
Submitted:
02 March 2024
Posted:
05 March 2024
You are already at the latest version
Abstract
Common rust (CR), caused by Puccina sorghi, is one of the major foliar diseases in maize, leading to quality deterioration and yield losses. To dissect the genetic architecture of CR resistance in maize, this study utilized the susceptible temperate inbred line Ye107 as the male parent crossed with three resistant tropical maize inbred lines (CML312, D39, and Y32) to generate 627 F7 recombinant inbred lines (RILs), aiming to identify maize disease-resistant loci and candidate genes for common rust. Phenotypic data exhibited good segregation between resistance and susceptibility, with varying degrees of resistance observed across different subpopulations. Significant genotype effects and genotype × environment interactions were noted with heritability ranging from 85.7% to 92.2%. Linkage analysis and genome-wide association analysis across three environments identified 20 QTLs and 62 significant SNPs. Among them, seven major QTLs explained 66% of the phenotypic variance. Comparison with six SNPs repeatedly identified across different environments revealed overlap between qRUST3-3 and Snp-203,116,453, and Snp-204,202,469. Haplotype analysis indicated two significantly different haplotypes for CR resistance for both of these SNPs. Based on LD decay plots, three co-located candidate genes, namely Zm00001d043536, Zm00001d043566, and Zm00001d043569, were identified within 20 kb up-stream and downstream of these two SNPs. Zm00001d043536 regulates hormone regulation, Zm00001d043566 controls stomatal opening and closure, related to trichome, and Zm00001d043569 is associated with plant disease immune responses. Additionally, we performed candidate gene screening for five additional SNPs repeatedly detected across different environments, resulting in the identification of five candidate genes. These findings contribute to the development of genetic resources for common rust resistance in maize breeding programs.
Keywords:
tropical maize
; multiparent populations
; common rust
; QTL
; GWAS
; candidate genes
1. Introduction
Maize common rust is caused by maize stalk rust Puccinia sorghi Schw during maize growth and development, which is widely distributed in tropical, subtropical and temperate growing areas. It develops easily at 15°C-25°C and 98% humidity; it reduces photosynthesis in leaf area and foliar failure by producing spots on the leaves, resulting in incomplete filling of the kernels and lower yields. Losses due to maize common rust have been reported to range from 12 to 75 per cent [1,2,3,4], and due to ecological and economic problems, breeding resistant plants is the best way to combat the disease and improve yields [5,6].
Breeding for disease resistant varieties begins with the identification of their resistance loci. Previous studies have shown that maize has qualitative and quantitative resistance to common rust [6,7,8]. Early studies were conducted to improve maize resistance by identifying dominant resistance (Rp) genes, but because dominant genes do not possess horizontal resistance, the loss of maize resistance is often accompanied by mutations in a specific P. sorghi race [5,9]. As a result, the focus of research on common rust resistance in maize has shifted to non-specific quantitative resistance.
Multiple studies have successfully identified quantitative trait loci (QTL) for resistance to common rust in temperate maize through linkage mapping, locating QTL on all 10 chromosomes of maize. One study identified a QTL in the 2.05-06 bin interval in a European dent maize population, explaining 25.5% of the phenotypic variance, consistently found across different genetic backgrounds. Another study found a QTL in the 7.01 bin interval, explaining 23.6% of the phenotypic variance, also observed in different genetic combinations. Yet another study discovered a QTL in the 3.04 bin interval (98mb), explaining 20% of the phenotypic variance, which overlapped with previous research. [7,10,11] These findings suggest the possibility of overlapping QTL regions for resistance to common rust in maize with different genetic backgrounds. Enhanced resistance to common rust in maize occurs when multiple partial resistance QTL are combined. Accumulating disease-resistant QTL can enhance plant resistance, underscoring the importance of identifying additional resistance QTL to further enhance maize resistance to common rust. [12,13,14,15,16,17,18].
Compared to linkage analysis, genome-wide association analysis(GWAS) offers higher resolution and is therefore widely utilized in plant breeding studies. However, this method often tends to generate false associations. Thus, to obtain accurate results, it is crucial to eliminate these false associations. Considering population structure is the most effective approach to reduce these false associations.[19,20,21,22,23,24,25,26]. For instance, a GWAS analysis was conducted on resistance to common rust in a population of 274 temperate maize inbred lines. Four SNPs were identified, located on chromosome 2 (59,014,463 bp), chromosome 3 (21,262,214 bp and 56,476,524 bp), and chromosome 8 (107,796,411 bp). Subsequently, four candidate genes (GRMZM2G437912, GRMZM2G031004, GRMZM2G409309, GRMZM2G089308) were selected based on these SNP associations. [27]. When GWAS and linkage analysis are combined, resulting SNP loci fall within QTL regions, often rendering these outcomes more reliable than those obtained by GWAS or linkage analysis alone. Based on the current research findings, the simultaneous application of these two methods can effectively elucidate the genetic mechanism of quantitative traits [28,29].
In recent years, tropical maize germplasm has been used in studies of common rust resistance loci due to its rich genetic variance base [5,9,12].However, utilizing a multi-parental population derived from common temperate parents at high generations (F7) for linkage mapping and genome-wide association analysis (GWAS) in maize common rust resistance has not been previously attempted. Crosses between temperate and tropical lines can maximize genetic diversity, provide clearer genetic backgrounds for each plant, enabling better tracking and understanding of the genetic mechanisms underlying disease susceptibility and resistance. The relative genetic stability of F7 generations can narrow the QTL range, thereby enhancing accuracy. Moreover, previous GWAS studies on this trait have predominantly utilized natural populations, including studies on maize common rust resistance using 296 tropical inbred lines and 282 diverse inbred lines, as well as GWAS analysis on 380 tropical and subtropical inbred lines. These studies have all adjusted GWAS models to account for population structure effects but have not explored the use of constructed populations to mitigate these effects. While natural populations offer time and cost benefits and directly reflect real ecological conditions, issues such as population structure effects, lack of genetic variation control, and environmental noise can compromise GWAS accuracy. This study addresses these challenges through population construction, ensuring more precise results.[30,31,32,33,34,35].
In this study, three tropical inbred parents (CML312, D39, and Y32) showing resistance to maize common rust were crossed with a common temperate susceptible backbone inbred parent, Ye107. A population with three F7 RIL subpopulations was constructed from these crosses. These three subpopulations were phenotyped in a multi-environmental test to assess their response to common rust and genotyped by sequencing (GBS) single nucleotide polymorphisms (SNPs). The main objectives of this study were to identify QTLs and SNP loci significantly associated with common rust in different environments and to screen candidate genes associated with common rust.
2. Results
2.1. Phenotypic Data on Common Rust Resistance in RILs
Three RIL subpopulations were investigated for their response to common rust in three different environments, and phenotypic data were recorded. Descriptive statistics data showed that the co-efficient of variation of common rust disease score in the three RIL populations in the three environments ranged from 29.4% to 49.7%, with a high degree of inter-sample variability (Table 1). The kurtosis and skewness of the common rust phenotypic data ranged from -1 to 1 in absolute values across environments, indicating a small degree of deviation, and that the data were normally distributed. The genotypic variance of the three populations and the genotypic variation due to environmental interactions were statistically significant, P < 0.05. The heritability of common rust disease classes was high in all three populations (85.7%-92.2%), indicating that the trait is more influenced by genotype and less by environment.
Pearson’s correlation analysis at P < 0.001 showed strong correlations (0.773-0.856) between populations in different environments (Figure 1B). The strong correlation indicated that the RILs responded consistently to common rust infection in different environments, which not only indicates the high heritability of the common rust disease reactions, but also reflects the high reliability of this experiment, providing a solid foundation for subsequent QTL/SNP mapping related to common rust resistance in maize.
2.2. QTL Mapping of Common Rust Resistance in Three RIL Populations
To identify QTLs for common rust resistance, we performed QTL mapping of three RIL subpopulations (Pop1, Pop2 and Pop3) in different environments. SNP markers with ≥ 10% deletions and loci with minimum allele frequencies below 5% were excluded from the analysis. The LOD threshold was set at ≥ 3. A total of 20 QTLs (Supplementary Table 1) located on chromosomes 1, 2, 3, 4, 6, and 8 were identified in the three populations. The Best Linear Un- biased Prediction (BLUP) information of three subpopulations also showed the QTLs with significant LOD values given in Figure 2 and Table 2.
The seven QTLs were distributed on chromosomes 2, 3, 4, and 6, and the LOD values ranged from 3.1 to 6.63; the rate of explanation of phenotypic variation (R2) ranged from 8% to 12% (Table 2). Among these QTLs, the effect value of qRUST3-3 had the highest value of 5.39; the phenotypic variation was 11%, and we concluded that this QTL had the potential to become the main QTL for maize resistance to common rust. qRUST6-1 exhibit the highest explained rate of phenotypic variance was 12%, and the additive effects of the three QTLs, qRUST2-1, qRUST3-1, and qRUST3-2, were negative, indicating that these three QTLs could negatively affect maize resistance to common rust.
2.3. SNP Characterization, and Population Structure
The heat map illustrating marker density across the ten maize chromosomes is shown in Figure 3. The numbers of SNPs on chromosomes 1 to 10 were 1,223,552, 65,803, 63,745, 73,660, 56,253, 46,108, 52,458, 48,899, 43,833, and 42,070, respectively. Chromosome 1 had the highest number of SNP markers and chromosome 10 had the lowest (Figure 3A). In the filtered SNP dataset, the average SNP deletion rate was 0.2 and the average minor allele frequency (MAF) was 0.5, which is applicable to subsequent genome-wide association studies (Figure 3B and C). The raw SNP dataset for each RIL population was used for linkage disequilibrium (LD) decay analysis. We calculated the LD decay for all the populations and found that the physical distance was about 20kb when the rate of decline of r2 levelled off (Figure 3D). Thus, 20kb was chosen as the criterion for screening candidates and searching for candidate genes in the 20kb interval upstream and downstream of the significant SNPs.
The phylogenetic tree shows that all the 627 RILs were categorized into three clusters (Figure 4). Overall, the phylogenetic tree, principal component analysis, and correlation heat map revealed that the kinship among the RILs were consistent and the population was divided into three clusters. The number of lines in the three clusters was 180, 223, and 224, respectively. The results of the first two principal component analyses validated the three clusters (Pop1, Pop2, and Pop3) identified by the phylogenetic tree. The small overlap in the center of the principal component analysis plot is due to the presence of the common parent Ye107 in all three populations.
