
Submitted:
01 March 2024
Posted:
01 March 2024
You are already at the latest version
Abstract
A heterograft copolymer with an alginate backbone, hetero-grafted by polymer pendant chains displaying different Lower Critical Solution Temperatures (LCSTs), combined with a pH-responsive Poly(2-vinyl pyridine)-b-poly(ethylene oxide) (P2VP-b-PEO) diblock copolymer forming micellar nanoparticles, was investigated in aqueous media at various pH. Due to its thermoresponsive side chains, the copolymer forms hydrogels with thermo–induced sol–gel transition, above a critical temperature, Tgel (thermo–thickening). However, by lowering pH of the medium in acidic regime, a remarkable increase of the elasticity of the formulation was observed. This effect was more pronounced in low temperatures (below Tgel) suggesting secondary physical crosslinking, which induces significant changes in the hydrogel thermoresponsiveness, transforming the sol–gel transition to soft gel–strong gel. Moreover, the onset of thermothickening shifted to lower temperatures followed by broadening of the transition zone, implying intermolecular interactions between the uncharged alginate backbone with the PNIPAM side chains, likely through H–bonding. The shear–thinning behavior of the soft gel in low temperatures provides injectability, which allows 3D–printing potential applications. Furthermore, the heterograft copolymer/nanoparticles composite hydrogel, encapsulating a model hydrophobic drug in the hydrophobic cores of the nanoparticles, was evaluated as pH-responsive drug delivery system. The presented tunable drug delivery system might be useful for biomedical potential applications.
Keywords:
heterograft copolymer
; hydrogel
; alginate
; thermo-responsive
; pH-responsive
; poly(2- vinyl pyridine)-b-polyethylene oxide
; diblock copolymer micelles
; drug delivery
1. Introduction
Hydrogels are three-dimensional (3D) network structures formed through chemical or physical cross-links or chain entanglements. Stimuli responsive hydrogels, also known as “smart” hydrogels, are materials that respond to various stimuli (pH, temperature, light, ionic strength, magnetic, redox, electric, chemical response) exhibiting reversible changes in their chemical and physical state [1,2,3,4]. Due to their soft texture, elasticity, biocompatibility, high swelling capability and similarity to body tissues, hydrogels are ideal candidates for use as biomaterials in a variety of applications [5,6] like delivery of drugs and other bioactive ingredients [7,8,9,10,11,12,13,14,15,16], 3D printing [17,18,19,20], tissue engineering, repair and regeneration [21,22,23,24], or shear-thinning injectable matrices for bioapplications [25,26].
Nowadays, various stimuli responsive hydrogels, originating from synthetic as well as natural resources, are extensively used as biomaterials in several bioapplications, including tissue engineering, drug delivery, etc. Mostly known examples of synthetic hydrogels as biomaterials include polymeric and composite systems. On the other hand, common biomaterials of natural origin are polysaccharide-based or protein-based biomaterials. Polysaccharides are non-synthetic polymers that can be isolated from a variety of natural sources like plants, animals, or algae. Some examples of polysaccharides are chitosan, hyaluronan, pullulan, dextran, xanthan, cellulose, pectin, guar gum, collagen, gelatin, agar, alginate, carrageenan and silk. This class of materials has gained scientific interest over synthetic polymers due to their exceptional advantages such as biocompatibility, biodegradability, hydrophilicity, environmentally friendly, low or non-toxicity.
The incorporation of thermo-sensitive groups in polysaccharides creates new “smart” materials with thermo-responsive behavior [27,28,29]. Interestingly, such materials can present thermo-induced sol to gel transition property, rendering them ideal candidates in use as potential injectable hydrogels [30]. In this respect, thermo–sensitive polymers like poly(N–isopropylacrylamide) (PNIPAM) are grafted onto or cross-linked with polysaccharide backbones. PNIPAM is one of the most extensively studied thermo-responsive polymer exhibiting a lower critical solution temperature (LCST) around 32 °C, close to physiological body temperature. Its polymer chains are hydrophilic and water soluble at temperatures below the LCST but above the LCST, they become hydrophobic and their solubility in water is not favorable anymore, leading to phase separation [31]. The LCST behavior of PNIPAM can be finely altered and tuned to desired values, simply by the addition of hydrophobic or hydrophilic monomers. Addition of more hydrophobic monomers like N-tert-butyl acrylamide (NtBAM) decreases the LCST [32,33] while the incorporation of more hydrophilic monomers such as acrylic acid, shifts LCST to higher temperatures [34]. Many interesting articles report the thermo-induced sol to gel transition of alginate polysaccharide grafted with PNIPAM chains [35,36,37,38,39,40]. Our group has long experience in the design and study of thermo–responsive graft copolymers comprising an alginate backbone and thermo-sensitive NIPAM–based homopolymers or copolymers [32,41,42]. It has been shown that such polymers, due to shear-thinning and thermo-thickening effects, can form hydrogels with injectability and self-healing capabilities, rendering them ideal applicants for use in fields like tissue engineering and therapeutic cargo delivery.
The incorporation of organic or inorganic particles into polymeric scaffolds like hydrogels has been proven to ameliorate the pharmacokinetic profiles of drug–loaded molecules as well as to improve their stability and increase local drug concentration, providing a drug reservoir at the site of administration [43,44,45]. Moreover, it has been reported that hydrogel/nanoparticle complex systems display improved rheological and mechanical properties, which in turn enhance their printability and injectability [38,46,47].
In this paper we report on a composite hydrogel constituted of an Alginate heterografted gelator, exhibiting thermothickening behavior through a sol-gel transition at Tgel of 34.5 °C, encapsulating polymeric nanoparticles, formed by spontaneous self-assembly of P2VP-b-PEO diblock copolymers. Thanks to the pH-dependent properties of P2VP (cationic/hydrophobic), the rheological behavior of the composite was focused on a pH range of 3.5-5.4, in the vicinity of its pKa. The data showed a remarkable influence of pH on the thermo-induced gelation of the alginate copolymer namely, elasticity enhancement, shift of the critical thermothickening temperature at lower values and broadening of the gelation transition zone. Surprisingly, the same behavior was observed for the gelator without nanoparticles. These results are attributed to secondary intermolecular interactions between the protonated repeating units of alginate backbone and the PNIPAM side chains. Finally, the composite was evaluated as pH dependent drug delivery system using Nile Red as model hydrophobic drug.
2. Materials and Methods
2.1. Materials
The synthesis of the heterograft copolymer ALG-g-P(NIPAM86-co-NtBAM14)-g-PNIPAM (denoted by ALG-g-HG ) has been previously described [42]. A linear diblock P2VP-b-PEO copolymer was created in our lab by anionic “living” polymerization and sequential addition of monomers, according to the literature [48]. The molecular characteristics of ALG-g-HG and P2VP-b-PEO copolymers are presented in Tables S1 and S2, respectively (see Supplementary Material). Nile Red was purchased from Sigma-Aldrich (St. Louis, MO, USA). Standard solutions of hydrochloric acid (HCl) and sodium hydroxide (NaOH) were purchased from Panreac (Chicago, IL, USA) and used as received, without further purification. Disodium hydrogen phosphate (Na2HPO4) and potassium dihydrogen phosphate (KH2PO4) were obtained by Carlo Erba (Milano, Italy) and Merck (Darmstadt, Germany), respectively, and used as received, for the preparation of two PB buffer solutions (10mM, pH 3.5 or pH 7.4). The organic solvent tetrahydrofuran (THF) was obtained from Sigma-Aldrich (St. Louis, MO, USA). An ELGA Medica-R7/15 device (ELGA Labwater, Woodridge, IL, USA) was used to produce ultrapure water.
2.2. Preparation of Aqueous Polymer Solutions and Rheology Study
Aqueous solutions of ALG-g-HG copolymer or hybrid ALG-g-HG/P2VP-b-PEO system were prepared and left under stirring at room temperature, until homogeneity. In all solutions, concentration of heterograft ALG-g-HG copolymer was set at 4 wt%, while the concentration of the P2VP-b-PEO micelles in the ALG-g-HG/P2VP-b-PEO hybrids was set at 4 wt%. The pH of the solutions was set at the desired values, using HCl (1 M) or NaOH (1 M). The rheological study of the ALG-g-HG heterograft copolymer and hybrid ALG-g-HG/P2VP-b-PEO system was performed using an AR-2000ex stress-controlled rheometer (TA Instruments, New Castle, DE, USA) equipped with a Peltier system for controlling the temperature and a solvent trap to avoid concentration changes owing to water evaporation. A cone–plate geometry was used (diameter 20 mm, angle 3o, truncation 111 μm) and the experiments were performed in the linear viscoelastic regime (LVR), determined by strain sweep tests at a frequency of 1 Hz.
2.3. Loading and Release of Nile Red from P2VP-b-PEO micelles
The P2VP-b-PEO micelles were prepared by the film hydration method. In brief, predetermined amounts of P2VP-b-PEO copolymer and the target model drug (Nile Red) were solubilized in THF. The theoretical (feed) loading (Lth %) in the polymeric micelles was equal to 9.98%, calculated by Equation 1:
where w0 and wm are the amounts (μg) of initially added probe (Nile Red) and amount of polymer micelles, respectively.
At a next step, THF was removed using a rotary evaporator, until a thin polymer/Nile Red film was formed. Afterwards, ultrapure water was added to solubilize the film and form the P2VP-b-PEO copolymer micelles, loaded with Nile Red (P2VP-b-PEO@Nile Red). Finally, the Nile Red loaded micelles were obtained in solid state using freeze drying. In order to determine the encapsulation efficiency (EE %) and the loading efficiency (LE %) of Nile Red in the P2VP-b-PEO copolymer micelles, two calibration curves were constructed in THF/PB buffer at pH 3.5 and THF/PB buffer at 7.4, where the volume ratio of THF to PB buffers was 60/40 v/v.
For these calibration curves, different concentrations of Nile Red (ranging from 0.5 to 15 μg/mL) in THF/PB buffer solutions were prepared and their absorbance values were recorded on a Hitachi U–2001 UV–VIS spectrophotometer (Schaumburg, IL, USA). Absorbances were determined at the lambda maximum (550 nm) and probe concentrations were quantified using molar absorption coefficients (ε) determined in each THF/PB buffered solution (pH 3.5; pH 7.4).
The loading efficiency (LE %) and encapsulation efficiency (EE %) of Nile Red were calculated using Equations 2 and 3, respectively:
where w, w0 and wm are the amounts (μg) of entrapped model drug, initially added probe (Nile Red) and amount of polymer micelles, respectively.
As for the drug release kinetics study, a given amount (~5 mg) of lyophilized model drug-loaded micellar powder (P2VP–b–PEO@Nile Red) was dispersed in 1 mL of PB buffer with pH 3.5 (stimulating the stomach pH) or 7.4 (which is specific for blood and colonic fluids) and then these solutions were transferred into dialysis membranes (12 kDa MWCO). These membranes were immersed in 5 mL PB buffer solutions of pH 3.5 or pH 7.4, in glass bottles, and kept under stirring (100 rpm) in a water bath at 37 °C, in the dark, to avoid photo-degradation of the model drug. After 120 hours of experiment, the dialysis membranes in 5 mL PB buffer solutions were subjected to sonication using an ultrasonic bath sonicator. All 5 mL PB samples were extracted at definite time intervals, replaced with newly added 5 mL of fresh PB solution in the glass bottles with the membranes. The extracted 5 mL samples were mixed with 7.5 mL of THF to achieve a mixture THF/PB with 60/40 ratio in volume, and then used to quantify spectrophotometrically the amount of the released Nile Red, at a wavelength of 550 nm. The amount of Nile Red from the micelles was spectrophotometrically quantified, based on their calibration curve, previously designed.
