
Submitted:
02 March 2024
Posted:
04 March 2024
You are already at the latest version
Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disease characterized by persistent deficits in social interaction and communication, repetitive movements, abnormal focusing on objects, or activity that can significantly affect the quality of life of the afflicted. Brain neuronal and glial cells have been implicated. It has a genetic component but can also be triggered by environmental factors or drugs. For example, prenatal exposure to valproic acid (VPA) or acetaminophen (APAP) or ingestion of propionic acid (PPA) can increase the risk of ASD. Recently, epigenetic influences on ASD have been brought to the forefront of investigations on the etiology, prevention, and treatment of this disorder. Epigenetics refers to DNA modifications that alter gene expression without making any changes to the DNA sequence. Although an increasing number of pharmaceuticals and environmental chemicals are being implicated in the etiology of ASD, here we specifically focus on the molecular influence of the above-mentioned drugs on epigenetic alterations in neuronal and glial cells and their potential connection to ASD. It is concluded that a better understanding of these phenomena can lead to more effective interventions in ASD.
Keywords:
Autism Spectrum Disorder
; Epigenetics
; Teratogen
; Valproic Acid
; Propionic Acid
; Acetaminophen
; Gliosis
; Glial Cells
; Neuroinflammation
1. Introduction
Autism spectrum disorder (ASD) is a complex and multifactorial neurodevelopmental disorder that occurs in all ethnic, racial, and socioeconomic groups. According to the latest Autism and Developmental Disabilities Monitoring (ADDM) report, the prevalence of ASD has been increasing over the years, such that currently 1 in 36 eight-year-old children in the US are diagnosed with the condition. Males outnumber females with a ratio of 4:1 [1]. The diagnosis of ASD in the DSM V requires the child to meet a panel of criteria such as persistent deficits in each of three areas of social communication and interaction: social-emotional reciprocity, nonverbal communicative behaviors, and processing of relationship, plus at least two of four types of restricted repetitive patterns of behaviors and interests [1]. In 1970, Bailey and colleagues first suggested a genetic component to ASD based on their twin studies [2]. Since then, a number of genes have been identified (discussed below). However, it is important to note that in this case the genetic predisposition does not necessarily translate into the phenotype without a triggering stimulus [3]. Thus, ASD may be considered a multifactorial disorder with components such as oxidative stress, neurotransmitter imbalance, immune system dysfunction, exposure to a xenobiotics, maternal infections, teratogenic infections, cholesterol metabolism, and exposure to heavy metals, and some drugs affecting the outcome[4,5,6].
Several different classes of pharmaceutical drugs, such as SSRI antidepressants [7], acetaminophen (Tylenol) [8], asthma medications [9], valproic acid [10], and opioids [11] have been implicated in the development of ASD. Additionally, various environmental compounds, such as endocrine-disrupting chemicals including bisphenol A (BPA) [12], phthalates [13], and propionic acid (an endogenous metabolite but also used in the manufacture of herbicides) [14], may also increase the chance of ASD. Propionic acid is frequently used in developing animal models of ASD.
For these reasons, it has been suggested that the multigenic condition of ASD may be dependent on epigenetic effects [15,16]. But what, precisely, is meant by epigenetics?
Epigenetics is a term that refers to DNA modifications that alter gene expression without making any changes to the genetic code. The full extent of epigenetics in modulating gene expression, especially in relation to neurological diseases, is gradually being revealed. Although, as stated above, an increasing number of chemicals are being implicated in ASD, in this review we will focus on the examples of prenatal exposure to valproic acid (VPA) and acetaminophen (APAP), and postnatal exposure to propionic acid (PPA). Both neuronal and glial contributions are considered. Following a brief discussion of the genetics of ASD, epigenetic effects of the above-mentioned drugs on phenotypic expression, as well as potential use of this knowledge in developing novel interventions, is discussed.
2. ASD – Genetics
Based on family studies, ASD is recognized as the most heritable neurodevelopmental disorder. Since the monozygotic twin study by Barley et al. (1970), which found a concordance of autism between 60-90% in monozygotic twins and 5-40% in dizygotic twins, the heritability and contribution of genetics to ASD has been verified [3,17,18,19]. A large population-based study conducted recently on more than 2 million individuals and 680,000 families across 5 countries (Denmark, Finland, Sweden, Israel, and Western Australia), estimated the heritability for ASD to be 80 % [20]. Also, Butler and colleagues compiled approximately 800 genes linked to ASD, arranged them in alphabetical order, and included high-resolution human chromosome ideograms to facilitate visualization of the specific location and arrangement of ASD-associated genes [21].
In reality, ASD consists of a group of heterogeneous genetic neurobehavioral disorders associated with developmental impairments in social communication skills and stereotypic, rigid, or repetitive behaviors. Novel gene-protein interactions with pathway and molecular function analyses have identified at least three functional pathways including chromatin modeling, Wnt, Notch and other signaling pathways and metabolic disturbances involving neuronal growth and dendritic spine profiles. An estimated 50 % of individuals with ASD have chromosome deletions or duplications (e.g., 15q11.2, BP1-BP2, 16p11.2 and 15q13.3), resulting in behavioral and psychiatric conditions [21]. Hence, chromosomal microarrays may be applied with high diagnostic yield [21,22]. In addition, pharmacogenetics testing may be used to guide the selection of medications in ASD, a technique which is also used in Down syndrome, Fragile X syndrome (FMR1 gene) and the PTEN gene mutation, which encodes a phosphatase associated with extreme macrocephaly[23] .
2.1. Epigenetics
Epigenetics, as mentioned above, is a term that refers to DNA modifications that alter gene expression without making any changes to the genetic backbone [24] . It is a process that regulates gene expression, through modifications of DNA bases and changes to DNA packaging in response to environmental factors and behavioral conditions [24]. Epigenetic changes are potentially reversible but factors regulating reversibility are not yet fully understood. Through epigenetics, genes can be fully silenced, under-expressed, or overexpressed [24] . Additionally, epigenetics relates to a causal chain linking genetics, environmental exposure, and disease development. Epigenetic changes occur often during an organism's lifetime and may be transmitted to the next generation [25]. Also, numerous factors may cause epigenetic changes, such as breastfeeding and maternal care, physical activity or inactivity, hyperglycemia, mitochondrial dysfunction, aging, and menopause [25] . We have previously hypothesized that epigenetic state may be influenced by pharmaceutical drugs [26], and have detected DNA methylation changes in a human cell line in response to the widely-used antidepressant, citalopram [27]. Epigenetic state may also be influenced by integrative medicine [28]. Moreover, epigenetics may contribute to the development of metabolic diseases such as diabetes [29,30].
DNA, with its phosphate groups, is negatively charged and packaged around a positively charged histone protein octamer that contains 2 copies of histone proteins H2A, H2B, H3 and H4. DNA loops twice around the histone octamer forming the functional unit of DNA, the nucleoprotein called the nucleosome [31,32,33]. DNA is thus packaged in a “beads on a string” pattern. H1 is the last histone protein that binds to the nucleosome and linker DNA, thereby stabilizing the chromatin fiber. Linker DNA is a double-stranded DNA situated in between two nucleosome cores that, in association with histone H1, holds the cores together. It was recently revealed that histone H1 binding to nucleosome arrays depends on linker DNA length and trajectory [34]. Thus, Linker DNA is considered as the string in the "beads and string model", on the chromatin. Chromatin is the condensed form in which DNA exists to fit in the nucleus. The aggregation of chromatin results in the formation of chromosomes. In its loose shape, chromatin is transcriptionally active and referred to as euchromatin as opposed to the highly condensed, transcriptionally inactive state called heterochromatin [32].
There are currently three well-understood factors regulating epigenetic expression:
- Modifications to histones that either make the chromatin available (euchromatin state) or unavailable (heterochromatin) for transcriptional processes [24,35,36]. In this context, three different mechanisms have been described. First, is histone methylation that usually silences DNA expression. Second is histone acetylation that relaxes DNA coiling, increasing its transcription. Third is the reverse process, histone deacetylation that removes an acetyl group and further tightens DNA coiling, thus decreasing gene expression.
- DNA methylation [37,38], is a reversible mechanism wherein methyl groups (–CH3) are delivered to cytosines positioned in CpG (5′ -cytosine-phosphate-guanosine-3′ ) nucleotides turning these cytosines into 5-methyl cytosines (5mC) [39]. When methylation occurs in cytosine-phosphate-guanine (CpG) islands in the gene promoter, interaction between the DNA and transcription factors is reduced and the gene is silenced [3,40]. In neural cells, either hypermethylation or hypomethylation of DNA can affect learning or memory. Indeed, dysregulated methylation has been linked to neurodevelopmental disorders such as ASD [39].
- Gene silencing may also occur via non-coding RNA (ncRNA), referring to RNA sequences that are transcribed but not translated, hence not leading to protein synthesis [41]. More than 89% of non-ribosomal RNA transcripts are non-coding [41]. After years of being considered as junk RNA, recent studies emphasize the crucial role of ncRNA in modulating the expression of the genome [42].
2.2. Epigenetics – ASD
The importance of epigenetic modulation in ASD is underscored by experimental models designed to study the impact of environmental exposure in a genetically predisposed subject [43]. Epigenetic changes via DNA methylation may impact several physiological and pathological processes such as transcriptional regulation, chromosomal stability, tissue-specific gene regulation and genome imprinting [3,40]. Abnormal DNA methylation patterns have been observed in various neurodegenerative and neurodevelopmental disorders such as Alzheimer’s disease (AD), depression, ADHD, and ASD.
The complexity of epigenetic mechanisms renders identification of the actual process in ASD challenging [18]. For example, Angelman syndrome (AS) is a neurodevelopmental imprinting disorder characterized by severe mental retardation, cognitive disability and lack of speech that has a high comorbidity with autism, thus considered nowadays a syndromic form of ASD [44,45,46]. In 3% to 5% of cases, AS is directly caused by a DNA methylation defect in the maternal SNRPN promoter silencing the UBE3A gene [45]. Thus, AS may be considered an epigenetic disorder. The phenotypic overlap between ASD and AS suggests possible implication of the UBE3A gene in ASD. Indeed, imprinting of this gene was one of the first identified parent-of-origin effects (referring to phenotype inheritance from the mother or father) in ASD. This provided evidence on how epigenetic mechanisms via DNA methylation can be responsible for the phenotypic manifestations in ASD [44,45,47].
2.3. Glial Cells -ASD
Although most studies on ASD have focused on neuronal pathological changes, including increased cell proliferation, accelerated neuronal differentiation, impaired synaptic development, and reduced neuronal spontaneous and synchronous activity, more and more research has found that glial cells may also be of both pathological and therapeutic potential.
Glial cells represent the largest population of cells in the human brain, as they outnumber the neurons 10-fold [48,49].These cells perform pivotal functions such as energetic support for neurons [50], regulation of neurotransmitters [51,52,53], formation of the blood-brain barrier (BBB) [54,55], detoxification [56,57], development and remodeling of synapses[58,59,60] , control of fluid/electrolyte homeostasis [61], neuroendocrine function [62] innate immunity response[63,64], control of metabolism [65,66], and myelination [67,68]. Thus, glial cells play a key role in maintaining homeostasis, disruption of which can lead to neurodevelopmental, neuropsychiatric, and neurodegenerative diseases [68,69,70,71,72,73]. In the central nervous system (CNS), 4 major subsets of glial cells (microglia, astrocytes, oligodendrocytes and synantocytes or NG2 cells) have been identified. A brief description of each with relevance to ASD is discussed below.
Microglia, constituting 10% -15% of all CNS cells, act in the innate immune response, and play a key role in neuroinflammation. They are considered the resident macrophages of the CNS, with a vital role in maintaining homeostasis due to their ability to remove debris, regulate neurogenesis, participate in formation and elimination of neuronal synapses, and control the number of neuronal precursor cells[74,75,76,77] .
As resident immune cells, microglia can polarize into different proinflammatory (M1) or anti-inflammatory (M2) phenotypes. M1 microglia activation leads to BBB dysfunction and vascular “leakage”, whereas M2 microglia act as protectors of the BBB. Under physiological conditions, microglia readily and continuously monitor the CNS microenvironment to quickly remove debris and provide repair at the damaged area. However, their overactivation is the primary cause of neuroinflammation and cellular death [74,76,78].