2.4. Genome-Wide Association Analysis of Three RIL Subpopulations
We employed 573,112 SNPs in four different environments for GWAS analysis. SNPs with minor allele frequency (MAF) ≥ 5% and r2 < 0.2 were used to identify significant SNPs. A total of 62 SNPs associated with common rust resistance were identified with -log10(P)>4.5 (Figure 5; Supplementary Table 2). These 62 SNPs were distributed on chromosomes 2, 3, 4, 5, 6, 7, 8, and 10. Seventeen significant SNPs were identified in the BLUP environment, 17 in the 21JH environment, 6 in the 21YS environment, and 22 in the 22YS environment. While only one SNP was distributed on chromosome 8, chromosome 3 had a higher distribution across all the environments.
Six SNPs were consistently identified across the three different environments. These SNPs, along with the 10 candidate genes screened and presented in Table 3. SNPs significantly associated with common rust resistance were detected on chromosomes 3 and 5 in the 21JH, BLUP, and 22YS environments, with the highest p-value of Snp-224,639,688 on chromosome 3 in all three environments, which exceeded to 5.7. Whereas, chromosomes 8 and 10 detected peaks associated with common rust resistance in BLUP, 21JH, and 21YS environments. We screened candidate genes 20kb upstream and downstream of the SNP site based on the results of linkage disequilibrium (LD) attenuation analysis. Among the 10 candidate genes screened, Zm00001d043536, Zm00001d043566, Zm00001d043569, Zm00001d044303, and Zm00001d015778 had functional annotations, while the remaining five candidate genes were not yet annotated.
2.5. Analysis of Consistent Loci Identified by GWAS and QTL Mapping
In order to determine whether the two different approaches could jointly screen for candidate genes that are clearly associated with common rust resistance, we compared the QTL results with the GWAS results. As shown in Table 4, Snp-203,116,453 and Snp-204,202,469 on chromosome 3 were found to fall within qRUST3-3 in GWAS. Then, a search within 20kb upstream and downstream of the above two significant loci revealed three co-located candidate genes (Zm00001d043536, Zm00001d043566 and Zm00001d043569) associated with common rust resistance.
We further analysed Snp-203,116,453 and the candidate gene Zm00001d043536 (Figure 6). The relative positions of the significant SNP locus and the candidate gene with the position of the significant SNP on chromosome 3 identified by GWAS are given in Figure 6A&B. This locus has two haplotypes, CC and TT, and the plants with the CC haplotype have significantly better performance in maize resistance to common rust (Figure 6C). The candidate gene Zm00001d043536 was identified for the first time in maize resistance to common rust, which might be due to the abundant variation in the tropical parents. We then compared the exon base sequences within the gene interval and found that the exon 2 region of the gene had three subversions common to tropical inbred lines and resulted in the transcription of two amino acids that were different from those of the temperate parent (Figure 6D). In RNA-seq expression, the gene was found to be expressed during growth of both young and mature leaves, which demonstrate its association with common rust resistance in maize (Figure 6E). [36].
Similarly, we analyzed another significant locus, Snp-204,202,469, and its candidate genes, Zm00001d043566 and Zm00001d043569, and the results are shown in Figure 7. Figure 7A and 7B show the locations of the significant locus and its candidate genes. We performed haplotype analysis of the locus and found that there were two haplotypes, GG and AA, the plants of the GG haplotype were significantly better in the resistance of maize to common rust (Figure 7C). Our study of the four parents revealed that there were also widespread base substitutions and resulting changes in amino acid translation in two candidate gene segments that differed in the tropical parents from the temperate parents (Figure 7D and F). The candidate gene Zm00001d043566 had the highest expression in the leaf of the plant(Figure 7E), and the candidate gene Zm00001d043569 had increasing expression in the leaf and internode after pollination (Figure 7G), which could be a proof that the two candidate genes are related to the resistance of maize to common rust.
3. Discussion
In this study, three tropical inbred lines were used as maternal parents, and temperate inbred lines were used as the common paternal parent to construct three F7 subpopulations, totaling 627 recombinant inbred lines (RILs). The phenotype analysis revealed a spectrum of susceptibility ranging from highly susceptible to highly resistant across all populations, indicating polygenic resistance in the selected materials. Significant genotype-environment interaction variance in the three populations underscores its importance in maize resistance to common rust. The high heritability observed across the populations can be attributed to the abundant genetic variation resulting from the hybridization of temperate and tropical germplasms.
Recent years, studies aiming to identify resistance loci against common rust disease in tropical maize have discovered new resistance loci and candidate genes, owing to the rich genetic variation present in tropical maize breeding populations. However, these studies have primarily focused on analyzing natural or early-generation populations, lacking investigations into resistance to common rust disease in tropical maize using high-generation populations. This study addresses this gap by employing a different approach through population construction, aiming to enhance the accuracy of linkage analysis and genome-wide association analysis results. [5,9,12]. Constructing high-generation populations offers several advantages compared to natural populations: (1) While natural populations effectively reflect real biodiversity and genetic backgrounds, their complex population structure can compromise the reliability of GWAS analysis. This issue can be addressed through design itself. During the formation of recombinant parents, the mixing of parental alleles can mitigate population structure within each population, thereby reducing the occurrence of false positive associations and enhancing GWAS resolution. (2) Environmental conditions experienced by individuals in natural populations are difficult to replicate. Whereas high-generation population construction allows for better environmental control by conducting multiple replicated experiments to improve phenotype accuracy and subsequently enhance GWAS resolution.
Using temperate material as the common parent, and hybridization with tropical material resulted in a broader genetic base compared to the tropical natural population. Additionally, while constructing high-generation populations may require more time compared to early-generation populations, they undergo more rounds of self-crossing, resulting in more fixed genotypes and narrower intervals of QTLs. For studies of this nature, the identification of genetic loci and the selection of candidate genes lay the groundwork for subsequent gene function validation. Therefore, enhancing the reliability of GWAS results and narrowing the QTL intervals are crucial components of improving precision. The results have disclosed the identification of overlapping QTLs and SNPs with prior studies, along with newly significant loci not previously identified. To enable a clear comparison of these findings, they have been succinctly summarized in Table 5.
Utilizing different populations to identify QTLs within the same genomic interval demonstrates the potential of these QTLs as major-effect QTLs. Previous studies conducted QTL mapping using five F3 early-generation populations, wherein qCR3-113 overlapped with the qRUST3-3 in this study, as detailed in Table 6. The previous study utilized early-generation populations (F3), resulting in a considerably large interval for qCR3-113 (111.4 Mb). In contrast, qRUST3-3, covered spans of the length (34.7 Mb) compared to the previous study, and the overlapping QTL in this study exhibits a higher LOD value (5.39) compared to the previous study (2.85) [5]. For studies aimed at identifying candidate genes, narrower intervals and increased LOD values represent a more accurate QTL, which underscores the advantage of the F7 population. In another study, 296 tropical maize inbred lines were utilized to identify QTLs distributed on chromosomes 1, 3, 5, 6, 8, and 10, among which the QTL on chromosome 6 (Rp 6.1) was found to be close to qRUST6-1 identified in our study, with a distance of 564,094 bp [9].
This study considered eliminating population structure and environmental noise to enhance GWAS resolution in population construction to achieve accuracy in SNP selection. The criteria for SNP selection were based on being "repeatedly screened in different environments" (Table 3). To determine whether the two co-located SNPs (Snp-203,116,453 and Snp-204,202,469) are associated with resistance to common rust disease in maize, haplotype analysis was conducted, indicating that both loci play a significant role in the target trait (Figure 6C and Figure 7C). Additionally, Snp-224,639,688 is 71,788 bp away from the qCR3-113 interval, and Snp-118,608,571 on chromosome 5 overlaps with the QTL qCR5-51 interval. While Kibe et al. also identified QTLs associated with resistance to common rust disease on chromosomes 8 and 10, the SNPs discovered in this study did not overlap with these regions [5]. The candidate gene GRMZM2G060540, which was identified through the screening of S3_147013779 in the study by Kibe et al. is of uncharacterized nature. Whereas, our study suggests three candidate genes (Zm00001d043536, Zm00001d043566, and Zm00001d043569) identified through co-located SNPs could provide a new direction of research on this stable QTL for common rust resistance in maize.
One of the three candidate genes, Zm00001d043536, encodes the heat stress transcription factor c1-b of the HSF family. HSF transcription factors regulate the expression of abscisic acid (ABA)[37], jasmonic acid (JA)[38], indole-3-acetic acid (IAA)[38], and other plant hormones[40], mediating gene activation under heat or other stress conditions to enhance plant stress tolerance. Thirty HSF proteins have been identified in maize. Zm00001d043566 encodes a member of the STICHEL-3 protein family. The STICHEL (STI) gene encodes a protein containing a domain with sequence similarity to the ATP-binding portion of the γ subunit of the DNA polymerase III of true bacteria [41], which has been shown to be associated with the regulation of trichome branching number in Arabidopsis [42]. In maize, this gene’s function has been correlated with the number of trichome branches through gene homology studies with Arabidopsis [43]. The Zm00001d043569 gene encodes the WRKY29 transcription factor, which has been shown in Arabidopsis thaliana to regulate ethylene biosynthesis and response [44]. Previous research indicates that excessive immune responses in plants can adversely affect growth and development. Plant-induced ethylene synthesis acts as a negative regulator of immune responses, alleviating their impact on plants [44].Furthermore, in GWAS, two annotated genes, Zm00001d044303 and Zm00001d015778, were identified near significant SNPs. Zm00001d044303, located near Snp-224,639,688 on chromosome 3, encodes a myosin protein crucial for cytokinesis and intracellular movement. Zm00001d015778, near Snp-118,608,571 on chromosome 5, encodes leucine repeats associated with plant innate immunity. These genes overlap with previously reported QTLs for common rust resistance [5].
In addition, among the genes screened in GWAS, two annotated genes Zm00001d044303 localized near Snp-224,639,688 was mapped on chromosome 3 in GWAS analysis. Snp-224,639,688 is only 0.71 Mb away from the QTL qCR3-113 reported by Kibe et al [5]. Zm00001d044303 encodes a myosin protein, which plays an important role in cytokinesis and intracellular movement, and is thus crucial for cell division and intracellular movement. Zm00001d015778, located near the Snp-118,608,571 on chromosome 5 overlapped with QTL qCR5-51 reported by Kibe et al. [5]. Zm0001d015778 encodes a segment of leucine repeats associated with innate immunity in plants. Innate immunity is the first line of defense against pathogen-associated molecular patterns (PAMPs) [46]. Since Kibe only reported QTLs and did not annotate the genes, the above three genes could be considered candidate genes. These candidate genes can serve as a valuable reference for genetic studies of common rust resistance in maize [5].
