhamala, S.; et al. Upregulation of IFN-Inducible and Damage-Response Pathways in Chronic Graft-versus-Host Disease. J Immunol 2016, 197, 3490–3503. [Google Scholar] [CrossRef] [PubMed]
- Betts, B.C.; Sagatys, E.M.; Veerapathran, A.; Lloyd, M.C.; Beato, F.; Lawrence, H.R.; Yue, B.; Kim, J.; Sebti, S.M.; Anasetti, C.; et al. CD4+ T cell STAT3 phosphorylation precedes acute GVHD, and subsequent Th17 tissue invasion correlates with GVHD severity and therapeutic response. J Leukoc Biol 2015, 97, 807–819. [Google Scholar] [CrossRef]
- Ziegler, J.A.; Lokshin, A.; Sepulveda, A.R.; Bedeir, A.; Lentzsch, S.; Mapara, M.Y. Role of STAT1 Expression during Graft-Versus-Host Disease (GVHD) in the Gastrointestinal Tract: Association with Lamina Propria Cell Infiltration and Tissue Cytokine/Chemokine Expression. Blood 2006, 108, 3175–3175. [Google Scholar] [CrossRef]
- Belle, L.; Agle, K.; Zhou, V.; Yin-Yuan, C.; Komorowski, R.; Eastwood, D.; Logan, B.; Sun, J.; Ghilardi, N.; Cua, D.; et al. Blockade of interleukin-27 signaling reduces GVHD in mice by augmenting Treg reconstitution and stabilizing Foxp3 expression. Blood 2016, 128, 2068–2082. [Google Scholar] [CrossRef]
- Calcaterra, C.; Sfondrini, L.; Rossini, A.; Sommariva, M.; Rumio, C.; Menard, S.; Balsari, A. Critical role of TLR9 in acute graft-versus-host disease. J Immunol 2008, 181, 6132–6139. [Google Scholar] [CrossRef]
- Suthers, A.N.; Su, H.; Anand, S.M.; Poe, J.C.; Rose, J.J.; Hakim, F.T.; Pavletic, S.Z.; Rizzieri, D.A.; Horwitz, M.E.; Yang, Y.; et al. Increased TLR7 Signaling of BCR-Activated B Cells in Chronic Graft-Versus Host Disease (cGVHD). Blood 2017, 130, 75–75. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Nogai, A.; Bereswill, S.; Plickert, R.; Fischer, A.; Loddenkemper, C.; Steinhoff, U.; Tchaptchet, S.; Thiel, E.; Freudenberg, M.A.; et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut 2010, 59, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Hashimoto, D.; Aoyama, K.; Matsuoka, K.; Karube, K.; Niiro, H.; Harada, M.; Tanimoto, M.; Akashi, K.; Teshima, T. Plasmacytoid dendritic cells prime alloreactive T cells to mediate graft-versus-host disease as antigen-presenting cells. Blood 2009, 113, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Sayed, A.A.; Han, P.; Tan, M.M.H.; Watt, E.; Constantinescu-Bercu, A.; Cocker, A.T.H.; Khoder, A.; Saputil, R.C.; Thorley, E.; et al. The role of CD8+ T-cell clones in immune thrombocytopenia. Blood 2023, 141, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Tesse, R.; Del Vecchio, G.C.; De Mattia, D.; Sangerardi, M.; Valente, F.; Giordano, P. Association of interleukin-(IL)10 haplotypes and serum IL-10 levels in the progression of childhood immune thrombocytopenic purpura. Gene 2012, 505, 53–56. [Google Scholar] [CrossRef]
- Hua, F.; Ji, L.; Zhan, Y.; Li, F.; Zou, S.; Chen, L.; Gao, S.; Li, Y.; Chen, H.; Cheng, Y. Aberrant frequency of IL-10-producing B cells and its association with Treg/Th17 in adult primary immune thrombocytopenia patients. Biomed Res Int 2014, 2014, 571302. [Google Scholar] [CrossRef]
