
Submitted:
29 February 2024
Posted:
01 March 2024
You are already at the latest version
Abstract
Metabolic-associated fatty liver disease (MASLD) is not only associated with traditional cardiovascular risk factors such as T2DM and obesity, but it is also an independent risk factor for the development of cardiovascular disease. MASLD has been shown to be independently related to endothelial dysfunction and atherosclerosis. MASLD is characterized by a chronic proinflammatory response that, in turn, may induce a prothrombotic state. Several mechanisms such as endothelial and platelet dysfunction, changes in the coagulation cascade, decreased fibrinolytic activity can contribute to induce the prothrombotic state. Platelets are players and addresses of metabolic dysregulation; obesity and insulin resistance are related to platelet hyperactivation. Furthermore, platelets can exert a direct effect on liver cells, particularly through the release of mediators from granules. Growing data in literature support the use of antiplatelet agent as a treatment for MASLD. The use of antiplatelets drugs seems to have potential clinical implication for the prevention of HCC patients with MASLD, since platelets contribute to fibrosis progression and cancer development. This review aims to summarize the main data on the role of platelets in the pathogenesis of MAFLD and its main complications such as cardiovascular events and the development of liver fibrosis. Furthermore, we will examine the role of antiplatelet therapy not only in the prevention and treatment of cardiovascular events but also as a possible anti-fibrotic and anti-tumor agent.
Keywords:
cancer
; NAFLD
; MASLD
; aspirin
; thromboxane
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of liver disease worldwide [1]. Recently, researchers and scientific societies moved towards changing the terminology. The novel nomenclature for a metabolic-associated fatty liver disease (MAFLD) has been proposed in 2020 by a group of experts to overcome the issues related to the old terminology [2]. The diagnosis of MAFLD is based on the presence of hepatic steatosis and at least one between these three conditions: type 2 diabetes mellitus (T2DM), obesity or metabolic dysregulation [2]. In 2023, a multi-society Delphi consensus statement proposed a new nomenclature about steatotic liver disease (SLD) [3]. SLD can be diagnosed histologically or by imaging, and it displays many potential aetiologies [3]. Metabolic dysfunction associated steatotic liver disease (MASLD) is defined as the presence of hepatic steatosis in conjunction with one cardiometabolic risk factor and no other discernible cause [3]. Persons with MASLD and steatohepatitis will be designated as metabolic dysfunction associated steatohepatitis (MASH) [3].
Recent evidence demonstrated that MASLD is not only associated with traditional cardiovascular risk factors such as T2DM [4] and obesity [5], but it is also an independent risk factor for the development of cardiovascular disease [6,7].
NAFLD has been shown to be independently related to endothelial dysfunction and atherosclerosis [8]. NAFLD is characterized by a chronic proinflammatory response that, in turn, may induce a prothrombotic state [9]. Several mechanisms such as endothelial and platelet dysfunction, changes in the coagulation cascade, decreased fibrinolytic activity can contribute to induce the prothrombotic state [10]. The “thrombophilic” state correlated with liver diseases may induce macrovascular and microvascular events; in particular, the formation of microthrombi in the hepatic venules can affect the course of liver disease [11]. The process of hepatic injury induced by the microthrombi in cirrhotic patients has been called “parenchymal extinction” [12,13]. The obliteration of hepatic and portal venules by microthrombi induce the disruption of the normal blood flow, thus leading to congestion, local ischemia and parenchymal injury [12,13]. The subsequent hepatocyte apoptosis induces the extinction of the parenchyma, which is replaced by fibrous septa [12].
Moreover, hepatic stellate cells can be activated directly by increased amounts of thrombin and coagulation proteases, even in the absence of intrahepatic thrombosis [11,13]. Furthermore, a growing body of evidence demonstrated that antithrombotic treatment not only prevents thrombotic events in patients with liver diseases, but also potentially slows down liver disease progression [14].
This review aims to summarize the role of platelets in the pathophysiology of patients with NAFLD and its complications, and to examine the evidence on the use of antiplatelet therapy in this category of patients.
2. Coagulation Cascade in Non-Alcoholic Fatty Liver Disease
There are currently conflicting data on the dysregulation of the coagulation cascade in patients with NAFLD, especially in the early stages. Kotronen et al performed a study on 54 subjects with NAFLD demonstrating that higher levels of coagulation factors VIII, XI, and XII were related to liver fat content [15]. Another study on 273 participants with histologically diagnosed NAFLD showed increased levels of fibrinogen, factor VIII, and von Willebrand factor (vWF) factor and decreased levels of antithrombin than healthy subjects [16]. Otherwise, levels of coagulation factors were not related to histological changes, and PAI-1 levels were the only parameter correlating with NAFLD severity [16]. Tripodi et al. demonstrated a significant prothrombotic state in 113 subjects with different stages of NAFLD, that was more evident in patients with more severe liver disease (cirrhosis) than in those with simple steatosis [17]. Many other works in NAFLD/NASH patients demonstrated higher levels of factors VII, VIII, IX, XII, vWF, and tissue factor (TF), probably related to higher C-reactive protein (CRP), plasminogen activator inhibitor 1 (PAI-1), and fibrinogen levels (9, 11, 13, 15). Otherwise, two works performed on biopsy-proven NAFLD patients argued that the prothrombotic dysregulation was more likely to be related to obesity or insulin resistance than to liver fat content [18,19]. Moreover, Assy et al. showed that some factors in the coagulation cascade are initially increased in NAFLD, but tend to decrease with liver disease progression, thus resulting in hemorrhagic diathesis [20]. Eventually, the concept of “rebalanced hemostasis” is actually well established in advanced forms of the disease that have progressed to cirrhosis [21].
3. Role of Platelet in Masld (NAFLD)
3.1. Platelet, Metabolism Dysregulation & Liver Disease
NAFLD/NASH often develops in the context of obesity, metabolic syndrome and T2DM [22] (Figure 1). NAFLD/NASH induces a chronic inflammatory state on the liver, which is characterized by a complex pathophysiology. Lipid species can induce an inflammatory response in the liver by activating infiltrating and resident immune cells. There is a relevant correlation between liver fat and markers of systemic inflammation and oxidative stress [23].
NAFLD patients are characterized by low levels of plasma endogenous secretory receptor for advanced glycation endproducts (esRAGE), interleukin (IL)-10 and adiponectin, and higher CD40 ligand, endogenous thrombin potential and oxidized low-density lipoproteins (LDL) [24]. The RAGE axis is involved in a wide spectrum of diseases, including T2DM, atherothrombosis, chronic renal failure, neurodegeneration, cancer and aging [25,26]. Circulating soluble forms of RAGE, arising from receptor ectodomain shedding and splice variant esRAGE secretion, can counteract RAGE-mediated pathogenesis, by acting as a decoy [25,26]. Several works showed that low levels of esRAGE can be useful as a biomarker of ligand-RAGE pathway hyperactivity and inadequate endogenous protective response [25,26].
Liver fat is directly related to CRP, isoprostanes, IL-6, intercellular adhesion molecule-1 (ICAM-1) and P-selectin levels [23]. ICAM-1 and P-selectin levels are significantly greater in subjects with liver steatosis and elevated ALT in comparison with those without steatosis [23,27]. In vitro studies demonstrated that exposure of hepatocytes to fatty-acids induces the expression of tumor necrosis factor (TNF)-α [28], IL-6, ICAM-1, [29] and isoprostanes, [30] through the nuclear factor-κB (NF-kB) [31]. Chronic hepatic activation of the NF-κB pathway can induce IL-6-mediated insulin resistance; TNF-α inhibition decreases liver fatty acid oxidation and insulin resistance by Kupffer cell activation [32,33].
In obese subjects, platelets are characterized by increased aggregability and activation [34] (Figure 2). The adipokine leptin provides a potential link between platelets, obesity and NAFLD. Leptin levels are related to NAFLD degree, and atherothrombotic events can be triggered in a platelet leptin receptor-dependent manner in those patients [35]. Indeed, leptin induces adenosine diphosphate (ADP)-related platelet aggregation at clinically relevant levels [36]. Thromboxane (TX)A2 release as well as hepatic TXA2 receptor expression are increased in NAFLD patients than healthy subjects [37]. In obese and insulin resistant patients, plasmatic levels of P-selectin are increased, and decrease after weight loss [38].
In obese patients, high levels of circulating platelet-derived microvesicles (PMVs) are directly related to BMI and waist circumference. In obese patients, PMVs are heterogeneous in size and distribution, with different amounts of molecules related to thrombosis and tumorigenesis [39]. Notably, weight reduction can decrease the circulating levels of PMVs [40]. PMVs may bear functional receptors from platelet membranes, thus exerting different effects. The exposure of phosphatidylserine is related to pro-thrombotic and inflammatory milieu [41]. PMVs can regulate the expression of cyclooxygenase-2 (COX-2) and prostacyclin (PGI2) in endothelial cells [42]. Moreover, PMVs are involved in monocytes and endothelial cells interaction by regulating the expression of ICAM-1 [43], and the recruitment of neutrophils by P-Selectin and IL-1 expression [44]. Furthermore, they can induce the production of pro-inflammatory molecules [e.g. CD40-ligand (CD40L), IL-1, IL-6 and TNF-α] [45], thus increasing the activation of the classic complement pathway [46].
Platelet hyper-activation is also observed in patients with hypercholesterolemia, together with higher expression of fibrinogen binding, P-selectin, superoxide anion and enhanced TXA2 production. Plasma from patients with high cholesterol levels is characterized by increased levels of platelet activation markers, such as CD40L, soluble P-selectin, platelet factor 4 (PF-4) and thromboglobulin [47]. Notably, triglycerides-rich particles can directly induce platelet activation [48].
In patients with insulin resistance, the adipokines resistin, leptin, PAI-1 and retinol binding protein 4 (RBP4) can dysregulate insulin receptor substrate-1 (IRS-1) expression in megakaryocytes, thus disrupting insulin signaling in platelets [49,50]. High glucose levels in diabetes are related to platelet hyperactivation, enhanced fibrinogen binding and TXA2 production (51-53). Platelets from obese, insulin-resistant subjects demonstrated an impaired response to nitric oxide (NO) and dysregulated cyclin guanosine monophosphate (cGMP)-dependent protein kinase (PKG) signaling system.
