Submitted:
03 March 2024
Posted:
04 March 2024
You are already at the latest version
Abstract
Background: Cell proliferation and the cell cycle are fundamental to tumor development, with mitosis playing a crucial role in cell division. Understanding the complexities of the cell cycle and mitosis is critical for cancer research, especially in the context of developing therapeutic strategies. Objective: This review highlights mitotic poisons as potential anti-cancer pharmacotherapeutics, particularly their structural and functional group diversity presenting opportunity for semi-synthesis of new derivatives with multi-target mechanisms relevant for cancer pharmacotherapeutics. Also, the review explores naturally occurring mitotic poisons and inhibitors of microtubule-dependent molecular motors as promising cancer pharmacotherapeutics. Methods: A comprehensive review of literature from 2004 to 2023 was conducted, covering studies on cell proliferation, mechanism of cell cycle regulation, mitosis, cytokinesis, and anti-cancer drug development. The published literature was collected primarily from PubMed, Scopus, Medline, Web of Science Core Collections, Google Scholar and other relevant scientific databases using keywords including but not limited to mitotic poisons and cancer, sources of mitotic poisons, mechanism of mitotic poisons, targets of mitotic poisons and structural diversity of mitotic poisons. The efficacy and limitations of various mitotic poisons and their impact on different cancer types were examined. Recent advancements in targeted therapeutic interventions for various cancers were also explored. Results: The effectiveness and limitations of mitotic poisons, including Taxanes, Epothilones, Colchicine, Vinca alkaloids, Berberine, Laulimalide, and Pseudolaric acid B, were highlighted. Drugs targeting mitotic kinases such as CDKs, Aurora kinases, PLK1, and WEE1 inhibitors were reported. Inhibiting kinesin motor proteins such as Eg5 and KIF15 to interfere with spindle formation and cell division were discussed, along with specific genomic alterations in KIF genes and the discovery of KIFC1 as potential for future research and drug development. Conclusion: Targeting microtubules and mitotic kinases, along with inhibiting kinesin motor proteins, show promise for cancer pharmacotherapeutics. The combination of targeted therapies, patient stratification based on biomarkers (such as low alpha-1 acid glycoprotein and high p63 expression), and exploration of alternative natural sources of mitotic poisons and analogues can significantly advance cancer pharmacotherapy to improve patient outcomes.
Keywords:
Cancer Pharmacotherapy
; Drug development
; Kinesin motor proteins
; Microtubules
; Mitotic kinases
; Mitotic poisons
1. Introduction
Cell proliferation remains a fundamental feature of all tumors, as cells undergo a series of ordered events collectively known as the cell cycle [1]. This cycle involves growth phases interspersed with S-phase, where DNA replication occurs, and mitosis, which encompasses nuclear and cytoplasmic division [2]. The progression through these events is strictly regulated by checkpoints that assess whether the conditions are appropriate for cell cycle continuation [3]. When a checkpoint cannot be satisfied, the cell cycle is arrested to resolve the issue, and if resolution is not possible, cells may either undergo senescence or cell death [4].
One particularly vulnerable phase of the cell cycle is mitosis, responsible for the equal partitioning of the duplicated genome into two daughter cells [2]. Mitotic cells are characterized by the presence of a bipolar spindle, a multimolecular machine that captures, aligns, and separates sister chromatid pairs [5]. The mitotic spindle is composed of sliding microtubule polymers, which are intrinsically polar structures with stable minus ends and dynamic plus ends [6]. These microtubules rapidly polymerize and depolymerize. The plus ends of spindle microtubules extend into the cytoplasm to facilitate chromosome capture, while the minus ends are focused at the two spindle poles formed by centrosomes [5].
Centrosome separation at the onset of mitosis is a crucial determinant of bipolar spindle formation, and the spindle assembly checkpoint coordinates mitotic events [5]. This checkpoint ensures that sister chromatids are not separated, and cell cleavage does not occur until all chromosomes are attached to microtubules emanating from opposite spindle poles [7]. Once the checkpoint is satisfied, the ubiquitin ligase anaphase-promoting complex is rapidly activated, leading to proteasome-dependent sister chromatid separation and destruction of cyclin B, the activating subunit of the mitotic kinase cyclin-dependent kinase 1 (CDK1) [8]. Subsequently, sister chromatids are pulled towards the spindle poles, culminating in cytokinesis, the physical separation of cells [5].
However, certain stress conditions, either extracellular or intracellular, can trigger chronic activation of the spindle assembly checkpoint, potentially leading to cell death during mitosis [9]. Alternatively, cells may exit mitosis without cytokinesis through a process known as mitotic slippage, which is caused by the progressive degradation of cyclin B during mitotic arrest [10]. Understanding the intricacies of the cell cycle, mitosis, and cytokinesis is of high importance in cancer research, as it opens routes for potential therapeutic approaches aimed at targeting and controlling cell proliferation in tumors [11].
Chemotherapeutic drugs targeting the cell cycle aim to inhibit tumor cell hyperproliferation and induce apoptosis. These drugs fall into three categories: (i) DNA synthesis inhibitors, (ii) DNA-damaging agents, and (iii) mitotic spindle inhibitors. Mitotic poisons, inhibit microtubule function, arresting mitotic cell cycle progression and inducing tumor cell death. However, their use is limited by severe side effects. Novel anti-mitotic drug targets focus on sparing microtubules while inhibiting mitosis. Research explores mitotic kinesins, kinases, and strategies to induce mitosis-associated cell death, offering promising mechanisms for developing next-generation anti-cancer drugs.
2. Mitotic Poisons
Cancer therapies often target pathways driving cell division, with microtubule-based mitotic spindle dynamics being an important key. Anti-microtubule drugs induce tumor cell death primarily by causing mitotic arrest, leading to apoptosis. Taxanes, epothilones, and Vinca alkaloids inhibit mitotic spindle dynamics, resulting in chromosomal misalignment and chronic activation of the spindle checkpoint. Prolonged treatment leads to mitotic slippage and tetraploid cell formation. Subsequent activation of p53 triggers a checkpoint response in G1, preventing further polyploidization. Apoptosis induced by these drugs requires spindle checkpoint activation and mitotic slippage [12].
Mitotic poisons are associated with resistance. Impaired mitotic spindle checkpoint function, either due to rare mutations or deregulated expression of checkpoint genes like MAD1 or MAD2, may enable escape from apoptosis. Overexpression of the oncogene Aurora-A or survivin in cancer cells could weaken the spindle checkpoint, leading to drug resistance. Changes in microtubule composition and dynamics, involving mutations in tubulin genes or altered expression of microtubule-associated proteins, contribute to resistance. Some tumors express mutant tubulin forms or overexpress specific tubulin isoforms, affecting microtubule dynamics. P-glycoprotein-mediated drug efflux, particularly for Taxanes and Vinca alkaloids, can reduce cellular drug concentrations, causing resistance. Epothilones, escaping P-glycoprotein efflux, show activity in taxol-resistant cells, offering potential alternatives. Ongoing research explores non-substrate microtubule-binding drugs to overcome drug efflux challenges [12].
Newer compounds target mitotic kinases and kinesin motor proteins, showing promise in preclinical models but limited success in clinical trials, particularly in solid tumors. Challenges include low mitotic cell numbers in solid tumors and the need for sustained drug presence. Mitotic poisons offer broad antitumor effects and immune activation. Future therapies may target specific cancer alterations or exploit new mechanisms like chromosome mis-segregation to improve clinical outcomes [13].
3. Targets of Mitotic Poisons
Mitotic poisons target key components of cell division including (i) microtubules, (ii) mitotic kinases, and (iii) kinesin motor proteins. Novel mitotic targets are emerging from genome-wide screens for molecules essential for cell cycle regulation. These targets, like Haspin kinase and the p58 isoform of cdk11 [12], offer promising approaches for selective inhibition. Haspin, crucial for sister chromatid cohesion, when ablated, triggers spindle checkpoint activation and mitotic arrest. Similarly, depletion of the p58 isoform of cdk11 leads to the formation of monopolar spindles with shortened microtubules, a phenotype rescuable by the p58 isoform, indicating its significance in mitotic centrosomes. These findings underscore the potential of novel targets in enhancing our understanding of cell cycle regulation and progression.