Recently, it has been proposed that many pathological phenotypes of ASD may be caused by microglial abnormalities. Specifically, single-cell data have shown differences in gene expression patterns between fetal and adult microglia, indicating that microglia play an important role in CNS development. More and more studies have found that damage to microglia plays a key role in ASD pathology. This is because many pathological phenotypes of ASD are believed to be caused by abnormalities in immune cells, specifically the microglia. Mouse models have determined that damage to microglia affects synaptic pruning, leading to deficits in social behavior. Thus, mechanisms related to microglial abnormalities in ASD may become a promising direction for preventing ASD and/or developing therapeutic drugs[78,79].
Astroglia or astrocytes are star-shaped cells that make up between 17 to 61 % of the cells in the human brain, depending on the region, and like microglia exhibit heterogeneous phenotypes in response to various insults, a process known as astrocyte reactivity [80] . Astrocytes perform a myriad of essential functions, including maintenance and accuracy of brain signaling, recycling of neurotransmitters, maintenance of the BBB, modulation of ionic environment, providing metabolic support for the neurons, and regulating sphingolipid and cholesterol metabolism, where disruption of the latter has been directly linked to ASD [80,81,82]
As alluded to earlier, risk factors for ASD include disturbed brain homeostasis, genetic predispositions, and inflammation during the prenatal period caused by viruses or bacteria. Another factor linked to the onset of ASD is the activation of the maternal immune system where gestational inflammation at embryonic day 13 or 15 in mice, induces the most profound symptoms. This is because during this period the neural tube has closed and progenitors are migrating and proliferating. [83] Many studies suggest involvement of glia in the pathology of autism, as evidenced by increased expression of glia-associated proteins in the brain. It is hypothesized that alterations of astroglial function leads to disrupted homeostasis of excitatory/inhibitory balance due to aberrant Ca2+ signaling, and eventual manifestation of ASD [83,84]. In sum, disruption of astrocyte function may affect proper neurotransmitter metabolism, synaptogenesis, and inflammation, leading to ASD. Thus, astroglia may represent presumptive targets for novel therapeutic strategies [83,85].
Oligodendrocytes (ODs), the myelinating cells of the brain, represent about 75% of all glial cells in the adult CNS. In addition to axonal myelination, ODs control extracellular potassium concentration, provide metabolic and trophic supply to myelin, secrete glial and brain-derived neurotrophic factors (GDNF and BDNF), and modulate axonal growth [86,87], all of which highlight their importance in the functioning of the CNS. The importance of ODs in the pathogenesis of neurodevelopmental and neurodegenerative disease have been verified [88]. It is of relevance to note that in the peripheral nervous system, neuroglia that are equivalent to ODs are referred to as Schwann cells [89].
Recent comparative genomic analyses of the causative genes of ASD in animal models have demonstrated that ODs may also contribute to the molecular mechanisms underlying social functioning, disturbance of which can lead to ASD [90]. Specifically, it was demonstrated that OD-lineage cells and myelination are altered in a murine model of ASD induced by the prenatal exposure to VPA [91] . OD importance in neurodegenerative diseases in general and Parkinson’s disease, in particular, has been lately documented [88]. Moreover, ODs also express toll-like receptors (TLRs), considered of significant importance in myelin formation [67,92,93].
NG2 cells are a subset of CNS glial cells, commonly referred to as synantocytes or neuron glial 2, or nerve/glial antigen 2 (NG2). These cells display a variety of features including (i) a complex stellate morphology; (ii) an almost uniform distribution in both gray and white matter areas; (iii) the ability to keep proliferating in the adult brain; (iv) ability to give rise to astrocytes and neurons that may be recruited to areas of lesion and (v) a tendency to intimately associated with neuronal cell bodies and dendrites; [86,94,95].
NG2 cells were considered to function solely as progenitors for oligodendrocyte ODs. However, now they are believed to have many other important functions, dysfunction of which can lead to pathologies such as demyelinating, neurovascular disruption, neuroinflammation, and neurodegeneration [96,97,98,99,100]. They have been proposed as targets in various neurological diseases due to their ability to receive synapses from neurons and affect neuronal plasticity[101].
A link between NG2 cells and ASD is suggested by several studies. ASD and severe macrocephaly are associated with germline mutations in the PTEN gene. This mutation occurs in 7 - 27% of patients with ASD and macrocephaly and may account for up to 5% of all ASD cases as macrocephaly is found in approximately 20 % of the ASD population. Indeed, PTEN mutation screening is recommended in all cases of ASD where the individual has a head circumference greater than 3 standard deviations above the mean for their age and sex [102]. PTEN is best known for dephosphorylating phosphatidyl-inositol (3,4,5)-triphosphate, inhibiting the PI3K/AKT/mTOR signaling pathway. The Ptenm3m4 mouse model exhibited an increased NG2 cell proliferation and accumulation of glia, with behavioral abnormalities like some individuals ASD[103,104,105] .
Now that we have described the relevant cell populations, we will turn our attention to specific chemical exposures that have been implicated in the etiology of ASD.
3. Valproic Acid (VPA)
Valproic Acid (VPA, 2-propyl-pentanoic acid), a broad-spectrum antiepileptic drug (AED), is used to treat many types of seizures. Despite its recognized role as an AED, it is also now used to treat bipolar disorder, and is approved for migraine prophylaxis and neuropathic pain. Its antiepileptic effect is mainly explained by its inhibitory action on the neuron: it increases GABA concentration within the neuron by inhibiting GABA transaminase (GABA-T) and blocking the voltage gated sodium channels. The pharmacodynamic effects of VPA are numerous and complex and range from diminishing the fast and transient inward Na+ currents, thus interfering with the mechanism of sustained and prolonged firing to a direct action on the neuron, reducing synaptic localization of glutamate-receptor subunits [106,107].
There is also evidence suggesting that VPA affects the enzymatic activity of the brain to exert a neuroprotective, anti-inflammatory and antioxidant effect in patients suffering from epilepsy and X-linked adrenoleukodystrophy [108]. Recently, it was shown that VPA exerts an inhibitory effect on histone deacetylation. This, together with its ability to block the voltage-gated Na+ and Ca++ channels, both of which are considered tumor markers, has prompted the suggestion of its use in breast, prostate, and other types of cancers. [109]
3.1. VPA Action in Glial Cells
In vivo and in vitro studies have confirmed VPA modulation of glial cell function. Thus, it has been shown that VPA selectively induces caspase-3 mediated apoptosis in rodent microglial cells. Although VPA does not induce apoptosis in microglia derived from adult human brains, it does decrease the expression of the microglial markers and dramatically reduces microglial phagocytosis. [110] VPA also alters gliogenesis and may result in an abnormal number of glial cells, hence increasing the risk of neurodevelopmental disorders [111]. It has been suggested that teratogenic actions of VPA may be mediated through changes in astrocyte generation [112]. VPA affects neuronal fate and microglial function via enhancing autophagic flux in mice after traumatic brain injury [113]. VPA exposure decreases neurogenic potential of outer radial glia in human brain organoids [114]. Indeed, because long-term VPA-induced microgliosis could be the result of altered microglia-astroglia crosstalk, it has been proposed that targeting this crosstalk could offer a novel intervention in neuroinflammatory mechanism in ASD [115].
3.2. VPA and Pregnancy
Valproic acid is a well-known teratogen. Reports suggesting its teratogenicity go back to as early as the 1980s in children whose mothers took VPA in the first trimester of pregnancy. The first documented effect was an increased rate of spina bifida (neural tube defect), whereas now the congenital malformations caused by VPA encompass all the major congenital anomalies including neural tube and heart defects, skeletal and limb abnormalities, cleft lip and palate, anomalies of the urinary tract and cranio-facial dysmorphism [10,116,117]. In addition to these physical anomalies, VPA may also affect the cognitive function and social behavior of the offspring, leading to ASD-like syndrome [116,117]. Numerous animal studies in rodents and non-human primates also confirm the teratogenic effects of VPA and its link to ASD [118,119]. Indeed, prenatal VPA exposure in rodents has been established as a reliable translational model to study the [118,119] pathophysiology of ASD [120]. In rare cases, in utero fetal exposure of VPA can cause the human fetal valproate spectrum disorder (FVSD), a disease that causes morphological defects with a distinct facial dysmorphism, congenital anomalies and behavioral alterations that affect language development and communication. Therefore, exposure to VPA in the first trimester of pregnancy is directly related to ASD [121].
3.3. VPA and ASD
VPA alters the developmental patterns of the embryo. Numerous studies suggest VPA involvement in the development of ASD in neonates[120,122]. In a rat study it was shown that VPA exposure even during early postnatal period may precipitate ASD-like behavior [123]. A population-based investigation in Denmark indicated that the use of VPA in pregnancy raised the chances of autism in neonates by 4.42% [10]. Bromley et al. (2019), found that 8.9% of neonates exposed to VPA have Asperger’s syndrome or ASD. As VPA is responsible for cognitive and behavioral alterations, numerous VPA rodent models of ASD have been studied. Rodents’ models of ASD developed by prenatal VPA exposure confirm behavioral abnormalities like those observed in ASD. More recently, a VPA model of ASD, using domestic chicks has been proposed [124].
The pathophysiology of VPA-induced ASD is not yet fully known. Nonetheless, numerous hypotheses have been presented. For example, it has been suggested that VPA’s effect on histone deacetylase (HDAC) leads to abnormal signaling pathways, synaptic dysfunction and neurogenesis deficits, all of which can cause ASD. HDAC refers to a class of enzymes that eliminate acetyl groups from histones and allow the histones to enclose the DNA more tightly, causing transient hyperacetylation in the brain, inhibiting transcription, leading to an increase in apoptotic cells and reducing cell proliferation in certain areas of the brain. Thus, it is believed that VPA impairment of histone acetylation of the ALDH1A1 gene (an epigenetic modification) results in downregulation of the RA-RARα pathway, responsible for autism-like synaptic and behavioral deficits [119].
On the other hand, murine autism models have shown that VPA, by disrupting the AMPK/SIRT1/PGC1α signaling pathway, causes a dysfunction in the amygdala’s interneurons accompanied by elevated ROS and caspase 3, neuroinflammation and eventual manifestation of ASD [125]. In another murine model it was shown that VPA upregulates miR134-5p, similar to what is observed in ASD. There has also been the suggestion that VPA disruption of the Ca2+/calmodulin-dependent protein kinase (CaMKII), protein kinase C (PKC), and protein kinase A (PKA) pathways in the hippocampus is responsible for the elevated risk of autism and cognitive impairments [126]. Moreover, low-dose VPA during pregnancy may result in neocortical dysgenesis due to increased neuronal projection in specific cortical layers, resulting in abnormal social behavior, cognitive changes, heightened pain sensitivity, and impaired locomotion, resembling autism symptoms [126].
4. Acetaminophen (APAP)
Acetaminophen, also called N-acetyl para-aminophenol (APAP) or paracetamol, is a widely used non-opioid analgesic and antipyretic drug. APAP acts on multiple biological cascades, including 1) increasing expression of the anti-apoptotic protein Bcl2 and decreasing the pro-apoptotic protein caspase-3; 2) reducing prostaglandin formation via competitive inhibition of the prostaglandin H2 synthase; 3) interfering with cannabinoid receptor signaling through its metabolite N-arachidonoyl-phentolamine; 4) a metabolite, N-acetyl-p-benzo-quinone-imine, depleting glutathione in the CNS [129,130,131].
4.1. APAP Action in Glial Cells
APAP possesses both antioxidant and anti-inflammatory effects, at least part of which is believed to be mediated through its interaction with glial cells in general and microglia, in particular. Thus, in an inflammatory mouse model where microglia are overactivated, APAP administration restored microglial function and reversed the cognitive impairment [132]. In another transgenic mouse model of AD, administration of APAP derivative, N,N'-diacetyl-p-phenylenediamine restored microglial phagocytosis and improved cognitive defects [133]. APAP also attenuated LPS-induced cognitive impairment through antioxidant and anti-inflammatory properties, as well as its ability to inhibit the mitochondrial permeability transition (MPT) pore and apoptotic pathway [134]. These properties are potentially responsible for the effectiveness of APAP in reducing the risk of amyotrophic lateral sclerosis [135]. However, Long-term APAP treatment impairs cognitive function and BDNF in adult rat brain [136].
