
Submitted:
28 February 2024
Posted:
29 February 2024
You are already at the latest version
Abstract
Peripheral and autonomic neuropathy are common disease manifestations in systemic amyloidosis. Neurofilament light chain (NfL), a neuron-specific biomarker, is released into the blood and cerebrospinal fluid after neuronal damage. This systematic review provides an overview on the value of NfL in early detection of neuropathy, central nervous system involvement, monitoring of neuropathy progression, and treatment effect in systemic amyloidosis. A literature search in PubMed, Embase and Web of Science was performed on 14-02-2024 for studies investigating NfL levels in patients with systemic amyloidosis and transthyretin gene variant (TTRv) carriers. Only studies containing original data were included. Included were twelve full-text articles and six abstracts describing 1604 participants: 298 controls and 1306 TTRv carriers or patients with or without polyneuropathy. Patients with polyneuropathy demonstrated higher NfL levels compared to healthy controls and asymptomatic carriers. Disease onset was marked by rising NfL levels. Following initiation of transthyretin gene-silencer treatment, NfL levels decreased and remained stable over an extended period. NfL is not an outcome biomarker, but an early and sensitive disease process biomarker for neuropathy in systemic amyloidosis. Therefore, NfL has potential to be used for early detection of neuropathy, monitoring treatment effect, and monitoring disease progression in patients with systemic amyloidosis.
Keywords:
systemic amyloidosis
; hereditary transthyretin amyloid
; immunoglobulin light chain amyloid
; transthyretin gene variant carrier
; biomarker
; neurofilament light chain
; polyneuropathy
; small fiber neuropathy
; autonomic neuropathy
Introduction
Transthyretin (ATTR) amyloidosis and immunoglobulin light chain (AL) amyloidosis are the two main types of systemic amyloidosis that can affect the nervous system [1]. ATTR amyloidosis can be hereditary (ATTRv), the result of deposition of variant transthyretin (TTRv), or acquired (ATTRwt), the result of deposition of wild-type TTR [1]. The peripheral nervous system is frequently affected in ATTRv and AL amyloidosis, leading to polyneuropathy and autonomic neuropathy [2,3]. However, leptomeningeal involvement can occur in ATTRv amyloidosis [4] and peripheral polyneuropathy may also occur in ATTRwt amyloidosis [5].
Establishing the presence of polyneuropathy in patients with systemic amyloidosis is crucial for early diagnosis and initiation of treatment. The presence and severity of polyneuropathy have prognostic implications for survival and quality of life. In addition, as polyneuropathy will progress over time, monitoring its course is important for assessing disease progression and treatment effect.
Polyneuropathy is confirmed by nerve conduction studies (NCS) showing axonal degeneration [6,7]. Although NCS are the most objective measure for the evaluation of polyneuropathy, NCS have limited sensitivity for axonal damage in early disease stages and are only able to measure large fiber neuropathy [6,8]. Small fiber neuropathy can be evaluated by quantitative sensory testing (QST). However, this non-invasive method is limited by its subjective nature [6,9]. The Sudoscan is another objective modality in the evaluation of small fiber neuropathy but is limited to the sympathetic C nerve fibers of the autonomic nervous system [10]. Another option to evaluate nerve involvement in systemic amyloidosis is to perform a sural nerve biopsy. However, this procedure carries the risk of permanent cutaneous anesthesia in the biopsied nerve area [11,12]. Lastly, a punch skin biopsy can be performed to detect amyloid and small nerve fiber loss [13,14,15,16,17].
In addition, the neuropathy impairment score (NIS), neuropathy impairment score lower limbs (NIS-LL), neuropathy impairment score upper limbs (NIS-UL), modified neuropathy impairment score +7 (mNIS +7) (includes NCS, QST, and autonomic endpoints), familial amyloidotic polyneuropathy (FAP) stage and polyneuropathy disability (PND) score were designated to assess polyneuropathy impairment in ATTRv amyloidosis [18]. As all these measures mentioned above assess damage to nerves and related nerve dysfunction, they all can be considered outcome markers. Outcome markers that are insensitive to tracking disease progression over shorter time intervals and, because the best outcome will not be a clear improvement but merely stabilization, they do not quickly provide useful information about a favorable treatment effect. This is in sharp contrast to a disease process biomarker that reflects the activity of the disease leading to the outcome. The earlier such a disease process biomarker improves or even normalizes, the less damage will be generated. Therefore, obtaining a disease process biomarker would signify a major gain in the neuropathy toolbox of the clinician.
Biomarkers are available and very helpful as disease process parameters for the detection and follow-up of cardiac disease (troponin T and NT-proBNP), liver disease (alkaline phosphatase, gamma-glutamyl transferase and bilirubin) and kidney disease (urea, creatinine, proteinuria and cystatin C) in amyloidosis [2,19]. Therefore, there is a clear need for an early and sensitive serum or plasma biomarker for polyneuropathy, for neuropathy progression and for assessing the effect of treatment on neuropathy in systemic amyloidosis. Recent research shows that neurofilament light chain (NfL), a neuron specific cytoskeletal protein released into the blood and cerebrospinal fluid during axonal damage [20,21], correlates with polyneuropathy and disease severity in systemic amyloidosis [22,23,24]. What is more, NfL levels normalize after treating vasculitis as cause of polyneuropathy, traumatic brain injury and stroke [25,26,27]. NfL thus has potential to behave as a disease process marker not only for disease progression, but also signifying a favorable treatment effect on polyneuropathy.
Concerning AL amyloidosis, little is known about the response of treatment in AL amyloidosis patients with polyneuropathy. NfL may serve as a valuable biomarker for assessing the presence of polyneuropathy. Treatment decisions can be informed by NfL results, as certain treatment options may be more advisable to avoid in the context of AL amyloidosis [28].
We performed a systematic search of current studies on NfL in systemic amyloidosis to evaluate the value of NfL in early detection of neuronal damage and monitoring polyneuropathy progression and treatment effect. Lastly, we evaluate if NfL has potential to be implemented in clinical practice in the near future.
Methods
Eligibility Criteria
Inclusion criteria were clinical studies of patients diagnosed with systemic amyloidosis and carriers of a variant in the TTR gene in which NfL levels were measured.
Exclusion criteria were (1) articles in languages other than English, and (2) studies concerning patients with Alzheimer’s disease.
Information Sources
PubMed, Embase and Web of Science were used.
Search Strategy
The following search query was used in PubMed, Embase and Web of Science: ((((amyloidosis) OR (amyloid neuropathy)) NOT (Alzheimer)) AND (neurofilament) OR (NfL)). References of the included articles were screened to find more articles.
Selection Process
A total number of 173 articles was found (PubMed 58, Embase 69 and Web of Science 46) on the search date 14 February 2023. See Figure 1.
First 62 duplicates were removed. Of the remaining 111 unique titles, 77 were excluded because the articles were written in languages other than English (N = 1), concerned animal studies (N = 10), or deemed irrelevant (N = 66), leaving 34 titles. After reading the abstracts, 5 additional publications were excluded because they were deemed irrelevant (N = 5), leaving 29 publications. Of these 29 publications, two were deemed irrelevant (N = 2), lacked original data (N = 7), or were written in a language other than Englisch (N = 2), leaving 12 articles and 6 relevant congress abstracts.
No new articles could be added after checking the reference lists of the publications.
Data Collection Process
Publications were categorized according to these topics in patients with amyloidosis:
Neurofilament light chain in relation to polyneuropathy and disease severity; Neurofilament light chain in an asymptomatic disease stage; Neurofilament light chain in relation to small fiber and autonomic neuropathy; Neurofilament light chain in relation to treatment; Confounders affecting neurofilament light chain levels.
Description of Cases
As not all cases will develop symptoms, cases can only in retrospect be described as presymptomatic after they have developed symptoms. Therefore, we chose to describe cases as asymptomatic instead of presymptomatic in this review, because asymptomatic describes the actual situation at the moment of evaluation.
Results
Results of Individual Studies
Our systematic review comprised a total of 1604 participants, including 1286 ATTRv amyloidosis patients with and without neurological symptoms along with 288 healthy controls and 20 AL amyloidosis patients with and without neurological symptoms along with 10 age- and sex-matched healthy controls. Currently, there are no published studies on NfL in ATTRwt amyloidosis. Table 1 provides a summary of the 18 studies that were included. Figure 2 displays the NfL levels per study and per patient group.
Neurofilament Light Chain in Relation to Polyneuropathy and Disease Severity
Nine studies compared serum or plasma NfL levels in ATTRv amyloidosis patients with polyneuropathy to concentrations in neurologically asymptomatic TTRv carriers, asymptomatic ATTRv patients or healthy controls [22,23,24,29,30,31,32,33,34]. One of these studies also compared serum NfL levels in AL amyloidosis patients with polyneuropathy to levels in AL amyloidosis without polyneuropathy or healthy controls [24]. All these studies found increased levels in patients with polyneuropathy compared to asymptomatic TTRv carriers, neurologically asymptomatic ATTRv patients or healthy controls. Details of these studies are shown in Table 1 and Figure 2.
Six studies found a correlation between NfL levels and disease severity measured by the (modified +7) Neuropathy Impairment Score (NIS) [22,23,31,32,33,34], six studies between NfL levels and disease severity assessed by the polyneuropathy disability (PND) score [23,24,29,32,33,35] and three studies between NfL and FAP stage [23,32,33]. However, one of the largest studies found no correlation between mNIS+7 score or PND score and plasma NfL levels [30]. Other studies also found no correlations between PND score or FAP stage and serum NfL [31,34] or NIS-LL scores and plasma NfL [36]. One study found that changes in plasma NfL correlated with mNIS+7 during treatment with patisiran [30] and another study showed that a sustained improvement in mNIS+7 score goes in parallel with maintained reduced plasma NfL levels after treatment with patisiran [37]. In contrast, Sato et al observed a significant decrease in serum NfL levels at one and two years after switch to patisiran whereas NIS scores did not change [38].
Neurofilament Light Chain in an Asymptomatic Disease Stage
Six studies [23,29,30,32,33,34] conducted receiver operating characteristics (ROC) analysis to assess the ability of NfL to distinguish between an asymptomatic stage and a symptomatic neuropathy stage. Details of these studies are shown in Table 1 and Table 2 and Figure 2.
First, Ticau et al [30] discriminated healthy controls and ATTRv amyloidosis patients with polyneuropathy based on a plasma NfL cutoff level of 37 pg/mL measured with the single-molecule array (Simoa) assay with a sensitivity of 84.9% and specificity of 96.4%. Second, Romano et al [32] established a serum NfL cutoff, which was almost the same as from Ticau et al, of 37.10 pg/mL for the transition from asymptomatic to symptomatic, with a sensitivity of 81.4% and specificity of 100% with the Ella assay. However, they used different analytical methods and sample types (serum or plasma). Third, Loser et al [23] concluded that serum NfL levels above 11.7 pg/mL measured with Simoa assay at both baseline and after 1-year follow-up could discriminate symptomatic from asymptomatic patients with 85.7% sensitivity and 100% specificity. No significant difference was found between healthy controls and asymptomatic ATTRv amyloidosis patients. Fourth, Maia et al [29] studied plasma NfL levels in two independent cohorts with Simoa assay. A NfL cutoff of 10.6 pg/mL discriminated between asymptomatic (PND 0) and early-stage patients (PND I) and between asymptomatic (PND 0) and symptomatic patients (PND ≥I) with a sensitivity of 92.3% and 96.2%, respectively, and specificity of 93.8% in both. In addition, comparing PND I with PND ≥ II in cohort #1 resulted in an optimal cutoff of 66.9 pg/mL with a sensitivity of 61.5% and specificity of 92.3% whereas the optimal cutoff was set on 75.7 pg/mL in cohort #2 with a sensitivity of 84.6% and specificity of 80%. Fifth, González-Moreno et al [34] established that a cutoff value of 93.6 pg/mL discriminates ATTRv amyloidosis patients from asymptomatic TTRv carriers with a sensitivity of 79% and a specificity of 87%. The high cutoff could be related to their use of a first-generation enzyme-linked immunosorbent assay (ELISA) to measure serum NfL levels. The NfL levels they found are higher compared to levels reported in studies using the Ella or Simoa assay. Sixth, Carroll et al. [33] found that baseline NfL levels greater than 64.5 pg/mL discriminated between a combined group of symptomatic patients and individuals who were at baseline asymptomatic but developed sensorimotor neuropathy (sensorimotor converters), and asymptomatic individuals with a sensitivity of 91.9% and a specificity of 88.5%. Asymptomatic individuals could only be discriminated from a combined group of sensory and sensorimotor converters or symptomatic patients by NfL levels above 88.9 pg/mL with a sensitivity 62.9% and specificity of 96.2%. However, an increase of 17% in NfL levels over 6 months could discriminate asymptomatic from sensory or sensorimotor converters with a sensitivity of 88.9% and specificity 80.0%.