3.1. Microtubules
Microtubules, composed of α- and β-tubulin dimers, form polymeric protein fibers in human cells. Thirteen protofilaments combine to create a hollow microtubule fiber, with α-tubulin at the minus end and β-tubulin at the plus end. The minus end is stabilized by a γ-tubulin cap [14]. Microtubules exhibit dynamic instability [15], constantly growing and decaying, crucial for functions like mitotic spindle activity. Tubulin dimers can attach or detach from both ends, with faster changes occurring at the plus end [16]. Microtubules play vital roles in maintaining cell shape, polarity, migration, and division [14]. They serve as tracks for protein motors (kinesins and dyneins) to transport molecules within a cell [17].
Microtubule-targeting substances fall into two categories: tubulin polymer stabilizers (e.g., Taxanes) and compounds causing tubulin fiber destabilization and disintegration (e.g., Vinca alkaloids). Tubulin inhibitors bind to five major sites: the binding site for Colchicine, Taxanes, Laulimalide, Epothilones, and Vinca alkaloids [18].
Microtubules are vital for various stages of mitosis, have become promising targets for antimitotic therapies [19]. Remarkably, plants have developed their own defensive blends of microtubule poisons to fight insects [10]. Compounds like Vinca alkaloids and Taxanes displayed clinical efficacy in treating diverse tumor types [20]. Studies have revealed that Paclitaxel has concentration-dependent effects on microtubules, suppressing their dynamic behavior at low concentrations and promoting polymerization at higher concentrations [21]. Upon administration, Paclitaxel disrupts the functioning of the mitotic spindle in dividing cells, causing a checkpoint-mediated mitotic arrest [9]. Nevertheless, the mechanism behind Paclitaxel-induced cell death remains intricate and varies across different cell lines and individual cells [10]. Some cells undergo mitosis-associated death, while others experience mitotic slippage followed by cell cycle arrest or apoptosis, and a few manage to continue into the next cell cycle [22]. The stochastic behavior of individual cells poses challenges in comprehending and predicting the drug’s effects. Despite its extensive clinical use, the exact mechanism of Paclitaxel’s action in tumors remains poorly understood. Recent studies employing time-lapse microscopy to compare the effects of taxanes on cancer cells in culture and xenografts in mice have provided valuable insights [23,24]. The findings suggest that taxanes may exhibit antitumor efficacy through mitosis-independent functions, influencing cells during the interphase. However, the precise mechanisms and whether they are intrinsic to the cells or involve the tumor microenvironment require further investigation. Notwithstanding their therapeutic potential, microtubule poisons, including Paclitaxel, have drawbacks like poorly tolerated side effects such as myelosuppression and peripheral neuropathy [19]. Neurotoxicity results from inhibition of essential neuron-specific microtubule functions and can lead to permanent damage [19]. To counter neurotoxicity, research efforts have focused on developing mitosis-specific drugs such as mitotic kinases inhibitors and kinesin motor proteins inhibitors. The goal is to find approaches that disrupt mitosis progression without impacting microtubule function. This shift in focus not only enhances treatment effectiveness but also addresses the pressing concern of side effects associated with current anti-microtubule drugs.
3.2. Mitotic Kinases
This primary anti-mitotic therapy category involves targeting mitotic kinases. Mitotic kinases, like CDK1, Aurora-A, and Plk1, play pivotal roles in regulating mitosis [25]. While their inhibition disrupts mitosis in cultured cells, clinical trials targeting these kinases have shown limited efficacy in solid cancers [26]. Several factors contribute to this limited success, including dose-limiting toxicities, a lack of biomarkers for patient stratification, poor compound specificity or uptake, and the scarcity of mitotic cells with low proliferation rates in solid tumors [27].
3.3. Kinesin motor proteins
Another group of mitosis-specific targets comprises the kinesin (KIF) family of microtubule motors. KIFs participate in various cellular processes, such as spindle assembly, chromosome alignment, and cytokinesis [28].
Mitotic kinesins, specifically KSP (kinesin spindle protein), emerge as promising targets for drug development. These proteins play crucial roles in proper chromosome alignment, segregation, and centrosome separation during mitosis. KSP/Eg5, a mitosis-specific kinesin, is essential for generating a bipolar spindle and ensuring correct sister chromatid segregation. Inhibiting KSP/Eg5 function leads to a monopolar spindle, activating the mitotic spindle checkpoint and causing cell cycle arrest in mitosis. Notably, mitotic kinesins, are considered highly druggable targets, offering potential for both competitive and allosteric inhibitors in drug development [12].
With 45 identified KIF genes in mammals, categorized into 14 families based on structure, there is some functional redundancy among KIFs, yet mutations in individual KIFs can lead to developmental abnormalities [29]. The clinical use of KIF inhibitors offers advantages like reduced neuron-related side effects, but it may also face challenges such as resistance to therapies.
4. Naturally Occurring Mitotic Poisons
Natural compounds derived from plants, microbes, slime molds, and the marine environment have been valuable sources of pharmacologically active molecules used in medicine [30]. Many successful drugs have been directly or indirectly derived from natural products, with secondary metabolites from plants being particularly important in drug design. These compounds have served as templates for the synthesis or semi-synthesis of novel substances to treat various diseases in humans [30]. The following (Table 1) and (Figure 1) are examples of various mitotic poisons obtained from natural sources.
4.1. Taxanes
Taxanes (Figure 2) are a group of diterpenes obtained from Geranylgeranyl pyrophosphate, and they encompass well-known drugs like Paclitaxel (also known commercially as Taxol), Initially isolated from the bark of the pacific yew tree Taxus brevifolia, and Docetaxel (marketed as Taxotere) a semisynthetic drug derived from the needles of the European yew tree Taxus baccata. The following eight genera of the Taxus species are typically recognized: Pacific or western yew (Taxus brevifolia), European or English yew (Taxus baccata), Japanese yew (Taxus cuspidata), Canadian yew (Taxus canadensis), Himalayan yew (Taxus wallichiana), Chinese yew (Taxus chinensis), Mexican yew (Taxus globosa), and Florida yew (Taxus floridana) [31].
Taxus species exhibit varied distributions of biologically active compounds like Paclitaxel, Baccatine III, and 10-deacetyl Paclitaxel. Paclitaxel is most abundant in Taxus cuspidata, whereas Taxus mairei has the lowest content. Similarly, 10-deacetylbaccatine III is highest in T. mairei and lowest in T. media. Taxanes show substantial variability within and among species, affected by factors like season and tree part. Notably, concentrations differ significantly between woody parts and needles. High inter- and intra-species variability underscores the complexity of taxane content in Taxus trees [31].
Taxanes are potent cytotoxic agents employed in the treatment of various solid tumor malignancies, such as breast cancer, non-small cell lung carcinoma (NSCLC), head and neck cancer, and ovarian cancer. Their mode of action involves binding to tubulin molecules, which leads to the halt of cell division and triggers the activation of the mitotic spindle checkpoint [33]. They not only block cell division but also inhibit androgen receptor signaling in prostate cancer. However, their effectiveness in neurodegenerative tauopathies is limited due to difficulties in crossing the blood-brain barrier. Research is ongoing to develop taxane analogs that can overcome this limitation, and some promising compounds have been identified [34].
While Taxanes have been successful in clinical applications, they do come with some side effects like neutropenia, diarrhea, nausea, vomiting, peripheral neuropathy, and fatigue. Additionally, due to their hydrophobic nature, they need to be dissolved in lipid-based solvents, which can cause adverse reactions. Researchers have been working on improving their solubility and delivery, exploring nanotechnology-based formulations such as ABI-007 (marketed as Abraxane) and CT-2103 (known as Xyotax). However, one concern is the potential development of resistance to Taxanes, which may arise from changes in drug transporter expression, variations in tubulin subtypes with reduced binding affinity, or acquired mutations in tubulin. Addressing these challenges remains crucial for optimizing the benefits of Taxanes in cancer treatment [33].