The tropical maize germplasm has long been recognized for its wide range of disease resistance and has been extensively utilized in the breeding of disease-resistant maize varieties. Historically, maize breeders introgressed the disease resistance genes from these tropical inbred lines to improve resistance in temperate germplasms. Ye107, a backbone inbred line of temperate origin, has played a crucial role in producing key corn varieties such as Yunrui8. CML312, selected from CIMMTY, is a high-quality tropical inbred line that has contributed to the development of disease-resistant hybrids such as’Yunrui2’. D39 is an excellent tropical inbred line which produced the hybrid 'Dedan5' which was inoculated and identified as highly resistant to rust (disease class 1) by the College of Plant Protection of Anhui Agricultural University (AAU). Y32, a high-quality inbred line selected from the classic tropical germplasm Suwan, has been instrumental in breeding a high-quality, stress-resistant hybrid, ‘Yunrui1’. However, as common rust resistance is a quantitative traits controlled by multiple minor genes, it is challenging for maize breeders to introgress minor resistance genes from tropical germplasms into the target maize germplasm. Many of these minor genes provide partial resistance and may not be sufficient to confer durable resistance, necessitating the introgression of multiple minor resistance loci. Nevertheless, advances in molecular marker techniques have refined the selection process for disease resistance. Hence, researchers can identify additional candidate loci associated with common rust resistance. The present study has certainly laid a strong foundation for developing common rust-resistant inbred lines and hybrids in maize.
4. Conclusions
In this study, we employed three F7 populations derived from 3 tropical inbreds crossed with common temperate parent, comprising 627 recombinant inbred lines, to elucidate the genetic architecture of maize resistance to common rust. Linkage mapping identified 20 QTLs among them 7 major QTLs explaining 66% of the phenotypic variance. GWAS was used to screen for SNPs that repeatedly appeared across six different environments and compared with QTL results. One QTL on chromosome 3 (qRUST3-3) overlapped with two of these SNPs. We conducted haplotype analysis on these two SNPs, revealing that one haplotype for each SNP exhibited significantly lower rust resistance phenotypes compared to the other haplotype. Using these two SNPs, we identified three candidate genes (Zm00001d043536, Zm00001d043566, Zm00001d043569). Our results should help elucidate the mechanism involved common rust resistance in future studies.
5. Materials and Methods
5.1. Experimental Materials and Field Experiment Design
In this study, the tropical maize inbred lines Y32, CML312, and D39 were used as female parents, and the temperate inbred line Ye107 was used as the male parent for hybridization, all three inbreds showed resistance to common rust. The F1 plants were self-pollinated for six generations through single seed descent method, and three RIL subpopulations were developed: Pop1 (CML312×Ye107), Pop2 (D39×Ye102) and Pop3 (Y32×Ye107) (Table 7). The three subpopulations pop1, pop2 and pop3 consisted of 180, 223 and 224 RILs, respectively, totalling 627 RILs. All RILs were planted in 2021 in Jinghong City (JH), Yunnan Province, and Yanshan County (YS), Wenshan Prefecture, Yunnan Province. In 2022, the experiment was replicated in YS County, Wenshan Prefecture, Yunnan Province. A randomized complete block design was used for the experiments. Each experimental plot was composed of a 4-meter-long row, with a row spacing of 0.70 meters and a plant spacing of 25 cm [47]. The trials were managed according to standard agronomic practices (Figure 8).
5.2. Common Rust Disease Evaluation
Three populations were evaluated for common rust response under sustained high natural disease pressure starting at week 2 after maize dispersal, and maize common rust incidence levels were surveyed at 7-day intervals for three times. Resistance scores were determined for each RIL based on the percentage of infected area to total area, and the scores were based on specific criteria listed in Table 8 [47].
5.3. Phenotypic Data Analysis
After preliminary processing of the phenotypic data from the three RIL subpopulations collected at two locations over two years, SPSS Statistics was used to analyze the data. First, descriptive statistical analyses, including mean, standard deviation, variance, skewness, kurtosis, and coefficient of variation, were calculated. Using SPSS Statistics. Kurtosis and skewness estimates confirms the frequency distribution is a normal distribution. Broad-spectrum heritability was calculated using the following method [48,49]:
where is the genetic variance, σge2 is the variance due to the environment × genotype interactions, σε2 is the residual, e represents the number of environments or locations, and r represents the number of replications per location. H2 identifies the degree of variation in a phenotypic trait; the more significant H2, the more the trait is influenced by the genotype and the less influenced by the environment.
Estimation of breeding values: The BLUP values for each trait in all environments were obtained for each inbred line using a linear mixed model in R (v.3.6.1) (http://www.r-project.org/) with the lme4 package. The formulae used for calculating BLUP values are given below [49]:
Where Yijk is the observed value of the jth repetition of the kth genotype in the ith environment, µ is the overall mean, Gk is the effect of the kth genotype, Ei is the effect of the ith environment, Rj(i) is the effect of the jth repetition nested in the ith environment, EGik is the effect of the interaction between the ith environment and the kth genotype, and εijk is the effect of experimental error.
Violin maps and correlation heat maps are plotted at the hiplot website (https://hiplot.com.cn/home/index.html).
5.4. DNA Extraction and Genotyping-by-Sequencing (GBS)
Genomic DNA was extracted from young maize leaves of three RIL subpopulations using the cetyltrimethylammonium bromide (CTAB) method [49]. The DNA was digested using restriction endonucleases Mspl and Pstl (New England Biolabs, Ipswich, MS, USA) and then ligated with a barcode adapter using T4 ligase (New England Biolabs). All ligated samples were mixed and purified using the QIAquick PCR Purification Kit (QIAGEN, Valencia, CA, USA). Primers complementary to the two adapters were used for PCR amplification. PCR products were then purified and quantified using the Qubit dsDNA HS Assay Kit (Life Technologies, Grand Island, NY, USA). PCR products between 200-300 bp in size were selected using the Egel system (Life Technologies, USA), and library concentrations were estimated using the Qubit 2.0 fluorometer and the Qubit dsDNA HS assay kit (Life Technologies, USA). GBS libraries were constructed, and sequencing was conducted based on the GBS protocol [51]. Sequencing was performed using an Ion Proton sequencer (Life Technologies, software version 5.10.1) and a P1v3 chip. Final reads were generated using TASSEL v5.0 (https://github.com/Euphrasiologist/GBS_V2_Tassel5, accessed 23 July 2023) [52]. Prior to TASSEL analysis, 80 polyadenylates (poly (A) bases) were appended to 30 ends of all sequencing reads [53]. Using the Genome Analysis Toolkit software, SNPs were identified by aligning to the maize reference genome, B73 (B73_V4, ftp://ftp.ensemblgenomes.org/pub/plants/release-37/fasta/zea_mays/DNA, accessed on 23 July 2023) [54]. A total of 573,112 high-quality SNPs were annotated using the ANNOVAR software tool (v2013-05-20) [55]. After mapping the filtered raw reads with the maize reference genome B73 (RefGen_v4) to discover SNPs, the filtering parameter for SNP data was set to MAF ≥ 0.05 to identify the high-quality SNPs.
5.5. QTL Mapping
We phenotyped the three populations and determined the best results based on whether the SNPs and QTLs overlapped in physical location, then carefully selected the populations and phenotypes with the best results and displayed the results on a graph. Pop2 (D39×Ye107) appeared to have an overlap of SNPs and QTLs. The allelic SNPs were then used to construct a genetic linkage map by JoinMap4 software[56]. Linkage groups were formed using a LOD threshold of ≥5.0. QTLs for CR were identified using the Composite Interval Mapping (CIM) method via Windows QTL Mapper v2.0 [57]. The LOD threshold was set based on 1000 random permutation tests with a significance level of p≤0.05 [58]. The results showed that QTL with LOD thresholds ≥3 were considered significant. The percentage of phenotypic variation (PVE) explained by a single QTL was calculated by the square of the partial correlation coefficient (R2).
5.6. Structure Analysis
We used Tassel 5.0 for phylogenetic tree analysis. R4.2.1 for principal component analysis and correlation heatmap plotting, whereas the principal component analysis was plotted with the scatterplot package; and the correlation heatmap was plotted with the GAPIT package.
PopLDdecay 3.40 software and perl scripts were used to assess linkage disequilibrium (LD) to determine the number of markers required for GWAS, detection efficiency and accuracy.The LD decay figure was drawn with default parameter.
5.7. Haplotype Analysis
We used Haploview v4.2 software for haplotype analysis of SNP loci. Box line plots were drawn using Origin2022.
5.8. Genome Wide Association Study
After Illumina NovaSeq6000 sequencing, BAM files were processed, and then GWAS analysis was performed using the mixed linear model (MLM) implemented in GEMMA software (https://www.xzlab.org/software.html, accessed on 26 August 2023). Parameters were set to plink-indep-pairwise 5050.2, -log10(P) > 4.5 [59], and MLM was used to adjust for population structure and individual relatedness. The threshold was set to 0.05, and Manhattan and QQ plots were generated [60].
5.9. Identification and Functional Annotation of Candidate Genes
Maize GDB (https://www.maizegdb.org/gbrowse, accessed 26 August 2023) and Maize Reference Genome B73 (RefGen_v4) were used to search for genes associated with common rust resistance in maize. The screened genes were considered as candidate genes involved in common rust resistance in maize and then the functions of the screened candidate genes on Maize GDB were annotated and compared using NCBI (https://www.ncbi.nlm.nih.gov/, accessed on 26 August 2023).
5.10. Candidate Gene Expression Analysis
We performed a query on a public database (http://ipf.sustech.edu.cn/pub/zmrna/) to retrieve information related to the expression of three co-localised candidate genes identified by association analysis. We obtained FPKM values for the candidate genes under the influence of maize common rust stress.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table 1. QTLs located in three RIL populations; Supplementary table2.Common rust significant SNP set.
Author Contributions
Conceptualization, X.F; data curation, J.L., M.L., X.Z., and S.L.; writing—original draft preparation, L.L.; writing—review and editing, F.J., B.Y., X.Y., R.K.S, B.I.; funding acquisition, X.F; All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by the National Natural Science Foundation of China (U2202204). The Yunnan Provincial Science and Technology Talent and Platform Program (202205AF150028).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank the authors for their help in the trial. We also thank the editors and reviewers for their valuable comments and time.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hooker, A.L. 7 - Corn and Sorghum Rusts. In Diseases, Distribution, Epidemiology, and Control; Roelfs, A.P., Bushnell, W.R., Eds.; Academic Press, 1985, pp. 207-236, ISBN 978-0-12-148402-6.
