
Submitted:
28 February 2024
Posted:
29 February 2024
You are already at the latest version
Abstract
Among the many Lysosomal Storage Disorders (LSDs) that would benefit from the establish-ment of novel cell models, either patient-derived or genetically engineered, is Mucopolysaccha-ridosis type II (MPS II). In fact, even though a specific therapeutic approach does exist for this disorder (Enzyme Replacement Therapy, ERT, with recombinant human IDS) and haematopoiet-ic stem cell transplantation (HSCT) may also hold promise whenever the disorder is diagnosed soon enough, both approaches have their own drawbacks.
Here we present our results on the establishment and characterization of two MPS II pa-tient-derived stem cell line(s) from deciduous baby teeth. To the best of our knowledge, this is the first time a stem cell population is isolated from LSD patient samples obtained from the oral cavity. Taking into account our results on the molecular and biochemical characterization of those cells and our first insights on their subcellular MPS phenotype, we consider these cells will be a valuable tool for both pathophysiological assessments and drugs screenings.
Ultimately, we believe patient-derived dental pulp stem cells (DPSC) may represent a feasible alternative to induced pluripotent stem cells (iPSC) in many labs with standard cell culture con-ditions and limited (human and economic) resources.
Keywords:
Mucopolysaccharidosis type II
; Disease Modelling
; in vitro models
; induced Pluripotent Stem Cells (iPSCs)
; Dental Pulp Stem Cells (DPSCs)
; SHED: Stem cells from Human Exfoliated Deciduous teeth (SHEDs)
1. Introduction
Mucopolysaccharidosis type II (MPS II; #MIM 309900; #ORPHA 579), also known as Hunter syndrome, is a rare genetic disorder that is inherited as an X-linked trait, with an incidence rate ranging from 0.38 to 1.09 per 100,000 live male births (reviewed in [1]). This disorder belongs to the group of lysosomal storage disorders (LSDs) and is caused by a deficiency of the lysosomal enzyme iduronate 2-sulphatase (IDS; EC 3.1.6.13), which catalyses the hydrolysis of 2-sulphate groups of dermatan sulphate (DS) and heparan sulphate (HS). Therefore, its deficit causes the pathological accumulation of these two glycosaminoglycans (GAGs), which translates into a multisystemic disease also affecting the brain, in at least two-thirds of cases (reviewed in [2]).
As many other LSDs, this disorder was first described more than 100 years ago, in 1917, by the Canadian physician Charles Hunter, from whom it got its most colloquial designation [3]. Since then, numerous advances were made regarding our understanding of this rare disease: it was shown to be a progressive and multi-systemic pathology, and its major causes were disclosed, both at biochemical [4] and molecular levels [5,6], with the disease mapping to the IDS gene (HGNC ID: 5389) on chromosome X, which encodes for the previously referred lysosomal enzyme. At a genetic level, for example, we now know that Hunter syndrome is characterized by a significant heterogeneity, as no highly recurring mutations have been reported so far, even though some variants seem to be slightly more frequent (reviewed in [7]). Also at the clinical level, much was discovered since the disorder was first reported. Indeed, what was once thought to be a single disease with two quite divergent clinical presentations, either severe or attenuated (depending on the length of survival and presence/absence of neurological disease), it is now known to be a continuum between the two forms, with disease severity linked to relative levels of IDS enzyme (reviewed in [8]). In general, MPS II clinical signs and symptoms include coarse facial features, skeletal deformities and joint stiffness, growth retardation, organomegaly as well as significant respiratory and cardiac impairments [9]. Neurological involvement has also been reported for at least two-thirds of cases [10,11,12]. Patients also present ENT (ear, nose, and throat) manifestations, sleep disturbances and obstructive apnea [13]. Visual symptoms may also be prominent [14]. According to the natural history of the disease, death occurs typically before adulthood for the most severe forms, while patients suffering from milder forms may usually survive until later in adult life [12] (reviewed in [2]). Also at a subcellular level, much was learnt on the disease pathophysiology. Increased accumulation of DS and HS was shown to impair numerous cellular functions including cell adhesion, endocytosis, intracellular trafficking, and intracellular ionic balance. It was also demonstrated to promote nitric oxide synthesis and trigger an inflammatory cascade, with numerous deleterious effects [15]. Yet, a full characterization of this cascade of secondary cellular events is still lacking (reviewed in [15]).
Currently, the standard of care for MPS II patients is enzyme replacement therapy (ERT), i.e, the intravenous administration of a functional recombinant version of the deficient enzyme. ERT has been shown to reduce urinary GAG levels and liver and spleen volumes in MPSII patients [16] (reviewed in [8]), with real-world data further suggesting that therapy may also improve other somatic cardio-respiratory parameters [17] (reviewed in [8]). Still, it does hold a number of limitations, namely, its inability to cross the blood brain barrier and act over the neurological symptoms, an issue which is common to virtually all ERT formulations developed so far, regardless of the LSD they apply to. To overcome the limitations of the ERT approach, some modifications to the traditional ERT protocol have actually been tested, including changes of the administration route, the introduction of modified fusion proteins, and the use of alternative hosts for enzyme production (reviewed in [2]). However, some challenges do persist, as the treatment’s inability to act over the neurological symptoms, its high cost, and life-long dependence [18].
Altogether, the need for deeper understanding of the pathophysiological mechanisms that underlie the disorder, and for more effective treatments to counteract it, justify the need for a cellular in vitro model that accurately recapitulates the disease phenotype in hard-to reach/hard to treat cells, such as those derived from the nervous and/or skeletal systems. Regarding neurons, in particular, the use of induced pluripotent stem cells (iPSC) to model neurogenetic disorders is well established, for example the function of cortical neurons from patient derived fibroblasts or blood cells is now well-documented and numerous studies in MPS II-derived iPSCs have already been published, with remarkable results and insights on the neuropathology of this disorder [19,20,21] (reviewed in [22]). However, there are notable drawbacks to using these de-differentiated, reprogrammed cells as in vitro models for molecular studies, mostly their high cost and time-consuming technology validation. That is why we are working with a completely different patient-derived cellular model: an alternative, less invasive and less laborious, source of stem cells, the so-called dental mesenchymal stem cells (DMSCs), which can be isolated from different sources in the oral cavity (reviewed in [22]).
Here we report the establishment of two independent MPS II-derived cultures of stem cells from human exfoliated deciduous teeth (SHED) and their subsequent characterization at molecular, biochemical and pathophysiological levels. The existence of this small population of dental pulp stem cells was first reported by Miura and co-workers in 2003, when SHEDs were first isolated from primary teeth that were lost due to the eruption of permanent teeth. That initial paper already provided remarkable insights into this particular population of DMSC, demonstrating their high proliferative capacity and their ability to differentiate into a variety of cell types including neural cells, adipocytes, and odontoblasts. Those authors have also attempted their in vivo transplantation, showing that SHEDs were able to induce bone formation, generate dentin, and survive in mouse brain [23]. Thanks to the efforts of many independent teams working on different fields, these highly proliferative cells are now fully characterized and their MSC expression profile ([24,25,26], reviewed in [27]). Soon enough, this DMSC population was considered to hold great promise for regenerative medicine approaches and other cell-based therapies. Indeed, numerous studies have shown that autologous transplantation of SHED, for example, may be a safe and promising approach for both dentin and pulp regeneration (reviewed in [28]). But their applications far exceed the orthodontic field, as SHEDs have also been tested for their regenerative potential against spinal cord injury [29,30,31], hypoxic–ischemic brain injury [32], ovariectomy-induced osteoporosis [33], liver fibrosis [34], and systemic lupus erythematosus [35]. And that potential lies not only on the cells themselves but also in the conditioned media one may collect from their culture, as elegantly shown by Murakami and co-workers in a recent study, where the authors immortalized a SHED cell line, and analysed the effects of its conditioned medium on cutaneous pressure ulcers [36].
In the LSD field, however, DMSCs (and SHEDs in particular) are mostly unknown and their potential unexplored. So, here we addressed the question of whether these cells could be used, not for therapeutic purposes, but for disease modelling, as they are well-known to retain the ability to differentiate into several cell lineages that represent disease-relevant cell types, not only for MPS II, but for LSDs in general (e.g chondrocytes, osteoblasts, and neuronal cell types; reviewed in [37]). To the best of our knowledge this was the first time that SHEDs were isolated from LSD patients.
Briefly, we have not only successfully confirmed the dental pulp stem cell identity of the established MPS II SHEDs, but also assessed their LSD phenotype. Regarding their stemness potential, the MPS II-derived SHED cell lines here described presented an expression pattern characteristic of a mesenchymal stem cell (MSC) line, with high expression levels of CD105, CD73 and CD90 and a weak but still detectable expression of the pluripotency genes Nanog, Oct4 and Sox2. They were also able to differentiate into endoderm, ectoderm and mesoderm. Also noteworthy, our results so far clearly demonstrate that the typical MPS subcellular phenotype is already present in the established MPS II-derived SHED cell lines. Overall, this is an original contribution to the LSD field, as it demonstrates the existence of an easy-accessible, non-invasive source of MSCs, which exhibits visible and measurable LSD subcellular phenotypes. Importantly, those can be collected from severely affected individuals (those who suffer from infantile forms of these disorders. It seems most valuable to use these methods in order to better establish patient-specific models to understand the cellular dynamic of the disease in the donor patients of such cells.
2. Results
2.1. Establishment of Primary SHED Cell Cultures from Patients and Controls
In this study, two unconventional MPS II samples were used to establish a novel patient-derived mesenchymal stem cell (MSC) line in house: stem cells obtained from normally shed deciduous teeth (SHED). The teeth included in the study were non-carious, had no previous restorations, and had no reports of prior trauma, even though one was surgically extracted (that from patient MPS II, 2.01). All other samples were spontaneously exfoliated teeth (namely, those obtained from patients MPS II, 2.02 and controls 01 and 02).
Upon reception of the biological sample, dental pulp was extracted, enzymatically digested, and left to grow in Poly-D-Lysine or Vitronectin-coated 12-well plates. Cells derived from the dental pulp were visible within 1-2 weeks. Then, in a process which took anywhere from two weeks to one month, we could observe a population of SHEDs, morphologically characterized by spindle-shape cells, similar to fibroblasts (Figure 1a), which initially formed small colonies (Figure 1b) that were left to grow until they reached sub-confluence (Figure 1c). Throughout the whole process, which involved the establishment of the primary cultures, their passage, freezing and thawing, SHED cells viability and morphology were checked using a light microscope and any relevant alternation noted. All established cell lines shared the same morphological features, regardless of whether they were derived from patients or controls.
2.2. The Established SHED Cell Lines Share a MSC Phenotype Identity
The MSC phenotype of the established SHED cultures was assessed through different protocols, namely qRT-PCR analysis of different stem cell markers and multi-lineage differentiation assays.