A study suggested "second-generation Taxoids" with modifications at the C-3′-phenyl and C-10 positions, rendering them significantly more potent against drug-resistant breast cancer cell lines compared to their parent drugs.[35]. In essence, new-generation Taxoids hold significant promise for further clinical development.
Furthermore, the search for other drugs that enhance microtubule polymerization has yielded promising candidates like Epothilones, Discodermolide, Sarcodictyins, Eleutherobin, and Laulimalide, some of which bind to unique sites on microtubules. These advances hold potential for enhancing cancer treatment and addressing neurodegenerative conditions [34].
The production of Paclitaxel poses challenges, with its industrial extraction from Taxus bark being ecologically demanding and yielding low quantities. Despite early total synthesis attempts, none were commercially viable due to low yields. Bristol-Myers Squibb’s semi-synthesis, using 10-deacetylbaccatin III as a precursor, became a commercially viable method. A more modern semisynthetic approach involves structurally similar taxanes, providing a fast and effective method, though it still relies on yew isolation. Biotechnological production in fungi, despite extensive research, has not reached industrial practice. Aspergillus fumigatus KU-837249 is a promising strain, producing 1.6 g of Paclitaxel per 1 L of medium; genetic engineering and cultivation modifications could enhance commercial viability [18].
4.2. Epothilones
Epothilones (Table 1 and Figure 1) are a family of novel Microtubule-Targeting Agents (MTAs) that stabilize microtubules, preventing cancer cells from dividing during mitosis. Epothilones A and B were initially discovered as antifungal agents produced by the bacterium Sorangium cellulolus [34]. They compete with paclitaxel for the same binding site on beta-tubulin and promote microtubule assembly, suggesting similarities with taxanes [34].
These compounds are distinct from taxanes in their binding modes to beta-tubulin, potentially explaining their effectiveness against taxane-resistant cancer cells. Another unique aspect of epothilones is their ability to maintain cytotoxic activity even in cells that overexpress P-glycoprotein, a protein responsible for drug efflux [34].
Currently, there are five epothilone-derived compounds undergoing clinical evaluation, with epothilone B and its lactam analog (Ixabepilone) being among the most advanced. They are being evaluated in phase III clinical trials for various cancer types [34].
While epothilones show promise as chemotherapeutic agents, it’s essential to address side effects. Epothilone D, for instance, demonstrated a higher therapeutic index compared to epothilone B, but severe central nervous system toxicities were reported in clinical trials. Ongoing research aims to optimize the efficacy and safety of these compounds in cancer treatment [34].
Epothilones, produced biotechnologically from Sorangium cellulosum, yield low quantities (1 mg/L) due to difficult genome manipulation and long bacterial doubling time [36]. Attempts to increase production include incorporating genome clusters into other organisms, with Myxococcus xanthus showing promising results of up to 23 mg/L [37]. Genetic modifications of Sorangium cellulosum, like introducing the vgb gene for better oxygen transmission and epoF gene for enhanced epothilone production, have increased yields by 58% and 122%, respectively [38].
Recent studies by Ye et al. (2019) utilized TALE-TF and CRISPR technologies to insert promoters for epothilone production, resulting in significant yield increases. TALE-TF augmented compound production by 2.89 and 1.12 times, while CRISPR enhanced production by 1.53 and 2.18 times compared to natural Sporangium cellulosum. These advancements offer promising methods for improving epothilone production efficiency.
4.3. Colchicine
Colchicine (Table 1 and Figure 1) is an alkaloid derived from plants like the Autumn crocus (Colchicum autumnale) and Glory Lilly (Gloriosa superba) found in Europe and North America. It has been used in medications approved by the US FDA since 2009. Colchicine is a water-soluble alkaloid with pale yellow needles or powder. It is primarily used as a secondary treatment for gout due to its anti-inflammatory properties [39].
The alkaloid is biosynthesized from phenylalanine and tyrosine through a series of chemical reactions. Colchicine’s mechanism of action involves preventing DNA synthesis and tubulin polymerization, effectively halting mitosis [39]. Colchicine binds permanently to tubulin, stabilizes microtubule formation, and induces cell cycle arrest and apoptosis [40]. However, its action is not very specific and can affect normal rapidly dividing cells, leading to toxicity. To address this, less toxic semisynthetic derivatives of colchicine, like colchicinamide and deacetylcolchicine, have been developed for the treatment of various cancers [40].
4.4. Vinca alkaloids
Vinblastine, Vincristine, Vindesine, and Vinorelbine (Table 1 and Figure 1) are Vinca alkaloids derived from various plant sources. The Vinca alkaloids, vincristine, and vinblastine, were the first plant-derived anticancer drugs to be used clinically [30]. They work by interfering with microtubular activity at low doses and causing cell cycle arrest and apoptosis at higher doses [41]. Semisynthetic derivatives like Vindesine, vinorelbine, and vinflunine were developed from vincristine and vinblastine [30]. Currently, these alkaloids are widely used for the treatment of different cancer types.
Vinblastine is extracted from Catharanthus roseus, belonging to the Apocynaceae family [41], was initially identified while exploring its anti-diabetic properties and is used to treat Hodgkin’s and non-Hodgkin lymphomas, breast cancer, and others [39]. Vincristine is a chemotherapy drug for leukemia, lymphoma, and more [39]. Vindesine is Derived from vinblastine, is effective against non-small cell lung cancer and relapsed juvenile chronic lymphocytic leukemia [39]. Vinorelbine suppresses cell proliferation by attaching to tubulin and is distinct from other alkaloids in its anticancer activity [39]. Additionally, Vincamine is a monoterpenoid indole alkaloid found in Vinca minor and is used for treating cerebrovascular diseases and circulatory disorders, particularly impacting brain vessels. It has effects on the central nervous and cardiovascular systems, promoting brain metabolism, improving memory and cognitive capacity, and enhancing immune function. Vincamine is also used for various other ailments, such as throat ailments, toothache, and wound healing [39].
4.5. Berberine
Berberine (Table 1 and Figure 1), extracted from the root and rhizome of plants such as Tinospora cordifolia, Berberis vulgaris, Berberis aquifolium, and Rhizoma coptidis, is a potent anticancer compound. Berberine induces apoptosis and cell cycle arrest at the G2/M phase in breast, colorectal, and liver cancer. Additionally, it inhibits anti-apoptotic proteins c-IAP1 and Bcl-2 while activating pro-apoptotic proteins like p21, p53, caspase-3, and caspase-9 [40].
4.6. Pseudolaric Acid B
Golden larch (Pseudolarix amabilis) is a tree known for its medicinal properties in traditional Chinese medicine. It produces a group of diterpenes called Pseudolaric acids, with Pseudolaric acid B (PAB) (Table 1 and Figure 1) being the most bioactive and showing promise as an anticancer agent. PAB inhibits microtubule polymerization, similar to other chemotherapy drugs [42]. Diterpenes, like those found in golden larch, are a diverse class of plant compounds with potential for drug discovery. Pseudolaric acids and other diterpenes have various biological activities, making them valuable sources of new biopharmaceuticals [42].
4.7. Laulimalide
Laulimalide (Table 1 and Figure 1) a natural compound discovered in 1999 alongside Isolaulimalide from the marine sponge Cacospongia mycofijiensis, influences microtubule polymerization. It binds to tubulin at a unique site between the two β-subunits, potentially advantageous in treating taxane-resistant tumors or in synergy with taxanes. Unlike taxanes, Laulimalide is not a P-glycoprotein substrate. In vitro, it increases microtubule polymerization, inhibiting cell proliferation in various cancer cell lines. However, in vivo experiments show lower efficacy against cancer cell proliferation and high systemic toxicity. Its instability in vivo could be mitigated by derivatization, yet derivatives have reduced biological activity. Despite interest, it hasn’t entered clinical use, primarily serving research purposes in understanding microtubule-associated processes [18].
Despite the success of naturally occurring mitotic poisons, their limitations, such as limited commercial production, solubility issues, resistance, and toxicity, they have led to the development of analogues, such as Cabazitaxel, which is currently undergoing clinical trials. These compounds continue to be promising candidates for the treatment of various malignancies [30].