Lau et al [36] followed six initially neurologically asymptomatic patients who developed polyneuropathy. During the study period, plasma NfL levels increased 1.5-fold from baseline over a follow-up period of 4.0 years [3.6 – 6.9]. They did not establish cutoff values for discriminating between no polyneuropathy and polyneuropathy. Their cohort, with only V122I-genopositive patients, likely had mild or subtle polyneuropathy, making a cutoff value hard to find due to small differences between asymptomatic and symptomatic patients in their study.
In addition, longitudinal data on serum NfL levels in twelve asymptomatic TTRv carriers and eight asymptomatic ATTRv patients have been presented [35]. Serum NfL increased over two years in asymptomatic ATTRv amyloidosis patients, but did not change in the asymptomatic TTRv carriers. Levels of serum NfL were also studied longitudinally in a group of seven TTRv carriers who progressed during a median follow-up of ten years from asymptomatic TTRv carriers to symptomatic ATTRv amyloidosis patients with NCS-confirmed polyneuropathy. In this group of TTRv carriers who developed polyneuropathy, the median baseline serum NfL level of 8.4 pg/mL rose to median 49.8 pg/mL upon the onset of initial symptoms (PND score I) and the serum NfL level had risen even further at the time polyneuropathy could be established by nerve conduction studies. Levels of serum NfL were already above the 95th reference percentile 5.5 years (range 3.0-7.6 years) before the onset of symptoms (PND I) [35].
Neurofilament Light Chain in Relation to Small Fiber and Autonomic Neuropathy
The axonal length-dependent polyneuropathy in amyloidosis is typically preceded by a small fiber neuropathy in the lower extremities [39,40]. Detecting small fiber neuropathy is thus desirable for early disease detection. The Sudoscan serves as an objective test for evaluating small fiber neuropathy limited to the sympathetic C nerve fibers of the autonomic nervous system [10]. In a study by Luigetti et al [31], a significant association was demonstrated between serum NfL values and Sudoscan values obtained from the feet. However, the participants in this study also exhibited (high-grade) polyneuropathy, which confounded this correlation. Therefore, at present it remains unknown whether NfL detects small fiber neuropathy in the absence of polyneuropathy. Details of this study are shown in Table 1 and Figure 2.
Autonomic dysfunction is also a common and early manifestation in ATTRv amyloidosis. Data on serum NfL levels in relation to cardiac autonomic neuropathy based on iodine-123-labeled meta-iodobenzylguanidine ([123I]mIBG) scintigraphy and other measures of autonomic neuropathy (Ewing Battery) have been presented [41]. In multivariate regression analysis polyneuropathy was the only independent predictor of serum NfL levels, in contrast to (cardiac) autonomic neuropathy. Details of this study are shown in Table 1 and Figure 2.
Neurofilament Light Chain in Relation to Treatment
Ten studies [23,30,33,35,36,37,38,42,43,44] also analyzed the effect of treatment. Details of these studies are shown in Table 1 and Table 3 and Figure 2.
Ticau et al [30] analyzed plasma NfL levels in a subset of ATTRv amyloidosis patients who participated in the phase 3, placebo-controlled study of patisiran (APOLLO-A). Patients treated with patisiran showed a significant decrease in plasma NfL levels at 9 months compared to baseline, that was maintained at 18 months. In contrast, patients in the placebo group showed an increase in plasma NfL levels at 9 months compared to baseline and this increase was maintained at 18 months. At the 18-month mark, patients receiving patisiran exhibited plasma NfL levels that were twice as low as those without treatment (placebo group). Additionally, after 18 months of patisiran treatment, mNIS+7 scores improved, and this improvement correlated with a decrease in plasma NfL levels. However, no correlation was observed between plasma NfL levels and mNIS+7 or PND score at baseline.
The patisiran Global open-label extension (OLE) study [37] revealed that the reduced levels of plasma NfL, along with improvements in clinical efficacy assessments, persisted for an additional period of 24 months. Unpublished data by Gilling [44], again showed the maintained reduction in plasma NfL levels at the 36-month mark of the open-label extension study. Patients who received placebo for 18 months in the APOLLO-A study [30] were switched to patisiran in the Global OLE study [37]. After 12 and 24 months of treatment with patisiran, plasma NfL levels decreased as compared to the APOLLO-A baseline and even significantly decreased as compared to the Global OLE baseline. After 24 months, these patients reached plasma NfL levels that were comparable to plasma NfL levels in the APOLLO-A patisiran group at 24 months in the Global OLE study. Data showed that this reduction persisted after 36 months (unpublished) [44].
Loser et al [23] studied serum NfL levels in a cohort of patients who were untreated but had previously received a liver transplant or were under treatment with tafamidis or patisiran. Serum NfL levels tended to increase during one-year follow-up in untreated symptomatic patients, all of whom had received a liver transplant in the past. Serum NfL levels in patients on treatment with either patisiran (n=4) or tafamidis (n=2) also showed a tendency to increase during one year of follow-up. In patients initiated on patisiran (n=2) during follow-up, serum NfL levels showed a tendency to decrease. All the patients in this study did not worsen neurologically during the follow-up period despite increasing serum NfL levels in some patients.
Sato et al [38] longitudinally analyzed changes in serum NfL levels of patients who switched from tafamidis to patisiran. They observed a significant reduction in serum NfL levels one year after switching to patisiran which maintained over two years. These findings are in line with the findings of Loser et al and Ticau et al [23,30,37]. Interestingly, there was no significant change in NIS scores during the same time period.
Lau et al [36] observed plasma NfL levels in patients who had polyneuropathy at baseline or developed it during follow-up. At the end of the follow-up period, part of the patients with polyneuropathy was on treatment with diflunisal, tafamidis, eplontersen, patisiran or revusiran. No sustained plasma NfL level changes were observed with treatment initiation or regimen changes and NIS scores did not correlate meaningfully with plasma NfL fluctuations.
Carroll et al [33] longitudinally evaluated the effect of TTR-gene silencer treatment on NfL levels in thirteen ATTRv amyloidosis patients. Levels of NfL decreased during treatment and the change in NfL positively correlated with the change in transthyretin levels over the same time interval.
Data on plasma NfL levels after 4 and 18 months after treatment initiation with patisiran or vutrisiran have been presented. Plasma NfL levels decreased at 4 months relative to baseline and this decrease was sustained at 18 months for both treatment regimens [42].
Data on serum NfL levels in patients with ATTRv amyloidosis and Coutinho Stage 1-2 polyneuropathy that were treated with eplontersen have also been presented. Patients receiving eplontersen throughout week 85 showed a trend of decreasing serum NfL levels [43].
Except for the study of Loser [23], the studies mentioned above show that NfL levels decrease, but do not normalize, after initiation of treatment with a TTR-gene silencer. A decrease in NfL levels within four months after treatment initiation with the TTR-gene silencer patisiran, is maintained for at least 36 months. Two studies included some patients in whom serum or plasma NfL levels were studied after initiation of treatment with a TTR-stabilizer [35,36], but there is insufficient data to draw conclusions about the effect of initiation of TTR-stabilizers on serum or plasma NfL levels. In patients already treated with a TTR-stabilizer before baseline, serum NfL levels remained stable after two years of follow-up [35]. Due to the limited number of patients undergoing TTR-stabilizer treatment in the other studies, these studies are not suitable for drawing conclusions on this matter.
Neurofilament Light Chain and Cerebral Manifestations in Hereditary ATTR Amyloidosis
NfL is a biomarker of axonal damage of both the central and peripheral nervous system. Increased blood levels of NfL have been reported in almost all neurodegenerative disorders among which sporadic (amyloid-beta) cerebral amyloid angiopathy and Alzheimer’s disease [20,45]. Cerebral involvement in ATTRv amyloidosis shows many neuropathological and imaging similarities with sporadic cerebral amyloid angiopathy [46,47]. It is likely that (subclinical) cerebral involvement in ATTRv amyloidosis causes increased blood levels of NfL. That cerebral involvement in ATTRv amyloidosis can potentially lead to increased NfL levels has been mentioned in several studies [29,32,37], but no studies have actually investigated this.
Confounders Affecting Neurofilament Light Chain Levels
Several influencing factors should be considered for the accurate interpretation of NfL levels in patients with ATTRv amyloidosis.
First, NfL is not specific to ATTRv amyloidosis-related polyneuropathy. Any cause of neuronal damage, whether cerebral or peripheral, may result in elevated levels of NfL [20]. Second, NfL levels increase with age [48]. NfL levels are expected to increase by 2.1% per year [49]. In individuals aged 60 years and older, there is an increase in the variability of NfL levels, possibly associated with subclinical comorbid pathology [49]. Eight studies included in this review took into account the effect of aging on NfL levels [22,23,24,29,32,33,34,36]. However, in all these studies the NfL increase due to polyneuropathy outweighed the increase associated with aging. Third, both serum creatinine and hemoglobin A1c (HbA1c) exhibit strong correlations with NfL levels, even after adjusting for age. Kidney function plays a crucial role in NfL clearance, and patients with elevated HbA1c levels may experience microvascular disease complications leading to NfL release [49]. Fourth, body mass index (BMI) can affect NfL levels. Individuals with a higher BMI have a larger volume of distribution leading to lower absolute NfL levels [50]. A study involving 1706 individuals without neurological disease, which assessed the predictive capacity of 52 demographic, lifestyle, comorbidity, anthropometric, or laboratory characteristics in explaining variability in serum NfL levels, did not identify additional independent predictors [49].
Discussion
The primary objective of this systematic review was to ascertain the value of NfL in the early detection of neuropathy and monitoring neuropathy progression and treatment effect in systemic amyloidosis. In addition, this review aimed to assess the feasibility of implementing NfL in clinical practice in the near future. There is substantial evidence for the use of NfL as marker of polyneuropathy and neuropathy severity in ATTRv amyloidosis. There is also substantial evidence supporting the use of NfL in monitoring disease progression and treatment effect of TTR-gene silencers. Some evidence supports the use of NfL in detecting neuropathy in a presymptomatic stage. However, in this context it is important to take into account that some evidence suggests that NfL is not suitable to detect small fiber neuropathy [51] and autonomic neuropathy [41]. Only one study evaluated NfL in AL amyloidosis. And no studies have been published on ATTRwt amyloidosis.
All available studies (Table 1) consistently show that NfL levels are increased in patients with ATTRv amyloidosis and polyneuropathy. The median levels are 4.3 to 15.4 times higher in patients with polyneuropathy compared to healthy controls, depending on the disease severity. The levels of NfL in patients with ATTRv amyloidosis with polyneuropathy are even higher than those observed for other peripheral nerve disorders, like chronic inflammatory demyelinating neuropathy [52,53] and Charcot-Marie-Tooth disease [21]. As ATTRv amyloidosis is a relatively rapidly progressive disease and, without treatment, fatal 7-12 years after the first disease manifestation [54], it can be hypothesized that this rate of progression contributes to higher NfL levels [22] even when the NIS score is lower than in, e.g. Charcot Marie Tooth disease.
Based on the combined NfL data from the current studies, we could construct a hypothetical course of NfL levels over time from asymptomatic TTRv carriers who progress to asymptomatic ATTRv amyloidosis patients without neurological symptoms to symptomatic ATTRv amyloidosis patients with polyneuropathy and subsequently receive treatment (Figure 3). Initially, the course of NfL levels in asymptomatic TTRv carriers resembles that of a healthy person [22,23,24,29,32,35,36]. However, when amyloid is deposited, transforming the asymptomatic TTRv carrier into an asymptomatic ATTRv amyloidosis patient, NfL levels start to rise more than can be expected by aging alone [33,35]. In the subsequent period, the first clinical manifestations emerge, polyneuropathy can be confirmed with NCS and levels of NfL continue to rise [22,23,24,29,30,32,33,35,36]. During treatment with a TTR-stabilizer, NfL levels either remain stable [35] or may increase in individual patients [38]. Whereas, levels of NfL decrease after initiation of a TTR-gene silencer and this decrease is sustained with extended treatment, up to 36 months, but levels do not normalize [30,33,37,38,42,43,44].