2.4. Release of Nile Red from ALG-g-HG/P2VP-b-PEO hydrogel Composite System
Two 4 wt% ALG-g-HG aqueous solutions with pH adjusted to 3.5 and 7.4 were prepared, as described in Section 2.2. 12 mg of Nile Red loaded P2VP–b–PEO micelles (P2VP–b–PEO@Nile Red) were added in 1.2 mL of the prepared ALG-g-HG aqueous solutions. The mixtures were transferred in two dialysis membranes (MWCO 12 kDa) which in turn were immediately submerged in 5 mL of phosphate buffer 10 mM of pH = 3.5 or 7.4 at 37 °C and shaken at 100 rpm in the dark. At specific timed intervals, the 5 mL of buffer was removed and renewed with 5 mL of fresh buffer. The cumulative release rate (%) of Nile Red was calculated in a similar way, as described in Section 2.3.
2.5. Techniques
Zeta potential and Dynamic light scattering (DLS) measurements of ALG-g-HG and P2VP-b-PEO aqueous solutions were obtained from a Malvern Nano Zetasizer analyzer (Malvern, UK) equipped with an He–Ne laser at 633 nm. The final polymer concentration of ALG-g-HG was set at 0.4 %wt while the concentration of P2VP-b-PEO was set at 0.1 %wt (keeping a 4:1 ratio of ALG-g-HG:P2VP-b-PEO). The desired pH values of the solutions were achieved adding HCl (1 M) or NaOH (1 M).
3. Results and Discussion
3.1. Rheological investigation at various pH
A hydrogel nanocomposite was designed, comprising a thermoresponsive Alginate-based gelator, encapsulating polymeric nanoparticles (NPs) as nano-carriers. Particularly, the gelator used is the heterograft copolymer constituted of an alginate backbone, grafted by two different types of thermoresponsive polymer chains (ALG-g-HG): (a) pure poly(N–isopropylacrylamide) (PNIPAM) homopolymer and (b) poly[(N–isopropylacrylamide)–co–(N–tert–butyl acrylamide)] (P(NIPAM–co–NtBAM)) random copolymer, displaying different lower critical solution temperatures (LCST) [42].The aqueous dispersion of 4 wt% ALG-g-HG, at pH 7.4, exhibits a sol-gel transition upon heating at Tgel=34.5 °C as determined by oscillatory shear experiments at the G’/G” crossover point (Figure S1a). The NPs formed by spontaneous self-assembly of 1 wt% P2VP-b-PEO block copolymers in aqueous media of pH 7.4, forming star-like micelles of core-shell structure with P2VP hydrophobic cores and PEO water-soluble shells. Note that P2VP is a pH-dependent cationic polyelectrolyte, exhibiting pKa at pH 5.0, above which becomes hydrophobic, driving hydrophobic association [49,50,51]. The hydrodynamic diameter of NPs at pH 7.4 was determined by dynamic light scattering at 23.56 nm (volume average, PDI 0.267) (Figure 1). Some bigger aggregates appearing in the volume-weighted size distribution are negligible in number as they disappear in the number-weighted distribution.
The final mixed aqueous solution under investigation contained 4 wt% ALG-g-HG and 1 wt% P2VP-b-PEO-based NPs. Nanocomposite hydrogels regulated at different pH were explored by oscillatory shear measurements to evaluate potential influence of pH to their thermal and mechanical response. The pH window was chosen in the narrow range of 3.5-5.5 which lies in the vicinity of pKa of P2VP in which its degree of ionization increases by lowering pH (see z-potential versus pH in SI, Figure S2). Figure 2 demonstrates the storage (G’) and loss (G”) modulus as a function of temperature at different pH values. The experiments were conducted in the linear viscoelastic regime by applying a successive heating/cooling ramp with a rate of 1 °C/min. In all cases the moduli exhibit strong temperature dependence revealing a thermothickening behavior which depends on the medium pH. At pH 5.4, a thermo-induced sol-gel transition can be observed, providing that the storage modulus increases sharply upon heating, surpassing the loss one at the critical Tgel determined at 34.1 °C. The phenomenon is reversible as revealed by the cooling procedure, with slight hysteresis. The gel-sol transition was shifted at 30.9 °C, a few degrees lower than that observed by heating, due to kinetic effects [29]. This thermothickening behavior is quite similar with the formulation of 4 wt% ALG-g-HG without NPs (Figure S1b). This implies that the presence of P2VP-b-PEO-based micelles at pH 5.4 does not affect the network structure formed by the physical crosslinking of the thermoresponsive side chains of the gelator. However, by lowering pH, remarkable changes on the characteristics of the hydrogel thermoresponsiveness can be observed. The main variation observed in the general thermal behavior of the formulations is that the sol-gel transition observed at pH 5.4 was transformed to a soft gel-strong gel transition at pH 3.5, since the G’/G” crosslinking disappeared and G’ predominates G’’ in the entire temperature range explored. At the intermediate pH 4.5 and 4.0, thermoresponsive behavior exhibits additional significant hysteresis effects. Particularly, at pH 4.5 the transition point Tgel in the heating procedure was shifted to lower values about 8 °C, notably more pronounced than the cooling procedure. While at pH 4.0, Tgel (heating) dropped to 20.2 °C that is about 14 °C lower from that at pH 5.4. Moreover, in the cooling procedure, the gel-sol transition did not occur, provided that the moduli although decreased upon lowering temperature, they never crossed each other, hence preserving the gel-like behavior at low temperatures.
We now focus on the data dealing with the heating process, which is related to potential biomedical applications. Figure 3 presents the temperature dependence of the elastic part of the complex modulus, G’ (Figure 3a), together with the loss tangent (G”/G’) at various pHs. By lowering pH two main effects can be observed: a) a decrease of the sol-gel transition (defined at tanδ=1, Figure 3b) and b) the elasticity of the formulation is expanded at lower temperatures and simultaneously increases. Provided that G’ reflects the degree of crosslinking, this behavior should be ascribed to additional crosslinking leading to the mechanical reinforcement of the hydrogel. This is also reflected in tanδ which decreases with pH alongside the temperature variation.
To better analyze qualitatively the data, we can distinguish three temperature zones as indicated in Figure 4: the low temperature zone, the high temperature zone and between them the transition zone. The latter zone is defined between the temperature at the onset of the abrupt increase of the elastic modulus G’ namely Tc,thermothickening and Tf, at the temperature where G’ becomes independent of T or increases slightly and smoothly. A number of factors characterizing the thermothickening phenomenon based on the elastic modulus G’, i.e., Tc,thermothickening, Tf, Tgel, ΔT(transition zone) and extend of the thermothickening effect exemplified by the ratio of G’ augmentation [G’(T=50)/G’(Tc,thermothickening)] are gathered in Table 1 at various pHs.
One of the important effects of pH concerns the transition zone. As indicated in Figure 5a, the onset of thermothickening is shifted to lower temperature more than 13 °C from pH 5.4 to 3.5. This causes the broadening of the transition zone ΔΤ from 9.4 to 19.5 degrees and the shifting of the sol-gel transition to lower temperatures. As it is known, the thermothickening effect is due to the thermal-induced hydrophobic interactions of the PNIPAM-based sticky side chains of the gelator, which are activated above their LCST, leading to intermolecular association. The fact that this association starts occurring at lower temperatures implies that the solubility of the side chains was obstructed by other interactions. Indeed, focusing in the low temperature zone we observe a remarkable increase of G’ more than two order of magnitude (Figure 3a and Figure 5b) which leads to gel-like behavior even at low temperatures below Tgel at pH 3.5. This suggests the formation of a network different than that induced by heating, which is formed from the interactions, responsible for the variations (Tc,thermothickening, Tgel, ΔT) observed in the transition zone. Moreover, these new interactions seem to contribute as well to the G’ augmentation in the high temperature zone (Figure 5b). Provided that the G’ augmentation is more pronounced in low temperatures it eventually weakens significantly the extend of the thermothickening effect, as reflected by the G’(T=50)/G’(Tc,thermothickening) ratio, from 110.5 to 3.5 (Table 1). This likely suggests that a smaller number of side chains are now available for hydrophobic association.
In order to elucidate the nature of the interactions, other than the hydrophobic one induced by heating, that contribute to the reinforcement of the network, we also explored the behavior of the ALG-g-HG without the presence of NPs. Figure S3 demonstrates the data from the temperature ramp experiments performed in the linear viscoelastic regime (strain amplitude 0.1%) at 1 Hz of 4 wt% ALG-g-HG at pH 4.5 and 3.5, keeping the same conditions with the composite formulations. For the sake of comparison these data have been plotted together with the data of Figure 3a and Figure S1b in Figure 6. Surprisingly, the behavior of the bare ALG-g-HG gelator is quite like those of the formulations incorporating the P2VP-b-PEO micellar NPs, since the data for the corresponding formulations at the same pH almost coincide. This suggests that the additional crosslinking of the ALG-g-HG, observed at low temperatures in the absence of PNIPAM hydrophobic association, must be attributed to the intermolecular interaction between the alginate backbone and the PNIPAM-based side chains. This is corroborated from the shift of the characteristics of the transition zone (Tc,thermothickening, Tgel) to lower temperatures discussed above. Note that gel-like behavior was not observed in pure alginate formulations of 4 wt % at the same pH range. At low pH, although the degree of ionization of the alginate backbone decreases, a number of negatively charged units persists. These units might interact with the positively charged units of P2VP lying within the micellar cores (Figure S2). It seems that this is not the case, which should be attributed to the thick PEO shells which protect the P2VP cores from contacts with the grafted alginate. However, some differentiation in Tc,thermothickening between the composite and the pure ALG-g-HG, i.e. shift at lower temperatures for the composite at pH<5, below the pKa of P2VP (Table S3), shows that ionic interactions between the charged units of P2VP and alginate should not be excluded.
In a previous paper [18] dealing with the thermothickening of ALG-g-P(NIPAM–co–NtBAM) hydrogels, it was shown that the ionic interactions between the negatively charged alginate units with the divalent cations of Ca2+ resulted to strong reinforcement of the network, as revealed by a significant increase of the elasticity below and above the transition temperature of thermothickening. Interestingly, the presence of the additional ionic interactions slightly affected the transition temperature, i.e. a few degrees lowering for the highest Ca2+ concentration. This means that the ionic interactions act independently and do not affect the hydrophobic ones exerted by the PNIPAM-based side chains, inducing thermothickening. The above corroborate our interpretation that the significant shift of the characteristics of the transition zone (Table 1 and Table S3) is due to the interactions between the alginate backbone and the side chains, the nature of which is likely of H-bonding, providing that at low pH most of the carboxylic units are in their protonated form.