- Pataky, J.K.; Tracy, W.F. Widespread Occurrence of Common Rust, Caused by Puccinia sorghi, on Rp-Resistant Sweet Corn in the Midwestern United States. Plant Dis. 1999, 83, 1177. [Google Scholar] [CrossRef]
- Dey, U.K.; Harlapur, S.I.; Dhutraj, D.N.; Suryawanshi, A.P.; Badgujar, S.L.; Jagtap, G.P.; Kuldhar, D.P. Spatiotemporal yield loss assessment in corn due to common rust caused by Puccinia sorghi Schw. African Journal of Agricultural Research 2012, 7, 5265–5269. [Google Scholar] [CrossRef]
- Dey, U.K.; Harlapur, S.I.; Dhutraj, D.N.; Suryawanshi, A.P.; Bhattacharjee, R. Integrated disease management strategy of common rust of maize incited by Puccinia sorghi Schw. African Journal of Microbiology Research 2015, 9, 1345–1351. [Google Scholar] [CrossRef]
- Kibe, M.; Nyaga, C.; Nair, S.K.; Beyene, Y.; Das, B.; M, S.L.; Bright, J.M.; Makumbi, D.; Kinyua, J.; Olsen, M.S.; et al. Combination of Linkage Mapping, GWAS, and GP to Dissect the Genetic Basis of Common Rust Resistance in Tropical Maize Germplasm. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Delaney, D.E.; Webb, C.A.; Hulbert, S.H. A Novel Rust Resistance Gene in Maize Showing Overdominance. Molecular Plant-Microbe Interactions® 1998, 11, 242–245. [Google Scholar] [CrossRef]
- Kerns, M.R.; Dudley, J.W.; Rufener, G.K. QTL for resistance to common rust and smut in maize. Maydica 1999, 44, 37–45. [Google Scholar]
- Wisser, R.J.; Balint-Kurti, P.J.; Nelson, R.J. The genetic architecture of disease resistance in maize: a synthesis of published studies. Phytopathology 2006, 96, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, J.; Mu, C.; Makumbi, D.; Xu, Y.; Mahuku, G. Combined linkage and association mapping reveal QTL for host plant resistance to common rust (Puccinia sorghi) in tropical maize. Bmc Plant Biol. 2018, 18, 310. [Google Scholar] [CrossRef]
- Lubberstedt, T.; Klein, D.; Melchinger, A.E. Comparative Quantitative Trait Loci Mapping of Partial Resistance to Puccinia sorghi Across Four Populations of European Flint Maize. Phytopathology 1998, 88, 1324–1329. [Google Scholar] [CrossRef]
- Brown, A.F.; Juvik, J.A.; Pataky, J.K. Quantitative Trait Loci in Sweet Corn Associated with Partial Resistance to Stewart's Wilt, Northern Corn Leaf Blight, and Common Rust. Phytopathology 2001, 91, 293–300. [Google Scholar] [CrossRef]
- Ren, J.; Li, Z.; Wu, P.; Zhang, A.; Liu, Y.; Hu, G.; Cao, S.; Qu, J.; Dhliwayo, T.; Zheng, H.; et al. Genetic Dissection of Quantitative Resistance to Common Rust (Puccinia sorghi) in Tropical Maize (Zea mays L.) by Combined Genome-Wide Association Study, Linkage Mapping, and Genomic Prediction. Front. Plant Sci. 2021, 12, 692205. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xu, D.; Dong, Y.; Li, F.; Bian, Y.; Li, L.; Luo, X.; Fei, S.; Li, L.; Zhao, C.; et al. Fine mapping and characterization of a major QTL for grain weight on wheat chromosome arm 5DL. Theor. Appl. Genet. 2022, 135, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, X.; Zhang, Y.; Jin, Y.; Xia, Z.; Xiang, M.; Huang, S.; Qiao, L.; Zheng, W.; Zeng, Q.; et al. Enhanced stripe rust resistance obtained by combining Yr30 with a widely dispersed, consistent QTL on chromosome arm 4BL. Theor. Appl. Genet. 2022, 135, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Li, H.; Liu, E.; He, K.; Song, Y.; Dong, C.; Wang, Z.; Zhang, X.; Zhou, Z.; Xu, Y.; et al. Evaluation and Identification of Resistance Lines and QTLs of Maize to Seedborne Fusarium verticillioides. Plant Dis. 2022, 106, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Johnmark, O.; Indieka, S.; Liu, G.; Gowda, M.; Suresh, L.M.; Zhang, W.; Gao, X. Fighting Death for Living: Recent Advances in Molecular and Genetic Mechanisms Underlying Maize Lethal Necrosis Disease Resistance. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Warburton, M.L.; Jeffers, D.; Smith, J.S.; Scapim, C.; Uhdre, R.; Thrash, A.; Williams, W.P. Comparative Analysis of Multiple GWAS Results Identifies Metabolic Pathways Associated with Resistance to A. flavus Infection and Aflatoxin Accumulation in Maize. Toxins 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Galiano-Carneiro, A.L.; Kessel, B.; Presterl, T.; Miedaner, T. Intercontinental trials reveal stable QTL for Northern corn leaf blight resistance in Europe and in Brazil. Theor. Appl. Genet. 2021, 134, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Shikha, K.; Shahi, J.P.; Vinayan, M.T.; Zaidi, P.H.; Singh, A.K.; Sinha, B. Genome-wide association mapping in maize: status and prospects. 3 Biotech 2021, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.M.; Lor, V.S.; Zhang, X.; Saengwilai, P.; Hanlon, M.T.; Klein, S.P.; Davis, J.L.; Borkar, A.N.; Depew, C.L.; Bennett, M.J.; et al. Transcription factor bHLH121 regulates root cortical aerenchyma formation in maize. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2075299176. [Google Scholar] [CrossRef]
- Qian, F.; Jing, J.; Zhang, Z.; Chen, S.; Sang, Z.; Li, W. GWAS and Meta-QTL Analysis of Yield-Related Ear Traits in Maize. Plants 2023, 12. [Google Scholar] [CrossRef]
- Yang, W.; Liu, X.; Yu, S.; Liu, J.; Jiang, L.; Lu, X.; Liu, Y.; Zhang, J.; Li, X.; Zhang, S. The maize ATP-binding cassette (ABC) transporter ZmMRPA6 confers cold and salt stress tolerance in plants. Plant Cell Rep. 2023, 43, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Xue, M.; Qin, F.; He, Y.; Zhou, Y. Natural polymorphisms in ZmIRX15A affect water-use efficiency by modulating stomatal density in maize. Plant Biotechnol. J. 2023, 21, 2560–2573. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, W.; Le L; Yu, J. ; Wu, Y.; Li, D.; Wang, Y.; Wang, H.; Lu, X.; Qiao, H.; et al. Integration of high-throughput phenotyping, GWAS, and predictive models reveals the genetic architecture of plant height in maize. Mol. Plant. 2023, 16, 354–373. [Google Scholar] [CrossRef]
- Okunlola, G.; Badu-Apraku, B.; Ariyo, O.; Agre, P.; Offernedo, Q.; Ayo-Vaughan, M. Genome-wide association studies of Striga resistance in extra-early maturing quality protein maize inbred lines. G3-Genes Genomes Genet. 2023, 13. [Google Scholar] [CrossRef]
- Zheng, Y.; Yuan, F.; Huang, Y.; Zhao, Y.; Jia, X.; Zhu, L.; Guo, J. Genome-wide association studies of grain quality traits in maize. Sci. Rep. 2021, 11, 9797. [Google Scholar] [CrossRef]
- Olukolu, B.A.; Tracy, W.F.; Wisser, R.; De Vries, B.; Balint-Kurti, P.J. A Genome-Wide Association Study for Partial Resistance to Maize Common Rust. Phytopathology 2016, 106, 745–751. [Google Scholar] [CrossRef]
- Yan, J.; Warburton, M.L.; Crouch, J.H. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Sci. 2011, 51, 433–449. [Google Scholar] [CrossRef]
- Cao, S.; Loladze, A.; Yuan, Y.; Wu, Y.; Zhang, A.; Chen, J.; Huestis, G.; Cao, J.; Chaikam, V.; Olsen, M.; et al. Genome-Wide Analysis of Tar Spot Complex Resistance in Maize Using Genotyping-by-Sequencing SNPs and Whole-Genome Prediction. Plant Genome 2017, 10. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; Mcmullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; Mcmullen, M.D.; Ware, D.; et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar] [CrossRef]
- Zhang, N.; Gibon, Y.; Wallace, J.G.; Lepak, N.; Li, P.; Dedow, L.; Chen, C.; So, Y.S.; Kremling, K.; Bradbury, P.J.; et al. Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol. 2015, 168, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.G.; Zhang, X.; Beyene, Y.; Semagn, K.; Olsen, M.; Prasanna, B.M.; Buckler, E.S. Genome-wide Association for Plant Height and Flowering Time across 15 Tropical Maize Populations under Managed Drought Stress and Well-Watered Conditions in Sub-Saharan Africa. Crop Sci. 2016, 56, 2365–2378. [Google Scholar] [CrossRef]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; Mcmullen, M.D.; Ware, D.; et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, L.; Li, Z.; Bi, Y.; Yin, X.; Guo, R.; Wang, J.; Zhang, Y.; Shaw, R.K.; Fan, X. Identification of Candidate QTLs and Genes for Ear Diameter by Multi-Parent Population in Maize. Genes 2023, 14. [Google Scholar] [CrossRef]
- Walley, J.W.; Sartor, R.C.; Shen, Z.; Schmitz, R.J.; Wu, K.J.; Urich, M.A.; Nery, J.R.; Smith, L.G.; Schnable, J.C.; Ecker, J.R.; et al. Integration of omic networks in a developmental atlas of maize. Science 2016, 353, 814–818. [Google Scholar] [CrossRef]
- Krattinger, S.G.; Kang, J.; Braunlich, S.; Boni, R.; Chauhan, H.; Selter, L.L.; Robinson, M.D.; Schmid, M.W.; Wiederhold, E.; Hensel, G.; et al. Abscisic acid is a substrate of the ABC transporter encoded by the durable wheat disease resistance gene Lr34. New Phytol. 2019, 223, 853–866. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, C.; Ren, X.; Li, Z.; Liu, C.; Qiao, X.; Shen, S.; Zhang, F.; Wan, F.; Liu, B.; et al. Inhibition of invasive plant Mikania micrantha rapid growth by host-specific rust (Puccinia spegazzinii). Plant Physiologyplant Physiology 2023, 192, 1204–1220. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Park, J.J.; Gang, D.R.; Hulbert, S.H. Characterization of a tryptophan 2-monooxygenase gene from Puccinia graminis f. sp. tritici involved in auxin biosynthesis and rust pathogenicity. Mol. Plant. Microbe. Interact. 2014, 27, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Rizwan, M.; Arif, M.S.; Ahmad, R.; Hasanuzzaman, M.; Ali, B.; Hussain, A. Approaches in Enhancing Thermotolerance in Plants: An Updated Review. J. Plant Growth Regul. 2020, 39, 456–480. [Google Scholar] [CrossRef]
- Ilgenfritz, H.; Bouyer, D.; Schnittger, A.; Mathur, J.; Kirik, V.; Schwab, B.; Chua, N.H.; Jurgens, G.; Hulskamp, M. The Arabidopsis STICHEL gene is a regulator of trichome branch number and encodes a novel protein. Plant Physiol. 2003, 131, 643–655. [Google Scholar] [CrossRef]
- Xi, A.; Yang, X.; Deng, M.; Chen, Y.; Shao, J.; Zhao, J.; An, L. Isolation and identification of two new alleles of STICHEL in Arabidopsis. Biochem. Biophys. Res. Commun. 2018, 499, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Pan, X.; Jing, Y.; Zhao, Y.; Duan, Y.; Yang, J.; Wang, B.; Liu, Y.; Shen, R.; Cao, Y.; et al. ZmSPL10/14/26 are required for epidermal hair cell fate specification on maize leaf. New Phytol. 2021, 230, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Lin, Q.; Lan, J.; Zhang, T.; Liu, X.; Miao, R.; Mou, C.; Nguyen, T.; Wang, J.; Zhang, X.; et al. WRKY Transcription Factor OsWRKY29 Represses Seed Dormancy in Rice by Weakening Abscisic Acid Response. Front. Plant Sci. 2020, 11, 691. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meng, H.; Tang, Y.; Zhang, Y.; He, Y.; Zhou, J.; Meng, X. Phosphorylation of an ethylene response factor by MPK3/MPK6 mediates negative feedback regulation of pathogen-induced ethylene biosynthesis in Arabidopsis. J. Genet. Genomics 2022, 49, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Dievart, A.; Gottin, C.; Perin, C.; Ranwez, V.; Chantret, N. Origin and Diversity of Plant Receptor-Like Kinases. Annu. Rev. Plant Biol. 2020, 71, 131–156. [Google Scholar] [CrossRef]
- Wang. Lecture Series on Knowledge of Maize Pests and Diseases (Ⅲ) Maize Pest and Disease Resistance Identification and Investigation Techniques. Crop J. 2005, 53–55.