The lack of normalization in NfL, in contrast to what is observed in patients after treatment of vasculitis as cause of polyneuropathy, stroke and traumatic brain injury [25,26,27], may have several explanations. First, despite the halt in disease progression, the axons already affected may gradually degenerate through a dying-back mechanism [55]. This smoldering axonal damage may cause ongoing leakage of NfL from neurons. Second, the existing amyloid deposits between axons may remain to be toxic to the neurons, subsequently leading to continuous release of NfL [56]. Third, despite a significant reduction in TTR levels due to TTR-gene silencer treatment, a residual quantity of TTR persists in the bloodstream which still could deposit on pre-existing amyloid deposits and consequently cause continuous, subtle nerve damage [57].
Apparently conflicting results concerning a relationship between NfL levels and measures of disease severity have been reported. Several studies found correlations between NfL levels and disease severity measured by the different NIS scores (NIS, NIS-LL, mNIS+7) [22,23,31,32,33,34], PND score and/or FAP stage [23,24,29,32,33,35], while one of the largest studies did not find a correlation between individual NfL levels and the mNIS+7 score [30]. Sato et al found that levels of NfL significantly decreased one and two years after initiation of patisiran, with no change in NIS values [38]. In contrast, Ticau et al reported a significant correlation between the decrease in NfL levels and improvement in mNIS+7 score after 18 months of treatment with patisiran [30]. The most likely explanation for these apparently contradictory results is that NfL reflects the active process of neuronal damage at a specific point in time, whereas PND and NIS scores reflect the overall burden of neurological impairment. PND and NIS scores can be considered outcome markers whereas NfL is a disease process biomarker. Consequently, it makes sense that these markers do not always correlate with each other.
Three different analytical technologies and either serum or plasma samples were used for measuring NfL in the studies included in this review. Three studies used the Ella assay [31,32,43], fourteen studies used the Simoa assay [22,23,24,29,30,31,35,36,37,38,41,42,44,58] and one study used a first generation ELISA [34]. Both the Ella and Simoa assays make use of ultrasensitive immunoassay technology and there is a good correlation between the outcomes of both assays. However, NfL levels are 17-24% higher when measured with the Ella assay as compared to levels measured with the Simoa assay [59,60,61]. The first generation ELISA used in one of the studies included in this review was reported to have a lower limit of detection of 33 pg/mL [34]. Many of the asymptomatic TTRv carriers included in this study had NfL levels below the lower limit of detection. Therefore, this ELISA lacks the sensitivity needed to detect early neuronal damage in asymptomatic TTRv carriers that transition to symptomatic patients. Eight studies included in this review used plasma [22,29,30,36,37,42,44,58] and ten studies used serum [23,24,31,32,33,34,35,38,41,43]. There are proportional and systematic differences between serum and plasma NfL measurements. Plasma NfL levels are approximately 10% lower than serum NfL levels [62,63], however results can be used interchangeably if standardized values are used [64]. The pre-analytical stability of NfL is good: concentrations of NfL in serum or plasma remain stable at room temperature when processing of samples is delayed up to 7 days [63] and concentrations of NfL remain stable in serum and plasma samples stored at -80°C for up to 20 and 16 years, respectively [65].
Clinical Implications
Current evidence supports implementation of NfL as early and sensitive serum or plasma biomarker for polyneuropathy, for neuropathy progression, and for assessing the effect of treatment on neuropathy in ATTRv amyloidosis.
NfL has added value compared to polyneuropathy impairment measures (e.g. FAP stage, PND and NIS scores) and NCS, which can be considered outcome measures, as NfL is a biomarker for the neuropathic disease process. In addition, NfL measurement is not burdensome to patients, it is reproducible and objective. The established cutoffs to discriminate asymptomatic TTRv carriers from ATTRv amyloidosis patients with polyneuropathy vary considerably and depend on the sample type (serum versus plasma) and the assay that was used (Table 2). Some cutoffs have limited sensitivity and therefore cannot be used to rule out the presence of polyneuropathy, other cutoffs lack specificity (Table 2). Center-specific cutoff values may be useful but have to be established for each particular center.
The best approach for incorporating NfL measurements into clinical practice seems to compare the measured NfL level with age-dependent reference values from one of two large online databases comprising individuals without a neurological disorder [48,66]. If the value exceeds the 95th percentile of normal, additional neurological examination and/ or vigilance for the onset of polyneuropathy is recommended [35]. Another useful approach could be to look at changes in NfL levels over time instead of absolute values at one moment. Carroll et al [33] showed that a relative increase of NfL over time could discriminate asymptomatic TTRv carriers from carriers that developed sensory or sensorimotor neuropathy with good sensitivity. This approach provided better discrimination than assessing a single NfL value, in particular for detection of neuronal damage in an early disease stage. This is also supported by the longitudinal data of Brunger et al [35]. It is relevant to detect neuronal damage, even in a presymptomatic stage because treatment with diflunisal can then be considered [67].
NfL is sensitive to tracking disease progression and treatment effect over short time intervals, thus providing added value compared to disease outcome measures in monitoring both disease progression and treatment effect. Increasing NfL levels indicate disease progression, while decreasing levels after initiation of treatment indicate a beneficial treatment effect. NfL measurements will likely serve as useful adjunct measurements in future clinical trials.
When implementing NfL in daily practice, several factors need to be considered. First, NfL levels should be measured using a reliable and sensitive immunoassay, e.g. the Ella or Simoa assay, both of which unfortunately are not widely available. However, also routine laboratory technologies, such as Lumipulse [68] allow straightforward, reliable and sensitive longitudinal quantification of serum and plasma NfL. Confounders such as the presence of other neurological diseases, renal insufficiency, diabetes mellitus with microvascular complications and aging should be taken into account when interpreting NfL levels. NfL is an early and sensitive marker for polyneuropathy, but there is some evidence that it does not detect small fiber neuropathy and autonomic neuropathy. Therefore, NfL cannot be used as an absolute marker of neuropathy onset in ATTRv amyloidosis.
Considerations for Future Research
NfL is a reliable and objective measure to detect neuronal damage in a presymptomatic stage in ATTRv amyloidosis [35]. Longitudinal investigation of a larger number of TTRv carriers with a variety of genotypes is warranted to specify the dynamics of NfL in TTRv carriers that progress to symptomatic ATTRv amyloidosis patients over time. There is a lack of studies investigating the use of NfL as marker for small fiber neuropathy, autonomic neuropathy and central nervous system involvement in ATTRv amyloidosis. Studying NfL in relation to intra-epidermal nerve fiber density (IENFD) would be of particular interest, as IENFD has shown to be a sensitive marker for early detection of ATTRv amyloidosis [69]. NfL bears potential as a marker to detect polyneuropathy in patients with ATTRwt amyloidosis and ATTRv amyloidosis patients with apparently only cardiomyopathy [58]. However, studies in these patients are currently lacking. Another research gap that merits investigation pertains to the role of NfL in AL amyloidosis. Despite the higher occurrence of this form of systemic amyloidosis compared to ATTRv amyloidosis, only one study has examined NfL in AL amyloidosis.
Limitations
Major limitation of this systemic review on NfL in systemic amyloidosis is the lack of a meta-analysis. This is attributed to extremely high clinical heterogeneity resulting from variations in NfL measurements (sample types and analytical methods), outcome variables, composition of study groups and composition of control groups.
Conclusion
NfL is not an outcome biomarker, but an early and sensitive disease process biomarker for neuropathy, particularly large fiber neuropathy, in systemic amyloidosis. Therefore, NfL has potential to be used for early detection of peripheral neuropathy and for monitoring treatment effect and disease progression in patients with systemic amyloidosis.
Author Contributions
Conceptualization, M.B., H.L.A.N., B.P.C.H.; methodology, M.B., H.L.A.N, B.P.C.H.; software, M.B.; validation, H.L.A.N; formal analysis, M.B.; investigation, M.B., H.L.A.N.; data curation, M.B., H.L.A.N., B.P.C.H.; writing—original draft preparation, M.B.; writing—review and editing, M.B., H.L.A.N., D.A., C.K., M.L., M.P., M.M.R., Y.S, B.P.C.H.; visualization, M.B.; supervision, H.L.A.N., B.P.C.H.; project administration, M.B., H.L.A.N., funding acquisition, not applicable.
Funding
This article concerns a non-funded study.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated or analyzed in support of this research.
Acknowledgments
The authors have no acknowledgements to share.
Conflicts of Interest
Chafic Karam has served as consultant and/or advisory board member for Acceleron, Alpine, Alexion, Alnylam, Annexon, Argenx, Astra Zeneca, Corino, Biogen, CSL Behring, Genentech, Ionis, Neuroderm, Novo Nordisk, Pfizer, Sanofi, UCB, Takeda, and Zai lab. He has received research funding from Argenx, Astra Zeneca, Ionis and Genzyme. The University Medical Center Groningen, which employs Hans Nienhuis, received consultancy and speaker fees from Pfizer and Alnylam and grant support from Pfizer.
