
Submitted:
29 February 2024
Posted:
01 March 2024
You are already at the latest version
Abstract
The development of saltiness enhancement peptides is important in reducing salt intake and promoting dietary health. Our study aimed to analyze and identify straw mushroom enzymatic peptides with salty taste enhancing effects. The taste-related peptides within the straw mush-room extract were isolated via ultrafiltration and identified using UPLC-Q-TOF-MS/MS. Sub-sequent, E-tongue and sensory analyses unveiled that the ultrafiltration fraction (500-2000 Da) of straw mushroom peptides exhibited a saltiness enhancement effect. The UPLC-Q-TOF-MS/MS analysis identified 220 peptides within the ultrafiltration fractions (500-2000 Da), which were then assessed for their interaction with the T1R1/T1R3 receptor. The investigation underscored the significant involvement of Asp223, Gln243, Leu232, Asp251, and Pro254 in the binding of peptides from triple enzymatically hydrolyzed straw mushrooms to T1R1/T1R3. Based on the binding energy and active site analysis, three peptides were selected for synthesis: DFNALPFK (-9.2 kcal/mol), YNEDNGIVK (-8.8 kcal/mol), and VPGGQEIKDR (-8.9 kcal/mol). Notably, 3.2 mmol of VPGGQEIKDR enhanced a 0.05% NaCl solution to the saltiness level of a 0.15% NaCl solution. Also, 0.8 mmol of YNEDNGIVK elevated a 0.05% NaCl solution to the saltiness of a 0.1% NaCl solution.
Keywords:
straw mushroom
; peptides from triple enzymatically hydrolyzed straw mushroom proteins
; molecular docking
; UPLC-Q-TOF-MS/MS
; T1R1/T1R3
1. Introduction
T Sodium Chloride (NaCl) is recognized as a nutrient in the human diet and is a widely favored flavor enhancer, while also providing an important source of the essential human nutrient, sodium [1,2]. Despite its role in vital physiological processes, excessive dietary sodium intake is predominantly associated with elevated blood pressure and hypertension, elevating the risks of gastric cancer, cardiovascular disease, and chronic kidney disease[3,4]. Consequently, the imperative action of reducing dietary salt intake is evident. Currently, the most effective method for salt reduction involves the utilization of saltiness peptides, or saltiness enhancement peptides, which is an urgently needed salt reduction strategy in both theoretical research and practical applications[5]. Saltiness enhancement peptides are characterized by their lack of inherent saltiness; however, in conjunction with sodium chloride, they significantly enhance the perception of saltiness in humans. This characteristic allows food products to maintain or even intensify their saltiness despite a reduction in sodium content when using saltiness enhancement peptides [6,7,8]. Furthermore, the composition of peptides, consisting of linked amino acids, enables them to supply the body with essential amino acids. Moreover, in comparison to other salts, saltiness enhancement peptides not only reduce irritation but also demonstrate a more pronounced saltiness enhancement effect [9].
In recent years, numerous studies have highlighted the saltiness enhancement properties of various food-derived peptides, such as saltiness enhancement peptides, umami peptides, Maillard-reacted peptides, and γ-glutamyl peptides [8]. Moore et al. discovered that various taste-modulating pyroglutamyl dipeptides, extracted from mushrooms, (e.g., pyroglutamylcysteine, pyroglutamylvaline, pyroglutamylaspartate, pyroglutamic acid, and pyroglutamylproline), exhibited notable saltiness enhancement effects at a concentration of 143 μmol/L in a mushroom broth model [10]. Yu et al. reported that grass carp skin collagen, subjected to enzymatic ultrafiltration followed by a Maillard reaction with glucose, yielded Maillard-reacted peptides with saltiness-enhancing properties[11]. Additionally, EDEGEQPRPF, a taste peptide isolated from commercial plain fermented soybean curd, was found to enhance saltiness perception[6]. These findings underscore the feasibility of extracting saltiness enhancement peptides from both plant and animal sources, thereby providing a theoretical foundation for the extraction of such peptides from straw mushrooms.
To investigate the mechanism underlying peptide-induced saltiness enhancement, it is essential to begin with an understanding of how humans perceive saltiness. Salt taste perception is triggered when sodium and chloride ions are present in the oral cavity[12]. Prior investigations have revealed that human saltiness receptors include epithelial sodium channels (ENaCs) and transient receptor potential vanilloid 1 (TRPV1)[11,13]. The salt taste transduction pathway sensitive to amiloride is associated with ENaCs, whereas the amiloride-insensitive effect is mediated by TRPV1[14,15]. Fu Yu et al. conducted molecular docking studies of collagen glycopeptides, produced through transglutaminase-induced glycosylation, with saltiness receptor proteins ENaC and TRPV1[5]. They observed that collagen glycopeptides had the capacity to enhance saltiness perception. However, these findings are not universally applicable since some individuals have salt taste receptors insensitive to amiloride, resulting in a chloride-dominated, amiloride-insensitive salty response [12]. Additionally, it is noteworthy that the salt receptor TRPV1 responds not only to sodium ions (Na+) but also to potassium ions (K+) and ammonium ions (NH4+) [16]. Consequently, there are inherent limitations in exploring the mechanism of saltiness enhancement when using either ENaC or TRPV1 receptors for molecular docking with flavor-presenting peptides.
This study employed umami taste receptors to initially screen taste-presenting peptides for their potential to enhance saltiness. Subsequently, the identified saltiness enhancement peptides were subjected to artificial sensory analysis for validation. To substantiate the effectiveness of molecular docking experiments umami taste receptors (T1R1/T1R3), extensive research has demonstrated the significant role of umami substances in saltiness enhancement. For instance, Xie et al. highlighted the impact of umami on enhancing saltiness at various concentrations[17]. Notably, ingredients such as hydrolyzed vegetable protein (HVP), yeast extracts, and specific amino acids like arginine, lysine, and taurine are recognized for increasing saltiness perception while concurrently reducing sodium chloride (NaCl) dependency [18]. Beyond these established flavor enhancers, umami peptides have been discerned as key contributors to saltiness enhancement. Xie et al. utilized molecular docking techniques to screen umami peptides from Ruditapes philippinarum and ham, identifying peptides such as KEMQKN, NGKET, RGEPNND, AHSVRFY, LSERYP, NRTF, TYLPVH, EV, AGAGTP, and GPAGAGPR for their ability to enhance saltiness[17]. Similarly, Shan et al. isolated taste peptides from yeast extracts and conducted molecular docking with the umami taste receptor proteins T1R1/T1R3, successfully identifying peptides that possess saltiness enhancement properties[18]. These discoveries collectively underscore the integral role of umami compounds in augmenting saltiness perception and suggest the potential of molecular docking with umami receptors as a viable technique for screening saltiness enhancement peptides.
The goal of the present investigation was to find out in which molecular weight range the straw mushroom enhancing peptides are mainly found, and to analyze and verify their saltiness enhancement effect. Subsequent, we isolated and identified peptides from Straw mushroom through sensory-guide analysis, combined with UPLC-Q-TOF-MS/MS. The identified taste peptides were then molecularly docked with the umami receptors T1R1 / T1R3 and analyzed for their mechanism of action. This study not only develops the saltiness enhancement possibility of straw mushroom peptides, but also provides a scientific basis for further development of saltiness enhancement peptides’ products.
2. Materials and Methods
2.1. Materials and Chemicals
T Dried straw mushrooms (bought from Qingyuan High Mountain Agricultural Products Whole Sale Department). Food-grade salt (Deep Well Rock Salt) was purchased from China National Salt Industry Group Co., Ltd (Beijing, China). Pectinase、cellulase、papain (bought from Yuan Ye Biological Ltd.). Purified water (purchased from Watsons). Three peptides DFNALPFX, YNEDNGIVK, VPGGQEIKDR were purchased from Hangzhou ALL PEPTIDE Biology Co., Ltd.
2.2. Extraction of Taste Peptides from Straw Mushrooms
The flavor peptide in straw mushrooms were extracted through step-by-step enzymatic hydrolysis. Firstly, the straw mushrooms were crushed and sieved. An appropriate amount of deionized water was added and the two were mixed, with a material-liquid ratio of 1:20. Then, the enzyme was incubated at 90 ℃ for 5 mins and allowed to cool to room temperature. The pH was adjusted to 3.5 and pectinase 0.5% was add for enzymatically hydrolysis at 50 °C for 1hour. Next, the pH was adjusted to 4.5 and cellulase 0.5% was added for further enzymatic hydrolysis at 50 °C for 1hour. Next, the pH was adjusted to 6.5 and 0.4% papain was added, and continued enzymatic hydrolysis at 50 °C for 2 hours. After the enzymatic hydrolysis was completed, the enzyme was inactivated at 90 °C for 5 mins. Once the samples reached the room temperature, the enzymatic hydrolysate was centrifuged at a speed rate of 7000r/min for 15minutes, and the supernatant was then stored for later use. The degree of hydrolysis (DH) of the supernatant after centrifugation was measured and combined with the solids’ content to determine the extent of enzymatic digestion.
The method chosen for determining the degree of hydrolysis is the formaldehyde titration method, which is calculated using the following formula:
where C is concentration of standard solutions of sodium hydroxide, mol/L;
is volume of sodium hydroxide standard solution consumed in the titration of the sample dilution to the end point by adding formaldehyde, ml;
is volume of sodium hydroxide standard solution consumed by adding formaldehyde titration to the end point in a blank test, ml;
m is weight of sample, g;
M is protein content of ingredients, g/100g;
V is total volume of enzymatic supernatant, ml.
2.3. Purification of the Peptides by Ultrafiltration
According to the reported method, the three samples were centrifuged at 4 ℃, 7000r/min for 15 min to obtain the supernatant[11]. The supernatant was put into the ultrafiltration membrane separately. The samples were separated using an ultrafiltration system equipped with 500Da and 2000Da ultrafiltration membranes. Fractions with molecular weight <500Da, 500-2000Da, >2000Da were collected.
2.4. Identification of the Taste Peptides
Dissolve the freeze-dried portion of the ultrafiltration separation with the strongest saltiness enhancement effect in deionized water. The fraction with the best saltiness enhancement effect will be continued in UPLC-Q-TOF-MS/MS for peptides analysis. A sample of 30mg was weighed 100 μL of 0.1% TFA was added and sonicated for 20min, then centrifuge 17000g to take the supernatant for later use, then rinsed the TIP 10 times with 50 μL 60% ACN/0.1% TFA, then washed the TIP 10 times with 10 μL 0.1% TFA, then inhaled and drained the TIP 20 times, drain the liquid, continued to wash the TIP with 10 μL 0.1% TFA 5 times, and finally washed the TIP with 10 μL 60% ACN, 0.1% TFA eluted the peptide to a new EP tube, which is then vacuum dried and tested on the machine using a preparative column (75 μm index 150mm, packed with Acclaim Pep Map RSLC C18, 2 μm, 100Å, nano Viper). The above treated peptides were dissolved with 20μL dissolved solution (0.1% formic acid, 5% acetonitrile), fully shaken and vortexed, centrifuged at 13500 rpm at 4 °C for 20 min, then transferred the supernatant to the sample tube, and pipette red 8μL for mass spectrometry identification. Nitrogen was used as an atomizer and auxiliary gas. TOF-MS/MS (Agilent 6530), were performed, and the mass range was m/z 100−1250 Da. PEAKS was used for data retrieval and the amino acid sequences of the peptides were obtained by matching them with the UniProt protein database. Data were analyzed based on the database NCBI_Volvariella_validated.fasta provided by Shanghai Research Dog Technology Co., Ltd.