- Knapp, S.J.; Ross, W.M.; Stroup, W.W. Exact Confidence Intervals for Heritability on a Progeny Mean Basis1. Crop Sci. 1983, 25, 192–194. [Google Scholar] [CrossRef]
- Alvarado, G.; Rodríguez, F.M.; Pacheco, A.; Burgueño, J.; Crossa, J.; Vargas, M.; Pérez-Rodríguez, P.; Lopez-Cruz, M.A. META-R: A software to analyze data from multi-environment plant breeding trials. The Crop Journal 2020, 8, 745–756. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic. Acids. Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. Plos One 2012, 7, e32253. [Google Scholar] [CrossRef]
- Li, C.; Guan, H.; Jing, X.; Li, Y.; Wang, B.; Li, Y.; Liu, X.; Zhang, D.; Liu, C.; Xie, X.; et al. Genomic insights into historical improvement of heterotic groups during modern hybrid maize breeding. Nat. Plants 2022, 8, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic. Acids. Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Van, J.W.; Wageningen, O.; B V, K. Software for the calculation of genetic linkage maps. In 2001.
- Zeng, Z.B. Precision mapping of quantitative trait loci. Genetics 1994, 136, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Vogt, F.; Shirsekar, G.; Weigel, D. vcf2gwas: Python API for comprehensive GWAS analysis using GEMMA. Bioinformatics 2022, 38, 839–840. [Google Scholar] [CrossRef]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef]
Figure 1.
Distribution of rust traits in three populations and their correlation.(A) Violin plots of the phenotypic distribution of the three populations(B) Heat map of correlation between three groups in three environments.
Figure 1.
Distribution of rust traits in three populations and their correlation.(A) Violin plots of the phenotypic distribution of the three populations(B) Heat map of correlation between three groups in three environments.
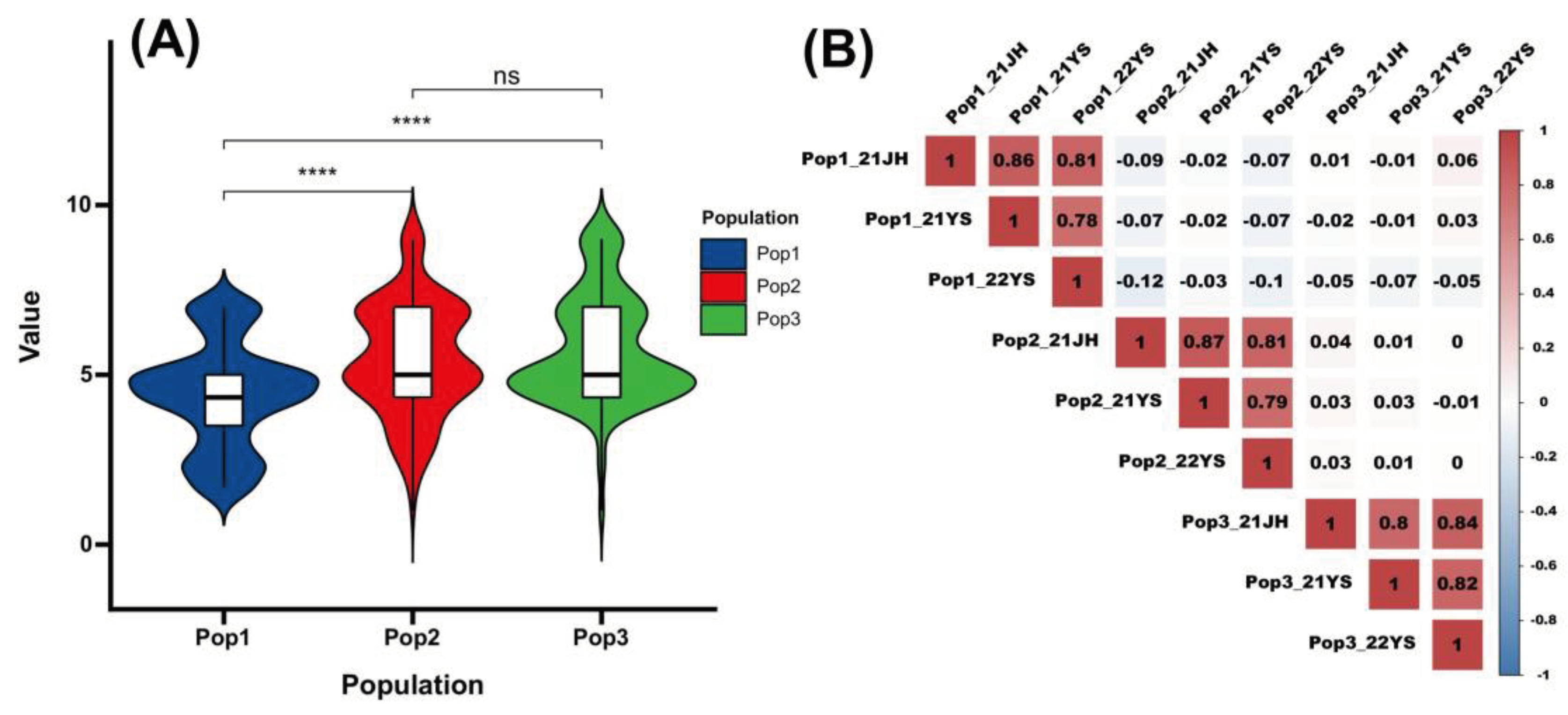
Figure 2.
Three populations Logarithm-of-odds (LOD) profiles in the BLUP environment. (A) Log-of-ood (LOD) profiles of Pop1 (CML312×Ye107); (B) Log-of-ood (LOD) profiles of Pop2 (D39×Ye107); (C) Log-of-ood (LOD) profiles of Pop3 (Y32×Ye107).
Figure 2.
Three populations Logarithm-of-odds (LOD) profiles in the BLUP environment. (A) Log-of-ood (LOD) profiles of Pop1 (CML312×Ye107); (B) Log-of-ood (LOD) profiles of Pop2 (D39×Ye107); (C) Log-of-ood (LOD) profiles of Pop3 (Y32×Ye107).
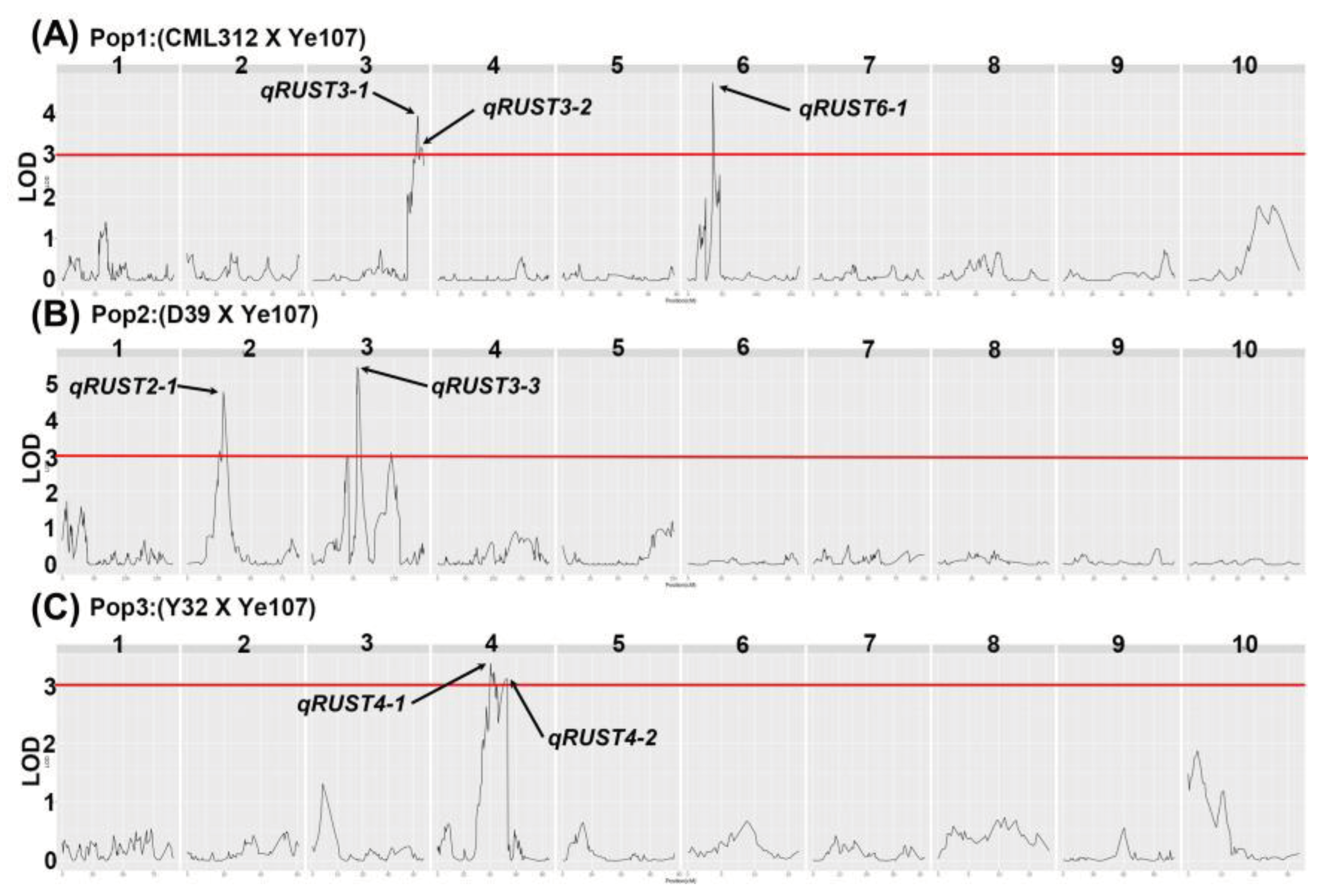
Figure 3.