Regarding the qRT-PCR analysis, total RNA was successfully extracted not only from cultured SHEDs but also from an iPSC cell line already available in the lab (INSAi002-A; derived from Fabry fibroblasts [35]). The extracted RNA samples were reversely transcribed into cDNA, and specific gene expression was assessed (Table 1). In summary, CD34 was not detected as expected for SHEDs. CD105, CD73 and CD90 were highly expressed and CD166, MHC I and CD117 showed strong to moderate expression. The pluripotency genes Nanog, Oct4 and Sox2, were also expressed, although weakly (Ct value > 35). Moreover, weak expression of MHC Class II was also detected in SHEDs. The expression levels observed for the iPSC cell line are also summarized in Table 1. Briefly, INSAi002-A iPSCs displayed high expression levels of CD105, CD73, CD90 and OCT-3/4, and moderate expression levels of MHC II, Nanog, and Sox-2. Absolute ΔCt values obtained calculated using GAPDH and β-actin as housekeeping genes are listed in Figure S1.
Further insights on the stem nature of the established SHED cell lines came from multi -lineage differentiation assays. In a first assessment, the Human Pluripotent Stem Cell Functional Identification Kit (R&D Systems, Minneapolis, Minnesota, USA) was used. This kit contains especially formulated media supplements and growth factors that can be used to differentiate human pluripotent stem cells into endoderm, ectoderm and mesoderm. It is a fast protocol, which relies on 2 to 4-days incubations with those media, and also includes an antibody to characterize each of the three primordial germ layers: Sox17 for endoderm; Otx2 for ectoderm and Brachyury for mesoderm. Not surprisingly, all established cell lines stained positive for each respective marker, thus confirming their capacity to differentiate into each of the three germ layers (Figure 2a).
2.3. The Established SHED Cell Lines Express Major NPC Markers
As a final characterization step of the established SHED cell lines, the DMSC early commitment to their so-called "neuronal fate" was also confirmed. This study was performed in primary SHED cells, before any neuronal differentiation protocol was attempted, and relied on the use of a commercially available kit. Briefly, their Neural Progenitor Cell (NPC) stage was analysed based on the expression of major NPC markers (Nestin, Sox1, Sox2 and PAX6), having a positive fluorescence pattern been observed for all four markers evaluated (Figure 2b). No significant differences were detected between healthy and MPS II cell lines in early passages (from passage 5 up to passage 7).
2.4. The Established MPS II-Derived SHED Cell Lines Display the Disease-Related Biochemical and Molecular IDS Defects
Whenever an MPS tooth was received in the laboratory, the only information available was the type of MPS from which the patient it belonged suffers. Therefore, as soon as its derived SHED cell line was established and the first vials stored, cell pellets were collected and used for mutational analysis and enzyme activity assays.
2.4.1. The Presence of Pathogenic IDS Variants Was Confirmed in the Established MPS II Patient-Derived SHED Cell Lines
For molecular analyses, DNA was successfully extracted from both MPS II patient-derived SHED cultures, and standard amplification and Sanger sequencing protocols were employed to analyse the 9 IDS exons plus their surrounding intronic regions, as previously reported [38]. Whenever standard procedures were not enough to identify the disease-causing variant, additional studies were performed, namely restriction fragment length polymorphism (RFLP) [38] (see Materials and Methods, Section 4.4.1.).
Briefly, Case 2.01 was confirmed to carry a rearrangement involving recombination between intron 7 of the IDS gene and sequences located distal of exon 3 in the IDS pseudogene (IDS-2) [GAATC>AGAGG (IDSP1> IDS)]. This recombination event had already been reported, and is known to cause a partial inversion of the IDS gene [39]. Conversely, Case 2.02 was shown to be hemizygous for a much straighforward to detect variant: the previously reported c.22C>T (p.R8*) nonsense mutation [39]. Both identified genotypes were in accordance with MPS II phenotype.
2.4.2. IDS Enzyme Activity Is Significantly Decreased in MPS II Patient-Derived SHED Cells
When the IDS activity was measured with the fluorescent substrate 4-methylumbelliferyl (4MU)-α-L-iduronate-2-sulphate in SHED cell lysates from both controls and the two independent MPS II patients, no activity could be detected in cases 2.01 and 2.02, whilst control Ctrl.01 and Ctrl.02 cells showed normal IDS activity (Figure 3a). The other lysosomal enzymatic assays performed where within the normal range in all the SHED lysates (Supplementary Figure S1).
2.5. MPS II Patient-Derived SHED Cells Exibit a Subcellular LSD Phenotype
Subsequently, the LSD phenotype of the established MPS II patient-derived phenotype was assessed. Briefly, we checked not only the primary GAG storage, which is known to trigger the whole MPS II pathophysiological cascade, but also the lysosomal localization pattern, to check whether it was altered.
2.5.1. GAG Accumulation Is Evident in MPS II Patient-Derived SHED Cells, Despite Their High Proliferation Rate
Decreased (or absent) IDS activity in MPS II patients is known to cause an intracellular and extracellular accumulation of two major GAGs, namely HS and DS. Thus, the levels of those two GAGs were measured by LC MS/MS, and both MPS II SHED cultures showed evident GAG accumulation when compared with control SHEDs (Figure 3b).
2.5.2. LAMP1 Staining Is Altered in Patient-Derived SHED Cells
Accumulation of DS and HS causes lysosomal hypertrophy and an increase in the number of lysosomes in cells. Therefore, any method that allows for an evaluation of the number, size and sub-cellular localization of these disease-relevant organelles, may provide relevant data on the health/disease status of a given cell line. Here we chose to perform a LAMP1 immunostaining protocol in control vs MPS II cells. LAMP1 is one of the most abundant lysosomal membrane proteins, nicely correlating with lysosomal dysfunction, as its overexpression reflects abnormal accumulation of lysosomes. Overall, the pathological phenotype was quite evident, with MPS II cells presenting a prominent LAMP1-positive perinuclear fluorescence when compared to controls (Figure 3c).
3. Discussion
Here we describe the establishment and characterization of two MPS II patient-derived SHED cell lines (named 2020TF-MPS2.01 and 2020TF-MPS2.02), while addressing their modelling potential for this severe condition by evaluating whether they display visible and measurable MPS subcellular features.
Briefly, we have successfully implemented a protocol developed by Goorha and Reiter [40] for the efficacious remote tooth collection and subsequent dental pulp extraction for growth and expansion of that particular subset of dental mesenchymal stem cells (DMSCs), and applied it in two independent MPS II cases and a significant number of controls (> 30). As originally reported by Goorha and Reiter, the process of growing these particular DMSC can take anywhere from 1 to 2 weeks and, at least in our hands, there seems to be no particular correlation between the size of the pulp, or the time it takes to arrive at the lab (as long as the 48/72h interval is ensured) and the time it takes for the first cells/colonies to become visible in the plate. Overall, the whole method is well-described in the publication we refer to, and it is not hard to implement in a lab with standard cell culture conditions, regardless of whether the operators had previous experience with other sorts of stem cells, namely iPSCs.
Technically, MSCs are classified as multipotent stem cells and not as pluripotent stem cells. Still, as we have already seen, they do present a positive expression pattern of OCT-3/4, Nanog, and Sox-2, which are standard pluripotency markers [41,42,43]. DMSCs in general, and SHEDs in particular, are already known to express those markers for quite a while, now. In fact, that characteristic was already reported in healthy SHEDs back in 2006 in an original paper by Kerkis et al. [24], where their stemness character was confirmed [25]. Nevertheless, the expression level of any of these markers, when compared with other commonly assessed MSCs markers is known to be weak. These data correlate nicely with our results, where all SHED cell lines presented with positive expression levels of these three pluripotency markers, but at a level which was significantly lower than that seen for specific MSCs markers (CD105, CD90, and CD73). They also seem to be in accordance with what we saw on the iPSC cell line we used as a control (INSAi002-A; derived from Fabry fibroblasts [44]): while positive, the levels of expression of OCT-3/4, Nanog, and Sox-2 were much lower in the established multipotent SHED cell lines, than in the truly pluripotent iPSC line, which was triggered to overexpress those markers, through a reprogramming protocol with the Yamanaka factors. Remarkably, no studies comparing the expression levels of stemness markers between DMSCs and iPSCs are available, at least that we are aware of. Therefore, these results become even more interesting.
Additionally, specific MSCs markers were also measured, in both healthy and disease-derived SHEDs, as well as in the iPSC line INSAi002-A and, overall, the results were in line to what would be expected, according to the literature: MSC markers (CD105, CD90, and CD73) were the ones that displayed higher expression levels, thus supporting the MSC phenotype of the established SHEDs. The two remaining markers assessed, CD34 and MHCII, are commonly described as absent in MSCs. They did, however, show positive expression, even though with significantly lower levels than those observed for CD105, CD90, and CD73; they were actually comparable with the ΔCt value observed for the pluripotency markers. And, while this result seems unaligned with MSC requirements, as they are reported in the bibliography, when we look at individual papers where SHED and DPSC expression patterns for these markers were assessed, this observation is actually common. For example, recently, positive expression levels of MHCII were reported in a commercially available DPSC line, and considered a normal aspect [45]. Additionally, there is already literature commenting on the possibility that the absence of expression of those markers may not be mandatory for a cell to be classified as MSC, once several MSCs have been shown to express, at least to some extent, both of them [34,35].
Adding up to our qRT-PCR results regarding the MSC phenotype are our results on the differentiation capacity of the established SHED cell lines into distinct cell types. Traditionally, one of the listed requirements to identify MSCs is their ability to differentiate into three different cell types: adipocytes, osteocytes and chondrocytes [48]. More recently, though, many authors have argued those requirements should be updated to include cells from the 3 germ layers: ectoderm, mesoderm, and endoderm [49]. That is why we chose to perform a novel (and faster) 4-days-long tri-lineage differentiation protocol, through which our MPS II patient-derived SHED cell lines were forced to differentiate into ectoderm, endoderm and mesoderm. Overall, the protocol worked precisely as anticipated, with the cells staining positive for all tested markers, regardless of the differentiation attempted.
Still, there was yet another assessment we considered to be relevant regarding overall nature of the established cell lines, regardless of their health/disease status. SHEDs have a behaviour similar to neuronal precursor cells. Indeed, there are now numerous publications providing evidence that SHEDs express neuronal and glial cell markers, owing to the neural crest-cell origin of the dental pulp [50] (reviewed in [51]). And in fact, staining of neuronal markers in SHEDs not subjected to any type of neurodifferentiation protocol, revealed a positive fluorescence pattern for all four markers evaluated: Nestin; Sox-1; Pax-6 and Sox-2, further validating the assumption that SHEDs may actually be classified as NPCs, as stated by several different authors.
Having extensively demonstrated the stemness capacity of all established SHED cell lines, and further characterized them as NPCs, we moved on to analyse whether they were able to mimic the primary defect underlying the MPS II phenotype in the patients from whom they were derived. Thus, a careful molecular characterization of their associated genotypes was performed, together with a quantification of each one’s defective enzyme.
Unsurprisingly, when the two established MPS II cell lines were molecularly characterized, both cases were shown to harbour pathogenic IDS variants. Case 2.01 was shown to be hemizygous for a complex rearrangement that results from a recombination event between intron 7 of the IDS gene and sequences located distal of exon 3 in the IDS pseudogene (IDS-2) [GAATC > AGAGG (IDSP1 > IDS)] and causes a partial inversion of the IDS gene [39]. Case 2.02 was shown to be hemizygous for the previously reported c.22C>T (p.R8*) nonsense mutation [52]. This mutation had already been reported in different populations, correlating either with severe or intermediate forms of the disease [52,53,54]. Altogether, both mutations may easily correlate with severe, early-onset phenotypes, as the ones presented by both patients included in this study (see Table 2).