2.5. Molecular Docking (MD) of the Identified Peptides and T1R1/T1R3
The PDB Protein Library contains X-ray crystal diffraction and nuclear magnetic resonance (NMR) structural data uploaded by scientists for a number of currently known proteins or nucleic acids[19]. The amino acid sequences of the T1R1/T1R3 protein dimers were obtained from the Uniprot website (T1R1, UniProtKB: Q7RTX1; T1R3, UniProtKB: Q7RTX0). The T1R1/T1R3 receptor model used in this study was obtained by homology modelling with the Modeller v9.19 program, using the crystal structure 5X2M as a template and optimizing the model for molecular dynamics with the Amber14 force field, as used by Song et al.[20]. 3D models of taste peptides identified by Q-Tof-MS/MS were built in PyMol, and these peptides were docked with the receptors T1R1/T1R3 in the program Autodockvina (The Scripps Research Institute Molecular Graphics Laboratory, La Jolla, CA, USA). Prior to molecular docking we pre-treated the protein receptor and straw mushroom peptides ligand, including dehydrogenation of the receptor ligand, prediction of possible docking sites for the ligand, and optimization of hydrogen bonding. The docking position in T1R1/T1R3 site (x, y, z=47.163, 35.635, 22.891). For each peptide docking with protein receptor T1R1/T1R3, 10 docking procedures were set up, and one peptide-protein conformation with the lowest binding energy was selected from the results, which was finally imported into pymol for further analysis.
2.6. Sensory Assessors
The Sun et al. sensory method was used as a baseline, with some modifications based on it[21]. Sixteen sensory personnel were recruited from our laboratory to form a sensory panel, including 4 males and 12 females, aged 21±2 years. None of them had oral diseases, their sense of taste was normal, and they were not usually alcohol and tobacco users. The training and evaluation of the sensory team was carried out according to the standard ISO 8586[22]. The sensory panel rated the different concentrations of reference organoleptic references Zhang et al. [23]. The following five flavors reference solutions were prepared to enable the sensory personnel to reach a consensus on the different taste intensities. The concentrations of the reference solutions were as follows: citric acid solution (0.430 mg/mL); sucrose solution (5.76 mg/mL), aqueous quinine sulphate solution (0.0325 mg/mL), sodium chloride solution (1.19 mg/mL), and sodium glutamate (0.595 mg/mL).
2.7. The Saltiness Was Compared by QDA Method
According to Shan et al. method, we performed quantitative descriptive analysis (QDA) of the saltiness and umami flavors of different samples [18]. In order to distinguish the taste differences between different samples, we referred to the standard GB/T 12315-2008[24]. After the panelists had tasted each sample, they were scored according to the perceived intensity of taste using a nine-point scale, where 1-3 was weak; 4-6 was moderate; and 7-9 was strong.
2.8. Comparing the Relative Saltiness Using the Two- Alternative Forced-Choice (2-AFC) Method
Control and test samples with the same salt concentration were evaluated using the 2-AFC design according to ISO-5495:2005[25] with a risk of error of 5% [26]. We used this method to analyze the saltiness of three ultrafiltration samples. In subsequent experiments, the sensory team was trained to familiarize themselves with assessing the salty intensity of the samples using the 2-AFC method. Referring to Sun et al., we set up six sets of salt solutions D1-D6 with different concentrations, namely D1: 2.03g/L, D2: 2.99g/L, D3: 4.39g/L, D4: 6.45g/L, D5: 9.48g/L, and D6: 13.94g/L[21]. The sensory personnel were required to evaluate one set of solutions at a time, and each set consisted of the same concentration of salt solution Each set of solutions consists of a salt solution of the same concentration (5 ml) and a solution to be tested (5 ml). After tasting, the sensory personnel must choose the saltier sample as the result of the experiment. As it was a two-choice test, the sensory personnel evaluated the samples in the order of the gradient from the lowest to the highest salt concentration. All experiments were conducted at 25±1°C and the sensory personnel were asked to rinse their mouths before evaluating the different groups of solutions to ensure the accuracy of the experiment.
2.9. Rating the Saltiness Intensity Using the General Labeled Magnitude Scale (gLMS)
Sensory personnel were required to specifically evaluate the intensity of saltiness in different samples using the general labelled magnitude scale (gLMS) method, a psychophysical evaluation tool that requires sensory personnel to grade the intensity of perceived taste intensity along a vertical axis labelled with adjectives[27]. The descriptors it contains range from barely detectable=1.38, weak=5.75, moderate =16.22, strong=33.11, very strong=50.12, and the strongest sensation of any type=100[28,29]. The experiment was conducted with only descriptors given to the sensory personnel, not numbers, and then the sensory personnel evaluated the results of the experiment based on the descriptors provided by the sensory evaluators. The descriptors provided by the sensory evaluators were then used to transform the experimental results into numbers and statistics, and the mean of the scores was used to indicate the intensity of the saltiness of the different samples. We used this method for a specific and quantitative sensory evaluation of the saltiness of different ultrafiltered samples.
Reference was made to the method of Sun et al., with slight modifications [21]. For the formal experiments, the sensory personnel were required to evaluate one group of solutions at a time by the gLMS method, and each group consisted of a salt solution with different concentrations (D1-D6) and a solution with different molecular weights of taste peptides of straw mushrooms added. Finally, the results of the salt solutions of the same concentration were put together and analyzed. The sensory personnel put the samples (5 ml) in their mouths to revolve them for 10 s and then spit them out, and then evaluated the intensity of the saltiness of the different samples using the gLMS method. In order to reduce the experimental error, each set of experiments was repeated three times and the final results were averaged.
Saltiness intensity was calculated according to the method of Sun et al[21]. The natural logarithmic mean ratings of assessors’ saltiness intensities were calculated, and the power function between the salt concentration and saltiness intensity was obtained.
lnI = lnk + nlnC
where I is the sensory intensity, in this case, the saltiness intensity of the salt solution; C is the stimulus level, in this case, the salt concentration; n is the power exponent; k is a proportionality constant.
2.10. Evaluation of the Saltiness Enhancement Effect of Synthetic Peptides and Their Threshold Determination
In order to determine the saltiness enhancement effect of the synthetic peptides, we prepared the synthetic peptides solution by referring to the method of Zhang et al. with slight adjustments [23]. 1 g of the synthetic peptide was dissolved in 0.5% NaCl solution to prepare a 1 g/L peptide-containing salt solution. The salt solution containing the synthetic peptides was subjected to sensory evaluation using the QDA method described previously. Among them, the 0.5% NaCl solution was assigned a score of 5 and served as a blank control group. In order to ensure the reasonableness of the results, we also measured the E-tongue of these solutions.
Threshold determination of synthetic peptides was determined by taste dilution (TD) analysis. In this test, we determined the taste thresholds of synthetic peptides in water and in 0.5% NaCl solution, respectively. The peptide solution was first gradually diluted with deionized water at a ratio of 1:1 (v/v). Sensory evaluation was tested using triangulation, whereby the sensory personnel were required to taste these diluted peptide solutions continuously until it was not possible to distinguish the difference in taste between the sample solution and the two cups of deionized water. The number of dilutions at this point is the TD value, and the results are averaged across all sensory personnel evaluations[30].
2.11. Dose-Response Evaluation of the Saltiness Taste for Synthetic Peptides Solutions with NaCl Solutions
With reference to Song et al.'s approach, we make some modifications [20]. To verify the saltiness enhancement effect of the synthetic peptides, we prepared a series of three synthetic peptide solutions at different concentrations (0, 0.2, 0.4, 0.8, 1.6 and 3.2 mM). They were each prepared with NaCl solution (0.5 g/L) in a 1:1 ratio to form a solution to be tested for the assessment of saltiness. We continued the QDA approach by having the sensory personnel rate the intensity of the salty flavor of these samples on a scale of 1-9. In this case, scores of 1, 5, and 9 represent the saltiness enhancement intensity of 0.5 g/L, 1 g/L, and 1.5 g/L NaCl solutions, respectively.
2.12. E-Tongue Analysis
Minor modifications based on the method of Yan et al[31]. The TZ-5000Z E-tongue system from insent Japan was used and the device was equipped with 6 sensors: AAE, CAO, CTO, COO, AE1 and GL1 for umami, sour, salty, bitter, astringent and sweet respectively. In order to make the experimental results more accurate, the whole testing process was carried out at an ambient temperature of about 25℃. Before the start of the experiment, the E-tongue was debugged in the following sessions: activation, initialization and calibration, etc. The samples were configured with a reference solution of 30 mM KCl and 0.3 mM tartaric acid. At the beginning of the experiment, the liquid to be measured was poured into the special beaker for the E-tongue, and the liquid to be measured was placed alternately with the cleaning liquid on the E-tongue autosampler. The data of each sample was repeated 4 times, the collection time was 120s, and the minimum error value of 3 times was taken as the measurement data of each sample.
2.13. Statistical Analysis
All experiments were repeated three times. Results are expressed as mean ± standard deviation. All data were statistically analyzed using SPSS Statistics 27.0.1 (SPSS Inc., Chicago, USA), and differences between groups were analyzed by one-way ANOVA using Duncan's post hoc test (p < 0.05). Plotting was performed using Origin 2023 (Origin Lab, Northampton, USA).
3. Results and Discussion
3.1. Solid Content and Degree of Hydrolysis of Straw Mushroom Peptides
Prior studies primarily employed single-enzyme or two-enzyme sequential approaches for the enzymatic digestion of plant proteins. Luo et al. employed a single-enzyme digestion method to extract chlorella proteins, while efficient extraction of soy protein active peptides from soybeans was achieved using a double-enzyme digestion method [32,33]. In this study, we selected the optimal enzyme combination among single-enzyme enzymatic digestion, double-enzyme enzymatic digestion, and triple-enzyme enzymatic digestion based on the analysis of solid content, peptide molecular weight distribution, E-tongue results, and salting effects of the straw mushroom's enzymatic supernatant. The chosen optimal enzymatic digestion method was subsequently employed for the following experimental steps. Three enzymes, namely pectinase, cellulase, and papain, were selected for the experiments. The enzyme combinations for each group during enzymatic digestion were as follows: papain; cellulase and papain; pectinase, cellulase, and papain.
Figure 1 illustrates the solids content and degree of hydrolysis in the supernatant after enzymatic digestion using various enzyme combinations. The solids content in the supernatant following single-enzyme digestion was 3.5%, which did not significantly differ from the solids content after double-enzyme digestion (3.6%). However, the solids content in the supernatant after triple-enzyme digestion reached 4.8%, signifying a notable difference compared to the other two groups and representing the highest solids content among the three enzymatic methods. In terms of the degree of hydrolysis, significant differences were observed among the three groups. The highest degree of hydrolysis was attained with triple-enzyme digestion at 50.1%, followed by double-enzyme digestion at 40.1%, with the lowest degree of hydrolysis recorded in single-enzyme digestion at 32.8%. These disparities may be attributed to the distinct properties of cellulase and pectinase. Pectin forms cross-links with cellulase, hemicellulose, lignin, and plant cell tissue proteins, resulting in strong bonding between adjacent fiber cells and inherent morphological traits[34]. As a result, the application of pectinase as a pretreatment on the raw material can partially degrade intercellular pectin, thereby easing fiber separation. On the other hand, cellulase is a highly efficient biocatalytic enzyme that degrades cellulase into reducing sugars [35,36]. Extensive research has demonstrated that the addition of cellulase can reduce lignocellulosic polymerization, increase pore space, and, when combined with papain, it can enhance the contact between papain and the substrate [35,37,38]. The concurrent use of pectinase and cellulase results in a more comprehensive degradation of the straw mushroom's cell wall, enabling papain to more effectively degrade straw mushroom proteins and yield an increased quantity of straw mushroom polypeptides. Figure 1 unambiguously demonstrates that the solids content and degree of hydrolysis in the product supernatant were highest following enzymatic hydrolysis using the triple-enzyme method, confirming its efficiency in enzymatic hydrolysis.
3.2. Free Amino Acid Content and Molecular Weight Distribution of Straw Mushroom Peptides
Amino acids serve not only as participants in human metabolism but also as pivotal flavor-contributing compounds, playing an integral role in the taste characteristics of food [39]. The contents of free amino acids in various flavor products following enzymatic digestion via distinct methods are tabulated in Table 1. Complete data concerning free amino acid content can be found in Table S1. Among these compounds, free amino acids are particularly essential as active constituents in the umami flavor of edible mushrooms. Based on their flavor attributes, amino acids can be categorized into four classes: umami, sweet, bitter, and tasteless. The content within each class has been quantified, with amino acids categorized as follows: umami (aspartic acid and glutamic acid), sweet (threonine, serine, glycine, alanine, and proline), bitter (valine, methionine, isoleucine, leucine, phenylalanine, histidine, and arginine), and tasteless (cysteine, tyrosine, and lysine)[40]. As illustrated in Table 1, triple enzymatic digestion yielded a higher quantity of free amino acids compared to the other treatments. This can be attributed to the synergistic action of cellulase and pectinase, which enhanced the degradation of plant cell walls, thereby releasing a greater quantity of free amino acids[34].
