
Submitted:
28 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
Chitinase 3-like 1 (also known as CHI3L1 or YKL-40)is a mammalian chitinase that has no enzymatic activity, but has the ability to bind to chitin, the polymer of N-acetylglucosamine (GlcNAc). Chitin is a component of fungi, crustaceans, arthropods including insects and mites, and parasites, but is completely absent from mammals, including humans and mice. In general, chitin-containing organisms produce CHI3L1 to protect the body from exogenous pathogen as well as hostile environments, and it was thought that it has a similar effect in mammals. However, recent studies have revealed that CHI3L1 plays a pro-inflammatory role by inducing anti-apoptotic activity in epithelial cells and macrophages.
Under chronic inflammatory conditions such as inflammatory bowel disease and chronic obstructive pulmonary disease, many groups already confirmed that the expression of CHI3L1 is significantly induced on the apical side of epithelial cells, and activates many downstream pathways involved in inflammation and carcinogenesis.
In this review article, we will summarize the expression of CHI3L1 under the chronic inflammatory conditions in various disorders and would like to discuss the potential roles of CHI3L1 in those disorders on various cell types.
Keywords:
inflammation
; pathogenesis
; inflammatory bowel disease
; asthma
; dysbiosis
1. Introductions
Inflammatory Bowel Disease (IBD) includes two major diseases, Ulcerative Colitis (UC) and Crohn’s Disease (CD), both of which are diseases associated with chronic inflammation that are difficult to cure even if they go into remission, and the patients must live with these chronic diseases for the rest of their lives most of cases [1,2,3]. Dysbiosis, an abnormal composition of the intestinal microflora, and complex reactivity of genetic, environmental, and immunological factors are involved in the development of IBD, and in particular, the abnormal interactions between host cells and potentially pathogenic bacteria are thought to play an important role in its pathogenesis as well as pathology [4].
In healthy individuals, microflora and intestinal epithelial cells are tightly separated by a mucin layer consisting of an inner and outer two-later structure [4]. In the status of dysbiosis, several changes occur in the interactions between the host and the resident microflora, resulting in the pathogenesis of intestinal inflammation induced by various factors such as increased epithelial permeability, decreased productions of antimicrobial peptides as well as mucosal mucins, altered cytokine balance, excess inflammatory cell infiltration, and endoplasmic reticulum stress [4,5,6]. Furthermore, some mammalian chitinases, of which expression is mainly induced in cells involved in innate immunity such as epithelial cells and macrophages under chronic inflammation, are also one of the factors that play a central role in host-microbial interactions [7,8,9].
Chitinases produced by mammals and bacteria are glycosidases that break down the glycosidic bonds of chitin, a polymer of N-acetyl glucosamine. Chitin is a major component of the exoskeleton and cell walls of various organisms, including arthropods, nematodes, and fungi. However, it’s existence in vertebrates (including mice and humans) has not been confirmed. Among the 131 types of Glycoside Hydrolases (GH), chitinases are broadly classified into the families of GH18 and GH19 based on differences in their amino acid sequences, three-dimensional structures, and catalytic actions [10]. The GH18 family chitinase is divided into two types: authentic (true) chitinases, which has enzymatic activity, and Chitinae-Like Proteins (CLP), which do not have enzymatic activities. The former includes Chitotriosidase (chitinase 1) and Acidic mammalian chitinase (AMCase), and the latter includes Chitinase 3-like 1 (CHI3L1 or YKL-40) [10]. It has been suggested that CLPs act as endogenous lectins that recognize specific sugar chains, such as chitin and chito-oligosaccharide, and regulate cell adhesion, migration, differentiation, and proliferation [11]. True chitinases and CLPs share structural similarities, but they have different catalytic activities due to differences in their amino acid structures. CHI3L1 completely loses enzymatic activity due to the replacement of the glutamic acid residue with leucin residue in the chiton-binding site in its multidomain structure, but it remains sufficient affinity for chitin [12,13]. Although many aspects of the in vivo functions of mammalian chitinases remain unclear, they are strongly involved in immune responses in various organs and tissues. In particular, CHI3L1 is strongly correlated with certain pathological conditions, including tissue damage responses and acute/chronic inflammation, and is likely to influence both innate and adaptive immunity [14].
Epithelial cells, in particular colonic epithelial cells (CECs), form a barrier mechanism as the first line of defense to avoid immune responses from external stimuli. In addition to absorbing nutrients and water, CECs protect intestinal tissues from many intestinal bacteria, and form a two-layered mucin layer on the outside. The mucin layers, together with CECs, form a strong barrier between intestinal bacteria and the lamina propria, and greatly contribute to the maintenance of homeostasis in the intestinal tract.
However, once this mucosal layer is thinner or lost, an imbalance between the host and intestinal flora, so called dysbiosis, occurs and resulting in inflammation of the intestinal tract [4,15]. In this review article, we would like to discuss the potential role of CHI3L1 expressed on epithelial cells during the inflammatory conditions in mice and humans.
2. Potential Biological Roles of CHI3L1
In 2004, Elias group reported an exciting fact that a formation of crystallin structures in lung tissues of a murine asthma model and identified that the crystals were AMCase, one of the true mammalian chitinases [16]. In this report the authors discovered that AMCase is induced by Interleukin (IL)-13-mediated T helper-2 (Th2)-specific inflammatory signaling pathway in lung epithelial cells and macrophages [16]. Later studies revealed that not only AMCase but also CHI3L1 levels I serum as well as lung tissues were significantly upregulated in the joint cohort study of the U.S. and France [17] suggesting that serum CHI3L1 produced by various cell types become a good parameter for inflammatory conditions. A huge numbers of reports are published regarding the association between CHI3L1 and inflammatory conditions over the past nearly three decades [18,19,20,21,22]. Over the past five years, the role of CHI3L1 in various types of inflammation has been rapidly elucidated as shown in Table 1 [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74]. We also have summarized the potential roles of CHI3L1 in the major four diseases in the following Section 2.1.
2.1. Increased Levels of CHI3L1 under the Major Inflammatory Disorders
2.1.1. Inflammatory Bowel Disease (IBD)
The serum as well as tissue levels of CHI3L1 are significantly elevated in patients with IBD including UC and CD, and the elevation of serum CHI3L1 is primarily associated with the severity, the extent of inflammation, and the existence of complications such as arthritis [7,18,19,20,21]. Interestingly, serum CHI3L1 levels become high in the CD patients with fibrosis, which appears in more severe cases suggesting the CHI3L1 as a possible inflammatory biomarker in IBD [22]. Our group previously reported that the CHI3L1 mRNA expression levels were increased in active UC and involved the region of CD compared with inactive UC, the uninvolved region of CD, and normal individuals [7]. Maily apical sides of CECs seem to produce CHI3L1 protein in the involved region of active CD patients [7]. Since the expression pattern of CHI3L1and bacterial biofilm formation are almost identical, it is easy to imagine the interaction between CHI3L1 expression and intestinal bacteria, in particular potentially pathogenic bacteria. In active phase of IBD, CHI3L1 is continuously secreted as a 40kDa protein from CECs and macrophages into the intestinal lumens, and therefore it is reasonable that not only serum but also fecal CHI3L1 seems to be a reliable biomarker for predicting the severity and activity of IBD [75,76].
Interestingly, fecal CHI3L1 expression levels were almost undetectable in healthy individuals and a non-significant step-wise increase in IBD patients under the remission phase (CRP<0.1), but the levels were significantly upregulated in IBD patients with dysplasia/adenocarcinoma compared with other adenoma or sporadic colorectal cancer patients, suggesting that fecal CHI3L1 levels might be a non-invasive and reliable biomarker for IBD-associated malignant changes of CECs under the remission phase of IBD [77].
2.1.2. Multiple Sclerosis (MS)
MS is an inflammatory demyelinating disease of the central nervous system and is characterized by multiple temporal and spatial occurrences. It is an intractable autoimmune disease that takes a chronic course and causes inflammation in the brain, spinal cord, and optic nerves, damaging nerve tissue. In 2010, Comabella et al. identified that CHI3L1 seems to be a prognostic biomarker for conversion to MS and development of disability utilizing the previously collected cerebrospinal fluid samples from MS patients by a mass spectrometry-based proteomic approach validating with ELISA (enzyme-linked immunosorbent assay) [78]. Positively associated elevation of CHI3L1 in MS samples was followed/reviewed by many other groups so far [27,28,29,30,31,32,33,34,35,79,80,81].
Of note, CHI3L1 expression in astrocytes positively associated with increased expression of representative proinflammatory cytokines, IL-1 and IL-6, and together with IL-6 family cytokine, Oncostain M, synergistically upregulated CHI3L1 expression, of which is required for both STAT3 and NF-κB binding elements of the promoter region of CHI3L1 [79]. Our group also previously proved that CHI3L1 and IL-6 synergistically activates STAT3 signaling pathway in intestinal epithelial cells in an NF-κB and MAPK-dependent manner [82], so it is extremely interesting that the same thing happens in brain tissue as well. In addition, dysregulated productions of TNF, another representative pro-inflammatory cytokine, and soluble TNF receptors type I and type II protein levels in CSF associate with specific clinical profiles and are useful for identifying at a very early stage in MS patients, that is very useful to the prediction of the MS disease outcome [32]. Overall, CHI3L1 seems to be regulated by the signaling pathways of pro-inflammatory cytokines and their receptors in MS as well as IBD [7,32,79,82,83].
2.1.3. Alzheimer’s Disease (AD)
AD, frequently occurring and the debilitating disorder of the central nervous system (CNS), is classically viewed as a progression neurodegenerative disorder resulting in intellectual capabilities, memory loss, and spatial disorientation [84]. The hallmarks of AD pathology are the deposition of amyloid beta (Aβ) containing plaques and neurofibrillary tangles composed of hyperphosphorylated tau protein in the brain [85]. In the longitudinal early-onset AD study (LEADS), results show that cerebrospinal fluid (CSF) biomarkers were correlated with each other including CHI3L1, and CSF CHI3L1 was associated with cognition and astrocytic changes during early onset of AD [41,86]. The CHI3L1 levels in CSF, also used as an astrocyte biomarker, increased very early in AD progression and mediated Aβ-induced tau phosphorylation and tau-induced neuronal injury. One study observed that CSF CHI3L1 levels were associated with tau pathology and the over-secreted CHI3L1 from astrocytes related to the accumulation of tau tangles in the living AD brains. These results suggest that CHI3L1 is an important mediator of key pathogenic events in the AD pathogenic cascade and contribute to the AD progression [87,88]. In cells and mice, the deletion of CHI3L1 alters the responses of glial inflammation, promotes microglial Aβ and astrocyte phagocytosis, and decreases amyloid plaque deposition, but glial activation and neuroinflammation may be dependent on context because that the deletion of CHI3L1 could be neuro-protective in AD, but destructive in acute inflammation. Thus, the up-regulation of CHI3L1 suppressed microglial Aβ and astrocyte phagocytosis and accelerated amyloid plaque formation, which contribute to the progression of AD [89]. The increase of CHI3L1 was also associated with cognitive dysfunction, and CHI3L1 plays a significant role in white matter neuroinflammation associated with cognitive decline in AD patients, which suggests that white matter CHI3L1 relates to cognitive impairment in the early onset of AD [39]. Recent studies confirmed that DNA variants in CHI3L1 could be associated with increased neuronal injury and inflammation, and CSF levels of CHI3L1 could lead to the increased risk of AD. Also, the CHI3L1 expression in both blood and CSF is positively associated with variants in CHI3L1 [36,90]. The suppression of CHI3L1 DNA variants may contribute to lower the levels of blood and CSF CHI3L1, which reduce the risk of AD development.
In conclusion, up-regulation of CHI3L1 both in blood and CSF can contribute to the progression of AD, and the implications of anti-CHI3L1 therapies may enhance treatment responses in future clinical trials.
2.1.4. Asthma, Chronic Obstructive Pulmonary Disease (COPD)
Recent studies have demonstrated that the concentration of serum CHI3L1, which relates to the severity of the disease, is upregulated in patients with COPD. The elevation of serum CHI3L1 may contribute to tissue inflammation and remodeling by activating alveolar macrophages, which are both the target and the source of CHI3L1 [11,45,48]. Similarly, circulating CHI3L1 levels also elevated in asthma patients compared with healthy controls and positively correlated with the severity of asthma [17,91].
It also has been demonstrated that a promoter -131C→G SNP (single nucleotide polymorphism) in CHI3L1 is associated with increased serum CHI3L1 levels and the severity of asthma. [92]. A novel intronic SNP, rs12141494, alters airway CHI3L1 expression to contribute to the severity of asthma and airway remodeling. Although this SNP, unlike promoter SNP rs4950928(-131C>G), was associated with CHI3L1 expression in the sputum, there was no association with asthma severity. Furthermore, the A allele of rs4950928 was associated with higher serum CHI3L1 levels and severer asthma after the control of risk genotype (CC). The A allele of rs12141494 have significantly higher CHI3L1 sputum levels, compared to the G allele [93,94,95]. These results suggest that CHI3L1 is an intermediate phenotype for asthma susceptibility, and DNA variants in CHI3L1 play important roles in the progression of severe asthma and airway remodeling. Thus, the inhibition of CHI3L1 DNA variants could contribute to the lower production of circulating CHI3L1 and decrease the risk of asthma and airway remodeling.
A prospective cohort design found that serum CHI3L1 level relates to the increase of the risks from moderate to severe asthma exacerbations and can be a predictor of moderate to severe asthma exacerbation. Furthermore, CHI3L1 is a signature of non-type 2 inflammation for NEA (non-eosinophilic asthma) patients and increased serum CHI3L1 levels are associated with NEA phenotypes [48]. Murine studies have found that CHI3L1 was induced by high-fat diet and Th2 inflammation (such as asthma) and contributes to the genesis of obesity. Serum CHI3L1 was also associated with persistent asthma in obese asthma patients. However, sputum CHI3L1 expression was associated with only truncal obesity in humans [95]. Thus, the inhibition of CHI3L1 or CHI3L1 pathways could provide potential therapeutic treatments for obesity-related asthma.
2.2. CHI3L1 Expression in Various Cell Types
The expression CHI3L1 expression was first identified in human synovial cells and articular cartilage chondrocytes, and osteosarcoma cells [96]. The authors identified soluble form of CHI3L1 levels in serum and synovial fluid were significantly higher in the patients with joint disease as compared to normal adults [96]. Their continuous studies of CHI3L1 in various inflammatory disorders as well as malignant diseases, it was revealed that CHI3L1 was produced by inflammatory cells and cancer cells by regulating cell proliferation, differentiation, and extracellular tissue remodeling [97]. Our group first reported that apical side of CECs as well as lamina propria cells strongly express CHI3L1 in several murine colitis models and IBD patients but completely absent in normal controls/individuals, suggesting that CHI3L1 is an inducible molecule under inflammatory conditions in the colon and plays a pathogenic role in colitis [7]. We also previously reported that CHI3L1 on CECs is further upregulated during the processes of colitis-associated cancer [77]. The CHI3L1 expression in HCT116 human CECs are observed mainly peri-nuclear and cytoplasm regions [Figure 1A] although the location was mainly restricted in nucleus with reduced expression of CD44 after stimulating with human TNFα recombinant protein for 48 hours [Figure 1B]. This restricted trans-nuclear localization was inhibited by combinational pan-chitinase inhibitors (caffeine and pentoxifylline) [Figure 1C], suggesting this nuclear localization of CHI3L1 after TNFα stimulation seems to be specifically controlled by chitinase activity. In the future, we plan to study the mechanism by which CHI3L1 trans-locates into the nucleus and whether there are changes in its biological function after the trans-location.