Abbreviations
| ATTRv | hereditary transthyretin amyloid |
| ATTRwt | wildtype transthyretin amyloid |
| AL | immunoglobulin light chain amyloid |
| APOLLO | phase 3 study of Patisiran for treatment of hereditary transthyretin amyloidosis with polyneuropathy |
| CADT | compound autonomic dysfunction test |
| CM | cardiomyopathy |
| ELISA | enzyme-linked immunosorbent assay |
| EMG | electromyography |
| ESC | electrochemical skin conductance |
| FAP | familial amyloidotic polyneuropathy |
| HELIOS | phase 3 open-label study of Vutrisiran in patients with hereditary transthyretin amyloidosis with polyneuropathy |
| IENFD | intra-epidermal nerve fiber density |
| mNIS+7 | modified neuropathy impairment score +7 |
| MRC | medical research council |
| NCS | nerve conduction studies |
| NfL | neurofilament light chain |
| NIS | neuropathy impairment score |
| NIS-LL | neuropathy impairment score lower limb |
| NIS-UL | neuropathy impairment score upper limb |
| Norfolk QOL-DN | Norfolk quality of life diabetic neuropathy |
| NT-proBNP | N-terminal pro-brain-type natriuretic peptide |
| OLE | open label extension |
| PND | polyneuropathy disability |
| PNP | polyneuropathy |
| QST | quantitative sensory testing |
| R-ODS | Rasch-built overall disability score |
| ROC | receiver operating characteristics |
| SFN-SIQ | small fiber neuropathy- symptom inventory questionnaire |
| Simoa | single-molecule array |
| TTR | transthyretin |
| TTRv | transthyretin gene variant |
References
- Buxbaum, J.N.; Dispenzieri, A.; Eisenberg, D.S.; Fändrich, M.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Westermark, P. Amyloid Nomenclature 2022: Update, Novel Proteins, and Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2022, 29, 213–219. [Google Scholar] [CrossRef]
- Hazenberg, B.P.C. Amyloidosis. A Clinical Overview. Rheum. Dis. Clin. North Am. 2013, 39, 323–345. [Google Scholar] [CrossRef]
- Shin, S.C.; Robinson-Papp, J. Amyloid Neuropathies. Mt. Sinai J. Med. 2012, 79, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.F.; Magalhães, R.; Freitas, J.; Taipa, R.; Pires, M.M.; Osório, H.; Dias, D.; Pessegueiro, H.; Correia, M.; Coelho, T. CNS Involvement in V30M Transthyretin Amyloidosis: Clinical, Neuropathological and Biochemical Findings. J. Neurol. Neurosurg. Psychiatry. 2015, 86, 159–167. [Google Scholar] [CrossRef]
- Campagnolo, M.; Cacciavillani, M.; Cipriani, A.; Salvalaggio, A.; Castellani, F.; Pilichou, K.; Briani, C. Peripheral Nerve Involvement in Wild-Type Transthyretin Amyloidosis. Neurol. Sci. 2023, 44, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Suhr, O.B.; Hund, E.; Obici, L.; Tournev, I.; Campistol, J.M.; Slama, M.S.; Hazenberg, B.P.; Coelho, T. First European Consensus for Diagnosis, Management, and Treatment of Transthyretin Familial Amyloid Polyneuropathy. Curr. Opin. Neurol. 2016, 29 Suppl 1, S14–26. [Google Scholar] [CrossRef]
- Gertz, M.A.; Comenzo, R.; Falk, R.H.; Fermand, J.P.; Hazenberg, B.P.; Hawkins, P.N.; Merlini, G.; Moreau, P.; Ronco, P.; Sanchorawala, V.; et al. Definition of Organ Involvement and Treatment Response in Immunoglobulin Light Chain Amyloidosis (AL): A Consensus Opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am. J. Hematol. 2005, 79, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Davies, J.L.; Litchy, W.J.; O’Brien, P.C. Longitudinal Assessment of Diabetic Polyneuropathy Using a Composite Score in the Rochester Diabetic Neuropathy Study Cohort. Neurology 1997, 49, 229–239. [Google Scholar] [CrossRef]
- Dyck, P.J.; Dyck, P.J.; Kennedy, W.R.; Kesserwani, H.; Melanson, M.; Ochoa, J.; Shy, M.; Stevens, J.C.; Suarez, G.A.; O’Brien, P.C. Limitations of Quantitative Sensory Testing When Patients Are Biased toward a Bad Outcome. Neurology. 1998, 50, 1213. [Google Scholar] [CrossRef]
- Casellini, C.M.; Parson, H.K.; Richardson, M.S.; Nevoret, M.L.; Vinik, A.I. Sudoscan, a Noninvasive Tool for Detecting Diabetic Small Fiber Neuropathy and Autonomic Dysfunction. Diabetes Technol. Ther. 2013, 15, 948–953. [Google Scholar] [CrossRef]
- Luigetti, M.; Romozzi, M.; Bisogni, G.; Cardellini, D.; Cavallaro, T.; Di Paolantonio, A.; Fabrizi, G.M.; Fenu, S.; Gentile, L.; Grandis, M.; et al. HATTR Pathology: Nerve Biopsy Results from Italian Referral Centers. Brain Sci. 2020, 10, 780. [Google Scholar] [CrossRef]
- Fernandes, A.; Coelho, T.; Rodrigues, A.; Felgueiras, H.; Oliveira, P.; Guimarães, A.; Melo-Pires, M.; Taipa, R. Clinicopathological Correlations of Sural Nerve Biopsies in TTR Val30Met Familial Amyloid Polyneuropathy. Brain Commun. 2019, 1, fcz032. [Google Scholar] [CrossRef]
- Ebenezer, G.J.; Liu, Y.; Judge, D.P.; Cunningham, K.; Truelove, S.; Carter, N.D.; Sebastian, B.; Byrnes, K.; Polydefkis, M. Cutaneous Nerve Biomarkers in Transthyretin Familial Amyloid Polyneuropathy. Ann. Neurol. 2017, 82, 44–56. [Google Scholar] [CrossRef]
- Masuda, T.; Ueda, M.; Misumi, Y.; Nomura, T.; Inoue, Y.; Isoguchi, A.; Kanenawa, K.; Tasaki, M.; Yamashita, T.; Sonoda, Y.; et al. Reduced Intraepidermal Nerve Fibre Density in Patients with Hereditary Transthyretin Amyloidosis. Amyloid. 2019, 26 (sup1), 79–80. [Google Scholar] [CrossRef]
- Leonardi, L.; Galosi, E.; Vanoli, F.; Fasolino, A.; Di Pietro, G.; Luigetti, M.; Sabatelli, M.; Fionda, L.; Garibaldi, M.; Alfieri, G.; et al. Skin Biopsy and Quantitative Sensory Assessment in an Italian Cohort of ATTRv Patients with Polyneuropathy and Asymptomatic Carriers: Possible Evidence of Early Non-Length Dependent Denervation. Neurol. Sci. 2022, 43, 1359–1364. [Google Scholar] [CrossRef]
- Leonardi, L.; Adam, C.; Beaudonnet, G.; Beauvais, D.; Cauquil, C.; Not, A.; Morassi, O.; Benmalek, A.; Trassard, O.; Echaniz-Laguna, A.; et al. Skin Amyloid Deposits and Nerve Fiber Loss as Markers of Neuropathy Onset and Progression in Hereditary Transthyretin Amyloidosis. Eur. J. Neurol. 2022, 29, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.; Labeyrie, C.; Cauquil, C.; Francou, B.; Eliahou, L.; Not, A.; Echaniz-Laguna, A.; Adam, C.; Slama, M.S.; Benmalek, A.; et al. Detailed Clinical, Physiological and Pathological Phenotyping Can Impact Access to Disease-Modifying Treatments in ATTR Carriers. J. Neurol. Neurosurg. Psychiatry 2023. [online ahead of print]. [Google Scholar] [CrossRef]
- Dyck, P.J.B.; González-Duarte, A.; Obici, L.; Polydefkis, M.; Wiesman, J.F.; Antonino, I.; Litchy, W.J.; Dyck, P.J. Development of Measures of Polyneuropathy Impairment in HATTR Amyloidosis: From NIS to MNIS + 7. J. Neurol. Sci. 2019, 405, 116424. [Google Scholar] [CrossRef]
- D’Ambrosio, V.; Ferraro, P.M.; Guglielmino, V.; Luigetti, M. Kidney Involvement in Hereditary Transthyretin Amyloidosis: Is There a Role for Cystatin C? Clin. Kidney J. 2022, 16, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as Biomarkers in Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Sandelius, Å.; Zetterberg, H.; Blennow, K.; Adiutori, R.; Malaspina, A.; Laura, M.; Reilly, M.M.; Rossor, A.M. Plasma Neurofilament Light Chain Concentration in the Inherited Peripheral Neuropathies. Neurology. 2018, 90, e518–e524. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Foiani, M.; Heslegrave, A.; Zetterberg, H.; Lunn, M.P.; Malaspina, A.; Gillmore, J.D.; Rossor, A.M.; Reilly, M.M. Plasma Neurofilament Light Chain Concentration Is Increased and Correlates with the Severity of Neuropathy in Hereditary Transthyretin Amyloidosis. J. Peripher. Nerv. Syst. 2019, 24, 314–319. [Google Scholar] [CrossRef]
- Loser, V.; Benkert, P.; Vicino, A.; Lim Dubois Ferriere, P.; Kuntzer, T.; Pasquier, J.; Maceski, A.; Kuhle, J.; Theaudin, M. Serum Neurofilament Light Chain as a Reliable Biomarker of Hereditary Transthyretin-Related Amyloidosis-A Swiss Reference Center Experience. J. Peripher. Nerv. Syst. 2023, 28, 86–97. [Google Scholar] [CrossRef]
- Louwsma, J.; Brunger, A.F.; Bijzet, J.; Kroesen, B.J.; Roeloffzen, W.W.H.; Bischof, A.; Kuhle, J.; Drost, G.; Lange, F.; Kuks, J.B.M.; et al. Neurofilament Light Chain, a Biomarker for Polyneuropathy in Systemic Amyloidosis. Amyloid. 2021, 28, 50–55. [Google Scholar] [CrossRef]
- Sanchez, J.D.; Martirosian, R.A.; Mun, K.T.; Chong, D.S.; Llorente, I.L.; Uphaus, T.; Gröschel, K.; Wölfer, T.A.; Tiedt, S.; Hinman, J.D. Temporal Patterning of Neurofilament Light as a Blood-Based Biomarker for Stroke: A Systematic Review and Meta-Analysis. Front. Neurol. 2022, 13, 841898. [Google Scholar] [CrossRef]
- Bischof, A.; Manigold, T.; Barro, C.; Heijnen, I.; Berger, C.T.; Derfuss, T.; Kuhle, J.; Daikeler, T. Serum Neurofilament Light Chain: A Biomarker of Neuronal Injury in Vasculitic Neuropathy. Ann. Rheum. Dis. 2018, 77, 1093–1094. [Google Scholar] [CrossRef]
- Zetterberg, H.; Hietala, M.A.; Jonsson, M.; Andreasen, N.; Styrud, E.; Karlsson, I.; Edman, A.; Popa, C.; Rasulzada, A.; Wahlund, L.-O.; et al. Neurochemical Aftermath of Amateur Boxing. Arch. Neurol. 2006, 63, 1277–1280. [Google Scholar] [CrossRef]
- Kokotis, P.; Manios, E.; Schmelz, M.; Fotiou, D.; Dialoupi, I.; Gavriatopoulou, M.; Roussou, M.; Lykka, A.; Dimopoulos, M.A.; Kastritis, E. Involvement of Small Nerve Fibres and Autonomic Nervous System in AL Amyloidosis: Comprehensive Characteristics and Clinical Implications. Amyloid. 2020, 27, 103–110. [Google Scholar] [CrossRef]
- Maia, L.F.; Maceski, A.; Conceição, I.; Obici, L.; Magalhães, R.; Cortese, A.; Leppert, D.; Merlini, G.; Kuhle, J.; Saraiva, M.J. Plasma Neurofilament Light Chain: An Early Biomarker for Hereditary ATTR Amyloid Polyneuropathy. Amyloid. 2020, 27, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Ticau, S.; Sridharan, G. V; Tsour, S.; Cantley, W.L.; Chan, A.; Gilbert, J.A.; Erbe, D.; Aldinc, E.; Reilly, M.M.; Adams, D.; et al. Neurofilament Light Chain as a Biomarker of Hereditary Transthyretin-Mediated Amyloidosis. Neurology 2021, 96, e412–e422. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Di Paolantonio, A.; Guglielmino, V.; Romano, A.; Rossi, S.; Sabino, A.; Servidei, S.; Sabatelli, M.; Primiano, G. Neurofilament Light Chain as a Disease Severity Biomarker in ATTRv: Data from a Single-Centre Experience. Neurol. Sci. 2022, 43, 2845–2848. [Google Scholar] [CrossRef]
- Romano, A.; Primiano, G.; Antonini, G.; Ceccanti, M.; Fenu, S.; Forcina, F.; Gentile, L.; Inghilleri, M.; Leonardi, L.; Manganelli, F.; et al. Serum Neurofilament Light Chain: A Promising Early Diagnostic Biomarker for Hereditary Transthyretin Amyloidosis? Eur. J. Neurol. 2024, 31, e16070. [Google Scholar] [CrossRef]
- Carroll, A.S.; Razvi, Y.; O’Donnell, L.; Veleva, E.; Heslegrave, A.; Zetterberg, H.; Vucic, S.; Kiernan, M.C.; Rossor, A.M.; Gillmore, J.D.; et al. Serum Neurofilament Light Chain in Hereditary Transthyretin Amyloidosis: Validation in Real-Life Practice. Amyloid 2024. [online ahead of print]. [Google Scholar] [CrossRef]