5. Mitotic Kinase Inhibitors
Mitotic kinase inhibitors (Table 2 and Figure 3) present valuable insights into potential targeted therapeutic interventions for gastric, liver, pancreatic, and colorectal cancers, offering a potential strategy to combat these deadly diseases.
Mitotic kinases are essential in the advancement of gastric cancer (GC) and present as potential targets for anti-cancer pharmacotherapeutics [43]. Several mitotic kinases have been used in GC including
- (i)
- Cyclin-Dependent Kinases (CDKs): are crucial regulators of the cell cycle and DNA damage response, and their overexpression is observed in various cancers, including GC [44]. A study conducted by Tang et al. unveiled that CDK2 regulates aerobic glycolysis in GC cell lines. Knocking down CDK2 led to decreased glycolytic mRNA levels and increased expression of SIRT5, a tumor suppressor involved in metabolic reprogramming [44].
- (ii)
- Aurora Kinases: are serine/threonine kinases that regulate G2/M phase transitions during cell division [45]. A study by Ding et al. revealed that Aurora B and cyclin B1 are co-expressed during the G2/M phase in GC cells. They found that Aurora B’s upregulation in the presence of high cyclin B1 levels is essential for G2/M phase transition, and it interacts with CREPT to modulate Cyclin B1 expression [46].
- (iii)
- Polo-Like Kinase 1 (PLK1): it is part of the PLK family, plays a vital role in cell cycle regulation, DNA synthesis, and p53 transactivation and is frequently overexpressed in multiple human cancers [47]. Dang et al. identified elevated PLK1 levels in GC cell lines with anti-miR-505 transfection. They demonstrated that miR-505 directly targets PLK1, thereby affecting its expression in GC cell lines [48]. Inhibition of PLK1 resulted in G2-phase cell cycle arrest and suppressed proliferation, migration, and apoptosis of GC cells. Moreover, PLK1 inhibition reduced the activation of the MEK/ERK signaling pathway [48]. Cai et al. also highlighted PLK1’s role in promoting GC cell migration, invasion, and epithelial-mesenchymal transition (EMT), indicating that PLK1 could be a potential target for therapeutic intervention in GC [49].
- (iv)
- Wee1-Like Protein Kinase (WEE1): this enzyme belongs to the Serine/Threonine protein kinase family and regulates the DNA damage checkpoint in the G2/M cell-cycle transition, enabling DNA repair before mitotic entry [50]. Wang et al. investigated WEE1’s tumorigenic role in GC and found high expression and secretion of WEE1 in GC cell lines. Silencing WEE1 through siRNA reduced cell viability in GC cell lines [51].
5.1. Mitotic Kinase Inhibitors Used against Gastric Cancers
- (i)
- Danusertib (Table 2 and Figure 3), previously known as PHA739358, is a potent pan-Aurora kinase inhibitor. Studies by Santo et al. indicated that Danusertib reduces the survival rate of human GC cell lines and inhibits the growth of GC cells, particularly in the G1 phase, along with reduced expression of CDK2 [52]. Kamran et al. demonstrated that Aurora A knockdown or inhibition with Alisertib (Figure 3) induced apoptosis in GC cells, reducing tumor volumes in mice [45].
- (ii)
- Volasertib (Table 2 and Figure 3), a selective PLK1 inhibitor, induces mitotic arrest and apoptosis in GC patients. Nokihara et al. confirmed the inhibitory effects of volasertib in Japanese patients with advanced GC, indicating its potential as a therapeutic option [53]. Lin et al. showed that AZD1775 (Table 2 and Figure 3), another PLK1 inhibitor, effectively targeted PLK1 in GC cells, inducing apoptosis and enhancing the inhibition of gastric tumor growth when combined with Olaparib [50].
- (iii)
- (iv)
- Liu et al. had proposed Procaterol (Table 2 and Figure 3), a β2-receptor agonist, as a potential treatment strategy for GC. They found that Procaterol suppressed cell viability and colony formation in GC cell lines and inhibited tumor growth in patient-derived gastric tumor xenografts by inhibiting CDK12 [54].
The following mitotic kinase inhibitors show promise as potential therapeutic options for liver, pancreatic, and colorectal cancers [43]. Further research and clinical trials are ongoing to explore their efficacy and safety in combination anti-cancer therapy.
5.2. Mitotic Kinase Inhibitors Used against Liver Cancer
CDK1 Inhibition (RO-3306, Metformin, Lycorine, Dihydroartemisinin) slows tumor growth. Aurora Kinase Inhibition (MLN8237, SNS-314) with sorafenib reduces tumor growth. PLK1 Inhibition (Volasertib, GSK461364, Rigosertib, Danusertib) targets p53-mutated cells and may enhance treatment with BIRC5 inhibitor [43].
5.3. Mitotic Kinase Inhibitors Used against Pancreatic Cancer
Aurora-A Inhibition (CCT137690, Danusertib) hinders cell growth and induces death. CDK1 Inhibition (oxadiazole-based Topsentin derivative, Dinaciclib) induces apoptosis and immunogenic cell death. WEE1 Inhibition (AZD1775) sensitizes p53-mutated cells to chemotherapy. TTK Inhibition (AZ3146) reduces cell proliferation [43].
5.4. Mitotic Kinase Inhibitors Used against Colorectal Cancer
PLK1 Inhibition (TAK-960) limits colony formation. Aurora Kinase Inhibition (ENMD-2076) induces G2/M arrest and apoptosis. CDK Inhibition (CP668863) may be a therapeutic agent for CRC. Mps1 Inhibition (CCT271850) induces apoptotic cell death in CRC cells [43].
6. Inhibitors of microtubule-dependent molecular motors
Two motor proteins, Eg5 (Kinesin-5) and KIF15 (Kinesin-12), play crucial roles in centrosome separation and spindle bipolarity during mitosis. Inhibiting these motor proteins can interfere with the proper formation and function of the mitotic spindle, leading to cell cycle arrest and cell death in rapidly dividing cancer cells [55].
In the late 1990s, researchers explored KIF-targeting antimitotics, discovering Monastrol as a promising inhibitor of Eg5/KIF11, essential for centrosome separation during cell division. Clinical trials with Eg5 inhibitors like Ispinesib and Filanesib were less effective than anticipated, but they yielded valuable insights for future therapies [56].
KIFC1, involved in centrosome clustering, presents a potential target for tumor cells with extra centrosomes. Genomic alterations in KIF genes, like KIF14 amplification in liver, breast, and lung cancers, and KIF5A amplification in glioblastomas, offer new avenues for research. Deletions in KIF9 and KIF15 in renal clear cell carcinomas could sensitize tumors to Eg5 inhibitors [56].
7. Future Perspective
From a medicinal chemistry standpoint, the structural and functional group diversity (Figure 4) observed among the mitotic poisons is indicative of the broad spectrum of target proteins and enzymes involved in these processes. This structural heterogeneity not only underscores the complexity of the cellular mechanisms associated with mitosis but also presents an opportunity to explore novel chemical classes of compounds. Moreover, it provides a rationale for repurposing existing drugs from different therapeutic indications by subjecting them to systematic screening against the identified target proteins and enzymes associated with mitotic processes. In this context, an intriguing example is the identification of the β-adrenoceptor agonist Procaterol as an inhibitor of tumor growth in gastric cancer. This discovery, reported by Liu et al., exemplifies the potential of repurposing existing drugs for anti-mitotic purposes [54]. The inhibition of CDK12 by Procaterol suggests a previously unrecognized therapeutic avenue for β-adrenoceptor agonists in cancer treatment. This finding not only expands our understanding of the pharmacological effects of known compounds but also emphasizes the importance of considering unconventional drug candidates in the pursuit of novel anti-mitotic agents.
The review mentions the potential of exploiting new mechanisms like chromosome mis-segregation for improving clinical outcomes. Understanding how cancer cells respond to alterations in mitotic spindle dynamics and developing drugs that target these processes could lead to novel treatment strategies with improved efficacy and reduced resistance. Novel targets identified from genome-wide screens, such as Haspin kinase and the p58 isoform of cdk11, offer promising avenues for selective inhibition and enhancing our understanding of cell cycle regulation and progression. Further exploration of these targets could lead to the development of more specific and effective drugs.