- Hassan, T.; Abdel Rahman, D.; Raafat, N.; Fathy, M.; Shehab, M.; Hosny, A.; Fawzy, R.; Zakaria, M. Contribution of interleukin 27 serum level to pathogenesis and prognosis in children with immune thrombocytopenia. Medicine (Baltimore) 2022, 101, e29504. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Xue, F.; Xu, J.; Fang, Z. Interleukin-27 rs153109 polymorphism and the risk for immune thrombocytopenia. Autoimmunity 2013, 46, 509–512. [Google Scholar] [CrossRef]
- Yamane, A.; Nakamura, T.; Suzuki, H.; Ito, M.; Ohnishi, Y.; Ikeda, Y.; Miyakawa, Y. Interferon-alpha 2b-induced thrombocytopenia is caused by inhibition of platelet production but not proliferation and endomitosis in human megakaryocytes. Blood 2008, 112, 542–550. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, S.; Li, X.; Wang, B.; Wang, X.; Ma, D.; Yang, L.; Peng, J.; Hou, M. Pathway of Toll-like receptor 7/B cell activating factor/B cell activating factor receptor plays a role in immune thrombocytopenia in vivo. PLoS One 2011, 6, e22708. [Google Scholar] [CrossRef]
- Chan, H.; Moore, J.C.; Finch, C.N.; Warkentin, T.E.; Kelton, J.G. The IgG subclasses of platelet-associated autoantibodies directed against platelet glycoproteins IIb/IIIa in patients with idiopathic thrombocytopenic purpura. Br J Haematol 2003, 122, 818–824. [Google Scholar] [CrossRef]
- Liu, Y.; Zuo, X.; Chen, P.; Hu, X.; Sheng, Z.; Liu, A.; Liu, Q.; Leng, S.; Zhang, X.; Li, X.; et al. Deciphering transcriptome alterations in bone marrow hematopoiesis at single-cell resolution in immune thrombocytopenia. Signal Transduct Target Ther 2022, 7, 347. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Guo, Z.; Ma, J.; Liu, F.; Gao, C.; Liu, S.; Wang, A.; Wu, R. STAT1 single nucleotide polymorphisms and susceptibility to immune thrombocytopenia. Autoimmunity 2015, 48, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ke, Y.; Cheng, Y.; Zhan, Y.; Wu, B. Aberrant Phosphorylation of STAT3 Protein of the CD4+ T Cells in Patients with Primary Immune Thrombocytopenia. Blood 2016, 128, 3740–3740. [Google Scholar] [CrossRef]
- Ward, F.J.; Hall, A.M.; Cairns, L.S.; Leggat, A.S.; Urbaniak, S.J.; Vickers, M.A.; Barker, R.N. Clonal regulatory T cells specific for a red blood cell autoantigen in human autoimmune hemolytic anemia. Blood 2008, 111, 680–687. [Google Scholar] [CrossRef]
- Sonneveld, M.E.; de Haas, M.; Koeleman, C.; de Haan, N.; Zeerleder, S.S.; Ligthart, P.C.; Wuhrer, M.; van der Schoot, C.E.; Vidarsson, G. Patients with IgG1-anti-red blood cell autoantibodies show aberrant Fc-glycosylation. Sci Rep 2017, 7, 8187. [Google Scholar] [CrossRef]
- Rangnekar, A.; Shenoy, M.S.; Mahabala, C.; Balanthimogru, P. Impact of baseline fluorescent antinuclear antibody positivity on the clinical outcome of patients with primary autoimmune hemolytic anemia. Hematol Transfus Cell Ther 2023, 45, 204–210. [Google Scholar] [CrossRef]
- Pattanakitsakul, P.; Sirachainan, N.; Tassaneetrithep, B.; Priengprom, T.; Kijporka, P.; Apiwattanakul, N. Enterovirus 71-Induced Autoimmune Hemolytic Anemia in a Boy. Clin Med Insights Case Rep 2022, 15, 11795476221132283. [Google Scholar] [CrossRef]