4.2. APAP and Pregnancy
APAP is the most frequently used FDA-approved over the counter (OTC) analgesic and antipyretic drug. It is used by the general population including pregnant and breastfeeding mothers for its safety profile. In fact, APAP has been widely prescribed during pregnancy for the symptomatic treatment of moderate to severe pain, fever, and headaches because it is considered a relatively safe drug within the recommended dosage range in different countries. However, it was classified as a class B drug in Pregnancy drugs categories, due to concerns about its safety profile during pregnancy, which prompted the FDA to relabel APAP packaging. While its hepatotoxic effects are well established, its impact on fetal brain development during the prenatal and postnatal periods are still to be fully elucidated.
In recent years, numerous studies have questioned the safety of this drug especially during the prenatal and early postnatal period. It has been reported that APAP might be harmful in the embryological phase as it disrupts hormonal balance and interferes with sex and thyroid hormone metabolism. These hormones are essential for neurogenesis, neural cell differentiation and migration, synaptogenesis, and myelination process. Indeed, it is a well-established fact that thyroid hormone deficiency severely affects brain maturation, causing mental retardation, intellectual deficits, and neurobehavioral impairments [137]. In sum, potential neurotoxic effects of APAP on the developing brain in the prenatal and postnatal period is a concern.
4.3. APAP and ASD
Various studies have addressed the biological effects of APAP on the developing brain. In-vivo animal studies suggest a causal relation between prenatal APAP exposure and altered behaviors in the offspring such as stereotyped behavior and hyperactivity [138,139]. These effects have been attributed to APAP’s reduction of BDNF levels in the striatum of rats [140], as well as alterations of dopamine metabolism [140,141]. Moreover, APAP may also cause oxidative stress and mitochondrial dysfunction, which can contribute to behavioral abnormalities [131].
An increasing number of human observational and animal studies [8,142,143,144,145] suggest that prenatal exposure to APAP is associated with an increased risk of ASD in offspring. This association is four times higher among boys [142,143]. Several animal studies also confirm the propensity to develop ASD-like symptoms, especially in male offspring following prenatal APAP exposure [146]. Prenatal APAP exposure has been associated with impairments in motor milestones, gross motor function, and autistic like symptoms of communication, externalizing behavior, internalizing behavior, sociability and emotionality [131,142,143,144].
Timing and duration of APAP exposure are two important factors to consider when studying its relation to neurodevelopmental impairments. In 2018, a systematic review by Bauer et al. focused on the adverse neurodevelopmental outcomes following APAP exposure. It was determined that the exposure during the second trimester of gestation as well as exposure for more than 28 days is responsible for a significantly higher risk of ASD, ADHD and intellectual retardation (lower IQ) [142,144]. This study suggested that the longer the exposure the higher the risk. A more recent study has verified that early postnatal exposure to APAP may also increase the risk of ASD in males [147]. Dosing is also critical in this context as at higher dosage, not only the neurons, but also glial cells would be exposed to higher oxidative stress [131].
It may thus be concluded that prenatal exposure to acetaminophen increases the risk of ASD particularly in genetically susceptible offspring.
5. Propionic Acid
Propionic acid (PPA) also known as propanoic acid, is a ubiquitous short chain fatty acid (SCFA) present in many processed foods as well as animal feedstocks. Biological processes regularly produce PPA or its conjugate base, propionate, through propionic coenzyme A as well as from cholesterol oxidation [148]. PPA is also a metabolic product of enteric bacteria that can cross into the CNS and accumulate in the cells and lead to intracellular acidification [149]. PPA exerts a myriad of effects, including disturbance of several neurotransmitters’ functions. It is now well-recognized that an imbalance between the excitatory and inhibitory neurotransmitters, namely glutamate and GABA, plays a crucial role in ASD pathology. This is further supported by the high percentage of comorbidity between epilepsy and ASD [150]. PPA also affects cell proliferation and differentiation and is considered a major neuroinflammatory mediator as it induces gliosis and neuro-inflammation through modulation of PTEN/AKT, a pathway intimately involved in ASD [151]. These and other neurobiological effects of PPA, including mitochondrial damage, have established it as a suitable inducer of experimental models for ASD [14,150,152,153,154].
5.1. PPA–Glial Cells–ASD
At the nuclear level, PPA stimulates the tumor necrosis factor alpha (TNF-α), a pro-inflammatory cytokine, gene expression and transcription, causing uncontrolled inflammation and gliosis, both of which have been implicated in ASD development [150]. Moreover, behavioral and associated morphological changes induced by PPA in the hippocampus and amygdala, brain regions that are associated with the regulation of social behavior and cognition, validate the PPA rodent model of ASD [150,154]. Specifically, morphological analyses of PPA-treated rat brains (hippocampus and amygdala) revealed reduced neuron number and an increase in the number of glial cells, particularly those of astrocytes and microglia. Moreover, pericapillary glia were most affected and axons were moderately demyelinated following PPA treatment [154].
5.2. Epigenetic Mechanisms in ASD
As discussed above, the role of epigenetics in ASD had been suggested long ago [44]. However, the exact epigenetic mechanisms that are involved in ASD pathophysiology are still to be fully elucidated [46]. Current understanding is that the epigenetic mechanisms implicated in neurological or neurodevelopmental disorders primarily involve DNA methylation and/or histone deacetylation [155] (Figure 1).
Regarding DNA methylation, several signatures and patterns are found in specific genomic regions impacting gene expression related to synaptic function, neuronal signaling and brain development and maturation. Hence, methylation of newly discovered genes like the oxytocin receptor (OXTR), methyl-CpG-binding protein 2 (MECP2), Reelin (RELN) and BDNF have been linked to ASD pathophysiology [156,157,158,159].
Specifically, MECP2 loss of function in neural cell lineages is the main cause of Rett syndrome, a neurodevelopmental disorder, mainly diagnosed in girls, that manifests with neurobehavioral, social and communication symptoms similar to those found in patients suffering from ASD. The clinical evolution of Rett syndrome (i.e., severe neurological and neurobehavioral regression, often one year after reaching the corresponding motor and verbal milestones) has suggested a probable inculpation of environmental factors and epigenetics in the development of this neurological disorder. The clinical correlation between these two neurodevelopmental entities has led researchers to further investigate the MECP2 gene as a genomic marker of ASD. It is well-known that abnormal MECP2 expression leads to ASD in humans and ASD-like symptoms in animal models. Epigenetic manipulation of this gene can in fact cause abnormal behaviors like those seen in ASD. A cohort study of patients with ASD revealed significantly increased levels of MECP2 methylation compared to controls in specific CpG islands [160]. Moreover, locus-specific methylation at the MECP2 promoter was positively correlated with the development of ASD-like symptoms such as increased repetitive behaviors, anxiety, and impaired social interactions [159] . Interestingly, targeting of the MECP2 gene in the hippocampus was sufficient to provoke neurobehavioral ASD-like symptoms, suggesting potential therapeutic benefit in targeting this gene product [161].
Additionally, recent findings have implicated oxytocin and oxytocin receptors (OXTR) in ASD pathophysiology. Oxytocin is a neuropeptide that is extremely important during birth, mother-child bonding, and emotional functioning, as well as the development of social behaviors [156,162]. OXTR methylation can influence oxytocin’s function in social interactions [163]. Similarly, genetic alterations of RELN may also contribute to ASD phenotype [157,158,164].
The histone deacetylation (HDAC) process is another major epigenetic mechanism that has been linked to ASD. This HDAC effect is manifested via modulation of genomic regions implicated in synaptic function, brain development and maturation. For instance, FMR1 gene has been associated with fragile X syndrome, a genetic condition that presents with ASD-like symptoms. Decrease in FMR1 transcription is associated with loss of the FMR1 protein that is necessary for brain maturation. HDAC, in this case decreases the amount of chromatin available for transcription at the FMR1 promoter and may contribute to ASD manifestation [165].
Therefore, HDAC inhibitors may offer promising therapeutic interventions in transcriptional activators of the FMR1 gene in such conditions as Fragile X syndrome and ASD [166]. Indeed, as discussed below, the utility of HDAC inhibitors as novel therapeutic intervention in ASD has been proposed.
5.3. Epigenetics-Glial Cells-ASD
It is important to note that epigenetic modulations may also extend to glial cells. That neuroinflammation may contribute to lifelong neurological disabilities including ASD has been amply verified [167]. Astrocytes and microglia, as mentioned earlier, play a pivotal role in neurodevelopment and predisposition to neurological diseases. It is now believed that a pathological glial-neuronal interplay increases the risk of ASD. Thus, it is hypothesized that fetal neuroinflammation, as well as parental stress, via gene-environment interactions (i.e., epigenetic mechanism) reprogram glial phenotypes, hence impacting neurodevelopment [167]. Similarly, neuroinflammation induced by activation of monocytes, macrophages, mast cells and microglia, which can lead to ASD's onset and development, may be modified epigenetically [168]. It was recently suggested that epigenetic modifications may be applied to reprogram Müller glia for retinal regeneration [169]. Interestingly, epigenetic influences may be responsible for accelerated biological aging of glial cells of patients with multiple sclerosis [170]. Finally, the potential impacts of the gut microbiome in the dysfunction of enteric and brain glia, as well as astrocytes, which, in turn, may affect neuronal functions in neurodevelopmental disorders including ASD should be considered [171] (Figure 2).
Hence, VPA, APAP, and PPA by virtue of their interaction with glial cells, may also epigenetically affect the function of these cells. For example, VPA may stimulate the proliferation of glial precursors during cortical gliogenesis [112], and prenatal VPA may activate spinal microglia leading to allodynia, where pain is felt by a stimulus that does not normally provoke pain [172], or prenatal APAP’s detrimental effect on attention-related behavior, may be at least partly mediated via glial cells [173,174] . PPA interaction with glial cells and epigenetic modification of these cells has also been documented [151]. However, in these cases the exact epigenetic mechanism is yet to be elucidated.
5.4. VPA–Epigenetics–ASD
VPA, by inducing HDAC, interacts with histones, changes the DNA stereo arrangement by reducing its molecular order and affecting histone H1 and H3 conformations. It alters the expression of transcription factors, thus leading to modulation of gene expression. All these epigenetic changes are associated with chromatin remodeling [175] (Fig.1).
5.5. APAP–Epigenetics–ASD
In contrast to VPA, the epigenetic effect of APAP is mediated via DNA methylation. This was demonstrated in 2017, where a prospective cohort study provided a potential causal link between APAP, neurodevelopmental disorders, and DNA methylation. APAP primarily affected the genes involved in oxidative stress, neural transmission, synaptogenesis, and olfactory sensory pathways [176]. It is noteworthy that altered olfactory response is evident in children with ASD and is more pronounced with increased autism severity. Indeed, it has been suggested that olfaction may provide a novel early non-verbal non-task-dependent ASD marker as well as serve in ASD intervention [177]. Moreover, recent investigations indicate that impairment in odor identification in ASD appears to be associated with mitochondrial dysfunction [178], another anomaly strongly linked to ASD [179] (Fig. 1).
5.6. PPA-Epigenetics-ASD
As alluded to earlier, PPA disruption of biogenesis, bioenergetic, and metabolism in neurons and glial cells has made it a suitable compound in modeling ASD. It is now well-recognized that epigenetic effects of PPA, like APAP, are mediated via DNA methylation [180,181]. Specifically, it was shown that PPA attenuates IL-6 cytokine production through downregulating expression of the epigenetic modifier DNA methyltransferase 1, and inhibition of DNA methylation in both mice and humans. [180] Moreover, PPA modification of mitochondrial morphology and dynamic may at least partially be mediated via DNA methylation [181,182] (Fig. 1).