Moreover, the inhibition of platelet activation by PGI2 and the activation of the cyclin adenosine monophosphate (cAMP)-dependent protein-kinase (PKA) pathways are dysregulated [54]. In that context, platelet activation signals are overexpressed including increase in free intracellular calcium and the release of platelet activation molecules such as PMVs [52,55].
High glucose levels are correlated with the release of pro-oxidant molecules, which can enhance cytosolic phospholipase A2 signaling, thus catalyzing arachidonic acid release and TXA2 generation. Aldose reductase pathway activation can enhance TXA2 biosynthesis amplified by exposure to collagen [52].
In diabetes, TXA2-mediated platelet hyper-activation is driven by protein kinase C (PKC)/p38 mitogen activated protein kinase (MAPK) pathway, and it also related to increased CD40L release [52,56]. CD40L belongs to the TNF superfamily, and it is increased in NAFLD platelet surface [57]. In animal studies on insulin resistance, the genetic or antibody mediated disruption of CD40L signaling have been shown to decrease the effects of diet on steatosis, adipose tissue accumulation and insulin resistance, acting on hepatic very low density lipoprotein (VLDL) secretion and genes regulating lipid balance [58].
Moreover, diabetes is related to the loss of function and damage of mitochondria in platelets, cytochrome c release, caspase-3 activation, thus inducing platelet apoptosis [59].
3.2. Platelet & Liver
A growing body of evidence demonstrated that platelets play an active and direct role in the pathogenesis of liver disease and inflammation. Activated platelets contribute to cytotoxic T lymphocyte (CTL)-mediated liver damage in a model of viral hepatitis [60]. Kupffer cells can recruit platelets to the liver in early and late stages of NAFLD/NASH [61]. In the early stages of NASH this involved hyaluronan and platelet CD44 [61]. Glycoprotein Ib platelet subunit alpha (GPIbα) is a platelet surface membrane glycoprotein, and it is involved in the interaction of platelets with Kupffer cells at late but not at early phases of NASH development [61]. Furthermore, there is no evidence for a role of platelet GPIIb/IIIa in NASH [61]. Once recruited at the liver level, platelets can release granules containing several molecules [61]. Alpha and delta (dense) granules may release in the microenvironment the pro-aggregatory factors ADP, serotonin and thrombin along with inflammatory cytokines, chemokines and growth factors [62,63]. Platelets can store and even synthesize IL-1, PAI-1 and tissue factor (TF). Platelets release factors can change gene expression in endothelial cells, leukocytes, stromal cells and fibroblasts thus directly participating to inflammation [64]. Patients and mice with NAFLD have increased blood levels of molecules present in granules from platelets. Thrombospondin (TSP-1) is present in platelets, but it is synthesized by hepatic stellate cells, Kupffer cells, endothelial cells, and adipocytes also, and it can exert a beneficial effect on NAFLD due to inhibition of genes promoting lipid production [65].
Malehmir et al demonstrated that several cytokines and platelet-secreted factors are decreased upon anti-GPIbα antibody treatment thus arguing that molecules contained in α granules yield an increase of immune cell attracting chemokines/cytokines [61]. The same authors argued that three main ligands of GPIbα (vWF, macrophage integrin-1 and P-selectin) are not relevant for NASH, but they reinforced the concept of the pro-inflammatory function of α-granules in intrahepatic immune cell attraction [61]. Selectins have been demonstrated to be involved in leukocyte recruitment to liver microvasculature upon inflammatory response [66]. Eventually, GPIbα can play a role in disease development independent of a ligand [67]. Notably, anti-GPIbα antibody treatment can exert a therapeutical anti-NASH effect (61, 68, 69).
Indeed, both ticagrelor and anti-GPIbα antibody treatment can partially revert fibrosis on liver [61]. Therefore, P2Y12 antagonist treatment, depletion of functional GPIbα or lack of α-granules not only abolish activation, accumulation and adhesion of platelets to the liver endothelium but can also decrease intrahepatic immune-cell recruitment, thus reducing liver damage and disease development [61].
In models of liver disease, platelets have been shown to regulate gene expression in hepatocyte and deliver genetic signals to target cells. In vitro study with hepatoblastoma cell line (HepG2) demonstrated the direct transfer of mRNA from platelets to hepatocytes by internalizing platelets. Platelets internalization has been also demonstrated in animal model after a partial hepatectomy, and it was related to hepatocyte proliferation. On the other hand, enzymatic removal of platelet-derived ribonucleic acid (RNA) decreases hepatocyte proliferation [70]. Moreover, micro-RNA (miRNA) can be transferred from platelets to hepatocytes through the release of microparticles. PMP bearing miR-25-3p induces hepatocyte proliferation by changing gene expression [71].
Platelets may cross-talk with hepatic stellate cells by some molecules with both pro- and anti-fibrotic effects [72]. Adenine nucleotides and hepatocyte growth factor contained in platelets granules display antifibrotic effects [73], and those beneficial effects are proved by the reduction of liver fibrosis after treatment with platelet-rich plasma (PRP) [74].
Activated platelets exert a profibrotic effects by inducing hepatic micro-thrombosis [75] and through TGF [76], platelet-derived growth factor subunit B (PDGF-B) [77], vWF [78], platelet-derived sphingosine-1-phosphate signaling [79] which increase collagen secretion by hepatic stellate cells [80], thus becoming myofibroblasts [81].
The contribution of platelets to liver inflammation was confirmed by immunohistochemical staining on liver biopsies, which demonstrated the accumulation of platelet and neutrophil extracellular traps (NET) in liver, with a correlation with NAFLD activity score. Circulating platelets from NAFLD subjects were demonstrated to have significant increase of inflammatory transcripts, while leukocytes did not [82].
4. Platelet and Predictive Score of Fibrosis
NAFLD patients are characterized by higher values of platelet distribution width (PDW) compared to healthy subjects [83,84]. Larger platelets display higher amounts of granules and adhesion receptors, thus leading to increased platelet activation [84]. PDW is directly correlated with platelet size thus reflecting the heterogeneity in platelet morphology, and it changes upon platelet activation [83]. Cao et al. demonstrated that platelet count and PDW are negatively related to the stage of fibrosis [85].
Moreover, lower platelet count and higher mean platelet volume (MPV) have been shown to be independent NAFLD predictors [86]. Several previous studies reported that steatosis was related to an increase in MPV values [86,87]. A recent Korean study showed a relevant correlation between the presence of NAFLD and higher MPV values in 628 obese volunteers [87]. A recent meta-analysis combining data from eight cross-sectional and cohort studies confirmed that NAFLD patients compared with controls had significantly increased MPV [88]. A study performed on 100 patients with biopsy-proven NAFLD demonstrated a significant stepwise increase in MPV levels from subjects with normal histology through patients with simple steatosis to those with NASH; MPV was significantly related to the histological features of NASH, such as steatosis, inflammation, ballooning, and fibrosis [89]. Those observations can be explained by the fact that an increased systemic inflammatory response with the release of cytokines induced by NAFLD can change the platelet size, with larger platelets consequently secreting more granules and prothrombotic factors [88,90].
The platelet count is based on the balance between the production and destruction of platelets. In chronic liver diseases, the increase in spleen volume induces the sequestration and the destruction of platelets [91]. In NAFLD patients, platelet count was negative related to the degree and the severity of liver fibrosis (92-95). Indeed, fibrosis arises around the central veins, and portal hypertension seems to be primarily the result of central vein occlusion in NAFLD progression to NASH [96]. Moreover, platelets play a role in the process of liver fibrosis by decreasing expression of the main fibrogenic cytokine TGF-𝛽 and by enhancing the expression of matrix metalloproteinases [95].
Shah et al. evaluated the diagnostic accuracy of different scoring system in 541 NAFLD patients [97]. The area under the receiver operating characteristic (AUROC) of the fibrosis index based on 4 Factors (FIB4) index, NAFLD fibrosis score, the aspartate aminotransferase to platelet ratio index (APRI) and AST-to-platelet ratio to evaluate the diagnostic performance for hepatic fibrosis stages ≥ stage 3 were 0.802, 0.768, 0.730 and 0.720, respectively [97]. The AUROC of the platelet count to detect NAFLD patients with hepatic fibrosis ≥ stage 3 was 0.774 [92]. Indeed, the platelet count shows almost the same diagnostic accuracy than other biomarkers of liver fibrosis [92]. Therefore, it would be interesting to establish a threshold or range of platelet count that could suggest evolution towards hepatic fibrosis in patients with NAFLD.
In the work of Yoneda et al., the most accurate platelet count threshold for the detection of severe fibrosis (stage 3–4) was 19.2 x 104 /μl, and that for the diagnosis of cirrhosis (stage 4) was 15.3 x 104/μl in patients with NAFLD [92]. Therefore, NAFLD patients with platelet counts of less than 19.2 x 104/μl should be closely monitored, because it is likely that they may progress to NASH [92].
In a recent work, MPV has been shown to be decreased in women with MASLD associated with obesity and closely related to early liver inflammation in NASH [98]. Specifically, platelet count and plateletcrit have been related to hepatic ballooning [98]. Duran-Bertran et al. presented a new predictive model by using MPV, ALT levels and the presence of DM and MS to predict MASLD in obese women, displaying an area under the ROC curve of 0.84 [98].
In conclusion, patients with hepatic steatosis and NAFLD are characterized by higher values of PDW and MPV, while low platelet counts would be indicative of progression towards hepatic fibrosis.
5. Antiplatelet Therapy in NAFLD
A growing body of evidence demonstrate how antiplatelet therapy can mitigate fibrosis development in NAFLD, thus identifying it as a potential antifibrotic agent (81, 99-102). In murine models, aspirin has been shown to exert an anti-inflammatory effect by limiting the activation of hepatic stellate cells through the inhibition of the pro-inflammatory enzyme COX-2 and by interfering with the PDGF signaling [81,102]. Fujita et al. performed a study on fisher 344 male rats fed with a choline-deficient, l-amino acid-defined (CDAA) diet or a high-fat high-calorie (HF/HC) diet, and treated with or without the antiplatelet agents, aspirin, ticlopidine or cilostazol for 16 weeks [103]. All three antiplatelet drugs significantly decreased liver steatosis, inflammation and fibrosis in the CDAA diet group [103]. Cilostazol was the most effective, and it reduced liver steatosis also in HF/HC diet group [103]. Cilostazol exerts its beneficial effect against NAFLD by suppressing MAPK activation induced by oxidative stress and PDGF by intercepting signal transduction from a Akt to c-Raf [103]. Those findings are consistent with the results of a recent study conducted on human patients showing the association between the use of acetylsalicylic acid with reduced liver steatosis and fibrosis [104].