As highlighted in the review, resistance to mitotic poisons remains a significant challenge in cancer treatment. Future therapies could focus on targeting specific alterations found in cancer cells, such as mutations in tubulin genes, overexpression of certain oncogenes like Aurora-A or survivin, and changes in microtubule composition. Efforts are ongoing to develop non-substrate microtubule-binding drugs that can overcome challenges such as drug resistance through cellular efflux mediated by P-glycoprotein. Researches to identify and optimize compounds that selectively inhibit P-glycoprotein functions without causing significant toxicity can potentially enhance the intracellular concentration of anticancer drugs, overcoming P-glycoprotein-mediated drug efflux. These drugs would offer advantage in circumventing resistance mechanisms associated with traditional mitotic poisons like Taxanes and Vinca alkaloids. Investigating synergistic combinations of existing anticancer drugs with P-glycoprotein inhibitors may help overcome resistance by simultaneously targeting cancer cells with the primary drug while inhibiting P-glycoprotein -mediated efflux. Furthermore, utilizing computational modeling and simulation to predict and understand P-glycoprotein substrate interactions can aid in the rational design of drugs less prone to efflux.
Mitotic poisons like Taxanes face challenges related to side effects and delivery methods. Research efforts aimed at improving drug delivery and developing novel formulations, such as nanotechnology-based approaches, could mitigate these challenges and enhance the therapeutic efficacy of mitotic poisons while minimizing adverse effects. Optimal treatment intensity involving frequent dosing and sustained exposure is crucial to induce cell death. Biomarkers, such as low alpha-1 acid glycoprotein benefiting from Filanesib or high p63 expression predicting response in bladder cancer to AZD4877, enable patient stratification for better outcomes. Combining EGFR suppression with Eg5 inhibitors may combat resistance, and targeting KIF15 alongside Eg5 may boost antitumor response [56]. KIF inhibitors hold promise, and understanding patient stratification and combination therapies can enhance their efficacy in treating cancer. The identification of KIFC1 as a potential target and genomic alterations in KIF genes open exciting possibilities for further natural products screening and drug development. Furthermore, inhibiting kinesin motor proteins like Eg5 and KIF15 provides potential strategies to interfere with spindle formation and cell division in cancer cells. The exploration of genomic alterations in KIF genes and the discovery of KIFC1 as a potential target offer exciting prospects for future research and drug development.
While currently available new compounds targeting mitotic kinases and kinesin motor proteins have shown promise in preclinical models, their limited success in clinical trials, particularly in solid tumors, underscores the need for expanded clinical trials. The combination of these targeted therapies, patient stratification based on biomarkers, and further investigation into natural compounds hold great potential in advancing cancer treatment and improving patient outcomes.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
AZ3146: TTK Inhibitor, AZD1775: Wee1-Like Protein Kinase Inhibitor, Bcl-2: B-cell lymphoma 2, c-IAP1: Cellular Inhibitor of Apoptosis Protein 1, CDKs: Cyclin-Dependent Kinases, CCT137690: Aurora-A Inhibitor, CCT271850: Mps1 Inhibitor, CRC: Colorectal Cancer, DNA: Deoxyribonucleic acid, EG5: Kinesin spindle protein, EGFR: Epidermal Growth Factor Receptor, FDA: U.S. Food and Drug Administration, G1: Gap 1 phase, G2: Gap 2 phase, G2/M: Gap 2/ Mitosis, GC: Gastric Cancer, KIF15: Kinesin family member 15, MAD1: Mitotic arrest deficient 1, MAD2: Mitotic arrest deficient 2, MEK/ERK: Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase, MTAs: Microtubule-Targeting Agents, NSCLC: Non-Small Cell Lung Carcinoma, PAB: Pseudolaric Acid B, p21: Cyclin-Dependent Kinase Inhibitor 1A, p53: Tumor Protein p53, PLK1: Polo-Like Kinase 1, RNA: Ribonucleic acid, S-phase: Synthesis phase, TAK-960: PLK1 Inhibitor, TTK: Dual Specificity Protein Kinase TTK, WEE1: Wee1-Like Protein Kinase.
References
- Weinberg, R., Chapter 8: pRb and control of the cell cycle clock. The Biology of Cancer, 2nd Edition, 2014: p. 275-329.
- Alberts, B., et al., Molecular Biology of the Cell 6th ed (New York, NY: Garland Science). 2014.
- Hartwell, L.H. and M.B. Kastan, Cell cycle control and cancer. Science, 1994. 266(5192): p. 1821-8. [CrossRef]
- Hanahan, D. and R.A. Weinberg, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646-74. [CrossRef]
- Maiato, H. and E. Logarinho, Mitotic spindle multipolarity without centrosome amplification. Nature Cell Biology, 2014. 16(5): p. 386-94. [CrossRef]
- Dumont, S. and T.J. Mitchison, Force and length in the mitotic spindle. Current Biology, 2009. 19(17): p. R749-61. [CrossRef]
- Musacchio, A. and E.D. Salmon, The spindle-assembly checkpoint in space and time. Nature reviews Molecular cell biology, 2007. 8(5): p. 379-393. [CrossRef]
- Pines, J., Mitosis: a matter of getting rid of the right protein at the right time. Trends in cell biology, 2006. 16(1): p. 55-63. [CrossRef]
- Brito, D.A. and C.L. Rieder, Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Current Biology, 2006. 16(12): p. 1194-1200. [CrossRef]
- Gascoigne, K.E. and S.S. Taylor, Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell, 2008. 14(2): p. 111-22. [CrossRef]
- Lara-Gonzalez, P., F.G. Westhorpe, and S.S. Taylor, The spindle assembly checkpoint. Current biology, 2012. 22(22): p. R966-R980. [CrossRef]
- Schmidt, M. and H. Bastians, Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Update, 2007. 10(4-5): p. 162-81. [CrossRef]
- Tischer, J. and F. Gergely, Anti-mitotic therapies in cancer. Journal of Cell Biology, 2019. 218(1): p. 10-11. [CrossRef]
- Chen, X., et al., Remote control of microtubule plus-end dynamics and function from the minus-end. Elife, 2019. 8. [CrossRef]
- Li, C., et al., Microtubule dynamic instability: the role of cracks between protofilaments. Soft Matter, 2014. 10(12): p. 2069-2080. [CrossRef]
- Strothman, C., et al., Microtubule minus-end stability is dictated by the tubulin off-rate. J Cell Biol, 2019. 218(9): p. 2841-2853. [CrossRef]