Table S2 reveals the distinct molecular mass distribution of peptides within the preparations obtained through different enzymatic digestion methods. The notable increase in the fraction with a molecular mass of less than 500 Da following triple enzymatic digestion can be attributed to the triple enzymatic digestion conditions' effectiveness in breaking down larger molecules into smaller ones. Notably, the molecular masses of amino acids typically fall below 500 Da. When considering this information alongside Table S2, it becomes evident that the most substantial increase in amino acid content after triple enzymatic digestion is observed when utilizing the three distinct extraction methods, consistent with the findings from the analysis of free amino acids. Moreover, the flavor of straw mushrooms is influenced by the presence of numerous flavor compounds [41]. A significant subset of these compounds comprises flavor-presenting peptides, which are oligopeptides with a molecular mass below 3000 Da[42]. As indicated by Table S2, the fractions below 3000 Da are the most abundant following enzymatic digestion using three types of enzymes.
3.3. Evaluation of the Saltiness Enhancement Effect of Enzyme Solution of Straw Mushroom.
To examine the potential saltiness enhancement effects of straw mushroom enzymatic peptides, we conducted sensory and E-tongue assessments on these peptides in 0.5% NaCl solution. The outcomes of our experiments are presented in Figure 2. Specifically, Figure 2(A) and (B) display the results of E-tongue and sensory evaluations of saltiness and umami for the three enzymatic supernatants from straw mushrooms. Sensory evaluation employed the previously described 9-point scale, with a score of 5 signifying detectable saltiness (as indicated by the dotted line in Figure 2(B)). Figure 2(A) reveals that the three enzyme digests of straw mushrooms exhibited no perceivable saltiness and a decreasing umami intensity in the sequence of 15.81, 13.81, and 13.21, respectively. Figure 2(B) further illustrates that these enzyme digests were devoiced of saltiness according to sensory assessors but still retained umami. Notably, saltiness enhancement peptides are a category of taste peptides that lack inherent saltiness but heighten the perception of saltiness[5]. Previous studies suggested that the presence of umami substances might intensify the perception of saltiness in a saline solution[43]. To assess whether the extracted straw mushroom peptides possess saltiness enhancement properties, we introduced the enzymatically digested supernatants of straw mushrooms into a 0.5% salt solution for an investigation of their saltiness-enhancing effects.
Figure 2(C) and (D) depict the results of E-tongue and sensory evaluations conducted on the three enzymatic supernatants of straw mushrooms following their addition to a 0.5% NaCl solution. In Figure 2(C), all three enzyme-digested supernatants of straw mushrooms exhibited saltiness enhancement in the 0.5% NaCl solution environment compared to the saltiness intensity of the 0.5% NaCl solution (denoted as 4.58 by the dotted line). This outcome aligns with the findings of Shan et al.[18]. Among these, the triple enzyme digestion demonstrated the most pronounced saltiness enhancement, elevating the saltiness value of the 0.5% NaCl solution from 4.58 to 6.49, consistent with sensory evaluation results. Figure 2(D) further indicates that sensory assessors considered the peptides from the triple enzymatic hydrolysis of straw mushrooms more effective in enhancing the saltiness value of 0.5% NaCl. It's noteworthy that in a 0.5% NaCl solution environment, the umami of the enzyme solution decreased, while saltiness was heightened.
In light of these experimental results, it becomes evident that triple enzyme enzymatic hydrolysis, characterized by a higher degree of hydrolysis, can yield more flavor-presenting peptides and offers superior saltiness enhancement effects. This suggests that triple enzyme digestion could be a promising method for preparing saltiness enhancement peptides. To delve deeper into the saltiness enhancement potential of straw mushroom enzyme peptides, we performed ultrafiltration on the triple enzyme digested supernatants using 500 Da and 2000 Da ultrafiltration membranes, resulting in three fractions: A1 (<500 Da), A2 (500-2000 Da), and A3 (>2000 Da). These fractions were subsequently freeze-dried and prepared for further analysis.
3.4. Comparison of Saltiness Intensity Using the 2-AFC
The data were modeled under a binomial distribution, with a minimum of 12 agreeing judgments required for significance (one-tailed binomial distribution, n=32, α=0.05).
As depicted in Figure 3, more than 12 evaluators deemed samples with these fractions saltier than control samples at all salt concentrations (D1-D6). This indicates that all three ultrafiltration fractions can enhance saltiness in the salt solution. Notably, most panelists, across various salt concentrations, found samples with these fractions to be saltier than the control sample. As the saltiness enhancement did not significantly differ among the fractions, in-depth analysis of gLMS scores is warranted[43].
3.5. Saltiness Intensity Evaluation Using the gLMS
The gLMS method is a psychophysical evaluation tool in which only descriptors, not numbers, are provided to the sensory evaluator during the experiment. The experimenter then performs numerical transformations and statistics on the results of the experiment based on the descriptors provided by the sensory evaluator, and the degree of saltiness is expressed as the mean of the scores[44]. For gLMS saltiness intensity evaluations, guided by the 2-AFC results that revealed a significant saltiness enhancement effect of the three ultrafiltration fractions, we further scrutinized the saltiness enhancement ability of these fractions. Figure 4 portrays the results. A repeated measures ANOVA was employed to assess the significance of saltiness intensity ratings. Figure 4(A) illustrates that fraction A2 significantly heightened saltiness intensity across all salt concentrations compared to the other two fractions. A1, on the other hand, substantially enhanced saltiness in D2, D3, and D6 salt solutions but exhibited no significant enhancement for D5. Conversely, A3 displayed no apparent saltiness enhancement in all six salt concentrations, and A2 had no discernible enhancement for D4. In summary, the enhancement effects of different ultrafiltration fractions at various salt concentrations on saltiness intensity varied,with A2 demonstrating significant saltiness enhancement across concentrations from D1 to D6[18].
Analyzing the gLMS results tentatively suggests that A2 is the most effective of the three ultrafiltration fractions in enhancing saltiness. Subsequently, saltiness intensity scores were logarithmically transformed to establish power functions correlating with salt concentration. The power functions, reflecting the relationship between the natural logarithm of salt concentration and the natural logarithm of saltiness intensity, are presented in Table S3 to help determine iso-saltiness concentrations among the fractions. The results reveal exponents ranging from 0.9207 to 1.0736, both less than the control. Consistent with Sun et al.'s findings, the ln-ln curves support that all three ultrafiltration fractions enhance saltiness at varying salt concentrations[43]. These curves demonstrate a more pronounced increase in the lower salt concentration range, while achieving a higher salt reduction percentage becomes challenging at higher concentrations. In salt solutions of D2, D3, and D4 concentrations, A2 exhibits the most prominent saltiness enhancement effect compared to the other fractions. Hence, we tentatively conclude that A2 (500-2000 Da) is particularly adept at enhancing saltiness.
3.6. Identification and Molecular Docking of Peptides
Through a comprehensive analysis of preceding experiments, it was evident that the A2 fraction (500-2000 Da) from enzymatic supernatants of straw mushrooms holds the most prominent saltiness enhancement effect. It's clear from this study that peptides derived from the triple enzymatic hydrolysis of straw mushrooms impart umami rather than saltiness. However, when introduced to a salt solution, they exhibit a saltiness-enhancing effect. This aligns with Xie et al.'s findings that substances with umami attributes can heighten saltiness in salt solutions[17]. To delve deeper into the saltiness enhancement effect of peptides from triple enzymatic hydrolyzed straw mushrooms, we subjected the A2 fraction to UPLC-Q-TOF-MS/MS analysis to acquire specific peptide information[17]. Building upon the research of Shan et al., which revealed that peptides possessing inherent umami characteristics but lacking saltiness could be initially selected through docking with umami receptors T1R1/T1R3, our study delved deeper into the saltiness enhancement potential of these screened peptides[18]. We subsequently conducted molecular docking experiments on the identified peptides to unravel the underlying molecular mechanisms. To investigate whether peptides derived from triple enzymatic hydrolysis of straw mushrooms possessed saltiness enhancement attributes, we pursued the identification of peptides within the A2 fraction and their molecular docking with T1R1/T1R3[45].
Peptide identification was performed via UPLC-Q-TOF-MS/MS. In this context, electrospray ionization in a high-resolution mass spectrometer was employed to dissociate peptides in A2 fraction samples, generating fragment ions. Amino acid sequences were determined by aligning these fragments with straw mushroom protein libraries on UniProt[46]. The identified peptides, with molecular weights of <2k Da and containing 5-10 amino acid residues, totaled 220. Subsequently, molecular docking simulations with T1R1/T1R3 were carried out to screen the peptides. Peptides with matching mass-to-charge ratios were initially screened based on their compatibility with b and y ions. Subsequently, eight potential taste peptides were selected based on molecular docking scores: DFNALPFK (DF8, mass 875.04, m/z 574.25), VPGGQEIKDR (VP10, mass 1097.5829, m/z 366.87), GVGPFDDDR (GV9, mass 976.425, m/z 489.22), SEHEENGYAV (SE10, mass 1133.46, m/z 567.75), YNEDNGIVK (YN9, mass 1050.50, m/z 526.26), IDNEPEFRWA (ID10, mass 1275.59, m/z 638.81), DKLHEGIK (DK8, mass 938.52, m/z 313.85), and IGDEAAENRN (IG10, mass 1254.42, m/z 544.75). Their MS/MS data is illustrated in Figure S1.
The use of molecular docking with taste peptides, employing T1R1/T1R3 receptors, offers an avenue to explore the taste properties of these peptides and introduces novel perspectives for taste peptide research[47]. The two-dimensional diagrams of the eight peptides are displayed in Figure 5, and the molecular docking scores are provided in Table 2. These scores demonstrate that all eight peptides can bind to the Venus flytrap (VFT) binding domains of T1R1/T1R3[48]. Throughout the docking process, the T1R1/T1R3 receptor structure remains fixed, while the peptide structure adapts its conformation. The docking process allows for diverse conformational changes of the peptides, with the binding model featuring the lowest binding energy (highest score) being selected[49]. The docking energies are ranked as follows: DF8 (-9.2 kcal/mol), VP10 (-8.9 kcal/mol), YN9 (-8.8 kcal/mol), GV9 (-8.7 kcal/mol), SE10 (-8.7 kcal/mol), ID10 (-8.6 kcal/mol), DK8 (-8.6 kcal/mol), and IG10 (-8.3 kcal/mol). As demonstrated by Liang et al., lower binding energies in molecular docking results indicate more stable conformations[45]. Notably, DF8 exhibits the lowest docking energy (highest score) with T1R1/T1R3. The molecular docking 2D diagrams illustrate that peptides from triple enzymatic hydrolyzed straw mushrooms chiefly engage in hydrogen bonding and hydrophobic interactions when binding to receptor T1R1/T1R3. However, the binding characteristics of different peptides to the receptor T1R1/T1R3 are not uniform, suggesting that taste peptide properties can't be exclusively determined based on binding energy[18]. Apart from determining the most stable binding conformation through binding energy, molecular docking studies offer insights into the peptide-receptor binding site[50,51]. A total of 45 amino acid residues in T1R1/T1R3 play pivotal roles in interactions with peptides from triple enzymatic hydrolyzed straw mushrooms. As depicted in Figure 6, the actively docked residues predominantly include Gln 227, Lys 215, Gln 223, Asn 223, Gln 243, Glu 226, Ser 167, Val 168, Glu 151, Lys 171, Lys 222, Glu 219, Tyr 242, Asn 262, Lys 176, and Ala 255. Notably, amino acids Asp, Glu, Ser, and His make the most substantial contributions to molecular interactions. In conclusion, the active sites highlighted in this molecular docking study serve as a foundational step for preliminary screening and prediction of unknown taste peptides in subsequent research. As supported by existing literature, protein receptors and peptide ligands in molecular docking establish connections through hydrogen bonding and peptide ligands, in addition to hydrogen bonding, also engage in van der Waals interactions, electrostatic interactions, alkyl interactions, and hydrogen interactions[52,53]. Consistent with our findings, the binding forces between the peptides from triple enzymatic hydrolyzed straw mushrooms in this study and the protein receptor's active residues can be observed in 2D diagrams, comprising primarily carbon-hydrogen bonding, conventional hydrogen bonding, charge attractions, followed by salt bridges, π-alkyl groups, π-anions, and alkyl groups.