6. Implications for Novel Interventions in ASD
Thus, it is evident that different epigenetic mechanisms (e.g., deacetylation vs methylation) may be mediating the action of the drugs implicated in ASD. This knowledge may lead to insights into targeted therapy via specific epigenetic manipulation to prevent or perhaps even reverse some of the detrimental consequences of ASD. Clearly, more work in this area is necessary. Nonetheless, hints of such interventions are already evident. For example, maternal treatment with butyrate, an HDAC inhibitor, was shown to ameliorate ASD-like traits, in a mouse model of ASD [182,183]. Moreover, the behavioral effects were accompanied by neurochemical observations where cerebellar cortex hypertrophy, as well as Purkinje cell firing, and long-term synaptic plasticity deficits were prevented by butyrate [183]. A recent study suggests that butyrate supplementation could be a possible therapeutic intervention for lead-induced neurotoxicity [184]. Curiously, a strong association between lead and ASD has also been indicated [185] . Thus, lead-induced ASD-like symptoms may also be targeted by butyrate. Another HDAC inhibitor, vorinostat, ameliorated impairments in sociability and cognitive memory in an Ash1L-deletion-induced ASD/ID mouse model [186]. In addition to butyrate and/other HDAC inhibitors, more and more studies are pointing to the potential utility of probiotics, or in some instances, psychobiotics, a combination of bee pollen and probiotics in ASD [187]. Moreover, dietary therapies composed of prebiotics (artichoke & Luteolin) and probiotics (yogurt & Lacticaseibacillus rhamnosus GG) provided improvements in oxidative stress and neuroinflammation, biochemical characteristics of ASD, in a rat model of ASD [188]. Hence, combination therapies aimed at correcting epigenetic influences, as well as use of probiotics may offer novel interventions in ASD.
Reversing or inhibiting DNA methylation, which may have as much of a contribution to ASD as HDAC, could provide further potential venues to prevent or treat ASD. Although currently, pharmacological tools for such interventions are not available, potential manipulation of this epigenetic mechanism via diet has been proposed [189]. Indeed, therapeutic potential of DNA hypomethylation in ameliorating erosive inflammatory arthritis has recently been indicated [190]. Thus, as our knowledge of epigenetic mechanism and its potential manipulation progresses, therapeutic interventions in neurodegenerative-neuropsychiatric and neurodevelopmental disorders including ASD becomes tangible.
7. Conclusion
ASD is a serious neurodevelopmental disorder involving both neuronal and glial cells in the CNS. Maternal usage of certain drugs (e.g., VPA and APAP) during pregnancy and early postnatal period may increase the risk of ASD in the offspring. Postnatal exposure to some chemicals such as propionic acid may have similar effects. ASD is more prevalent in males and may be caused, at least partially, by two epigenetic mechanisms consisting of HDAC or DNA methylation. Interestingly, different drugs may affect epigenetics via different mechanisms. Hence, VPA affects HDAC, whereas APAP and PPA affect DNA methylation. Hints of potential intervention in ASD via manipulation of epigenetics, particularly via HDAC inhibition, are already surfacing. It is anticipated that with further progress in our understanding of this phenomenon, more selective ways to prevent or treat ASD will become available.
Acknowledgements
ABC was supported in part by NIA/NIH R25 AG047843-01, YT was supported in part by NIH/NIAAA R03 AA022479 and NIH/NIGMS (2 SO6 GM08016-39), MA was supported in part by grants from the National Institute of Environmental Health Sciences (NIEHS) R01ES10563 and R01ES07331.
References
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a Strongly Genetic Disorder: Evidence from a British Twin Study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.Y.; Kabiraj, G.; Ahmed, M.A.; Adam, M.; Mannuru, S.P.; Ramesh, V.; Shahzad, A.; Chaduvula, P.; Khan, S. A Systematic Review of the Link Between Autism Spectrum Disorder and Acetaminophen: A Mystery to Resolve. Cureus 2022, 14, e26995. [Google Scholar] [CrossRef] [PubMed]
- Benachenhou, S.; Laroui, A.; Dionne, O.; Rojas, D.; Toupin, A.; Çaku, A. Cholesterol Alterations in Fragile X Syndrome, Autism Spectrum Disorders and Other Neurodevelopmental Disorders. In International Review of Neurobiology; Elsevier, 2023; Vol. 173, pp. 115–139. ISBN 978-0-443-13729-7. [Google Scholar] [CrossRef]
- Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Interaction of Heavy Metal Lead with Gut Microbiota: Implications for Autism Spectrum Disorder. Biomolecules 2023, 13, 1549. [Google Scholar] [CrossRef] [PubMed]
- Andalib, S.; Emamhadi, M.R.; Yousefzadeh-Chabok, S.; Shakouri, S.K.; Høilund-Carlsen, P.F.; Vafaee, M.S.; Michel, T.M. Maternal SSRI Exposure Increases the Risk of Autistic Offspring: A Meta-Analysis and Systematic Review. Eur. Psychiatry 2017, 45, 161–166. [Google Scholar] [CrossRef]
- Ji, Y.; Azuine, R.E.; Zhang, Y.; Hou, W.; Hong, X.; Wang, G.; Riley, A.; Pearson, C.; Zuckerman, B.; Wang, X. Association of Cord Plasma Biomarkers of In Utero Acetaminophen Exposure With Risk of Attention-Deficit/Hyperactivity Disorder and Autism Spectrum Disorder in Childhood. JAMA Psychiatry 2020, 77, 180–189. [Google Scholar] [CrossRef]
- Gidaya, N.B.; Lee, B.K.; Burstyn, I.; Michael, Y.; Newschaffer, C.J.; Mortensen, E.L. In Utero Exposure to β-2-Adrenergic Receptor Agonist Drugs and Risk for Autism Spectrum Disorders. Pediatrics 2016, 137, e20151316. [Google Scholar] [CrossRef]
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal Valproate Exposure and Risk of Autism Spectrum Disorders and Childhood Autism. JAMA 2013, 309, 1696. [Google Scholar] [CrossRef]
- Rubenstein, E.; Young, J.C.; Croen, L.A.; DiGuiseppi, C.; Dowling, N.F.; Lee, L.-C.; Schieve, L.; Wiggins, L.D.; Daniels, J. Brief Report: Maternal Opioid Prescription from Preconception Through Pregnancy and the Odds of Autism Spectrum Disorder and Autism Features in Children. J. Autism Dev. Disord. 2019, 49, 376–382. [Google Scholar] [CrossRef]
- Hansen, J.B.; Bilenberg, N.; Timmermann, C.A.G.; Jensen, R.C.; Frederiksen, H.; Andersson, A.-M.; Kyhl, H.B.; Jensen, T.K. Prenatal Exposure to Bisphenol A and Autistic- and ADHD-Related Symptoms in Children Aged 2 And5 Years from the Odense Child Cohort. Environ. Health 2021, 20, 24. [Google Scholar] [CrossRef]
- Kim, J.I.; Lee, J.; Lee, K.-S.; Lee, Y.A.; Shin, C.H.; Hong, Y.-C.; Kim, B.-N.; Lim, Y.-H. Association of Phthalate Exposure with Autistic Traits in Children. Environ. Int. 2021, 157, 106775. [Google Scholar] [CrossRef]
- El-Ansary, A.K.; Bacha, A.B.; Kotb, M. Etiology of Autistic Features: The Persisting Neurotoxic Effects of Propionic Acid. J. Neuroinflammation 2012, 9, 661. [Google Scholar] [CrossRef]
- Williams, L.A.; LaSalle, J.M. Future Prospects for Epigenetics in Autism Spectrum Disorder. Mol. Diagn. Ther. 2022, 26, 569–579. [Google Scholar] [CrossRef]
- LaSalle, J.M. Epigenomic Signatures Reveal Mechanistic Clues and Predictive Markers for Autism Spectrum Disorder. Mol. Psychiatry 2023, 28, 1890–1901. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182. [Google Scholar] [CrossRef] [PubMed]
- Taleb, A.; Lin, W.; Xu, X.; Zhang, G.; Zhou, Q.-G.; Naveed, M.; Meng, F.; Fukunaga, K.; Han, F. Emerging Mechanisms of Valproic Acid-Induced Neurotoxic Events in Autism and Its Implications for Pharmacological Treatment. Biomed. Pharmacother. 2021, 137, 111322. [Google Scholar] [CrossRef] [PubMed]
- Ghirardi, L.; Kuja-Halkola, R.; Butwicka, A.; Martin, J.; Larsson, H.; D’Onofrio, B.M.; Lichtenstein, P.; Taylor, M.J. Familial and Genetic Associations between Autism Spectrum Disorder and Other Neurodevelopmental and Psychiatric Disorders. J. Child Psychol. Psychiatry 2021, 62, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Genovese, A.; Butler, M.G. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes 2023, 14, 677. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.S.; Wassman, E.R.; Baxter, A.L.; Hensel, C.H.; Martin, M.M.; Prasad, A.; Twede, H.; Vanzo, R.J.; Butler, M.G. Chromosomal Microarray Analysis of Consecutive Individuals with Autism Spectrum Disorders Using an Ultra-High Resolution Chromosomal Microarray Optimized for Neurodevelopmental Disorders. Int. J. Mol. Sci. 2016, 17, 2070. [Google Scholar] [CrossRef]
- Dhaliwal, N.; Choi, W.W.Y.; Muffat, J.; Li, Y. Modeling PTEN Overexpression-Induced Microcephaly in Human Brain Organoids. Mol. Brain 2021, 14, 131. [Google Scholar] [CrossRef]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the Human Lifespan. Front. Cell Dev. Biol. 2014, 2. [Google Scholar] [CrossRef]
- Csoka, A.B.; Szyf, M. Epigenetic Side-Effects of Common Pharmaceuticals: A Potential New Field in Medicine and Pharmacology. Med. Hypotheses 2009, 73, 770–780. [Google Scholar] [CrossRef]
- Kanherkar, R.R.; Getachew, B.; Ben-Sheetrit, J.; Varma, S.; Heinbockel, T.; Tizabi, Y.; Csoka, A.B. The Effect of Citalopram on Genome-Wide DNA Methylation of Human Cells. Int. J. Genomics 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Kanherkar, R.R.; Stair, S.E.; Bhatia-Dey, N.; Mills, P.J.; Chopra, D.; Csoka, A.B. Epigenetic Mechanisms of Integrative Medicine. Evid. Based Complement. Alternat. Med. 2017, 2017, 1–19. [Google Scholar] [CrossRef] [PubMed]
- A. Alhazzaa, R.; Heinbockel, T.; B. Csoka, A. Diabetes and Epigenetics. In Biochemistry; Anwar, M., Farooq, Z., Ahmad Rather, R., Tauseef, M., Heinbockel, T., Eds.; IntechOpen, 2022; Vol. 35, ISBN 978-1-83880-993-5. [Google Scholar]
- Alhazzaa, R.A.; McKinley, R.E.; Getachew, B.; Tizabi, Y.; Heinbockel, T.; Csoka, A.B. Epigenetic Changes Induced by High Glucose in Human Pancreatic Beta Cells. J. Diabetes Res. 2023, 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Minchin, S.; Lodge, J. Understanding Biochemistry: Structure and Function of Nucleic Acids. Essays Biochem. 2019, 63, 433–456. [Google Scholar] [CrossRef] [PubMed]
- Paro, R.; Grossniklaus, U.; Santoro, R.; Wutz, A. Biology of Chromatin. In Introduction to Epigenetics; Learning Materials in Biosciences; Springer International Publishing: Cham, 2021; pp. 1–28. ISBN 978-3-030-68669-7. [Google Scholar]
- Ghannam, J.Y.; Wang, J.; Jan, A. Biochemistry, DNA Structure. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Dombrowski, M.; Engeholm, M.; Dienemann, C.; Dodonova, S.; Cramer, P. Histone H1 Binding to Nucleosome Arrays Depends on Linker DNA Length and Trajectory. Nat. Struct. Mol. Biol. 2022, 29, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Grant, P.A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell. Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA Methylation in Human Diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Liu, R.; Wu, J.; Guo, H.; Yao, W.; Li, S.; Lu, Y.; Jia, Y.; Liang, X.; Tang, J.; Zhang, H. Post-Translational Modifications of Histones: Mechanisms, Biological Functions, and Therapeutic Targets. MedComm 2023, 4, e292. [Google Scholar] [CrossRef]
- Rasmi, Y.; Shokati, A.; Hassan, A.; Aziz, S.G.-G.; Bastani, S.; Jalali, L.; Moradi, F.; Alipour, S. The Role of DNA Methylation in Progression of Neurological Disorders and Neurodegenerative Diseases as Well as the Prospect of Using DNA Methylation Inhibitors as Therapeutic Agents for Such Disorders. IBRO Neurosci. Rep. 2023, 14, 28–37. [Google Scholar] [CrossRef]