- Ferro, L.S., et al., Kinesin and dynein use distinct mechanisms to bypass obstacles. Elife, 2019. 8. [CrossRef]
- Škubník, J., et al., Mitotic poisons in research and medicine. Molecules, 2020. 25(20): p. 4632. [CrossRef]
- Jordan, M.A. and L. Wilson, Microtubules as a target for anticancer drugs. Nature reviews cancer, 2004. 4(4): p. 253-265. [CrossRef]
- Schiff, P.B. and S.B. Horwitz, Taxol stabilizes microtubules in mouse fibroblast cells. Proceedings of the National Academy of Sciences of the United States of America, 1980. 77(3): p. 1561-5. [CrossRef]
- Jordan, M.A., et al., Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proceedings of the National Academy of Sciences of the United States of America, 1993. 90(20): p. 9552-9556. [CrossRef]
- Shi, J., J.D. Orth, and T. Mitchison, Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer research, 2008. 68(9): p. 3269-3276. [CrossRef]
- Janssen, A., et al., Intravital FRET imaging of tumor cell viability and mitosis during chemotherapy. PloS one, 2013. 8(5). [CrossRef]
- Orth, J.D., et al., Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer research, 2011. 71(13): p. 4608-4616. [CrossRef]
- Malumbres, M., Physiological relevance of cell cycle kinases. Physiological reviews, 2011. [CrossRef]
- Salmela, A.-L. and M.J. Kallio, Mitosis as an anti-cancer drug target. Chromosoma, 2013. 122: p. 431-449. [CrossRef]
- Mitchison, T., The proliferation rate paradox in antimitotic chemotherapy. Molecular biology of the cell, 2012. 23(1): p. 1-6. [CrossRef]
- Vicente, J.J. and L. Wordeman, Mitosis, microtubule dynamics and the evolution of kinesins. Experimental cell research, 2015. 334(1): p. 61. [CrossRef]
- Hirokawa, N. and Y. Tanaka, Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Experimental cell research, 2015. 334(1): p. 16-25. [CrossRef]
- Dehelean, C.A., et al., Plant-Derived Anticancer Compounds as New Perspectives in Drug Discovery and Alternative Therapy. Molecules, 2021. 26(4). [CrossRef]
- Nižnanský, Ľ., et al., Natural Taxanes: From Plant Composition to Human Pharmacology and Toxicity. Int J Mol Sci, 2022. 23(24). [CrossRef]
- Hao, D.C., X.-J. Gu, and P.G. Xiao, 3 - Taxus medicinal resources: a comprehensive study, in Medicinal Plants, D.C. Hao, X.-J. Gu, and P.G. Xiao, Editors. 2015, Woodhead Publishing. p. 97-136. [CrossRef]
- V Rao, C., C. D Kurkjian, and H.J.C.D.T. Y Yamada, Mitosis-targeting natural products for cancer prevention and therapy. 2012. 13(14): p. 1820-1830. [CrossRef]
- Mukhtar, E., V.M. Adhami, and H. Mukhtar, Targeting Microtubules by Natural Agents for Cancer Therapy. Molecular Cancer Therapeutics, 2014. 13(2): p. 275-284. [CrossRef]
- Ojima, I. and M. Das, Recent advances in the chemistry and biology of new generation taxoids. J Nat Prod, 2009. 72(3): p. 554-65. [CrossRef]
- Ye, W., et al., An Easy and Efficient Strategy for the Enhancement of Epothilone Production Mediated by TALE-TF and CRISPR/dcas9 Systems in Sorangium cellulosum. Front Bioeng Biotechnol, 2019. 7: p. 334. [CrossRef]
- Lau, J., et al., Optimizing the heterologous production of epothilone D in Myxococcus xanthus. Biotechnol Bioeng, 2002. 78(3): p. 280-8. [CrossRef]
- Ye, W., et al., A new approach for improving epothilone B yield in Sorangium cellulosum by the introduction of vgb epoF genes. J Ind Microbiol Biotechnol, 2016. 43(5): p. 641-50. [CrossRef]
- Dhyani, P., et al., Anticancer potential of alkaloids: a key emphasis to colchicine, vinblastine, vincristine, vindesine, vinorelbine and vincamine. Cancer Cell International, 2022. 22(1): p. 206. [CrossRef]
- Iqbal, J., et al., Plant-derived anticancer agents: A green anticancer approach. Asian Pacific Journal of Tropical Biomedicine, 2017. 7(12): p. 1129-1150. [CrossRef]
- Siddiqui, A.J., et al., Plants in Anticancer Drug Discovery: From Molecular Mechanism to Chemoprevention. BioMed Research International, 2022. 2022: p. 5425485. [CrossRef]
- Mafu, S., et al., Biosynthesis of the microtubule-destabilizing diterpene pseudolaric acid B from golden larch involves an unusual diterpene synthase. Proceedings of the National Academy of Sciences of the United States of America, 2017. 114(5): p. 974-979. [CrossRef]
- Javed, A., et al., Therapeutic Potential of Mitotic Kinases’ Inhibitors in Cancers of the Gastrointestinal System. Future Pharmacology, 2022. 2(3): p. 214-237. [CrossRef]
- Tang, Z., et al., CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer. Cancer Science, 2018. 109(8): p. 2590-2598. [CrossRef]
- Kamran, M., et al., Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis, 2017. 6(2): p. e298. [CrossRef]
- Ding, L., et al., CREPT/RPRD1B associates with Aurora B to regulate Cyclin B1 expression for accelerating the G2/M transition in gastric cancer. Cell Death Dis, 2018. 9(12): p. 1172. [CrossRef]
- Wakida, T., et al., The CDK-PLK1 axis targets the DNA damage checkpoint sensor protein RAD9 to promote cell proliferation and tolerance to genotoxic stress. eLife, 2017. 6: p. e29953. [CrossRef]
- Dang, S.C., et al., MicroRNA-505 suppresses gastric cancer cell proliferation and invasion by directly targeting Polo-like kinase-1. Onco Targets Ther, 2019. 12: p. 795-803. [CrossRef]
- Cai, X.P., et al., PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am J Transl Res, 2016. 8(10): p. 4172-4183.
- Lin, X., et al., Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J Exp Clin Cancer Res, 2018. 37(1): p. 129. [CrossRef]
- Wang, C.H., et al., Tazarotene-induced gene 1 interacts with Polo-like kinase 2 and inhibits cell proliferation in HCT116 colorectal cancer cells. Cell Biol Int, 2021. 45(11): p. 2347-2356. [CrossRef]
- Santo, L., et al., AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene, 2010. 29(16): p. 2325-36. [CrossRef]
- Nokihara, H., et al., Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs, 2016. 34(1): p. 66-74. [CrossRef]
- Liu, H., et al., CDK12 and PAK2 as novel therapeutic targets for human gastric cancer. Theranostics, 2020. 10(14): p. 6201-6215. [CrossRef]
- Dumas, M.E., et al., Dual inhibition of Kif15 by oxindole and quinazolinedione chemical probes. Bioorganic & Medicinal Chemistry Letters, 2019. 29(2): p. 148-154. [CrossRef]
- Chandrasekaran, G., P. Tátrai, and F. Gergely, Hitting the brakes: targeting microtubule motors in cancer. British Journal of Cancer, 2015. 113(5): p. 693-698. [CrossRef]
Figure 1.
Chemical structures of some naturally occurring mitotic poisons.
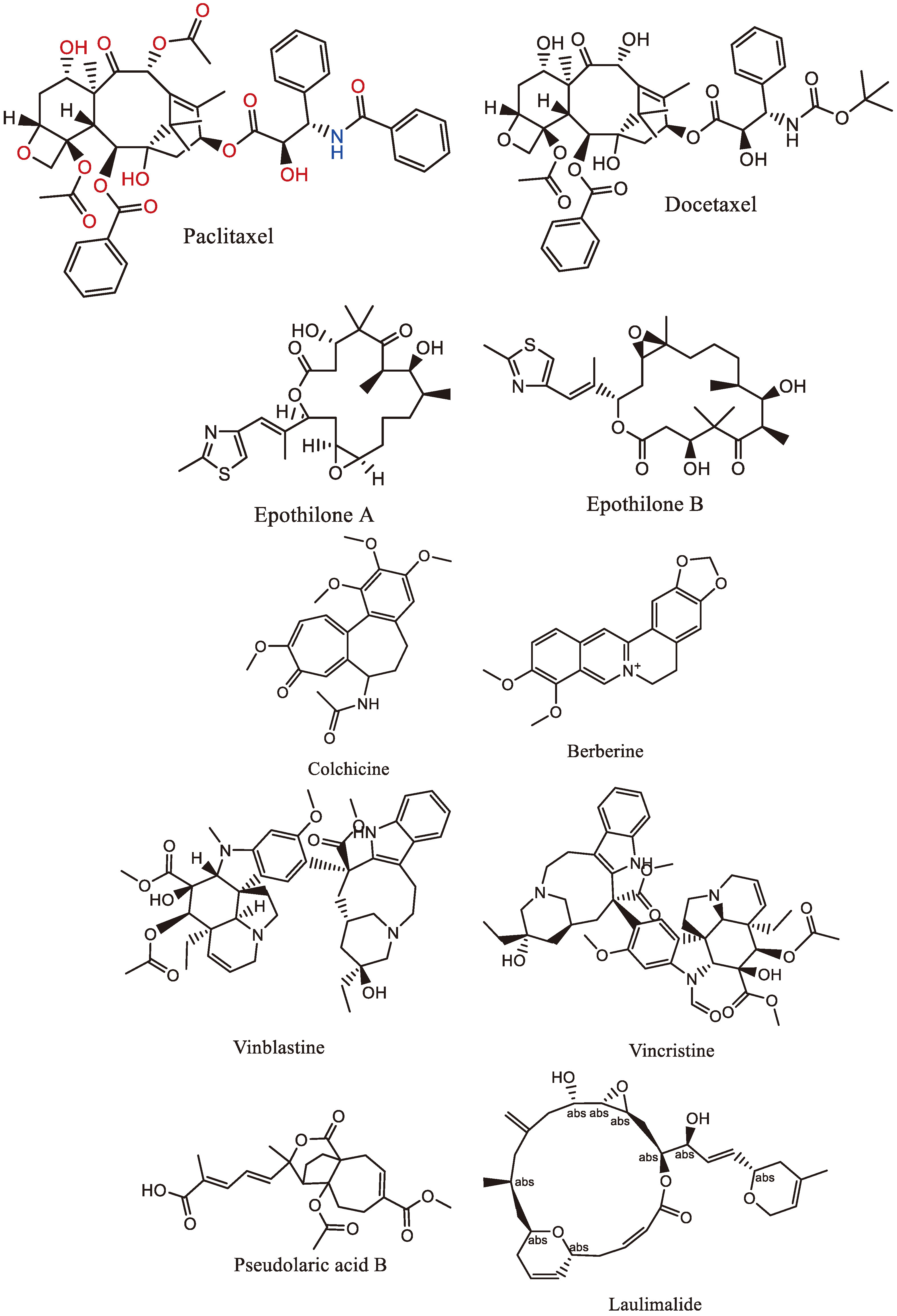
Figure 2.