- González-Moreno, J.; Gragera-Martínez, Á.; Rodríguez, A.; Borrachero-Garro, C.; García-Garrido, S.; Barceló, C.; Manovel-Sánchez, A.; Ribot-Sansó, M.A.; Ibargüen-González, L.; Gomila, R.; et al. Biomarkers of Axonal Damage to Favor Early Diagnosis in Variant Transthyretin Amyloidosis (A-ATTRv). Sci. Rep. 2024, 14, 581. [Google Scholar] [CrossRef]
- Brunger, A.F.; Berends, M.; Bijzet, J.; Van Der Zwaag, P.; Kroesen, B.J.; Teunissen, C.; In ’T Veld, S.; Drost, G.; Lange, F.; Gans, R.; et al. Neurofilament Light Chain, an Early Biomarker for Polyneuropathy in Hereditary ATTR Amyloidosis. In Ann. Rheum. Dis., Proceedings of the European Congress of Rheumatology, Copenhagen, Denmark, 01-06-2022 to 04-06-2022; p. 2022. [CrossRef]
- Lau, K.H.V.; Prokaeva, T.; Zheng, L.; Doros, G.; Kaku, M.C.; Spencer, B.; Berk, J.; Sanchorawala, V. Neurofilament Light Chain Kinetics as a Biomarker for Polyneuropathy in V122I Hereditary Transthyretin Amyloidosis. Amyloid 2023. [online ahead of print]. [Google Scholar] [CrossRef]
- Ticau, S.; Aldinc, E.; Polydefkis, M.; Adams, D.; Coelho, T.; Ueda, M.; Hale, C.; Vest, J.; Nioi, P. Treatment Response and Neurofilament Light Chain Levels with Long-Term Patisiran in Hereditary Transthyretin-Mediated Amyloidosis with Polyneuropathy: 24-Month Results of an Open-Label Extension Study. Amyloid 2023. [online ahead of print]. [Google Scholar] [CrossRef]
- Sato, M.; Mochizuki, Y.; Takahashi, Y.; Takasone, K.; Aldinc, E.; Ticau, S.; Jia, G.; Sekijima, Y. Neurofilament Light Chain as a Biomarker for Monitoring Response to Change in Treatment in Hereditary ATTR Amyloidosis. Amyloid. 2023, 30, 351–352. [Google Scholar] [CrossRef]
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-Related Familial Amyloidotic Polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kincaid, J.C. The Molecular Biology and Clinical Features of Amyloid Neuropathy. Muscle & nerve. 2007, 36, 411–423. [Google Scholar]
- Berends, M.; Nienhuis, H.L.A.; Brunger, A.; Bijzet, J.; Van Der Zwaag, P.A.; Hazenberg, B.P.C.; Noordzij, W.; Slart, R.H.J. Serum Neurofilament Light Chain (SNfL) in Relation to Myocardial Sympathetic Neuronal Damage Based on 123I-Meta-Iodobenzylguanidine (MIBG) Scintigraphy in Hereditary Transthyretin (ATTRv) Amyloidosis. In Eur. J. Nucl. Med. Mol. Imaging, Proceedings of the 35th Annual Congress of the European Association of Nuclear Medicine, Barcelona, Spain, 15-10-2022 to 19-10-2022; Springer: New York, United States, 2022. [Google Scholar] [CrossRef]
- Luigetti, M.; Aldinc, E.; Ticau, S.; Polydefkis, M.; Nienhuis, H.; Karam, C.; Ajroud-Driss, S.; Sekijima, Y.; Waddington-Cruz, M.; Barnes, J.; et al. NfL Levels Significantly Decrease in Response to Treatment with Patisiran or Vutrisiran in hATTR Amyloidosis with Polyneuropathy. In J. Peripher. Nerv. Syst., Proceedings of the 13th Annual Meeting of the Italian Association for the Study of the Peripheral Nervous System, Naples, Italy, 25-05-2023 to 27-05-2023; Wiley: New Jersey, United States, 2023. [Google Scholar] [CrossRef]
- Conceicao, I.; Polydefkis, M.; Obici, L.; Adams, D.; Gillmore, J.; Masri, A.; Brannagan, T.; Coelho, T.; Jung, S.; Wessman, P.; et al. Neurofilament Light Chain as a Potential Biomarker in Patients with Hereditary ATTR-Polyneuropathy in NEURO-TTRansform. In Eur. J. Neurol., Proceedings of the 9th Congress of the European Academy Neurology, Budapest, Hungary, 01-07-2023 to 04-07-2023; Wiley: New Jersey, United States, 2023. [Google Scholar]
- Gilling, K.; Aldinc, E.; Ticau, S.; Polydefkis, M.; Adams, D.; Reilly, M.; Nioi, P. P-48 Neurofilament Light Chain as a Biomarker in Hereditary Transthyretin-Mediated Amyloidosis: 36-Month Data from the Patisiran Global Open-Label Extension. In Clin. Neurophysiol., Proceedings of the Congress for Clinical Neuroscience with Advanced Training Academy of the German Society for Clinical Neurophysiology and Functional Neuroimaging, Hamburg, Germany, 02-03-203 to 04-03-2023; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar] [CrossRef]
- Cheng, X.; Su, Y.; Wang, Q.; Gao, F.; Ye, X.; Wang, Y.; Xia, Y.; Fu, J.; Shen, Y.; Al-Shahi Salman, R.; et al. Neurofilament Light Chain Predicts Risk of Recurrence in Cerebral Amyloid Angiopathy-Related Intracerebral Hemorrhage. Aging (Albany. NY). 2020, 12, 23727–23738. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Yazaki, M.; Oguchi, K.; Ezawa, N.; Yoshinaga, T.; Yamada, M.; Yahikozawa, H.; Watanabe, M.; Kametani, F.; Ikeda, S.I. Cerebral Amyloid Angiopathy in Posttransplant Patients with Hereditary ATTR Amyloidosis. Neurology. 2016, 87, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; Sousa, L.; Pinto, M.; Reis, I.; Rodrigues, A.; Oliveira, P.; Melo-Pires, M.; Coelho, T. Neuropathology of Central Nervous System Involvement in TTR Amyloidosis. Acta Neuropathol. 2023, 145, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Vermunt, L.; Otte, M.; Verberk, I.M.W.; Killestein, J.; Lemstra, A.W.; van der Flier, W.M.; Pijnenburg, Y.A.L.; Vijverberg, E.G.B.; Bouwman, F.H.; Gravesteijn, G.; et al. Age- and Disease-Specific Reference Values for Neurofilament Light Presented in an Online Interactive Support Interface. Ann. Clin. Transl. Neurol. 2022, 9, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; Sotirchos, E.S.; Smith, M.D.; Lord, H.-N.; DuVal, A.; Mowry, E.M.; Calabresi, P.A. Contributors to Serum NfL Levels in People without Neurologic Disease. Ann. Neurol. 2022, 92, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Abdelhak, A.; Kuhle, J.; Green, A.J. Challenges and Opportunities for the Promising Biomarker Blood Neurofilament Light Chain. JAMA Neurol. 2023, 80, 542–543. [Google Scholar] [CrossRef]
- Baka, P.; Steenken, L.; Escolano-Lozano, F.; Steffen, F.; Papagianni, A.; Sommer, C.; Pogatzki-Zahn, E.; Hirsch, S.; Protopapa, M.; Bittner, S.; et al. Studying Serum Neurofilament Light Chain Levels as a Potential New Biomarker for Small Fiber Neuropathy. Eur. J. Neurol. 2024. [online ahead of print]. [Google Scholar] [CrossRef]
- Luigetti, M.; Primiano, G.; Basile, V.; Vitali, F.; Pignalosa, S.; Romano, A.; Sabino, A.; Marino, M.; Di Santo, R.; Ciasca, G.; et al. Serum Neurofilament and Free Light Chain Levels in Patients Undergoing Treatment for Chronic Inflammatory Demyelinating Polyneuropathy. Int. J. Mol. Sci. 2024, 25. [Google Scholar] [CrossRef]
- van Lieverloo, G.G.A.; Wieske, L.; Verhamme, C.; Vrancken, A.F.J.; van Doorn, P.A.; Michalak, Z.; Barro, C.; van Schaik, I.N.; Kuhle, J.; Eftimov, F. Serum Neurofilament Light Chain in Chronic Inflammatory Demyelinating Polyneuropathy. J. Peripher. Nerv. Syst. 2019, 24, 187–194. [Google Scholar] [CrossRef]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.G.; Ikeda, S.I.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of Transthyretin-Related Hereditary Amyloidosis for Clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon Degeneration: Molecular Mechanisms of a Self-Destruction Pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary Transthyretin Amyloidosis: A Model of Medical Progress for a Fatal Disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef]
- Butler, J.S.; Chan, A.; Costelha, S.; Fishman, S.; Willoughby, J.L.S.; Borland, T.D.; Milstein, S.; Foster, D.J.; Gonçalves, P.; Chen, Q.; et al. Preclinical Evaluation of RNAi as a Treatment for Transthyretin-Mediated Amyloidosis. Amyloid. 2016, 23, 109–118. [Google Scholar] [CrossRef]
- Ticau, S.; Sridharan, G.; Tsour, S.; Cantley, W.; Chan, A.; Gilbert, J.A.; Erbe, D.; Vest, J.; Fitzgerald, K.; Vaishnaw, A.; et al. Neurofilament Light Chain May Serve As a Biomarker of Neuropathy in hATTR Amyloidosis with Cardiomyopathy. In J. Card. Fail. Proceedings of the Heart Failure Society of America's Annual Scientific Meeting 2020, Online Meeting, 13-12-2020 to 07-12-2020; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of Three Analytical Platforms for Quantification of the Neurofilament Light Chain in Blood Samples: ELISA, Electrochemiluminescence Immunoassay and Simoa. Clin. Chem. Lab. Med. 2016, 54, 1655–1661. [Google Scholar] [CrossRef]
- Andreasson, U.; Gobom, J.; Delatour, V.; Auclair, G.; Noam, Y.; Lee, S.; Wen, J.; Jeromin, A.; Arslan, B.; Maceski, A.; et al. Assessing the Commutability of Candidate Reference Materials for the Harmonization of Neurofilament Light Measurements in Blood. Clin. Chem. Lab. Med. 2023, 61, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.; Viel, S.; Perret, M.; Brocard, G.; Casey, R.; Lombard, C.; Laurent-Chabalier, S.; Debouverie, M.; Edan, G.; Vukusic, S.; et al. Comparison of Simoa(TM) and Ella(TM) to Assess Serum Neurofilament-Light Chain in Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Lee, H.; Kim, Y.S.; Choi, H.; Lee, K.-Y.; Lee, Y.J.; Lee, E.-H.; Hwang, M.; Park, H.; Koh, S.-H. Comparing Neurofilament Light Chain Levels in Serum and Plasma. Dement. Neurocogni. disord. 2023, 22, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Altmann, P.; Ponleitner, M.; Rommer, P.S.; Haslacher, H.; Mucher, P.; Leutmezer, F.; Petzold, A.; Wotawa, C.; Lanzenberger, R.; Berger, T.; et al. Seven Day Pre-Analytical Stability of Serum and Plasma Neurofilament Light Chain. Sci. Rep. 2021, 11, 11034. [Google Scholar] [CrossRef] [PubMed]
- Rübsamen, N.; Willemse, E.A.J.; Leppert, D.; Wiendl, H.; Nauck, M.; Karch, A.; Kuhle, J.; Berger, K. A Method to Combine Neurofilament Light Measurements From Blood Serum and Plasma in Clinical and Population-Based Studies. Front. Neurol. 2022, 13, 894119. [Google Scholar] [CrossRef] [PubMed]
- Schubert, C.R.; Paulsen, A.J.; Pinto, A.A.; Merten, N.; Cruickshanks, K.J. Effect of Long-Term Storage on the Reliability of Blood Biomarkers for Alzheimer’s Disease and Neurodegeneration. J. Alzheimers. Dis. 2022, 85, 1021–1029. [Google Scholar] [CrossRef]
- Benkert, P.; Meier, S.; Schaedelin, S.; Manouchehrinia, A.; Yaldizli, Ö.; Maceski, A.; Oechtering, J.; Achtnichts, L.; Conen, D.; Derfuss, T.; et al. Serum Neurofilament Light Chain for Individual Prognostication of Disease Activity in People with Multiple Sclerosis: A Retrospective Modelling and Validation Study. Lancet. Neurol. 2022, 21, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA. 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Coppens, S.; Lehmann, S.; Hopley, C.; Hirtz, C. Neurofilament-Light, a Promising Biomarker: Analytical, Metrological and Clinical Challenges. Int. J. Mol. Sci. 2023, 24, 11624. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.; Labeyrie, C.; Cauquil, C.; Francou, B.; Eliahou, L.; Not, A.; Echaniz-Laguna, A.; Adam, C.; Slama, M.S.; Benmalek, A.; et al. Detailed Clinical, Physiological and Pathological Phenotyping Can Impact Access to Disease-Modifying Treatments in ATTR Carriers. J. Neurol. Neurosurg. Psychiatry 2023, 0, 1–11. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Flowchart of database search and selection of studies.