- Martínez-Iglesias, O.; Carrera, I.; Carril, J.C.; Fernández-Novoa, L.; Cacabelos, N.; Cacabelos, R. DNA Methylation in Neurodegenerative and Cerebrovascular Disorders. Int. J. Mol. Sci. 2020, 21, 2220. [Google Scholar] [CrossRef]
- Kimura, R.; Nakata, M.; Funabiki, Y.; Suzuki, S.; Awaya, T.; Murai, T.; Hagiwara, M. An Epigenetic Biomarker for Adult High-Functioning Autism Spectrum Disorder. Sci. Rep. 2019, 9, 13662. [Google Scholar] [CrossRef] [PubMed]
- Boivin, V.; Deschamps-Francoeur, G.; Couture, S.; Nottingham, R.M.; Bouchard-Bourelle, P.; Lambowitz, A.M.; Scott, M.S.; Abou-Elela, S. Simultaneous Sequencing of Coding and Noncoding RNA Reveals a Human Transcriptome Dominated by a Small Number of Highly Expressed Noncoding Genes. RNA N. Y. N 2018, 24, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, Y.-X.; Gu, L.-J.; Cheng, Y. Understanding Autism Spectrum Disorders with Animal Models: Applications, Insights, and Perspectives. Zool. Res. 2021, 42, 800–824. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.U.; Beaudet, A.L.; Madduri, N.; Bacino, C.A. Autism in Angelman Syndrome: Implications for Autism Research. Clin. Genet. 2004, 66, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Beygo, J.; Grosser, C.; Kaya, S.; Mertel, C.; Buiting, K.; Horsthemke, B. Common Genetic Variation in the Angelman Syndrome Imprinting Centre Affects the Imprinting of Chromosome 15. Eur. J. Hum. Genet. EJHG 2020, 28, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Madaan, M.; Mendez, M.D. Angelman Syndrome. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Ryan, N.M.; Heron, E.A. Evidence for Parent-of-Origin Effects in Autism Spectrum Disorder: A Narrative Review. J. Appl. Genet. 2023, 64, 303–317. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef]
- Shi, J.; Huang, S. Comparative Insight into Microglia/Macrophages-Associated Pathways in Glioblastoma and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 25, 16. [Google Scholar] [CrossRef]
- Kim, J.D.; Copperi, F.; Diano, S. Microglia in Central Control of Metabolism. Physiology 2024, 39, 5–17. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Novikov, N.I.; Brazhnik, E.S.; Kitchigina, V.F. Pathological Correlates of Cognitive Decline in Parkinson’s Disease: From Molecules to Neural Networks. Biochem. Mosc. 2023, 88, 1890–1904. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. [Google Scholar] [CrossRef]
- Fernandes, V.M.; Auld, V.; Klämbt, C. Glia as Functional Barriers and Signaling Intermediaries. Cold Spring Harb. Perspect. Biol. 2024, 16, a041423. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.-M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.-H.; Chang, C.-L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Fiacco, T.A.; McCarthy, K.D. Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions. J. Neurosci. 2018, 38, 3–13. [Google Scholar] [CrossRef]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The Tripartite Glutamatergic Synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef]
- Rasia-Filho, A.A.; Calcagnotto, M.E.; Von Bohlen Und Halbach, O. Glial Cell Modulation of Dendritic Spine Structure and Synaptic Function. In Dendritic Spines; Rasia-Filho, A.A., Calcagnotto, M.E., Von Bohlen Und Halbach, O., Eds.; Advances in Neurobiology; Springer International Publishing: Cham, 2023; Vol. 34, pp. 255–310. ISBN 978-3-031-36158-6. [Google Scholar]
- Channels and Transporters in Astrocyte Volume Regulation in Health and Disease. Cell. Physiol. Biochem. 2022, 56, 12–30. [CrossRef] [PubMed]
- Clayton, R.W.; Lovell-Badge, R.; Galichet, C. The Properties and Functions of Glial Cell Types of the Hypothalamic Median Eminence. Front. Endocrinol. 2022, 13, 953995. [Google Scholar] [CrossRef]
- Chen, X.; Holtzman, D.M. Emerging Roles of Innate and Adaptive Immunity in Alzheimer’s Disease. Immunity 2022, 55, 2236–2254. [Google Scholar] [CrossRef] [PubMed]
- Kofler, J.; Wiley, C.A. Microglia: Key Innate Immune Cells of the Brain. Toxicol. Pathol. 2011, 39, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Ebling, F.J.P.; Lewis, J.E. Tanycytes and Hypothalamic Control of Energy Metabolism. Glia 2018, 66, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.A.; Huang, N.; Xie, Y.; LiCausi, F.; Li, S.; Li, Y.; Sheng, Z.-H. Oligodendrocytes Enhance Axonal Energy Metabolism by Deacetylation of Mitochondrial Proteins through Transcellular Delivery of SIRT2. Neuron 2021, 109, 3456–3472.e8. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like Receptors 2 and 4 Differentially Regulate the Self-Renewal and Differentiation of Spinal Cord Neural Precursor Cells. Stem Cell Res. Ther. 2022, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Wies Mancini, V.S.B.; Mattera, V.S.; Pasquini, J.M.; Pasquini, L.A.; Correale, J.D. Microglia-derived Extracellular Vesicles in Homeostasis and Demyelination/Remyelination Processes. J. Neurochem. 2024, 168, 3–25. [Google Scholar] [CrossRef]
- Rahman, S.; Alzarea, S. Glial Mechanisms Underlying Major Depressive Disorder: Potential Therapeutic Opportunities. In Progress in Molecular Biology and Translational Science; Elsevier, 2019; Vol. 167, pp. 159–178. ISBN 978-0-12-818855-2. [Google Scholar]
- Scuderi, C.; Verkhratsky, A.; Parpura, V.; Li, B. Neuroglia in Psychiatric Disorders. In Astrocytes in Psychiatric Disorders; Li, B., Parpura, V., Verkhratsky, A., Scuderi, C., Eds.; Advances in Neurobiology; Springer International Publishing: Cham, 2021; Vol. 26, pp. 3–19. ISBN 978-3-030-77374-8. [Google Scholar]
- Hanslik, K.L.; Marino, K.M.; Ulland, T.K. Modulation of Glial Function in Health, Aging, and Neurodegenerative Disease. Front. Cell. Neurosci. 2021, 15, 718324. [Google Scholar] [CrossRef]
- Zhu, H.; Guan, A.; Liu, J.; Peng, L.; Zhang, Z.; Wang, S. Noteworthy Perspectives on Microglia in Neuropsychiatric Disorders. J. Neuroinflammation 2023, 20, 223. [Google Scholar] [CrossRef] [PubMed]
- Shared and Disease-Specific Glial Gene Expression Changes in Neurodegenerative Diseases. Nat. Aging 2023, 3, 246–247. [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, Z.; Lu, G.; Chan, W.; Zhang, Y.; Wong, G.C. Microglia Activation, Classification and Microglia-Mediated Neuroinflammatory Modulators in Subarachnoid Hemorrhage. Neural Regen. Res. 2022, 17, 1404. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Martins, S.; Ferreira, P.A.; Cardoso, A.M.S.; Guedes, J.R.; Peça, J.; Cardoso, A.L. The Old Guard: Age-Related Changes in Microglia and Their Consequences. Mech. Ageing Dev. 2021, 197, 111512. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, Z. The Impact of Microglia on Neurodevelopment and Brain Function in Autism. Biomedicines 2024, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-X.; Kim, G.H.; Tan, J.-W.; Riso, A.E.; Sun, Y.; Xu, E.Y.; Liao, G.-Y.; Xu, H.; Lee, S.-H.; Do, N.-Y.; et al. Elevated Protein Synthesis in Microglia Causes Autism-like Synaptic and Behavioral Aberrations. Nat. Commun. 2020, 11, 1797. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-Astrocyte Omnidirectional Signaling in Neurological Health and Disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef]
- Peltier, M.R.; Behbodikhah, J.; Renna, H.A.; Ahmed, S.; Srivastava, A.; Arita, Y.; Kasselman, L.J.; Pinkhasov, A.; Wisniewski, T.; De Leon, J.; et al. Cholesterol Deficiency as a Mechanism for Autism: A Valproic Acid Model. J. Investig. Med. 2024, 72, 80–87. [Google Scholar] [CrossRef]
- Gzielo, K.; Nikiforuk, A. Astroglia in Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 11544. [Google Scholar] [CrossRef]
- Allen, M.; Huang, B.S.; Notaras, M.J.; Lodhi, A.; Barrio-Alonso, E.; Lituma, P.J.; Wolujewicz, P.; Witztum, J.; Longo, F.; Chen, M.; et al. Astrocytes Derived from ASD Individuals Alter Behavior and Destabilize Neuronal Activity through Aberrant Ca2+ Signaling. Mol. Psychiatry 2022, 27, 2470–2484. [Google Scholar] [CrossRef] [PubMed]
- Vakilzadeh, G.; Martinez-Cerdeño, V. Pathology and Astrocytes in Autism. Neuropsychiatr. Dis. Treat. 2023, Volume 19, 841–850. [Google Scholar] [CrossRef]
- Michalski, J.-P.; Kothary, R. Oligodendrocytes in a Nutshell. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, J. Neuronal Activity and Remyelination: New Insights into the Molecular Mechanisms and Therapeutic Advancements. Front. Cell Dev. Biol. 2023, 11, 1221890. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Pérez-Acuña, D.; Rhee, K.H.; Lee, S.-J. Changes in Oligodendroglial Subpopulations in Parkinson’s Disease. Mol. Brain 2023, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.; Tadi, P. Histology, Schwann Cells. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2024. [Google Scholar]
- Usui, N. Possible Roles of Deep Cortical Neurons and Oligodendrocytes in the Neural Basis of Human Sociality. Anat. Sci. Int. 2024, 99, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Graciarena, M.; Seiffe, A.; Nait-Oumesmar, B.; Depino, A.M. Hypomyelination and Oligodendroglial Alterations in a Mouse Model of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 12, 517. [Google Scholar] [CrossRef]
- Bsibsi, M.; Nomden, A.; Van Noort, J.M.; Baron, W. Toll-like Receptors 2 and 3 Agonists Differentially Affect Oligodendrocyte Survival, Differentiation, and Myelin Membrane Formation. J. Neurosci. Res. 2012, 90, 388–398. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like Receptors in the Pathogenesis of Neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Hill, R.A.; Nishiyama, A. NG2 Cells (Polydendrocytes): Listeners to the Neural Network with Diverse Properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef]
- Kirdajova, D.; Anderova, M. NG2 Cells and Their Neurogenic Potential. Curr. Opin. Pharmacol. 2020, 50, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-P.; Zhao, J.; Li, S. Roles of NG2 Glial Cells in Diseases of the Central Nervous System. Neurosci. Bull. 2011, 27, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Gallo, V. NG 2-glia and Their Functions in the Central Nervous System. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a Common Denominator for Neuroinflammation, Blood–Brain Barrier Alteration, and Oligodendrocyte Precursor Response in EAE, Plays a Role in Dendritic Cell Activation. Acta Neuropathol. (Berl.) 2016, 132, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Q.; Yang, Q.; Gu, H.; Yin, Y.; Li, Y.; Hou, J.; Chen, R.; Sun, Q.; Sun, Y.; et al. NG2 Glia Regulate Brain Innate Immunity via TGF-Β2/TGFBR2 Axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Geng, P.; Zhao, X.; Wang, Q.; Liu, C.; Guo, C.; Dong, W.; Jin, X. The NG2-Glia Is a Potential Target to Maintain the Integrity of Neurovascular Unit after Acute Ischemic Stroke. Neurobiol. Dis. 2023, 180, 106076. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, A.; Tascio, D.; Jabs, R.; Boehlen, A.; Domingos, C.; Skubal, M.; Huang, W.; Kirchhoff, F.; Henneberger, C.; Bilkei-Gorzo, A.; et al. Dysfunction of NG2 Glial Cells Affects Neuronal Plasticity and Behavior. Glia 2023, 71, 1481–1501. [Google Scholar] [CrossRef]
- Carter, M.; Scherer, S. Autism Spectrum Disorder in the Genetics Clinic: A Review. Clin. Genet. 2013, 83, 399–407. [Google Scholar] [CrossRef]