Accordingly, when IDS enzyme activity was measured in the lysate of MPS II patients’ SHEDs, it was shown to drop to zero.
While the results so far already testify on the overall potential of patient-derived SHED cell lines to accurately express a measurable LSD phenotype, the ones we will now focus on, further highlight the uniqueness of this cell model.
Our results regarding MPS II-related primary storage in patient-derived SHEDs, for example, are worth of additional discussion. Briefly, when we quantified DS and HS in healthy and diseased SHED cell lysates by butanolysis derivatization [55], significant differences were observed between patients and controls, with both MPS II samples showing GAG accumulation (Figure 3b). This is particularly relevant when compared with the results other authors have achieved with MPS II iPSC-derived cell lines. Currently, there are numerous reports on the generation of MPS II human iPSC lines. Still, not all papers evaluated the LSD phenotype they present. Instead, most publications focus only on the iPSC generation and characterization protocol, already well establish to validate an iPSC line: they report the method of reprogramming, present proof on the established cell line(s) pluripotency and differentiation potential; assess its identity compared to the cells they were reprogrammed from, as well as their karyotype to confirm it remains normal; double-check the presence of the original disease-causing variant, and rule out mycoplasma contamination. There is, however, an original publication by Kobolák and co-workers ([20]), where numerous analyses were performed, not only in MPS-derived iPSC but also, and perhaps most importantly, in neural precursor cells (NPCs) and terminally differentiated neurons (TD) generated from them. Briefly, they used iPSC lines generated from three independent MPS II patients, a healthy control, and a carrier, to generate NPCs and TD neuronal cells, and compared results amongst all those lines. Curiously, all the three MPS II NPC cultures analysed in that work showed lower total GAG levels (p < 0.05) compared to either control or carrier cell lines. For the hallmark accumulation to (finally) be observed, the authors had to promote the terminal differentiation of those MPS II iPSC derived NPCs into cortical neural cells. Only then a marked GAG accumulation could be detected, even though for two of the three MPS II cell lines alone. The third patient-derived MPS II neuronal cell line did not differ significantly from the control cell line [20] (reviewed in [22]). This is significantly different from our own observation in MPS II SHED cell lines, where GAG storage was quite evident. And, while we cannot find the reasoning for this discrepant observations, it is (quite) obvious they further highlight the disease modelling potential of this simple and easily accessible type of stem cells.
Similar results were obtained regarding LAMP1 staining. Again, the MPS II SHEDs we established showed obvious differences compared to the controls, a pattern that is in accordance with previous reports from other teams that observed increased LAMP1 levels in lysosomal storage disease model animals, as well as in human patients. For MPS II in particular, Morimoto and co-worker have recently reported increased Lamp1-staining in the brain of (untreated) MPS II mice. Importantly, when those mice were treated with the recombinant enzyme idursulfatase, irrespective of the dosing regimen, that intensity decreased in most regions of the brain. Those observations further support the assumption that abnormal Lamp1 staining in MPS II correlates with lysosomal dysfunction [56]. Altered staining patterns for both LAMP1 and LAMP2 were also observed in MPS II iPS-derived neural stem cells [57]. Remarkably, however, this pattern was not seen in all MPS II-derived neural stem cells reported so far. Indeed, in the original publication by Kobolák et al. [20], to which we have already referred to, significant differences between patient and control-derived cells, were only observed in TD neuronal cells, where indeed MPS II samples showed cell-type specific differences in their LAMP2 staining patterns, with many more LAMP2+ vacuoles in GFAP + astrocytes than in MAP2+ neurons [20]. Again, these significant differences between the novel, naturally-occurring stem cell model here reported (patient-derived SHEDs) and its iPSC-derived equivalent (NPCs), further highlight the advantage of establishing this type of cell lines.
It is also worth reinforcing that there are many other DMSC, which may be collected from the oral cavity (Figure 4). We have focused attention on SHEDs, because they may be collected in a non-invasive way in paediatric patients. However, other sources may be considered, particularly for adult patients with milder forms of the disorders, who tend to be diagnosed later in life. A good example is the use of adult human third molar teeth, from where DMSC may also be isolated. While there are slight variations in the protocols described in the literature for the isolation of DPSC from this source, the overall method is not significantly different from the one here reported for SHED cell culture.
Overall, this type of sample would allow for a significant increase on the number of eligible patients’, because their recruitment platform would be much larger than the current one: it would move from children who are currently losing their baby teeth, to virtually any patient, regardless of his/her age. The fact is that the surgical removal of human third molars (also known as wisdom teeth), is the most common surgical procedure in the orthodontist field, also adds to the interest of implementing this protocol, and asking for this type of samples. This picture is probably even more prominent in individuals who suffer from MPS, particularly from the skeletal forms of these disorders. In fact, amongst some of the most common and obvious orofacial abnormalities in MPS patients, are maxillomandibular abnormalities. GAG accumulation in soft tissues, cartilage, and bones and secondary cellular responses to accumulated GAGs are probably the culprit to abnormalities in orofacial soft tissues, orofacial bones, and teeth [58]. That is why MPS patients are frequently subjected to teeth removal surgeries, among other orofacial interventions.
Finally, it is also worth mentioning that, while we recognize the therapeutic potential any stem cell line may eventually hold, particularly in a field where HSCT is a feasible and recommended approach for a few disorders, depending on the severity of the phenotype and the age of the patient, our goal with this work was never to either establish or characterize SHED cell lines for therapeutic purposes. Still, since the cells we isolated present the same genetic defect harboured by their donor, only after gene editing (e.g. CRISPR) would they be suitable for transplantation. Plus, as these cells are isolated from naturally exfoliated baby teeth, one would only have access to that sample when these patients started losing their teeth, i.e., at about age 6 or later. And that is certainly late for HSCT approaches, as it is well documented for other MPS (namely MPS I with CNS affection) that HSCT only works in children who are less than 2.5 years of age. Additionally, it is also worth mentioning that the protocol we used here was not suitable to establish clinical-grade stem cells.
Altogether, our results show that this patient-derived sample is a much faster and economical way to establish a stem cell model, and it also holds the potential to display disease-relevant sub-cellular features. Thus, patient-derived SHEDs may be assessed not only to allow a better understanding of the pathophysiological mechanisms underlying the disorder, but also to evaluate the potential impact of novel therapeutic approaches in vitro.
4. Materials and Methods
4.1. Materials

Table 3.
Antibodies.
| Antibody | Dilution | Company Cat # and RRID | ||
|---|---|---|---|---|
| Primary Antibodies | Germ Layer Markers | Goat Anti-Human Brachyury Polyclonal Antibody, unconjugated (Mesoderm) | 1:10 | R&D Systems Cat# AF2085, RRID:AB_2200235 |
| Goat Anti-Human Sox17 Polyclonal Antibody, unconjugated (Endoderm) | 1:10 | R&D Systems Cat# AF1924, RRID:AB_355060 | ||
| Goat Anti-Human Otx2 Polyclonal Antibody, unconjugated (Ectoderm) | 1:10 | R&D Systems Cat# AF1979, RRID:AB_2157172 | ||
| Neural Stem Cell Markers | Mouse Anti-Human Nestin Monoclonal Antibody | 1:49 | Thermo Fisher Scientific Cat# MA1-110 (A24345) | |
| Rabbit Anti-Human PAX6 | 1:49 | Thermo Fisher Scientific Cat#(A24340) | ||
| Goat Anti-Human SOX1 | 1:49 | Thermo Fisher Scientific Cat#(A24347) | ||
| Rabbit Anti-Human SOX2 | 1:49 | Thermo Fisher Scientific Cat#(A24339) | ||
| LAMP1 Staining | Mouse Anti-Human LAMP-1 Monoclonal Antibody | 1:200 | Santa Cruz Biotechnology, Inc. Cat# sc-20011 |
|
| Secondary Antibodies | Alexa Fluor 488 Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody | 1:200 | Thermo Fisher Scientific Cat# A-11055, RRID:AB_2534102 | |
| Alexa Fluor™ 488 Donkey Anti-Mouse; for use with anti- Nestin | 1:249 | Thermo Fisher Scientific Cat#(A24350) | ||
| Alexa Fluor™ 488 Donkey Anti-Goat; for use with anti-SOX1 | 1:249 | Thermo Fisher Scientific Cat#(A24349) | ||
| Alexa Fluor™ 555 Donkey Anti-Rabbit; for use with anti-PAX6 or anti-SOX2 | 1:249 | Thermo Fisher Scientific Cat#(A24342) | ||
| Alexa Fluor™ 594 Donkey Anti-Rabbit; for use with anti-PAX6 or anti-SOX2 | 1:249 | Thermo Fisher Scientific Cat#(A24343) |
4.2. Patients and Samples
4.2.1. Patient 1 (Case MPS 2.01)
Patient 1 was the first child of a non-consanguineous couple. While there was no access to a complete family tree, family history records included a reference to the mother's learning difficulties and another to a disabled maternal cousin (son of a maternal great aunt), who died in childhood without diagnosis.
At 22-months, initial signs of developmental delay, mostly regarding language, were reported, accompanied by other clinical features, which included wide-based gait with knee flexion and motor agitation. It was also at this age that a first reference was made to a “rough face” in the records of the objective examination of paediatric consultations.
By then, the patient’s skeletal and somatic features included coarse facies, stiff joints and post-natal macroglossia. Some cardiac manifestations were also detected, namely interventricular communication (IVC) and patent ductus arteriosus (PDA); both solved by now. At 35 months, thickening of the aortic valve was detected. Additionally, moderate aortic insufficiency and left ventricular hypertrophy were also referenced. ENT manifestations were also present, including chronic nasal obstruction, even though no recurrent otitis or hearing deficit have been detected so far. Finally, a mild psychomotor development retardation was also reported.
Upon objective examination at the metabolic and genetic consultation (also at 35 months of age), the child displayed relatively understandable language and showed important emotional lability. His dysmorphic features (namely coarse face, abundant and thickened eyebrows, macroglossia apparent, abnormal posture with knee flexion and wide-based gait and limitation in elbow extension, slight pectus carinatum with far apart nipples and preputial ring punctiform) were considered suggestive of MPS and the child was refer-enced to molecular and biochemical diagnosis. No evidence of hepatosplenomegaly or hernias. Regarding evolution of weight gain and head circumference, there was a positive crossing of percentiles of the various parameters, currently having: height at P85-97, weight at P>97 (with BMI at P>99 – P>3SD) and head circumference at P>97.
4.2.2. Patient 2 (Case MPS 2.02)
Patient 2 is a 6-year old male child, diagnosed at 2 years old with MPS type II. The child presented with multisystemic involvement, which included not only several classical musculoskeletal features of the disease (inguinal hernias, claw hands and low stature) but also a series of other symptoms, which included hypertrichosis, hepatomegaly and cardiac involvement (minimal mitral regurgitation). Later, he underwent otorhinolaryngological surgery to perform bilateral myringotomy and tubes with adenoidectomy. Currently, his inguinal hernias have already been corrected. Recently (June, 2022), his global psychomotor development was assessed and considered normal. Nevertheless, he developed an orthopaedic condition with joint restrictions, which requires physiotherapy.