The association between acetylsalicylic acid and histological features of NAFLD has been further investigated in a prospective study conducted on a cohort of patients with histological diagnosis of NAFLD, followed by an extensive follow-up [50]. It has been observed that daily intake of acetylsalicylic acid is associated with a 46% reduction in the risk of developing fibrosis compared to non-users. Notably, this effect is specific to the molecule itself and not to the class of drugs, as other NSAIDs, lacking antifibrotic action, do not reduce the incidence of advanced fibrosis [50].
A cross-sectional analysis using data from the National Health and Nutrition Examination Survey III (NHANES III) specifically showed that the effect is linked to acetylsalicylic acid, unlike ibuprofen [104]. This study highlighted how aspirin use is significantly associated with lower fibrosis indices (e.g. FIB4, APRI), especially in individuals at risk of chronic liver diseases, with a consistent negative association across different types of chronic liver diseases (chronic viral hepatitis, suspected alcoholic liver disease and NASH) [104].
The possibility of a correlation between regular aspirin intake and a reduction in the prevalence of NAFLD was demonstrated also by Shen and colleagues in a study of the general population in the United States involving over 11000 adults [105]. Regular aspirin use, compared to not using it, was significantly related to a 38% lower probability of developing NAFLD [105]. The probability of developing NAFLD among men was less than half in those who reported occasional use of aspirin and two-thirds in those who used it regularly [105]. Aspirin use and NAFLD were not found to be correlated in women and younger participants, possibly because women have higher urinary excretion of aspirin and its metabolites than men, and younger subjects have higher aspirin clearance than older subjects [105]. Therefore, it can be argued that younger or female patients may require higher doses of aspirin to achieve a pharmacological effect against NAFLD. However, the lack of information on exact doses of aspirin made it difficult to evaluate dose-dependent associations between aspirin and NAFLD. Furthermore, the diagnosis of NAFLD was based on ultrasound but was not confirmed by liver biopsy; therefore, some participants may have been misclassified as having or not having NAFLD. According to the findings, taking aspirin regularly may lead to a lower risk of NAFLD, especially in men or older adults [105]. However, as a cross-sectional study, it was not possible to assess whether the use of aspirin could prevent the incidence or reduce the progression of NAFLD [105].
It also been proven that aspirin use was associated with significantly lower liver fibrosis indexes among U.S. adults with suspected chronic liver disease [104]. According to a systematic review and meta-analysis, there is a protective relationship between antiplatelet therapy and the prevalence of advanced liver fibrosis in patients with NAFLD [106].
In the work of Simon et al, longer duration of aspirin use was associated with progressively reduced risk for incident advanced fibrosis, and this association was similar for any age and sex [50].
In a prospective study of patients with bioptically proven NAFLD, daily use of aspirin was associated with less severe histological characteristics of NAFLD and NASH and a lower risk of progression towards advanced fibrosis [50]. Compared to non-regular use, daily use of aspirin has been associated with significantly lower odds of NASH [odds ratio adjusted (aOR), 0.68; 95% confidence interval (CI), 0.37-0.89] and fibrosis (aOR, 0.54; 95% CI, 0.31-0.82) [50]. Daily consumers of aspirin had a significantly lower risk of developing advanced fibrosis than nonhabitual consumers [hazard ratio adjusted (aHR) [107], 0.63; 95% CI. 0.43-0.85] [50]. This relationship appeared to depend on duration, with the greatest benefit found with at least 4 years of aspirin use (aHR, 0.50; 95% CI, 0.35-0.73) [50].
Several molecular mechanisms have been suggested to explain the possible efficacy of antiplatelet therapy against NAFLD and its progression to NASH. Aspirin may stimulate NO and prostacyclin production through the expression of endothelial NO synthase and vascular endothelial growth factor, resulting in antioxidant activity [108]. Moreover, aspirin can inhibit the production of TNF-α, which causes inflammation and liver fibrosis [109]. Furthermore, aspirin reduces the expression of PDGF, thus decreasing hepatic inflammation, steatosis and fibrosis [110]. Antiplatelet therapy can stimulate the insulin signaling pathway and improving IR by PDGF-induced activation of the mitogen-activated protein kinase pathway [111,112]. Eventually, aspirin-induced lipoxins in the liver regulate cytokine-chemokine axes in liver cells [113]. Malehmir et al demonstrated that several cytokines and platelet-secreted factors are decreased upon anti-GPIbα antibody treatment [61]. Moreover, P2Y12 antagonist treatment, as well as depletion of functional GPIbα or lack of α-granules can decrease activation, accumulation and adhesion of platelets to the liver endothelium, thus ameliorating liver damage and attenuating fibrosis development [61].
Furthermore, in presence of hyperlipidemia and metabolic dysregulation, many of the biological substrates and conditions, such as IR and pro-inflammatory cytokines, are involved in the pathogenesis of both NAFLD and atherosclerosis [114]. Aspirin has been shown to improve both atherosclerosis and NAFLD simultaneously [115]; this mainly occurs through the following two pathways: activating catabolic lipid metabolism (decreased lipid accumulation in HepG2 cells, aorta and liver) and reducing inflammation (decrease in NF-κB and TNF-α) [115].
6. Antiplatelet Therapy and Cancer
Hepatocellular carcinoma (HCC) incidence is increasing most rapidly than any cancer, with an age-adjusted annual increase of 3,8% and 2,8% in men and women in the U.S., respectively [116,117]. Several epidemiological studies confirmed the increasing incidence and risk of NAFLD-associated HCC [118,119]. The increasing rates of NAFLD-associated HCC is also demonstrated by changes in liver transplantation (LT) indications [120]. The European Liver Transplant Registry database from 2002-2016 showed that 8,4% of transplant patients were for NASH in 2016 (compared to 1,3% in 2002), 39% of whom had HCC [121].
Pharmaceutical inhibition of platelet function by using antiplatelet treatment have been previously shown to correlate with better outcome in cancer patients (122-124). Therefore, the potential cancer chemo-preventive role of antiplatelet drugs in NAFLD/NASH patients, especially aspirin and other NSAIDs, represents an intriguing research area [125].
A recent meta-analysis by AISF (Associazione Italiana per lo Studio del Fegato) HCC Special Interest Group including 20 studies demonstrated that patients treated with antiplatelet therapy show a 40% reduced risk of HCC incidence and a halved risk for all-cause mortality in patients with HCC treated with either curative or palliative strategies [126]. In this meta-analysis patients with a clear indication for antiplatelet therapy were evaluated, but there was no specific evaluation for subgroups of patients nor for the NAFLD/NASH field specifically.
Another study analyzing prospective data of the National Institutes of Health– AARP Diet and Health Study cohort demonstrated that aspirin users had statistically significant reduced risk of HCC incidence and mortality due to chronic liver disease (CLD) compared to those who did not use aspirin [127]; otherwise, users of non-aspirin nonsteroidal anti-inflammatory drugs (NSAIDs) had a reduced risk of mortality due to CLD but did not have lower risk of HCC incidence in comparison with those who did not use non-aspirin NSAIDs [127]. This finding may reflect differences in COX-inhibitory effects of aspirin and non-aspirin NSAIDs, because aspirin irreversibly inhibits and modifies both isoforms of COX, the constitutive COX1, which is expressed in most normal tissues, and the inducible COX2, which is highly expressed in response to a broad spectrum of proinflammatory stimuli, including those that mediate hepatic carcinogenesis [127]; moreover, while aspirin irreversibly inhibits COX isoenzymes, NSAIDs do so reversibly, with transient effects that may reduce anti-fibrotic activity [127].
Lee et al. evaluated 35,898 cirrhotic patients dividing them into two groups (those treated with aspirin for at least 84 days vs controls without treatment) [128]. Daily aspirin use was independently associated with a reduced risk of HCC (three-year HR 0.57; 95% CI 0.37-0.87; p = 0.0091; five-year HR 0.63, 95% CI 0.45-0.88; p = 0.0072) inversely correlated with the treatment duration [3-12 months: HR 0.88 (95% CI 0.58-1.34); 12-36 months: HR 0.56 (0.31-0.99); and ≥ 36 months: HR 0.37 (0.18-0.76)] [128]. Overall mortality rates were significantly lower among aspirin users compared with untreated controls [three-year HR 0.43 (0.33-0.57); five-year HR 0.51 (0.42-0.63)] [128]. The incidence of GI bleeding was not increased in daily aspirin users compared to untreated cirrhotic patients without a previous history of GI bleeding or with a previous history of GI bleeding [128].
Furthermore, Choi et al. evaluated LT recipients with pre-transplant HCC and divided them into two groups according to the use of antiplatelet agents for > 90 days or not [129]. The 5-year cumulative incidences of HCC recurrence and HCC-specific death were similar between the antiplatelet and non-antiplatelet groups [129]. All-cause and non-HCC deaths were also similar between the two groups [129].
A recent study compared the outcomes between 33,484 patients with NAFLD who continuously received a daily dose of aspirin for 90 days or more, and 55,543 subjects who had not received antiplatelet therapy, by using Taiwan's National Health Insurance Research Database [130].
The 10-year cumulative incidence of HCC in the treated group was significantly lower than that in the untreated group (0.25% [95% CI, 0.19-0.32%] vs. 0.67% [95% CI, 0.54-0.81%]; P < 0.001) [130]. Aspirin therapy was significantly associated with a reduced HCC risk (aHR 0.48 [95% CI, 0.37-0.63]; P < 0.001) [130]. In older (age > 55 years) patients with serum ALT elevation (high risk patients), the 10-year cumulative incidence of HCC in the treated group was significantly lower than that in the untreated group (3.59% [95% CI, 2.99-4.19%] vs. 6.54% [95% CI, 5.65-7.42%]; P < 0.001) [130]. Moreover, HCC risk was significantly lower by using aspirin for ≥ 3 years (aHR 0.64 [95% CI, 0.44-0.91]; P = 0.013), when compared with short-term use (< 1 year) [130].