Chemical structures of representative naturally occurring taxanes [32].
Figure 2.
Chemical structures of representative naturally occurring taxanes [32].
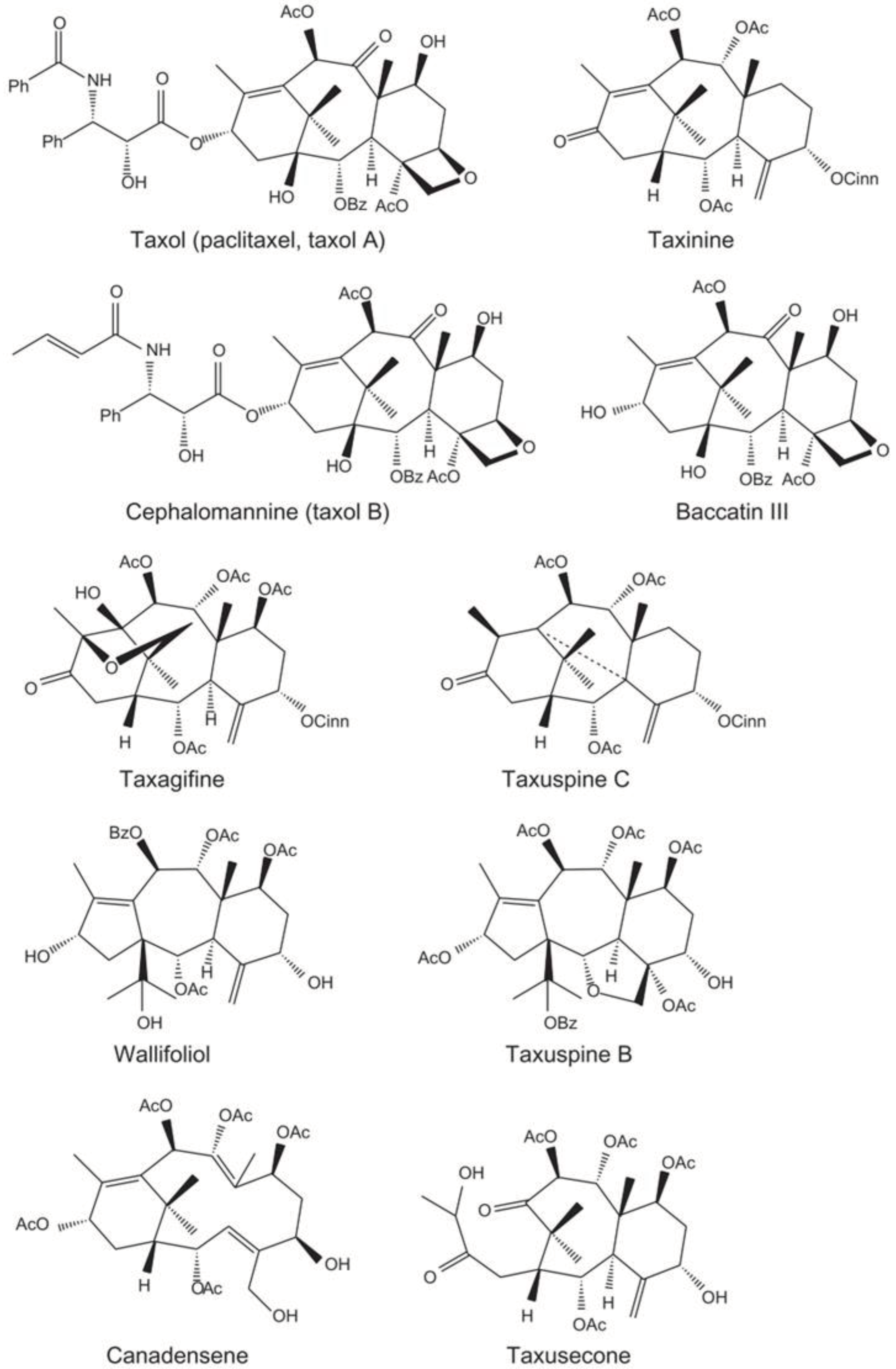
Figure 3.
Chemical structures of mitotic kinases inhibitors.
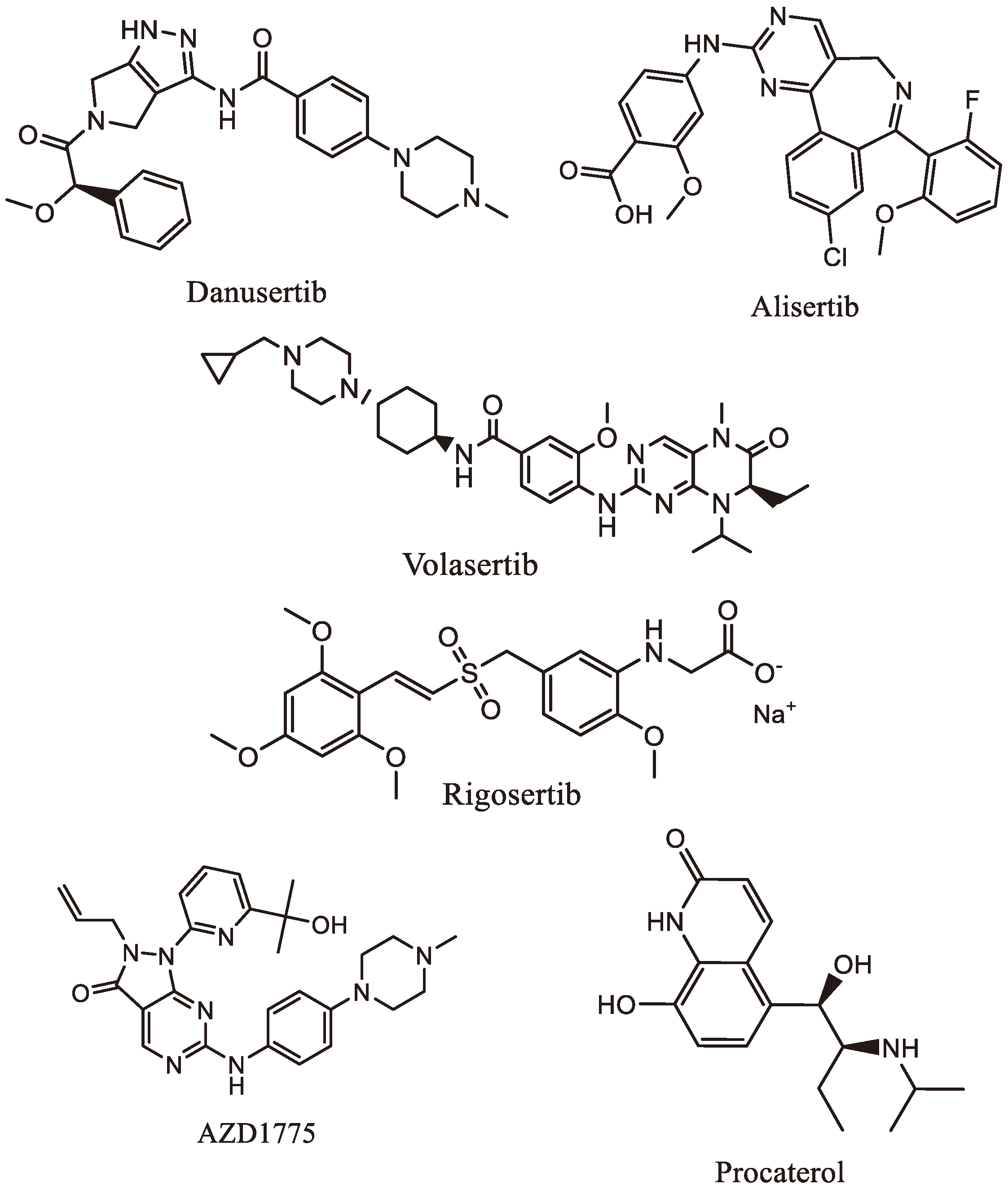
Figure 4.