It has been reported that CHI3L1 expression is highly upregulated with cancer-infiltrating macrophages, so-called TAM (tumor-associated macrophages) [98]. CHI3L1 specifically promotes macrophage recruitment and tumor angiogenesis in colon cancer [99]. Alternatively activated macrophages (M2 Mφ) but not classically activated macrophages (M1 Mφ) generally express mammalian chitinases, including CHI3L1, CHI3L3 (YM1), and CHI3L4 (YM2) under the activation of MAPK pathway [100].
Brain tumor such as glioma highly express CHI3L1 by interacting with CD44 on the surface of glioma stem cells (GSCs) that results in activating Akt and β-catenin signaling cascade [101]. These activation in turn upregulates CD44 expression in a pro-mesenchymal feedback loop [101]. The CHI3L1 expression seems to alter the state of GSCs to support tumor growth and also regulate cellular plasticity leading to a targetable glioblastoma vulnerability [102]. This result suggests that a blockade of CHI3L1 by specific antibody may serve as one of the helpful therapeutic strategies for malignant brain tumors including glioblastoma.
3. CHI3L1-Mediated Host-Microbial Interactions
3.1. CHI3L1 as an Enhancer of Bacterial Adhesion and Invasion on Colonic Epithelial Cells
The interplay between the intestinal microbiome and the gastrointestinal (GI) tract has been extensively presented as bidirectional [103]: reciprocal signaling occurs between the bacterial flora and the mucosal immune system, thus modulating gut homeostasis. Despite inter-individual differences, inappropriate interactions between enteric microorganisms and host cells disrupt the intestinal immune balance, leading to acute and chronic inflammatory outcomes.
The pathophysiology of IBD is an example of how the proliferation of bacteria, primarily commensal, and gastrointestinal dysbiosis can play a pivotal role in the initiation and/or perpetuation of chronic disorders.
It has been predicted that the altered expression of specific receptor(s) on host intestinal epithelial cells might enhance the interaction with bacterial components under inflammatory conditions [104]. Among these molecules, CHI3L1 has been targeted as a potential enhancer of bacterial adhesion and invasion on/into CECs [7,105].
Although CHI3L1 doesn’t possess any enzymatic activity, it retains the ability to bind to chitin, β-1, 4 N-acetylglucosamine (GlcNAc), and chito-oligosaccharide, and therefore named as chitinase-like proteins (CLPs). Microbial chitinases, which are generally associated with chitinolytic activity for nutritious purposes, have been recently linked to bacterial virulence. Although mammals do not synthesize chitin, L. Pneumophila and V. Cholera chitinases have been found responsible for promoting bacterial colonization of lungs and intestine, respectively [106]. It’s conceivable that the presence of a chitin-binding motif on bacterial chitinases favors bacterial adherence to the surface of host epithelial cells under inflammatory conditions [107]. This has been confirmed for both CBP21 of Serratia Marcescens and ChiA of AIEC which interact with CHI3L1 to attach to intestinal epithelial cells [8].
The post-translational structure of CHI3L1 presents an N-glycosylated protein with two molecules of GlcNAc at the 60th asparagine residue in human [108]. It’s noteworthy how the extent of glycosylation of both host and microbiomes changes in the context of bacterial infection. The resulting glycome becomes an expression of highly complex glycosylated ligands which serve as receptors and primary sites of contact for bacteria [109]. In particular N-linked surface glycoproteins expressed on host cells are likely to be a target for bacterial chitinases. For instance, S. Typhimurium links to sugar compounds on apical host cells with high specificity, thus showing preference among the glycosylated moieties.
Moreover, alterations of the glycome occur before bacterial entry which proves them to be a direct consequence of host-microbial interaction [110]. Accordingly, it’s possible to infer that N-glycosylation of host CHI3L1 is one of the critical steps to bacterial binding. Glycosylation of epithelial cells highly depends on the integrity of the sub- and supra-mucosal environment. Flaws in mucus glycosylation can yield a degraded mucus layer and less efficient segregation between host and intact bacteria [111,112] [Figure 2]. In addition, glycosylated CHI3L1 plays a key role for host-microbial interactions since the mutation in CHI3L1 60th or 68th asparagine residue in human or mice, respectively, result in the reduction of bacterial adhesion to colonic epithelial cells [9] [Figure 2].
Mucosal disruption is a typical finding in IBD cases as well as in the context of a systemic inflammatory response [113]. Specifically, it has been documented that CHI3L1 is overexpressed in the colon tissue upon bacterial colonization of severe burn injuries. Again, the interruption of the mucosal barrier promotes bacterial contact with the underlying epithelium, thus accounting for increased chitinase levels. It’s liable that the presence of host ligands for microbial chitinases (which contains chitin-binding proteins) also modulates the transcriptional patterns of bacteria. This may account for the release of virulence factors and the internalization of pathogenic microbes.
Following adhesion, the invasion of bacteria into the colonic epithelium is the end result of complex cellular mechanisms involving both sides of the equation. The release of pro-inflammatory cytokines, primarily TNF-α, IL-1β, and IL-6, from invasive commensal bacteria fosters a comparable expression on host cells. To this extent, it has been established that these proinflammatory factors, especially TNF-α, can induce the expression of CHI3L1 mRNA and late secretion of CHI3L1 protein. In turn, CHI3L1 can activate the NF-κB signaling pathway which produces the same pro-inflammatory cytokines. This feedback loop further highlights how the host immune system and the microbiota are intrinsically related.
Another key step to bacterial penetration is the polarization of macrophages. The CHI3L1-driven M2 transition is part of a compensated anti-inflammatory response. However, in a dysregulated environment, the M2 presence hinders the pro-inflammatory defenses owing to poor antigenic properties. This leads to an equally poor bacterial clearance because the engulfed pathogens reside internalized within the mucosa. Interestingly bacteria, like Staphylococcus. Aureus, exploit this mechanism to evade immune recognition [114]. Others, such as adherent invasive Escherichia coli (AIEC), keep replicating within macrophages. This data indicates how commensal bacteria can both start and uphold enteric inflammation.
3.2. Interactions between CHI3L1 and Bacterial Chitinase (ChiA) in Escherichia coli
E. coli is one of the major representatives of commensal bacteria producing bacterial chitinase. Particularly, AIEC is normally present in the intestinal flora of healthy individuals but shows high levels of virulence in CD patients. This finding suggests that AIEC strains display pathogenicity in susceptible hosts via increased adhesion to host cells. The disrupted mucus layer, typical of IBD, and the CHI3L1 upregulation make enteric epithelial cells accessible to AIEC strains [Figure 2]. Indeed, AIEC’s primary interaction consists in binding the host CHI3L1 via the bacterial chitinase, ChiA. Particularly, it has been recorded that ChiA overexpression occurs in AIEC-strains rather than non-AIEC strains, thus accounting for microbe-specific features [115].
To firmly adhere to intestinal epithelial cells, type I pili and flagella are usually required. However, it has been demonstrated that ChiA expression strongly affects the invading ability of AIEC. By comparing ChiA knockout and wild-type AIEC (reference strain LF82), it’s clear how the level of bacterial virulence decreases in the former compared to the latter [9]. Interestingly, ChiA does not change the gross structure of the microbe. This observation suggests that ChiA is critical in AIEC adherence as much as its membrane extensions. Similarly to mammalian chitinases, the genotype of ChiA can influence the rate of invasion of E. coli into host CECs. The presence of polymorphisms in the ChiA-chitin-binding domains allows clustering of E. coli strains according to their relative pathogenicity, which is measured in terms of adhesiveness to CECs [9].
Multiple factors compromise the host microenvironment and favor bacterial access to intestinal epithelium. First of all, host macrophages release pro-inflammatory cytokines in conditions of chronic inflammation. It is noteworthy how inflammatory cytokines, such as TNFα, IL-1β, and IL-6 foster a greater expression of CHI3L1 and, thus, a wider site of contact for bacterial chitinases [116]. Additionally, AIEC induces submucosal macrophages to yield pro-inflammatory mediators, thus fueling a vicious cycle of inflammation. To this extent, the main bacterial advantage may consist in promoting intestinal permeability, by increasing CHI3L1 expression, and intra-macrophage replication within the submucosal space.
The post-translational N-glycosylation of CHI3L1 is crucial for an efficient host-microbe interaction [9] [Figure 2]. In addition, the expression of glycosylated moieties, namely CEACAM6, results necessary for AIEC adhesion. Similarly to CHI3L1, CEACAM6 serves as a binding receptor for the bacterial appendices and is expressed upon TNF-α stimulation following AIEC infection [104]. This data confirms how commensal bacteria can sustain colonization by exploiting modification of host cells by glycation [9,104].
3.3. Potential Role of CHI3L1 as an Inducer of Intestinal Dysbiosis
The alteration of the enteric microbiota is associated with a wide variety of gastrointestinal diseases. Intestinal dysbiosis may present as the source, the result, and, most frequently, the sustainer of chronic inflammatory states [4,15]. The composition of gut microbiota is modulated by several factors, some of which are unmodifiable such as the immune system, the enteric mucosa and the microbiome. This finding is supported by the pathophysiology of IBD, which usually presents mucus disruption, immune dysregulation and dysbiosis [4,15].
Any imbalance among the bacterial taxa can lead to reduced microbial diversity and predominance of pathogenic strains. These favor disease development and severity, by impairing intestinal homeostasis and promoting immunosuppression and cancer cell growth. In this context, the host-microbial interaction plays a central role. It has been demonstrated that CHI3L1 enhances bacterial adhesion in susceptible hosts. Interestingly, it’s possible that CHI3L1 preferentially engages pathogenic (e.g., S. typhimurium) and potentially pathogenic (e.g., AIEC) strains rather than non-pathogenic ones (e.g., DH5α) [9,13,117]. This mechanism would reinforce the extent of microbial penetration within colonic epithelium and further contribute to intestinal dysbiosis. In addition, the formation of a bacterial biofilm on the surface of CECs is related to the pathogenic transition of certain bacterial strains. This finding suggests that the loss of mucosal protection and the increased intestinal permeability induce the intramucosal replication of intact bacteria that are normally excluded from colonic tissue. Altogether these events contribute to shaping intestinal flora and affecting immune tolerance.
Mice remain some of the best animal models to investigate changes in the microbiota presentation. It has been shown that, following bacterial infection, chemically-induced colitis or immune deficiency, mice enteric flora develops a lower number of total commensal bacterial as well as a reduced richness in resident strains with respect to normal controls [118]. This data underlies how different sources of inflammation can account for intestinal dysbiosis.
IBD is one of the most representative cases of chronic intestinal dysbiosis. Despite the multifactorial nature of the disease, the alteration in the microbiota composition is rather relevant. Most of the bacterial phyla in a healthy intestinal flora are Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria. In IBD patients Bacteroidetes and Proteobacteria are more abundant whereas Firmicutes are reduced [119]. Moreover, the microbial richness diminishes with evidence of predominant strains and clusters, such as Enterobacteriaceae and Bilophila for Proteobacteria and Faecalibacterium prausnitzii for Firmicutes. This background might lead to metabolic changes that affect the whole gut homeostasis. In addition, the amount of mucus-degrading bacteria, such as R. gnavus and R. torque, is significantly higher in IBD with respect to normal controls [120]. Thus, contributing to reduced mucus protection and increased epithelial exposure to commensal bacteria.
Overall, the data above suggests the potential role of host CHI3L1 in shaping the intestinal biome and favoring the penetration of potentially pathogenic bacteria in normal flora under inflammatory conditions. This evidence suggests a prospective therapeutic target for the treatment of IBD by inhibiting CHI3L1 expression, in the attempt to exclude the entry route of invasive species from the aftermath of intestinal dysbiosis.
4. Association between CHI3L1 and Chronic Inflammation
4.1. CHI3L1-Associated Chronic Inflammation in Animal Models
Previous research has revealed that CHI3L1 is involved in various inflammatory diseases, and CHI3L1 plays a significant role in inflammatory conditions. The involvement of CHI3L1 across such a broad spectrum of diseases highlights its biological significance in understanding the pathogenesis of chronic inflammation. Using animal models has allowed us to assess the significance of CHI3L1 in numerous chronic inflammations listed in Table 2 [60,77,82,95,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137]. Intriguingly, beyond the chronic inflammations delineated in Section 2.1, emerging evidence indicates the modulation of cardiovascular diseases, liver injuries, kidney diseases, systemic musculoskeletal disorders, and obesity by CHI3L1.
Tsantilas et al. employed ApoE knockout (KO) mice subject to a high-fat diet to investigate the atherosclerosis plaque rupture mechanism, revealing CHI3L1 plays a regulatory role in plaque size and vulnerability [131]. These findings elucidated that CHI3L1 expression is induced by pro-inflammatory cytokines, such as IL-1β, and IL-6, originating from smooth muscle cells within the plaque. Moreover, CHI3L1 KO resulted in smooth muscle cell apoptosis and the formation of larger, unstable, or ruptured plaques [131]. Notably, CHI3L1 induction extends beyond pro-inflammatory cytokines, as demonstrated by Lee et al., who unveiled that CHI3L1 facilitates oxidative stress and chronic inflammation in the alcohol liver injury rats model [133]. CHI3L1 was found to upregulate the expression of iNOS (inducible nitric oxide synthase), COX-2 (cyclooxygenase-2), TNF-α, IL-1β, and chemokines such as MIP-1α and MIP-1β in the liver. Additionally, CHI3L1 KO rats in the alcohol injury model exhibited suppressed levels of ICAM-1, suggesting the involvement of CHI3L1 in neutrophil-mediated inflammation in the liver [133]. CHI3L1 is widely acknowledged as a potent inducer of Th2-type immune responses [137]. However, numerous studies also suggest that CHI3L1 also exerts a robust induction of pro-inflammatory cytokines, particularly in certain chronic inflammatory contexts, by modulating IL-6 production or STAT3-mediated signaling activation [82,123].
Collectively, these findings underline the collaborative roles of CHI3L1 with various cell types in modulating inflammatory responses and establishing feedback loops across diverse chronic inflammatory contexts. Therefore, the animal models of chronic inflammation have been valuable in elucidating the significant impact of CHI3L1 and have advanced our development of novel therapeutic strategies.