Tan et al. performed a metanalysis on 147 283 participants by including 19 studies evaluating patients with hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol-related liver disease (ALD) or NASH that were administered at least one NSAID or antiplatelet therapy for a defined period of time and were followed for at least 6 months [131]. Aspirin use reduced the risk of HCC incidence (HR: 0.51, 95% CI: 0.36-0.72); and improved liver-related mortality (OR: 0.32, 95% CI: 0.15-0.70), with a small increased risk of gastrointestinal bleeding events (OR: 1.32, 95% CI: 1.08-1.94) [131]. With respect to HCC recurrence following treatment, analysis of all aspirin and NSAID treatment was associated with a decreased risk of HCC recurrence (HR: 0.80, 95% CI: 0.75-0.86) [131]. By stratified analysis, only the non-aspirin NSAID group showed significant risk reduction (HR: 0.73, 95% CI: 0.63-0.84) [131].
Therefore, there is evidence that the use of antiplatelet drugs (both aspirin and others) is correlated with a reduced incidence of HCC. Regarding the type of drug (aspirin vs other NSAIDs) there are non-univocal data, in some cases conflicting [127,131], which deserve studies based on specific molecular mechanisms.
Molecular Mechanisms
Antiplatelet drugs, in particular aspirin, have been investigated as cancer preventatives, particularly in the context of colorectal cancer [125,132]. A recent survey demonstrated that among subjects who had previously taken aspirin, 1.9% had taken it for cancer prevention [133]. Participants with a personal or family history of cancer were more likely to perceive aspirin as necessary for cancer prevention [133]. Otherwise, concerns about taking aspirin at higher doses and its side effects, such as gastrointestinal bleeding, were common [133].
The beneficial effects of antiplatelet therapy on cancer can be explained by the involvement of TX-dependent platelet activation and cyclooxygenase-2-driven inflammatory response in the early stages of carcinogenesis [134,135] (Figure 3). Joharatnam-Hogan et al measured urinary 11-dehydro-thromboxane B2 (U-TXM), a biomarker of in vivo platelet activation, in patients with breast, colorectal, gastro-oesophageal and prostate cancers after radical therapy [136]. Aspirin 100 mg daily decreased U-TXM similarly across all tumour types; however, aspirin 300 mg daily provided no additional suppression of U-TXM compared with 100 mg [136].
The beneficial effects of antiplatelet drugs against the development of HCC in patients with NAFLD can be divided into two main mechanisms. On the one hand, the slowdown in the progression towards liver fibrosis and NASH (as previously described), and on the other a possible direct effect of platelets and their mediators on carcinogenesis mechanisms.
A recent work by Ma et al. demonstrated that the inhibition of the P2Y12 receptor on platelets can promote tumor growth via CD40L in mice with NAFLD [137]; however, there are conflicting data on this mechanism in the literature. Indeed, in the context of NAFLD and HCC, platelets can exert both pro-hepatocarcinogenesis function by driving NASH pathology, and anti-tumor activity against established tumors by driving CD8+ T cell responses (122-124, 138, 139).
Previous studies showed that platelets can enhance adaptive immunity [138,139], and activated platelets can release several molecules from α-granules, such as CD40L (Table 1). Several studies in literature showed CD40L to exert a role in anti-tumor immunity, and platelets are the main source of circulating CD40L [140]. Platelet-derived CD40L is greater released in both NAFLD mouse models and patients with NASH [137]. Moreover, TGF-β released from platelets can impair T cell anti-tumor function [141].
A recent study in mice by Vogt et al showed that intratumoral co-stimulation with CD40L-expressing dendritic cells (DC) significantly improves vaccination with murine (m) alpha-fetoprotein (AFP)-transduced DC in pre-established HCC in vivo [142]. Combined therapy induced an early and strong Th1-shift in the tumor environment as well as higher tumor apoptosis, leading to synergistic tumor regression of HCC [142].
Different platelet inhibition strategies can affect platelet CD40L release differently, causing opposite effects on HCC tumor growth in NAFLD [137]. Actually, Santilli et al previously showed TX-dependent CD40L release in patients with T2DM, showing dose-dependent inhibition of CD40L circulating levels upon aspirin administration and after slow recovery after aspirin withdrawal [56].
Indeed, COX inhibition fails to block platelet CD40L release depending on internal Ca2+ and protein kinase C but not involving tyrosine kinases, extracellular signal-regulated kinase (ERK) or p38, and COXs are downstream of ERK and p38 during platelet activation [143]. Therefore, P2Y12 inhibition related beneficial effect on HCC development in NAFLD may not be found using COX or phosphodiesterase 3 (PDE3) inhibitors [137]. Blocking platelet aggregation while sparing CD40L release using non-P2Y12 inhibitors such as aspirin may be more suitable for HCC patients with NAFLD [61].
Recent data demonstrated an IL-12 dependent increase of CD40L production in bone marrow megakaryocytes in NAFLD models [137,144]. Particularly, macrophages can induce an increase of IL-12 in the liver and CD40L production from bone marrow megakaryocytes [137]. A recent work demonstrated that the expansion of hepatic dendritic cells in NASH is induced by increased production of dendritic cell progenitors inside the bone marrow and enhanced liver recruitment [145]. Moreover, megakaryocytes harbored in lung can produce higher CD40L amount [137,145]. Intriguing, the lung can potentially contribute to liver tumor regulation in NAFLD by modulating platelets and CD40L release.
Moreover, data from a recent study on obese patients suggested that in NAFLD patients CD40L protein production is induced in megakaryocytes rather than NAFLD or tumors causing a transfer of CD40L pre-mRNA or mRNA into platelets [146].
Eventually, CD40L is related to a strong CD8+ T cells responses through CD40 licensing of dendritic cells [147]. Platelet-derived CD40L can increase CD8+ T cell activation [137] and their recruitment to liver in NAFLD [61]. Notably, to depletion of CD8+ T cells has been showed to decreased antiplatelet beneficial effect on HCC development in NAFLD, thus suggesting that the anti-HCC platelet function is at least in part mediated by CD8+ T cells [137,148].
7. Implications for Antiplatelet Therapy in Primary Prevention
The use of antiplatelet therapy in primary prevention has been scaled down in recent years on the basis of 3 large trials which highlighted a greater risk of bleeding complications compared to a benefit in terms of reduction of cardiovascular events (149-151). Subsequent works and expert opinions reworked this type of evidence, thereby suggesting that in patients at higher cardiovascular risk (e.g. T2DM patients) the benefit of antiplatelet therapy exceeds the bleeding risk [152]. Nowadays, several international guidelines report different types of indications for the use of antiplatelet therapy in primary prevention, thus creating heterogeneity in prescription methods [153].
The recent “DCRM Multispecialty Practice Recommendations for the management of diabetes, cardiorenal, and metabolic diseases” argued that the use of aspirin is not generally recommended for patients without a history of ASCVD or other risk factors [154]. The Task Force believes prescribing aspirin for patients with two or more cardiovascular risk factors (i.e., advanced age, elevated non-HDL-C, elevated LDL-C, low HDL-C, diabetes, hypertension, CKD, cigarette smoking, family history of ASCVD, elevated CAC score > 100) may benefit those who are not at increased risk of bleeding [154].
Nowadays, the prescription of antiplatelet therapy in patients with MASLD is based mainly (solely) on the evaluation of cardio-metabolic comorbidities, with no reference to the degree of hepatic alteration; this is due to the lack of strong evidence and adequate studies.
A recent work randomized two thousand four hundred participants with NASH to a strategy with polypill containing aspirin, atorvastatin, hydrochlorothiazide, and valsartan vs a control group with the standard or care, with a 5-years follow-up [144]. As expected, the adjusted relative risk of major cardiovascular events in participants with NASH (0.35, 95% CI 0.17-0.74) was under half that for participants without NASH (0.73, 95% CI 0.49-1.00), even if the difference did not reach statistical significance [155]. In this study, NAFLD patients demonstrated a significant decrease in liver enzymes after 60 months of follow-up (intragroup -12.0 IU/L, 95% CI -14.2 to -9.6) [155].
Furthermore, growing evidence in the literature refers to an inter-individual variability in response and efficacy to the use of antiplatelet therapy, particularly in some categories of subjects with specific comorbidities [156,157]. More specifically, antiplatelet therapy can be less effective in patients with a faster recovery of platelet COX-1 activity during the 24-hour dosing interval [157].
A recent study by Simeone et al. showed that patients with accelerated COX-1 recovery are characterized by reduced platelet glycoprotein (GP)Ibα shedding, and this can contribute to higher thrombopoietin (TPO) production and higher rates of newly formed PLT, escaping aspirin inhibition over 24 hours [158]. Intriguing, NAFLD and visceral obesity have been identified among clinical markers, together with younger age and higher TPO/GC ratio, predictive for the likelihood of faster COX-1 recovery and suboptimal aspirin response [158].
8. Conclusions
Growing data in literature support future research on the use of an antiplatelet agent as a new treatment for MASLD. Nowadays, there are limited data from prospective studies on the effects of aspirin in patients with MASLD. Most studies available on the association between antiplatelet drugs and NAFLD prevalence are observational. Prospective studies and randomized and controlled clinical trials are necessary to determine if antiplatelet therapy can prevent or delay the onset or progression of NAFLD, whether this effect is restricted to certain population subgroups, and what type and dose range of antiplatelet agent is most effective. The use of antiplatelets drugs seems to have potential clinical implication for the prevention of HCC patients with MASLD, since platelets contribute to fibrosis progression and cancer development. Therefore, MASLD patients may require anti-platelet agents to prevent cardiovascular diseases, fibrosis progression and cancer development.
Conflicts of Interest
authors declare no conflict of interest.