3.2. Self-healing and Injectability
As it is known, this kind of physically crosslinked hydrogels exhibit finite linear viscoelastic regime, the extent of which depends on the network structure. Above a certain critical strain (γc) a gel-sol transition occurs due to the mechanical disruption of the physical crosslinks. This γc was determined at 51% conducting a strain sweep test at 37 °C on the formulation of pH 4.0 (Figure S4), where a reinforced network was observed.
To evaluate the self-healing of this hydrogel, successive oscillatory, time sweep experiments were performed by applying stepwise strains below and above γc. Particularly, the hydrogel was subjected to strain amplitudes at 1% and 300% for 600 s at each step, at 37 °C. As seen in Figure 7, the formulation exhibits gel-like behavior (G’>G”) at 1% strain which turns instantly to sol (G’<G”) upon applying 300% strain. The hydrogel was recovered instantly in the third step by switching the strain amplitude again to 1%, within the linear regime. The hydrogel recovery was reproducible as shown in the fourth and fifth steps. The above results confirmed the self-healing ability of the hydrogel since it is recovered after sudden network disruption.
Concerning the hydrogel performance (e.g. 3D printing), injectability is one of the critical properties of hydrogels as carriers of payloads. This property can be easily afforded when the thermoresponsive hydrogel exhibits sol-gel transition above room temperature, since it flows below the gelation temperature. This is the case of all the formulations for pH>4.0, while for the system at lower pH (pH 3.5), a gel-like behavior is valid even at room temperature. In the latter case shear thinning behavior is demanded. Hence, shear flow experiments were conducted to evaluate the response of the hydrogels to shear. Figure 8 displays the apparent shear viscosity (η/Pa.s) as a function of shear rate (increasing/decreasing) obtained at 20 °C for all pHs. Obviously, the viscosity increased by lowering pH in any shear rate examined, exhibiting shear thinning except for that at pH 5.4 where a Newtonian behavior was observed. Notably, the shear thinning is perfectly reversible practically without hysteresis since the viscosity values were recovered by decreasing shear rate. Moreover, the extent of shear thinning augments when the viscosity increases by lowering pH. For instance, the viscosity drops more than 500 folds by progressively increasing the shear rate from 0.01 to 100 s-1 for the formulation of pH 3.5, exhibiting the highest elasticity, while at pH 4.0, the viscosity decrease is about 200 times lower.
Finally, the response of viscosity to shear was evaluated by sudden changes of the applied shear rate as demonstrated in Figure 9. Specifically, the formulation of the highest elasticity (pH 3.5) was subjected to successive stepwise changes of shear rates from 0.01 (approaching situation at rest) to 20 s-1 (situation during injection). As observed, the viscosity drops about two orders of magnitude, and it is recovered promptly and reproducibly. In conclusion, the results of the experiments demonstrated in Figure 7, Figure 8 and Figure 9 showed that this kind of hydrogels exhibit reversible shear thinning, responding perfectly to sudden changes of shear and self-healing properties, meeting the requirements of 3D printable hydrogels.
3.3. Evaluation as drug delivery system
In order to evaluate the effectiveness of the nanocomposite hydrogel as pH-controlled drug delivery system, the micellar P2VP-b-PEO NPs were loaded with the hydrophobic Nile Red which was encapsulated in their hydrophobic cores and its release kinetics was conducted at the physiological temperature at two different pH.
It was found that the P2VP-b-PEO@Nile Red micelles displayed Nile Red loading efficiency LE=1.79% (w/w) for a theoretical (feed) loading Lth= 9.98% (w/w) and encapsulation efficiency EE=17.93% (w/w).
Nile Red-loaded P2VP-b-PEO micelles (P2VP-b-PEO@Nile Red) and ALG-g-HG/ /P2VP-b-PEO@Nile Red nanocomposite hydrogels were studied to evaluate the Nile Red release. Figure 10 presents the cumulative release of Nile Red versus time for both nanocarrier systems, at 37 °C and at two different buffer solutions of pH 3.5 (similar to the pH of gastric acid in the human stomach) and pH 7.4 (simulates the pH of blood and pH of healthy tissues). From the release study of the P2VP-b-PEO@Nile Red micelles, it appears that the release process is highly influenced by the pH of the medium. The micelles in buffer pH 3.5 release a higher amount of Nile Red (16% of Nile Red is released at 120 hours) than the micelles in buffer pH 7.4 (4% of Nile Red is released at 120 hours). This is due to the protonation of the P2VP moieties of the P2VP-b-PEO polymer that makes them less hydrophobic, leading to micelles with a less dense P2VP core. This way, the Nile Red molecules can escape easier from the hydrophobic P2VP micelle cores.
Table S4 in Supplementary Material summarizes the particle size distributions of the P2VP-b-PEO NPs at three different pH values of the aqueous medium. As observed when the pH was adjusted to a lower value of pH 3.5, the micellar diameter was substantially decreased to ~3.5 nm. At such acidic environment, the degree of ionization of P2VP blocks increases by protonation, leading to disassembly of the micelles into smaller associates or even unimers. These results are in good agreement with those previously reported for copolymers with similar PEO/P2VP(P4VP) hydrophilic/hydrophobic balance [52,53,54]. Thus, one would expect acceleration of the Nile Red release from the P2VP-b-PEO micelles at pH 3.5. where the P2VP blocks are partially charged (Figure S2). However, only 16% of Nile Red was achieved at 120 hours, at pH 3.5. This probably could be attributed by the fact that Nile Red is a molecule of high hydrophobicity which could also form π–π stacking interactions with the PVP blocks, making it firmly retained in the polymer micelles.
At pH 7.4, an initial burst release can be observed, which may imply that some Nile Red molecules are weakly bound within the micelle cores and that a fraction of the encapsulated Nile Red is located at the core/corona interface of the micelles and therefore it is more accessible and prone to release. In this pH, high above the pKa of P2VP at pH 5.0, the P2VP moieties are hydrophobic. The low release 4% of Nile Red in buffer pH 7.4 could be attributed to the highly hydrophobic nature of Nile Red and possible strong, non-covalent interactions of Nile Red with the P2VP hydrophobic micellar cores causing a more firmly Nile Red entrapment in the P2VP-b-PEO micelles. These findings are in agreement with the pH-dependent release profiles of various hydrophobic drugs from poly(ethylene glycol)-b-poly(2-vinylpyridine) micelles. It should be mentioned that the release kinetics of hydrophobic drugs is remarkably influenced by their chemical structure except of the size and the degree of protonation (controlled by pH) of the P2VP blocks forming the hydrophobic cores [54].
Inspired by the study of Chroni et al. [55], which proved that ultrasound has significant influence on the release profile of curcumin (CUR) from CUR-loaded nanocarriers, we proceeded to a similar study. So, after the 120 hours of the release experiments, the samples were subjected to ultrasonication. It can be clearly seen that for pH 3.5, after 8 hours under ultrasound stimulation, a high release of Nile Red occurred (release rate increased from 16% to 31% at 128 hours (total experiment hours). After further sonication, 16 more hours (to reach 144 total experiment hours), Nile Red release increased further to 35%. The increase of Nile Red release from the micelles submerged in the PB buffer of pH 7.4, was marginal, showing a rise of only 2% after 24 hours of sonication (from 4% at 120 hours, the release of Nile Red reached a 6% at 144 hours). This release resistance under sonication confirms the strong attractive interactions between Nile Red and the vinyl pyridine moieties of the micellar cores (see also reference [56]), which justifies the observed release kinetics.
When the P2VP-b-PEO@Nile Red micelles were loaded in the ALG-g-HG hydrogel, it can be seen, that for both PB buffer solutions at pH 3.5 and pH 7.4, Nile Red release is lower, around 6% and 3.3%, suggesting noticeable slowdown of the Nile Red release rate which is attributed to the high viscosity of the hydrogel medium [46]. Importantly, this effect was more pronounced at pH 3.5. This behavior is related to the pH-induced alteration of the network structure formed by the ALG-g-HG gelator. As already discussed in Figure 2 and Figure 3, the network is reinforced by decreasing pH as reflected to G’ augmentation, due to additional crosslinking induced by H-bonding. Particularly, the complex viscosity η* increased remarkably with the pH reduction at 37 °C, where the release experiments were conducted, as demonstrated in Figure S5. This effect might justify the deceleration of the Nile Red release rate due to the decrease of drug diffusion within the higher viscosity hydrogel medium.
4. Conclusions
The pH dependent properties of LCST-type thermo-responsive alginate-based hydrogels incorporating poly(2-vinyl pyridine)-b-polyethylene oxide (P2VP-b-PEO) micellar nanoparticles were explored by rheology. Due to the thermo-responsive side chains, the ALG-g-HG copolymer (4 wt%) self-assembles upon heating, exhibiting thermothickening behavior above a sol–gel transition Tgel. Interestingly, the study at low pH range just below 5.0 revealed a notable increase of the elasticity of the polymer solution occurring even at low temperatures below Tgel. This effect transformed the sol-gel to a soft gel–strong gel transition. It was proved that this was not due to the interactions with the protonated P2VP (pH<pKa) of the diblock copolymer, provided that this effect was similar with the formulation in the absence of P2VP-b-PEO copolymer. Importantly, the onset of thermothickening shifted notably to lower temperatures followed by broadening of the transition zone, implying secondary intermolecular interactions between the protonated repeating units of the alginate backbone with the PNIPAM side chains, likely through H–bonding.
Evaluating the ALG-g-HG/P2VP-b-PEO composite hydrogel as drug delivery system, it was found that the release of a hydrophobic model drug (Nile Red) is affected by the pH of the medium. At pH 3.5, the release of Nile Red was decelerated, due to the additional H-bonding that increases the crosslinking density of the hydrogel network and in turn the viscosity of the hydrogel medium. Moreover, the shear–thinning behavior in combination with the thermo-thickening characteristics of the material, make it a good candidate for potential applications such as 3D–printable composite carrier of payloads in biomedicine.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Molecular characteristics of the ALG-g-HG heterograft copolymer; Table S2: Molecular characteristics of the P2VP-b-PEO copolymer; Table S3: Characteristic factors of thermoresponsiveness at various pH for the pure ALG-g-HG copolymer; Table S4: The hydrodynamic diameters of P2VP-b-PEO polymeric micelles obtained from DLS analyses; Figure S1: Storage (G’) and loss (G”) modulus as a function of temperature of ALG-g-HG solutions (a) 5 wt% and (b) 4 wt%; Figure S2: Zeta potential of aqueous P2VP-b-PEO and ALG-g-HG solutions at different pH; Figure S3: Storage (G’) and loss (G”) modulus as a function of temperature of 4 wt% ALG-g-HG hydrogels at (a) pH 4.5; (b) pH 3.5; Figure S4: Oscillatory strain sweep data at 1 Hz of the 4 wt% ALG-g-HG hydrogel at pH 4 and 37 °C; Figure S5: pH dependence of complex viscosity (1Hz) at 37 °C.