4.2. CHI3L1-Associated Chronic Inflammation in Human
Numerous evidence has highlighted the significance of CHI3L1 in various human inflammatory conditions, as described in Table 1. Numerous studies have elucidated a robust correlation between CHI3L1 expression levels and the severity as well as prognostic outcomes of diseases, including asthma, atopic dermatitis, and interstitial lung disease [138,139,140]. This collective body of research underlines the pivotal role of CHI3L1 in the pathophysiology of inflammatory disorders, providing valuable insights into its potential as a diagnostic and prognostic biomarker.
The pathogenesis of CHI3L1 in chronic inflammation exhibits disease-specific variations [Figure 3]. In pulmonary disorders such as asthma or Hermansky-Pudlak syndrome (HPS), a predominant Th2 immune response orchestrates chronic inflammatory processes [127,129]. In asthma, this immune activation is initiated as dendritic cells engage with allergens and pathogens presented on epithelial cells, subsequently inducing Th2 cell differentiation. Moreover, damaged epithelial cells contribute to Th2 cell activation by secreting IL-25 and IL-33, which also stimulate innate lymphoid cells type 2 (ILC2) [141]. The resultant Th2-mediated inflammation not only manifests as eosinophilia and mast cell degranulation but also fosters the polarization of M0 macrophages towards the M2 phenotype. Notably, IL-13 emerges as a critical mediator in inducing CHI3L1 expression in both epithelial cells and M2 macrophages. Consequently, elevated CHI3L1 expression in epithelial cells exacerbates pathogen infiltration, establishes a feedback loop, and perpetuates chronic inflammation [127] [Figure 3].
In contrast to pulmonary conditions, IBD presents a distinct chronic inflammatory profile modulated by CHI3L1. Our previous investigations revealed a notable increase in CHI3L1 expression within colonic epithelial cells during the course of chronic inflammation [77]. Additionally, our research demonstrated the pivotal role of CHI3L1 in facilitating pathogenic bacterial adhesion to colonic epithelial cells [82]. In this context, bacterial interaction with Toll-like receptor 4 (TLR4) on epithelial cells initiates the production of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, via MyD88, TRIF, and MAPK signaling pathways [142]. These cytokines further stimulate the upregulation of CHI3L1 expression in epithelial cells. Moreover, CHI3L1 exerts direct effects on the human colorectal cancer SW480 cell line by activating the NF-κB and MAPK pathways, consequently upregulating the expression of pro-inflammatory cytokines such as IL-8, TNF-α, and CCL2 (C-C motif chemokine ligand 2). Activation of these signaling cascades facilitates the production of pro-inflammatory mediators, promoting macrophage recruitment and enhancing angiogenesis within the tumor microenvironment, thereby fostering tumor growth [99,142].
CHI3L1 has been identified as a potent modulator of immune responses, particularly through its stimulation of macrophage and neutrophil activity, as well as its influence on immune checkpoints, thereby establishing a conducive environment for tumor growth. Notably, CHI3L1 has been shown to drive the polarization of macrophages towards the M2 phenotype, commonly referred to as TAMs (tumor-associated macrophages), within the tumor microenvironment [143,144,145]. TAMs are recognized for their pivotal roles in various aspects of tumor progression, including tumor development, neo-angiogenesis modulation, immune suppression, and metastasis [146]. Moreover, recent findings by Taifour et al. have underscored the role of CHI3L1 in inducing NETosis, thereby facilitating the exclusion of T cells and promoting the establishment of triple-negative breast cancer tumors [147]. These observations collectively highlight the multifaceted involvement of CHI3L1 in shaping the inflammatory tumor microenvironment and influencing cancer progression.
5. Therapeutic Potentials of CHI3L1-Blockers/Inhibitors for Various Diseases
5.1. Anti-CHI3L1 Antibody
Given the pivotal role of CHI3L1 in chronic inflammation, targeting this protein for therapeutic purposes has gathered significant interest. Choi et al. investigated the inhibitory potential of 2-({3-[2-(1-cyclohexen-1-yl)ethyl]-6, 7-dimethoxy-4-oxo-3,4-dihydro-2-quinazolinyl}sulfanyl)-N-(4-ethylphenyl) butanamide (K284-6111) in an AD mouse model [123]. Their study revealed that K284-6111 binds to CHI3L1, thereby inhibiting its interaction with the receptor for advanced glycation end products (RAGE). This interaction led to the suppression of NF-κB activation and NF-κB-related neuro-inflammatory gene expressions. Oral administration of K284-6111 resulted in a reduction of Aβ1–42-induced β-secretase activity and Aβ generation, as well as decreased levels of neuro-inflammatory cytokines and amyloidogenic proteins. Consequently, K284-6111 exhibited memory recovery effects in the Morris water maze test [123]. Furthermore, K284-6111 was evaluated in a phthalic anhydride (5% PA)-induced atopic dermatitis animal model, where topical application attenuated dermatitis severity, epidermal hyperplasia, inflammatory cell infiltration, and release of inflammatory cytokines [148]. In addition to small molecules like K284-6111, humanized antibodies targeting CHI3L1 have been developed and tested for their efficacy.
CHI3L1 antibodies have earned considerable interest among researchers for their potential application in cancer therapy. In the context of chronic inflammation, CHI3L1 plays a crucial role in fostering tumor-associated inflammation and shaping the tumor microenvironment [Figure 3]. Yang et al. developed polyclonal neutralizing CHI3L1 antibodies (nCHI3L1 Abs) and assessed their efficacy in lung, pancreas, and colon cancer allograft models [149]. Their findings demonstrated that nCHI3L1 Abs effectively inhibited tumor growth and metastasis in orthotopic lung, pancreatic, and colon cancer models. Additionally, in vitro studies confirmed the ability of nCHI3L1 Abs to suppress the AKT, β-catenin, and NF-κB signaling pathways [149]. Furthermore, Yu et al. reported similar findings, wherein their anti-CHI3L1 antibody exhibited efficacy in reducing lung tumor growth and metastasis by inhibiting M2 polarization [150].
Moreover, CHI3L1 exerts a significant regulatory influence on immune checkpoints. In a melanoma lung metastasis mice model, Ma et al. demonstrated that CHI3L1 upregulates the expression of programmed death-ligand 1 (PD-L1) on activated macrophages while concurrently suppressing the expression of Inducible T-cell Co-Stimulator (ICOS), ICOS Ligand, and CD28 on T cells and antigen-presenting cells [151,152]. Their anti-CHI3L1 antibody (so called FRG), PD-1 antibody, and CTLA-4 antibodies exhibited substantial anti-tumor effects and displayed additive responses in metastasis models. Intriguingly, in vitro studies confirmed synergistic cytotoxic effects on tumor cells, while significantly enhanced anti-tumor responses were observed in vivo tumor models treated with bispecific antibodies targeting both FRG and PD-1. Similar effects were confirmed with bispecific antibodies targeting both FRG and CTLA-4 [151,152].
In recent clinical practice, immune checkpoint inhibitors (ICIs) have become commonplace. Nevertheless, with the utilization of ICIs, a noTable 43% of patients report experiencing immune-related adverse events (irAEs) [153]. Considering that the most prominent complications associated with ICIs encompass chronic immune-related adverse events like dermatitis, hepatitis, arthritis, and colitis, and recognizing CHI3L1 as a significant immune checkpoint modulator, the concurrent targeting of CHI3L1 using bispecific antibodies may represent a prospective solution [154,155].
5.2. Methylxanthine Derivatives Including Caffeine as a Chitinase Inhibitor
Methylxanthines, including caffeine, pentoxifylline, theophylline and allosamidin, are a group of alkaloids which are derived from the purine-based xanthine [156]. Interestingly, it has been demonstrated through the use of drug screening tools that several methylxanthine derivatives potentially work as chitinase inhibitors [157]. Allosamidin, a chitinase inhibitor produced by Streptomyces, showed higher affinity against fungal chitinase as compared to caffeine, pentoxifylline, and theophylline [156]. Based on X-ray diffraction analysis showed that all the three methylxanthine derivatives listed above have a common binding position for family 18 chitinases just like as allosamidin and working as a pan-chitinase inhibitor.
Based on the characteristic feature of methylxanthine derivatives as pan-chitinase inhibitor, our group compared the influence of CHI3L1 m RNA expression levels in SW480, a human colon epithelial cells, after treatment with caffeine, pentoxifylline, theophylline [156]. As a result, all the three methylxanthine derivatives directly down regulated the CHI3L1 mRNA expression levels in SW480 cells with a dose-dependent manner [157]. Since CHI3L1 is induced on epithelial cells and macrophages during inflammatory conditions as well as inflammation-associated cancer states by activating several important signaling pathways, including AKT and β-catenin, thus methylxanthine derivatives have potential effects for anti-inflammatory and anti-cancer through the inhibition of CHI3L1. In fact, oral caffeine administration at the concentration of 2.5 mM efficiently prevents onset of a murine model of acute colitis by DSS (dextran sulfate sodium) [158]. The caffeine-mediated anti-inflammatory effect was exerted by suppressing CHI3L1 and AMCase but not chitinase -1 as determined by quantitative PCR of colonic tissue after induction of DSS-acute colitis [158]. However, a paradoxical effect of caffeine was also identified that low-dose (0.17 mM) caffeine with 10% sucrose (but not fructose) cause apparent carcinogenic change on CECs in a murine model of chronic colitis [159]. Therefore, we need to carefully determine the influence of methylxanthine derivatives as a potential anti-inflammatory therapeutic strategy in inflammatory conditions.
5.3. Chitin Microparticles and Chito-Oligosaccharides
Chitin is a polymer of GlcNAc, which is produced by fungi, crustaceans, and insects [160,161]. Chitin is the second most abundant polysaccharide in nature next to cellulose [160]. In addition, chitin is a valuable biological resource that is estimated to be synthesized on earth in an amount of 100 billion tons per year. However, chitin is difficult to utilize because it cannot be dissolved in ordinary solvents, but only be broken down by enzymatic active true chitinases. Interestingly, chitin has different biological effects depending on its size: Large chitin particles (diameter >100 μm) are non-functional, medium chitin particles (40-70 μm) are pro-inflammatory function, and chitin micro particles (CMPs: 1-10 μm) are believed to be anti-inflammatory as well as regulatory functions by stimulating IL-10 production [162,163]. Both host CHI3L1 and bacterial chitin-binding proteins are characterized by their ability to bind to chitin [7]. These findings had promoted us to propose a “Trimetric“ Interaction model that the interaction of CHI3L1 and CBP is mediated by exogenous/endogenous chitin orchitinlike oligosaccharides, forming a CHI3L1/chitin/CBP trimeric complex [Figure 4]. However, our published data [9,166] now suggest that glycosylated (60th asparagine in humanand 68th asparagine in mouse) CHI3L1 can directly interact with CBP, promoting us to proposean alternative mechanism called “dimeric” interaction theory [Figure 2 and Figure 4]. In addition, we recently found AIEC LF82 strain attachment on colonic epithelial cells was abolished whenthese cells were treated with N-glycosylation inhibitor (tunicamycin) or or engineered tooverexpress mutant CHI3L1 lacking 68th asparagine (N68P mutant), a site of N-glycosylationfor CHI3L1 [9] [Figure 2]. These results raise a possibility that N-glycosylated CHI3L1 is essential for the interaction between CECs and potentially pathogenic bacteria under inflammatory conditions.
Furthermore, CMPs inhibit, rather than enhance, the interaction of CECs and pathogenic and efficiently modulate intestinal inflammation in vivo [164]. As mentioned previously, chitin particles play different biological roles depending on their size. Since chitin is unstable, it is difficult to generate small chitin of the same size, which has hampered investigators’ abilities to dissect the biological role of chitin more closely and accurately, in particular in vivo. To overcome this problem, water-soluble and equal-sized chito-oligosaccharide (CHOS) nano-particles (1-10 nm in size) must be more useful [165,166] for in vivo studies as therapeutic strategy for inflammatory disorders including IBD and COPD since CHOS is the very end-product of chitin and does not be dissected anymore.
6. Conclusions
As a result of many research reports to date, it has become clear that the expression of CHI3L1 on epithelial cells is deeply involved in the process of chronic inflammation and carcinogenesis. Therefore, inhibiting CHI3L1 expression is expected to be a new prevention and treatment strategy for chronic inflammation as well as inflammation-associated cancer. CHI3L1 expression is positively associated with increased angiogenesis and metastasis in highly malignant tumors such as colorectal cancer, lung cancer, and glioblastoma. It is expected that it could also contribute to the treatment of cancer by inhibiting CHI3L1 expression with multiple ways including anti-CHI3L1 specific antibody, methylxanthine derivatives and chitin microparticles.
Acknowledgments
I am grateful to Ms. Kori Aiken and Mr. Rumi Shibuta for her professional English edits and secretarial help in preparing this manuscript, respectively. I also thank Mr. Koji Matuda in Keyence Cooperation for taking fluorescence images.
Grant Supports
This work has been supported by grants from the Japanese Society for the Promotion of Science (18K07987 and 21K07996).