References
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005, 112, 2735–2752. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Tilg, H.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of incident diabetes mellitus: an updated meta-analysis of 501 022 adult individuals. Gut. 2021, 70, 962–969. [Google Scholar] [CrossRef]
- Li, L.; Liu, D.W.; Yan, H.Y.; Wang, Z.Y.; Zhao, S.H.; Wang, B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obes Rev. 2016, 17, 510–519. [Google Scholar] [CrossRef]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; et al. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: an updated systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef]
- Lee, H.H.; Lee, H.A.; Kim, E.J.; Kim, H.Y.; Kim, H.C.; Ahn, S.H.; et al. Metabolic dysfunction-associated steatotic liver disease and risk of cardiovascular disease. Gut. 2023. [CrossRef] [PubMed]
- Boccatonda, A.; Andreetto, L.; D'Ardes, D.; Cocco, G.; Rossi, I.; Vicari, S.; et al. From NAFLD to MAFLD: Definition, Pathophysiological Basis and Cardiovascular Implications. Biomedicines 2023, 11, 883. [Google Scholar] [CrossRef]
- Targher, G.; Chonchol, M.; Miele, L.; Zoppini, G.; Pichiri, I.; Muggeo, M. Nonalcoholic fatty liver disease as a contributor to hypercoagulation and thrombophilia in the metabolic syndrome. Semin Thromb Hemost. 2009, 35, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Northup, P.G.; Sundaram, V.; Fallon, M.B.; Reddy, K.R.; Balogun, R.A.; Sanyal, A.J.; et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008, 6, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Northup, P.G.; Argo, C.K.; Shah, N.; Caldwell, S.H. Hypercoagulation and thrombophilia in nonalcoholic fatty liver disease: mechanisms, human evidence, therapeutic implications, and preventive implications. Semin Liver Dis. 2012, 32, 39–48. [Google Scholar] [CrossRef]
- Wanless, I.R.; Wong, F.; Blendis, L.M.; Greig, P.; Heathcote, E.J.; Levy, G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995, 21, 1238–1247. [Google Scholar]
- Anstee, Q.M.; Wright, M.; Goldin, R.; Thursz, M.R. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis. 2009, 13, 117–126. [Google Scholar] [CrossRef]
- Lisman, T.; Kamphuisen, P.W.; Northup, P.G.; Porte, R.J. Established and new-generation antithrombotic drugs in patients with cirrhosis - possibilities and caveats. J Hepatol. 2013, 59, 358–366. [Google Scholar] [CrossRef]
- Kotronen, A.; Joutsi-Korhonen, L.; Sevastianova, K.; Bergholm, R.; Hakkarainen, A.; Pietiläinen, K.H.; et al. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int. 2011, 31, 176–183. [Google Scholar] [CrossRef]
- Verrijken, A.; Francque, S.; Mertens, I.; Prawitt, J.; Caron, S.; Hubens, G.; et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014, 59, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Fracanzani, A.L.; Primignani, M.; Chantarangkul, V.; Clerici, M.; Mannucci, P.M.; et al. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J Hepatol. 2014, 61, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lallukka, S.; Luukkonen, P.K.; Zhou, Y.; Isokuortti, E.; Leivonen, M.; Juuti, A.; et al. Obesity/insulin resistance rather than liver fat increases coagulation factor activities and expression in humans. Thromb Haemost. 2017, 117, 286–294. [Google Scholar] [CrossRef]
- Potze, W.; Siddiqui, M.S.; Boyett, S.L.; Adelmeijer, J.; Daita, K.; Sanyal, A.J.; et al. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J Hepatol. 2016, 65, 980–987. [Google Scholar] [CrossRef]
- Assy, N.; Bekirov, I.; Mejritsky, Y.; Solomon, L.; Szvalb, S.; Hussein, O. Association between thrombotic risk factors and extent of fibrosis in patients with non-alcoholic fatty liver diseases. World J Gastroenterol. 2005, 11, 5834–5839. [Google Scholar] [CrossRef] [PubMed]
- Zanetto, A.; Campello, E.; Senzolo, M.; Simioni, P. The evolving knowledge on primary hemostasis in patients with cirrhosis: A comprehensive review. Hepatology. 2023. [CrossRef]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Fricker, Z.P.; Pedley, A.; Massaro, J.M.; Vasan, R.S.; Hoffmann, U.; Benjamin, E.J.; et al. Liver Fat Is Associated With Markers of Inflammation and Oxidative Stress in Analysis of Data From the Framingham Heart Study. Clin Gastroenterol Hepatol. 2019, 17, 1157–1164. [Google Scholar] [CrossRef]
- Santilli, F.; Blardi, P.; Scapellato, C.; Bocchia, M.; Guazzi, G.; Terzuoli, L.; et al. Decreased plasma endogenous soluble RAGE, and enhanced adipokine secretion, oxidative stress and platelet/coagulative activation identify non-alcoholic fatty liver disease among patients with familial combined hyperlipidemia and/or metabolic syndrome. Vascul Pharmacol. 2015, 72, 16–24. [Google Scholar] [CrossRef]
- Santilli, F.; Vazzana, N.; Bucciarelli, L.G.; Davì, G. Soluble forms of RAGE in human diseases: clinical and therapeutical implications. Curr Med Chem. 2009, 16, 940–952. [Google Scholar] [CrossRef]
- Vazzana, N.; Santilli, F.; Cuccurullo, C.; Davì, G. Soluble forms of RAGE in internal medicine. Intern Emerg Med. 2009, 4, 389–401. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Scala, L.; Zoppini, G.; Zenari, L.; Falezza, G. Non-alcoholic hepatic steatosis and its relation to increased plasma biomarkers of inflammation and endothelial dysfunction in non-diabetic men. Role of visceral adipose tissue. Diabet Med. 2005, 22, 1354–1358. [Google Scholar]
- Chávez-Tapia, N.C.; Rosso, N.; Uribe, M.; Bojalil, R.; Tiribelli, C. Kinetics of the inflammatory response induced by free fatty acid accumulation in hepatocytes. Ann Hepatol. 2013, 13, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.W.; Hsu, Y.C.; Lee, T.F.; Lin, Y.; Chiu, Y.T.; Yang, K.C.; et al. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Gardemann, A.; Keilhoff, G.; Peter, D.; Wiswedel, I.; Schild, L. Prevention of free fatty acid-induced lipid accumulation, oxidative stress, and cell death in primary hepatocyte cultures by a Gynostemma pentaphyllum extract. Phytomedicine. 2012, 19, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010, 59, 347–357. [Google Scholar] [CrossRef]
- Beavers, C.J.; Heron, P.; Smyth, S.S.; Bain, J.A.; Macaulay, T.E. Obesity and Antiplatelets-Does One Size Fit All? Thromb Res. 2015, 136, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin Mol Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef]
- Corsonello, A.; Perticone, F.; Malara, A.; De Domenico, D.; Loddo, S.; Buemi, M.; et al. Leptin-dependent platelet aggregation in healthy, overweight and obese subjects. Int J Obes Relat Metab Disord. 2003, 27, 566–573. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.; Mao, J.; Li, H.; Wang, M.; Zhang, H.; et al. Genistein Ameliorates Non-alcoholic Fatty Liver Disease by Targeting the Thromboxane A(2) Pathway. J Agric Food Chem. 2018, 66, 5853–5859. [Google Scholar] [CrossRef]
- Russo, I.; Traversa, M.; Bonomo, K.; De Salve, A.; Mattiello, L.; Del Mese, P.; et al. In central obesity, weight loss restores platelet sensitivity to nitric oxide and prostacyclin. Obesity (Silver Spring). 2010, 18, 788–797. [Google Scholar] [CrossRef]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; et al. Platelet-Derived Microparticles From Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk With Cancer and Endothelial Cells. Front Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Horigome, H.; Tanaka, K.; Nakata, Y.; Ohkawara, K.; Katayama, Y.; et al. Impact of weight reduction on production of platelet-derived microparticles and fibrinolytic parameters in obesity. Thromb Res. 2007, 119, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Burnouf, T.; Goubran, H.A.; Chou, M.L.; Devos, D.; Radosevic, M. Platelet microparticles: detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014, 28, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest. 1997, 99, 2118–2127. [Google Scholar] [CrossRef]
- Smith, C.W.; Marlin, S.D.; Rothlein, R.; Toman, C.; Anderson, D.C. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989, 83, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Forlow, S.B.; McEver, R.P.; Nollert, M.U. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000, 95, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes. EMBO Mol Med. 2013, 5, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Braig, D.; Nero, T.L.; Koch, H.G.; Kaiser, B.; Wang, X.; Thiele, J.R.; et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun. 2017, 8, 14188. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; et al. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr Metab Cardiovasc Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef]
- Yamazaki, M.; Uchiyama, S.; Xiong, Y.; Nakano, T.; Nakamura, T.; Iwata, M. Effect of remnant-like particle on shear-induced platelet activation and its inhibition by antiplatelet agents. Thromb Res. 2005, 115, 211–218. [Google Scholar] [CrossRef]
- Gerrits, A.J.; Gitz, E.; Koekman, C.A.; Visseren, F.L.; van Haeften, T.W.; Akkerman, J.W. Induction of insulin resistance by the adipokines resistin, leptin, plasminogen activator inhibitor-1 and retinol binding protein 4 in human megakaryocytes. Haematologica. 2012, 97, 1149–1157. [Google Scholar] [CrossRef]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; et al. Daily Aspirin Use Associated With Reduced Risk For Fibrosis Progression In Patients With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol. 2019, 17, 2776–2784. [Google Scholar] [CrossRef]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; et al. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N Engl J Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J Clin Invest. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [PubMed]
- Watala, C. Blood platelet reactivity and its pharmacological modulation in (people with) diabetes mellitus. Curr Pharm Des. 2005, 11, 2331–2365. [Google Scholar] [CrossRef]