Author Contributions
Conceptualization, C.T.; methodology, Z.I and A.I.; validation, Z.I and A.I.; investigation, Z.I and A.I.; data curation, Z.I and A.I.; writing—original draft preparation, C.T, Z.I and A.I.; writing—review and editing, C.T, Z.I and A.I.; supervision, C.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chassenieux, C.; Tsitsilianis, C. Recent trends on pH/thermo-responsive self assembling hydrogels: From polyions to peptide-based polymeric gelators. Soft Matter 2016, 12, 1344–1359. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Choudhury, A.R. Stimuli-Responsive Polysaccharide-Based Smart Hydrogels and Their Emerging Applications. Ind. Eng. Chem. Res. 2023, 62, 841–866. [Google Scholar] [CrossRef]
- Trivedi, J.; Chourasia, A. Sodium Salt of Partially Carboxymethylated Sodium Alginate-Graft-Poly(Acrylonitrile): II Superabsorbency, Salt Sensitivity and Swelling Kinetics of Hydrogel, H-Na-PCMSA-g-PAN. Gels 2023, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Lupu, A.; Gradinaru, L.M.; Rusu, D.; Bercea, M. Self-Healing of Pluronic® F127 Hydrogels in the Presence of Various Polysaccharides. Gels 2023, 9, 719. [Google Scholar] [CrossRef] [PubMed]
- Koetting, M.C.; Peters, J.T.; Steichen, S.D.; Peppas, N.A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Engin. R 2015, 93, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Manna, K.; Pal, S. Recent Advances in Various Stimuli-Responsive Hydrogels: From Synthetic Designs to Emerging Healthcare Applications. Mater. Chem. Front. 2022, 6, 2338–2385. [Google Scholar] [CrossRef]
- Afzal, S.; Maswal, M.; Ahmad Dar, A. Rheological behavior of pH responsive composite hydrogels ofchitosan and alginate: Characterization and its use in encapsulation of citral. Colloids Surf. B Biointerfaces 2018, 169, 99–106. [Google Scholar] [CrossRef]
- Diego, S.; Lima, D.S.; Tenório-Neto, E.T.; Lima-Tenório, M.K.; Guilherme, M.R.; Scariot, D.B.; Nakamura, C.V.; Muniz, E.C.; Rubira, A.F. pH-responsive alginate-based hydrogels for protein delivery. J. Mol. Liq. 2018, 262, 29–36. [Google Scholar] [CrossRef]
- Lin, X.; Guan, X.; Wu, Y.; Zhuang, S.; Wu, Y.; Du, L.; Zhao, J.; Rong, J.; Zhao, J.; Tu, M. An Alginate/Poly(N-Isopropylacrylamide)-Based Composite Hydrogel Dressing with Stepwise Delivery of Drug and Growth Factor for Wound Repair. Mater. Sci. Eng. C 2020, 115, 111123. [Google Scholar] [CrossRef]
- Ilgin, P.; Ozay, H.; Ozay, O. Synthesis and characterization of pH responsive alginate based-hydrogels as oral drug delivery carrier. J. Polym. Res. 2020, 27, 251. [Google Scholar] [CrossRef]
- Chatterjee, S.; Chi-leung Hui, P. Review of Applications and Future Prospects of Stimuli-Responsive Hydrogel Based on Thermo-Responsive Biopolymers in Drug Delivery Systems. Polymers 2021, 13, 2086. [Google Scholar] [CrossRef] [PubMed]
- Santhamoorthy, M.; Vy Phan, T.T.; Ramkumar, V.; Raorane, C.J.; Thirupathi, K.; Kim, S.-C. Thermo-sensitive Poly (N-Isopropylacrylamide-Co-Polyacrylamide) Hydrogel for PH-Responsive Therapeutic Delivery. Polymers 2022, 14, 4128. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.; Gani, A. Alginate-Based pH-Sensitive Hydrogels Encoated with Chitosan as a Bioactive Cargo Carrier with Caffeic Acid as a Model Biomolecule. ACS Food Sci. Technol. 2022, 2, 667–672. [Google Scholar] [CrossRef]
- Suhail, M.; Ullah, H.; Vu, Q.L.; Khan, A.; Tsai, M.-J.; Wu, P.-C. Preparation of pH-Responsive Hydrogels Based on Chondroitin Sulfate/Alginate for Oral Drug Delivery. Pharmaceutics 2022, 14, 2110. [Google Scholar] [CrossRef]
- Peng, X.; Peng, Q.; Wu, M.; Wang, W.; Gao, Y.; Liu, X.; Sun, Y.; Yang, D.; Peng, Q.; Wang, T.; Chen, X.-Z.; Liu, J.; Zhang, H.; Zeng, H. A PH and Temperature Dual-Responsive Microgel-Embedded, Adhesive, and Tough Hydrogel for Drug Delivery and Wound Healing. ACS Appl. Mater. Interfaces 2023, 15, 19560–19573. [Google Scholar] [CrossRef] [PubMed]
- Aleena Mir, A.; Kumar, A.; Alam, J.; Riaz, U. Synthesis and characterization of pH-responsive conducting polymer/ Na-alginate/gelatin based composite hydrogels for sustained release of amoxicillin drug. Int. J. Biol. Macromol. 2023, 252, 126015. [Google Scholar] [CrossRef]
- Murab, S.; Gupta, A.; Włodarczyk-Biegun, M.K.; Kumar, A.; van Rijn, P.; Whitlock, P.; Han, S.S.; Agrawal, G. Alginate Based Hydrogel Inks for 3D Bioprinting of Engineered Orthopedic Tissues. Carbohydr. Polym. 2022, 296, 119964. [Google Scholar] [CrossRef]
- Saravanou, S.F.; Ioannidis, K.; Dimopoulos, A.; Paxinou, A.; Kounelaki, F.; Varsami, S.M.; Tsitsilianis, C.; Papantoniou, I.; Pasparakis, G. Dually Crosslinked Injectable Alginate-Based Graft Copolymer Thermoresponsive Hydrogels as 3D Printing Bioinks for Cell Spheroid Growth and Release. Carbohydr. Polym. 2023, 312, 120790. [Google Scholar] [CrossRef]
- Saravanou, S.F.; Tsitsilianis, C.; Pasparakis, G. Harnessing the Interplay of Triple Cross-Linked Hydrogels toward Multiresponsive Alginate-Based Injectable Gels for 3D Printing Bioapplications. ACS Macro Lett. 2023, 12, 1614–1622. [Google Scholar] [CrossRef]
- Gialouri, A.; Saravanou, S.F.; Loukelis, K.; Chatzinikolaidou, M.; Pasparakis, G.; Bouropoulos, N. Thermoresponsive Alginate-GraftpNIPAM/Methyl Cellulose 3D-Printed Scaffolds Promote Osteogenesis In Vitro. Gels 2023, 9, 984. [Google Scholar] [CrossRef]
- Knipe, J.M.; Peppas, N.A. Multi-Responsive Hydrogels for Drug Delivery and Tissue Engineering Applications. Regen. Biomater. 2014, 1, 57–65. [Google Scholar] [CrossRef]
- Kumar, A.; I Matari, I.A.; Han, S.S. 3D Printable Carboxylated Cellulose Nanocrystal-Reinforced Hydrogel Inks for Tissue Engineering. Biofabrication 2020, 12, 025029. [Google Scholar] [CrossRef]
- Bellotti, E.; Schilling, A.L.; Little, S.R.; Decuzzi, P. Injectable thermoresponsive hydrogels as drug delivery system for the treatment of central nervous system disorders: A review. J. Control. Release 2021, 329, 16–35. [Google Scholar] [CrossRef]
- Ghandforoushan, P.; Alehosseini, M.; Golafshan, N.; Castilho, M.; Dolatshahi-Pirouz, A.; Hanaee, J.; Davaran, S.; Orive, G. Injectable hydrogels for cartilage and bone tissue regeneration: A review. Int. J. Biol. Macromol. 2023, 246, 125674. [Google Scholar] [CrossRef]
- Qian Huang, Q.; Zhou1, Y.; Fu, Z.; Zhu, J. Preparation of an injectable hydrogel reinforced by graphene oxide and its application in dye wastewater treatment. J. Mater. Sci. 2023, 58, 3117–3133. [Google Scholar] [CrossRef]
- Tanga, S.; Aucamp, M.; Ramburrun, P. Injectable Thermoresponsive Hydrogels for Cancer Therapy: Challenges and Prospects. Gels 2023, 9, 418. [Google Scholar] [CrossRef]
- Fan, R.; Cheng, Y.; Wang, R.; Zhang, T.; Zhang, H.; Li, J.; Song, S.; Zheng, A. Thermosensitive Hydrogels and Advances in Their Application in Disease Therapy. Polymers 2022, 14, 2379. [Google Scholar] [CrossRef]
- Huang, Y.; Zeng, W.; Li, P.; Liao, Q.; Xia, A.; Zhu, X.; Zhu, X. Poly(N-isopropylacrylamide-co-N-tert-butylacrylamide)-graph-polydopamine as a Thermoresponsive Surface with Adjustable Transformation Temperature for Efficient Microalgal Cell Adhesion and Detachment. ACS Appl. Polym. Mater. 2022, 4, 8180–8192. [Google Scholar] [CrossRef]