Phenotypic diversity in the atlas (A) Density of chromosome-specific SNPs in the 0.1 Mb genomic interval. The number of SNPs is indicated on a green to red scale. (B) Distribution of minor allele frequency of the SNPs. (C) Distribution of the frequency of missing genotypes. (D) Whole-genome LD in the entire panel based on 627 maize RILs.
Figure 3.
Phenotypic diversity in the atlas (A) Density of chromosome-specific SNPs in the 0.1 Mb genomic interval. The number of SNPs is indicated on a green to red scale. (B) Distribution of minor allele frequency of the SNPs. (C) Distribution of the frequency of missing genotypes. (D) Whole-genome LD in the entire panel based on 627 maize RILs.
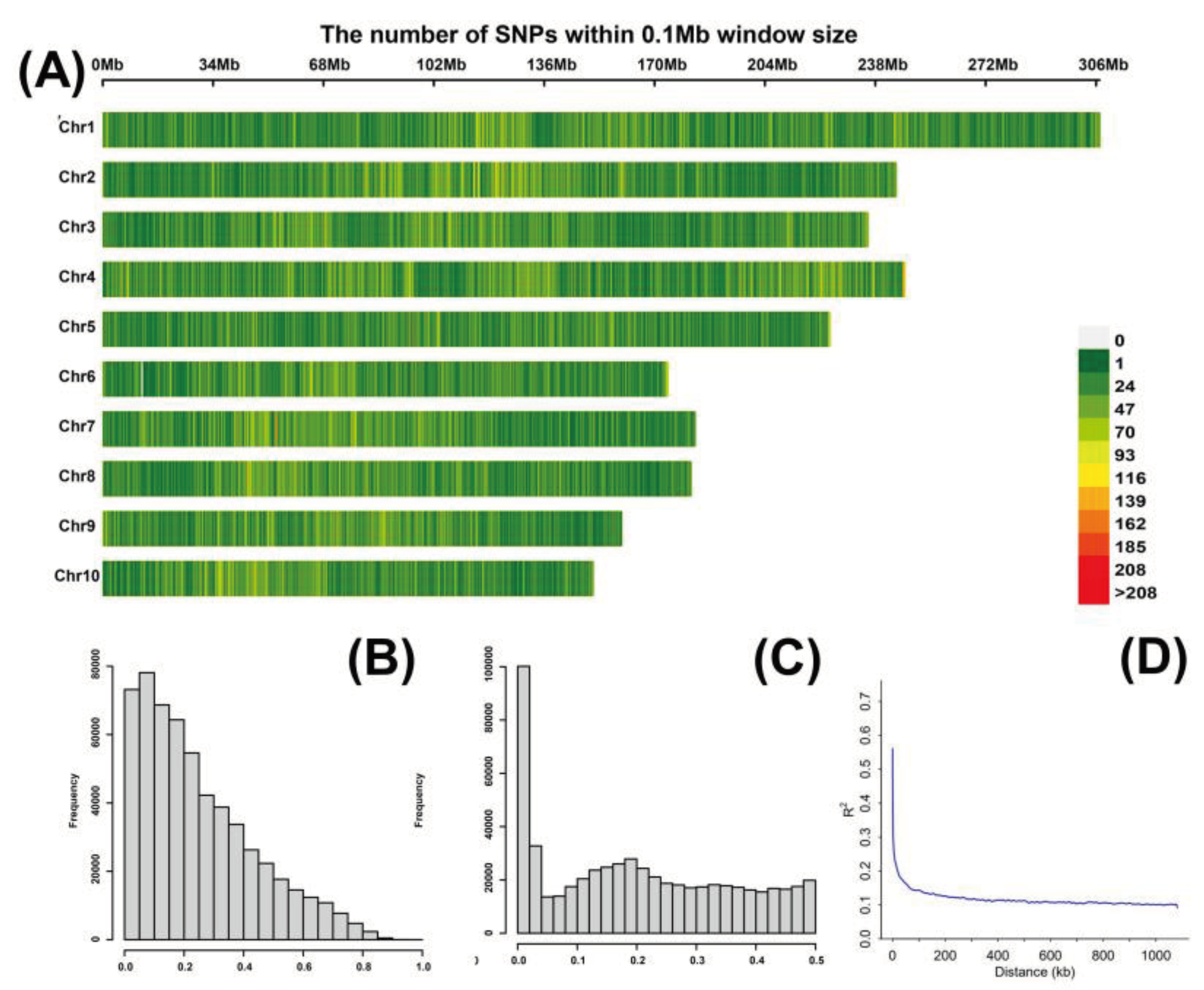
Figure 4.
Genetic diversity analysis (A) Phylogenetic tree of three populations (B) Principal component analysis of 627 RILs (C) Correlation heat map of 627 RILs.
Figure 4.
Genetic diversity analysis (A) Phylogenetic tree of three populations (B) Principal component analysis of 627 RILs (C) Correlation heat map of 627 RILs.
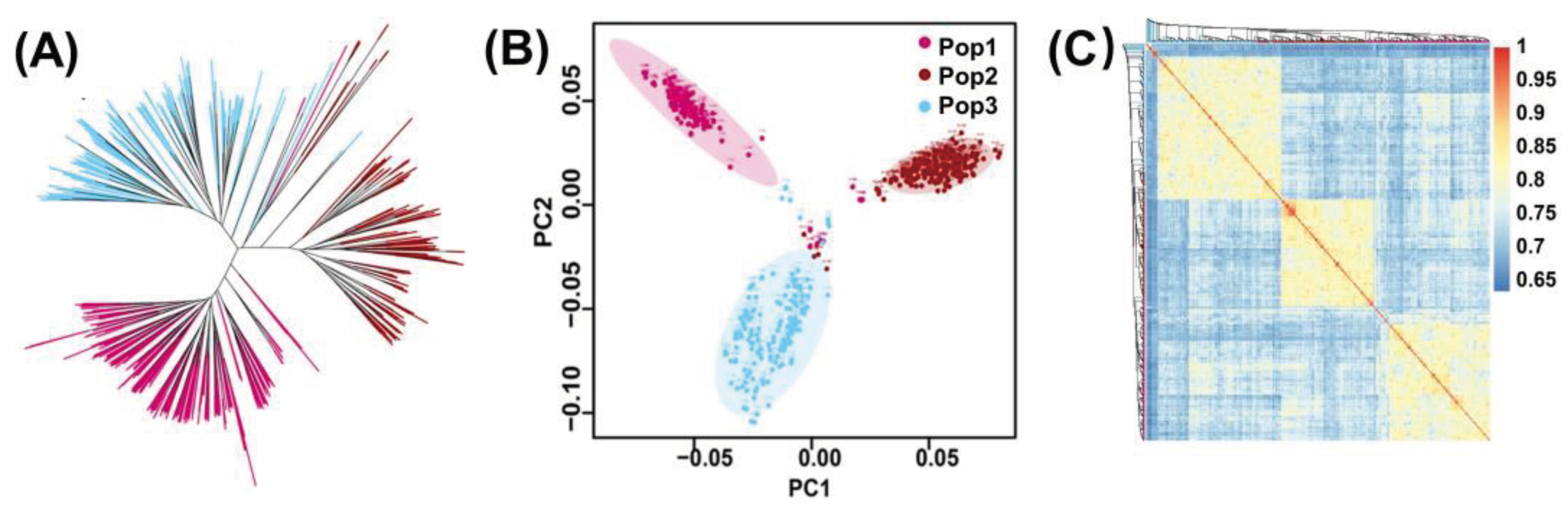
Figure 5.
GWAS analyses in the three RIL populations. (A) Manhattan and Q-Q plots of the BLUP environment for common rust resistance; (B) Manhattan and Q-Q plots for the 21JH environment for common rust resistance; (C) Manhattan and Q-Q plots for the 21YS environment for common rust resistance; (D) Manhattan and Q-Q plots for the 22YS environment for common rust resistance.
Figure 5.
GWAS analyses in the three RIL populations. (A) Manhattan and Q-Q plots of the BLUP environment for common rust resistance; (B) Manhattan and Q-Q plots for the 21JH environment for common rust resistance; (C) Manhattan and Q-Q plots for the 21YS environment for common rust resistance; (D) Manhattan and Q-Q plots for the 22YS environment for common rust resistance.