- Anfossi, G.; Russo, I.; Massucco, P.; Mattiello, L.; Doronzo, G.; De Salve, A.; et al. Impaired synthesis and action of antiaggregating cyclic nucleotides in platelets from obese subjects: possible role in platelet hyperactivation in obesity. Eur J Clin Invest. 2004, 34, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Marchisio, M.; Lanuti, P.; Boccatonda, A.; Miscia, S.; Davì, G. Microparticles as new markers of cardiovascular risk in diabetes and beyond. Thromb Haemost. 2016, 116, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Davì, G.; Consoli, A.; Cipollone, F.; Mezzetti, A.; Falco, A.; et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J Am Coll Cardiol. 2006, 47, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; et al. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis. 2010, 209, 585–591. [Google Scholar] [CrossRef]
- Poggi, M.; Engel, D.; Christ, A.; Beckers, L.; Wijnands, E.; Boon, L.; et al. CD40L deficiency ameliorates adipose tissue inflammation and metabolic manifestations of obesity in mice. Arterioscler Thromb Vasc Biol. 2011, 31, 2251–2260. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation. 2014, 129, 1598–1609. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology. 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019, 25, 641–655. [Google Scholar] [CrossRef]
- Heijnen, H.; van der Sluijs, P. Platelet secretory behaviour: as diverse as the granules … or not? J Thromb Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Taus, F.; Meneguzzi, A.; Castelli, M.; Minuz, P. Platelet-Derived Extracellular Vesicles as Target of Antiplatelet Agents. What Is the Evidence? Front Pharmacol. 2019, 10, 1256. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Bai, J.; Xia, M.; Xue, Y.; Ma, F.; Cui, A.; Sun, Y.; et al. Thrombospondin 1 improves hepatic steatosis in diet-induced insulin-resistant mice and is associated with hepatic fat content in humans. EBioMedicine 2020, 57, 102849. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Merz, C.N.; Brewer, H.B., Jr.; Hunninghake, D.B.; et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004, 110, 227–239. [Google Scholar] [CrossRef]
- Werner, M.; Driftmann, S.; Kleinehr, K.; Kaiser, G.M.; Mathé, Z.; Treckmann, J.W.; et al. All-In-One: Advanced preparation of Human Parenchymal and Non-Parenchymal Liver Cells. PLoS One. 2015, 10, e0138655. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J Clin Invest. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Karimian, G.; Adelmeijer, J.; Giepmans, B.N.; Porte, R.J.; Lisman, T. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood. 2015, 126, 798–806. [Google Scholar] [CrossRef]
- Xu, Y.; Li, W.; Liang, G.; Peng, J.; Xu, X. Platelet microparticles-derived miR-25-3p promotes the hepatocyte proliferation and cell autophagy via reducing B-cell translocation gene 2. J Cell Biochem. 2020, 121, 4959–4973. [Google Scholar] [CrossRef]
- Kurokawa, T.; Ohkohchi, N. Platelets in liver disease, cancer and regeneration. World J Gastroenterol. 2017, 23, 3228–3239. [Google Scholar] [CrossRef]
- Ikeda, N.; Murata, S.; Maruyama, T.; Tamura, T.; Nozaki, R.; Kawasaki, T.; et al. Platelet-derived adenosine 5'-triphosphate suppresses activation of human hepatic stellate cell: In vitro study. Hepatol Res. 2012, 42, 91–102. [Google Scholar] [CrossRef]
- Salem, N.A.; Hamza, A.; Alnahdi, H.; Ayaz, N. Biochemical and Molecular Mechanisms of Platelet-Rich Plasma in Ameliorating Liver Fibrosis Induced by Dimethylnitrosurea. Cell Physiol Biochem. 2018, 47, 2331–2339. [Google Scholar] [CrossRef]
- Zaldivar, M.M.; Pauels, K.; von Hundelshausen, P.; Berres, M.L.; Schmitz, P.; Bornemann, J.; et al. CXC chemokine ligand 4 (Cxcl4) is a platelet-derived mediator of experimental liver fibrosis. Hepatology. 2010, 51, 1345–1353. [Google Scholar] [CrossRef]
- Mahmoud, N.I.; Messiha, B.A.S.; Salehc, I.G.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Interruption of platelets and thrombin function as a new approach against liver fibrosis induced experimentally in rats. Life Sci. 2019, 231, 116522. [Google Scholar] [CrossRef] [PubMed]
- Kinnman, N.; Francoz, C.; Barbu, V.; Wendum, D.; Rey, C.; Hultcrantz, R.; et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest. 2003, 83, 163–173. [Google Scholar] [CrossRef]
- Joshi, N.; Kopec, A.K.; Ray, J.L.; Cline-Fedewa, H.; Groeneveld, D.J.; Lisman, T.; et al. Von Willebrand factor deficiency reduces liver fibrosis in mice. Toxicol Appl Pharmacol. 2017, 328, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, T.; Maceyka, M.; Spiegel, S. Sphingosine kinase and sphingosine-1-phosphate in liver pathobiology. Crit Rev Biochem Mol Biol. 2017, 52, 543–553. [Google Scholar] [CrossRef]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; et al. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.W.; Chung, J.; Sverdlov, D.Y.; et al. Extrahepatic platelet-derived growth factor-β, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology. 2014, 147, 1378–1392. [Google Scholar] [CrossRef]
- Miele, L.; Alberelli, M.A.; Martini, M.; Liguori, A.; Marrone, G.; Cocomazzi, A.; et al. Nonalcoholic fatty liver disease (NAFLD) severity is associated to a nonhemostatic contribution and proinflammatory phenotype of platelets. Transl Res. 2021, 231, 24–38. [Google Scholar] [CrossRef]
- Chauhan, A.; Adams, D.H.; Watson, S.P.; Lalor, P.F. Platelets: No longer bystanders in liver disease. Hepatology. 2016, 64, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic Alempijevic, T.; Stojkovic Lalosevic, M.; Dumic, I.; Jocic, N.; Pavlovic Markovic, A.; Dragasevic, S.; et al. Diagnostic Accuracy of Platelet Count and Platelet Indices in Noninvasive Assessment of Fibrosis in Nonalcoholic Fatty Liver Disease Patients. Can J Gastroenterol Hepatol. 2017, 2017, 6070135. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhao, C.; Shen, C.; Wang, Y. Cytokeratin 18, alanine aminotransferase, platelets and triglycerides predict the presence of nonalcoholic steatohepatitis. PLoS One. 2013, 8, e82092. [Google Scholar] [CrossRef]
- Ozhan, H.; Aydin, M.; Yazici, M.; Yazgan, O.; Basar, C.; Gungor, A.; et al. Mean platelet volume in patients with non-alcoholic fatty liver disease. Platelets. 2010, 21, 29–32. [Google Scholar] [CrossRef]
- Shin, W.Y.; Jung, D.H.; Shim, J.Y.; Lee, H.R. The association between non-alcoholic hepatic steatosis and mean platelet volume in an obese Korean population. Platelets. 2011, 22, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Madan, S.A.; John, F.; Pitchumoni, C.S. Nonalcoholic Fatty Liver Disease and Mean Platelet Volume: A Systemic Review and Meta-analysis. J Clin Gastroenterol. 2016, 50, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Potze, W.; Siddiqui, M.S.; Sanyal, A.J. Vascular Disease in Patients with Nonalcoholic Fatty Liver Disease. Semin Thromb Hemost. 2015, 41, 488–493. [Google Scholar] [CrossRef]
- Papanas, N.; Symeonidis, G.; Maltezos, E.; Mavridis, G.; Karavageli, E.; Vosnakidis, T.; et al. Mean platelet volume in patients with type 2 diabetes mellitus. Platelets. 2004, 15, 475–478. [Google Scholar] [CrossRef]
- Schmidt, K.G.; Rasmussen, J.W.; Bekker, C.; Madsen, P.E. Kinetics and in vivo distribution of 111-In-labelled autologous platelets in chronic hepatic disease: mechanisms of thrombocytopenia. Scand J Haematol. 1985, 34, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Fujii, H.; Sumida, Y.; Hyogo, H.; Itoh, Y.; Ono, M.; et al. Platelet count for predicting fibrosis in nonalcoholic fatty liver disease. J Gastroenterol. 2011, 46, 1300–1306. [Google Scholar] [CrossRef]
- Park, K.S.; Lee, Y.S.; Park, H.W.; Seo, S.H.; Jang, B.G.; Hwang, J.Y.; et al. Factors associated or related to with pathological severity of nonalcoholic fatty liver disease. Korean J Intern Med. 2004, 19, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, H.; Hashimoto, E.; Yatsuji, S.; Tokushige, K.; Shiratori, K. Hyaluronic acid levels can predict severe fibrosis and platelet counts can predict cirrhosis in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2006, 21, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Fierbinteanu-Braticevici, C.; Dina, I.; Petrisor, A.; Tribus, L.; Negreanu, L.; Carstoiu, C. Noninvasive investigations for non alcoholic fatty liver disease and liver fibrosis. World J Gastroenterol. 2010, 16, 4784–4791. [Google Scholar] [CrossRef]
- Nakamura, S.; Konishi, H.; Kishino, M.; Yatsuji, S.; Tokushige, K.; Hashimoto, E.; et al. Prevalence of esophagogastric varices in patients with non-alcoholic steatohepatitis. Hepatol Res. 2008, 38, 572–579. [Google Scholar] [CrossRef]
- Shah, A.G.; Lydecker, A.; Murray, K.; Tetri, B.N.; Contos, M.J.; Sanyal, A.J. Comparison of noninvasive markers of fibrosis in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2009, 7, 1104–1112. [Google Scholar] [CrossRef]
- Duran-Bertran, J.; Rusu, E.C.; Barrientos-Riosalido, A.; Bertran, L.; Mahmoudian, R.; Aguilar, C.; et al. Platelet-associated biomarkers in nonalcoholic steatohepatitis: Insights from a female cohort with obesity. Eur J Clin Invest. 2023, 54, e14123. [Google Scholar] [CrossRef]
- Liu, Y.; Nong, L.; Jia, Y.; Tan, A.; Duan, L.; Lu, Y.; et al. Aspirin alleviates hepatic fibrosis by suppressing hepatic stellate cells activation via the TLR4/NF-κB pathway. Aging (Albany NY). 2020, 12, 6058–6066. [Google Scholar] [CrossRef]
- Li, C.J.; Yang, Z.H.; Shi, X.L.; Liu, D.L. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J Gastroenterol. 2017, 23, 6412–6419. [Google Scholar] [CrossRef]
- Börgeson, E.; Johnson, A.M.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Kim, J.K.; Lee, J.I.; Kang, S.H.; Kim, D.Y.; An, S.H.; et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut. 2009, 58, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Endo, H.; Takahashi, H.; et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut. 2008, 57, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.G.; Feldbrügge, L.; Tapper, E.B.; Popov, Y.; Ghaziani, T.; Afdhal, N.; et al. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment Pharmacol Ther. 2016, 43, 734–743. [Google Scholar] [CrossRef]
- Shen, H.; Shahzad, G.; Jawairia, M.; Bostick, R.M.; Mustacchia, P. Association between aspirin use and the prevalence of nonalcoholic fatty liver disease: a cross-sectional study from the Third National Health and Nutrition Examination Survey. Aliment Pharmacol Ther. 2014, 40, 1066–1073. [Google Scholar] [CrossRef]