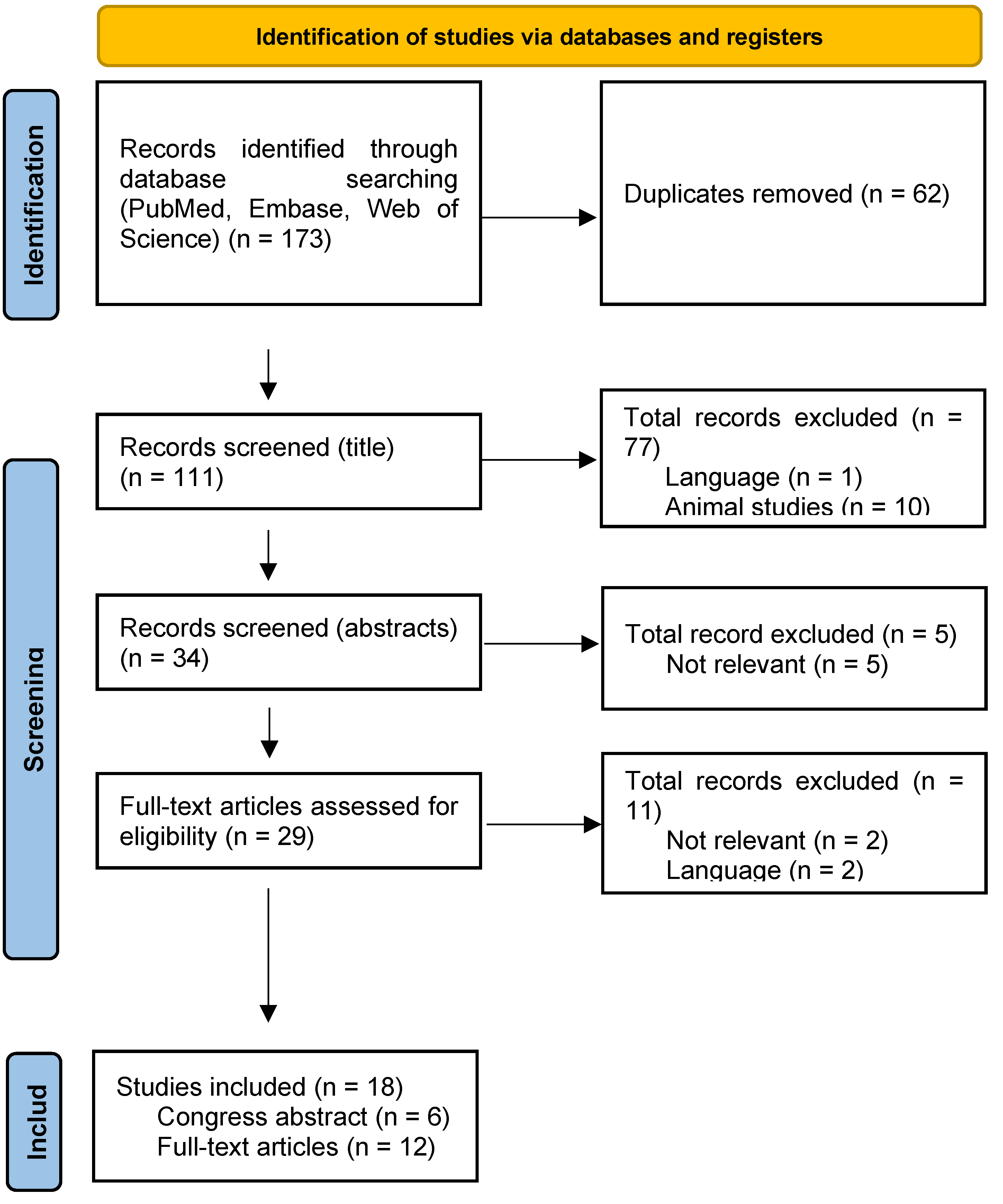
Figure 2.
Neurofilament light chain levels in all studies. (A) neurofilament light chain levels in healthy controls, TTRv carriers, ATTRv patients without neuropathy and ATTRv patients with neuropathy with or without treatment. (B) Neurofilament light chain levels in ATTRv patients with neuropathy and with treatment. a: Five patients used diflunisal and one patient used tafamidis but it was not specified to which studygroup these patients belonged. b: One patient used diflunisal but switched to tafamidis during the follow-up period. c: Two patients used inotersen. d: Patients used a TTR-stabilizer or a TTR-gene silencer, but it was unspecified which one. e: Six of the eight untreated patients had received a liver transplantation in the past and two of the four patients treated with patisiran had received a liver transplantation in the past. f: All (six) untreated patients had received a liver transplantation in the past and two of the six patients treated with patisiran had received a liver transplantation in the past. g: One patient used diflunisal and switched to tafamidis during the follow-up period and one patient used diflunisal and switched to eplontersen during the follow-up period. ATTRv: hereditary transthyretin amyloid; B: baseline; CM: cardiomyopathy; FAP: familial amyloid polyneuropathy; FU: follow-up; m: month; MIBG: meta-iodobenzylguanidine scintigraphy; NfL: neurofilament light chain; PND: polyneuropathy disability; PNP: polyneuropathy; TTRv: transthyretin gene variant; y: year.
Figure 2.
Neurofilament light chain levels in all studies. (A) neurofilament light chain levels in healthy controls, TTRv carriers, ATTRv patients without neuropathy and ATTRv patients with neuropathy with or without treatment. (B) Neurofilament light chain levels in ATTRv patients with neuropathy and with treatment. a: Five patients used diflunisal and one patient used tafamidis but it was not specified to which studygroup these patients belonged. b: One patient used diflunisal but switched to tafamidis during the follow-up period. c: Two patients used inotersen. d: Patients used a TTR-stabilizer or a TTR-gene silencer, but it was unspecified which one. e: Six of the eight untreated patients had received a liver transplantation in the past and two of the four patients treated with patisiran had received a liver transplantation in the past. f: All (six) untreated patients had received a liver transplantation in the past and two of the six patients treated with patisiran had received a liver transplantation in the past. g: One patient used diflunisal and switched to tafamidis during the follow-up period and one patient used diflunisal and switched to eplontersen during the follow-up period. ATTRv: hereditary transthyretin amyloid; B: baseline; CM: cardiomyopathy; FAP: familial amyloid polyneuropathy; FU: follow-up; m: month; MIBG: meta-iodobenzylguanidine scintigraphy; NfL: neurofilament light chain; PND: polyneuropathy disability; PNP: polyneuropathy; TTRv: transthyretin gene variant; y: year.
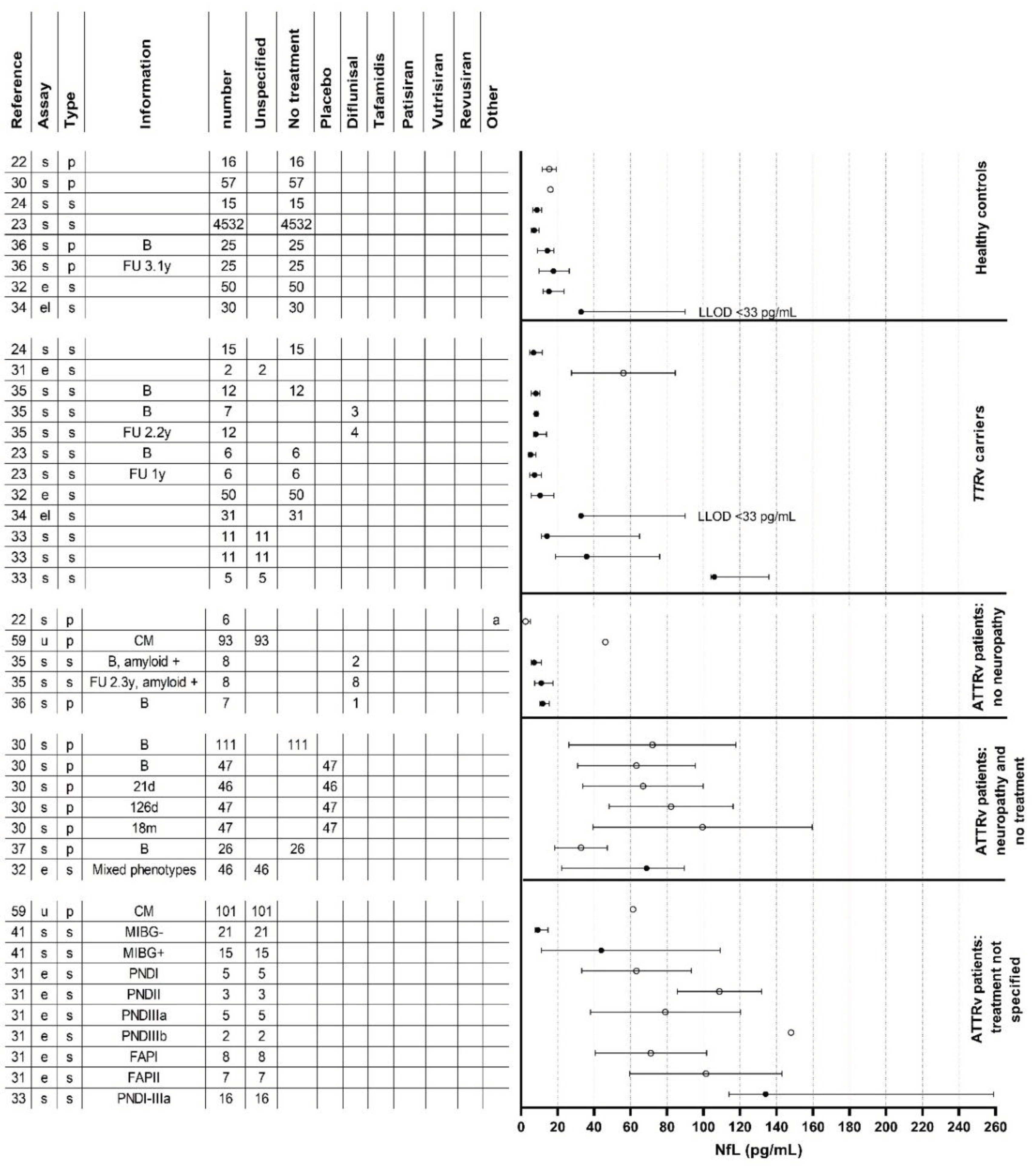
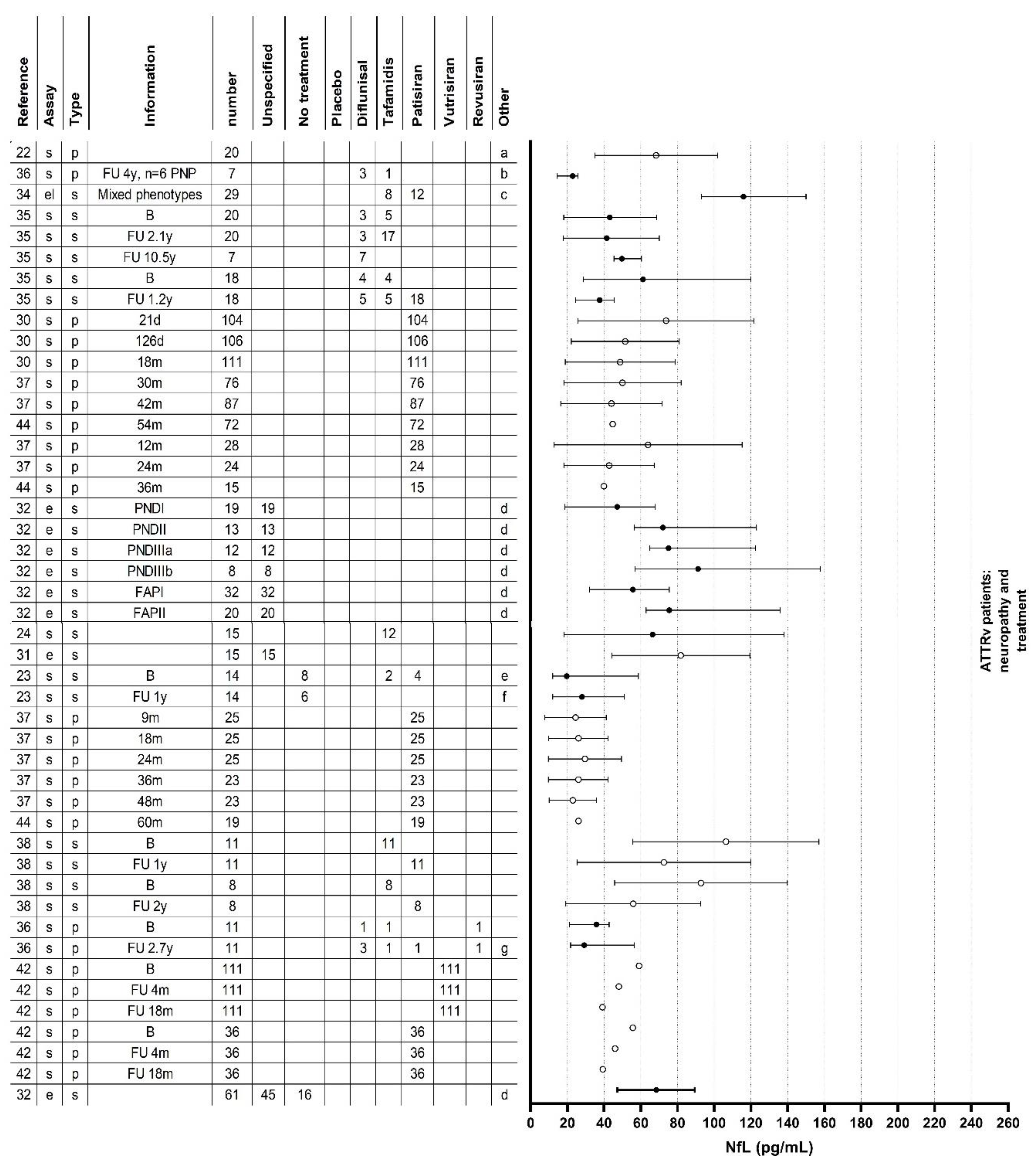
Figure 3.