- Barbier, L.; Protat, M.; Pipart, P.; Marcellan, A.; Tran, Y.; Hourdet, D. Sol/gel transition of thermoresponsive Hyaluronan: From liquids to elastic and sticky materials. Carbohydr. Polym. 2023, 310, 120715. [Google Scholar] [CrossRef]
- Eskens, O.; Villani, G.; Amin, S. Rheological Investigation of Thermoresponsive Alginate-Methylcellulose Gels for Epidermal Growth Factor Formulation. Cosmetics 2021, 8, 3. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution Properties of Poly(N–isopropylacrylamide). J. Macromol. Sci. Part A – Chem. 1969, 2, 1441–1455. [Google Scholar] [CrossRef]
- Iatridi, Z.; Saravanou, S.F.; Tsitsilianis, C. Injectable self-assembling hydrogel from alginate grafted by P(N-isopropylacrylamide-co-N-tert-butylacrylamide) random copolymers. Carbohydr. Polym. 2019, 219, 344–352. [Google Scholar] [CrossRef]
- Safakas, K.; Saravanou, S.-F.; Iatridi, Z.; Tsitsilianis, C. Alginate-g-PNIPAM-based Thermo/shear-responsive Injectable Hydrogels: Tailoring the Rheological Properties by Adjusting the LCST of the Grafting Chains. Int. J. Mol. Sci. 2021, 22, 3824. [Google Scholar] [CrossRef]
- Durand, A.; Hourdet, D. Thermoassociative graft copolymers based on poly(N-isopropylacrylamide): Effect of added co-solutes on the rheological behaviour. Polymer 2000, 41, 545–557. [Google Scholar] [CrossRef]
- Leal, D.; De Borggraeve, W.; Encinas, M.V.; Matsuhiro, B.; Muller, R. Preparation and characterization of hydrogels based on homopolymeric fractions of sodium alginate and PNIPAAm. Carbohydr. Polym. 2013, 92, 157–166. [Google Scholar] [CrossRef]
- Martinez-Gomez, F.; Encinas, M.V.; Matsuhiro, B.; Pavez, J. Preparation and swelling properties of homopolymeric alginic acid fractions/poly(N-isopropyl acrylamide) graft copolymers. J. Appl. Polym. Sci. 2015, 132, 42398. [Google Scholar] [CrossRef]
- Lencina, S.M.M.; Ciolino, A.E.; Andreucetti, N.A.; Villar, M.A. Thermoresponsive hydrogels based on alginate-g-poly(N-isopropylacrylamide) copolymers obtained by low doses of gamma radiation. Eur. Polym. J. 2015, 68, 641–649. [Google Scholar] [CrossRef]
- Liu, M.; Song, X.; Wen, Y.; Zhu, J.-L.; Li, J. Injectable Thermoresponsive Hydrogel Formed by Alginate-g-Poly(N-isopropylacrylamide) Releasing Doxorubicin-Encapsulated Micelles as Smart Drug Delivery System. ACS Appl. Mater. Interfaces 2017, 9, 35673–35682. [Google Scholar] [CrossRef]
- Guo, H.; de Magalhaes Goncalves, M.; Ducouret, G.; Hourdet, D. Cold and Hot Gelling of Alginate-graft-PNIPAM: A Schizophrenic Behavior Induced by Potassium Salts. Biomacromolecules 2018, 12, 576–587. [Google Scholar] [CrossRef]
- Chalanqui, M.J.; Pentlavalli, S.; McCrudden, C.; Chambers, P.; Ziminska, M.; Dunne, N.; McCarthy, H.O. Influence of alginate backbone on efficacy of thermo-responsive alginate-g-P(NIPAAm) hydrogel as a vehicle for sustained and controlled gene delivery. Mater. Sci. Eng. C 2019, 95, 409–421. [Google Scholar] [CrossRef]
- Lencina, M.M.S.; Iatridi, Z.; Villar, M.A.; Tsitsilianis, C. Thermoresponsive hydrogels from alginate-based graft copolymers. Eur. Polym. J. 2014, 61, 33–44. [Google Scholar] [CrossRef]
- Safakas, K.; Saravanou, S.-F.; Iatridi, Z.; Tsitsilianis, C. Thermo-Responsive Injectable Hydrogels Formed by Self-Assembly of Alginate-Based Heterograft Copolymers. Gels 2023, 9, 236. [Google Scholar] [CrossRef]
- Wei, L.; Cai, C.; Lin, J.; Chen, T. Dual-drug delivery system based on hydrogel/micelle composites. Biomaterials 2009, 30, 2606–2613. [Google Scholar] [CrossRef]
- Lu, C.; Mikhail, A.S.; Wang, X.; Brook, M.A.; Allen, C. Hydrogels Containing Core Cross-Linked Block Co-Polymer Micelles. J. Biomater. Sci. Polym. Ed. 2012, 23, 1069–1090. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Parvathy, J.; Nair, A.S. A novel composite matrix based on polymeric micelle and hydrogel asa drug carrier for the controlled release of dual drugs. Carbohydr. Polym. 2016, 136, 1118–1127. [Google Scholar] [CrossRef]
- Theodorakis, N.; Saravanou, S.-F.; Kouli, N.-P.; Iatridi, Z.; Tsitsilianis, C. pH/Thermo-Responsive Grafted Alginate-Based SiO2 Hybrid Nanocarrier/Hydrogel Drug Delivery Systems. Polymers 2021, 13, 1228. [Google Scholar] [CrossRef]
- Ertugral-Samgar, E.G.; Ozmen, A.M.; Gok, O. Thermo-Responsive Hydrogels Encapsulating Targeted Core–Shell Nanoparticles as Injectable Drug Delivery Systems. Pharmaceutics 2023, 15, 2358. [Google Scholar] [CrossRef]
- Karanikolas, A.; Tsolakis, P.; Bokias, G.; Tsitsilianis, C. Stimuli-responsive poly(ethylene oxide)-b-poly(2-vinylpyridine)-b- poly(ethylene oxide) triblock copolymers and complexation with poly(acrylic acid) at low pH. Europ. Phys. J. E. 2008, 27, 335–343. [Google Scholar] [CrossRef]
- Martin, T.J.; Prochazka, K.; Munk, P.; Webber, S.E. pH-Dependent Micellization of Poly(2-vinylpyridine)-block-poly(ethylene oxide). Macromolecules 1996, 29, 6071. [Google Scholar] [CrossRef]
- Förster, S.; Abetz, V.; Muller, A.H.E. Polyelectrolyte Block Copolymer Micelles. In: Schmidt, M. (Eds.), Polyelectrolytes with Defined Molecular Architecture II. Adv. Polym. Sci. 2004, 166, 173. [Google Scholar] [CrossRef]
- Vamvakaki, M.; Papoutsakis, L.; Katsamanis, V.; Afchoudia, T.; Fragouli, P.G.; Iatrou, H.; Hadjichristidis, N.; Armes, S.P.; Sidorov, S.; Zhirov, D.; Zhirov, V.; Kosylev, M.; Bronstein, L.; Anastasiadis, S.H. Micellization in pH-sensitive amphiphilic block copolymers in aqueous media and the formation of metal nanoparticles. Faraday Discuss. 2005, 128, 129. [Google Scholar] [CrossRef]
- Gohy, J.-F.; Mores, S.; Varshney, S.K.; Zhang, J.-X.; Jérôme, R. pH-Dependence of the morphology of micelles formed by poly(2-vinylpyridine)-block-poly(ethylene oxide) copolymers in water. e-Polymers 2002, 2, 290–297. [Google Scholar] [CrossRef]
- Borchert, U.; Lipprandt, U.; Bilang, M.; Kimpfler, A.; Rank, A.; Peschka-Süss, R.; Schubert, R.; Lindner, P.; Förster, S. pH-Induced Release from P2VP-PEO Block Copolymer Vesicles. Langmuir 2006, 22, 5843–5847. [Google Scholar] [CrossRef] [PubMed]
- Brewer, K.; Bai, F.; Blencowe, A. pH-Responsive Poly(ethylene glycol)-b-poly(2-vinylpyridine) Micelles for the Triggered Release of Therapeutics. Pharmaceutics 2023, 15, 977. [Google Scholar] [CrossRef] [PubMed]
- Chroni, A.; Mavromoustakos, T.; Pispas, S. Curcumin-Loaded PnBA-b-POEGA Nanoformulations: A Study of Drug-Polymer Interactions and Release Behavior. Int. J. Mol. Sci. 2023, 24, 4621. [Google Scholar] [CrossRef] [PubMed]
- Popescu, M.-T.; Tsitsilianis, C. Controlled Delivery of Functionalized Gold Nanoparticles by pH-Sensitive Polymersomes. ACS Macro Lett. 2013, 2, 222–225. [Google Scholar] [CrossRef]
Figure 1.
(a) Volume and (b) Number average particle size distributions of 0.075 wt% P2VP-b-PEO aqueous solution at pH 7.4 and at 25 °C.
Figure 1.
(a) Volume and (b) Number average particle size distributions of 0.075 wt% P2VP-b-PEO aqueous solution at pH 7.4 and at 25 °C.
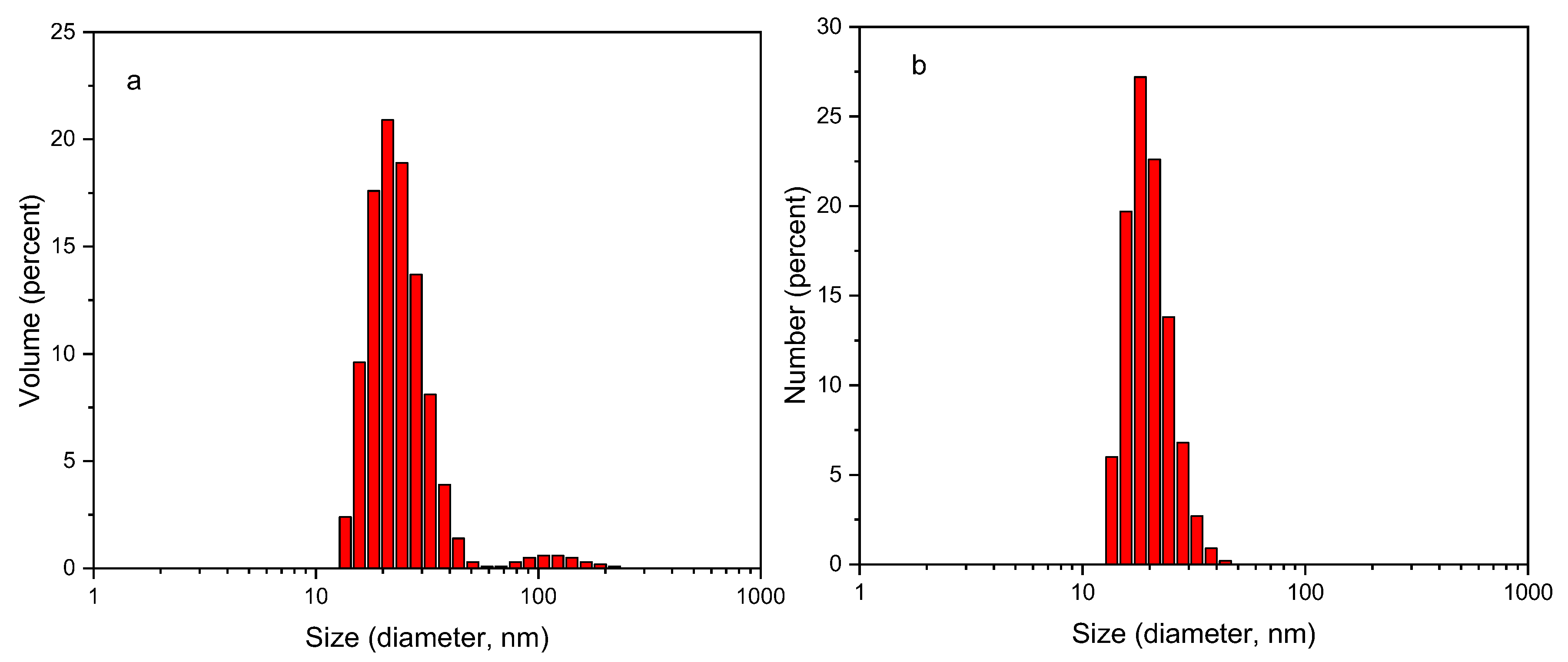
Figure 2.
Storage (G’) and loss (G”) modulus as a function of temperature of the ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels at (a) pH 5.4; (b) pH 4.5; (c) pH 4.0 and (d) pH 3.5.
Figure 2.
Storage (G’) and loss (G”) modulus as a function of temperature of the ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels at (a) pH 5.4; (b) pH 4.5; (c) pH 4.0 and (d) pH 3.5.