4.2.3. Biological Samples
Deciduous teeth (i.e., “baby” teeth) from two independent MPS II patients and from an equal number of controls.
Briefly, the process of sample collection was performed as follows: all subjects included in this study, either WT controls or MPS II patients, received a tooth collection kit, which included a parafilm-sealed Falcon tube filled with adequate transport media accompanied by return instructions, a biohazard bag, plus a pre-filled delivery form. Also included in the kit was an informed consent form to be filled by the participant’s legal representative, a summary of the project and its objectives and a flyer with major recommendations and frequently asked questions (See Figure S2). The families were instructed to store the Falcon tube in the refrigerator (4 °C) and to place the tooth in it, virtually as soon as it fell (within 20 minutes), and send it to the lab within 24 hours.
While we received more than 50 deciduous teeth and established over 30 independent control cell lines from them, most of the results here reported were obtained through the analyses of only two of those controls (here termed control 01 and 02). An exception was made for enzyme activity and LC-MS/MS assays, where a significantly higher number of controls was included, to account for inter-individual differences.
In general, the deciduous teeth used in this work were spontaneously exfoliated teeth. However, for patient 1 (MPS II, 2.01), a surgically extracted deciduous tooth was received.
4.3. Cell Culture
4.3.1. Establishment of Primary SHED Cell Cultures
The whole protocol for the establishment of primary SHED cell cultures derived from that published by Goorha and Reiter, in 2017 [40] for remote tooth collection of exfoliated teeth and subsequent production of DPSC cultures for differentiation or storage, with minor alterations. In detail, upon every tooth reception, and after a careful inspection of the transport medium to discard possible microbial contamination, teeth were broke open under sterile conditions, using a sterilized hammer. The dental pulp was then pulled out and placed into pre-heated washing media [saline solution, with PenStrep (50 U/mL) fungizone (1,25 µg/mL)] in a small petri dish, where it was minced into smaller fragments. After a brief centrifugation, the minced pulp was resuspended in fresh DPSC culture medium [DMEM/F12 (1:1) with 20% FBS, PenStrep (50 U/mL) and fungizone (50 U/mL) ] containing 3 mg/mL collagenase (Gibco, Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 1–4 mg/mL dispase (Neutral Protease Grade II, Roche, Basel, Switzerland) and incubated for 1 hour at 37 °C. Following enzymatic digestion, samples were centrifuged at 448× g during 5 min, resuspended in 1 mL of DPSC culture medium and seeded in a single well of a poly-D-lysine (Gibco, Thermo Fisher Scientific, Waltham, Massachusetts, USA)- or vitronectin (Thermo Fisher Scientific, Waltham, Massachusetts, USA)-coated 12-well tissue culture plate. The plate was then left to incubate overnight in a standard 37 °C incubator, with 5% CO2. The following day, supernatant was removed from the well and briefly centrifuged to spin down the floating, unattached cells. The resulting pellet was resuspended in fresh DPSC culture media and seeded in another coated well from the same plate. Cultures were washed with the previously described washing solution and media changed twice a week, up until cells reached sub-confluence (80–90%).
4.3.2. Passage, Freezing and Thawing of SHED Cell Cultures
Again, in accordance with the original protocol developed by Goorha and Reiter [40], SHED cultures were passaged at a ratio of 1:3, according to the following rationale: one part was sub-cultured for passage, and the remaining two were frozen for future applications. In higher passages, when there was no need to assure the maximum possible number of stored vials to ensure the preservation of the cell line, parts 2 and 3 were frequently used to generate pellets for subsequent analyses. For cell detachment, Accutase (Grisp, Lda., Portugal) was used. Early passage cells were frozen for long term storage in liquid nitrogen, in culture media supplemented with 15% DMSO (Sigma-Aldrich, St. Louis, Missouri, USA), according to standard freezing protocols.
4.4. Molecular Analyses
4.4.1. Genotype Assessment by PCR and Sanger Sequencing
Genomic DNA was automated extracted and purified from cell pellets on a BioRobot EZ1 instrument (QIAGEN, Germantown, MD, USA) using the EZ1 DNA Tissue Kit (QIAGEN, Germantown, MD, USA). Previously reported primers [38] covering all IDS exons and their surrounding intronic regions, as well as its promoter region, were used to sequence the gene of interest. Each PCR reaction was carried out using approximately 40 ng of genomic DNA, 1X the PCR reaction mix ImmoMix™ Red (Bioline, London, UK) and 0.25 µM of each primer. For some particular fragments, betaine and/or DMSO were also used to enhance the PCR amplification of the target region (conditions available upon request). Then, samples were heated to 95 °C for 7 min, followed by 35 cycles of denaturation, annealing and extension. The final extension was completed by 5 min at 72 °C. The amplified fragments were purified with illustra ExoStar™ 1-Step (GE Healthcare Life Sciences, Buckinghamshire, UK) and sequenced using a BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on an ABI PRISM 3130xl Genetic Analyser (Applied Biosystems, Foster City, CA, USA). Results were analysed with the sequence analysis software FINCHTV, version 1.3.1. Sequencing profiles were compared with the IDS reference sequence ENST00000340855.11 (https://www.ensembl.org) using the Clustal Omega multiple sequence alignment bioinformatic tool (https://www.ebi.ac.uk/Tools/msa/clustalo/). The presence of the variants found in both patients was also confirmed at cDNA level. Briefly, total RNA was extracted from independent cell pellets using GRS Total RNA-Blood & Cultured Cells Kit (GRiSP, Porto, Portugal). 1 ng of RNA were reverse transcribed using the Ready-To-Go You-Prime First-Strand Beads (GE Healthcare Life Sciences, Chicago, Illinois, USA), according to the manufacturer’s instructions.
Importantly, only after fully genotyping the patients, were the results confirmed by the clinicians, who had access to the patients’ clinical file and molecular results from previous analyses.
4.4.2. qRT-PCR Analyses
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was performed for MSCs’ related genes sequences from Bio Rad® (Bio-Rad Laboratories, Hercules, California, USA) to confirm the DPSC/MSC phenotype identity of the established cell lines: CD34 (qHsaCID0007456), CD90/THY1 (qHsaCED0036661), CD73/NT5E (qHsaCID0036556), CD105/ENG (qHsaCID0010800), CD166/ALCAM (qHsaCID0037887), CD117/c-kit (qHsaCID0008692), SOX2 (qHsaCED0036871), OCT-3/4/POU5F1 (qHsaCED0038334), MHC Class I/HLA-A (qHsaCED0037388), MHC Class II/HLA-DRA (qHsaCED0037296) and housekeeping genes: β-actin (qHsaCED0036269), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (qHsaCED0038674).
Total RNA was extracted as described in the previous section, and reverse transcribed using the same kits. Nevertheless, for qRT-qPCR analyses, lower amounts of total RNA were used for cDNA synthesis (0.5–1 µg). qPCR was performed in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, California, USA) apparatus using the iTAQTM SYBR® Green Supermix (Bio-Rad Laboratories, Hercules, California, USA). All plates were designed to contain duplicates of targeted human genes as well as a negative control. Recommended PrimePCR cycling protocol was employed in all cases: 95 °C for 2 min (activation), 40 cycles comprising 95 °C for 5 s (denaturation), −60 °C for 30 s (annealing), and 65–95 °C (0.5 °C increments), 5 s/step (melt curve). The number of cycles for each well was recorded. Data was processed using BioRad CFX® Manager Software 3.1 (Bio-Rad Laboratories, Hercules, California, USA). Fold differences were calculated using the standard ΔCq method with GAPDH and β-actin as housekeeping genes.
4.5. Biochemical Analyses
4.5.1. Fluorometric IDS Enzyme Assay
Control (WT) and MPS II-derived SHEDs were used in the determination of IDS enzyme activity. Briefly, cell homogenates were prepared by sonication, and their protein concentration determined using the Pierce™ BCA Protein Assay Kit (TermoFisher Scientific, Waltham, Massachusetts, USA) and measured by spectrophotometer (VICTOR® NivoTM Plate Reader, PerkinElmer Inc, Waltham, Massachusetts, USA). Then, the IDS enzyme activity was assayed using fluorogenic substrate 4-methylumbelliferyl (4MU)-α-L-iduronate-2-sulphate, according to the method described by Voznyi et al. [59], and the fluorescence was measured in the fluorimeter (VICTOR® NivoTM Plate Reader, PerkinElmer Inc, Waltham, Massachusetts, USA). The enzymatic activity was determined according to a calibration curve done with 4-methylumbelliferyl (4MU), and normalized to 1mg/ml of protein.
4.5.2. Other Enzymatic Assays for Additional Lysosomal Enzymes
Other lysosomal enzyme activities were also measured in control (WT) and MPS II-derived SHEDs, namely beta-galactosidase (GLB1, E.C. 3.2.1.23, the enzyme deficient in either GM1-gangliosidosis or MPS IVB); beta-glucuronidase (GUSB, E.C. 3.2.1.31, the enzyme deficient in MPS VII); hexosaminidase A (HEXA, E.C. 3.2.1.52, the enzyme deficient in GM2-gangliosidosis), alpha-N-acetyl-glucosaminidase (NAGLU, E.C. 3.2.1.50, the enzyme deficient in MPS IIIB) and alpha-galactosidase (GLA, E.C. 3.2.1.22, the enzyme deficient in Fabry Disease). All measurements relied on the breakdown of specific 4MU-substrates, except for that of ARSB, which relied on the use of an artificial chromogenic, substrate 4-nitrocatechol sulfate [60,61]. The enzymatic activity was determined according to a calibration curve done with 4-methylumbelliferyl (4MU), and normalized to 1mg/ml of protein.
4.5.3. Glycosaminoglycans Quantification by LC MS/MS
GAGs were quantified by simultaneous analysis of DS and HS in WT and MPS II-derived SHED homogenates, by LC-MS/MS, after butanolysis reaction, according to the method recently described by Forni and co-workers [55,62]. While initially described to perform HS and DS analysis in urine samples, this method was adapted to quantify the same compounds in cell homogenates. Briefly, cell homogenates were prepared by sonication, and their protein concentration determined with the same method described in Section 4.5.1. Each individual cell homogenate was divided into two different sample tubes, one for HS and another for DS. Samples were then dried under a stream of nitrogen and 75 µL of 3N HCl in N-butanol added to each vial. For HS measurements, samples were incubated for 60 min at 90 °C. For DS measurements, on the other hand, samples were heated for 25 min at 65 °C. After those incubations, samples were cooled back to room temperature for 10min and dried under a stream of nitrogen. 100 µL of a 30:70 water/acetonitrile (v/v) solution were then added to each HS tube, and 250 µL to each DS tube and briefly vortexed. Finally, the DS samples were combined with their respective HS counterparts and vortexed again. Finally, dimers derived from butanolysis reactions were chromatographed on a HPLC using a gradient of acetonitrile and water (LC column: Gemini® 3 µm C6-Phenyl 110 Å, 100 x 2 mm, from Phenomenex) and detected on a triple quadrupole mass spectrometer (API4000 QTRAP from Sciex). Samples were quantified by interpolation from the calibration curve (prepared to cover a concentration range from 0.39 to 50 µg/mL for HS and from 1.56 to 100 µg/mL do DS using seven different levels) and reported in mg/mL. Then, HS and DS were normalized to protein concentration and finally reported as mg/g uCr.