3.7. Effect of Synthetic Peptides on Saltiness in Salt Solutions
We selected three peptides from the molecular docking results based on their lowest binding energies and robust interactions with receptor proteins for synthesis: DF8, YN9, and VP10. To investigate whether the peptide has a saltiness enhancement effect, the extracted peptide was added to a salt solution of the same concentration, and the peptide-salt solution system was used to assess the saltiness enhancement effect of peptides[54].To ascertain whether these synthesized peptides possess saltiness enhancement properties, they were introduced into a 0.5% salt solution for sensory and E-tongue evaluations. The results, presented in Figure 7, demonstrate that all three peptides indeed can enhance saltiness in the solution. Among them, VP10 exhibited the most potent effect, followed by DF8, and YN9. This observation aligns with the E-tongue results, substantiating that VP10 wields the most substantial saltiness enhancement effect. Furthermore, a notable correlation exists between the E-tongue outcomes and sensory evaluations, underscoring the utility of the E-tongue in assessing peptide-induced saltiness enhancements[55].
We established the taste thresholds for the synthetic peptides using previously defined methods. The taste characteristics and threshold data for the synthetic peptides are outlined in Table 3. We examined the taste thresholds for these peptides in both pure water and a 0.5% NaCl solution. Beyond the mere realm of taste, peptides may contribute to a broader flavor profile[56]. Evidently, sour and astringent notes could be perceived in the identified peptides. This sourness may originate from residual free amino acids and acetate remnants during peptide synthesis[20]. Research indicates that umami amino acids, such as Aspartic Acid (Asp), can evoke sourness or umami if found in a free state[23]. It's worth noting that DFNALPFK and VPGGQEIKDR were characterized by umami or a faint umami taste. Notably, in pure water, DFNALPFK displayed the lowest threshold at 0.38 mM, consistently mirroring the molecular docking scores. Higher scores corresponded to lower thresholds, while lower scores indicated comparatively higher thresholds. In both pure water and the 0.5% NaCl solution, all three synthetic peptides demonstrated lower thresholds in the saline environment, affirming their saltiness enhancement effects. Among these, DFNALPFK exhibited the lowest threshold in the 0.5% NaCl solution, signifying its most pronounced saltiness enhancement effect. Notably, VPGGQEIKDR displayed saltiness characteristics and is likely a peptide with saltiness attributes.
3.8. Dose-Feedback Saltiness Enhancement Effects of Synthetic Peptides
The figure clearly displays significant variations in saltiness intensity as the concentration of synthetic peptides increases. Notably, within the concentration range of 0.4-0.8 mmol, there is a sharp rise in saltiness intensity in the NaCl solution. However, when the concentration of synthetic peptides surpasses a critical threshold (0.8-1.6 mmol), the effectiveness of saltiness enhancement begins to decline. From Table 3, it can be seen that the synthetic peptide has an acidic flavor, and apart from the fact that the peptide itself contains acidic amino acid residues, this may be related to residual chemical reagents such as sodium acetate introduced during the peptide synthesis process[20,57].Consequently, sensory assessors may not perceive the changing saltiness. Figure 8 also reveals that in the 0.4-0.8 mmol concentration range, both DF8 and VP10 exert a significant saltiness enhancement effect on the NaCl solution. However, as the concentration reaches the critical range of 0.8-1.6 mmol, the saltiness enhancement effect of DF8 and YN9 starts to decline, while VP10 continues to augment the saltiness intensity of the solution. Notably, adding 0.8 mmol of YN9 results in a 0.05% NaCl solution achieving the saltiness intensity of a 0.1% NaCl solution. Furthermore, incorporating 3.2 mmol of VP10 elevates a 0.05% salt solution to the saltiness level of a 0.15% salt solution. This study's results align with Xie et al.'s findings, where peptides from ham enhanced a 0.3% salt solution to the saltiness of a 0.4-0.6% salt solution[17]. Similarly, a peptide extracted from curd (EDGEQPRPF) at a concentration of 0.4 mg/ml intensified a 50 mM NaCl solution to the saltiness of a 63 mM NaCl solution[6]. In summary, this study underscores that peptides derived from the triple enzymatic hydrolysis of straw mushrooms can indeed augment the saltiness of salt solutions. However, the quantity of added peptides should be carefully managed to achieve the optimal saltiness enhancement effect[5].
5. Conclusions
In this study, we conducted a comprehensive exploration into the flavor peptides extracted from straw mushrooms through a three-enzyme enzymatic method. Our research not only focused on the extraction process but also delved into the mechanisms by which these peptides contribute to flavor enhancement using molecular docking techniques. Ultimately, we identified three peptides that were synthesized based on the outcomes of molecular docking and examined their saltiness-enhancing properties. Our investigation revealed that peptides obtained from triple enzymatic hydrolysis of straw mushrooms exhibited umami qualities but lacked saltiness. However, when introduced to a 0.5% NaCl solution, these peptides displayed reduced umami and increased saltiness. This observation led us to speculate that peptides from triple enzymatic hydrolyzed straw mushrooms might indeed possess saltiness enhancement attributes. To further understand the molecular mechanisms at play, we initiated an assessment of the saltiness enhancement potential of straw mushroom enzymatic solutions with varying molecular weights in diverse salt solution concentrations. The results pinpointed the 500-2000 Da range of straw mushroom enzymatic peptides as having the most substantial saltiness enhancement effect. We selected eight peptides from triple enzymatically hydrolyzed straw mushrooms, primarily based on molecular docking binding energy scores, which held promise for saltiness enhancement. Among these peptides, DFNALPFK, YNEDNGIVK, and VPGGQEIKDR emerged as the top candidates for enhancing saltiness based on an analysis of the molecular docking active sites and their amino acid residues. Subsequently, we synthesized these three peptides to validate their saltiness enhancement properties. All three peptides exhibited saltiness enhancement effects, with VPGGQEIKDR demonstrating the most potent impact. Remarkably, a mere 3.2 mM of VPGGQEIKDR elevated a 0.05% NaCl solution to the saltiness intensity of a 0.15% NaCl solution. Moreover, the addition of 0.8 mM of YN9 enabled the 0.05% NaCl solution to attain the saltiness intensity of a 0.1% NaCl solution. This study, on the one hand, uncovers the saltiness enhancement attributes of peptides from triple enzymatically hydrolyzed straw mushrooms and emphasizes the importance of controlled dosages for optimal saltiness enhancement. In the long term, it provides novel insights into the study of saltiness enhancement peptides derived from straw mushrooms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: MS/MS spectra of DFNALPFK (m/z 574.25, A), VPGGQEIKDR (m/z 366.87, B), GVGPFDDDR (m/z 489.22, C), SEHEENGYAV (m/z 567.75, D), YNEDNGIVK (m/z 526.26, E), IDNEPEFRWA (m/z 638.81, F), DKLHEGIK (m/z 313.85, G), and IGDEAAENRN (m/z 544.75, H). The y and b ions refer to fragment ions of each peptide.; Table S1: Free amino acid content of straw mushroom enzymatic digest; Table S2: Results of the peptide molecular weight distribution; Table S3: The parameters of the power functions and the relationship between the natural logarithmic salt concentration and the natural logarithmic intensity of saltiness; Table S4: The dose-response relationship of three synthetic peptides on saltiness intensity of 0.5g/L NaCl solution;
Author Contributions
Yunpeng Cheng: Data analysis, methodology, initial draft writing. Jingyi Wangzhang: Methodology and supervision. Min sun: Methodology and supervision. Tao Feng: Methodology and supervision. Qian Liu: Data analysis. Linyun Yao: Data analysis. Chi-Tang Ho: Methodology and conceptualization. Shiqing Song: Manuscript review and editing. Chuang Yu: Manuscript review and editing.
Funding
This work was supported by the Natural Science Foundation of Shanghai (21ZR1462400); and National Natural Science Foundation of China (32102123).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Acknowledgments
A special appreciation to the volunteers for their participation in sensory experiment.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jimenez-Maroto, L.A., T. Sato, and S.A. Rankin, Saltiness potentiation in white bread by substituting sodium chloride with a fermented soy ingredient. Journal of Cereal Science, 2013. 58(2): p. 313-317. [CrossRef]
- Pujols, K.D., et al., Low-sodium roasted peanuts: effects of salt mixtures (NaCl,KCl and glycine) on consumer perception and purchase intent. International Journal of Food Science & Technology, 2019. 54(9): p. 2754-2762. [CrossRef]
- D’Elia, L., et al., Habitual salt intake and risk of gastric cancer: A meta-analysis of prospective studies. Clinical Nutrition, 2012. 31(4): p. 489-498. [CrossRef]
- Messerli, et al., Salt and heart disease: a second round of "bad science"? The Lancet, 2018.
- Le, B., et al., Salt taste receptors and associated salty/salt taste-enhancing peptides: A comprehensive review of structure and function. Trends in Food Science & Technology, 2022. 129: p. 657-666. [CrossRef]
- Chen, Y.P., et al., Saltiness-Enhancing Peptides Isolated from the Chinese Commercial Fermented Soybean Curds with Potential Applications in Salt Reduction. J Agric Food Chem, 2021. 69(35): p. 10272-10280. [CrossRef]
- Schindler, A., et al., Discovery of salt taste enhancing arginyl dipeptides in protein digests and fermented fish sauces by means of a sensomics approach. J Agric Food Chem, 2011. 59(23): p. 12578-88. [CrossRef]
- Ueda, Y., et al., Characteristic flavor constituents in water extract of garlic. Agricultural and Biological Chemistry, 1990. 54(1): p. 163-169.
- Xiang, F., Research progress on food salt reduction strategies. Food and fermentation industry, 2023: p. 1-10.
- Moore, A., C.R. Luckett, and J.P. Munafo, Jr., Taste-Active Dipeptides from Hydrolyzed Mushroom Protein Enhance Saltiness. J Agric Food Chem, 2021. 69(40): p. 11947-11959. [CrossRef]
- Yu, B., et al., Maillard-reacted peptides from glucosamine-induced glycation exhibit a pronounced salt taste-enhancing effect. Food Chem, 2022. 374: p. 131776. [CrossRef]
- Son, M. and T.H. Park, The bioelectronic nose and tongue using olfactory and taste receptors: Analytical tools for food quality and safety assessment. Biotechnology Advances, 2017: p. S0734975017301763. [CrossRef]
- Shigemura and Noriatsu, Angiotensin II Modulates Salty and Sweet Taste Sensitivities. Journal of Neuroscience, 2013. 33: p. 6267-6277.
- A, T.S., et al., Quantitative proteomics and SWATH-MS to elucidate peri-receptor mechanisms in human salt taste sensitivity. Food Chemistry, 2018. 254: p. 95-102.
- Niki, M., et al., Gustatory signaling in the periphery: detection, transmission, and modulation of taste information. Biological & Pharmaceutical Bulletin, 2010. 33(11): p. 1772-7. [CrossRef]
- Guo, M., et al., Collagen Glycopeptides from Transglutaminase-Induced Glycosylation Exhibit a Significant Salt Taste-Enhancing Effect. J Agric Food Chem, 2023. 71(22): p. 8558-8568. [CrossRef]
- Xie, X., et al., The enhancement and mechanism of the perception of saltiness by umami peptide from Ruditapes philippinarum and ham. Food Chem, 2023. 405(Pt A): p. 134886. [CrossRef]
- Shan, Y., et al., Decoding of the Saltiness Enhancement Taste Peptides from the Yeast Extract and Molecular Docking to the Taste Receptor T1R1/T1R3. J Agric Food Chem, 2022. 70(47): p. 14898-14906.