Hypothetical course of neurofilament light chain in a TTRv carrier who develops ATTRv amyloidosis with polyneuropathy. Hypothetical course of NfL levels in a TTRv carrier who develops ATTRv amyloidosis with polyneuropathy. NfL levels start to rise when amyloid is deposited, transforming a asymptomatic TTRv carrier into an asymptomatic ATTRv patients. At some point polyneuropathy can be confirmed by nerve conduction studies and the ATTRv patient experiences symptoms. The treatment approach adopted can influence the direction of NfL levels, leading to elevation, stabilization or reduction. Given the novelty of NfL as a biomarker in systemic amyloidosis and the evolving landscape of treatment modalities, uncertainties persist regarding the long-term course of NfL levels. Gray solid line: healthy control/ asymptomatic TTRv carrier; black solid line: asymptomatic ATTRv patient; red solid/ dotted line: symptomatic ATTRv patient without treatment; blue solid/ dotted line: symptomatic ATTRv patient on a TTR-stabilizer; green solid/ dotted line: symptomatic ATTRv patient on a TTR-gene silencer. NfL: neurofilament light chain; NCS: nerve conduction studies; ATTRv: hereditary transthyretin amyloid; Simoa: single-molecule array.
Figure 3.
Hypothetical course of neurofilament light chain in a TTRv carrier who develops ATTRv amyloidosis with polyneuropathy. Hypothetical course of NfL levels in a TTRv carrier who develops ATTRv amyloidosis with polyneuropathy. NfL levels start to rise when amyloid is deposited, transforming a asymptomatic TTRv carrier into an asymptomatic ATTRv patients. At some point polyneuropathy can be confirmed by nerve conduction studies and the ATTRv patient experiences symptoms. The treatment approach adopted can influence the direction of NfL levels, leading to elevation, stabilization or reduction. Given the novelty of NfL as a biomarker in systemic amyloidosis and the evolving landscape of treatment modalities, uncertainties persist regarding the long-term course of NfL levels. Gray solid line: healthy control/ asymptomatic TTRv carrier; black solid line: asymptomatic ATTRv patient; red solid/ dotted line: symptomatic ATTRv patient without treatment; blue solid/ dotted line: symptomatic ATTRv patient on a TTR-stabilizer; green solid/ dotted line: symptomatic ATTRv patient on a TTR-gene silencer. NfL: neurofilament light chain; NCS: nerve conduction studies; ATTRv: hereditary transthyretin amyloid; Simoa: single-molecule array.
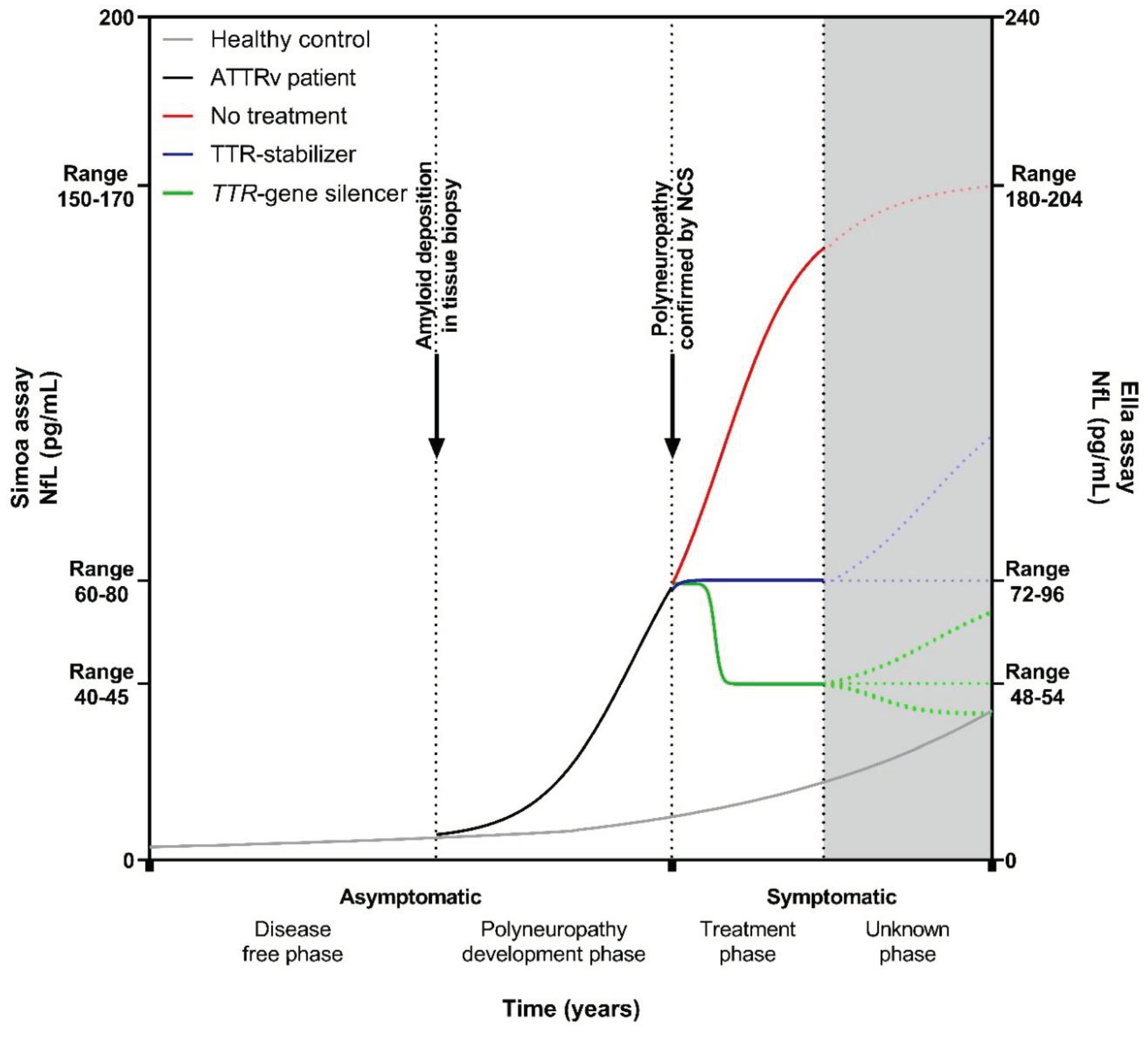

Table 1.
Study overview: neurofilament light chain levels and correlations with disease characteristics.
Table 1.
Study overview: neurofilament light chain levels and correlations with disease characteristics.
| Study (ref) | Comparisons between groups | Number of subjects | Fold increase in median NfL | NfL and correlation with disease characteristics | NfL and no correlation with disease characteristics |
|---|---|---|---|---|---|
| Full-text articles | |||||
| Kapoor et al. 2019 [22] | Healthy controls vs ATTRv no neuropathy |
16 6 |
0.2 (15.5 vs 2.5)* | NIS scale, CMTES-R | |
| Healthy controls vs ATTRv-PNP |
16 20 |
4.4 (15.5 vs 68.4) | |||
| ATTRv no neuropathy vs ATTRv-PNP |
6 20 |
27.4 (2.5 vs 68.4)* | |||
| Maia et al. 2020 [29] | Healthy controls vs TTRv carriers |
16 16 |
- | PND score | |
|
TTRv carriers vs ATTRv-PNP |
16 16 |
- | |||
| Healthy controls vs ATTRv-PNP PND I |
16 13 |
4.8 | |||
| Healthy controls vs ATTRv-PNP PND ≥II |
16 13 |
15.4 | |||
| Louwsma et al. 2021 [24] | Healthy controls vs TTRv carriers |
15 15 |
0.8 (8.8 vs 6.9) | PND score, sural nerve amplitude in ATTRv patients, troponin T in ATTRv patients with PNP | Sural nerve amplitude in TTRv carriers, digit 5 ulnar nerve amplitude, NT-proBNP, creatinine |
| Healthy controls vs ATTRv-PNP |
15 15 |
7.5 (8.8 vs 66.4) | |||
|
TTRv carriers vs ATTRv-PNP |
15 15 |
9.6 (6.9 vs 66.4) | |||
| ATTRv-PNP PND I vs ATTRv-PNP PND ≥I |
15 15 |
5.6 (21 vs 116) | |||
| Healthy controls vs AL no neuropathy |
10 10 |
1.7 (13.6 vs 22.7) | Troponin T in AL patients with and without PNP | NT-proBNP, creatinine | |
| Healthy controls vs AL-PNP |
10 10 |
11 (13.6 vs 149) | |||
| AL no neuropathy vs AL-PNP |
10 10 |
6.6 (22.7 vs 149) | |||
| Ticau et al. 2021 [30] | Healthy controls vs ATTRv-PNP (all) baseline |
57 189 |
4.3 (16.3 vs 69.4)* | Change in mNIS+7 after 18 months of treatment with patisiran | mNIS+7 at baseline and PND score at baseline |
| ATTRv-PNP patisiran 18 months vs ATTRv-PNP placebo 18 months |
111 47 |
2.0 (48.8 vs 99.5)* | |||
| Healthy controls vs ATTRv-PNP patisiran 18 months |
57 111 |
3.0 (16.3 vs 48.8)* | |||
| Healthy controls vs ATTRv-PNP placebo 18 months |
57 47 |
6.1 (16.3 vs 99.5)* | |||
| Luigetti et al. 2022 [31] | Healthy controls vs TTRv carriers and ATTRv-PNP |
26 17 |
4.5 (18 vs 81.8)* | NIS scale, Sudoscan values from feet, interventricular septum thickness, Norfolk QOL-DN | FAP stage, PND score, CADT |
| Loser at el. 2022 [23] |
TTRv carriers vs ATTRv-PNP |
6 14 |
B: 3.6 (5.4 vs 19.7) FU 1 year: 3.7 (7.5 vs 28.0) |
B and t1: PND score, FAP stage, R-ODS, SFN-SIQ, Norfolk-QOL-DN, NIS, NIS-UL, NIS-LL, ESC feet, ESC hands, NCS motor sum score, NCS sensory sum score. | CADT, handgrip right, handgrip left, |
| Sato et al. 2023 [38] | ATTRv-PNP tafamidis vs ATTR-PNP patisiran one year | 11 11 |
0.7 (106.4 vs 72.6)* | NIS score one and two years after treatment switch | |
| ATTRv-PNP tafamidis vs ATTR-PNP patisiran two years | 8 8 |
0.6 (92.8 vs 55.9)* | |||
| Lau et al. 2023 [36] | Healthy controls vs ATTRv no neuropathy |
25 7 |
0.8 (14.5 vs 11.9) | Creatinine | NIS-LL subscore, NT-proBNP, troponin I |
| Healthy controls vs ATTRv-PNP |
25 11 |
2.5 (14.5 vs 35.9) | |||
| ATTRv no neuropathy vs ATTRv-PNP |
7 11 |
3.0 (11.9 vs 35.9) | |||
| ATTRv no neuropathy vs ATTRv-PNP |
7 6 |
FU 4 years: 1.5 | |||
| Ticau et al. 2023 [37] | ATTRv-PNP baseline vs ATTRv-PNP patisiran 52 months |
111 87 |
0.6 (72.0 vs 44.1)* | Change in mNIS+7 and Norfolk QOL-DN sustained after 24 months additional patisiran treatment | |
| ATTRv-PNP patisiran Global OLE baseline vs ATTRv-PNP patisiran 24 months Global OLE |
111 87 |
0.9 (48.8 vs 44.1)* | |||
| ATTRv-PNP patisiran 30 months vs ATTRv-PNP placebo 18 months → 12 months patisiran Global OLE |
76 28 |
1.3 (50.1 vs 64.0)* | |||
| ATTRv-PNP patisiran 42 months vs ATTRv-PNP placebo 18 months → 24 months patisiran Global OLE |
87 24 |
1.0 (44.1 vs 42.8)* | |||
| ATTRv-PNP baseline vs ATTRv-PNP patisiran 18 months Phase II OLE |
26 25 |
0.8 (32.9 vs 26.1)* | |||
| ATTRv-PNP baseline vs ATTRv-PNP patisiran 48 months Global OLE |
26 23 |
0.7 (32.9 vs 23.0)* | |||
| Romano et al. 2024 [32] | Healthy controls vs TTRv carriers |
5 50 |
0.7 (17.7 vs 13.1) | PND score, NIS score, FAP stage |
|
| Healthy controls vs ATTRv-PNP |
5 61 |
4.2 (17.7 vs 74.0) | |||
|
TTRv carriers vs ATTRv-PNP |
50 61 |
5.6 (13.1 vs 74.0) | |||
| González-Moreno et al. 2024 [34] | Healthy controls vs TTRv V30M carriers |
30 31 |
Incalculable (<33 vs <33) |
NIS score | FAP stage |