Mitotic poisons.
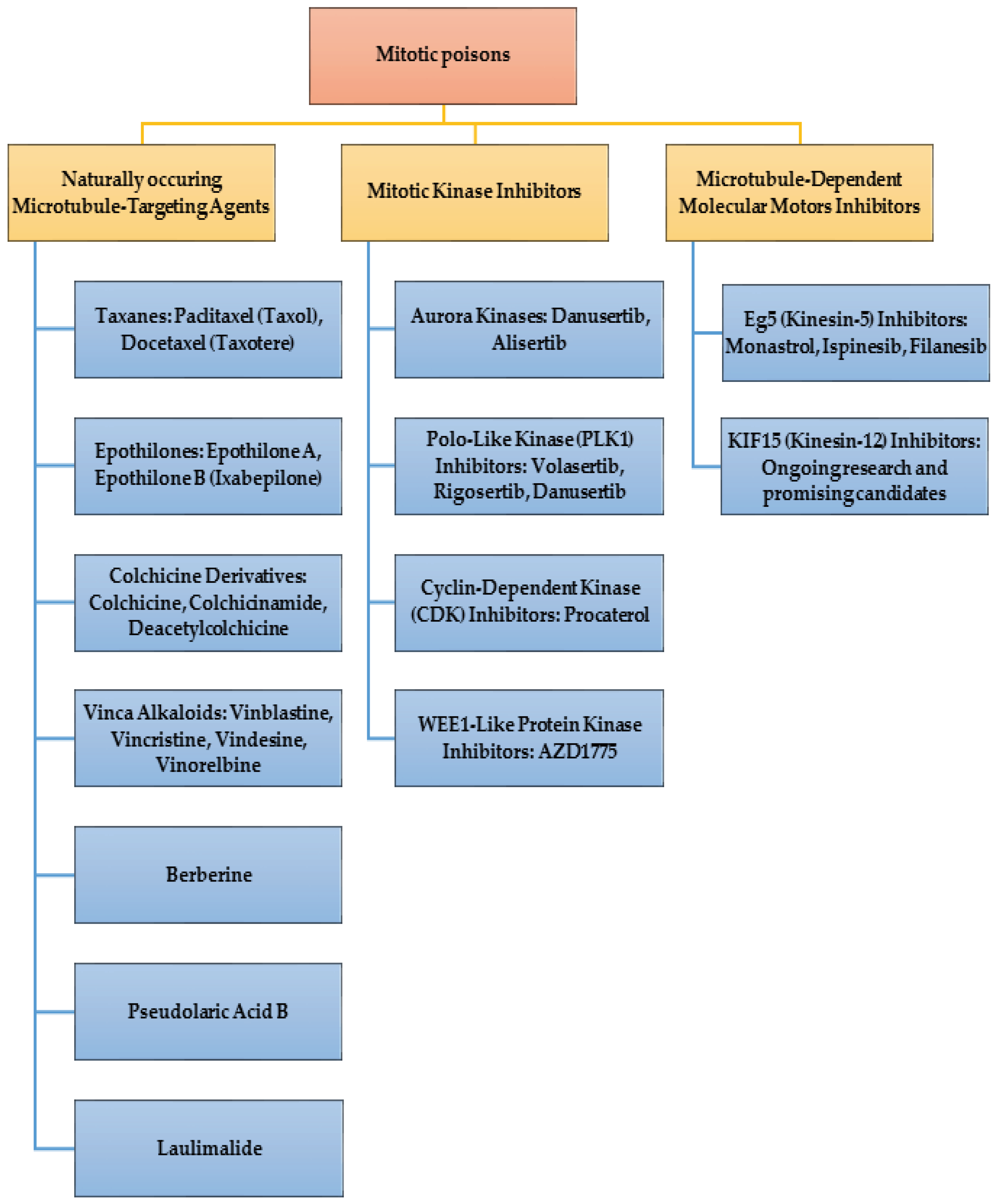

Table 1.
Some naturally occurring mitotic poisons.
| Molecule | Chemical class | Activity/Target | Source | Organism |
|---|---|---|---|---|
| Paclitaxel | Taxanes |
They enhance microtubule polymerization. Binding to tubulin leads to the halt of cell division and triggering the activation of the mitotic spindle checkpoint. They inhibit androgen receptor signaling in prostate cancer. |
Plant | Taxus brevifolia |
| Docetaxel | Semi-synthetic | Taxus baccata | ||
| Epothilone A | Lactones | Microtubule-Targeting Agents, that stabilize microtubules, preventing cancer cells from dividing during mitosis | Bacteria | Sorangium cellulolus |
| Epothilone B | ||||
|
Vinblastine Vincristine Vindesine Vinorelbine |
Alkaloids |
They work by interfering with microtubular activity at low doses and causing cell cycle arrest and apoptosis at higher doses. | Plant | Catharanthus roseus |
| Pseudolaric acid B | Diterpene | Inhibits microtubule polymerization | Plant | Pseudolarix amabilis |
| Laulimalide | Macrolide | Inhibits microtubule polymerization | Marine sponge | Cacospongia mycofijiensis |
| Colchicine | Alkaloid | Prevents DNA synthesis and tubulin polymerization, effectively halting mitosis | Plant |
Colchicum autumnale Gloriosa superba |
Table 2.
Some mitotic kinase inhibitors, their targets and effects.
| Molecule | Target/Activity | Effects |
|---|---|---|
| Danusertib | Aurora Kinase inhibitor |
1. Reduces the survival rate of human Gastric Cancer (GC) cell lines and inhibits their growth. 2. Hinders cell growth and induces cell death in pancreatic cancer. |
| Volasertib | Selective Polo-Like Kinase (PLK1) inhibitor | Induces mitotic arrest and apoptosis in GC patients. |
| AZD1775 |
Polo-Like Kinase (PLK1) inhibitor Wee1-Like Protein Kinase (WEE1) inhibitor |
1. Induces apoptosis and enhancing the inhibition of gastric tumor growth. 2. Sensitizes p53-mutated cells to chemotherapy in pancreatic cancer. |
| Procaterol |
Cyclin-Dependent Kinase (CDK12) inhibitor. β2-receptor agonist. |
Suppresses cell viability and colony formation in GC cell lines and inhibits tumor growth |
| RO-3306 | CDK1 Inhibitors |
Show promise as potential therapeutic options for liver cancer |
| Lycorine | ||
| MLN8237 | Aurora Kinase Inhibitors | |
| SNS-314 | ||
| GSK461364 | PLK1 Inhibitors | |
| Rigosertib | ||
| CCT137690 | Aurora Kinase inhibitor | Hinders cell growth and induces death in pancreatic cancer cells. |
| Dinaciclib | CDK1 Inhibitor | Induces apoptosis and immunogenic cell death in pancreatic cancer. |
| ENMD-2076 | Aurora Kinase inhibitor | Induces G2/M arrest and apoptosis in Colorectal Cancer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