- Zhang, J., et al., Identification and virtual screening of novel umami peptides from chicken soup by molecular docking. Food Chem, 2023. 404(Pt A): p. 134414. [CrossRef]
- Song, S., et al., Identification of novel umami peptides from Boletus edulis and its mechanism via sensory analysis and molecular simulation approaches. Food Chem, 2023. 398: p. 133835. [CrossRef]
- Sun, X., et al., The enhancement of the perception of saltiness by umami sensation elicited by flavor enhancers in salt solutions. Food Res Int, 2022. 157: p. 111287. [CrossRef]
- ISO, Sensory analysis -General guidance for the selection, training and monitoring of assessors. 2012.
- Zhang, N., et al., Sensory-Guided Analysis of Key Taste-Active Compounds in Pufferfish (Takifugu obscurus). J Agric Food Chem, 2019. 67(50): p. 13809-13816. [CrossRef]
- GB/T12315, Sensory analysis-Methodology-Ranking. 2008.
- ISO, Sensory analysis — Methodology — Paired comparison test. ISO, 2005.
- Blackman, J., A. Saliba, and L. Schmidtke, Sweetness acceptance of novices, experienced consumers and winemakers in Hunter Valley Semillon wines. Food Quality and Preference, 2010. 21(7): p. 679-683. [CrossRef]
- Zhang Huimin, D.Y., Zhang Ruifen, et al., Modification effects of different inhibitors on bitter melon powder flavour perception. Food Science, 2018. 39(10): p. 298-303.
- Green, B.G., et al., Evaluating the 'Labeled Magnitude Scale' for Measuring Sensations of Taste and Smell. Chemical Senses, 1996. 21(3): p. 323-334. [CrossRef]
- L.M. Bartoshuka, V.B. Duffya,b, K. Fasta, B.G. Greena,c, J. Prutkina, D.J. Snydera, Labeled scales (e.g., category, Likert, VAS) and invalid across-group comparisons: what we have learned from genetic variation in taste. Food Quality and Preference, 2003. [CrossRef]
- Zhao, J., et al., Isolation, identification and characterization of taste peptides from fermented broad bean paste. Food Funct, 2022. 13(16): p. 8730-8740.
- Yan, F., et al., Small Peptides Hydrolyzed from Pea Protein and Their Maillard Reaction Products as Taste Modifiers: Saltiness, Umami, and Kokumi Enhancement. Food and Bioprocess Technology, 2021. 14(6): p. 1132-1141. [CrossRef]
- Sun, H., Z. Chen, and Z. Zhou, Optimization of Soybean Protein Isolate Extraction by Response Surface Method and Complex Enzymatic Hydrolysis for Preparing Active Peptides. China Condiment, 2022. 46(3): p. 82-87.
- Luo, B., Protein Extraction from Chlorella and Peptide Preparation by Enzyme Hydrolysis Food Industry, 2023. 44(8): p. 95-98.
- Caffall, K.H. and D. Mohnen, The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr Res, 2009. 344(14): p. 1879-900. [CrossRef]
- Xu, X., et al., Lignocellulose degradation patterns, structural changes, and enzyme secretion by Inonotus obliquus on straw biomass under submerged fermentation. Bioresour Technol, 2017. 241: p. 415-423. [CrossRef]
- Zhao, G., et al., Effects of applying cellulase and starch on the fermentation characteristics and microbial communities of Napier grass (Pennisetum purpureum Schum.) silage. J Anim Sci Technol, 2021. 63(6): p. 1301-1313. [CrossRef]
- Feyisa, M.M. and P. Yadav, Role of Live Microbes for Fermentation and Enhancement of Feeding Value of Wheat Straw as Animal Fodder. The Asia Journal of Applied Microbiology, 2021. 8. [CrossRef]
- Silvestre, T., et al., Performance of dairy cows fed normal- or reduced-starch diets supplemented with an exogenous enzyme preparation. J Dairy Sci, 2022. 105(3): p. 2288-2300. [CrossRef]
- Gu,Z and Y. Yang, Research progress in flavor components of edible fungus. Food Industry Science and Technology, 2013. 34(5): p. 363-367.
- Yamaguchi, S., et al., Measurement of the relative taste intensity of some L-α-amino acids and 5'-nucleotides. Journal of Food Science, 2010. 36(6): p. 846-849.
- Xu, X.S.Z.S., S, Effects of Different Treatments on the Release of Flavor Substances from Straw Mushroom (Volvariella volvacea). Food Science, 2017. 39(12): p. 107-111.
- Adcock, S.A. and J.A. Mccammon, Molecular dynamics: survey of methods for simulating the activity of proteins. Chemical Reviews, 2006. 106(5): p. 1589. [CrossRef]
- Sun, X., et al., The enhancement of the perception of saltiness by umami sensation elicited by flavor enhancers in salt solutions. Food Research International, 2022. 157. [CrossRef]
- Zhang, H., et al., Taste Modification of Bitter Melon (Momordica charantia) Powder by Different Bitterness Inhibitors. Food Science, 2018. 39(10): p. 298-303.
- Liang, L., et al., Characteristics of umami peptides identified from porcine bone soup and molecular docking to the taste receptor T1R1/T1R3. Food Chem, 2022. 387: p. 132870.
- Liu, H., L.T. Da, and Y. Liu, Understanding the molecular mechanism of umami recognition by T1R1-T1R3 using molecular dynamics simulations. Biochem Biophys Res Commun, 2019. 514(3): p. 967-973. [CrossRef]
- Dang, Y., et al., Comparison of umami taste peptides in water-soluble extractions of Jinhua and Parma hams. LWT - Food Science and Technology, 2015. 60(2): p. 1179-1186. [CrossRef]
- Su, G., et al., Effect of transglutaminase on taste characteristics of pea protein hydrolysates through altering the composition of amino acids and peptides. Food Bioscience, 2023. 56. [CrossRef]
- Chang, R., et al., Ion-exchange purification, nano-HPLC–MS/MS identification and molecular dynamics simulation of novel umami peptides from fermented grain wine (Huangjiu). Journal of Food Composition and Analysis, 2024. 125.
- A, Y.Z., et al., Isolation, characterization and molecular docking of novel umami and umami-enhancing peptides from Ruditapes philippinarum. Food Chemistry, 2020.
- Li, C., et al., A rapid selection strategy for umami peptide screening based on machine learning and molecular docking. Food Chem, 2023. 404(Pt A): p. 134562. [CrossRef]
- Wang, W., et al., Identification and comparison of umami-peptides in commercially available dry-cured Spanish mackerels (Scomberomorus niphonius). Food Chem, 2022. 380: p. 132175. [CrossRef]
- Zhou, H., et al., Design, virtual screening, molecular docking and molecular dynamics studies of novel urushiol derivatives as potential HDAC2 selective inhibitors. Gene, 2017. 637: p. 63-71. [CrossRef]
- Chen, S., J. Chang, and M. Sun, Study on the Salt-reducing Effect of Agaricus bisporus Extract on Three Kinds of Broth. Science and Technology of Food Industry, 2023. 44(15): p. 69-77.
- Wang, K., et al., Evaluation of eight kinds of flavor enhancer of umami taste by an electronic tongue. Food Sci Nutr, 2021. 9(4): p. 2095-2104. [CrossRef]
- Moon, S.Y. and E.C.Y. Li-Chan, Changes in aroma characteristics of simulated beef flavour by soy protein isolate assessed by descriptive sensory analysis and gas chromatography. Food Research International, 2007. 40(10): p. 1239-1248. [CrossRef]
- Zhuang, M., et al., Sequence, taste and umami-enhancing effect of the peptides separated from soy sauce. Food Chem, 2016. 206: p. 174-81. [CrossRef]
Figure 1.
Results of solids content and the degree of hydrolysis (DH) after treatment with three enzymatic methods. The line matches the left y-axis (Solids content) and the bar matches the right y-axis (DH). Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the solids content and DH in the supernatant obtained using different enzymatic methods (p>0.05).
Figure 1.
Results of solids content and the degree of hydrolysis (DH) after treatment with three enzymatic methods. The line matches the left y-axis (Solids content) and the bar matches the right y-axis (DH). Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the solids content and DH in the supernatant obtained using different enzymatic methods (p>0.05).
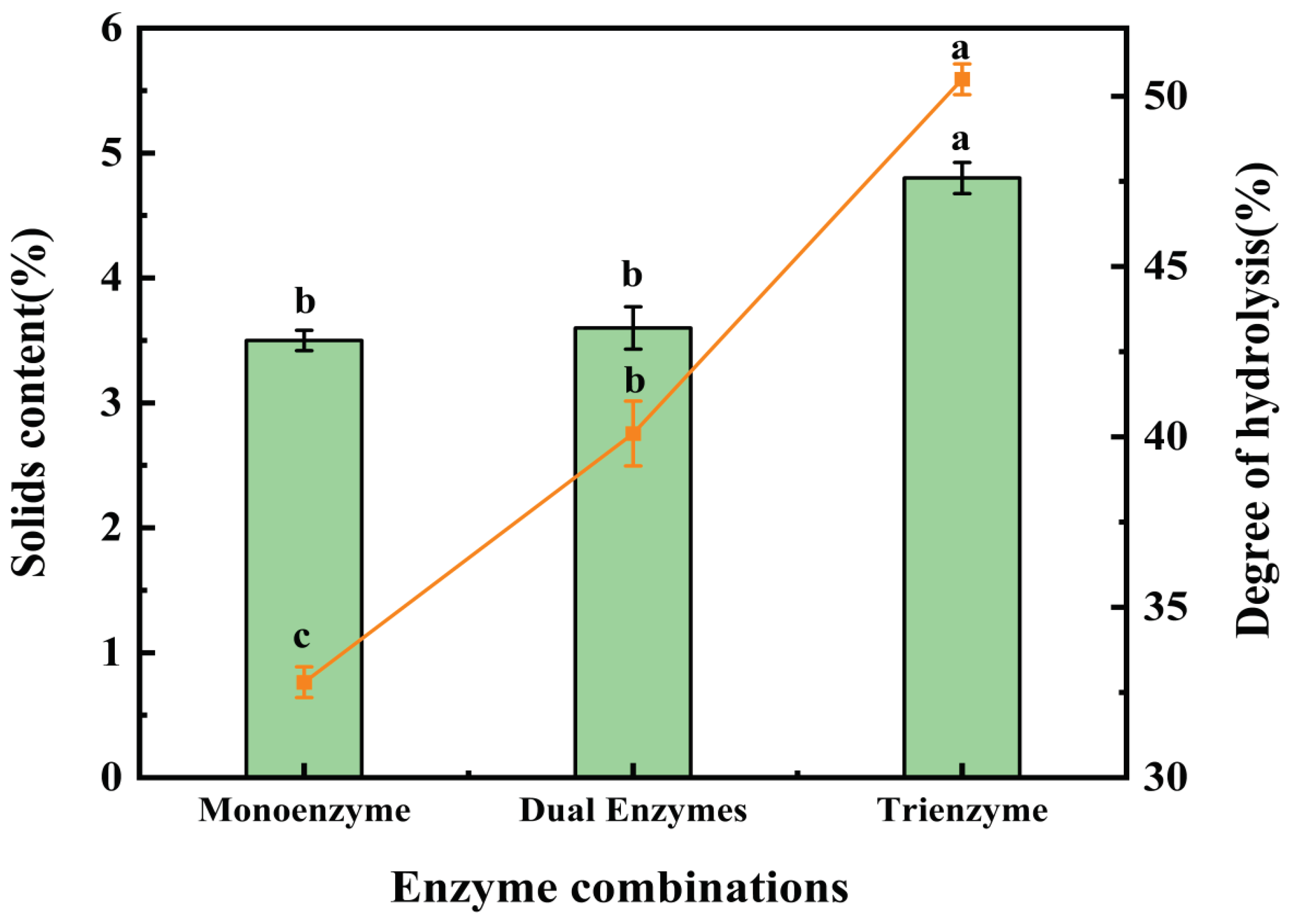
Figure 2.