4.5.4. LAMP1 Immunostaining
LAMP1 immunostaining was performed as previously reported in Encarnação et al., [63]. Briefly, WT and MPS II-derived SHEDs were seeded (approximately 25000 cells/well) in μ-slide 8-well chambers (Nunc, Roskilde, Denmark), fixed with 4% PFA/PBS for 30 min, quenched with 0.05 M NH4Cl, permeabilized in ice-cold methanol for 10 min, and blocked with 5% BSA (Sigma-Aldrich, St. Louis, Missouri, USA), according to standard procedures. Coverslips were washed three times in PBS and once in water, mounted (FluoroshieldTM with DAPI, Sigma-Aldrich, St. Louis, Missouri, USA) and examined by fluorescence microscopy (Leica TCS-SPE confocal microscope; Leica, Wetzlar, Germany). Spectral detection adjusted for the emission of DAPI and Alexa 488fluorochromes using the 405-, 488-, and 546-laser lines, respectively. Digital images were analysed using ImageJ version 2.0.0. Details on the antibodies used and their respective dilutions are listed in Table 3.
4.6. Multi-Lineage Differentiation Protocol
The multi-lineage differentiation capacity of the established SHED cell lines was assessed with a straightforward protocol to promote SHEDs differentiation into the three germ layers.
Briefly, control and patient-derived SHEDs (px5-px7) were plated onto 24-well plates (8000 viable cells/cm2) and cultured in standard media until reaching a confluency of 70–80% of culture surface. The Human Pluripotent Stem Cell Functional Identification Kit (R&D Systems, Minneapolis, Minnesota, USA) was then used to differentiate into endoderm and ectoderm, according to the manufacturer’s instructions. For mesoderm differentiation, however, a slight alternation to the original protocol was performed, by adding the CHIR99021 supplement (Stemcell Technologies, Vancouver, Canada), a WNT pathway activator, to the Differentiation Base Media Supplement (50X), which is part of the kit Stem Cell Functional Identification, as previously reported [44]. For the immunocytochemistry, antibodies against OTX2, BRACHYURY, and SOX17 were used as markers for ectoderm, mesoderm and endoderm, respectively. Briefly, seeded SHEDs were fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, Missouri, USA) for 10 min, incubated with PBST with 1% BSA (Sigma-Aldrich, St. Louis, Missouri, USA) for 30 min and stained by standard immunofluorescence procedures. Cells were analysed on DM400 M fluorescence microscope (Leica, Wetzlar, Germany). Antibodies are listed in Table 3.
4.7. Data analysis and Statistics
Statistical analysis was performed using GraphPad PrismVR (version 9, GraphPad Software, La Jolla, California, USA). Results were presented as mean ± standard error of the mean (SEM). Comparisons between groups were performed by one-way analysis of variance followed by Tukey’s multiple comparisons test. Differences were considered statistically significant when P ≤ 0.05. Significant results between groups were presented using the symbol (*). Significance results are also indicated according to P values with one, two, three or four of the symbols (*) corresponding to 0.01< P ≤ 0.05, 0.001 < P ≤ 0.01, 0.0001 < P ≤ 0.001 and P ≤ 0.0001, respectively.
5. Conclusions
Here we present our results on the establishment and characterization of two MPS II patient-derived stem cell lines from deciduous teeth. Quite remarkably, hallmark LSD features namely the presence of storage (GAGs, in this particular case) and of an abnormal LAMP1 staining pattern, are already quite evident in these cells. This is particularly relevant since those exact same features were not reported by other groups in MPS II-derived iPSCs [19]. Instead, iPSCs seem to need further differentiation in order to show the storage phenotype, which usually becomes evident only in NPCs or NSCs [19]. Altogether, this is a completely new observation in the field and we believe it holds potential to set a new trend for investigating not only the subcellular pathology of virtually any LSD, but also any gene expression changes that may occur in these pathologies. It may also allow for reliable genotype-phenotype correlations, as soon as significant samples from the same disorder are collected and their derived cell lines established.
Overall, this method relies on a non-invasive, cost-effective approach that can be set as a routine in any lab with standard cell culture conditions. By incredibly diminishing the costs associated with the establishment of a pluripotent cell line (which would, otherwise rely in expensive, laborious and time-consuming iPSC generation and characterization protocols), this protocol may allow a significant number of laboratories to establish their first LSD-derived stem cell lines. The fact that some of the major LSD pathological hallmarks are already evident in the SHED state, even before any differentiation protocol is attempted, further validates the modelling potential of these dental-derived mesenchymal stem cell cultures for this challenging group of disorders.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Absolute ΔCt values obtained by qRT-PCR of several markers, including CD105, CD73, CD90 and CD166 (MSCs markers), Sox-2, OCT-3/4, and Nanog (pluripotency markers), CD117, CD34, MHC I and MHCII, in SHEDs from patients and controls, and also iPSCs derived from Fabry fibroblasts. Differences were calculated using GAPDH and β-actin as housekeeping genes. Figure S1. Enzyme activity measurements for additional lysosomal enzymes not directly involved in MPS II pathology are not significantly different level between control and MPS SHEDs. (a) β-galactosidase (GLB1, E.C. 3.2.1.23, the enzyme deficient in either GM1-gangliosidosis or MPS IVB); (b) α-galactosidase (GLA, E.C. 3.2.1.22, the enzyme deficient in Fabry Disease); (c) β-glucuronidase (GUSB, E.C. 3.2.1.31, the enzyme deficient in MPS VII) and (d) total hexosaminidase [includes hexosaminidase A (HEXA, E.C. 3.2.1.52, the enzyme deficient in GM2-gangliosidosis)].
Author Contributions
Conceptualization, M.F.C. and S.A.; cell culture: S.C., J.I.S, D.R. and M.F.C.; molecular analysis: S.C. and MF.C.; qRT-PCR: S.C., J.I.S, L.M. (Luciana Moreira) and A.J.D; immunofluorescence/immunostaining: S.C., A.J.D., H.D. and M.E..; LC-MS/MS: M.F.C, J.I.S and H.R.; enzyme activities: S.C., M.F.C and. P.G.; statistical analysis, S.C., M.F.C., J.I.S., L.M., P.G. and H.R.; clinical characterization: L.D., S.F., C.S., E.M.; resources, M.F.C., S.A., M.E. and O.A.; data curation, S.C., J.I.S., M.E., L.M.; writing—original draft preparation, S.C. and M.F.C.; writing—review and editing, S.A., L.M. (Luciana Moreira) ; critical review: O.A., L.M. (Liliana Matos), M.J.P.; figures and tables: S.C. and M.C.F.; supervision, L.P.A., S.A..; project administration, M.F.C. and P.G..; funding acquisition, M.F.C., M.E., O.A and S.A.. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by national funds through FCT – Fundação para a Ciência e a Tecnologia, I.P., within the scope of the project EXPL/BTM-SAL/0659/2021 (DOI 10.54499/EXPL/BTM-SAL/0659/2021). This work was also partially supported by Sanfilippo Children's Foundation (2019DGH1656/ SCF2019I&D) and the Portuguese Society for Metabolic Disorders, SPDM (Bolsa SPDM de apoio à investigação Dr. Aguinaldo Cabral 2018; 2019DGH1629/ SPDM2018I&D). Some specific tasks also benefited from additional funds: LAMP1 staining was performed according to the method implemented under the scope of the ongoing FCT project EXPL/BTM-TEC/1477/2021, and using its associated reagents; similarly, the differentiation into the three germ layers protocol was performed using a kit acquired and implemented under the scope of the another FCT project, previously conducted in the lab: PTDC/BIM-MEC/4762/2014.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the National Institute of Health Dr. Ricardo Jorge, INSA, I.P. (date of approval: 28/06/2020).
Informed Consent Statement
Written informed consent had been obtained from the subjects who provided their samples for SHED cell lines’ establishment and, whenever necessary/adequate, subsequent differentiation into disease-relevant cell lines.
Data Availability Statement
Additional files are made available online along with the manuscript.