References
- Podolsky, D.K. Inflammatory bowel disease (1). N. Engl. J. Med. 1991, 325, 928–37. [Google Scholar] [CrossRef]
- Podolsky, D.K. Inflammatory bowel disease (2). N. Engl. J. Med. 1991, 325, 1008–1016. [Google Scholar] [CrossRef]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef]
- Ke, X.; You, K.; Pichaud, M.; Haiser, H.J.; Graham, D.B.; Viamakis, H.; Porter, J.A.; Xavier, R.J. Gut bacterial metabolites modulate endoplasmic reticulum stress. Genome Biol. 2021, 22, 292. [Google Scholar] [CrossRef]
- You, K.; Wang, L.; Chou, C.H.; Liu, K.; Nakata, T.; Jaiswal, A.; Yao, J.; Lefkovith, A.; Omar, A.; Perrigoue, J.G.; et al. QRICH1 dictates the outcome of ER stress through transcriptional control of proteostasis. Science 2021, 371, eabb6896. [Google Scholar] [CrossRef]
- Mizoguchi, E. Chitinase 3-like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterology 2006, 130, 398–411. [Google Scholar] [CrossRef]
- Kawada, M.; Chen, C.C.; Arihiro, A.; Nagatani, K.; Watanabe, T.; Mizoguchi, E. Chitinase 3-like-1 enhances bacterial adhesion to colonic epithelial cells through the interaction with bacterial chitin-binding protein. Lab Invest 2008, 88, 883–895. [Google Scholar] [CrossRef]
- Low, D; Tran, H.T.; Lee, I.A.; Dreux, N.; Kamba, A.; Reinecker, H.C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-binding domains of Escherichia coli ChA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology 2013, 145, 602-612. [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active enZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acid Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Lee, C.G.; Dela Cruz, C.S.; Ma, B.; Ahangari, F.; Zhou, Y.; Halaban, R.; Sznol, M.; Elias, J.A. Chitinase-like proteins in lung injury, repair, and metastasis. Proc. Am. Thora. Soc. 2012, 9, 57–61. [Google Scholar] [CrossRef]
- Houston, D.R.; Recklies, A.D.; Krupa, J.C.; van Aalten, D.M. Structure and ligand-induced conformational change of the 39-kDa glycoprotein from human articular chondrocytes. J. Biol. Chem. 2003, 278, 30206–30212. [Google Scholar] [CrossRef]
- Kawada, M.; Hachiya, Y.; Arihiro, A.; Mizoguchi, E. Role of mammalian chitinases in inflammatory conditions. Keio J. Med. 2007, 56, 21–27. [Google Scholar] [CrossRef]
- Hamid, R.; Khan, M.A.; Ahmad, M.; Ahmad, M.M.; Abdin, M.Z; Musarrat. J.; Javed, S. Chitinases: an update. J. Pharm. Bioallied Sci. 2013, 5, 21-29. [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Zhu, Z.; Zheng, T.; Homer, R.J.; Kim, Y.K.; Chen, N.Y.; Cohn, L.; Hamid, Q.; Elias, J.A. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science 2004, 304, 1678–1682. [Google Scholar] [CrossRef]
- Chupp, G.I.; Lee, C.G.; Jarjour, N.; Shim, Y.M.; Holm, C.T.; He, S.; Dziura, J.D.; Reed, J.; Coyle, A.J.; Kiener, P.; et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N. Eng. J. Med. 2007, 357, 2016–2027. [Google Scholar] [CrossRef]
- Blazevic, N.; Rogic, D.; Pelajic, S.; Miler, M.; Glancic, G.; Ratkajec, V.; Vrkolina, N.; Bakula, D.; Hrabar, D.; Pavic T. YKL-40 as a biomarker in various inflammatory diseases: A review. Biochem. Med. (Zagreb). 2024, 34: 010502. [CrossRef]
- Zhao, T.; Su, Z.; Li, Y.; Zhang, X.; You, Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal Transduct. Target Ther. 2020, 5, 201. [Google Scholar] [CrossRef]
- Koutroubakis, I.E.; Petinaki, E.; Dimoulios, P.; Vardas, E.; Roussomoustakaki, M.; Maniatis, A.N.; Kouroumalis, E.A. Increased serum levels of YKL-40 in patients with inflammatory bowel disease. Int. J. Colorectal Dis. 2003, 18, 254–259. [Google Scholar] [CrossRef]
- Vind, I,; Johansen, J.S.; Price, P.A.; Munkholm, P. Serum YKL-40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scand. J. Gastroenterol. 2003, 38, 599-605. [CrossRef]
- Punzi, L.; Podswiadek, M.; D’Inca, R.; Zaninotto, M.; Bernardi, D.; Pienani, M.; Sturniolo, G.C. Serum human cartilage glycoprotein 39 as a marker of arthritis associated with inflammatory bowel disease. Ann. Rheum. Dis. 2003, 62, 1224–1226. [Google Scholar] [CrossRef]
- Erzin, Y.; Uzun, H.; Karatas. A.; Celik, A.F. Serum YJK-40 as a marker of disease activity and structure formation in patients with Crohn’s disease. J. Gastroenterol. Hepatol. 2008, 23: e357-362. [CrossRef]
- Pieczarkowski, S.; Kowalska-Deptuch, K.; Kwinta, P.; Wedrychowicz, A.; Tomaski, P.; Stochel-Gaudyn, A.; Fyderek, K. Serum concentration of fibrosis markers in children with inflammatory bowel disease 2020, 60, 61-74. [CrossRef]
- Douadi, C.; Vazeille, E.; Chambon, C.; Hebraud, M.; Fargeas, M.; Dodel, M.; Coban, D.; Pereira, B.; Birer, A.; Sauvanet, P.; et al. Anti-TNF agents restrict adherent-invasive Escherichia coli replication within macrophages through modulation of chitinase 3-like 1 in patients with Crohn’s disease. J. Crohns Colitis 2022, 16, 1140–1150. [Google Scholar] [CrossRef]
- Deutschmann, C.; Roggenbuck, D.; Schierack, P. The loss of tolerance to CHI3L1-A putative role in inflammatory bowel disease? Clin. Immunol. 2019, 199, 12–17. [Google Scholar] [CrossRef]
- Akesson, J.; Hojjati, S.; Hellberg, S.; Reffetseder, J.; Khademi, M.; Rynkowski, R.; Kockum, I.; Altafini, C.; Lubovac-Pilav, Z.; Mellergard, J.; et al. Proteomics reveal biomarkers for diagnosis, disease activity and long-term disability outcomes in multiple sclerosis. Nat. Commun. 2023, 14, 6903. [Google Scholar] [CrossRef]
- Talaat, F.; Abdelatty, S.; Ragaie, C.; Dahshan, A. Chitinase-3-like 1-protein in CFS: a novel biomarker for progression in patients with multiple sclerosis. Neurol. Sci. 2023, 44, 3243–3252. [Google Scholar] [CrossRef]
- Ahmad, I.; Wergeland, S.; Overland, E.; Bϕ, L. An association of chitinase-3 like-protein-1 with neural deterioration in multiple sclerosis. A.S.N. Neuro. 2023, 15, 17590914231198980. [Google Scholar]
- Lamancova, P.; Urban, P.; Maslankova, J.; Rabajdova, M.; Marekova, M. Correlation of selected serum protein levels with the degree of disability and NEDA-3 status in multiple sclerosis phenotypes. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 3933–3941. [Google Scholar]
- Donder, A.; Ozdemir, H.H. Serum YKL-40 levels in patients with multiple sclerosis. Arq. Neuropsiquiatr. 2021, 79, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Pezzini, F.; Pucci, M.; Rossi, S.; Facchiano, F.; Marastoni, D.; Montagnana, M.; Lippi, G.; Reynolds, R.; Calabrese, M. Changes in cerebrospinal fluid balance of TNF and TNF receptors in Naïve multiple sclerosis patients: Early involvement in compartmentalized intrathecal inflammation. Cells 2021, 10, 1712. [Google Scholar] [CrossRef] [PubMed]
- Cubas-Nunez, L.; Gil-Perotin, S.; Castillo-Villalba, J.; Lopez, V.; Tarazona, L.S.; Gasque-Rubio, R.; Carratala-Bosca, S.; Alcala-Vicente, C.; Perez-Miralles, F.; Lassmann, H.; et al. Potential role of CHI3L1+ astrocytes in progression in MS. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e972. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.R.; Sφndergaard, H.B.; Buheit, S.; von Essen, M.R.; Christensen, J.R.; Enevold, C.; Sellebjerg, F. Multiplex assessment of cerebrospinal fluid biomarkers in multiple sclerosis. Mult. Scle.r. Relat. Disord. 2020, 45, 102391. [Google Scholar] [CrossRef] [PubMed]
- Picon, C.; Tejeda-Velarde, A.; Fernandez-Velasco, J.I.; Comabella, M.; Alvarez-Lafuente, R.; Quintane, E.; de la Maza, S.S.; Monreal E, Villarrubia, N.; Alvarez-Carmeno, J.C.; et al. Identification of the immunological changes appearing in the CSF during the early immunofluorescence process occurring in multiple sclerosis. Front. Immunol. 2021, 12, 685139. [CrossRef]
- Neumann, A.; Ohlei, O.; Kucukali, F.; Bos, I.; Timsina, J.; Vos, S.; Prokopenko, D.; Tijms, B.M.; Andreasson, U.; Blennow, K.; et al. Multivariate GWAS of Alzheimer’s disease CSF biomarker profiles implies GRIN2D in synaptic functioning. Genome. Med. 2023, 15, 79. [Google Scholar] [CrossRef]
- Connolly, K.; Lehoux, M.; O’Rourke, R.; Assetta, B.; Erdemir, G.A.; Elias, J.A.; Lee, C.G.; Huang, Y.W. Potential role of chitinase-3-like protein (CHI3L1/YKL-40) in neurodegeneration and Alzheimer’s disease. Alzheimers Dement. 2023, 19, 9–24. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Castrogiovanni, P.; Imbesi, R.; Nunnari, G.; Rosa, M.D. Postsynaptic damage and microglial activation in AD patients could be linked CXCR4/CXCL12 expression levels. Brain Res. 2020, 1749, 147127. [Google Scholar] [CrossRef]
- Moreno-Rodriguez, M.; Perez, S.E.; Nadeem, M.; Malek-Ahmadi, M.; Mufson, E.J. Frontal cortex chitinase and pentraxin neuroinflammatory alterations during the progression of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 58. [Google Scholar] [CrossRef]
- Wang, L.; Gao, T.; Cai, T.; Li, K.; Zheng, P.; Liu, J. Alzheimer’s Disease Neuroimaging initiative. Cerebrospinal fluid levels of YKL-40 in prodromal Alzheimer’s disease. Neurosci. Lett. 2020, 715, 134658. [CrossRef]
- Lleo, A., Alcolea, D.; Martinez-Lage, P.; Scheitens, P.; Parnetti, L.; Poirier, J.; Simonse, A.H.; Verbeek, M.M.; Rosa-Neto, P.; Slot, R.E.R.; et al. Longitudinal cerebrospinal fluid biomarker trajectories along the Alzheimer’s disease continuum in the BIOMARKAPD study. Alzheimers Dement. 2019, 15, 742-753. [CrossRef]
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal fluid biomarkers for understanding multiple aspect of Alzheimer’s disease pathogenesis. Cell Mol. Life Sci. 2019, 76, 1833–1863. [Google Scholar] [CrossRef] [PubMed]
- Nordengen, K.; Kirseborn, R.E.; Henjum, K.; Selnes, P.; Gisladottir, B.; Wettergreen, M.; Torsetnes, S.B.; Grontvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflammation 2019, 16, 46. [Google Scholar] [CrossRef]
- Pan, R.; Zhu, X.; Zhou, Y.; Ding, L.; Cul, Y. Diagnostic value of YKL-40 for patients with asthma: A meta-analysis. Allergy Asthma Proc. 2021; 42, e167. [CrossRef]
- Paplinska-Goryca, M.; Misiukiewicz-Stepien, P.; Proboszcz, M.; Nejman-Gryz, P.; Gorska, K.; Krenke, R. The expression of TSLP, IL-33, and IL-17A in monocyte derived dendritic cells from asthma and COPD patients are related to epithelial-macrophage interactions. Cells 2020, 9: 1944. [CrossRef]
- Hubner, K.; Karwelat, D.; Pietsch, E.; Beinborn, I.; Winterberg, S.; Bedenberder, K.; Benedikter, B.; Schmeck, B.; Vollmeister, E. NF-κB-mediated inhibition of microRNA-149-5p regulates Chitinase-3-like 1 expression in human airway epithelial cells. Cell Signal. 2020, 67, 109498. [Google Scholar] [CrossRef] [PubMed]
- Knihtila, H.; Kotaniemi-Syrjanen, A.; Pelkonen, A.S.; Savinko, T.; Malmberg, L.P.; Makela, M.J. Serum chitinase-like protein YKL-40 is linked to small airway function in children with asthmatic symptoms. Pediatr. Allergy Immunol. 2019, 30, 803–809. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, X.; Liu, Y.; Zhang, L.; Wang, J.; Hansbro, P.M.; Wang, L.; Wang, G.; Hsu, A.C.Y. Chitinase-like protein YKL-40 correlates with inflammatory phenotypes, anti-asthma responsiveness and future exacerbations. Respir. Res. 2019, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Yoosuf, N.; Maciejewski, M.; Ziemek, D.; Jelinsky, S.A.; Folkersen, L.; Muller. M.; Sahistrom, P.; Vivar, N,; Catrina, A.; Berg, L.; et al. Early prediction of clinical response to anti-TNF treatment using multi-omics and machine learning in rheumatoid arthritis. Rheumatology (Oxford). 2022, 61, 1680-1689. [CrossRef]
- Parlak, E.; Laloglu, E. Analysis of Chitinase-3-like protein 1, IL-1-alpha, and IL-6 as novelinflammatory biomarker for COVID-19. J. Interferon Cytokine Res. 2022, 42, 536–541. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, R.; Sciorati, C.; Lore, N.I.; Capobianco, A.; Tresoldi, C.; Cirillo, D.M.: Ciceri, F.; Rovere-Querini, P.; Manfredi, A.A. Chitinase- 3-like protein-1 at hospital admission predicts COVID-19 outcome: a prospective cohort study. Sci Rep 2022, 12, 7606. [CrossRef]
- Kimura, Y.; Nakai, Y.; Shin, J.; Hara, M.; Takeda, Y.; Kubo, S.; Jeremiah, S.S.; Ino, Y.; Akiyama, T.; Moriyama, K.; et al. Identification of serum prognostic biomarkers of severe COVID-19 using a quantitative proteomic approach. Sci Rep 2021, 11, 20638. [Google Scholar] [CrossRef]
- Hammoudeh, S.M.; Hammoudeh, A.M.; Bhamidimarri, P.M.; Al Safar, H.; Mahboub, B.; Kunstner, A.; Busch, H.; Halwani, R.; Hamid, Q.; Rahmani, M.; et al. Systems immunology analysis reveals the contribution of pulmonary and extrapulmonary tissues to the immunopathogenesis of severe COVID-19 patients. Front. Immunol. 2021, 12, 595150. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Xu, J.; Luo, H.; Tan, N.; Chen, H.; Cheng, R.; Pan, J.; Han, Y.; Liu, D.; Xi, H.; et al. Direct antiviral agent treatment leads to rapid and significant fibrosis regression after HCV eradication. J Viral Hepat 2021, 28, 1284–1292. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ratziu, V.; Boursier, J.; Francque, S.; Bedossa, P.; Majd, Z.; Cordonnier, G.; Sudrik, F.B.; Darteil, R.; Liebe, R.; et al. A blood-based biomarker panel (NIS4) for non-invasive diagnosis of non-alcoholic steatohepatitis and liver fibrosis: a prospective derivation and global validation study. Lancet Gastroenterol. Hepatol. 2020, 5, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhong, M.; Wang, W.; Li, Y.H. Chi3l1 regulates APAP-induced liver injury by promoting macrophage infiltration. Eur. Rev. Med. Pharmacol. Sci. 2019; 23, 4996-5003. [CrossRef]