The substrate in (A) and (B) is the enzymatic supernatant after enzymatic digestion using the three enzymatic methods described above; The substrate in C and D is the enzymatic supernatant added in the environment of 0.5% NaCl solution. The orange bar in the graph is the score for umami and the green bar is the score for saltiness. The dotted line in (C) indicates the score for the E-tongue saltiness of 0.5% NaCl and the dotted line in (D) indicates the score for the sensory saltiness of 0.5% NaCl. Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the E-tongue value and sensory score in the supernatant obtained using different enzymatic methods (p>0.05).
Figure 2.
The substrate in (A) and (B) is the enzymatic supernatant after enzymatic digestion using the three enzymatic methods described above; The substrate in C and D is the enzymatic supernatant added in the environment of 0.5% NaCl solution. The orange bar in the graph is the score for umami and the green bar is the score for saltiness. The dotted line in (C) indicates the score for the E-tongue saltiness of 0.5% NaCl and the dotted line in (D) indicates the score for the sensory saltiness of 0.5% NaCl. Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the E-tongue value and sensory score in the supernatant obtained using different enzymatic methods (p>0.05).
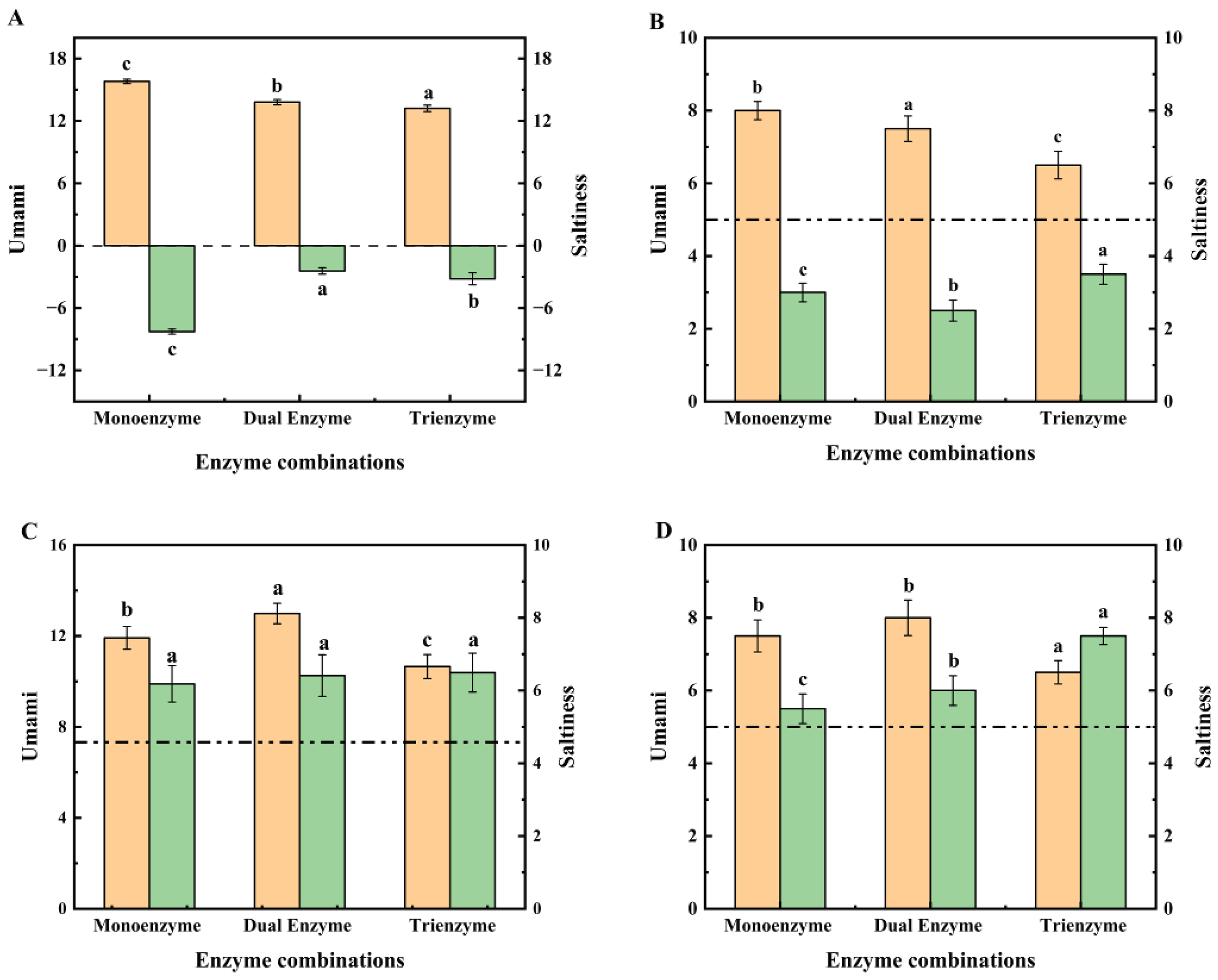
Figure 3.
The number of assessors indicating that sample with different ultrafiltration fractions carrier was saltier in a 2-AFC test (n=32). The dotted line (nmin= 12) represents the number of minimum agreeing judgments necessary to establish a saltier perception in a paired comparison test at α = 0.05 level when the total number of assessors was 32.
Figure 3.
The number of assessors indicating that sample with different ultrafiltration fractions carrier was saltier in a 2-AFC test (n=32). The dotted line (nmin= 12) represents the number of minimum agreeing judgments necessary to establish a saltier perception in a paired comparison test at α = 0.05 level when the total number of assessors was 32.
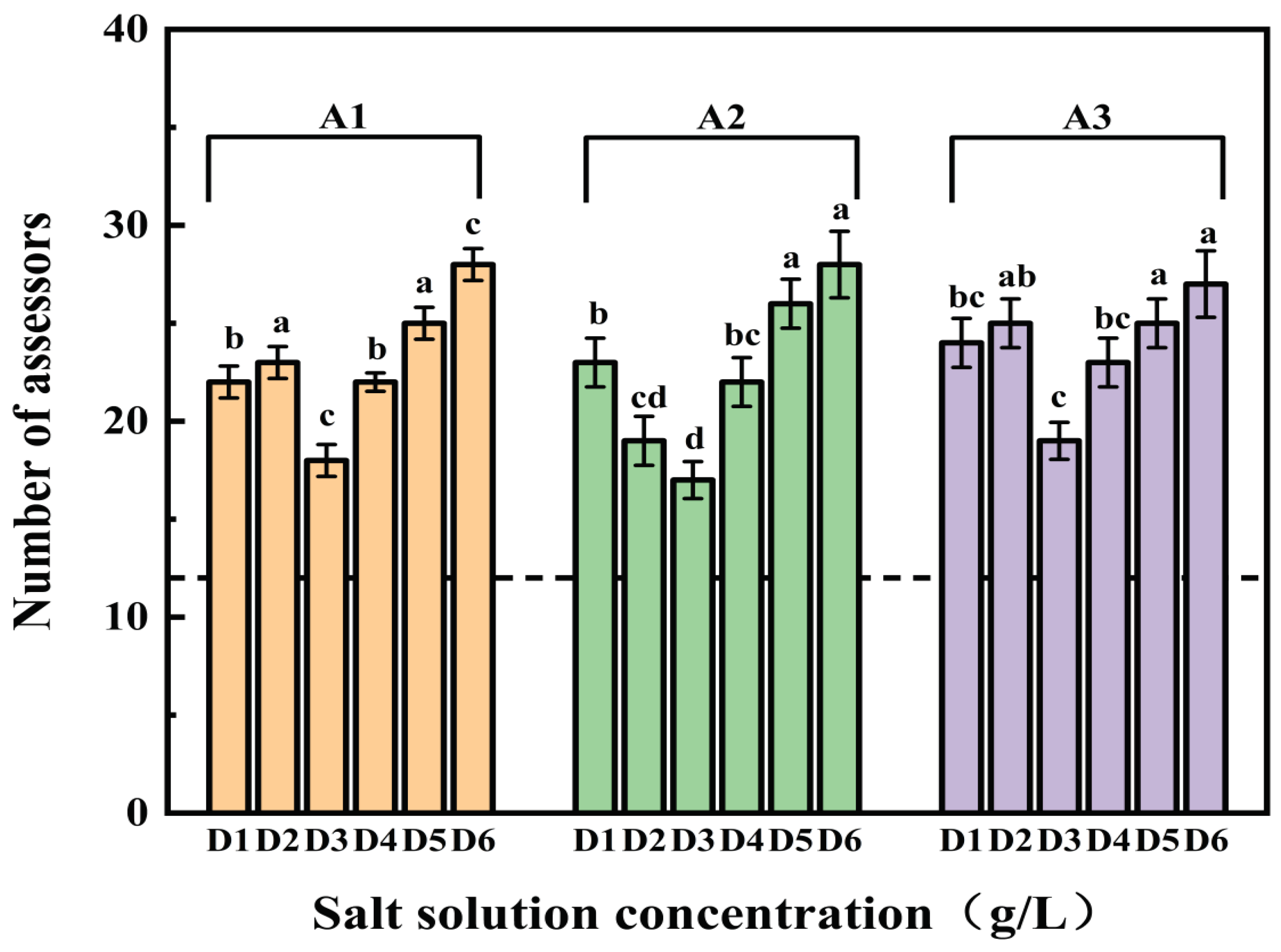
Figure 4.
The gLMS scores(A) and the change of saltiness intensity(B) under 3 ultrafiltration fractions. Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the gLMS scores in different ultrafiltration fractions (p>0.05). number of assessors indicating that sample with different ultrafiltration fractions carrier was saltier in a 2-AFC test (n=32). The dotted line (nmin= 12) represents the number of minimum agreeing judgments necessary to establish a saltier perception in a paired comparison test at α = 0.05 level when the total number of assessors was 32.
Figure 4.
The gLMS scores(A) and the change of saltiness intensity(B) under 3 ultrafiltration fractions. Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the gLMS scores in different ultrafiltration fractions (p>0.05). number of assessors indicating that sample with different ultrafiltration fractions carrier was saltier in a 2-AFC test (n=32). The dotted line (nmin= 12) represents the number of minimum agreeing judgments necessary to establish a saltier perception in a paired comparison test at α = 0.05 level when the total number of assessors was 32.