Acknowledgments
The authors wish to thank the many different people, who greatly contributed to the overall success of this project: 1) the patients and their families, for agreeing to participate in this study and gently providing us their baby teeth right after they fell; 2) the numerous families, who have donated control (WT) teeth for the extraction of stem cells, aiming to help us further understand a rare genetic disorder, from which most of them had never heard before; 3) everyone who contributed to disseminate this project and spread our ‘call for volunteers’ appeal, from patient associations (namely Associação Sanfilippo Portugal; Associação Portuguesa de Doenças do Lisossoma, APL; and RD-Portugal) to science communicators, journalists and opinion-makers (namely Cristina Nobre Soares, Luís Aguiar-Conraria, Ana Stilwell, Isabel Stilwell, Vera Gouveia Barros and Marco Neves) and, finally, 4) the Portuguese company Little Drops of Water, who gently offered each one of our volunteers a tiny hand painted resin figure of the Tooth Fairy.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of Mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Dʹavanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Hunter, C. A Rare Disease in Two Brothers. Proc. R. Soc. Med. 1917, 10, 104–116. [Google Scholar] [CrossRef]
- Bach, G.; Eisenberg, F.; Cantz, M.; Neufeld, E.F. The Defect in the Hunter Syndrome: Deficiency of Sulfoiduronate Sulfatase. Proc. Natl. Acad. Sci. U. S. A. 1973, 70, 2134–2138. [Google Scholar] [CrossRef]
- Wilson, P.J.; Meaney, C.A.; Hopwood, J.J.; Morris, C.P. Sequence of the Human Iduronate 2-Sulfatase (IDS) Gene. Genomics 1993, 17, 773–775. [Google Scholar] [CrossRef]
- Wilson, P.J.; Morris, C.P.; Anson, D.S.; Occhiodoro, T.; Bielicki, J.; Clements, P.R.; Hopwood, J.J. Hunter Syndrome: Isolation of an Iduronate-2-Sulfatase CDNA Clone and Analysis of Patient DNA. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 8531–8535. [Google Scholar] [CrossRef]
- Froissart, R.; Da Silva, I.M.; Maire, I. Mucopolysaccharidosis Type II: An Update on Mutation Spectrum. Acta Paediatr. Int. J. Paediatr. 2007, 96, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Horgan, C.; Jones, S.A.; Bigger, B.W.; Wynn, R. Current and Future Treatment of Mucopolysaccharidosis (MPS) Type II: Is Brain-Targeted Stem Cell Gene Therapy the Solution for This Devastating Disorder? Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The Natural Course and the Impact of Therapies of Cardiac Involvement in the Mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Parini, R.; Harmatz, P.; Giugliani, R.; Fang, J.; Mendelsohn, N.J. The Effect of Idursulfase on Growth in Patients with Hunter Syndrome: Data from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2013, 109, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van Der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis Type II (Hunter Syndrome): A Clinical Review and Recommendations for Treatment in the Era of Enzyme Replacement Therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef]
- Tomanin, R.; Zanetti, A.; D’Avanzo, F.; Rampazzo, A.; Gasparotto, N.; Parini, R.; Pascarella, A.; Concolino, D.; Procopio, E.; Fiumara, A.; et al. Clinical Efficacy of Enzyme Replacement Therapy in Paediatric Hunter Patients, an Independent Study of 3.5 Years. Orphanet J. Rare Dis. 2014, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Moreira, G.A.; Kyosen, S.O.; Patti, C.L.; Martins, A.M.; Tufik, S. Prevalence of Obstructive Sleep Apnea in Patients with Mucopolysaccharidosis Types I, II, and VI in a Reference Center. Sleep Breath. 2014, 18, 791–797. [Google Scholar] [CrossRef]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Muñoz, V.; Muenzer, J. Recognition and Diagnosis of Mucopolysaccharidosis II (Hunter Syndrome). Pediatrics 2008, 121. [Google Scholar] [CrossRef]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lampe, C.; Bosserhoff, A.K.; Burton, B.K.; Giugliani, R.; de Souza, C.F.; Bittar, C.; Muschol, N.; Olson, R.; Mendelsohn, N.J. Long-Term Experience with Enzyme Replacement Therapy (ERT) in MPS II Patients with a Severe Phenotype: An International Case Series. J. Inherit. Metab. Dis. 2014, 37, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Botha, J.; Harmatz, P.; Giugliani, R.; Kampmann, C.; Burton, B.K. Evaluation of the Long-Term Treatment Effects of Intravenous Idursulfase in Patients with Mucopolysaccharidosis II (MPS II) Using Statistical Modeling: Data from the Hunter Outcome Survey (HOS). Orphanet J. Rare Dis. 2021, 16, 1–14. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21, 1–30. [Google Scholar] [CrossRef]
- Rybová, J.; Ledvinová, J.; Sikora, J.; Kuchař, L.; Dobrovolný, R. Neural Cells Generated from Human Induced Pluripotent Stem Cells as a Model of CNS Involvement in Mucopolysaccharidosis Type II. J. Inherit. Metab. Dis. 2018, 41, 221–229. [Google Scholar] [CrossRef]
- Kobolák, J.; Molnár, K.; Varga, E.; Bock, I.; Jezsó, B.; Téglási, A.; Zhou, S.; Lo Giudice, M.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the Neuropathology of Lysosomal Storage Disorders through Disease-Specific Human Induced Pluripotent Stem Cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef]
- Hong, J.; Cheng, Y.S.; Yang, S.; Swaroop, M.; Xu, M.; Beers, J.; Zou, J.; Huang, W.; Marugan, J.J.; Cai, X.; et al. IPS-Derived Neural Stem Cells for Disease Modeling and Evaluation of Therapeutics for Mucopolysaccharidosis Type II. Exp. Cell Res. 2022, 412, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Santos, J.I.; Moreira, L.; Gonçalves, M.; David, H.; Matos, L.; Encarnação, M.; Alves, S.; Coutinho, M.F. Neurological Disease Modeling Using Pluripotent and Multipotent Stem Cells: A Key Step towards Understanding and Treating Mucopolysaccharidoses. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Miura, M.; Gronthos, S.; Zhao, M.; Lu, B.; Fisher, L.W.; Robey, P.G.; Shi, S. SHED: Stem Cells from Human Exfoliated Deciduous Teeth. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5807–5812. [Google Scholar] [CrossRef]
- Kerkis, I.; Kerkis, A.; Dozortsev, D.; Stukart-Parsons, G.C.; Gomes Massironi, S.M.; Pereira, L. V.; Caplan, A.I.; Cerruti, H.F. Isolation and Characterization of a Population of Immature Dental Pulp Stem Cells Expressing OCT-4 and Other Embryonic Stem Cell Markers. Cells Tissues Organs 2006, 184, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.T.J.; Gronthos, S.; Shi, S. Critical Reviews in Oral Biology & Medicine: Mesenchymal Stem Cells Derived from Dental Tissues vs. Those from Other Sources: Their Biology and Role in Regenerative Medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [CrossRef]
- Pivoriuunas, A.; Surovas, A.; Borutinskaite, V.; Matuzevicčius, D.; Treigyte, G.; Savickiene, J.; Tunaitis, V.; Aldonyte, R.; Jarmalavicčiuute, A.; Suriakaite, K.; et al. Proteomic Analysis of Stromal Cells Derived from the Dental Pulp of Human Exfoliated Deciduous Teeth. Stem Cells Dev. 2010, 19, 1081–1093. [Google Scholar] [CrossRef]
- Rodríguez-Lozano, F.J.; Bueno, C.; Insausti, C.L.; Meseguer, L.; Ramírez, M.C.; Blanquer, M.; Marín, N.; Martínez, S.; Moraleda, J.M. Mesenchymal Stem Cells Derived from Dental Tissues. Int. Endod. J. 2011, 44, 800–806. [Google Scholar] [CrossRef]
- Yoshida, S.; Tomokiyo, A.; Hasegawa, D.; Hamano, S.; Sugii, H.; Maeda, H. Insight into the Role of Dental Pulp Stem Cells in Regenerative Therapy. Biology (Basel). 2020, 9, 1–24. [Google Scholar] [CrossRef]
- Taghipour, Z.; Karbalaie, K.; Kiani, A.; Niapour, A.; Bahramian, H.; Nasr-Esfahani, M.H.; Baharvand, H. Transplantation of Undifferentiated and Induced Human Exfoliated Deciduous Teeth-Derived Stem Cells Promote Functional Recovery of Rat Spinal Cord Contusion Injury Model. Stem Cells Dev. 2012, 21, 1794–1802. [Google Scholar] [CrossRef]
- Nicola, F.; do, C.; Marques, M.R.; Odorcyk, F.; Arcego, D.M.; Petenuzzo, L.; Aristimunha, D.; Vizuete, A.; Sanches, E.F.; Pereira, D.P.; Maurmann, N.; et al. Neuroprotector Effect of Stem Cells from Human Exfoliated Deciduous Teeth Transplanted after Traumatic Spinal Cord Injury Involves Inhibition of Early Neuronal Apoptosis. Brain Res. 2017, 1663, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Nishii, T.; Osuka, K.; Nishimura, Y.; Ohmichi, Y.; Ohmichi, M.; Suzuki, C.; Nagashima, Y.; Oyama, T.; Abe, T.; Kato, H.; et al. Protective Mechanism of Stem Cells from Human Exfoliated Deciduous Teeth in Treating Spinal Cord Injury. J. Neurotrauma 2024, 41. [Google Scholar] [CrossRef]