- Teratani, Y. Chitinase 3-like-1 expression is upregulated under inflammatory conditions in human oral epithelial cells. Kurume Med. J. 2023, 68, 221–228. [Google Scholar] [CrossRef]
- Duruk, G., Laloglu, E. Relationship between dental caries and YKL-40 levels in saliva. J. Clin. Pediatr. Dent. 2022, 46, 137-142. [CrossRef]
- Laucyte-Cibulskiene, A.; Ward, L.J.; Ebert, T.; Tosti, G.; Tucci, C.; Hernandez, L.; Kautzky-Willer, A.; Herrenro, M.T.; Kautky-Willer, A.; Herrero, M.T.; et al. Role of GDF-15, YKL-40 and MMP9 in patients with end-stage kidney disease: focus on sex-specific associations with vascular outcomes and all-cause mortality. Biol. Sex Differ. 2021, 12, 50. [Google Scholar] [CrossRef]
- Puthumana, J.; Thiessen-Philbrook, H.; Xu, L.; Coca, S.G.; Garg, A.X.; Himmelfarb, J.; Bhatraju, P.K.; Ikizler, T.A.; Siew, E.D.; Ware, L.B.; et al. Biomarkers of inflammation and repair in kidney disease progression. J. Clin. Invest. 2021, 131, e139927. [Google Scholar] [CrossRef]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson.; A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; et al. Association of multiple plasma biomarker concentrations with progression of prevalent diabetic kidney disease: Findings from the chronic renal insufficiency cohort (CRIC) study. J. Am. Soc. Nephhrol. 2021, 32, 115-126. [CrossRef]
- Greenberg, J.H.; Abraham, A.G.; Xu, Y.; Schelling, J.R.; Feiman, H.I.; Sabbisetti, W.S.; Gonzalez, M.C.; Coca, S.; Schrauben, S.J.; Waikar, S.S.; et al. Plasma biomarkers of tubular injury and inflammation are associated with CKD progression in children. J Am. Soc. Nephrol. 2020, 31, 1067–1077. [Google Scholar] [CrossRef]
- Malhotra, R.; Katz. R.; Jotwani, V.; Ambrosius, W.; Raphael, K.L.; Haley, W.; Rastogi, A.; Cheung, A.K.; Freedman, B.I.; Punzi, H.; et al. Urine markers of kidney tubule cell injury and kidney function decline in SPRINT trial participants with CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 349–358. [CrossRef]
- Wallentin, L.; Eriksson, N.; Oiszowka, M.; Grammer, T.B.; Hagstrom, E.; Held, C.; Keleber, M.E.; Koenig, W.; Marz, W.; Stewart, R.A.H.; et al. Plasma proteins associated with cardiovascular death in patients with chronic coronary heart disease: A retrospective study. PLoS Med. 2021, 18, e1003513. [Google Scholar] [CrossRef]
- Folkersen, L.; Gustafsson, S.; Wang, Q.; Hansen, D.H.; Hedman, A.K.; Schork, A.; Page, K.; Zhernakova, D.V.; Wu, Y.; Peters, J.; et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30, 931 individuals. Nat. Metab. 2020, 2, 1135–1148. [Google Scholar] [CrossRef]
- Arain, F.; Abraityte, A.; Bogdanova, M.; Solberg, O.G.; Micheisen, A.E.; Lekva, T.; Aakhus, S.; Holm, S.; Halvorsen, B.; Finsen, A.V.; et al. YKL-40 (Chitinase-3-like protein 1) serum levels in aortic stenosis. Circ. Heart Fail. 2020, 13, e006643. [Google Scholar] [CrossRef]
- Kim, E.G.; Kim, M.N.; Hong, J.Y.; Lee, J.W.; Kim, S.Y.; Kim, K.W.; Lee, C.G.; Elias, J.A.; Song, T.W.; Sohn, M.Y. Chitinase 3-like 1 contributes to food allergy via M2 macrophage polarization. Allergy Asthma Immunol. Res. 2020, 12, 1012–1028. [Google Scholar] [CrossRef]
- Sianipar, I.R.; Sestramita, S.; Pradnjaparamita, T.; Yunir, E.; Harbuwono. D S.; Soewondo, P.; Tahapary, D.L. The role of intestinal-fatty acid binding proteins and chitinase-3-like protein 1 across the spectrum of dysglycemia. Diabetes Metab. Syndr. 2022, 16, 102366. [CrossRef]
- Omidian, M.; Mahmoudi, M.; Javanbakht, M.H.; Eshraghian, M.R.; Abshirini, M.; Daneshzad, E.; Hasani, H.; Alvandi, E.; Djalai, M. Effects of vitamin D supplementation on circulatory YKL-40 and MCP-1 biomarkers associated with vascular diabeteic complications; A randomized, placebo-controlled, double-blind clinical trial. Diabetes Metab. Syndr. 2019, 13, 2873–2877. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, I.T.; Cetin, B.A.; Koroglu, N.; Mathyk, B.A.; Erdem, B. Inflammatory marker YKL-40 levels in intrahepatic cholestasis of pregnancy. Gynecol. Endocrinol. 2019, 35, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Bulanik, M.; Sagsoz, N.; Sayan, C.D.; Yeral, M.I.; Kisa, U. Comparison of serum Ykl-40 and ischemia modified albumin levels between pregnant women with hyperemesis gravidarum and normal pregnant women. Med. Arch. 2019, 73, 97–100. [Google Scholar] [CrossRef]
- Sobkowiak, P.; Narozna, B.; Wojsyk-Banaszak, I.; Breborowicz, A.; Szczepankiewicz, A. Expression of proteins associated with airway fibrosis differs between children with allergic asthma and allergic rhinitis. Int. Immunopathol. Pharmacol. 2021, 35, 2058738421990493. [Google Scholar] [CrossRef] [PubMed]
- Permain, J.; Appleton, L.; Ho, S.S.C.; Coffey, M.; Ooi, C.Y.; Keenan, J.I.; Day, A.S. Children with cystic fibrosis have elevated levels of fecal chitinase-3-like-1. J. Pediatr. Gastroenterol. Nutr. 2022, 75, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Topcu, D.B.; Tugcu, G.; Er, B.; Polat, S.E.; Hizal, M.; Yalcin, E.E.; Ersoz, D.D.; Coplu, L.; Ozcelik, U.; Kiper, N.; et al. Increased plasma YKL-40 level and chitotriosidase activity in cystic fibrosis patients. Inflammation 2022, 45, 627–638. [Google Scholar] [CrossRef]
- Buisson, A.; Vazeille, E.; Minet-Quinard, R.; Goutte, M.; Bouvier, D.; Goutorbe, F.; Pereira, B.; Barnich, Bommelaer, G. Faecal chitinase 3-like 1 is a reliable marker as accurate as faecal calprotectin in detecting endoscopic activity in adult patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2016, 43, 1069-1079. [CrossRef]
- Aomatsu, T.; Imaeda, H.; Matumoto, K.; Kimura, E.; Yoden, A.; Tamai, H.; Fujiyama, Y.; Mizoguchi, E.; Andoh, A. Faecal chitinase 3-like -1: a novel biomarker of disease activity in paediatric inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 34, 941–948. [Google Scholar] [CrossRef]
- Low, D.; Subramaniam, R.; Lin, L.; Aomatsu, T.; Mizoguchi, A.; Ng, A.; DeGruttola, A.K.; Lee, C.G.; Elias, J.A.; Andoh, A.; et al. Chitinase 3-like 1 induces survival and proliferation of intestinal epithelial cells during chronic inflammation and colitis-associated cancer by regulating S100A9. Oncotarget 2015, 6, 36535–36550. [Google Scholar] [CrossRef]
- Comabella, M.; Fernandez, M.; Martin, R.; Rivera-Vallve, S.; Borras, E.; Chiva, C.; Julia, E.; Rovira, A.; Canto, E.; Alvarez-Cermeno, J.C.; et al. Celebrospinal fluid chitinase 3-like 1 levels are assocaited with conversion to multiple sclerosis. Brain 2010; 133, 1082-1093. [CrossRef]
- Bhardwaj, R.; Yester, J.; Singh, S.K.; Biswas, D.D.; Surace, M.J.; Waters, M.R.; Hauser, K.F.; Yao, Z.; Boyce, B.F.; Kordula, T. RelB/p50 complexes regulate cytokine-induced YKL-40 expression. J. Immunol. 2015, 194, 2862–2870. [Google Scholar] [CrossRef]
- Burman, J.; Raininko, R.; Blennow, K.; Zetterberg, H.; Axeisson, M.; Malmestrom, C. YKL-40 is a CSF biomarker of intrathecal inflammation in secondary progressive multiple sclerosis. J. Neuroimmunol. 2016, 292, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Dichev, V.; Kazakova, M.; Sarafian, V. YKL-40 and neuron-specific enolase in neurodegeneration and neuroinflammation. Rev. Neurosci. 2020, 31, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Lee, I.A.; Low, D.; Kamba, A.; Mizoguchi, A.; Shi, H.N.; Lee, C.G.; Elias, J.A.; Mizoguchi, E. Chitinase 3-like 1 synergistically activates IL-6 mediated STAT3 phosphorylation in intestinal epithelial cells in murine models of infectious colitis. Inflamm. Bowel Dis. 2014, 20, 835–846. [Google Scholar] [CrossRef]
- Eurich, K.; Segawa, M.; Toei-Shimizu, S.; Mizoguchi, E. Potential role of chitinase 3-like 1 in inflammation-associated carcinogenic changes of epithelial cells. World J. Gastroenterol. 2009, 15, 5249–5259. [Google Scholar] [CrossRef]
- Russo, C.; Valle, M.S.; Casabona, A.; Malaguarnera, L. Chitinase Signature in the Plasticity of Neurodegenerative Diseases. Int J Mol Sci. 2023, 24, 6301. [Google Scholar] [CrossRef]
- Temmerman, J.; Engelborghs, S.; Bjerke, M.; D’haeseleer, M. Cerebrospinal fluid inflammatory biomarkers for disease progression in Alzheimer’s disease and multiple sclerosis: a systematic review. Front Immunol. 2023, 14, 1162340. [Google Scholar] [CrossRef]
- Dage, J.L.; Eloyan, A.; Thangarajah, M.; Hammers, D.B.; Fagan, A.M.; Gray, J.D.; Schindler, S.E.; Snoddy, C.; Nudelman, K.N.H.; Faber, K.M.; et al. Cerebrospinal fluid biomarkers in the Longitudinal Early-onset Alzheimer’s Disease Study. Alzheimers Dement. 2023, 19, S115–S125. [Google Scholar] [CrossRef]
- Pelkmans, W.; Shekari, M.; Brugulat-Serrat, A.; Sánchez-Benavides, G.; Minguillón, C.; Fauria, K.; Molinuevo, J.L.; Grau-Rivera, O.; González Escalante, A.; Kollmorgen, G.; et al. Astrocyte biomarkers GFAP and YKL-40 mediate early Alzheimer’s disease progression. Alzheimers Dement. 2024, 20, 483–493. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry. 2022, 27, 4781–4789. [Google Scholar] [CrossRef]
- Lananna, B.V.; McKee, C.A.; King, M.W.; Del-Aguila, J.L.; Dimitry, J.M.; Farias, F.H.G.; Nadarajah, C.J.; Xiong, D.D.; Guo, C.; Cammack, A.J.; et al. Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci. Transl. Med. 2020, 12, eaax3519. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dobricic, V.; Ohlei, O.; Bos, I.; Vos, S.J.B.; Prokopenko, D.; Tijms, B.M.; Andreasson, U.; Blennow, K.; Vandenberghe, R.; et al. TMEM106B and CPOX are genetic determinants of cerebrospinal fluid Alzheimer’s disease biomarker levels. Alzheimers Dement. 2021, 17, 1628–1640. [Google Scholar] [CrossRef]
- Kim, M.A.; Shin, Y.S.; Pham le, D.; Park, H.S. Adult asthma biomarkers. Curr. Opin. Allergy Clim. Immunol. 2014;14.49-54. [CrossRef]
- Ober, C.; Tan, Z.; Sun, Y.; Possick, J.D.; Pan, L.; Nicolae, R.; Radford, S.; Parry, R.R.; Heinzmann, A.; Deichmann, K.A.; et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N. Engl. J. Med. 2008, 358, 1682–91. [Google Scholar] [CrossRef]
- Gomez, J.L.; Crisafi, G.M.; Holm, C.T.; Meyers, D.A.; Hawkins, G.A.; Bleecker, E.R.; Jarjour, N.; Severe Asthma Research Program (SARP) Investigators.; Cohn, L.; Chupp, G.L. Genetic variation in chitinase 3-like 1 (CHI3L1) contributes to asthma severity and airway expression of YKL-40. J. Allergy Clin. Immunol. 2015, 136, 51-58. [CrossRef]
- James, A.J.; Reinius, L.E.; Verhoek, M.; Gomes, A.; Kupczyk, M.; Hammar, U.; Ono, J.; Ohta, S.; Izuhara, K.; Bel, E.; et al. Increased YKL-40 and Chitotriosidase in Asthma and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit Care Med. 2016, 193, 131–42. [Google Scholar] [CrossRef]
- Ahangari, F.; Sood, A.; Ma, B.; Takyar, S.; Schuyler, M.; Qualls, C.; Dela Cruz, C.S.; Chupp, G.L.; Lee, C.G.; Elias, J.A. Chitinase 3-like-1 regulates both visceral fat accumulation and asthma-like Th2 inflammation. Am. J. Respir. Cri.t Care Med. 2015, 191, 746–57. [Google Scholar] [CrossRef]
- Johansen, J.S.; Johansen, H.S.; Price, P.A. A new biochemical marker for joint injury. Analysis of YKL-40 in serum and synovial fluid. Br. J. Rheumatol. 1993, 32, 949-955. [CrossRef]
- Johansen, J.S.; Achultz, N.A.; Jensen, B.V. Plasma YKL-40: a potential new cancer biomarker? Future Oncol. 2009, 5, 1065–1082. [Google Scholar] [CrossRef]
- Subramaniam, R.; Mizoguchi, A.; Mizoguchi, E. Mechanistic roles of epithelial and immune cell signaling during the development of colitis-associated cancer. Cancer Res. Front 2016, 2, 1–21. [Google Scholar] [CrossRef]
- Kawada, M.; Seno, H.; Kanda, K.; Nakanishi, Y.; Akitake, R.; Komekado, H.; Kawada, K.; Sakai, Y.; Mizoguchi, E.; Chiba, T. Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene 2021, 31, 3111–3123. [Google Scholar] [CrossRef]
- Ray, A.L.; Castillo, E.F.; Morris, K.T.; Nofchissey, R.A.; Weston, L.L.; Samedi, V.G.; et al. Blockade of MK2 is protective in inflammation-associated colorectal cancer development. Int. J. Cancer 2016, 138, 770–775. [Google Scholar] [CrossRef]
- Zhao, T.; Zeng, J.; Xu, Y.; Su, Z.; Chong, Y.; Ling, T.; Xu, H.; Shi, H.; Zhu, M.; Mo, Q.; et al. Chitinase-3 like-protein-1 promotes glioma progression via the NF-κB signaling pathway and tumor microenvironment reprogramming. Theranostics 2022, 12, 6989–7008. [Google Scholar] [CrossRef]
- Guetta-Terrier, C.; Karambizi, D.; Akosman, B.; Zepecki, J.P.; Chen, J.S.; Kamle, S.; Fajardo, J.E.; Fiser, A.; Singh, R.; Toms, S.A.; et al. Chi3l1 is a modulator of glioma stem cell states and a therapeutic target in glioblastoma. Cancer Res. 2023, 83, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Sleisenger and Fordtran’s Gastrointestinal and Liver, Disease.; Pathophysiology/ diagnosis/management, edited by Mark, Feldman.; et al., Elsevier 2020.