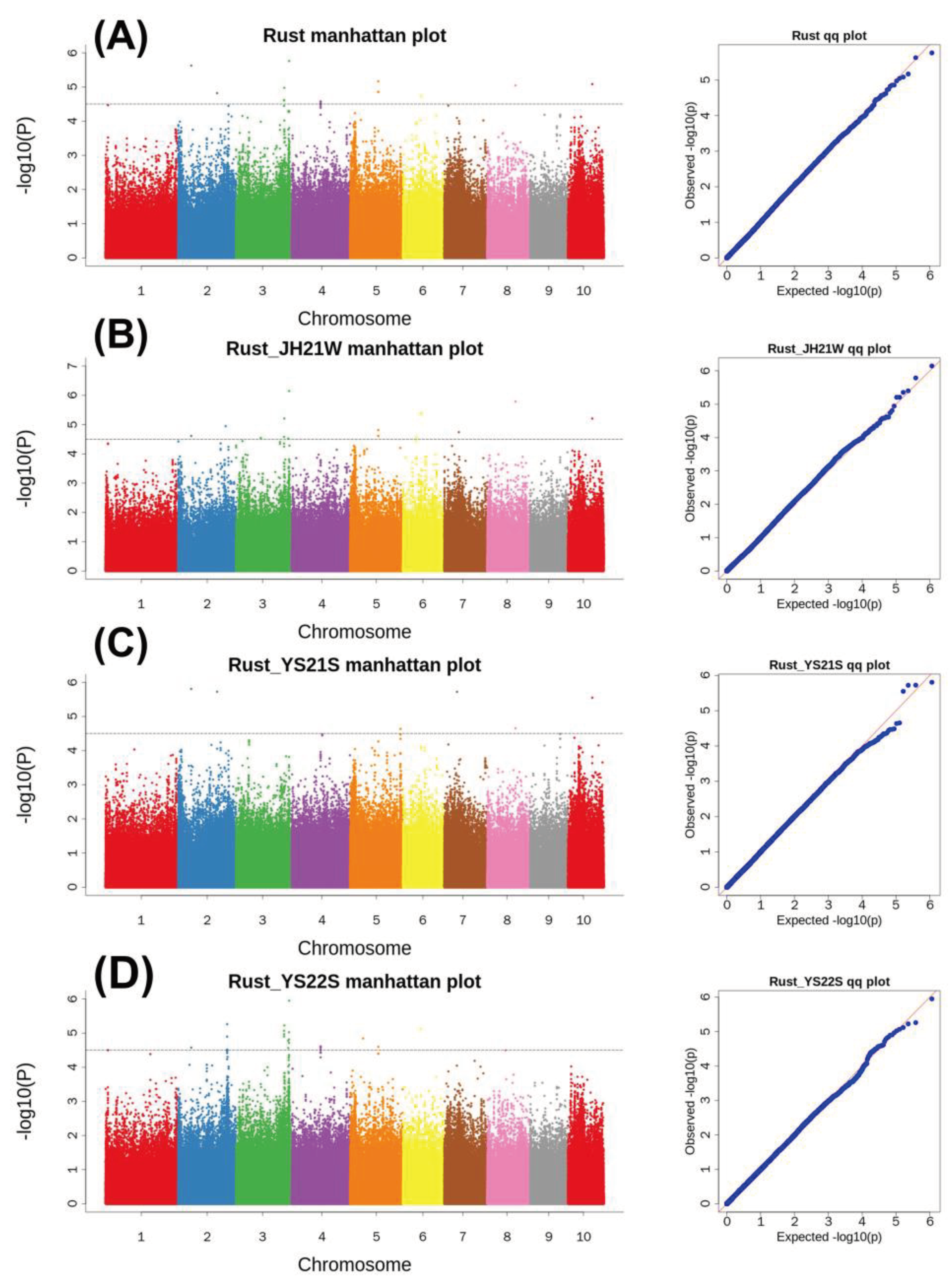
Figure 6.
Common rust based on the identification of Snp-203,116,453 candidate genes (A) Relative positions of Snp and candidate genes; (B) Positions of significant Snp in GWAS; (C) Differences between the two haplotypes in the overall resistance to common rust phenotype, with****indicating p<0.0001 (D) Candidate genes due to base reversal amino acid changes in the candidate gene (e) Expression levels of Zm00001d043536 in various tissues. (DAP: Days After Pollination DAS: Days After Sowing).
Figure 6.
Common rust based on the identification of Snp-203,116,453 candidate genes (A) Relative positions of Snp and candidate genes; (B) Positions of significant Snp in GWAS; (C) Differences between the two haplotypes in the overall resistance to common rust phenotype, with****indicating p<0.0001 (D) Candidate genes due to base reversal amino acid changes in the candidate gene (e) Expression levels of Zm00001d043536 in various tissues. (DAP: Days After Pollination DAS: Days After Sowing).
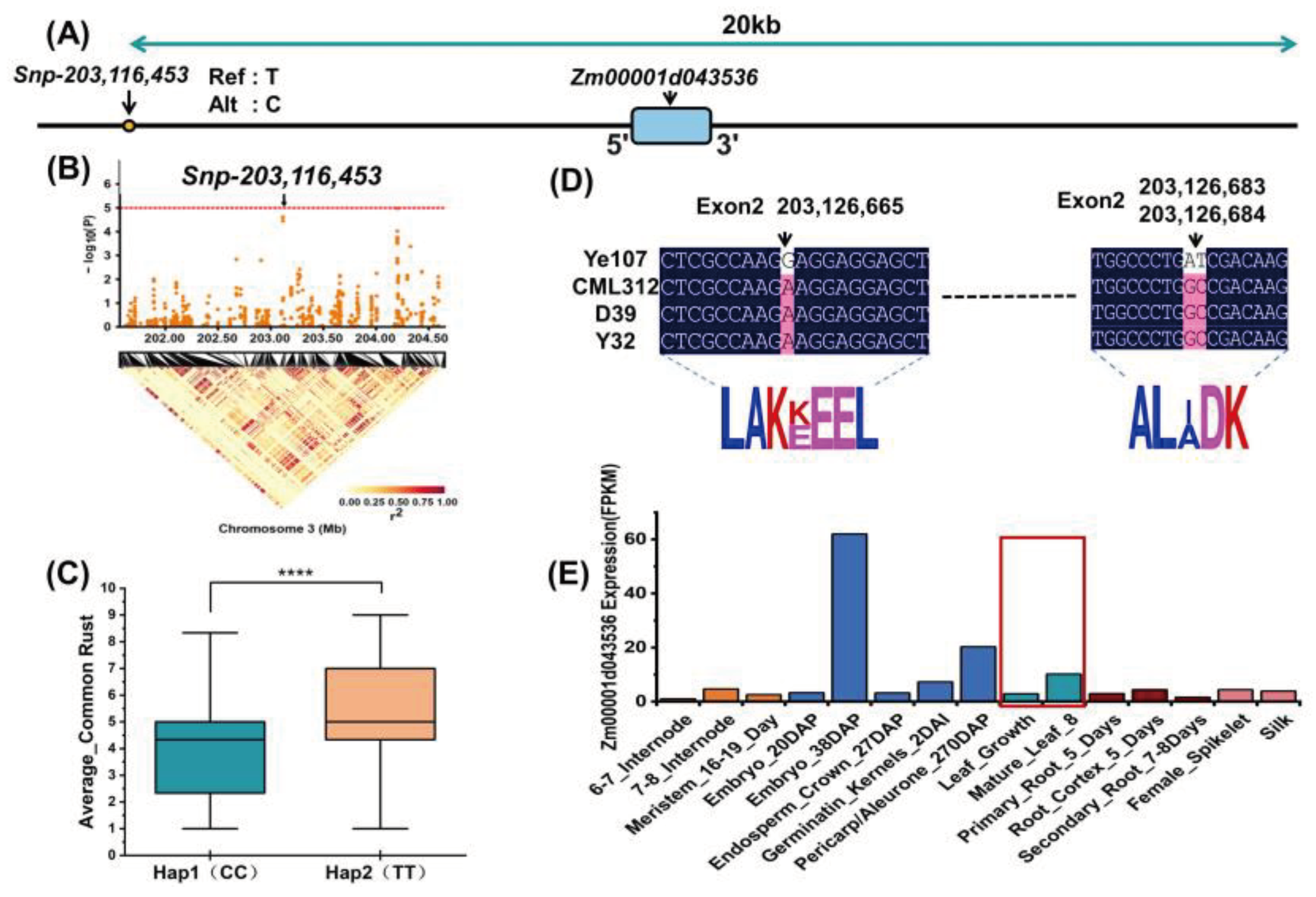
Figure 7.
Common rust based on the identification of Snp-204,202,469 candidate genes (A) Position of significant Snp in GWAS (B) Relative position of Snp and candidate genes (C) Difference between the two haplotypes in overall resistance to common rust phenotypes, with****indicating p<0.0001 (D) Candidate genes Amino acid changes due to subversion in Zm00001d043566 (E) Expression levels of the Zm00001d043566 gene in various tissues (F) Amino acid changes due to subversion in the candidate gene Zm00001d043569 (G) Expression of Zm00001d043569 in leaf and internode before and after pollination. (DAP: Days After Pollination DAS: Days After Sowing).
Figure 7.
Common rust based on the identification of Snp-204,202,469 candidate genes (A) Position of significant Snp in GWAS (B) Relative position of Snp and candidate genes (C) Difference between the two haplotypes in overall resistance to common rust phenotypes, with****indicating p<0.0001 (D) Candidate genes Amino acid changes due to subversion in Zm00001d043566 (E) Expression levels of the Zm00001d043566 gene in various tissues (F) Amino acid changes due to subversion in the candidate gene Zm00001d043569 (G) Expression of Zm00001d043569 in leaf and internode before and after pollination. (DAP: Days After Pollination DAS: Days After Sowing).