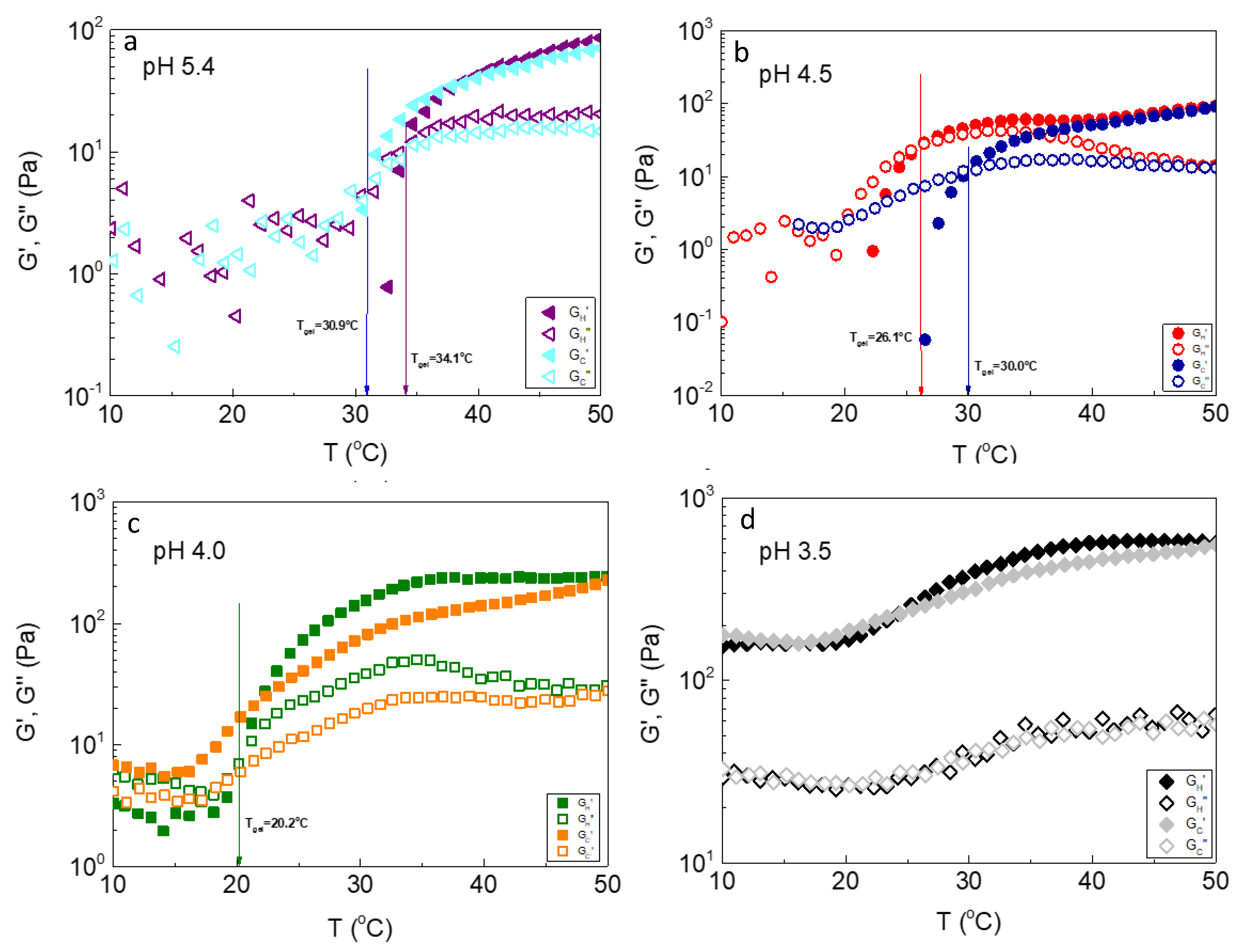
Figure 3.
(a) Storage (G’) modulus and (b) tanδ as a function of temperature of the ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels, at various pHs: pH 3.5 (black symbols); pH 4.0 (green symbols); pH 4.5 (red symbols); pH 5.4 (purple symbols).
Figure 3.
(a) Storage (G’) modulus and (b) tanδ as a function of temperature of the ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels, at various pHs: pH 3.5 (black symbols); pH 4.0 (green symbols); pH 4.5 (red symbols); pH 5.4 (purple symbols).
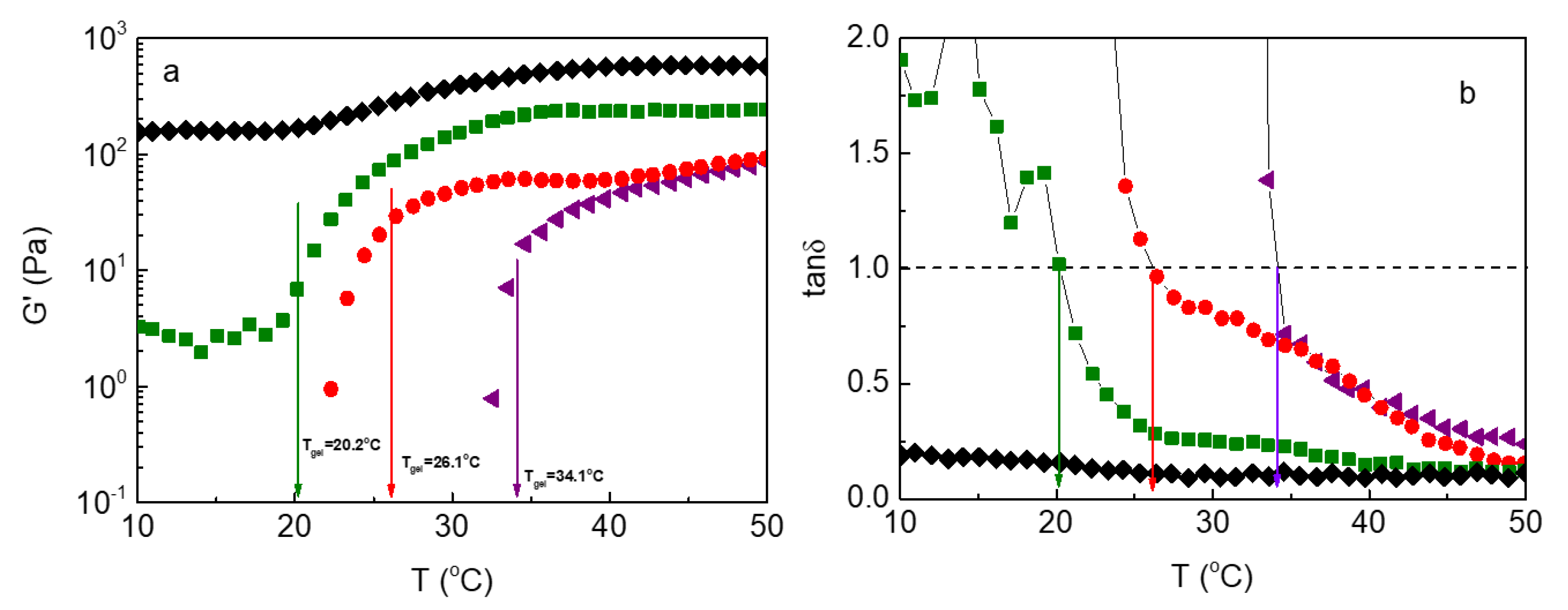
Figure 4.
The three temperature zones designated in the storage (G’) and loss (G”) modulus versus temperature plot of an ALG-g-HG/P2VP-b-PEO nanocomposite hydrogel.
Figure 4.
The three temperature zones designated in the storage (G’) and loss (G”) modulus versus temperature plot of an ALG-g-HG/P2VP-b-PEO nanocomposite hydrogel.
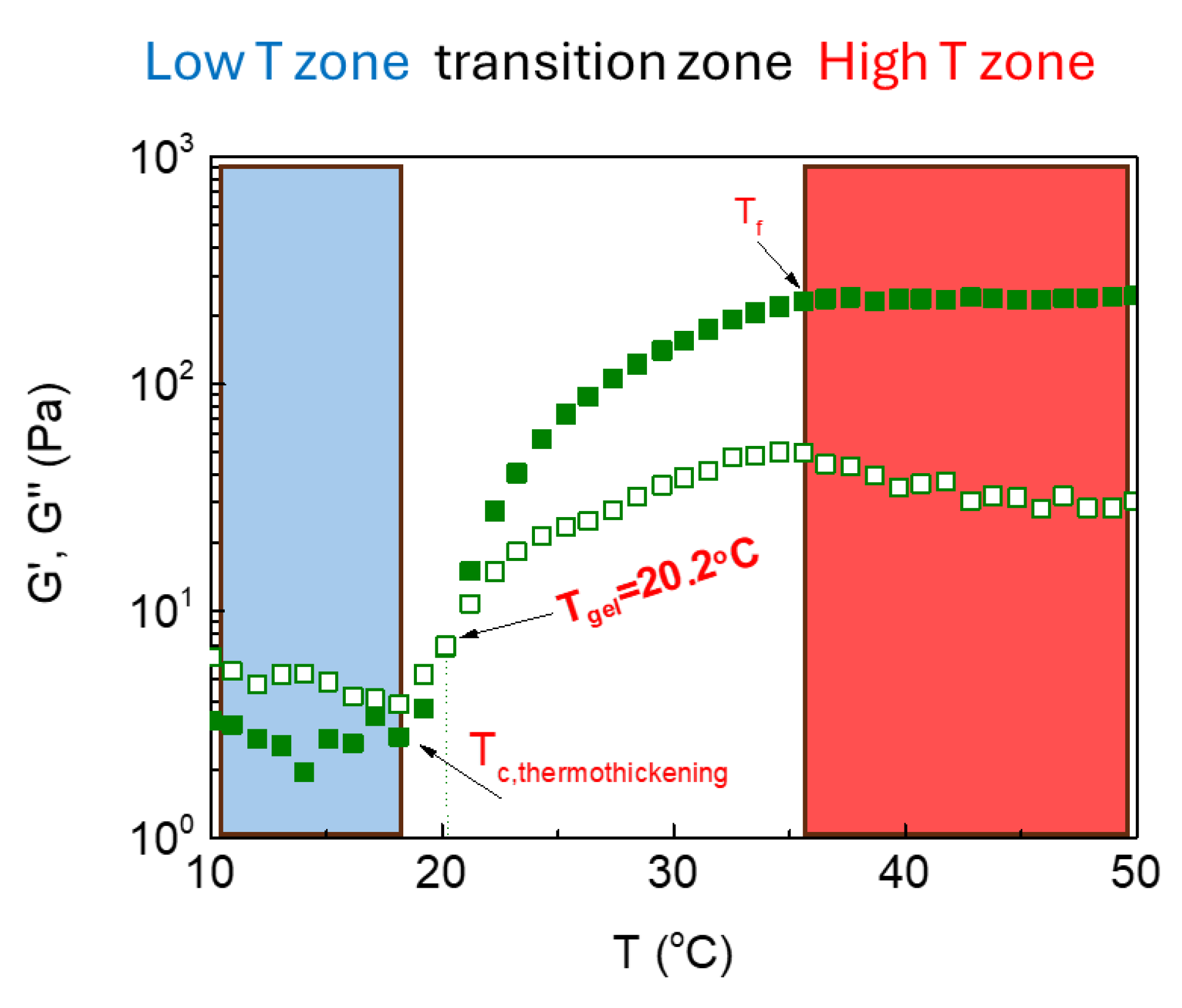
Figure 5.
pH dependence of (a) Tc,thermothickening and (b) Storage modulus (G’) at 15 and 50 °C, as obtained from Figure 3a, for the ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels.
Figure 5.
pH dependence of (a) Tc,thermothickening and (b) Storage modulus (G’) at 15 and 50 °C, as obtained from Figure 3a, for the ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels.

Figure 6.
Storage modulus (G’) as a function of temperature of the pure ALG-g-HG hydrogels (open symbols) and ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels (solid symbols), at various pHs: pH 3.5 (black symbols); pH 4.5 (blue symbols); pH 5.4 (purple symbols).