- Barnich, N.; Carvalho, F. A.; Glasser, A. L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J. F.; et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Invest. 2007, 117, 1566-74. [CrossRef]
- Tran, H.T.; Barnich, N.; Mizoguchi, E.; et al. Potential role of chitinases and chitin-binding proteins in host-microbial interactions during the development of intestinal inflammation. Histol. Histopathol. 2011, 26, 1453–64. [Google Scholar]
- Chaudhuri, S.; Bruno,J.C.; Alonzo, F. 3rd.; Xayarath, B.; Cianciotto, N.P.; Freitag, N.E. Contribution of chitinases to Listeria monocytogenes pathogenesis. Appl. Enviro. Microbiol. 2010, 76, 7302–7305. [CrossRef]
- Tanaka, H.; Akutsu, H.; Yabuta, I.; Hara, M.; Sugimoto, H.; Ikegami, T.; Watanabe, T.; Fujiwara, T. A novel chitin-binding mode of the chitin-binding domain of chitinase A1 from Bacillus circulans WL-12 revealed by solid-state NMR. FEBS Lett. 2018, 592, 3173–3182. [Google Scholar] [CrossRef]
- Fusetti, F.; Pijning, T.; Kalk, K.H.; Bos, E.; Dijkstra, B.W. Crystal structure and carbohydrate-binding properties of the human cartilage glycoprotein-39. J. Bio. Chem. 2003, 278, 37753–37760. [Google Scholar] [CrossRef]
- Moran, A. P.; Gupta, A.; Joshi, L. Sweet-talk: role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut 2011, 10: 1412-25. [CrossRef]
- Park, D.; Arabyan, N.; Williams, C.C.; Song, T.; Mitra, A.; Weimer, B.C.; Maverakis, E.; Lebrilla, C.B. Salmonella typhimurium enzymatically landscapes the host Intestinal epithelial cell (IEC) surface glycome to increase invasion. Mo.l Cell Proteo. 2016, 15, 3653–3664. [Google Scholar] [CrossRef]
- Johansson Malin, E.V. Mucus layers in inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 2124–31. [CrossRef]
- Hansson Gunnar, C.; and Malin Ev, J. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut microbes 2010, 1, 51-54. [CrossRef]
- Bohr, S.; Patel, S.J.; Vask, R.; Shen, K.; Golberg, A.; Berthiaume, F.; Yarmush, M.L. The Role of CHI3L1 (Chitinase-3-Like-1) in the Pathogenesis of Infections in Burns in a Mouse Model. PLoS ONE 2015, 10, e0140440. [Google Scholar] [CrossRef]
- Siwczak, F.; Cseresnyes, Z.; Hassan, M.I.A.; Aina, K.O.; Caristedt, S.; Sigmund, A.; Groger, M.; Surewaard, B.G.J.; Werz, O.; Figge, M.T. et al. Human macrophage polarization determines bacterial persistence of Staphylococcus aureus in a liver-on-chip-based infection model. Biomaterials 2022, 287, 121632. [CrossRef]
- Bonet-Rossinyol, Q.; Camprubi-Font, C.; Lopez-Siles, M.; Martinez-Medina, M. Identification of differences in gene expression implicated in the adherent-invasive Escherichia coli phenotype during in vitro infection of intestinal epithelial cells. Front. Cell. Infect. Microbiol. 2023, 13, 1228159. [Google Scholar] [CrossRef]
- Bringer, M.A.; Billard, E.; Glasser, A.L.; Colombel, J.F.; Darfeuille-Michaud, A. Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-α secretion. Lab. Invest. 2012, 92, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Tsunoda, T.; Mizoguchi, E.; Okada, T.; Sudo, T.; Kawahara, A.; Akiba, J.; Akagi, Y. Chitinase 3-like 1, carcinoembryonic antigen-related cell adhesion molecule 6, and ectopic claudin-2 in the carcinogenic process of ulcerative colitis. Anticancer Res. 2022, 42, 4119–4127. [Google Scholar] [CrossRef]
- Nell, S.; Suerbaum, S.; Josenhans, C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat. Rev. Microbiol. 2010, 8, 564–577. [Google Scholar] [CrossRef]
- Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3464. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cui, W.; Li, X. Interaction between commensal bacteria, immune response and the intestinal barrier in inflammatory bowel disease. Front. Immunol. 2021, 12, 761981. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Long, X.-B.; Cao, P.-P.; Wang, N.; Liu, Y.; Cui, Y.-H.; Huang, S.-K.; Liu, Z. Clara cell 10-kD protein suppresses Chitinase 3-like 1 expression associated with eosinophilic chronic rhinosinusitis. Am. J. Respir. Cri.t Care Med. 2010, 181, 908–916. [Google Scholar] [CrossRef]
- Zeng, X.; Cheung, S.K.K.; Shi, M.; Or, P.M.Y.; Li, Z.; Liu, J.Y.H.; Ho, W.L.H.; Liu, T.; Lu, K.; Rudd, J.A.; et al. Astrocyte-specific knockout of YKL-40/Chi3l1 reduces Aβ burden and restores memory functions in 5xFAD mice. J. Neuroinflamm. 2023, 20, 290. [Google Scholar] [CrossRef]
- Choi, J.Y.; Yeo, I.J.; Kim, K.C.; Choi, W.R.; Jung, J.-K.; Han, S.-B.; Hong, J.T. K284-6111 Prevents the amyloid beta-induced neuroinflammation and impairment of recognition memory through inhibition of NF-κB-mediated CHI3L1 expression. J. Neuroinflamm. 2018, 15, 224. [Google Scholar] [CrossRef]
- Anwar, M.M.; Fathi, M.H. Early Approaches of YKL-40 as a Biomarker and Therapeutic Target for Parkinson’s Disease. Neurodegener. Dis. Manag. 2023, 13, 85–99. [Google Scholar] [CrossRef]
- Birmpili, D.; Charmarke Askar, I.; Pham-Van, L.D.; Kuntzel, T.; Spenlé, C.; Riou, A.; Bagnard, D. Toward a combination of biomarkers for molecular characterization of multiple sclerosis. Int. J. Mol. Sci. 2022, 23, 14000. [Google Scholar] [CrossRef]
- Kwak, E.J.; Hong, J.Y.; Kim, M.N.; Kim, S.Y.; Kim, S.H.; Park, C.O.; Kim, K.W.; Lee, C.G.; Elias, J.A.; Jee, H.M.; et al. Chitinase 3-like 1 drives allergic skin inflammation via Th2 immunity and M2 macrophage activation. Clin. Exp. Allergy 2019, 49, 1464–1474. [Google Scholar] [CrossRef]
- Lee, C.G.; Hartl, D.; Lee, G.R.; Koller, B.; Matsuura, H.; Da Silva, C.A.; Sohn, M.H.; Cohn, L.; Homer, R.J.; Kozhich, A.A.; et al. Role of breast regression protein 39 (BRP-39)/Chitinase 3-like-1 in Th2 and IL-13-Induced tissue responses and apoptosis. J. Exp. Med. 2009, 206, 1149–1166. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Hartl, D.; Kang, M.-J.; Dela Cruz, C.S.; Koller, B.; Chupp, G.L.; Homer, R.J.; Zhou, Y.; Cho, W.-K.; Elias, J.A.; et al. Role of breast regression protein-39 in the pathogenesis of cigarette smoke-induced inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 44, 777–786. [Google Scholar] [CrossRef]
- Zhou, Y.; He, C.H.; Herzog, E.L.; Peng, X.; Lee, C.-M.; Nguyen, T.H.; Gulati, M.; Gochuico, B.R.; Gahl, W.A.; Slade, M.L.; et al. Chitinase 3-like-1 and its receptors in Hermansky-Pudlak Syndrome-associated lung disease. J. Clin. Invest. 2015, 125, 3178–3192. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Kim, K.C.; Park, M.H.; Seo, Y.; Park, H.; Park, M.H.; Chang, J.; Hwang, D.Y.; Han, S.B.; Kim, S.; et al. Atherosclerosis is exacerbated by Chitinase-3-like-1 in amyloid precursor protein transgenic mice. Theranostics 2018, 8, 749–766. [Google Scholar] [CrossRef]
- Tsantilas, P.; Lao, S.; Wu, Z.; Eberhard, A.; Winski, G.; Vaerst, M.; Nanda, V.; Wang, Y.; Kojima, Y.; Ye, J.; et al. Chitinase 3 like 1 Is a Regulator of Smooth Muscle Cell Physiology and atherosclerotic lesion stability. Cardiovasc. Res. 2021, 117, 2767–2780. [Google Scholar] [CrossRef]
- Kim, M.; Chang, J.Y.; Lee, D.W.; Kim, Y.R.; Son, D.J.; Yun, J.; Jung, Y.S.; Lee, D.H.; Han, S.; Hong, J.T. Chitinase 3 like 1 deficiency ameliorates lipopolysaccharide-induced acute liver injury by inhibition of M2 macrophage polarization. Mol. Immunol. 2023, 156, 98–110. [Google Scholar] [CrossRef]
- Lee, D.H.; Han, J.H.; Lee, Y.S.; Jung, Y.S.; Roh, Y.S.; Yun, J.S.; Han, S.B.; Hong, J.T. Chitinase-3-like-1 deficiency attenuates ethanol-induced liver injury by inhibition of sterol regulatory element binding protein 1-dependent triglyceride synthesis. Metabolism 2019, 95, 46–56. [Google Scholar] [CrossRef]
- Kim, A.D.; Kui, L.; Kaufmann, B.; Kim, S.E.; Leszczynska, A.; Feldstein, A.E. Myeloid-specific deletion of Chitinase-3-like 1 protein ameliorates murine diet-induced steatohepatitis progression. J. Mol. Med. 2023, 101, 813–828. [Google Scholar] [CrossRef]
- Pizano-Martínez, O.; Yañez-Sánchez, I.; Alatorre-Carranza, P.; Miranda-Díaz, A.; Ortiz-Lazareno, P.C.; García-Iglesias, T.; Daneri-Navarro, A.; Vázquez-Del Mercado, M.; Fafutis-Morris, M.; Delgado-Rizo, V. YKL-40 expression in CD14+ liver cells in acute and chronic injury. World J. Gastroenterol. 2011, 17, 3830–3835. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Szychlinska, M.A.; Tibullo, D.; Malaguarnera, L.; Musumeci, G. Expression of CHI3L1 and CHIT1 in osteoarthritic rat cartilage model. A Morphological Study. Eur J. Histochem. 2014, 58, 2423. [Google Scholar] [CrossRef] [PubMed]
- Verheijden, G.F.; Rijnders, A.W.; Bos, E.; Coenen-de Roo, C.J.; van Staveren, C.J.; Miltenburg, A.M.; Meijerink, J.H.; Elewaut, D.; de Keyser, F.; Veys, E.; et al. Human cartilage glycoprotein-39 as a candidate autoantigen in rheumatoid arthritis. Arthritis Rheum. 1997, 40, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Park, H.-J.; Lim, S.; Koo, J.-H.; Lee, H.-G.; Choi, J.O.; Oh, J.H.; Ha, S.-J.; Kang, M.-J.; Lee, C.-M.; et al. Regulation of Chitinase-3-like-1 in T cell elicits Th1 and cytotoxic responses to inhibit lung metastasis. Nat. Commun. 2018, 9, 503. [Google Scholar] [CrossRef]
- Gomez, J.L.; Crisafi, G.M.; Holm, C.T.; Meyers, D.A.; Hawkins, G.A.; Bleecker, E.R.; Jarjour, N.; Cohn, L.; Chupp, G.L. Genetic Variation in Chitinase 3-like 1 (CHI3L1) contributes to asthma severity and airway expression of YKL-40. Journal of Allergy and Clin. Immunol. 2015, 136, 51-58.e10. [CrossRef]