| Healthy controls vs symptomatic ATTRv V30M |
30 29 |
Incalculable (<33 vs 116) |
|||
|
TTRv V30M carriers vs symptomatic ATTRv V30M |
31 29 |
Incalculable (<33 vs 116) |
|||
| Carroll et al. 2024 [33] | Asymptomatic (PND 0) vs symptomatic (PND ≥ I) |
11 16 |
9.4 (14.3 vs 134) | Baseline: PND score, FAP stage, NIS, NIS-LL, CMTSS, CMTES, CMTNS, MRC scores | eGFR, creatinine, Baseline: Norfolk-QOL-DN |
| Abstracts | |||||
| Ticau et al. 2020 [58] | Healthy controls vs ATTRv-CM no neuropathy |
53 93 |
3.3 (16.3 vs 54.1)* | PND score |
Cardiomyopathy |
| Healthy controls vs ATTRv-CM PND >0 |
53 101 |
3.8 (16.3 vs 61.4)* | |||
| Healthy controls vs ATTRv-PNP APOLLO |
53 193 |
4.3 (16.3 vs 69.4)* | |||
| ATTRv-CM no neuropathy vs ATTRv-CM PND >0 |
93 101 |
1.3 (46.2 vs 61.4)* | |||
| ATTRv-CM no neuropathy vs ATTRv-PNP APOLLO |
93 193 |
1.5 (46.2 vs 69.4)* | |||
| ATTRv-CM PND >0 vs ATTRv-PNP APOLLO |
101 193 |
1.1 (61.4 vs 69.4)* | |||
| Brunger et al. 2022 [35] |
TTRv carriers vs ATTRv no neuropathy |
12 8 |
0.9 (8.2 vs 7.1) | PND score |
|
|
TTRv carriers vs ATTRv-PNP TTR-stabilizer |
12 20 |
5.3 (8.2 vs 43.2) | |||
|
TTRv carriers vs ATTRv-PNP patisiran |
12 18 |
7.5 (8.2 vs 61.2) | |||
|
TTRv carriers and ATTRv no neuropathy vs TTRv carrier who developed PNP baseline |
20 7 |
1.1 (7.6 vs 8.40) | |||
| ATTRv-PNP TTR-stabilizer Vs ATTRv-PNP patisiran |
20 18 |
1.4 (43.2 vs 61.2) | |||
| ATTRv-PNP TTR-stabilizer vs TTRv carrier who developed PNP PND ≥I |
20 7 |
1.2 (43.2 vs 49.8) | |||
| ATTRv-PNP patisiran vs TTRv carrier who developed PNP PND ≥I |
18 7 |
0.8 (61.2 vs 49.8) | |||
| Berends et al. 2022 [41] | [123I]mIBG-scintigraphy negative TTRv carriers and ATTRv patients vs [123I]mIBG-scintigraphy positive TTRv carriers and ATTRv patients |
22 16 |
4.8 (9.2 vs 44.0) | NCS, PND score, NT-proBNP, troponin T, late heart-to-mediastinum ratio, wash-out rate, Ewing battery tests, [123I]mIBG-scintigraphy | |
| Conçeicao et al. 2023 [43] | ATTRv-PNP eplontersen vs ATTRv-PNP inotersen until week 35 followed by eplontersen |
144 24 |
|||
| Luigetti et al. 2023 [42] | ATTRv-PNP patisiran baseline vs ATTRv-PNP patisiran 4 months |
36 36 |
0.8 (55.7 vs 46.0)* | ||
| ATTRv-PNP patisiran baseline vs ATTRv-PNP patisiran 18 months |
36 36 |
0.7 (55.7 vs 39.3)* | |||
| ATTRv-PNP vutrisiran baseline vs ATTRv-PNP vutrisiran 4 months |
111 111 |
0.8 (59.1 vs 48.1)* | |||
| ATTRv-PNP vutrisiran baseline vs ATTRv-PNP vutrisiran 18 months |
111 111 |
0.7 (59.1 vs 39.2)* | |||
| ATTRv-PNP patisiran baseline vs ATTRv-PNP vutrisiran baseline |
36 111 |
1.1 (55.7 vs 59.1)* | |||
| ATTRv-PNP patisiran 18 months vs ATTRv-PNP vutrisiran 18 months |
36 111 |
1.0 (39.3 vs 39.2)* | |||
| Gilling et al. 2023 [44] | ATTRv-PNP placebo baseline vs ATTRv-PNP placebo → patisiran 36 months |
47 15 |
(63.2 vs 40.0)* | ||
| ATTRv-PNP patisiran baseline vs ATTRv-PNP patisiran 18months + Global OLE patisiran 36 months |
111 72 |
(72.0 vs 44.8)* | |||
| ATTRv-PNP Phase II OLE patisiran 24 months + Global OLE 36 months | 19 | 26.1* | |||
AL: immunoglobulin light chain amyloid; ATTRv: hereditary transthyretin amyloid; CADT: compound autonomic dysfunction test; CM: cardiomyopathy; CMTES: Charcot-Marie-Tooth symptom and examination subscore; CMTNS: Charcot-Marie-Tooth Neuropathy Score version 2; CMTSS: Charcot-Marie-Tooth symptom subscore; EMG: electromyography; ESC: electrochemical skin conductance; FAP: Familial amyloid polyneuropathy; mNIS+7: modified Neuropathy Impairment Score +7; MRC: Medical Research Council power score; NCS: nerve conduction studies; NIS: Neuropathy Impairment Score; NIS-LL: Neuropathy Impairment score- lower limbs; NIS-UL: Neuropathy Impairment Score- upper limbs; Norfolk QOL-DN: Norfolk quality of life diabetic neuropathy; NT-proBNP: N-terminal pro-brain-type natriuretic peptide; [123I]mIBG-scintigraphy: iodine-123 labelled meta-iodobenzylguanidine scintigraphy; OLE: open-label extension; PND: Polyneuropathy Disability; PNP: polyneuropathy; R-ODS: Rasch-built Overall Disability Score; SFN-SIQ: Small fiber Neuropathy-Symptom Inventory Questionnaire; t1: first timepoint of follow-up; TTRv: transthyretin gene variant; V30M: TTRVal30Met p.(Val50Met). *Data are expressed as mean values.
Table 2.
Proposed neurofilament light chain cutoff levels.
| Study (ref) |
Source type |
Assay | NfL cutoff level (pg/mL) |
Disease stage | Sensitivity (%) |
Specificity (%) |
|---|---|---|---|---|---|---|
| Maia et al. 2020 [29] | Plasma | Simoa | 10.6 | PND 0 and PND ≥ I |
96.2 | 93.8 |
| 10.6 | PND 0 and PND I |
92.3 | 93.8 | |||
| 66.9 | PND I and PND ≥ II (Cohort #1) |
61.5 | 92.3 | |||
| 75.7 | PND I and PND ≥ II (Cohort #2) |
84.6 | 80.0 | |||
| Ticau et al. 2021 [30] | Plasma | Simoa | 37 | Healthy controls and ATTRv-PNP |
84.9 | 94.4 |
| Loser et al. 2022 [23] | Serum | Simoa | 11.7 | Asymptomatic and symptomatic |
85.7 | 100 |
| Romano et al. 2024 [32] | Serum | Ella | 37.0 | Healthy controls and ATTRv-PNP |
81.4 | 98.0 |
| 37.0 | Healthy controls and PND I |
63.2 | 98.0 | |||
| 37.1 | Asymptomatic carriers and symptomatic ATTRv patients |
81.4 | 100 | |||
| 37.1 | Asymptomatic carriers and PND I | 63.2 | 100 | |||
| 57.70 | PND I and PND ≥ II |
82.4 | 73.7 | |||
| González-Moreno et al. 2024 [34] | Serum | ELISA | 93.55 | Asymptomatic V30M TTRv carriers and ATTRv V30M patients |
79 | 87 |
| 92.6 | Healthy controls and ATTRv V30M patients |
79 | 80 | |||
| Carroll et al. 2024 [33] | Serum | Simoa | 52.2 | PND ≤ I and PND > II |
100 | 55.5 |
| 64.5 | Asymptomatic patients and symptomatic patients or sensorimotor converters |
92.0 | 88.5 | |||
| 88.9 | Asymptomatic patients and symptomatic patients and all converters |
62.9 | 96.2 |
ATTRv: hereditary transthyretin amyloid; ELISA: enzyme-linked immunosorbent assay; Ella: name of a microfluidic cartridge-based immunoassay platform; NfL: neurofilament light chain; PND: polyneuropathy disability; PNP: polyneuropathy; Simoa: single-molecule array; TTRv: transthyretin gene variant; V30M: TTRVal30Met p.(Val50Met).
Table 3.
Effect of treatment on neurofilament light chain levels.
| Study (ref) | Effect of no treatment on NfL levels | Effect of treatment on NfL levels |
|---|---|---|
| Ticau et al. 2021 [30] | Patisiran: ↓ | |
| Loser et al. 2022 [23] | No treatment: ↑ |
Tafamidis or patisiran: ↑ Initiation of patisiran: ↓ |
| Sato et al. 2023 [38] | After one- and two- years with patisiran: ↓ Tafamidis: ↑ |
|
| Ticau et al. 2023 [30] | Placebo: ↑ | Patisiran: ↓ |
| Brunger et al. 2022 [35] | No treatment and no neuropathy: ↑ | Diflunisal/tafamidis: = Patisiran: ↓ |
| Conçeicao et al. 2023 [43] | Eplontersen week 85: trend ↓ | |
| Luigetti et al. 2023 [42] | No treatment: ↑ | Patisiran or vutrisiran: ↓ |
| Gilling et al. 2023 [44] | Patisiran: ↓ Placobo → 36 months patisiran: ↓ to similar levels as patients continuously on patisiran. |
|
| Carroll et al. 2024 [33] | TTR-gene silencer: ↓ (n = 8) and ↑ (n = 4) |
↑: increase; ↓: decrease; =: stable; NfL: neurofilament light chain.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