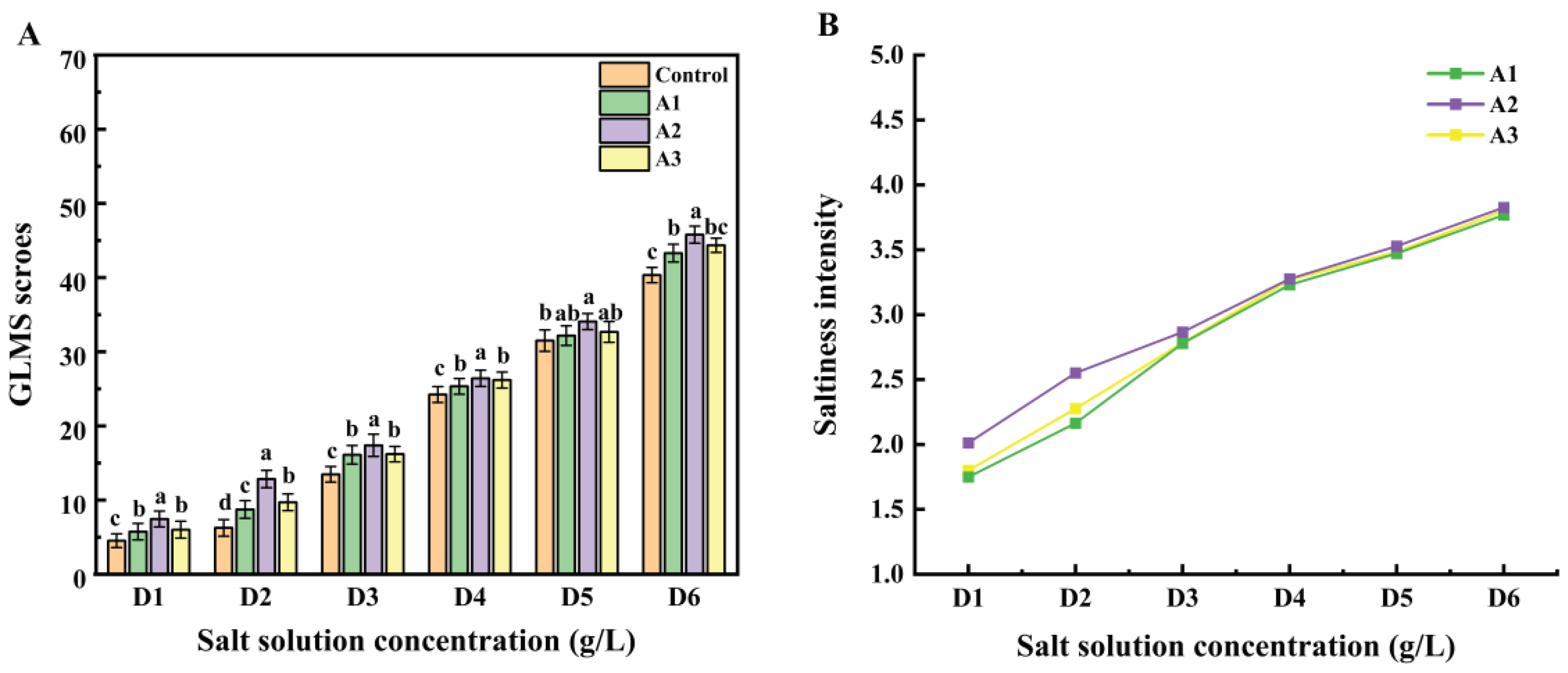
Figure 5.
Statistics of the active sites of eight taste peptides interacting with the umami receptor T1R1/T1R3.
Figure 5.
Statistics of the active sites of eight taste peptides interacting with the umami receptor T1R1/T1R3.
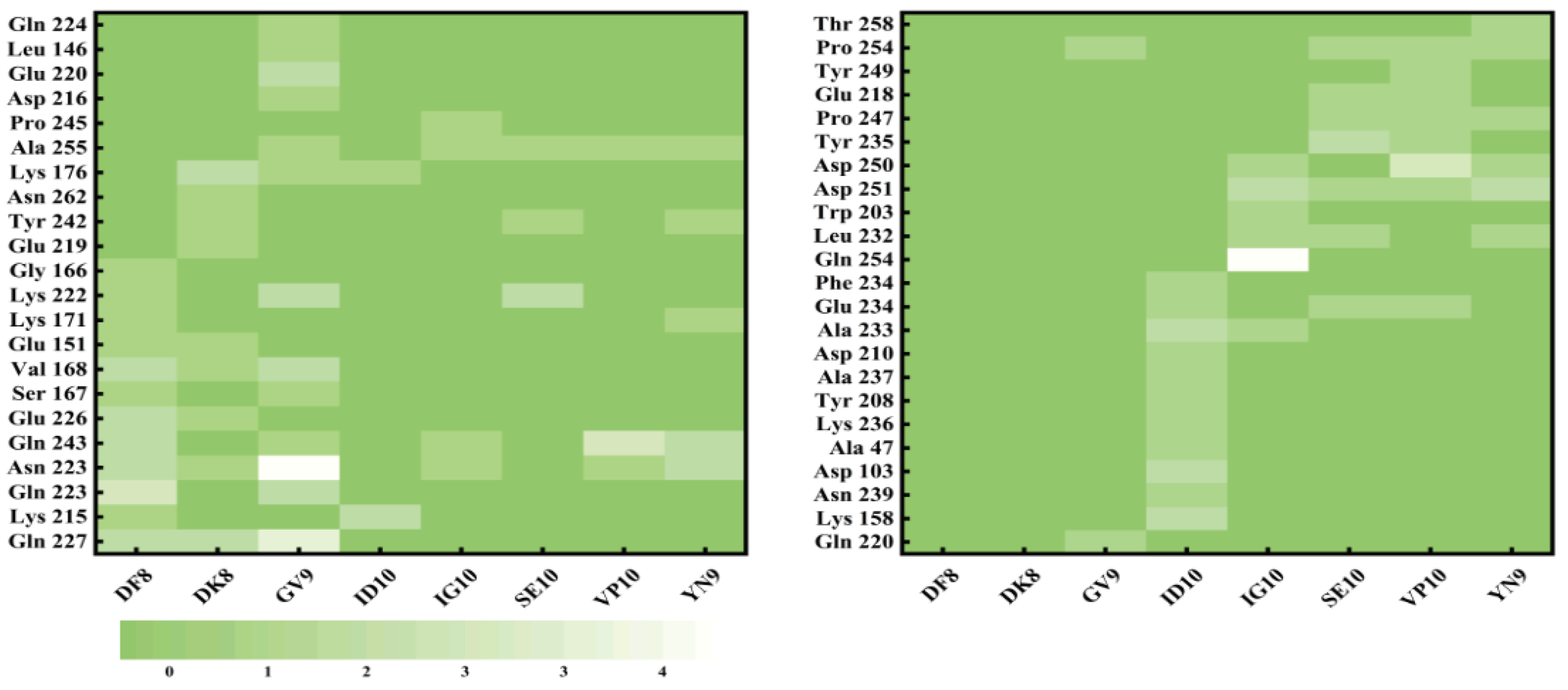
Figure 6.
The 2D diagram of the docking of DFNALPFK (A), DKLHEGIK (B), GVGPFDDDR(C), IDNEPEFRWA(D), IGDEAAENRV(E), SEHEENGYAV(F), VPGGQEIKDR(G), YNEDNGIVK(H) with T1R1/T1R3.
Figure 6.
The 2D diagram of the docking of DFNALPFK (A), DKLHEGIK (B), GVGPFDDDR(C), IDNEPEFRWA(D), IGDEAAENRV(E), SEHEENGYAV(F), VPGGQEIKDR(G), YNEDNGIVK(H) with T1R1/T1R3.
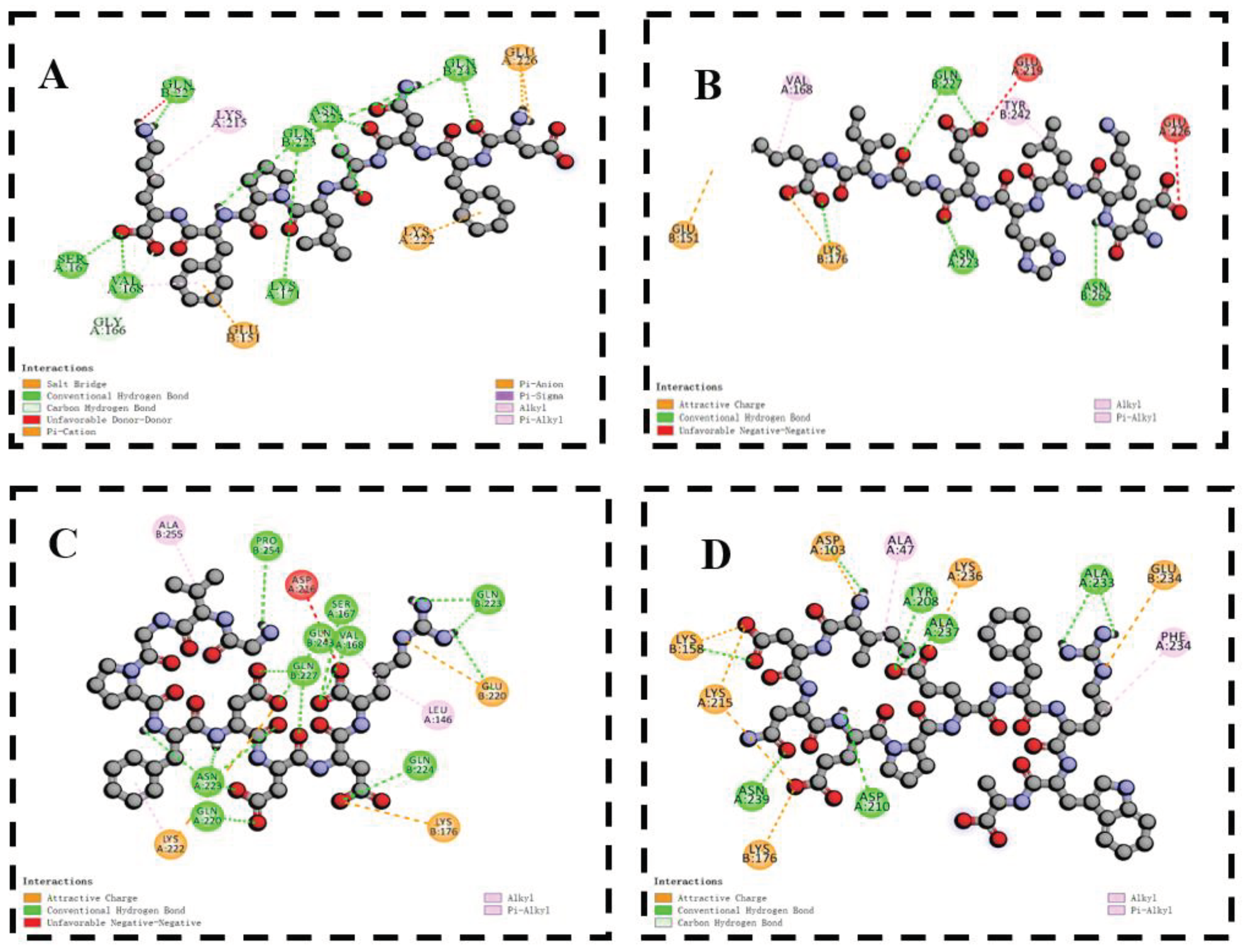
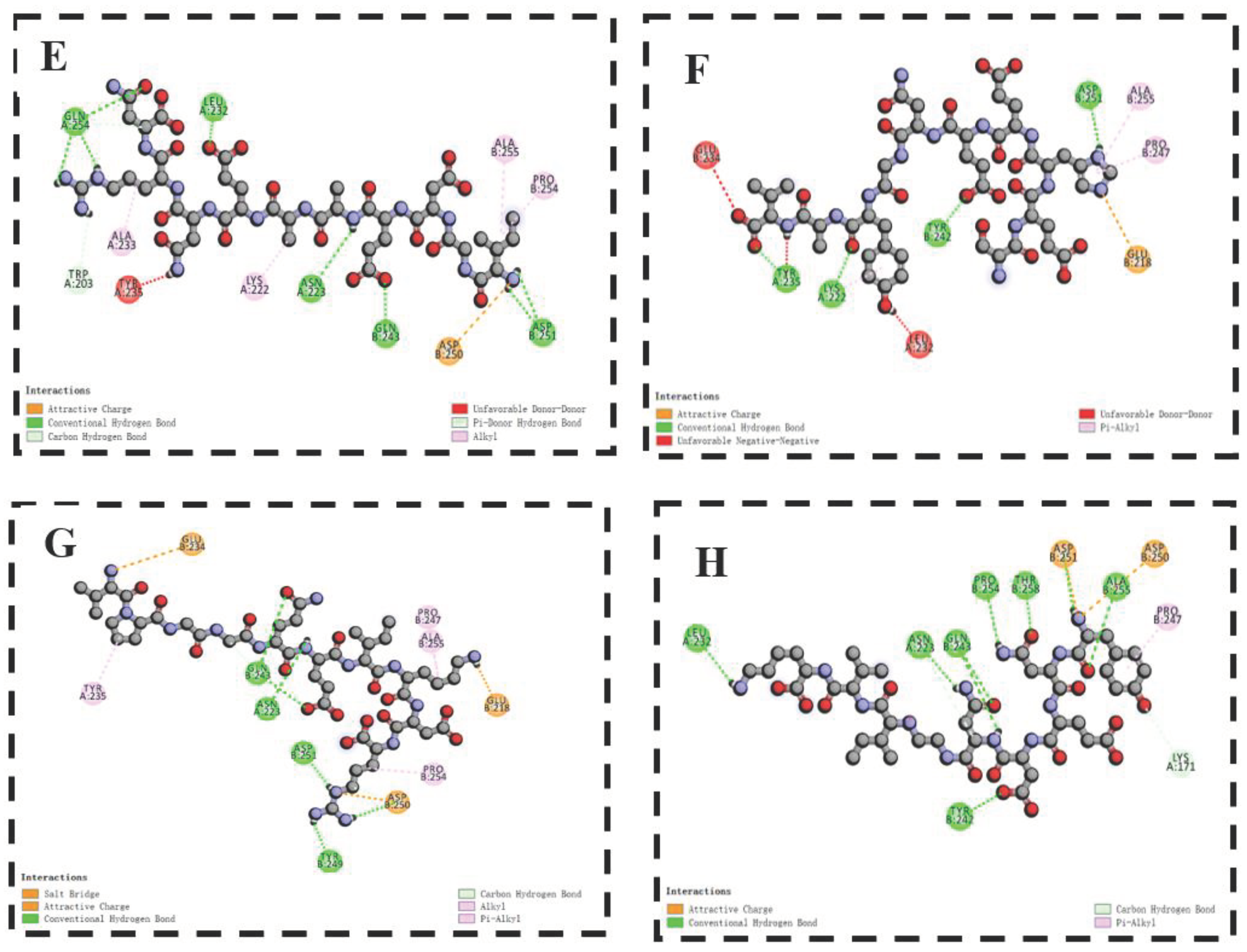
Figure 7.