- Tilot, A.K.; Gaugler, M.K.; Yu, Q.; Romigh, T.; Yu, W.; Miller, R.H.; Frazier, T.W.; Eng, C. Germline Disruption of Pten Localization Causes Enhanced Sex-Dependent Social Motivation and Increased Glial Production. Hum. Mol. Genet. 2014, 23, 3212–3227. [Google Scholar] [CrossRef]
- Lee, H.; Thacker, S.; Sarn, N.; Dutta, R.; Eng, C. Constitutional Mislocalization of Pten Drives Precocious Maturation in Oligodendrocytes and Aberrant Myelination in Model of Autism Spectrum Disorder. Transl. Psychiatry 2019, 9, 13. [Google Scholar] [CrossRef]
- Sarn, N.; Thacker, S.; Lee, H.; Eng, C. Germline Nuclear-Predominant Pten Murine Model Exhibits Impaired Social and Perseverative Behavior, Microglial Activation, and Increased Oxytocinergic Activity. Mol. Autism 2021, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, G.; Sula, A.; Miles, A.J.; Ng, L.C.T.; Torella, R.; Pryde, D.C.; DeCaen, P.G.; Wallace, B.A. Valproic Acid Interactions with the NavMs Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. 2019, 116, 26549–26554. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Ismail, F. A Comprehensive Review on Pharmacological Applications and Drug-Induced Toxicity of Valproic Acid. Saudi Pharm. J. 2023, 31, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Kukal, S.; Paul, P.R.; Bora, S.; Singh, A.; Kukreti, S.; Saso, L.; Muthusamy, K.; Hasija, Y.; Kukreti, R. Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile. Molecules 2021, 27, 104. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Ricci, E.; Ceraldi, R.; Nigro, A.; Bonofiglio, D.; Lanzino, M.; Morelli, C. From HDAC to Voltage-Gated Ion Channels: What’s Next? The Long Road of Antiepileptic Drugs Repositioning in Cancer. Cancers 2022, 14, 4401. [Google Scholar] [CrossRef]
- Gibbons, H.M.; Smith, A.M.; Teoh, H.H.; Bergin, P.M.; Mee, E.W.; Faull, R.L.M.; Dragunow, M. Valproic Acid Induces Microglial Dysfunction, Not Apoptosis, in Human Glial Cultures. Neurobiol. Dis. 2011, 41, 96–103. [Google Scholar] [CrossRef]
- Mony, T.J.; Lee, J.W.; Kim, S.S.; Chun, W.; Lee, H.J. Early Postnatal Valproic Acid Exposure Increase the Protein Level of Astrocyte Markers in Frontal Cortex of Rat. Clin. Psychopharmacol. Neurosci. 2018, 16, 214–217. [Google Scholar] [CrossRef]
- Lee, H.J.; Dreyfus, C.; DiCicco-Bloom, E. Valproic Acid Stimulates Proliferation of Glial Precursors during Cortical Gliogenesis in Developing Rat. Dev. Neurobiol. 2016, 76, 780–798. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, Y.; Li, Z.; Ye, L.; Lu, Q.; Zhou, Y.; Yuan, Y.; Jiang, T.; Xie, L.; Liu, Y.; et al. Valproic Acid Affects Neuronal Fate and Microglial Function via Enhancing Autophagic Flux in Mice after Traumatic Brain Injury. J. Neurochem. 2020, 154, 284–300. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.; Yin, H.; Du, Z.; Xie, R.; Yang, L.; Cai, Y.; Wang, L.; Zhang, D.; Li, X.; Liu, T.; et al. Valproic Acid Exposure Decreases Neurogenic Potential of Outer Radial Glia in Human Brain Organoids. Front. Mol. Neurosci. 2022, 15, 1023765. [Google Scholar] [CrossRef] [PubMed]
- Traetta, M.E.; Uccelli, N.A.; Zárate, S.C.; Gómez Cuautle, D.; Ramos, A.J.; Reinés, A. Long-Lasting Changes in Glial Cells Isolated From Rats Subjected to the Valproic Acid Model of Autism Spectrum Disorder. Front. Pharmacol. 2021, 12, 707859. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, A.; Greenhalgh, T. Sodium Valproate in Pregnancy: What Are the Risks and Should We Use a Shared Decision-Making Approach? BMC Pregnancy Childbirth 2018, 18, 200. [Google Scholar] [CrossRef]
- Ornoy, A. Valproic Acid in Pregnancy: How Much Are We Endangering the Embryo and Fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, Q.; Yan, T.; Zhang, Y.; Xu, H.; Yu, H.; Tu, Z.; Guo, X.; Jiang, Y.; Li, X.; et al. Maternal Valproic Acid Exposure Leads to Neurogenesis Defects and Autism-like Behaviors in Non-Human Primates. Transl. Psychiatry 2019, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, M.; Cheng, B.; Wang, S.; Xiao, L.; Zhu, J.; Wu, Q.; Lai, X.; Zhang, Q.; Chen, J.; et al. Valproic Acid Induces Autism-Like Synaptic and Behavioral Deficits by Disrupting Histone Acetylation of Prefrontal Cortex ALDH1A1 in Rats. Front. Neurosci. 2021, 15, 641284. [Google Scholar] [CrossRef]
- Zarate-Lopez, D.; Torres-Chávez, A.L.; Gálvez-Contreras, A.Y.; Gonzalez-Perez, O. Three Decades of Valproate: A Current Model for Studying AutismSpectrum Disorder. Curr. Neuropharmacol. 2024, 22, 260–289. [Google Scholar] [CrossRef]
- RESERVED, I.U.-A.R. Orphanet: Fetal Valproate Spectrum Disorder. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=1906 (accessed on 15 December 2023).
- Sharma, A.R.; Batra, G.; Saini, L.; Sharma, S.; Mishra, A.; Singla, R.; Singh, A.; Singh, R.S.; Jain, A.; Bansal, S.; et al. Valproic Acid and Propionic Acid Modulated Mechanical Pathways Associatedwith Autism Spectrum Disorder at Prenatal and Neonatal Exposure. CNS Neurol. Disord. - Drug Targets 2022, 21, 399–408. [Google Scholar] [CrossRef]
- Mony, T.J.; Lee, J.W.; Dreyfus, C.; DiCicco-Bloom, E.; Lee, H.J. Valproic Acid Exposure during Early Postnatal Gliogenesis Leads to Autistic-like Behaviors in Rats. Clin. Psychopharmacol. Neurosci. 2016, 14, 338–344. [Google Scholar] [CrossRef]
- Matsushima, T.; Izumi, T.; Vallortigara, G. The Domestic Chick as an Animal Model of Autism Spectrum Disorder: Building Adaptive Social Perceptions through Prenatally Formed Predispositions. Front. Neurosci. 2024, 18, 1279947. [Google Scholar] [CrossRef]
- Zahedi, E.; Sadr, S.S.; Sanaeierad, A.; Roghani, M. Valproate-Induced Murine Autism Spectrum Disorder Is Associated with Dysfunction of Amygdala Parvalbumin Interneurons and Downregulation of AMPK/SIRT1/PGC1α Signaling. Metab. Brain Dis. 2023, 38, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, T.; Hattori, S.; Fujimura, K.; Shibata, S.; Miyakawa, T.; Takahashi, T. In Utero Exposure to Valproic Acid throughout Pregnancy Causes Phenotypes of Autism in Offspring Mice. Dev. Neurosci. 2023, 45, 223–233. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, Y.-J.; Kim, S.J.; Lee, J.D.; Kim, S.; Ko, M.J.; Kim, J.-W.; Shin, C.Y.; Kim, K.-B. Metabolomics Profiling of Valproic Acid-Induced Symptoms Resembling Autism Spectrum Disorders Using 1H NMR Spectral Analysis in Rat Model. J. Toxicol. Environ. Health A 2022, 85, 1–13. [Google Scholar] [CrossRef]
- Guerra, M.; Medici, V.; Weatheritt, R.; Corvino, V.; Palacios, D.; Geloso, M.C.; Farini, D.; Sette, C. Fetal Exposure to Valproic Acid Dysregulates the Expression of Autism-Linked Genes in the Developing Cerebellum. Transl. Psychiatry 2023, 13, 114. [Google Scholar] [CrossRef]
- Tripathy, D.; Grammas, P. Acetaminophen Inhibits Neuronal Inflammation and Protects Neurons from Oxidative Stress. J. Neuroinflammation 2009, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.Z.; Swan, S.H.; Kriebel, D.; Liew, Z.; Taylor, H.S.; Bornehag, C.-G.; Andrade, A.M.; Olsen, J.; Jensen, R.H.; Mitchell, R.T.; et al. Paracetamol Use during Pregnancy — a Call for Precautionary Action. Nat. Rev. Endocrinol. 2021, 17, 757–766. [Google Scholar] [CrossRef]
- Wu, K.; Lu, W.; Yan, X. Potential Adverse Actions of Prenatal Exposure of Acetaminophen to Offspring. Front. Pharmacol. 2023, 14, 1094435. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.; Morelli, G.; Rastogi, M.; Savardi, A.; Fumagalli, A.; Petretto, A.; Bartolucci, M.; Varea, E.; Catelani, T.; Contestabile, A.; et al. Rescuing Over-Activated Microglia Restores Cognitive Performance in Juvenile Animals of the Dp(16) Mouse Model of Down Syndrome. Neuron 2020, 108, 887–904.e12. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, M.; Nam, G.; Kim, M.; Kang, J.; Choi, B.J.; Jeong, M.S.; Park, K.H.; Han, W.H.; Tak, E.; et al. N, N ′-Diacetyl- p -Phenylenediamine Restores Microglial Phagocytosis and Improves Cognitive Defects in Alzheimer’s Disease Transgenic Mice. Proc. Natl. Acad. Sci. 2019, 116, 23426–23436. [Google Scholar] [CrossRef]
- Zhao, W.-X.; Zhang, J.-H.; Cao, J.-B.; Wang, W.; Wang, D.-X.; Zhang, X.-Y.; Yu, J.; Zhang, Y.-Y.; Zhang, Y.-Z.; Mi, W.-D. Acetaminophen Attenuates Lipopolysaccharide-Induced Cognitive Impairment through Antioxidant Activity. J. Neuroinflammation 2017, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Kwak, S.G.; Park, J.-S.; Park, D. The Effectiveness of Nonsteroidal Anti-Inflammatory Drugs and Acetaminophen in Reduce the Risk of Amyotrophic Lateral Sclerosis? A Meta-Analysis. Sci. Rep. 2020, 10, 14759. [Google Scholar] [CrossRef] [PubMed]
- Lalert, L.; Tantarungsee, N.; Chotipinit, T.; Ji-au, W.; Srikiatkhachorn, A.; Maneesri-le Grand, S. Long-Term Paracetamol Treatment Impairs Cognitive Function and Brain-Derived Neurotrophic Factor in Adult Rat Brain. Sci. Pharm. 2023, 91, 11. [Google Scholar] [CrossRef]
- Bernal, J. Thyroid Hormones in Brain Development and Function. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., Kalra, S., Kaltsas, G., Kapoor, N., Koch, C., Kopp, P., Korbonits, M., Kovacs, C.S., Kuohung, W., Laferrère, B., Levy, M., McGee, E.A., McLachlan, R., New, M., Purnell, J., Sahay, R., Shah, A.S., Singer, F., Sperling, M.A., Stratakis, C.A., Trence, D.L., Wilson, D.P., Eds.; MDText.com, Inc.: South Dartmouth (MA), 2000. [Google Scholar]
- Klein, R.M.; Motomura, V.N.; Debiasi, J.D.; Moreira, E.G. Gestational Paracetamol Exposure Induces Core Behaviors of Neurodevelopmental Disorders in Infant Rats and Modifies Response to a Cannabinoid Agonist in Females. Neurotoxicol. Teratol. 2023, 99, 107279. [Google Scholar] [CrossRef]
- Bührer, C.; Endesfelder, S.; Scheuer, T.; Schmitz, T. Paracetamol (Acetaminophen) and the Developing Brain. Int. J. Mol. Sci. 2021, 22, 11156. [Google Scholar] [CrossRef]
- Kwok, J.; Luedecke, E.; Hall, H.A.; Murray, A.L.; Auyeung, B. Analgesic Drug Use in Pregnancy and Neurodevelopment Outcomes: An Umbrella Review. Neurosci. Biobehav. Rev. 2022, 136, 104607. [Google Scholar] [CrossRef] [PubMed]
- Blecharz-Klin, K.; Wawer, A.; Jawna-Zboińska, K.; Pyrzanowska, J.; Piechal, A.; Mirowska-Guzel, D.; Widy-Tyszkiewicz, E. Early Paracetamol Exposure Decreases Brain-Derived Neurotrophic Factor (BDNF) in Striatum and Affects Social Behaviour and Exploration in Rats. Pharmacol. Biochem. Behav. 2018, 168, 25–32. [Google Scholar] [CrossRef]
- Andrade, C. Use of Acetaminophen (Paracetamol) during Pregnancy and the Risk of Autism Spectrum Disorder in the Offspring. J. Clin. Psychiatry 2016, 77, e152-154. [Google Scholar] [CrossRef]
- Alemany, S.; Avella-García, C.; Liew, Z.; García-Esteban, R.; Inoue, K.; Cadman, T.; López-Vicente, M.; González, L.; Riaño Galán, I.; Andiarena, A.; et al. Prenatal and Postnatal Exposure to Acetaminophen in Relation to Autism Spectrum and Attention-Deficit and Hyperactivity Symptoms in Childhood: Meta-Analysis in Six European Population-Based Cohorts. Eur. J. Epidemiol. 2021, 36, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.Z.; Kriebel, D.; Herbert, M.R.; Bornehag, C.-G.; Swan, S.H. Prenatal Paracetamol Exposure and Child Neurodevelopment: A Review. Horm. Behav. 2018, 101, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Avella-Garcia, C.B.; Julvez, J.; Fortuny, J.; Rebordosa, C.; García-Esteban, R.; Galán, I.R.; Tardón, A.; Rodríguez-Bernal, C.L.; Iñiguez, C.; Andiarena, A.; et al. Acetaminophen Use in Pregnancy and Neurodevelopment: Attention Function and Autism Spectrum Symptoms. Int. J. Epidemiol. 2016, 45, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.H.; Rafikian, E.E.; Hamblin, P.B.; Strait, M.D.; Yang, M.; Pearson, B.L. Sex-Specific Neurobehavioral and Prefrontal Cortex Gene Expression Alterations Following Developmental Acetaminophen Exposure in Mice. Neurobiol. Dis. 2023, 177, 105970. [Google Scholar] [CrossRef]