- Yamagata, M.; Yamamoto, A.; Kako, E.; Kaneko, N.; Matsubara, K.; Sakai, K.; Sawamoto, K.; Ueda, M. Human Dental Pulp-Derived Stem Cells Protect against Hypoxic-Ischemic Brain Injury in Neonatal Mice. Stroke 2013, 44, 551–554. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Liu, S.; Liu, D.; Chen, C.; Xu, X.; Chen, X.; Shi, S. Transplantation of SHED Prevents Bone Loss in the Early Phase of Ovariectomy-Induced Osteoporosis. J. Dent. Res. 2014, 93, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Yamaza, T.; Alatas, F.S.; Yuniartha, R.; Yamaza, H.; Fujiyoshi, J.K.; Yanagi, Y.; Yoshimaru, K.; Hayashida, M.; Matsuura, T.; Aijima, R.; et al. In Vivo Hepatogenic Capacity and Therapeutic Potential of Stem Cells from Human Exfoliated Deciduous Teeth in Liver Fibrosis in Mice. Stem Cell Res. Ther. 2015, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, S.; Yamaza, T. A New Target of Dental Pulp-Derived Stem Cell-Based Therapy on Recipient Bone Marrow Niche in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Katahira, Y.; Murakami, F.; Inoue, S.; Miyakawa, S.; Sakamoto, E.; Furusaka, Y.; Watanabe, A.; Sekine, A.; Kuroda, M.; Hasegawa, H.; et al. Protective Effects of Conditioned Media of Immortalized Stem Cells from Human Exfoliated Deciduous Teeth on Pressure Ulcer Formation. Front. Immunol. 2023, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Victor, A.K.; Reiter, L.T. Dental Pulp Stem Cells for the Study of Neurogenetic Disorders. Hum. Mol. Genet. 2017, 26, R166–R171. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Mangas, M.; Prata, M.; Ribeiro, G.; Lopes, L.; Ribeiro, H.; Pinto-Basto, J.; Lima Reis, M.; Lacerda, L. Molecular Characterization of Portuguese Patients with Mucopolysaccharidosis Type II Shows Evidence That the IDS Gene Is Prone to Splicing Mutations. J. Inherit. Metab. Dis. 2006, 29, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Lagerstedt, K.; Karsten, S.L.; Carlberg, B.M.; Kleijer, W.J.; Tönnesen, T.; Pettersson, U.; Bondeson, M.L. Double-Strand Breaks May Initiate the Inversion Mutation Causing the Hunter Syndrome. Hum. Mol. Genet. 1997, 6, 627–633. [Google Scholar] [CrossRef]
- GOORHA, S.; REITER, L.T. Culturing and Neuronal Differentiation of Human Dental Pulp Stem Cells. Curr. Protoc. Hum. Genet. 2017, 2017, 21.6.1–21.6.10. [Google Scholar] [CrossRef]
- Pesce, M.; Schöler, H.R. Oct-4: Gatekeeper in the Beginnings of Mammalian Development OCT-4: THE REGULATOR OF TOTIPOTENCY IN THE. Stem Cells 2001, 19, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The Homeoprotein Nanog Is Required for Maintenance of Pluripotency in Mouse Epiblast and ES Cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent Cell Lineages in Early Mouse Development Depend on SOX2 Function. Genes Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.J.; Ribeiro, D.; Santos, R.; Moreira, L.; Bragança, J.; Amaral, O. Induced Pluripotent Stem Cell Line (INSAi002-A) from a Fabry Disease Patient Hemizygote for the Rare p.W287X Mutation. Stem Cell Res. 2020, 45, 101794. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.M.; Sousa, A.C.; Caseiro, A.R.; Pedrosa, S.S.; Pinto, P.O.; Branquinho, M. V.; Amorim, I.; Santos, J.D.; Pereira, T.; Mendonça, C.M.; et al. Dental Pulp Stem Cells and Bonelike® for Bone Regeneration in Ovine Model. Regen. Biomater. 2019, 6, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Grau-vorster, M.; Laitinen, A.; Nystedt, J.; Vives, J. HLA-DR Expression in Clinical-Grade Bone Marrow-Derived Multipotent Mesenchymal Stromal Cells : A Two-Site Study. Stem Cell Res. Ther. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- McNiece, I. Subsets of Mesenchymal Stromal Cells. Cytotherapy 2007, 9, 301–302. [Google Scholar] [CrossRef]
- Chai, Y.; Jiang, X.; Ito, Y.; Bringas, P.; Han, J.; Rowitch, D.H.; Soriano, P.; Mcmahon, A.P.; Sucov, H.M. Fate of the Mammalian Cranial Neural Crest during Tooth and Mandibular Morphogenesis. 2000, 1679, 1671–1679. [Google Scholar] [CrossRef]
- Kashyap, R. SHED - Basic Structure for Stem Cell Research. J. Clin. Diagnostic Res. 2015, 9. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Cooper, A.; Heptinstall, L.E.; Hatton, C.E.; Thornley, M.; Wraith, J.E. Mutation Analysis in 57 Unrelated Patients with MPS II (Hunter’s Disease). Arch. Dis. Child. 1998, 79, 237–241. [Google Scholar] [CrossRef]
- Lualdi, S.; Tappino, B.; Di Duca, M.; Dardis, A.; Anderson, C.J.; Biassoni, R.; Thompson, P.W.; Corsolini, F.; Di Rocco, M.; Bembi, B.; et al. Enigmatic in Vivo Iduronate-2-Sulfatase (IDS) Mutant Transcript Correction to Wild-Type in Hunter Syndrome. Hum. Mutat. 2010, 31, 1261–1285. [Google Scholar] [CrossRef]
- Filocamo, M.; Bonuccelli, G.; Corsolini, F.; Mazzotti, R.; Cusano, R.; Gatti, R. Molecular Analysis of 40 Italian Patients with Mucopolysaccharidosis Type II: New Mutations in the Iduronate-2-Sulfatase (IDS) Gene. Hum. Mutat. 2001, 18, 164–165. [Google Scholar] [CrossRef]
- Forni, G.; Malvagia, S.; Funghini, S.; Scolamiero, E.; Mura, M.; Della Bona, M.; Villanelli, F.; Damiano, R.; la Marca, G. LC-MS/MS Method for Simultaneous Quantification of Heparan Sulfate and Dermatan Sulfate in Urine by Butanolysis Derivatization. Clin. Chim. Acta 2019, 488, 98–103. [Google Scholar] [CrossRef]
- Morimoto, H.; Kida, S.; Yoden, E.; Kinoshita, M.; Tanaka, N.; Yamamoto, R.; Koshimura, Y.; Takagi, H.; Takahashi, K.; Hirato, T.; et al. Clearance of Heparan Sulfate in the Brain Prevents Neurodegeneration and Neurocognitive Impairment in MPS II Mice. Mol. Ther. 2021, 29, 1853–1861. [Google Scholar] [CrossRef]
- Hong, J.; Cheng, Y.S.; Yang, S.; Swaroop, M.; Xu, M.; Beers, J.; Zou, J.; Huang, W.; Marugan, J.J.; Cai, X.; et al. IPS-Derived Neural Stem Cells for Disease Modeling and Evaluation of Therapeutics for Mucopolysaccharidosis Type II. Exp. Cell Res. 2022, 412, 113007. [Google Scholar] [CrossRef]
- de Bode, C.J.; Dogterom, E.J.; Rozeboom, A.V.J.; Langendonk, J.J.; Wolvius, E.B.; van der Ploeg, A.T.; Oussoren, E.; Wagenmakers, M.A.E.M. Orofacial Abnormalities in Mucopolysaccharidosis and Mucolipidosis Type II and III : A Systematic Review. JIMD Rep. 2022, 63, 621–629. [Google Scholar] [CrossRef]
- VOZNYI, Y.N.; KEULEMANS, J.L.M.; VAN DIGGELEN, O.P. A Fluorimetric Enzyme Assay for the Diagnosis of MPS II (Hunter Disease). J. Inherit. Metab. Dis. 2001, 24, 675–680. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.B.; Gaggioli, D.; Casentini, C. Hurler-like Phenotype: Enzymatic Diagnosis in Dried Blood Spots on Filter Paper. Clin. Chem. 2001, 47, 2098–2102. [Google Scholar] [CrossRef]
- Civallero, G.; Michelin, K.; de Mari, J.; Viapiana, M.; Burin, M.; Coelho, J.C.; Giugliani, R. Twelve Different Enzyme Assays on Dried-Blood Filter Paper Samples for Detection of Patients with Selected Inherited Lysosomal Storage Diseases. Clin. Chim. Acta 2006, 372, 98–102. [Google Scholar] [CrossRef]
- Forni, G.; Malvagia, S.; Funghini, S.; Scolamiero, E.; Mura, M.; Bona, M. Della; Villanelli, F.; Damiano, R.; la Marca, G. Data in Support for the Measurement of Heparan Sulfate and Dermatan Sulfate by LC–MS/MS Analysis. Data Br. 2018, 21, 2398–2404. [Google Scholar] [CrossRef]
- Encarnação, M.; Coutinho, M.F.; Cho, S.M.; Cardoso, M.T.; Ribeiro, I.; Chaves, P.; Santos, J.I.; Quelhas, D.; Lacerda, L.; Leão Teles, E.; et al. NPC1 Silent Variant Induces Skipping of Exon 11 (p.V562V) and Unfolded Protein Response Was Found in a Specific Niemann-Pick Type C Patient. Mol. Genet. Genomic Med. 2020, 8, 1–13. [Google Scholar] [CrossRef]
Figure 1.
Optical microscope images (4×) of an MPS II-derived SHED primary culture. Undifferentiated SHEDs have a spindle-shape similar to fibroblasts. (a) Fusiform format; (b) Small colonies formation (CFU-F); (c) Sub-confluence. Cells were observed in a Leica DMIL inverted contrasting microscope (Leica Microsystems, Wetzlar, Germany) with 4× magnification.
Figure 1.
Optical microscope images (4×) of an MPS II-derived SHED primary culture. Undifferentiated SHEDs have a spindle-shape similar to fibroblasts. (a) Fusiform format; (b) Small colonies formation (CFU-F); (c) Sub-confluence. Cells were observed in a Leica DMIL inverted contrasting microscope (Leica Microsystems, Wetzlar, Germany) with 4× magnification.

Figure 2.
Immunostaining of MPSII-derived SHEDs. (a) Confirmation of 3 germ layer differentiation capacity: Mesoderm (top), Endoderm (middle), Ectoderm (down). (b) Confirmation of NPC stage of SHEDs: Nestin and Sox-1 (top line); Pax-6 and Sox-2 (down line). Scale bar: 50 µm. Images were acquired in a DM400 M fluorescence microscope (Leica, Wetzlar, Germany).
Figure 2.
Immunostaining of MPSII-derived SHEDs. (a) Confirmation of 3 germ layer differentiation capacity: Mesoderm (top), Endoderm (middle), Ectoderm (down). (b) Confirmation of NPC stage of SHEDs: Nestin and Sox-1 (top line); Pax-6 and Sox-2 (down line). Scale bar: 50 µm. Images were acquired in a DM400 M fluorescence microscope (Leica, Wetzlar, Germany).
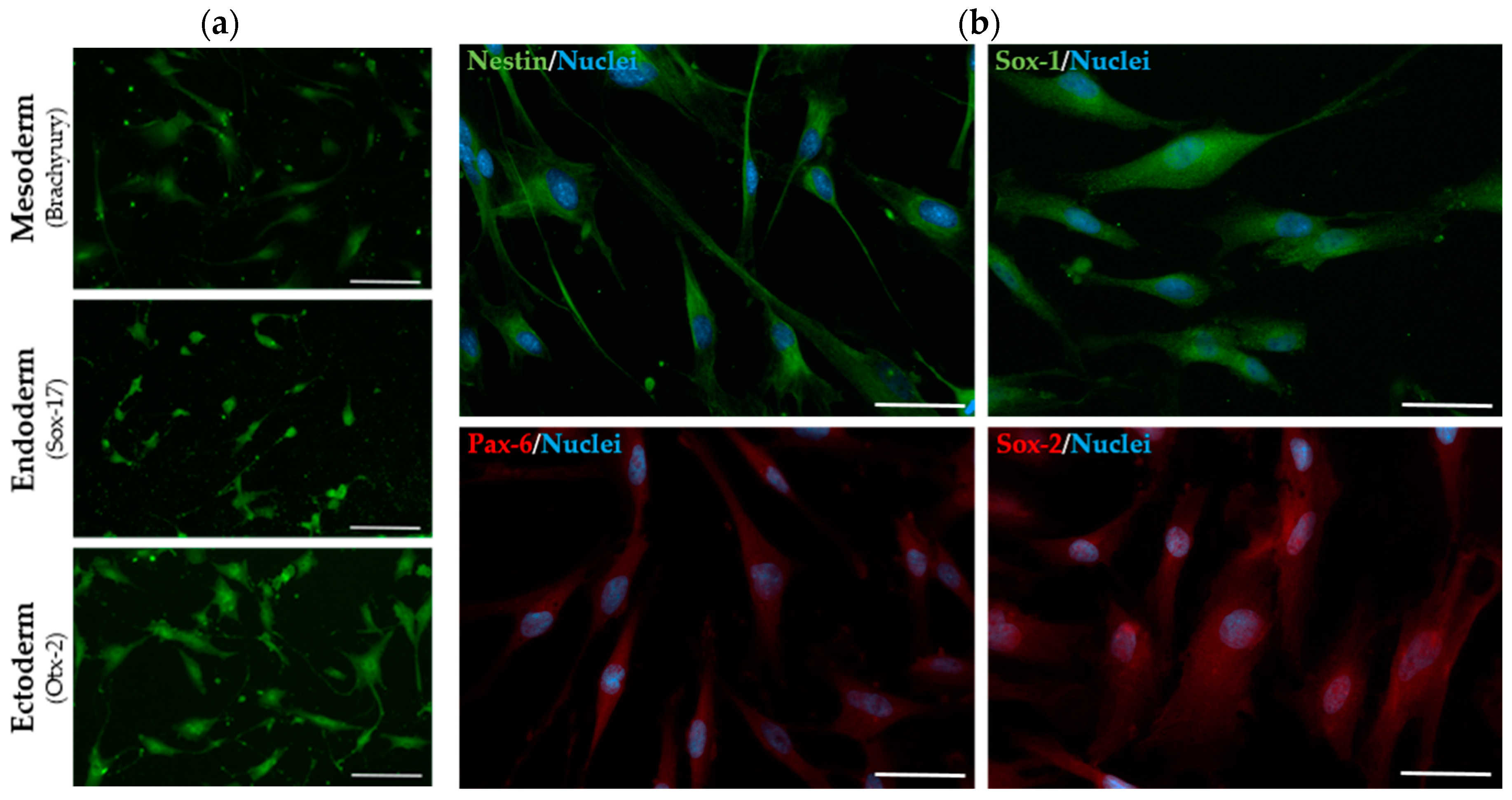
Figure 3.
Biochemical profile of SHEDs. (a) IDS enzyme activity. Data are presented as mean ± SEM (n = 3) *p < 0.05. (b) HS and DS levels of control- and MPS II-derived SHEDs. (c) Representative confocal images of control (left) and MPS II- derived SHEDs (middle and right) immunostained for LAMP1. Scale bar: 25 µm. The outlined regions highlight the different lysosomal positioning in controls vs patient-derived SHEDs, with a more pronounced LAMP1 staining in the perinuclear region of MPS II SHEDs. Images were acquired in a TCS-SPE confocal microscope (Leica, Wetzlar, Germany).
Figure 3.