The Saltiness enhancement characteristics of synthetic peptides. The bar matches the left y-axis (E-tongue) and the line matches the right y-axis (Sensory evaluation). Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the E-tongue and Sensory evaluation in different ultrafiltration fractions (p>0.05).
Figure 7.
The Saltiness enhancement characteristics of synthetic peptides. The bar matches the left y-axis (E-tongue) and the line matches the right y-axis (Sensory evaluation). Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the E-tongue and Sensory evaluation in different ultrafiltration fractions (p>0.05).
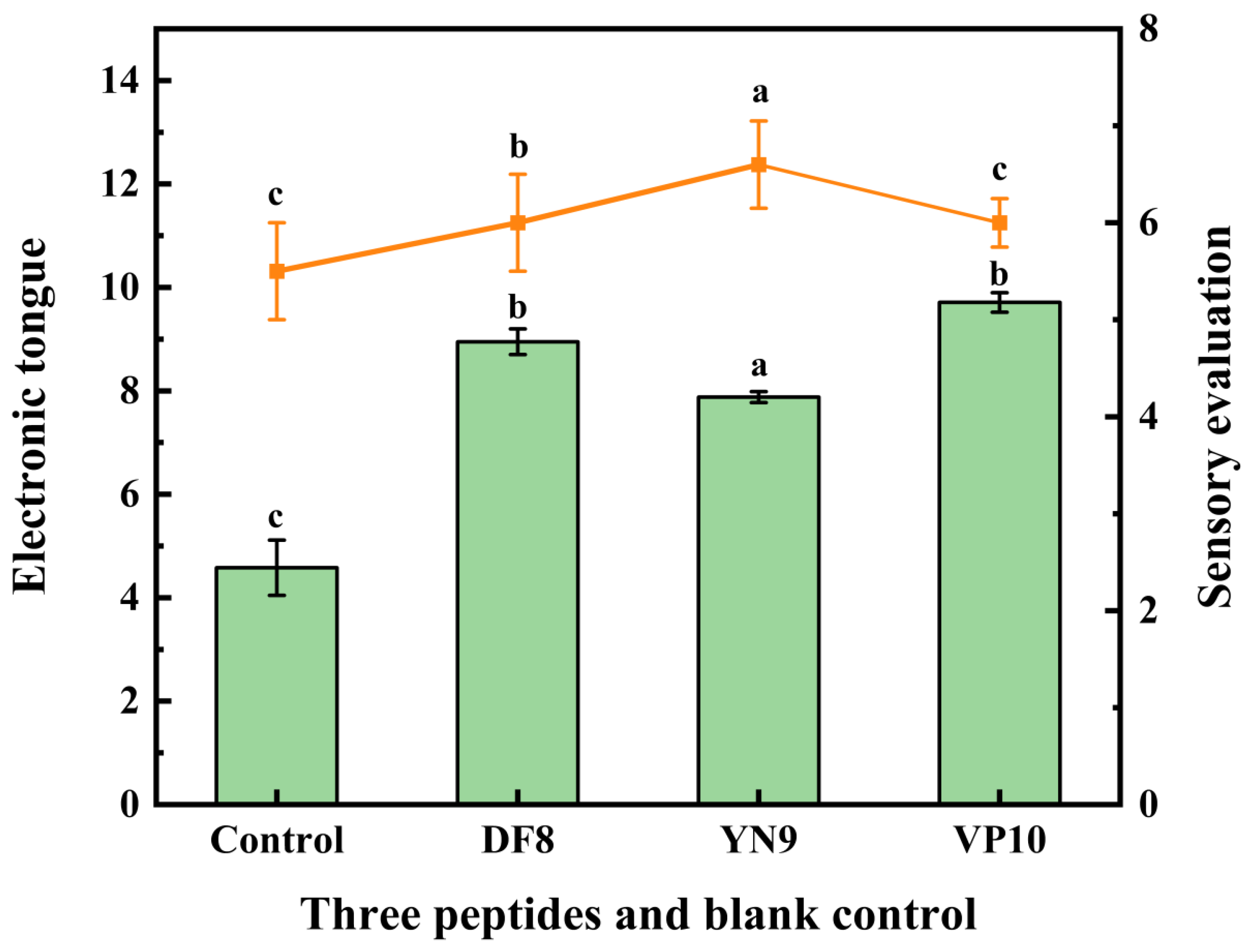
Figure 8.
Dose-feedback results for three synthetic peptides. The green line represents VP10, the blue line represents YN9 and the orange line represents DF8.
Figure 8.
Dose-feedback results for three synthetic peptides. The green line represents VP10, the blue line represents YN9 and the orange line represents DF8.
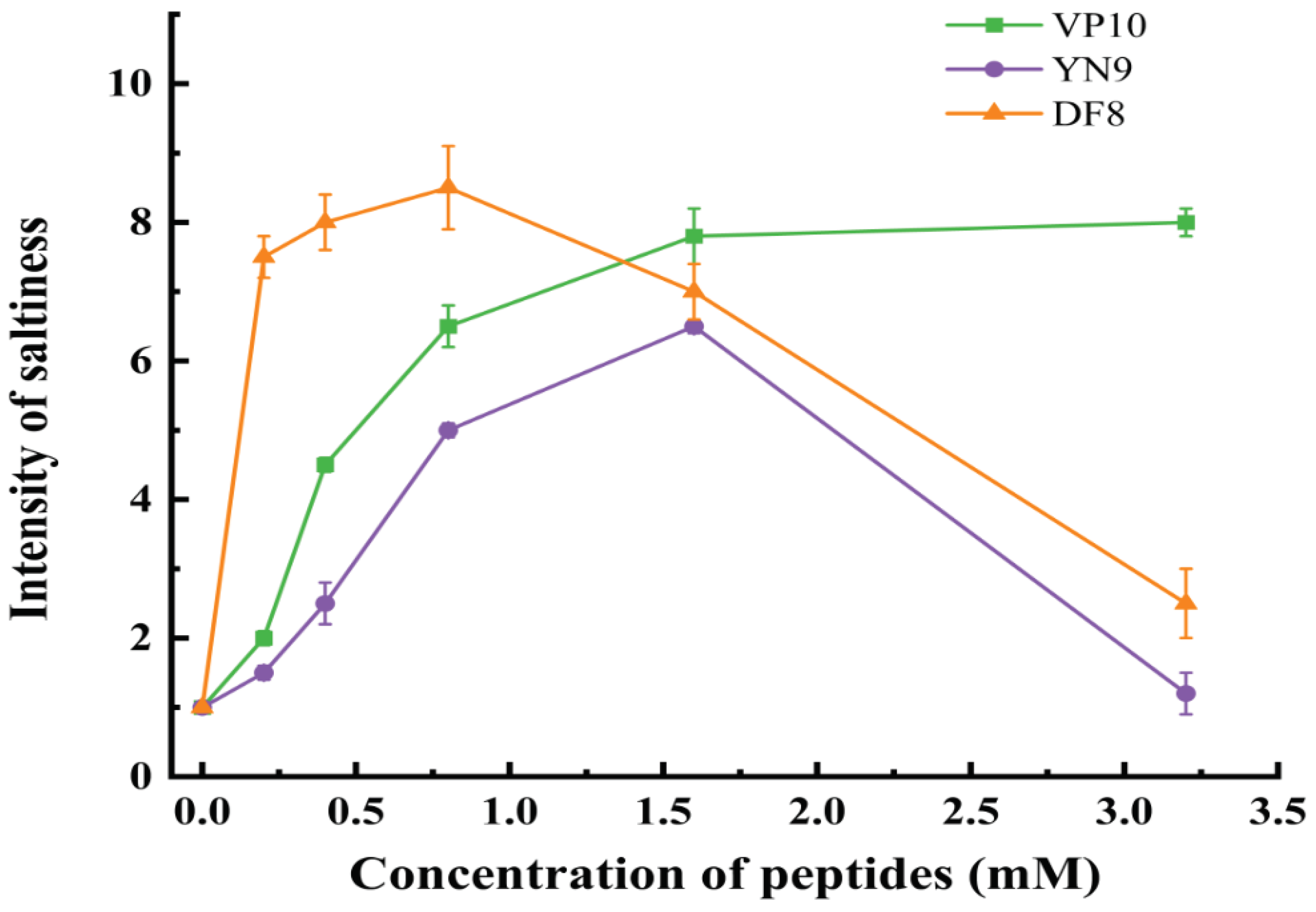

Table 1.
Results of the free amino acid content in the supernatant.
| Amino acids |
Content(mg/ml) | ||
|---|---|---|---|
| Monoenzyme | Dual Enzyme | Trienzyme | |
| Bitterness | 1.13±0.01a | 0.88±0.01b | 1.06±0.02c |
| Sweetness | 0.52±0.01b | 0.51±0.01b | 0.61±0.02a |
| umami | 0.52±0.02b | 0.53±0.02b | 0.65±0.01a |
| tasteless | 0.38±0.01b | 0.34±0.01b | 0.41±0.02a |
| Total | 2.55±0.01c | 2.26±0.01b | 2.74±0.02a |
* Using a one-way ANOVA, the same letter of the marker indicates that there is no statistical difference between the amounts of the free amino acid content in the supernatant obtained using different enzymatic methods (p>0.05).
Table 2.
Results of the peptide molecular weight distribution.
| Peptide | Length | affinity (kcal/mol) | |
|---|---|---|---|
| 1 | DFNALPFK | 8 | -9.2 |
| 2 | VPGGQEIKDR | 10 | -8.9 |
| 3 | YNEDNGIVK | 9 | -8.8 |
| 4 | IDNEPEFRWA | 10 | -8.6 |
| 5 | GVGPFDDDR | 9 | -8.7 |
| 6 | SEHEENGYAV | 10 | -8.7 |
| 7 | DKLHEGIK | 8 | -8.6 |
| 8 | IGDEAAENRN | 10 | -8.3 |
Table 3.
Taste attributes and threshold values of synthetic peptide.
| Peptides | Taste attribute | Threshold value (mM) | |
|---|---|---|---|
| water | 0.5%NaCl | ||
| DFNALPFK | sore, astringency, umami | 0.38±0.03b | 0.22±0.02c |
| YNEDNGIVK | sore, astringency | 0.45±0.02b | 0.33±0.05b |
| VPGGQEIKDR | sore, astringency, weak umami | 0.56±0.04a | 0.41±0.03a |
* Note: The values within the same column followed by different superscript letters are significantly different (p ≤ 0.05).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