- Bittker, S.S.; Bell, K.R. Postnatal Acetaminophen and Potential Risk of Autism Spectrum Disorder among Males. Behav. Sci. 2020, 10, 26. [Google Scholar] [CrossRef]
- Glasgow, A.M.; Chase, P.H. Effect of Propionic Acid on Fatty Acid Oxidation and Ureagenesis. Pediatr. Res. 1976, 10, 683–686. [Google Scholar] [CrossRef]
- Zhang, D.; Jian, Y.-P.; Zhang, Y.-N.; Li, Y.; Gu, L.-T.; Sun, H.-H.; Liu, M.-D.; Zhou, H.-L.; Wang, Y.-S.; Xu, Z.-X. Short-Chain Fatty Acids in Diseases. Cell Commun. Signal. 2023, 21, 212. [Google Scholar] [CrossRef]
- Hekim Medical Company, Istanbul, Turkey; Dogan, M.; Albayrak, Y.; Department of Psychiatry, Tekirdag Namik Kemal University Faculty of Medicine, Tekirdag, Turkey; Erbas, O.; Department of Physiology, Demiroglu Bilim University Faculty of Medicine, Istanbul, Turkey. Torasemide Improves the Propionic Acid-Induced Autism in Rats: A Histopathological and Imaging Study. ALPHA PSYCHIATRY 2023, 24, 22–31. [Google Scholar] [CrossRef]
- Abdelli, L.S.; Samsam, A.; Naser, S.A. Propionic Acid Induces Gliosis and Neuro-Inflammation through Modulation of PTEN/AKT Pathway in Autism Spectrum Disorder. Sci. Rep. 2019, 9, 8824. [Google Scholar] [CrossRef]
- Frye, R.E.; Melnyk, S.; MacFabe, D.F. Unique Acyl-Carnitine Profiles Are Potential Biomarkers for Acquired Mitochondrial Disease in Autism Spectrum Disorder. Transl. Psychiatry 2013, 3, e220–e220. [Google Scholar] [CrossRef]
- Macfabe, D. Autism: Metabolism, Mitochondria, and the Microbiome. Glob. Adv. Health Med. 2013, 2, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Lobzhanidze, G.; Lordkipanidze, T.; Zhvania, M.; Japaridze, N.; MacFabe, D.F.; Pochkidze, N.; Gasimov, E.; Rzaev, F. Effect of Propionic Acid on the Morphology of the Amygdala in Adolescent Male Rats and Their Behavior. Micron Oxf. Engl. 1993 2019, 125, 102732. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-E.J.; McDougle, C.J.; Hooker, J.M.; Zürcher, N.R. Epigenetics of Autism Spectrum Disorder: Histone Deacetylases. Biol. Psychiatry 2022, 91, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Pobbe, R.L.H.; Pearson, B.L.; Blanchard, D.C.; Blanchard, R.J. Oxytocin Receptor and Mecp2308/Y Knockout Mice Exhibit Altered Expression of Autism-Related Social Behaviors. Physiol. Behav. 2012, 107, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Chen, Y.; Guidotti, A.; Grayson, D.R. Epigenetic Regulation of RELN and GAD1 in the Frontal Cortex (FC) of Autism Spectrum Disorder (ASD) Subjects. Int. J. Dev. Neurosci. 2017, 62, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.; Stoccoro, A.; Cagiano, R.; Nicolì, V.; Ricciardi, R.; Tancredi, R.; Trovato, R.; Santorelli, F.M.; Calderoni, S.; Muratori, F.; et al. Correlation among Maternal Risk Factors, Gene Methylation and Disease Severity in Females with Autism Spectrum Disorder. Epigenomics 2022, 14, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Ghahfarroki, M.S.; Lorigooini, Z.; Shahrani, M.; Amini-Khoei, H. Umbelliprenin via Increase in the MECP2 and Attenuation of Oxidative Stress Mitigates the Autistic-like Behaviors in Mouse Model of Maternal Separation Stress. Front. Pharmacol. 2024, 14, 1300310. [Google Scholar] [CrossRef] [PubMed]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Banker, S.M.; Gu, X.; Schiller, D.; Foss-Feig, J.H. Hippocampal Contributions to Social and Cognitive Deficits in Autism Spectrum Disorder. Trends Neurosci. 2021, 44, 793–807. [Google Scholar] [CrossRef]
- Maud, C.; Ryan, J.; McIntosh, J.E.; Olsson, C.A. The Role of Oxytocin Receptor Gene (OXTR) DNA Methylation (DNAm) in Human Social and Emotional Functioning: A Systematic Narrative Review. BMC Psychiatry 2018, 18, 154. [Google Scholar] [CrossRef]
- Chen, X.; Nishitani, S.; Haroon, E.; Smith, A.K.; Rilling, J.K. OXTR Methylation Modulates Exogenous Oxytocin Effects on Human Brain Activity during Social Interaction. Genes Brain Behav. 2020, 19, e12555. [Google Scholar] [CrossRef]
- Scala, M.; Grasso, E.A.; Di Cara, G.; Riva, A.; Striano, P.; Verrotti, A. The Pathophysiological Link Between Reelin and Autism: Overview and New Insights. Front. Genet. 2022, 13, 869002. [Google Scholar] [CrossRef]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of Fragile X Syndrome and Fragile X-related Disorders. Dev. Med. Child Neurol. 2019, 61, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Dolskiy, A.A.; Pustylnyak, V.O.; Yarushkin, A.A.; Lemskaya, N.A.; Yudkin, D.V. Inhibitors of Histone Deacetylases Are Weak Activators of the FMR1 Gene in Fragile X Syndrome Cell Lines. BioMed Res. Int. 2017, 2017, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Frasch, M.G.; Yoon, B.-J.; Helbing, D.L.; Snir, G.; Antonelli, M.C.; Bauer, R. Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins. Biology 2023, 12, 914. [Google Scholar] [CrossRef] [PubMed]
- Lampiasi, N.; Bonaventura, R.; Deidda, I.; Zito, F.; Russo, R. Inflammation and the Potential Implication of Macrophage-Microglia Polarization in Human ASD: An Overview. Int. J. Mol. Sci. 2023, 24, 2703. [Google Scholar] [CrossRef] [PubMed]
- Si, T.-E.; Li, Z.; Zhang, J.; Su, S.; Liu, Y.; Chen, S.; Peng, G.-H.; Cao, J.; Zang, W. Epigenetic Mechanisms of Müller Glial Reprogramming Mediating Retinal Regeneration. Front. Cell Dev. Biol. 2023, 11, 1157893. [Google Scholar] [CrossRef]
- Kular, L.; Klose, D.; Urdánoz-Casado, A.; Ewing, E.; Planell, N.; Gomez-Cabrero, D.; Needhamsen, M.; Jagodic, M. Epigenetic Clock Indicates Accelerated Aging in Glial Cells of Progressive Multiple Sclerosis Patients. Front. Aging Neurosci. 2022, 14, 926468. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Martin, M.; Zhou, J.-R.; Thiagalingam, S. Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases. Genes 2023, 14, 896. [Google Scholar] [CrossRef]
- Imado, E.; Sun, S.; Abawa, A.R.; Tahara, T.; Kochi, T.; Huynh, T.N.B.; Asano, S.; Hasebe, S.; Nakamura, Y.; Hisaoka-Nakashima, K.; et al. Prenatal Exposure to Valproic Acid Causes Allodynia Associated with Spinal Microglial Activation. Neurochem. Int. 2022, 160, 105415. [Google Scholar] [CrossRef]
- Thiele, K.; Solano, M.E.; Huber, S.; Flavell, R.A.; Kessler, T.; Barikbin, R.; Jung, R.; Karimi, K.; Tiegs, G.; Arck, P.C. Prenatal Acetaminophen Affects Maternal Immune and Endocrine Adaptation to Pregnancy, Induces Placental Damage, and Impairs Fetal Development in Mice. Am. J. Pathol. 2015, 185, 2805–2818. [Google Scholar] [CrossRef] [PubMed]
- Woodbury, M.L.; Geiger, S.D.; Schantz, S.L. The Relationship of Prenatal Acetaminophen Exposure and Attention-Related Behavior in Early Childhood. Neurotoxicol. Teratol. 2024, 101, 107319. [Google Scholar] [CrossRef] [PubMed]
- De Campos Vidal, B.; Mello, M.L.S. Sodium Valproate (VPA) Interactions with DNA and Histones. Int. J. Biol. Macromol. 2020, 163, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Gervin, K.; Nordeng, H.; Ystrom, E.; Reichborn-Kjennerud, T.; Lyle, R. Long-Term Prenatal Exposure to Paracetamol Is Associated with DNA Methylation Differences in Children Diagnosed with ADHD. Clin. Epigenetics 2017, 9, 77. [Google Scholar] [CrossRef]
- Rozenkrantz, L.; Zachor, D.; Heller, I.; Plotkin, A.; Weissbrod, A.; Snitz, K.; Secundo, L.; Sobel, N. A Mechanistic Link between Olfaction and Autism Spectrum Disorder. Curr. Biol. 2015, 25, 1904–1910. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, G.; Shen, Y.; Ou, J.; Liu, Y.; Huang, L.; Zeng, Y.; Lin, J.; Liu, R.; Wu, R.; et al. Odor Identification Impairment in Autism Spectrum Disorder Might Be Associated with Mitochondrial Dysfunction. Asian J. Psychiatry 2022, 72, 103072. [Google Scholar] [CrossRef]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef]
- Wang, J.; Blaze, J.; Haghighi, F.; Kim-Schulze, S.; Raval, U.; Trageser, K.J.; Pasinetti, G.M. Characterization of 3(3,4-Dihydroxy-Phenyl) Propionic Acid as a Novel Microbiome-Derived Epigenetic Modifier in Attenuation of Immune Inflammatory Response in Human Monocytes. Mol. Immunol. 2020, 125, 172–177. [Google Scholar] [CrossRef]
- Buchanan, E.; Mahony, C.; Bam, S.; Jaffer, M.; Macleod, S.; Mangali, A.; Van Der Watt, M.; De Wet, S.; Theart, R.; Jacobs, C.; et al. Propionic Acid Induces Alterations in Mitochondrial Morphology and Dynamics in SH-SY5Y Cells. Sci. Rep. 2023, 13, 13248. [Google Scholar] [CrossRef]
- Stein, R.A.; Riber, L. Epigenetic Effects of Short-Chain Fatty Acids from the Large Intestine on Host Cells. microLife 2023, 4, uqad032. [Google Scholar] [CrossRef]
- Cristiano, C.; Hoxha, E.; Lippiello, P.; Balbo, I.; Russo, R.; Tempia, F.; Miniaci, M.C. Maternal Treatment with Sodium Butyrate Reduces the Development of Autism-like Traits in Mice Offspring. Biomed. Pharmacother. 2022, 156, 113870. [Google Scholar] [CrossRef]
- Li, Y.; Liu, A.; Chen, K.; Li, L.; Zhang, X.; Zou, F.; Zhang, X.; Meng, X. Sodium Butyrate Alleviates Lead-Induced Neuroinflammation and Improves Cognitive and Memory Impairment through the ACSS2/H3K9ac/BDNF Pathway. Environ. Int. 2024, 184, 108479. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Interaction of Heavy Metal Lead with Gut Microbiota: Implications for Autism Spectrum Disorder. Biomolecules 2023, 13, 1549. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Aljazi, M.B.; Wu, Y.; He, J. Vorinostat, a Histone Deacetylase Inhibitor, Ameliorates the Sociability and Cognitive Memory in an Ash1L-Deletion-Induced ASD/ID Mouse Model. Neurosci. Lett. 2021, 764, 136241. [Google Scholar] [CrossRef] [PubMed]
- Alonazi, M.; Ben Bacha, A.; Al Suhaibani, A.; Almnaizel, A.T.; Aloudah, H.S.; El-Ansary, A. Psychobiotics Improve Propionic Acid-Induced Neuroinflammation in Juvenile Rats, Rodent Model of Autism. Transl. Neurosci. 2022, 13, 292–300. [Google Scholar] [CrossRef]
- Alsubaiei, S.R.M.; Alfawaz, H.A.; Bhat, R.S.; El-Ansary, A. Nutritional Intervention as a Complementary Neuroprotective Approach against Propionic Acid-Induced Neurotoxicity and Associated Biochemical Autistic Features in Rat Pups. Metabolites 2023, 13, 738. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.; Kaliszewska, A.; Uceda, S.; Reiriz, M.; Arias, N. Targeting DNA Methylation in the Adult Brain through Diet. Nutrients 2021, 13, 3979. [Google Scholar] [CrossRef]
- Swarnkar, G.; Semenkovich, N.P.; Arra, M.; Mims, D.K.; Naqvi, S.K.; Peterson, T.; Mbalaviele, G.; Wu, C.-L.; Abu-Amer, Y. DNA Hypomethylation Ameliorates Erosive Inflammatory Arthritis by Modulating Interferon Regulatory Factor-8. Proc. Natl. Acad. Sci. 2024, 121, e2310264121. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram depicting direct influence of the drugs on epigenetics, leading to autism spectrum disorder (ASD). The two prominent epigenetic mechanisms, which affect the gene expression without changing the genetic sequence, involve histone deacetylation, which results in chromatin remodeling, or DNA methylation. Interestingly, valproic acid may exert its epigenetic effects via histone deacetylation, whereas acetaminophen and propionic acid affect DNA methylation. Regardless of the epigenetic mechanism, however, the drug may increase the chance of ASD development.