- Thongtan, T.; Deb, A.; Vutthikraivit, W.; Laoveeravat, P.; Mingbunjerdsuk, T.; Islam, S.; et al. Antiplatelet therapy associated with lower prevalence of advanced liver fibrosis in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Indian J Gastroenterol. 2022, 41, 119–126. [Google Scholar] [CrossRef]
- Karahan, O.I.; Kurt, A.; Yikilmaz, A.; Kahriman, G. New method for the detection of intraperitoneal free air by sonography: scissors maneuver. J Clin Ultrasound. 2004, 32, 381–385. [Google Scholar] [CrossRef]
- Murohara, T.; Horowitz, J.R.; Silver, M.; Tsurumi, Y.; Chen, D.; Sullivan, A.; et al. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998, 97, 99–107. [Google Scholar] [CrossRef]
- Sánchez de Miguel, L.; de Frutos, T.; González-Fernández, F.; del Pozo, V.; Lahoz, C.; Jiménez, A.; et al. Aspirin inhibits inducible nitric oxide synthase expression and tumour necrosis factor-alpha release by cultured smooth muscle cells. Eur J Clin Invest. 1999, 29, 93–99. [Google Scholar] [CrossRef]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- Prattali, R.R.; Barreiro, G.C.; Caliseo, C.T.; Fugiwara, F.Y.; Ueno, M.; Prada, P.O.; et al. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in growth hormone treated animals. FEBS Lett. 2005, 579, 3152–3158. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef]
- Clària, J.; Planagumà, A. Liver: the formation and actions of aspirin-triggered lipoxins. Prostaglandins Leukot Essent Fatty Acids 2005, 73, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Kozakova, M.; Højlund, K.; Flyvbjerg, A.; Favuzzi, A.; Mitrakou, A.; et al. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology. 2009, 49, 1537–1544. [Google Scholar] [CrossRef]
- Han, Y.M.; Lee, Y.J.; Jang, Y.N.; Kim, H.M.; Seo, H.S.; Jung, T.W.; et al. Aspirin Improves Nonalcoholic Fatty Liver Disease and Atherosclerosis through Regulation of the PPARδ-AMPK-PGC-1α Pathway in Dyslipidemic Conditions. Biomed Res Int. 2020, 2020, 7806860. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; El-Serag, H.B. Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin Gastroenterol Hepatol. 2015, 13, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology. 2018, 155, 1828–1837. [Google Scholar] [CrossRef]
- Mittal, S.; Sada, Y.H.; El-Serag, H.B.; Kanwal, F.; Duan, Z.; Temple, S.; et al. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin Gastroenterol Hepatol. 2015, 13, 594–601. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol. 2019, 17, 748–755. [Google Scholar] [CrossRef]
- Haldar, D.; Kern, B.; Hodson, J.; Armstrong, M.J.; Adam, R.; Berlakovich, G.; et al. Outcomes of liver transplantation for non-alcoholic steatohepatitis: A European Liver Transplant Registry study. J Hepatol. 2019, 71, 313–322. [Google Scholar] [CrossRef]
- Lee, M.; Chung, G.E.; Lee, J.H.; Oh, S.; Nam, J.Y.; Chang, Y.; et al. Antiplatelet therapy and the risk of hepatocellular carcinoma in chronic hepatitis B patients on antiviral treatment. Hepatology. 2017, 66, 1556–1569. [Google Scholar] [CrossRef]
- Lee, T.Y.; Hsu, Y.C.; Tseng, H.C.; Yu, S.H.; Lin, J.T.; Wu, M.S.; et al. Association of Daily Aspirin Therapy With Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. JAMA Intern Med. 2019, 179, 633–640. [Google Scholar] [CrossRef]
- Simon, T.G.; Duberg, A.S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N Engl J Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef]
- Santilli, F.; Boccatonda, A.; Davì, G. Aspirin, platelets, and cancer: The point of view of the internist. 2016, 34:11-20. Eur J Intern Med. 2016, 34, 11–20. [Google Scholar] [CrossRef]
- Lai, Q.; De Matthaeis, N.; Finotti, M.; Galati, G.; Marrone, G.; Melandro, F.; et al. The role of antiplatelet therapies on incidence and mortality of hepatocellular carcinoma. Eur J Clin Invest. 2023, 53, e13870. [Google Scholar] [CrossRef]
- Sahasrabuddhe, V.V.; Gunja, M.Z.; Graubard, B.I.; Trabert, B.; Schwartz, L.M.; Park, Y.; et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst. 2012, 104, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Hsu, C.Y.; Yen, T.H.; Wu, T.H.; Yu, M.C.; Hsieh, S.Y. Daily Aspirin Reduced the Incidence of Hepatocellular Carcinoma and Overall Mortality in Patients with Cirrhosis. Cancers (Basel) 2023, 15, 2946. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Min, E.K.; Lee, J.G.; Joo, D.J.; Kim, M.S.; Kim, D.G. Antiplatelet Drugs on the Recurrence of Hepatocellular Carcinoma after Liver Transplantation. Cancers (Basel) 2022, 14, 5329. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Hsu, Y.C.; Ho, H.J.; Lin, J.T.; Chen, Y.J.; Wu, C.Y. Daily aspirin associated with a reduced risk of hepatocellular carcinoma in patients with non-alcoholic fatty liver disease: a population-based cohort study. EClinicalMedicine 2023, 61, 102065. [Google Scholar] [CrossRef]
- Tan, R.Z.H.; Lockart, I.; Abdel Shaheed, C.; Danta, M. Systematic review with meta-analysis: The effects of non-steroidal anti-inflammatory drugs and anti-platelet therapy on the incidence and recurrence of hepatocellular carcinoma. Aliment Pharmacol Ther. 2021, 54, 356–367. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Aspirin, platelet inhibition and cancer prevention. Platelets. 2018, 29, 779–785. [Google Scholar] [CrossRef]
- Lloyd, K.E.; Hall, L.H.; Ziegler, L.; Foy, R.; Green, S.M.C.; MacKenzie, M.; et al. Acceptability of aspirin for cancer preventive therapy: a survey and qualitative study exploring the views of the UK general population. BMJ Open. 2023, 13, e078703. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. Cyclooxygenase Inhibitors and Cancer: The Missing Pieces. J Pharmacol Exp Ther. 2023, 386, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Boccatonda, A.; Davì, G.; Cipollone, F. The Coxib case: Are EP receptors really guilty? Atherosclerosis. 2016, 249, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Joharatnam-Hogan, N.; Hatem, D.; Cafferty, F.H.; Petrucci, G.; Cameron, D.A.; Ring, A.; et al. Thromboxane biosynthesis in cancer patients and its inhibition by aspirin: a sub-study of the Add-Aspirin trial. Br J Cancer. 2023, 129, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Fu, Q.; Diggs, L.P.; McVey, J.C.; McCallen, J.; Wabitsch, S.; et al. Platelets control liver tumor growth through P2Y12-dependent CD40L release in NAFLD. Cancer Cell. 2022, 40, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.R.; et al. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med. 2005, 11, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Elzey, B.D.; Tian, J.; Jensen, R.J.; Swanson, A.K.; Lees, J.R.; Lentz, S.R.; et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity. 2003, 19, 9–19. [Google Scholar] [CrossRef]
- Vonderheide, R.H. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007, 13, 1083–1088. [Google Scholar] [CrossRef]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef]
- Vogt, A.; Sadeghlar, F.; Ayub, T.H.; Schneider, C.; Möhring, C.; Zhou, T.; et al. Alpha-Fetoprotein- and CD40Ligand-Expressing Dendritic Cells for Immunotherapy of Hepatocellular Carcinoma. Cancers (Basel) 2021, 13, 3375. [Google Scholar] [CrossRef]
- Hermann, A.; Rauch, B.H.; Braun, M.; Schrör, K.; Weber, A.A. Platelet CD40 ligand (CD40L)--subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets. 2001, 12, 74–82. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; David, E.; Ramadori, P.; Pfister, D.; Safran, M.; Li, B.; et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med. 2021, 27, 1043–1054. [Google Scholar] [CrossRef]
- Ezzaty Mirhashemi, M.; Shah, R.V.; Kitchen, R.R.; Rong, J.; Spahillari, A.; Pico, A.R.; et al. The Dynamic Platelet Transcriptome in Obesity and Weight Loss. Arterioscler Thromb Vasc Biol. 2021, 41, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017, 551, 340–345. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Brotons, C.; Coppolecchia, R.; Cricelli, C.; Darius, H.; Gorelick, P.B.; et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): a randomised, double-blind, placebo-controlled trial. Lancet. 2018, 392, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N Engl J Med. 2018, 379, 1529–1539. [Google Scholar] [PubMed]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; et al. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. N Engl J Med. 2018, 379, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Simeone, P. Aspirin in primary prevention: the triumph of clinical judgement over complex equations. Intern Emerg Med. 2019, 14, 1217–1231. [Google Scholar] [CrossRef]
- Li, X.Y.; Li, L.; Na, S.H.; Santilli, F.; Shi, Z.; Blaha, M. Implications of the heterogeneity between guideline recommendations for the use of low dose aspirin in primary prevention of cardiovascular disease. Am J Prev Cardiol. 2022, 11, 100363. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, Y.; Anderson, J.E.; Bakris, G.L.; Ballantyne, C.M.; Beckman, J.A.; Bhatt, D.L.; et al. DCRM Multispecialty Practice Recommendations for the management of diabetes, cardiorenal, and metabolic diseases. J Diabetes Complications. 2022, 36, 108101. [Google Scholar] [CrossRef] [PubMed]
- Merat, S.; Jafari, E.; Radmard, A.R.; Khoshnia, M.; Sharafkhah, M.; Nateghi Baygi, A.; et al. Polypill for prevention of cardiovascular diseases with focus on non-alcoholic steatohepatitis: the PolyIran-Liver trial. Eur Heart J. 2022, 43, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Rocca, B.; De Cristofaro, R.; Lattanzio, S.; Pietrangelo, L.; Habib, A.; et al. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: implications for aspirin "resistance". J Am Coll Cardiol. 2009, 53, 667–677. [Google Scholar] [CrossRef]
- Rocca, B.; Santilli, F.; Pitocco, D.; Mucci, L.; Petrucci, G.; Vitacolonna, E.; et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012, 10, 1220–1230. [Google Scholar] [CrossRef]
- Simeone, P.; Liani, R.; Tripaldi, R.; Ciotti, S.; Recchiuti, A.; Abbonante, V.; et al. Reduced platelet glycoprotein Ibα shedding accelerates thrombopoiesis and COX-1 recovery: implications for aspirin dosing regimen. Haematologica. 2023, 108, 1141–1157. [Google Scholar] [CrossRef]
Figure 1.