Biochemical profile of SHEDs. (a) IDS enzyme activity. Data are presented as mean ± SEM (n = 3) *p < 0.05. (b) HS and DS levels of control- and MPS II-derived SHEDs. (c) Representative confocal images of control (left) and MPS II- derived SHEDs (middle and right) immunostained for LAMP1. Scale bar: 25 µm. The outlined regions highlight the different lysosomal positioning in controls vs patient-derived SHEDs, with a more pronounced LAMP1 staining in the perinuclear region of MPS II SHEDs. Images were acquired in a TCS-SPE confocal microscope (Leica, Wetzlar, Germany).
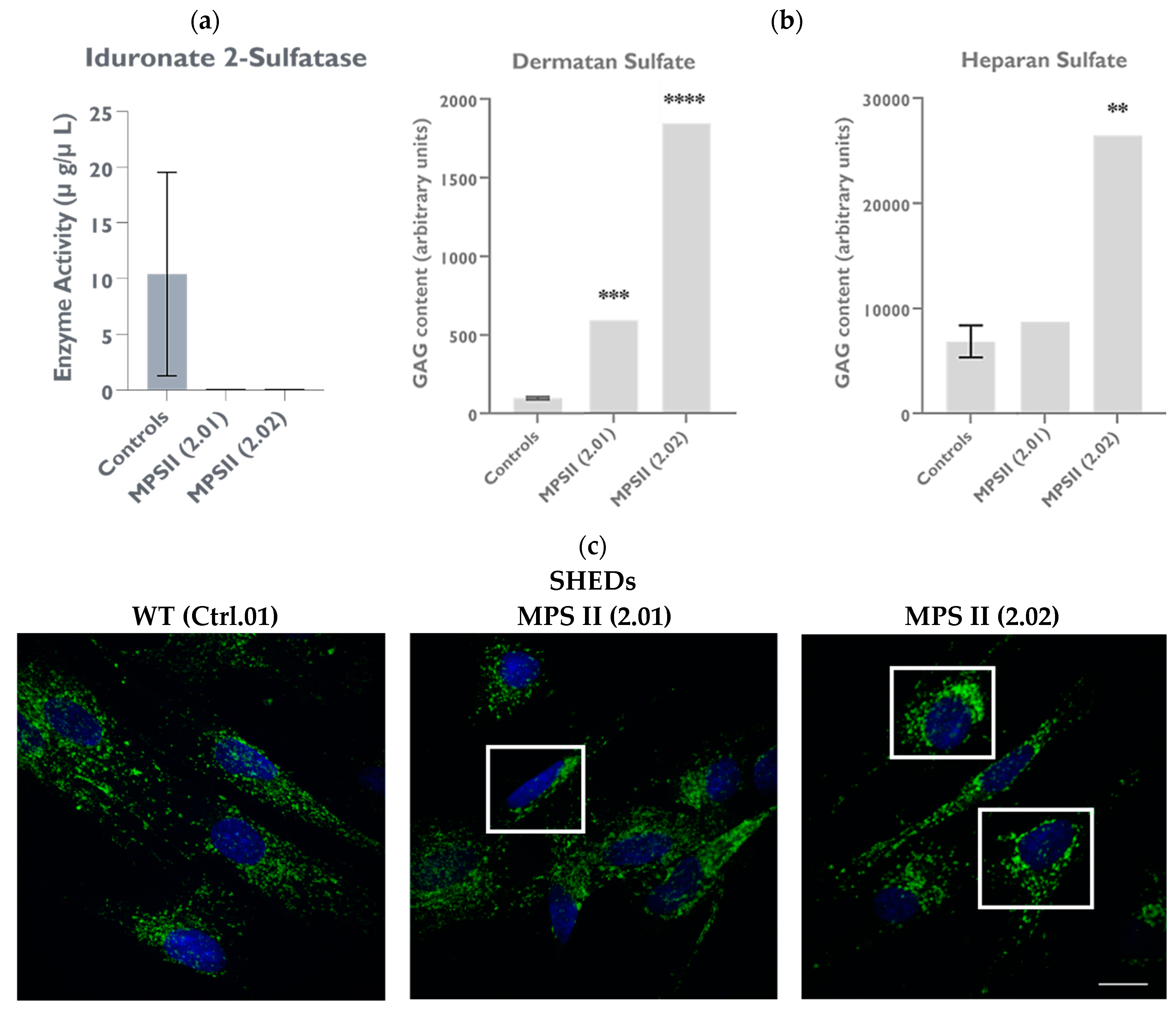
Figure 4.
Schematic drawing illustrating the different sources of dental mesenchymal stem cells (DMSCs) in the oral cavity. Abbreviations: GMSCs, gingiva-derived MSCs; DFSCs, dental follicle stem cells; SHED, stem cells from exfoliated deciduous teeth; SCAPs, stem cells from the apical papilla; DPSCs, dental pulp stem cells; PDLSCs, periodontal ligament stem cells (reproduced from [22]).
Figure 4.
Schematic drawing illustrating the different sources of dental mesenchymal stem cells (DMSCs) in the oral cavity. Abbreviations: GMSCs, gingiva-derived MSCs; DFSCs, dental follicle stem cells; SHED, stem cells from exfoliated deciduous teeth; SCAPs, stem cells from the apical papilla; DPSCs, dental pulp stem cells; PDLSCs, periodontal ligament stem cells (reproduced from [22]).
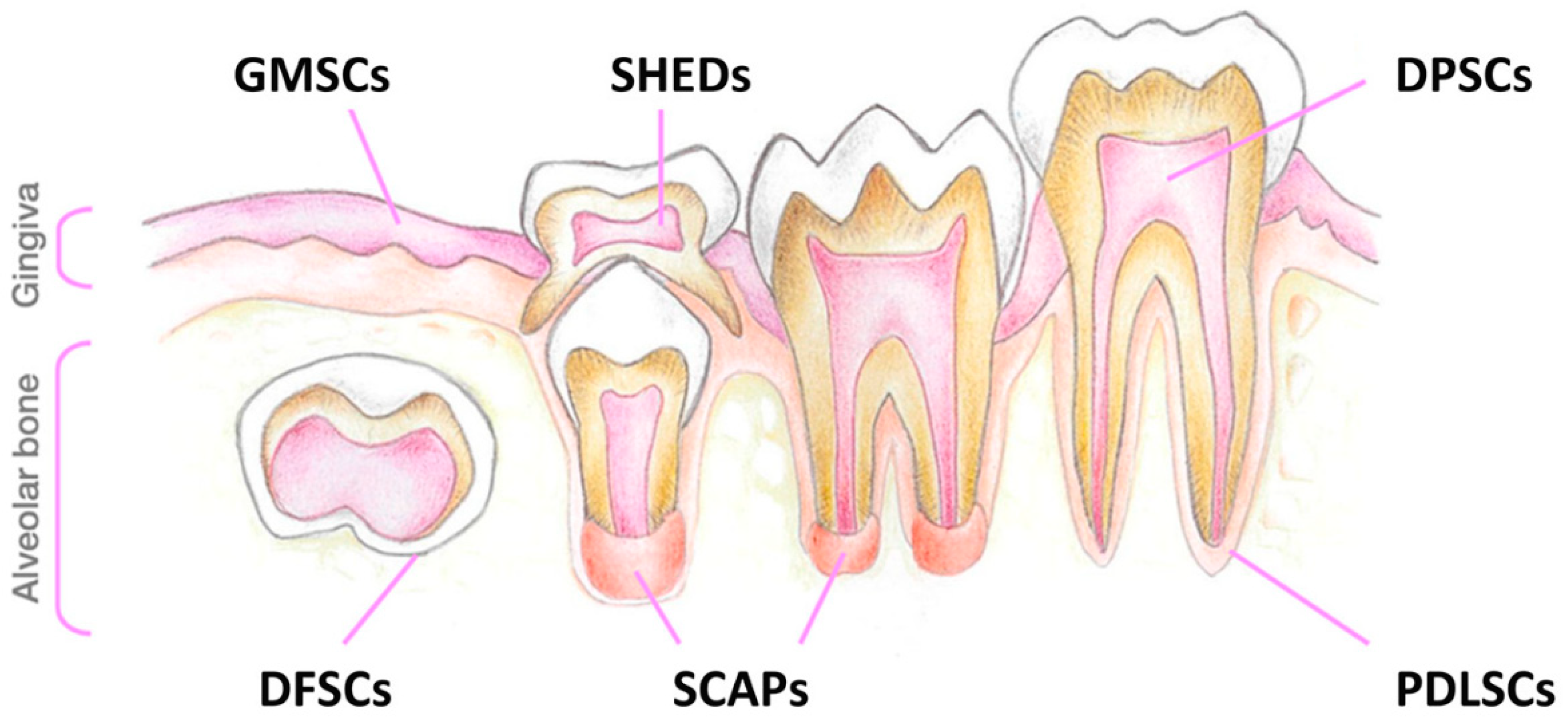
Table 1.
Relative expression levels of several markers, including CD105, CD73, CD90 and CD166 (MSCs markers), Sox-2, OCT-3/4, and Nanog (pluripotency markers), CD117, CD34, MHC I and MHCII, in SHEDs from patients and controls, and also iPSCs derived from Fabry fibroblasts. Differences were based in qRT-PCR results and calculated using the standard ΔCt methods, with GAPDH and β-actin as housekeeping genes.
Table 1.
Relative expression levels of several markers, including CD105, CD73, CD90 and CD166 (MSCs markers), Sox-2, OCT-3/4, and Nanog (pluripotency markers), CD117, CD34, MHC I and MHCII, in SHEDs from patients and controls, and also iPSCs derived from Fabry fibroblasts. Differences were based in qRT-PCR results and calculated using the standard ΔCt methods, with GAPDH and β-actin as housekeeping genes.
| Marker | SHEDs (2.01) | SHEDs (2.02) | iPSCs (INSAi002-A) |
|---|---|---|---|
| CD105 | *** | *** | *** |
| CD73 | *** | *** | *** |
| CD90 | *** | *** | *** |
| CD166 | *** | *** | |
| MHC I | *** | *** | |
| Sox-2 | * | * | ** |
| Oct-3/4 | * | * | *** |
| Nanog | * | * | ** |
| CD117 | ** | ** | |
| CD34 | * | * | * |
| MHC II | * | ** | ** |
***strong expression; **moderate expression; * weak expression.
Table 2.
Summary of the most relevant clinical data from each MPS II patient, including age of diagnosis, symptoms and age of starting treatment.
Table 2.
Summary of the most relevant clinical data from each MPS II patient, including age of diagnosis, symptoms and age of starting treatment.
| Case | Age at Diagnosis (Years) | Symptoms | Age of Starting Treatment (Years) |
|---|---|---|---|
| 2.01 | 3 | Coarse facies, Stiff Joints, etc. Post-natal macroglossia Mild psychomotor development retardation; Interventricular communication (IVC) and patent ductus arteriosus (PDA)1; Moderate aortic insufficiency and left ventricular hypertrophy; Hydrocele; Chronic nasal obstruction without recurrent otitis or hearing deficit |
|
| 2.02 | 2 | Inguinal Hernias; Claw Hands; Low stature; Hypertrichosis; Hepatomegaly; Cardiac involvement. |
4 |
1 Solved by now.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