Figure 6.
Storage modulus (G’) as a function of temperature of the pure ALG-g-HG hydrogels (open symbols) and ALG-g-HG/P2VP-b-PEO nanocomposite hydrogels (solid symbols), at various pHs: pH 3.5 (black symbols); pH 4.5 (blue symbols); pH 5.4 (purple symbols).
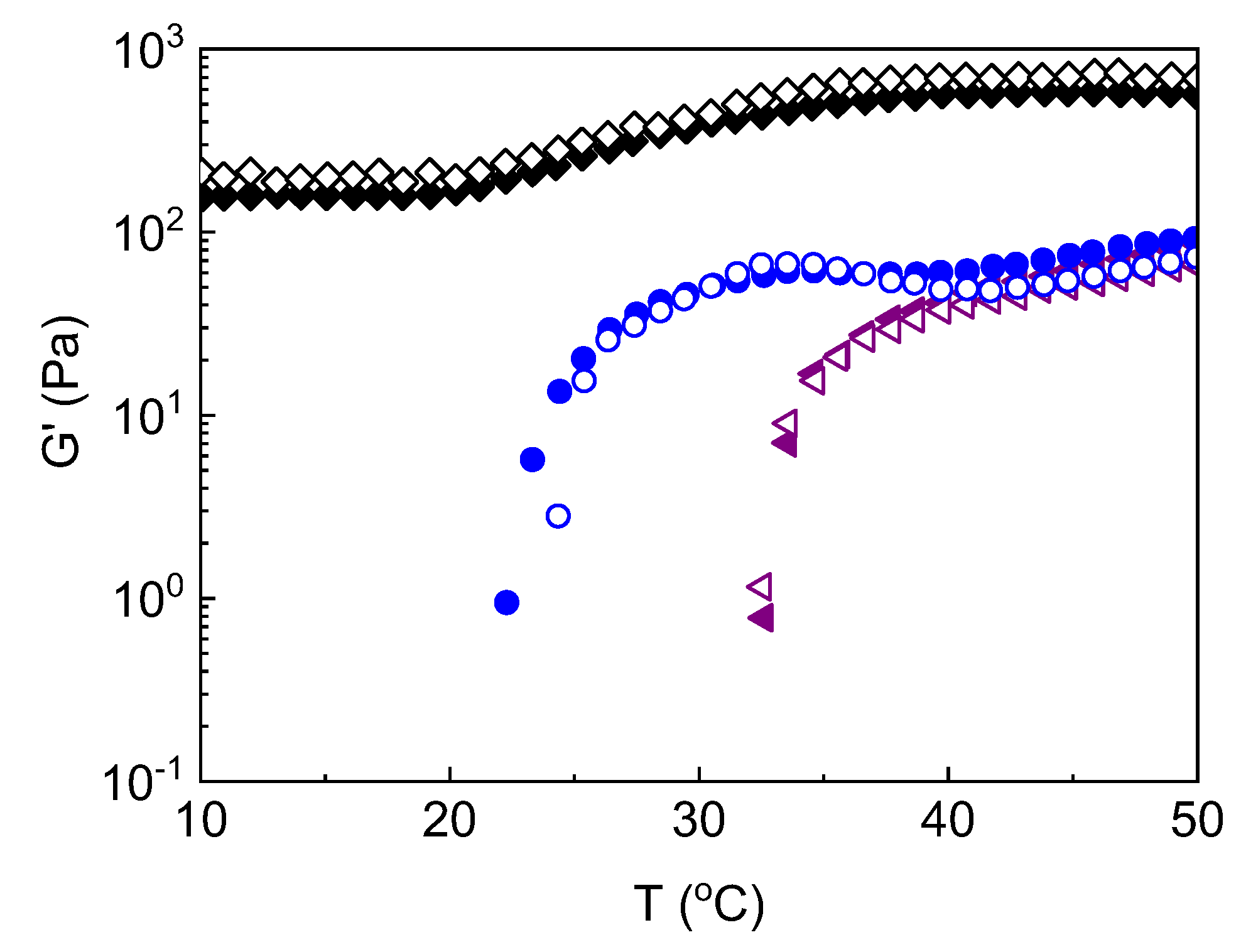
Figure 7.
Time dependence of G’ (solid symbols) and G” (open symbols), subjected to consecutive variations of strain amplitude, for the ALG-g-HG/P2VP-b-PEO composite hydrogel at pH 4.0 and 37 °C.
Figure 7.
Time dependence of G’ (solid symbols) and G” (open symbols), subjected to consecutive variations of strain amplitude, for the ALG-g-HG/P2VP-b-PEO composite hydrogel at pH 4.0 and 37 °C.
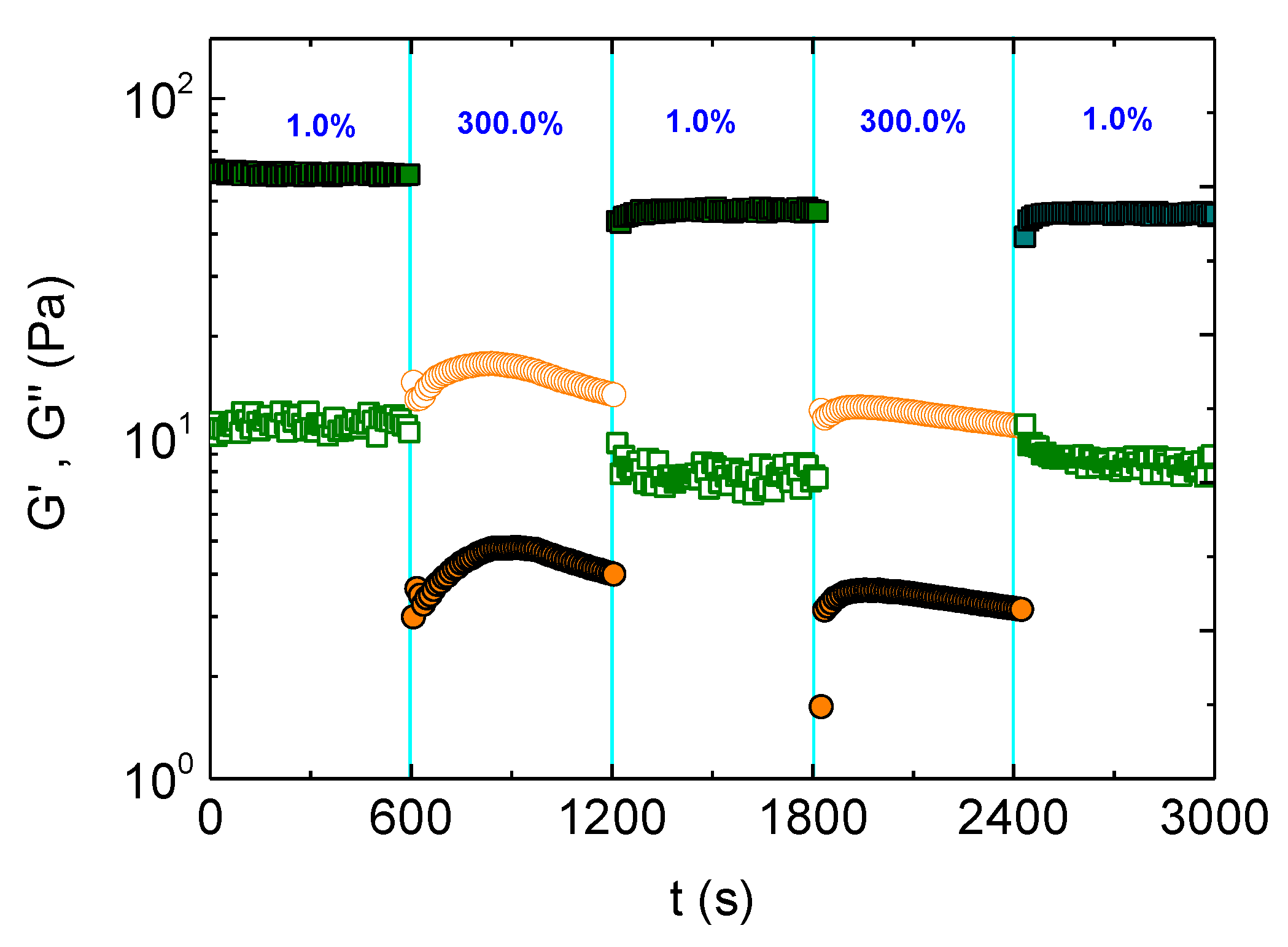
Figure 8.
Apparent shear viscosity as a function of shear rate (increasing (open symbols)/decreasing (solid symbols)) obtained at 20 °C for the ALG-g-HG/P2VP-b-PEO composite hydrogel at all pHs: pH 3.5 (black symbols); pH 4.0 (green symbols); pH 4.5 (red symbols); pH 5.4 (purple symbols).
Figure 8.
Apparent shear viscosity as a function of shear rate (increasing (open symbols)/decreasing (solid symbols)) obtained at 20 °C for the ALG-g-HG/P2VP-b-PEO composite hydrogel at all pHs: pH 3.5 (black symbols); pH 4.0 (green symbols); pH 4.5 (red symbols); pH 5.4 (purple symbols).
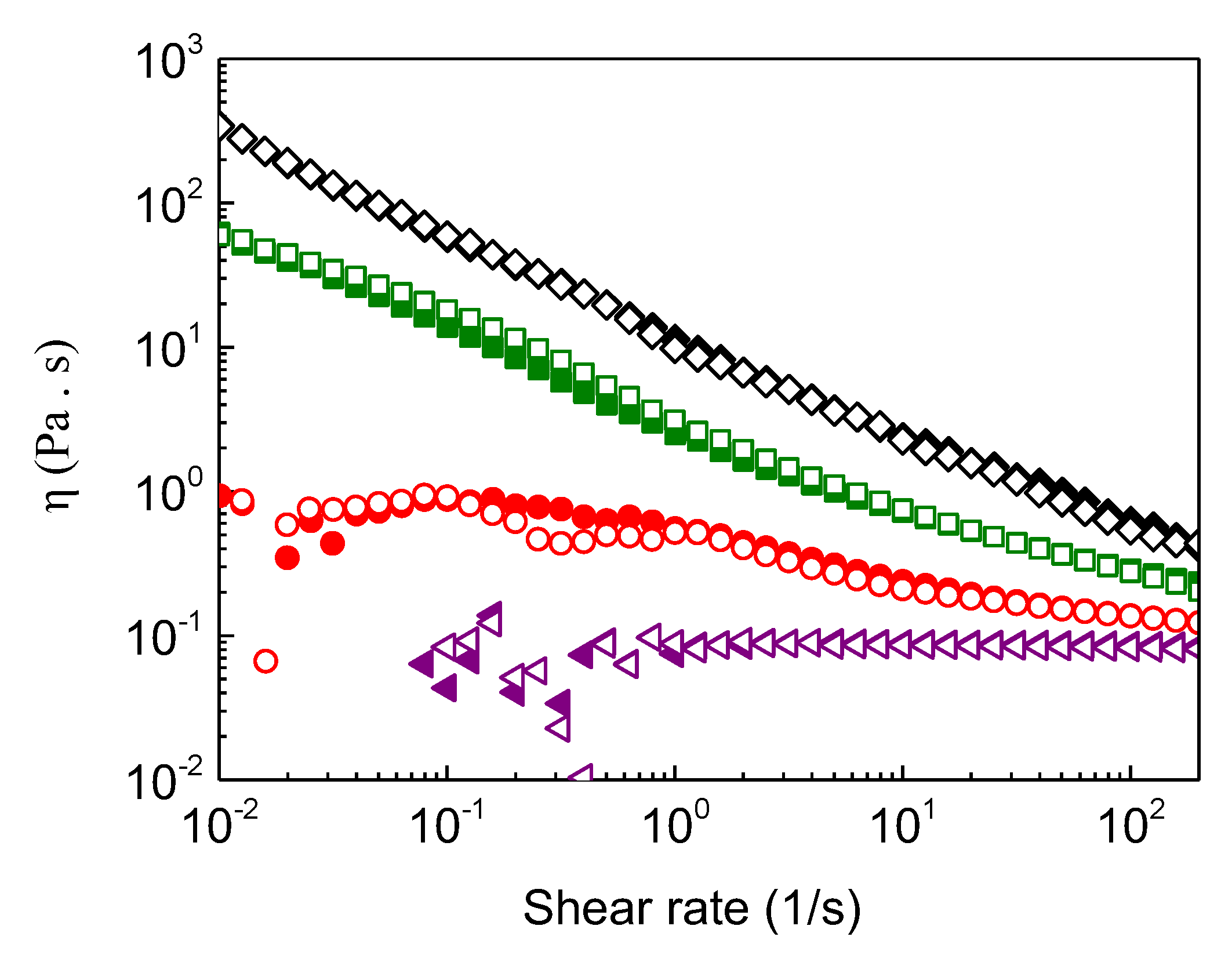
Figure 9.