- Gao, Y.; Jin, H.; Nan, D.; Yu, W.; Zhang, J.; Yang, Y.; Hou, R.; Qin, R.; Hao, H.; Sun, Y.; et al. The Role of T Follicular Helper Cells and T Follicular Regulatory Cells in the Pathogenesis of Autoimmune Hemolytic Anemia. Sci Rep 2019, 9, 19767. [Google Scholar] [CrossRef] [PubMed]
- Toriani-Terenzi, C.; Fagiolo, E. IL-10 and the cytokine network in the pathogenesis of human autoimmune hemolytic anemia. Ann N Y Acad Sci 2005, 1051, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, E.; Elgohary, T.; Ibrahim, H. Naturally occurring regulatory T cells and interleukins 10 and 12 in the pathogenesis of idiopathic warm autoimmune hemolytic anemia. J Investig Allergol Clin Immunol 2011, 21, 297–304. [Google Scholar]
- Wang, S.; Qin, E.; Zhi, Y.; Hua, R. Severe autoimmune hemolytic anemia during pegylated interferon plus ribavirin treatment for chronic hepatitis C: a case report. Clin Case Rep 2017, 5, 1490–1492. [Google Scholar] [CrossRef]
- Xie, Y.; Shao, F.; Lei, J.; Huang, N.; Fan, Z.; Yu, H. Case report: A STAT1 gain-of-function mutation causes a syndrome of combined immunodeficiency, autoimmunity and pure red cell aplasia. Front Immunol 2022, 13, 928213. [Google Scholar] [CrossRef]
- Ciullini Mannurita, S.; Goda, R.; Schiavo, E.; Coniglio, M.L.; Azzali, A.; Fotzi, I.; Tondo, A.; Tintori, V.; Frenos, S.; Sanvito, M.C.; et al. Case Report: Signal Transducer and Activator of Transcription 3 Gain-of-Function and Spectrin Deficiency: A Life-Threatening Case of Severe Hemolytic Anemia. Front Immunol 2020, 11, 620046. [Google Scholar] [CrossRef]
- Poulet, F.M.; Penraat, K.; Collins, N.; Evans, E.; Thackaberry, E.; Manfra, D.; Engstrom, L.; Geissler, R.; Geraci-Erck, M.; Frugone, C.; et al. Drug-induced hemolytic anemia and thrombocytopenia associated with alterations of cell membrane lipids and acanthocyte formation. Toxicol Pathol 2010, 38, 907–922. [Google Scholar] [CrossRef]
- Greenberg, S.A. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep 2010, 12, 198–203. [Google Scholar] [CrossRef]
- Zampieri, S.; Ghirardello, A.; Iaccarino, L.; Tarricone, E.; Gambari, P.F.; Doria, A. Anti-Jo-1 antibodies. Autoimmunity 2005, 38, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Fasth, A.E.; Dastmalchi, M.; Rahbar, A.; Salomonsson, S.; Pandya, J.M.; Lindroos, E.; Nennesmo, I.; Malmberg, K.J.; Soderberg-Naucler, C.; Trollmo, C.; et al. T cell infiltrates in the muscles of patients with dermatomyositis and polymyositis are dominated by CD28null T cells. J Immunol 2009, 183, 4792–4799. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, K.A.; Throm, A.A.; Pingel, J.T.; Saucier, N.; Zaher, H.S.; French, A.R. Expansion of a novel population of NK cells with low ribosome expression in juvenile dermatomyositis. Front Immunol 2022, 13, 1007022. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Tang, L.; Zhang, L.; Ren, Y.; Peng, H.; Xiao, Y.; Xu, J.; Mao, D.; Liu, L.; Liu, L. Identification of Biomarkers Associated With CD4(+) T-Cell Infiltration With Gene Coexpression Network in Dermatomyositis. Front Immunol 2022, 13, 854848. [Google Scholar] [CrossRef] [PubMed]