- Salomon, J.; Matusiak, Ł.; Nowicka-Suszko, D.; Szepietowski, J.C. Chitinase-3-Like Protein 1 (YKL-40) reflects the severity of symptoms in atopic dermatitis. Journal of Immunol. Res. 2017, 2017, e5746031. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, Y.; Peng, Q.; Shu, X.; Wang, G.; Wu, X. Serum YKL-40 Level Is Associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin. Rheumatol. 2019, 38, 1655–1663. [Google Scholar] [CrossRef]
- Hamilton, D.; Lehman, H. Asthma phenotypes as a guide for current and future biologic therapies. Clinic. Rev. Allerg. Immunol. 2020, 59, 160–174. [Google Scholar] [CrossRef]
- Kamba, A.; Lee, I.-A.; Mizoguchi, E. Potential Association between TLR4 and Chitinase 3-like 1 (CHI3L1/YKL-40) signaling on colonic epithelial cells in inflammatory bowel disease and colitis-associated cancer. Curr. Mol. Med. 2013, 13, 1110–1121. [Google Scholar] [CrossRef]
- Huang, J.; Gu, Z.; Xu, Y.; Jiang, L.; Zhu, W.; Wang, W. CHI3L1 (Chitinase 3 Like 1) upregulation ss associated with macrophage signatures in esophageal cancer. Bioengineered 12, 7882–7892. [CrossRef]
- Chen, A.; Jiang, Y.; Li, Z.; Wu, L.; Santiago, U.; Zou, H.; Cai, C.; Sharma, V.; Guan, Y.; McCarl, L.H.; et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated iImmune suppression in glioblastoma. J. Clin. Invest. 2021, 131. [Google Scholar]
- Zhao, T.; Zeng, J.; Xu, Y.; Su, Z.; Chong, Y.; Ling, T.; Xu, H.; Shi, H.; Zhu, M.; Mo, Q.; et al. Chitinase-3 like-protein-1 promotes glioma progression via the NF-κB signaling pathway and tumor microenvironment reprogramming. Theranostics 2022, 12, 6989–7008. [Google Scholar] [CrossRef] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The role of macrophages in cancer development and therapy. Cancers (Basel) 2021, 13, 1946. [Google Scholar] [CrossRef]
- Taifour, T.; Attalla, S.S.; Zuo, D.; Gu, Y.; Sanguin-Gendreau, V.; Proud, H.; Solymoss, E.; Bui, T.; Kuasne, H.; Papavasiliou, V.; et al. The tumor-derived cytokine Chi3l1 induces neutrophil extracellular traps that promote T cell exclusion in triple-negative breast cancer Immunity 2023, 56, 2755-2772.e8. [CrossRef]
- Jeon, S.H.; Lee, Y.S.; Yeo, I.J.; Lee, H.P.; Yoon, J.; Son, D.J.; Han, S.-B.; Hong, J.T. Inhibition of Chitinase-3-like-1 by K284-6111 reduces atopic skin inflammation via repressing lactoferrin. Immune. Netw. 2021, 21, e22. [Google Scholar] [CrossRef]
- Yang, P.-S.; Yu, M.-H.; Hou, Y.-C.; Chang, C.-P.; Lin, S.-C.; Kuo, I.-Y.; Su, P.-C.; Cheng, H.-C.; Su, W.-C.; Shan, Y.-S.; et al. Targeting protumor factor Chitinase-3-like-1 secreted by Rab37 vesicles for cancer immunotherapy. Theranostics 2022, 12, 340–361. [Google Scholar] [CrossRef]
- Yu, J.E.; Yeo, I.J.; Son, D.J.; Yun, J.; Han, S.-B.; Hong, J.T. Anti-Chi3L1 antibody suppresses lung tumor growth and metastasis through inhibition of M2 polarization. Mol. Oncol. 2022, 16, 2214–2234. [Google Scholar] [CrossRef]
- Ma, B.; Akosman, B.; Kamle, S.; Lee, C.-M.; He, C.H.; Koo, J.S.; Lee, C.G.; Elias, J.A. CHI3L1 Regulates PD-L1 and Anti–CHI3L1–PD-1 antibody elicits synergistic antitumor responses. J Clin. Invest. 131, e137750. [CrossRef]
- Ma, B.; Kamle, S.; Akosman, B.; Khan, H.; Lee, C.-M.; Lee, C.G.; Elias, J.A. CHI3L1 enhances melanoma lung metastasis via regulation of T cell co-stimulators and CTLA-4/B7 axis. Front. Immunol. 2022, 13, 1056397. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-Checkpoint Inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Som, A.; Mandaliya, R.; Alsaadi, D.; Farshidpour, M.; Charabaty, A.; Malhotra, N.; Mattar, M.C. Immune checkpoint inhibitor-induced colitis: A Comprehensive Review. World J. Clin. Cases 2019, 7, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.A.; Kamba, A.; Low, D.; Mizoguchi, E. Novel methylxanthine derivative-mediated anti-inflammatory effects in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Rao, F.V.; Andersen, O.A.; Vora, K.A.; Demartino, J.A.; van Aalten, D.M. Methylxanthine drugs are chitinase inhibitors: investigation of inhibition and binding modes. Chem. Biol. 2005, 12, 973–980. [Google Scholar] [CrossRef]
- Lee, I.A.; Low, D.; Kamba, A.; Llado, V.; Mizoguchi, E. Oral caffeine administration ameliorates acute colitis by suppressing chitinase 3-like 1 expression in intestinal epithelial cells. J. Gastroenterol. 2014, 49, 1206–1216. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Sadanaga, T.; Okada, T.; Minagawa, T.; Akiba, J. Does caffeine have a double-edged sword role in inflammation and carcinogenesis in the colon? Intest. Res. 2023, 21, 306–317. [Google Scholar] [CrossRef]
- Debono, M.; Gordee, R.S. Antibiotics that inhibit fungal cell wall development. Annu. Rev. Microbiol. 1994, 48, 471–497. [Google Scholar] [CrossRef]
- Hakala, B.E.; White, C.; Recklies, A.D. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J. Biol. Chem. 1993, 268, 25803–25810. [Google Scholar] [CrossRef]
- Lee, C.G.; Da, Silva, C.A.; Lee, J.Y.; Hartl, D.; Elias, J. A. Chitin regulation of immune responses: an old molecule with new roles. Curr. Opin. Immunol. 2008, 20, 1-6. [CrossRef]
- Lee, C.G.; Da, Silva, C.A.; Dela, Cruz, C.S.; Ahangari, F.; Ma, B.; Kang, M. J.; He, C. H.; Takyar, S.; Elias, J. A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 2011, 73, 479-450. [CrossRef]
- Nagatani, K.; Wang, S.; Llado, V.; Lau, C.W.; Li, Z.; Mizoguchi, A.; Nagler, C.R.; Shibata, Y.; Reinecker, H.C.; Mora, J.R.; et al. Chitin microparticles for the control of intestinal Inflammation. Inflamm. Bowel Dis. 2012, 18, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Letzel, M.; Synstad, B.; Eijsink, V.G.H.; Peter-Katalinic, J.; Peter, M.G. Libraries of chitin-oligosaccharides of mixed acetylation patterns and their interactions with chitinases. Adv. Chitin Sci. 2000, 4, 545–552. [Google Scholar]
- Cederkvist, F.H.; Parmer, M.P.; Varum, K.M.; Eijsink, V.G.H.; Sorlie, M. Inhibition of a family 18 chitinase by chitooligosaccharides. Carbohydr. Polym. 2008, 74, 41–49. [Google Scholar] [CrossRef]
Figure 1.
Nuclear translocation of CHI3L1 after TNFα stimulation in colonic epithelial cells: Human colonic epithelial cell line HCT116 cells have been cultured for 48 hours on 3 dimensional Matri-gel without stimulation (A), with 50ng/ml TNFα stimulation (B), or with 50 ng/ml TNFa stimulation with chitinase inhibitors (mixture of 2.5 mM caffeine and 25mM pentoxifylline) (C). The cells were stained with anti-CHI3L1 antibody (shown as red) and anti-CD44 (shown as green) followed by CF594 anti-rabbit IgG and CF488 anti-mouse IgG, respectively. Nucleus are stained by DAPI (shown as blue) and analyzed by fluorescence microscope BZ-X800 (Keyence, Osaka, Japan) at 100x objective lens with oil.
Figure 1.
Nuclear translocation of CHI3L1 after TNFα stimulation in colonic epithelial cells: Human colonic epithelial cell line HCT116 cells have been cultured for 48 hours on 3 dimensional Matri-gel without stimulation (A), with 50ng/ml TNFα stimulation (B), or with 50 ng/ml TNFa stimulation with chitinase inhibitors (mixture of 2.5 mM caffeine and 25mM pentoxifylline) (C). The cells were stained with anti-CHI3L1 antibody (shown as red) and anti-CD44 (shown as green) followed by CF594 anti-rabbit IgG and CF488 anti-mouse IgG, respectively. Nucleus are stained by DAPI (shown as blue) and analyzed by fluorescence microscope BZ-X800 (Keyence, Osaka, Japan) at 100x objective lens with oil.
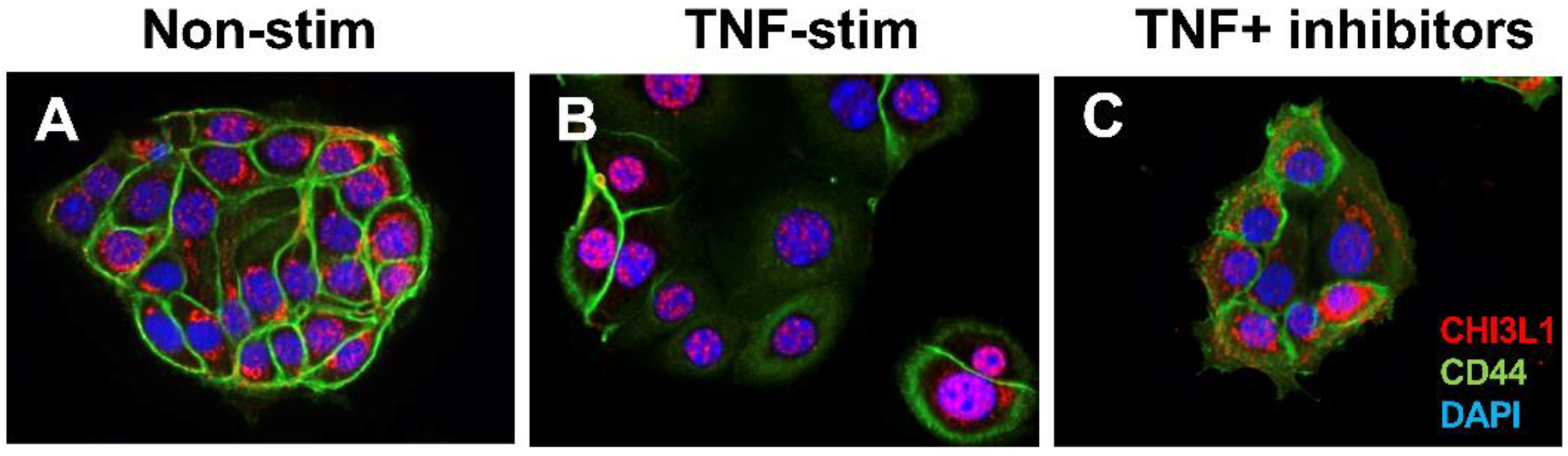
Figure 2.
Intestinal microflora and host colonic epithelial cells (CEC) interactions in healthy individuals versus inflammatory bowel disease (IBD) patients: In healthy individuals, CEC is protected by two (outer- and inner-) mucus layers, and intestinal bacteria cannot access CECs. In contrast, in IBD patients, disruption of these mucus layers causes the chitin-binding protein of potentially pathogenic bacteria to bind to glycosylated CHI3L1 expressed on CEC, allowing these bacteria to adhere and invade. Non-glycosylated mutant form of CHI3L1 shows less bacterial binding in vitro.
Figure 2.
Intestinal microflora and host colonic epithelial cells (CEC) interactions in healthy individuals versus inflammatory bowel disease (IBD) patients: In healthy individuals, CEC is protected by two (outer- and inner-) mucus layers, and intestinal bacteria cannot access CECs. In contrast, in IBD patients, disruption of these mucus layers causes the chitin-binding protein of potentially pathogenic bacteria to bind to glycosylated CHI3L1 expressed on CEC, allowing these bacteria to adhere and invade. Non-glycosylated mutant form of CHI3L1 shows less bacterial binding in vitro.
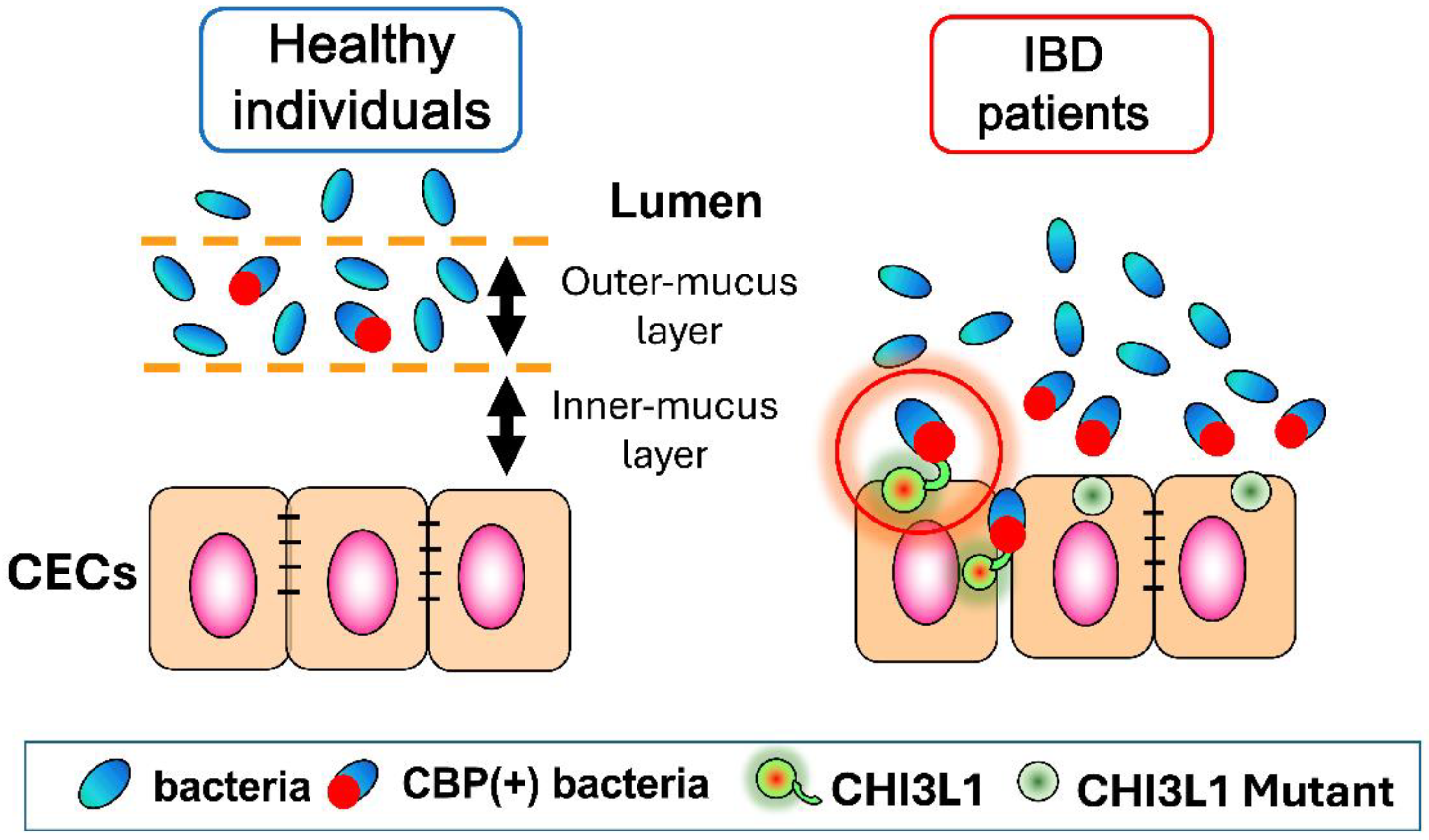
Figure 3.
Conceptual representation of the putative effects of CHI3L1 in chronic inflammation within epithelial cells: Bacterial binding to Toll-like receptor 4 (TLR4) on epithelial cells triggers the production of pro-inflammatory cytokines mediated by MyD88, TRIF, and MAPK signaling pathways. This inflammatory cascade results in the upregulation of CHI3L1 expression in epithelial cells. Additionally, innate lymphoid cells type 2 (ILC2) and Th2 cells initiate a Th2 inflammation in response to epithelial cell inflammation. Th2 inflammation further promotes the polarization of M0 macrophages towards the M2 phenotype. IL-13 plays a crucial role in inducing CHI3L1 expression in both epithelial cells and M2 macrophages. Elevated CHI3L1 expression in epithelial cells enhances bacterial adhesion and sustains chronic inflammation. Moreover, chronic inflammation mediated by CHI3L1 leads to tumorigenesis, contributing to the establishment of a tumor-promoting microenvironment. Created with BioRender.com.
Figure 3.