Time dependence of apparent shear viscosity subjected to consecutive variations of shear rates from 0.01 (□) to 20 s-1 (o) for the ALG-g-HG/P2VP-b-PEO composite hydrogel at pH 3.5 and 20 °C.
Figure 9.
Time dependence of apparent shear viscosity subjected to consecutive variations of shear rates from 0.01 (□) to 20 s-1 (o) for the ALG-g-HG/P2VP-b-PEO composite hydrogel at pH 3.5 and 20 °C.
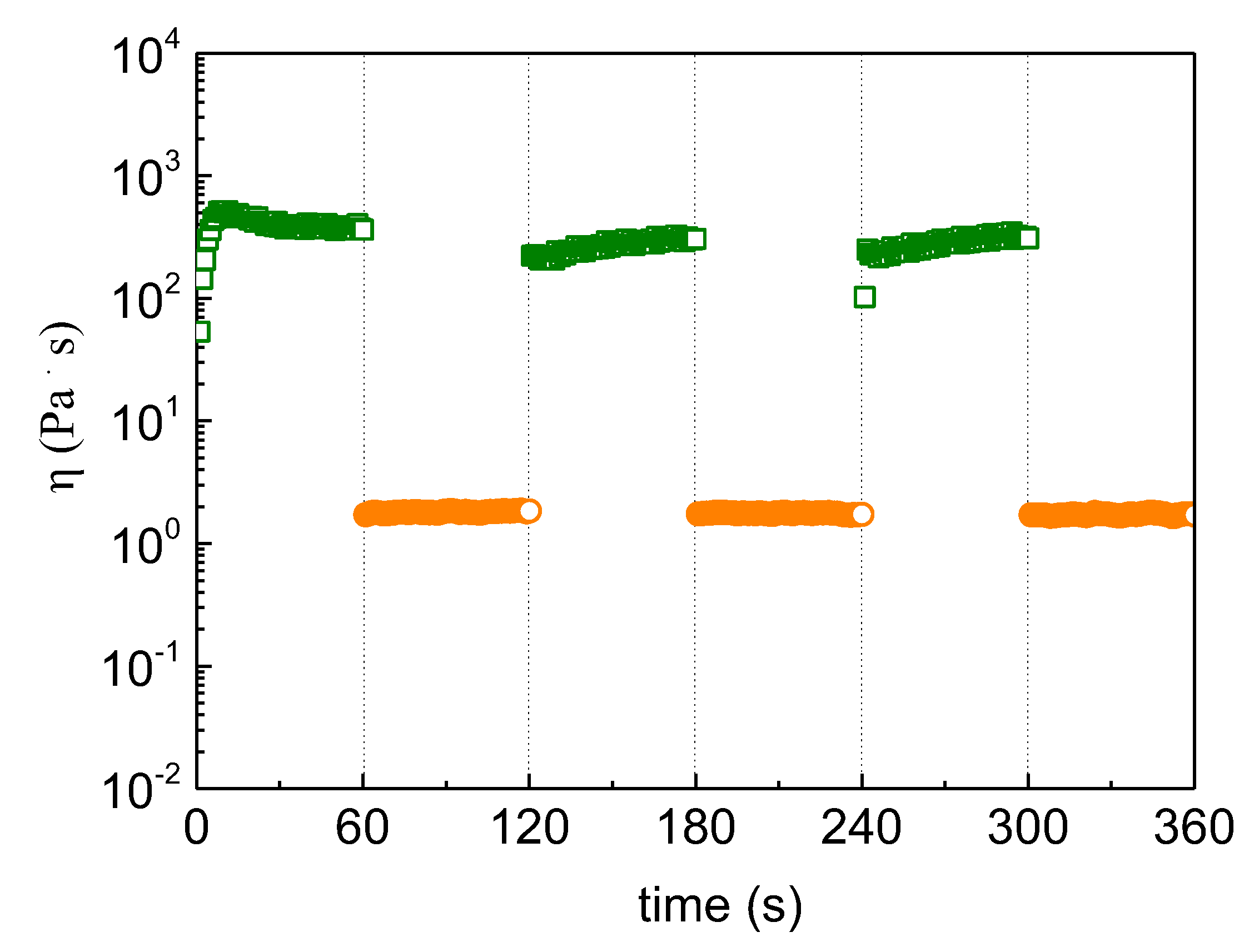
Figure 10.
Release of: Nile Red from P2VP-b-PEO@Nile Red micelles and ALG-g-HG/P2VP-b-PEO@Nile Red nanocomposite hydrogels at pH 3.5 and pH 7.4 buffer solutions and at 37 °C.
Figure 10.
Release of: Nile Red from P2VP-b-PEO@Nile Red micelles and ALG-g-HG/P2VP-b-PEO@Nile Red nanocomposite hydrogels at pH 3.5 and pH 7.4 buffer solutions and at 37 °C.
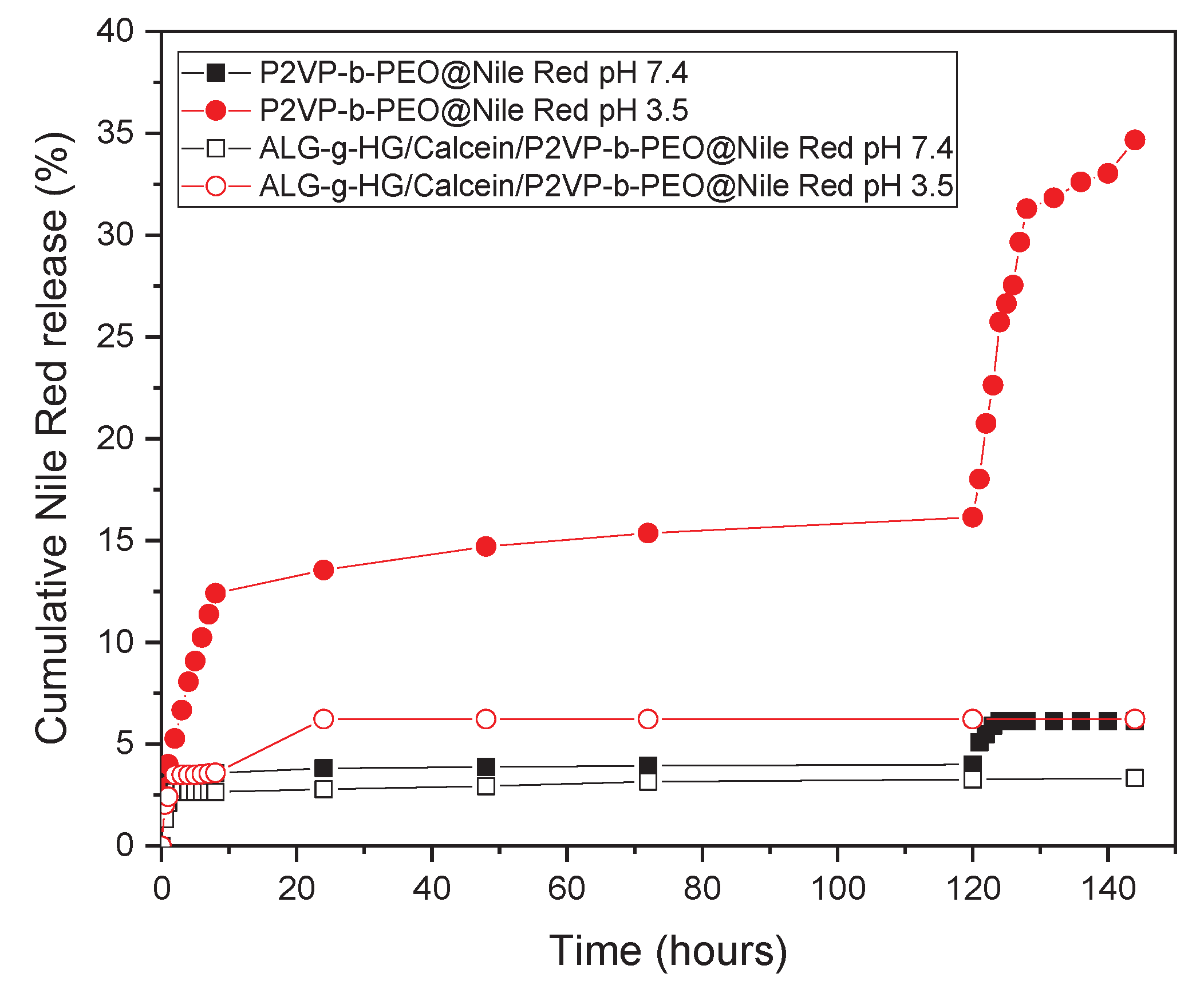

Table 1.
Characteristic factors of thermoresponsiveness at various pH.
| ALG-g-HG/P2VP-b-PEO | |||||
|---|---|---|---|---|---|
| pH | Tc,thermothickening (°C) | Tgel (°C) | Tf (°C) | ΔT=Tf-Tc, thermothickening (°C) | G’(T=50)/G’(Tc, thermothickening) |
| 5.4 | 32.6 | 34.1 | 41.7 | 9.4 | 86.2/0.78=110.5 |
| 4.5 | 22.3 | 26.1 | 40.8 | 18.5 | 92.3/0.95=97.2 |
| 4.0 | 18.1 | 20.2 | 36.6 | 18.5 | 244.6/2.8=87.3 |
| 3.5 | 19.2 | -- | 38.7 | 19.5 | 565.2/160.7=3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