Summary figure on the role of platelets in patients with MASLD (NAFLD). Platelets are players and addresses of metabolic dysregulation; obesity and insulin resistance are related to platelet hyperactivation. Furthermore, platelets can exert a direct effect on liver cells, particularly through the release of mediators from granules. Furthermore, platelets are included in the majority of clinical scores linked to liver disease and risk of evolution towards cirrhosis.
Figure 1.
Summary figure on the role of platelets in patients with MASLD (NAFLD). Platelets are players and addresses of metabolic dysregulation; obesity and insulin resistance are related to platelet hyperactivation. Furthermore, platelets can exert a direct effect on liver cells, particularly through the release of mediators from granules. Furthermore, platelets are included in the majority of clinical scores linked to liver disease and risk of evolution towards cirrhosis.
Figure 2.
Obesity and insulin resistance are correlated with platelet hyper-activation. Upon activation, platelets release some molecules and mediators harbored in their granules such as 5-HT, CD40L and TXA2. Those molecules are involved in determining a pro-thrombotic and pro-coagulant state. Moreover, many pathways are involved in direct liver damage by HSC activation, ER stress, lipotoxicity and oxidative stress. Eventually, there is a cross-talking between platelets and immune cells, thus inducing a dysregulated immune response involved in liver damage and in determining coagulative disorder. Abbreviations: chemokine (C-X-C motif) ligand 4, CXCL4; 5-hydroxytryptamine, 5-HT; CD40 ligand, CD40L; thromboxane A2, TXA2; von Willebrand factor, vWF; plasminogen activator inhibitor 1, PAI-1; hepatic stem cell, HSC; endoplasmic reticulum, ER.
Figure 2.
Obesity and insulin resistance are correlated with platelet hyper-activation. Upon activation, platelets release some molecules and mediators harbored in their granules such as 5-HT, CD40L and TXA2. Those molecules are involved in determining a pro-thrombotic and pro-coagulant state. Moreover, many pathways are involved in direct liver damage by HSC activation, ER stress, lipotoxicity and oxidative stress. Eventually, there is a cross-talking between platelets and immune cells, thus inducing a dysregulated immune response involved in liver damage and in determining coagulative disorder. Abbreviations: chemokine (C-X-C motif) ligand 4, CXCL4; 5-hydroxytryptamine, 5-HT; CD40 ligand, CD40L; thromboxane A2, TXA2; von Willebrand factor, vWF; plasminogen activator inhibitor 1, PAI-1; hepatic stem cell, HSC; endoplasmic reticulum, ER.

Figure 3.
Summary figure on the anti-cancer mechanisms of antiplatelet therapy (aspirin). Aspirin can block COX-2 both at the platelet level and in tumor cells. At the platelet level, COX blockade reduces platelet activation and the release of mediators that may be involved in tumorigenesis (e.g. TGF-β, VEGF, TXA2). At the tumor cell level, COX-2 blockade appears to be able to reduce cell proliferation and epithelial-mesenchymal transition. Abbreviations: thromboxane A2, TXA2; interleukin, IL; platelet-factor-4, PF-4; vascular endothelial growth factor, VEGF; platelet-derived growth factor, PDGF; transforming growth factor β, TGF-β; cyclooxygenase, COX; prostaglandin E, PGE; prostaglandin E receptor, EP; epitelio-mesenchimal transition, EMT.
Figure 3.
Summary figure on the anti-cancer mechanisms of antiplatelet therapy (aspirin). Aspirin can block COX-2 both at the platelet level and in tumor cells. At the platelet level, COX blockade reduces platelet activation and the release of mediators that may be involved in tumorigenesis (e.g. TGF-β, VEGF, TXA2). At the tumor cell level, COX-2 blockade appears to be able to reduce cell proliferation and epithelial-mesenchymal transition. Abbreviations: thromboxane A2, TXA2; interleukin, IL; platelet-factor-4, PF-4; vascular endothelial growth factor, VEGF; platelet-derived growth factor, PDGF; transforming growth factor β, TGF-β; cyclooxygenase, COX; prostaglandin E, PGE; prostaglandin E receptor, EP; epitelio-mesenchimal transition, EMT.
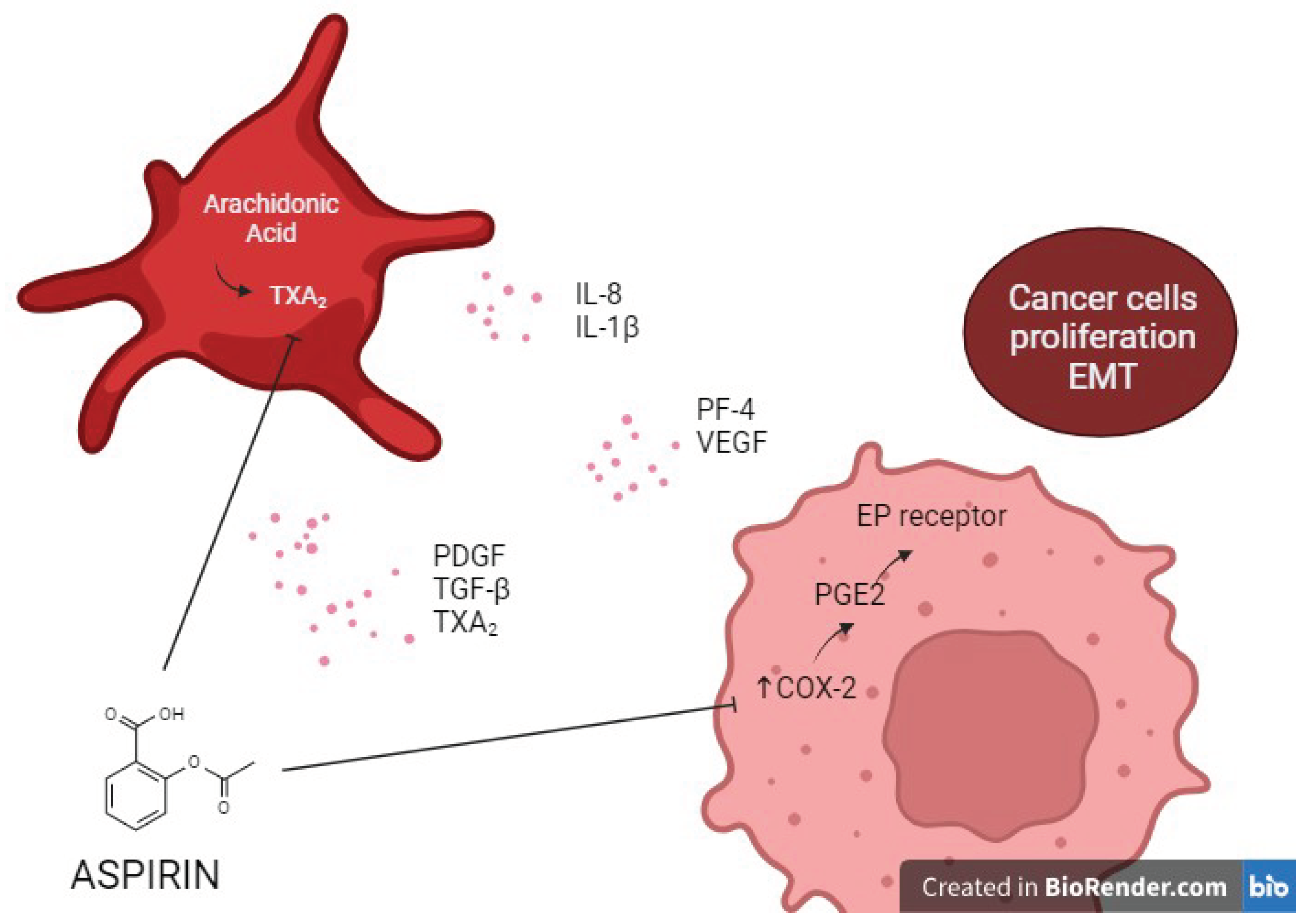

Table 1.
Comparison between different evidences on the role of the CD40-CD40L pathway on carcinogenesis.
Table 1.
Comparison between different evidences on the role of the CD40-CD40L pathway on carcinogenesis.
| CD40 pathway ANTI-TUMOR ACTIVITY | CD40 pathway PRO-TUMOR ACTIVITY |
|---|---|
| CD40L is related to a strong CD8+ T cells responses through CD40 licensing of dendritic cell (61, 122, 124, 136, 137, 138, 146). | Platelet-derived CD40L is greater released in both NAFLD mouse models and patients with NASH [136] |
| Co-stimulation with CD40L-expressing dendritic cells (DC) significantly improves vaccination by inducing an early and strong Th1-shift in the tumor environment as well as higher tumor apoptosis [141]. | In NAFLD patients CD40L protein production is induced in megakaryocytes rather than NAFLD or tumors causing a transfer of CD40L pre-mRNA or mRNA into platelets [145]. |
| Platelet-derived CD40L can increase CD8+ T cell activation and their recruitment to liver in NAFLD (61, 122, 124, 136, 137, 138, 146). | IL-12 dependent increase of CD40L production in bone marrow megakaryocytes in NAFLD models [136,143] |
| Megakaryocytes harbored in lung can produce higher CD40L amount [136,144] | |
| Inhibition of the P2Y12 receptor on platelets can promote tumor growth via CD40L in mice with NAFLD [136] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