Conceptual representation of the putative effects of CHI3L1 in chronic inflammation within epithelial cells: Bacterial binding to Toll-like receptor 4 (TLR4) on epithelial cells triggers the production of pro-inflammatory cytokines mediated by MyD88, TRIF, and MAPK signaling pathways. This inflammatory cascade results in the upregulation of CHI3L1 expression in epithelial cells. Additionally, innate lymphoid cells type 2 (ILC2) and Th2 cells initiate a Th2 inflammation in response to epithelial cell inflammation. Th2 inflammation further promotes the polarization of M0 macrophages towards the M2 phenotype. IL-13 plays a crucial role in inducing CHI3L1 expression in both epithelial cells and M2 macrophages. Elevated CHI3L1 expression in epithelial cells enhances bacterial adhesion and sustains chronic inflammation. Moreover, chronic inflammation mediated by CHI3L1 leads to tumorigenesis, contributing to the establishment of a tumor-promoting microenvironment. Created with BioRender.com.
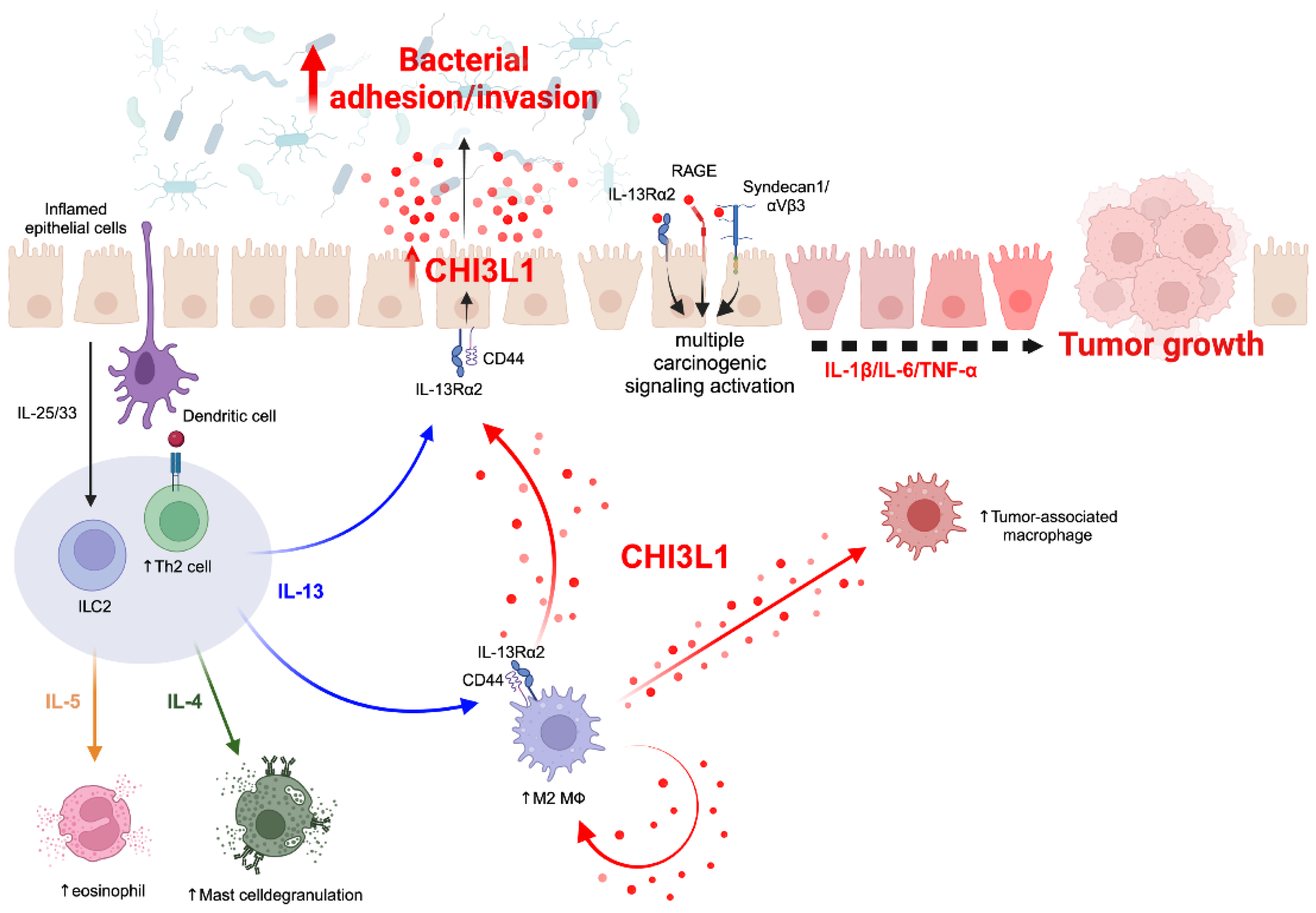
Figure 4.
Schematic representation on putative trimeric- and dimeric-interaction theories: Our current hypothesis is based on a dimeric interaction theory (a direct interaction of host N-glycosylated CHI3L1 protein and bacterial chitin-binding protein) but not a trimeric interaction theory (chitin or chitin-like oligosaccharides link CHI3L1 protein and chitin-binding protein together make those trimeric complex).
Figure 4.
Schematic representation on putative trimeric- and dimeric-interaction theories: Our current hypothesis is based on a dimeric interaction theory (a direct interaction of host N-glycosylated CHI3L1 protein and bacterial chitin-binding protein) but not a trimeric interaction theory (chitin or chitin-like oligosaccharides link CHI3L1 protein and chitin-binding protein together make those trimeric complex).
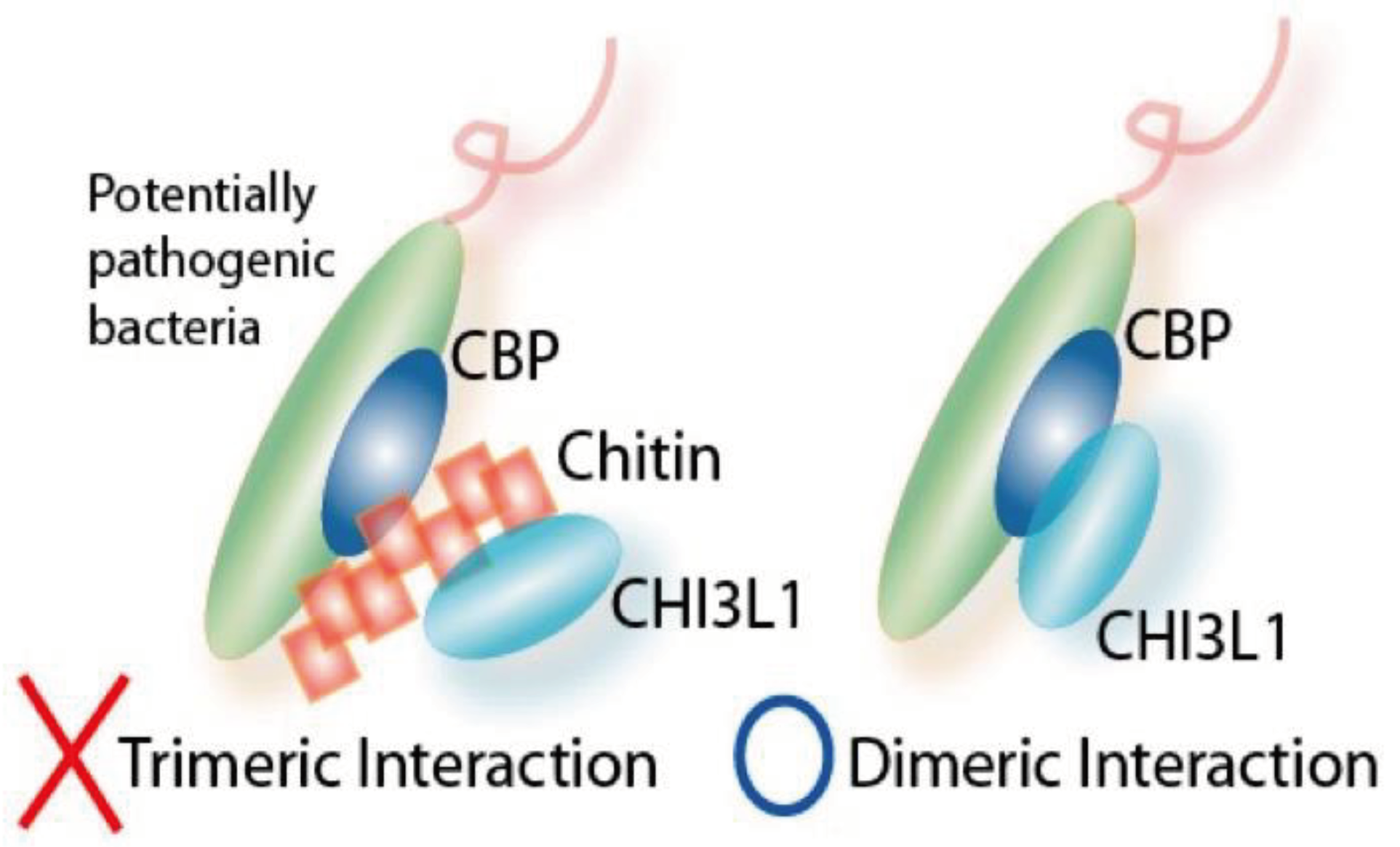

Table 1.
Increased levels of CHI3L1 under inflammatory conditions in the human referred within the past five years.
Table 1.
Increased levels of CHI3L1 under inflammatory conditions in the human referred within the past five years.
| Disease | Samples | General Overview | Ref. |
|---|---|---|---|
| IBD | Serum, CECs, Stool, Mφ, Peripheral MDMs, |
|
[23,24,25,26] |
| MS | CSF, Serum, Tissue |
|
[27,28,29,30,31,32,33,34,35] |
| AD | CSF, Tissue |
|
[36,37,38,39,40,41,42,43] |
| Asthma, COPD | Serum, BECs, |
|
[44,45,46,47,48] |
|
|
|
[49] |
| COVID-19 | Serum, Tissue, |
|
[50,51,52,53] |
| Liver Diseases | Serum, Tissue |
|
[54,55,56] |
| Oral Disease | Tissue, Saliva |
|
[57,58] |
| Kidney Disease | Serum, Urine |
|
[59,60,61,62,63] |
| Heart Disease | Serum, |
|
[64,65,66] |
| Food Allergy | Serum |
|
[67] |
| Diabetes Mellitus | Serum |
|
[68,69] |
| Pregnancy | Serum, Urine |
|
[70,71] |
| Rhinitis | Serum |
|
[72] |
| Cystic Fibrosis | Stool, Serum, |
|
[73,74] |
Abbreviations: AD, Alzheimer’s disease; AIEC, adherent invasive Escherichia coli; BECs, bronchial epithelial cells; APAPS, Acetaminophen; CECs, colonic epithelial cells; CF, cystic fibrosis; CHC, chronic hepatitis C; CHD, coronary heart disease; CHI3L1, chitinase 3-like 1; CF, cystic fibrosis; CKD, chronic kidney disease; CSF, cerebrospinal fluid protein; COPD, chronic obstructive pulmonary disease; COVID-19, Corona virus infectious disease, emerging in 2019; eGFR, estimated glomerular filtration rate; ESKD, end-stage kidney disease; GWAS, genome-wide association studies; IBD, inflammatory bowel disease; ICU, intensive care unit; MCI, mild cognitive impairment; LS-OCMB, lipid-specific oligoclonal IgM bands; MDMs, monocyte derived-macrophages; moDCs, monocyte derived dendritic cells; MS, multiple sclerosis; Mφ, macrophages; NASH, non-alchoholic steatohepatitis; RA, rheumatoid arthritis; SARS-CoV2, Severe acute respiratory syndrome coronavirus 2 of the genus Batacoronavirus; TLR, Toll-like receptor; TNF, tumor necrosis factor, TNF-R1, TNF-receptor type I.
Table 2.
CHI3L1-associated chronic inflammation in animal models.
| Disease | Model | Features | Ref. |
|---|---|---|---|
| Eosinophilic Chronic Rhinosinusitis | CC10 WT/KO mice with OVA sensitization |
|
[121] |
| Alzheimer’s disease | 5xFAD mice model Aβ1–42-induced AD mice model |
|
[122,123] |
| Parkinson’s disease | LPS-induced PD rats model |
|
[124] |
| Multiple sclerosis | EAE-PLP mice model |
|
[125] |
| Atopic dermatitis | Filaggrin mutated mice with OVA sensitization |
|
[126] |
| Asthma | BRP-39 WT/KO mice with OVA sensitization |
|
[127] |
| COPD | BRP-39 WT/KO mice with cigarette smoke |
|
[128] |
| Hermansky-Pudlak syndrome | Hps1 mutation mice with Bleomycin |
|
[129] |
| Atherosclerosis | ApoE-/- mice with high-fat diet |
|
[130,131] |
| Liver sepsis | LPS-induced mice model |
|
[132] |
| Alcoholic liver injury | CHI3L1 WT/KO mice with the Lieber-DeCarli ethanol liquid diet |
|
[133] |
| NASH | CHI3L1 WT/KO mice (CreLyz) with choline-deficient high-fat diet |
|
[134] |
| Chronic liver injury | CCl4 i.p. injection rats model |
|
[135] |
| Obesity | CHI3L1 WT/KO mice with HFD |
|
[95] |
| CKD | Ischemia/reperfusion injury mice model with microaneurysm clip |
|
[60] |
| Osteoarthritis | Osteoarthritis rats model with ACLT |
|
[136] |
| Rheumatic arthritis | RA mice model with HC gp-39 (CHI3L1) i.p. injection |
|
[137] |
| IBD | CHI3L1 WT/KO with AOM/DSS CHI3L1 WT/KO with S.typhimurium/AIEC inoculation |
|
[77,82] |
Abbreviations: Aβ, amyloid-beta; ACLT, anterior cruciate ligament transection; AD, Alzheimer’s disease; AIEC, adherent invasive Escherichia coli; ApoE, apolipoprotein E; AOM, azoxymethane; BAL, bronchoalveolar lavage; BRP-39, breast regression protein-39; CC10, clara cell 10kD; Ccl2, C-C motif chemokine ligand 2; CCl4, carbon tetrachloride; CHI3L1, chitinase 3-like 1; CKD, chronic kidney disease; CRTH2, chemoattractant receptor homologous molecule on T helper type 2 cells; COPD, chronic obstructive pulmonary disease; CSF, cerebrospinal fluid; DSS, dextran sulphate sodium; EAE, experimental autoimmune encephalomyelitis; ECRS, eosinophilic chronic rhinosinusitis; HC gp-39, human cartilage glycoprotein-39; HFD, high-fat diet; HPS, Hermansky-Pudlak syndrome; IL-13Rα2, interleukin-13 receptor alpha 2; i.p., intraperitoneal; KD, knock down; LPS, lipopolysaccharides; MS, multiple sclerosis; NASH, non-alcoholic steatohepatitis; OVA, ovalbumin; PD, Parkinson’s disease; PLP, proteolipid protein; RA, rheumatoid arthritis; RAGE, receptor for advanced glycation end products; STAT3, signal transducer and activator of transcription 3; S.typhimurium, Salmonella typhimurium.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