Figure 1.
Schematic diagram depicting direct influence of the drugs on epigenetics, leading to autism spectrum disorder (ASD). The two prominent epigenetic mechanisms, which affect the gene expression without changing the genetic sequence, involve histone deacetylation, which results in chromatin remodeling, or DNA methylation. Interestingly, valproic acid may exert its epigenetic effects via histone deacetylation, whereas acetaminophen and propionic acid affect DNA methylation. Regardless of the epigenetic mechanism, however, the drug may increase the chance of ASD development.
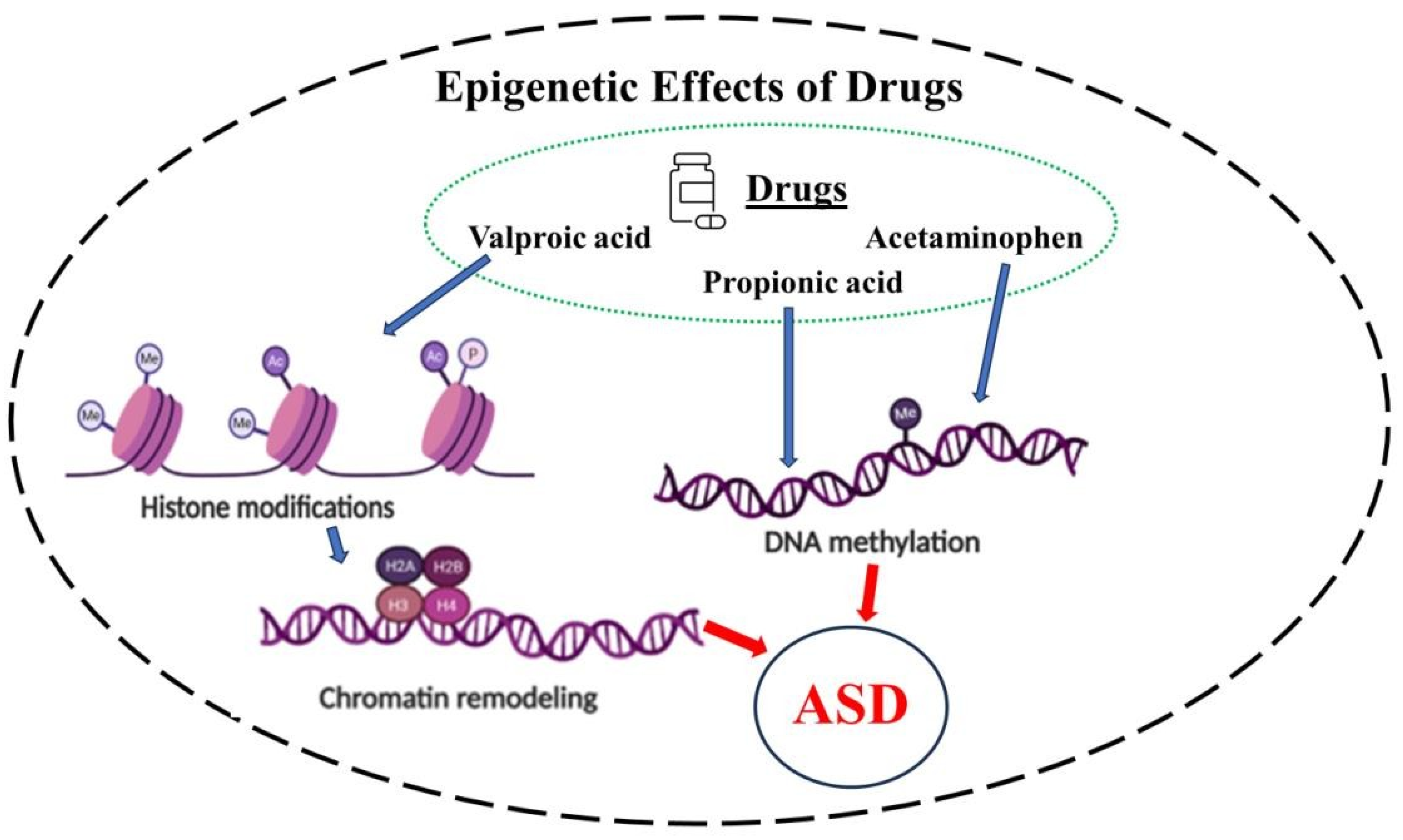
Figure 2.
Schematic diagram depicting how environmental triggers such as drugs, heavy metals, or dysbiosis in the gut microbiota may result in autism spectrum disorder (ASD). This can be due to the individual’s genetic predisposition or may be brought about indirectly via epigenetic effects. The outcome is a disturbance of homeostasis, which can also involve glial dysregulation.
Figure 2.
Schematic diagram depicting how environmental triggers such as drugs, heavy metals, or dysbiosis in the gut microbiota may result in autism spectrum disorder (ASD). This can be due to the individual’s genetic predisposition or may be brought about indirectly via epigenetic effects. The outcome is a disturbance of homeostasis, which can also involve glial dysregulation.
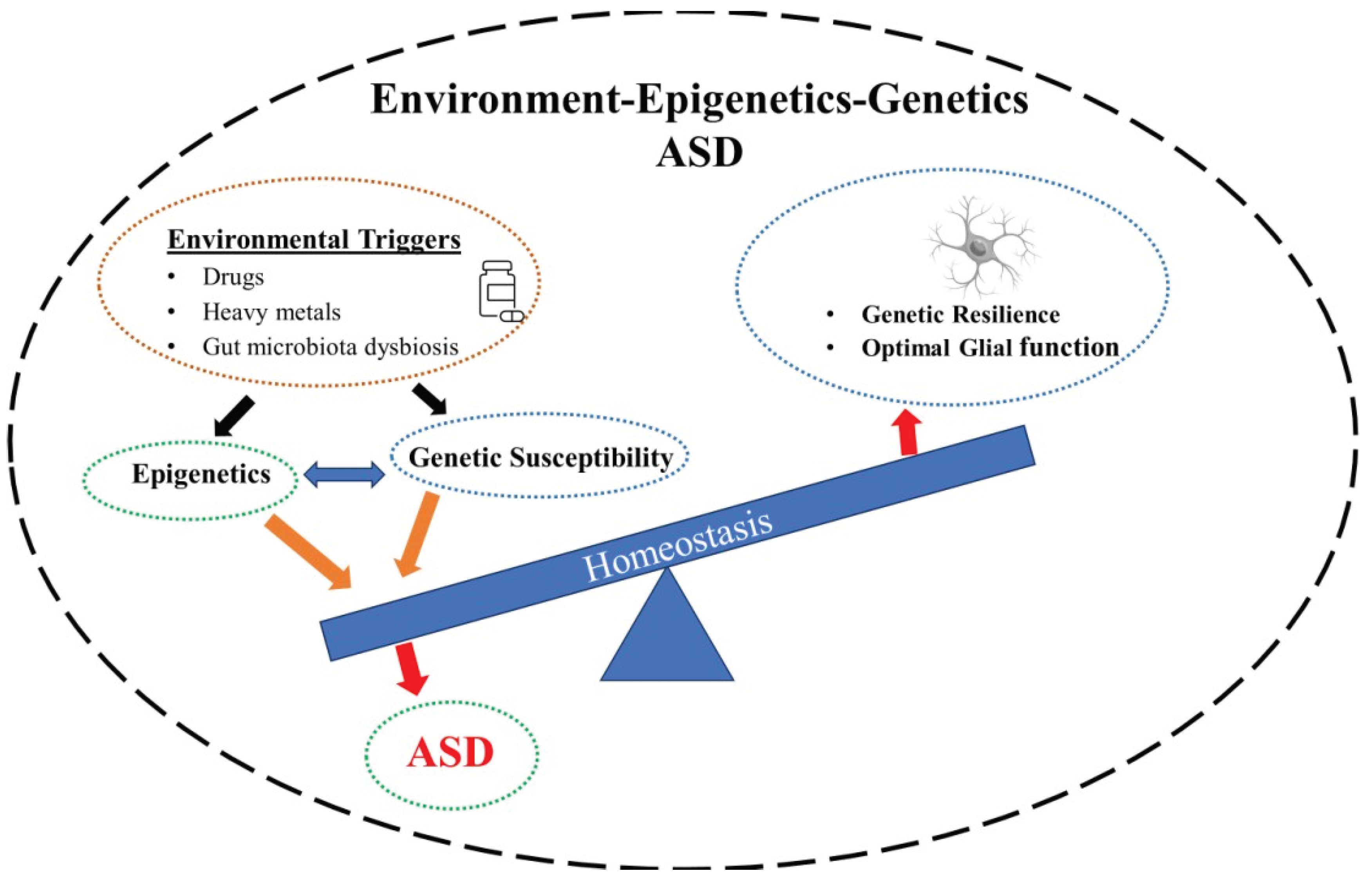
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