- Houtman, M.; Ekholm, L.; Hesselberg, E.; Chemin, K.; Malmstrom, V.; Reed, A.M.; Lundberg, I.E.; Padyukov, L. T-cell transcriptomics from peripheral blood highlights differences between polymyositis and dermatomyositis patients. Arthritis Res Ther 2018, 20, 188. [Google Scholar] [CrossRef]
- Piper, C.J.M.; Wilkinson, M.G.L.; Deakin, C.T.; Otto, G.W.; Dowle, S.; Duurland, C.L.; Adams, S.; Marasco, E.; Rosser, E.C.; Radziszewska, A.; et al. CD19(+)CD24(hi)CD38(hi) B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-alpha. Front Immunol 2018, 9, 1372. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Higgs, B.W.; Morehouse, C.; Walsh, R.J.; Kong, S.W.; Brohawn, P.; Zhu, W.; Amato, A.; Salajegheh, M.; White, B.; et al. Relationship between disease activity and type 1 interferon- and other cytokine-inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun 2012, 13, 207–213. [Google Scholar] [CrossRef]
- Kishi, T.; Chipman, J.; Evereklian, M.; Nghiem, K.; Stetler-Stevenson, M.; Rick, M.E.; Centola, M.; Miller, F.W.; Rider, L.G. Endothelial Activation Markers as Disease Activity and Damage Measures in Juvenile Dermatomyositis. J Rheumatol 2020, 47, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Szodoray, P.; Alex, P.; Knowlton, N.; Centola, M.; Dozmorov, I.; Csipo, I.; Nagy, A.T.; Constantin, T.; Ponyi, A.; Nakken, B.; et al. Idiopathic inflammatory myopathies, signified by distinctive peripheral cytokines, chemokines and the TNF family members B-cell activating factor and a proliferation inducing ligand. Rheumatology (Oxford) 2010, 49, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xia, L.; Lu, J. Pilot study of interleukin-27 in pathogenesis of dermatomyositis and polymyositis: associated with interstitial lung diseases. Cytokine 2012, 60, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Li, C.K.; Varsani, H.; Holton, J.L.; Gao, B.; Woo, P.; Wedderburn, L.R. MHC Class I overexpression on muscles in early juvenile dermatomyositis. J Rheumatol 2004, 31, 605–609. [Google Scholar] [PubMed]
- Nombel, A.; Fabien, N.; Coutant, F. Dermatomyositis With Anti-MDA5 Antibodies: Bioclinical Features, Pathogenesis and Emerging Therapies. Front Immunol 2021, 12, 773352. [Google Scholar] [CrossRef]
- Ladislau, L.; Suarez-Calvet, X.; Toquet, S.; Landon-Cardinal, O.; Amelin, D.; Depp, M.; Rodero, M.P.; Hathazi, D.; Duffy, D.; Bondet, V.; et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain 2018, 141, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.T.; Cho, M.L.; Park, Y.E.; Yoo, W.H.; Kim, J.H.; Oh, H.J.; Kim, D.S.; Baek, S.H.; Lee, S.H.; Lee, J.H.; et al. Expression of TLR2, TLR4, and TLR9 in dermatomyositis and polymyositis. Clin Rheumatol 2010, 29, 273–279. [Google Scholar] [CrossRef]
- Bellutti Enders, F.; van Wijk, F.; Scholman, R.; Hofer, M.; Prakken, B.J.; van Royen-Kerkhof, A.; de Jager, W. Correlation of CXCL10, tumor necrosis factor receptor type II, and galectin 9 with disease activity in juvenile dermatomyositis. Arthritis Rheumatol 2014, 66, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