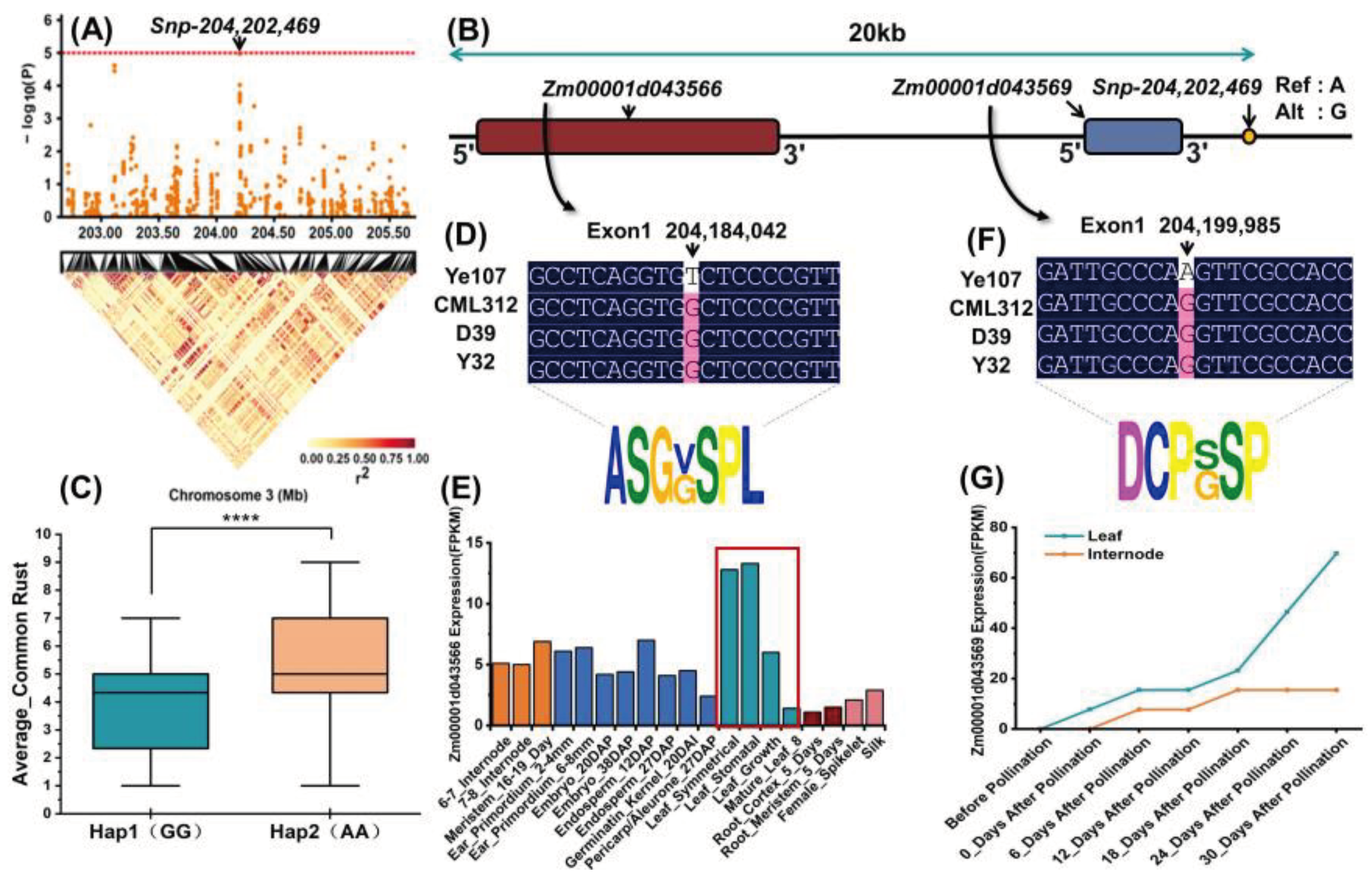
Figure 8.
The population structure is shown in the schematic diagram. CML312, D39, Y32 and Ye107 are the four parents and Ye107 is the susceptible parent. The F1 generation produced from the cross of these four parents underwent six consecutive rounds of self-crossing to obtain the F7 generation.
Figure 8.
The population structure is shown in the schematic diagram. CML312, D39, Y32 and Ye107 are the four parents and Ye107 is the susceptible parent. The F1 generation produced from the cross of these four parents underwent six consecutive rounds of self-crossing to obtain the F7 generation.

Table 1.
Statistical analysis of Common Rust phenotype of three RILs populations.
| Populations | Environments | Means | StandardDeviation | Skewness | Kurtosis | Coefficient ofVariation (%) | Variance components | PopulationHeritability (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pop1 | 21JH | 4.700 | 2.066 | -0.330 | -0.324 | 44.0 | 3.182* | 0.219* | 0.218 | 85.7 | ||
| 21YS | 4.322 | 2.147 | 0.218 | -0.428 | 49.7 | |||||||
| 22YS | 5.000 | 1.831 | 0.221 | 0.059 | 36.6 | |||||||
| Pop2 | 21JH | 5.789 | 1.705 | 0.201 | 0.123 | 29.4 | 2.377* | 0.382* | 0.057 | 90.6 | ||
| 21YS | 5.439 | 1.871 | 0.248 | -0.133 | 34.4 | |||||||
| 22YS | 5.964 | 1.596 | 0.406 | 0.143 | 26.5 | |||||||
| Pop3 | 21JH | 5.759 | 1.644 | -0.121 | -0.151 | 28.6 | 2.494* | 0.177* | 0.036 | 92.2 | ||
| 21YS | 5.268 | 1.817 | 0.166 | -0.379 | 34.5 | |||||||
| 22YS | 5.359 | 1.876 | -0.332 | -0.018 | 35.1 | |||||||
Table 2.
QTL information for maize common rust traits in a BLUP environment.
| QTL | Chr | Position(cM) | Mapping Interval(cM) | LOD | Additive_Effect | R2(%) |
|---|---|---|---|---|---|---|
| qRUST2-1 | 2 | 28.49 | 25.05-31.32 | 4.71 | -0.48 | 0.09 |
| qRUST3-1 | 3 | 103.72 | 101.71-103.72 | 3.92 | -0.59 | 0.1 |
| qRUST3-2 | 3 | 106.73 | 105.73-108.28 | 3.17 | -0.54 | 0.08 |
| qRUST3-3 | 3 | 54.96 | 54.41-59.02 | 5.39 | 0.7 | 0.11 |
| qRUST4-1 | 4 | 40.43 | 40.12-43.27 | 3.37 | 0.46 | 0.08 |
| qRUST4-2 | 4 | 52.39 | 51.39-53.39 | 3.1 | 0.45 | 0.08 |
| qRUST6-1 | 6 | 36.39 | 36.39-38.39 | 4.71 | 0.92 | 0.12 |
Table 3.
Distribution of significant SNPs and candidate genes consistently identified by GWAS in different environments.
Table 3.
Distribution of significant SNPs and candidate genes consistently identified by GWAS in different environments.
| Environment | SNP | Chr | p-BLUP | p-21JH | p-21YS | p-22YS | Candidate Gene | Gene Annotation |
|---|---|---|---|---|---|---|---|---|
| BLUP 21JH 22YS | Snp-203,116,453 | 3 | 4.618 | 4.580 | - | 5.066 | Zm00001d043536 | Heat stress transcription factor C-1b |
| Snp-204,202,469 | 3 | 4.978 | 5.208 | - | 5.223 | Zm00001d043566 | Protein STICHEL-like 3 | |
| Zm00001d043567 | - | |||||||
| Zm00001d043568 | - | |||||||
| Zm00001d043569 | WRKY-transcription factor 29 | |||||||
| Snp-224,639,688 | 3 | 5.763 | 6.145 | - | 5.949 | Zm00001d044303 | IQ_motif_EF-hand-BS | |
| Snp-118,608,571 | 5 | 5.169 | 4.812 | - | 4.596 | Zm00001d015778 | Leucine-rich repeat | |
| BLUP 21JH 21YS | Snp- 118,876,904 | 8 | 5.046 | 5.787 | 4.654 | - | Zm00001d010519 | - |
| Snp-102,507,767 | 10 | 5.084 | 5.206 | 5.548 | - | Zm00001d025070 | - | |
| Zm00001d025071 | - |
Table 4.
Consistent loci detected in two different mapping approaches.
| QTL/SNP | Chr | Position | Candidate Gene | Gene Annotation |
|---|---|---|---|---|
| qRUST3-3 | 3 | 172,823,884-210,543,887 | Zm00001d043536 | Heat stress transcription factorC-1b |
| Snp-203,116,453 | 3 | 203,116,453 | Zm00001d043566 | Protein STICHEL-like 3 |
| Snp-204,202,469 | 3 | 204,202,469 | Zm00001d043569 | WRKY-transcription factor 29 |
Table 5.
Comparison of QTL and significant SNPs for common rust resistance in maize in this study with previous studies.
Table 5.
Comparison of QTL and significant SNPs for common rust resistance in maize in this study with previous studies.
| Chr | This study | Previous study | |||
|---|---|---|---|---|---|
| QTL/Snp | Position | QTL/Snp | Position | reference | |
| 2 | qRUST2-1 | 125,535,857-125,535,857 | - | - | - |
| 3 | qRUST3-1 | 19,468,979-21,766,539 | - | - | - |
| 3 | qRUST3-2 | 17,098,052-18,118,650 | - | - | - |
| 3 | qRUST3-3 | 172,823,884-210,543,887 | qCR3-113 | 113,425,715-224,567,900 | [5] |
| 5 | qRUST4-1 | 121,288,117-128,564,645 | - | - | - |
| 5 | qRUST4-2 | 94,866,787-94,866,787 | - | - | - |
| 6 | qRUST6-1 | 99,941,104-110,962,870 | - | - | - |
| 3 | Snp-203,116,453 | 203,116,453 | qCR3-113 | 113,425,715-224,567,900 | [5] |
| 3 | Snp-204,202,469 | 204,202,469 | qCR3-113 | 113,425,715-224,567,900 | [5] |
| 3 | Snp-224,639,688 | 224,639,688 | - | - | - |
| 5 | Snp-118,608,571 | 118,608,571 | qCR5-51 | 51,355,494-186,678,634 | [5] |
| 8 | Snp-118,876,904 | 118,876,904 | - | - | - |
| 10 | Snp-102,507,767 | 102,507,767 | - | - | - |
Table 6.
Comparison of chromosome 3 QTL and SNPs with previous studies.
| Chr | This study | Distance(bp) | (Kibe et al., 2020)[5] | ||||||
| 3 | Ye107 × D39(F7) | CZL0618 × LaPostaSeqC7-F71-1-2-1-1B(F3) | |||||||
| QTL/Snp | Pos | LOD | QTL/Snp | Pos | LOD | ||||
| qRUST3-3 | 172,823,884 ~ 210,543,887 |
37.63Mb | 5.39 | - | qCR3-113 | 113,425,715 ~ 224,567,900 |
111.14Mb | 2.85 | |
| Snp-203,116,453 | 203,116,453 | - | 56,102,674 | S3_147013779 | 147,013,779 | - | |||
| Snp-204,202,469 | 204,202,469 | - | 57,188,690 | ||||||
Table 7.
Maize parental lines used in developing RIL subpopulations.
| Parents | Pedigree | Ecological type |
Rust resistance | Symptoms scale of CR |
|---|---|---|---|---|
| Ye107 | Derived from US hybrid DeKalb XL80 | Temperate | Susceptible | 9 |
| CML312 | S89500-F2-2-2-1-1-B*5-2-1-6-1 | Tropical | Resistant | 3 |
| D39 | Selected from Suwan1 | Tropical | Highly Resistant | 1 |
| Y32 | Suwan 1-SC9-S8-346-2(Kei 8902)-3-4-4-6 | Tropical | Highly Resistant | 1 |
*CR = Common Rust.
Table 8.
Common rust disease scale used for screening the RILs of Multi-parent populations.
| Scale | Reaction Category | Symptoms |
|---|---|---|
| 1 | highly resistant | no or very few rust spots on the leaves, or lesion area less than 6% of the total leaf area |
| 3 | resistant | a small number of spots on the leaves, or lesion area comprising 6% to 25% of the total leaf area |
| 5 | moderately resistant | number of spots on leaves or lesion area covering 26% to 50% of the total leaf area |
| 7 | susceptible | number of spots on leaves or area of damage comprising 51% to 75% of the total leaf area |
| 9 | highly susceptible | large lesion area on leaves or 76% to 100% of leaf death |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



