
Submitted:
07 March 2024
Posted:
08 March 2024
You are already at the latest version
Abstract
An original approach has been proposed for designing a nanofibrous (NFs) layer using UV-cured polyvinylpyrrolidone (PVP) as a matrix, incorporating mesoporous graphene carbon (MGC) nanopowder both inside and outside the fibres, creating a sandwich-like structure. This architecture is intended to selectively adsorb and detect acetic acid vapours, which are known to cause health issues in exposed workers. The nanocomposite MGC-PVP-NFs layer was fabricated through electrospinning deposition onto interdigitated microelectrodes (IDEs) and stabilised under UV-light irradiation. To enhance the adhesion of MGC onto the surface of the nanocomposite polymeric fibres, the layer was dipped in a suspension of polyethylenimine (PEI) and MGC. The resulting structure demonstrated promising electrical and sensing properties, including rapid responses, high sensitivity, good linearity, reversibility, repeatability, and selectivity towards acetic acid vapours. Initial testing was conducted in a laboratory using a bench electrometer, followed by validation in a portable sensing device based on consumer electronic components (by ARDUINO®). This portable system was designed to provide a compact, cost-effective solution with high sensing capabilities. Under room temperature and ambient air conditions, both laboratory and portable tests exhibited favourable linear responses, with detection limits of 0.16 and 1 ppm, respectively.
Keywords:
acetic acid detection
; electrospun nanocomposite nanofibers
; mesoporous graphene
; selective sensor
; portable sensing tool
1. Introduction
Currently, polymer nanocomposites represent one of the most significant areas of focus in polymer chemistry and nanotechnology research, including coating and printing [1,2,3], smart packaging [4], advanced electronics [5], energy storage and conversion devices [6] biomedical tools [7], drug delivery vehicles, antimicrobial materials [8,9] and sensor technology [10]. Polymer nanocomposites have sparked considerable interest in sensor applications over the past few decades, emerging as pivotal components in sensor system design. Hence, given the demand for innovation in sensors and the transition from traditional to high-performance, portable and nanoscale systems, the scientific community has extensively investigated the development of advanced combinations of nanomaterials and polymer matrices for sensor applications. This interest is largely aroused by the multitude of advantages these materials offer, such as their outstanding electrical and mechanical properties, sensitivity and simplicity of manufacturing [11]. Actually, the exceptional blend of versatility, processability, functionalization, biocompatibility, scalability and customised properties renders nanocomposite polymers highly appealing for sensor nanotechnology, facilitating diverse applications. These sensors can be synthesised on a large scale through cost-effective methods, rendering them economically feasible for mass production. Additionally, they can be imbued with a diverse array of functional groups, additives, or nanoparticles to confer specific properties, such as conductivity, biocompatibility, or responsiveness to distinct external stimuli.
In this study, a selective sensor to acetic acid vapours based on the combination of non-conductive polymer nanofibers (polyvinylpyrrolidone, PVP) with nanopowder of mesoporous graphitized carbon (MGC) is described. As far as authors know, there is limited literature on specific acetic acid vapour sensors [12,13,14,15,16,17,18,19,20,21,22,23,24,25], and they are not commonly found among commercially available sensor options. So far, the rarity of commercial sensors specifically designed to detect acetic acid vapours (for instance based on electrochemical cells [26] or colorimetric sensing technology [https://www.draeger.com/en-us_us/Products/Short-term-Tubes?s=202]) could be attributed to several factors. The use of acetic acid sensors is often limited to specific industries or applications where the detection of acetic acid vapours is critical, such as in chemical manufacturing (e.g., paints, adhesives, plastics, and textile finishes, cleaners), food processing (e.g., flavouring, preservation, acidification, sanitising) or pharmaceutical production (e.g., solvent in synthesis, pH adjustment, drug delivery systems, excipient in formulations). This specialisation restricts their widespread adoption compared to sensors for more commonly monitored gases. Additionally, detecting acetic acid vapours accurately and selectively can be technically challenging due to factors such as interference from other gases, VOCs cross-reactivity, variability in concentration levels, and the need for high sensitivity and specificity. Overall, while acetic acid sensors play a vital role in certain niche applications, their limited demand, technical challenges, cost considerations, and regulatory factors may contribute to their specialised status compared to sensors for more commonly monitored gases.
Additionally, acetic acid is considered an environmentally friendly chemical since it is biodegradable and produced from renewable sources according to sustainable routes [27]. On the other hand, its vapours are hazardous with potential health risks (respiratory irritation, eye irritation, skin burns, and other health problems), overall for people exposed to high concentrations over prolonged periods.
Occupational exposure to acetic acid can occur through inhalation, skin contact, or eye contact. Acetic acid is corrosive to the skin and eyes, and the Occupational Safety and Health Administration (OSHA) has established standards for exposure to it. In Europe, the indicative occupational exposure limit value (IOELVs, from Commission Directive 2017/164/EU) for acetic acid is 10 ppm (25 mg/m3) over an 8-hour work shift and the short term exposure limit (STEL) is 20 ppm (50 mg/m3) [28]. Main symptoms of exposure to acetic acid vapours at this level may include irritation of the eyes, nose, and throat. At concentrations of 100 ppm, individuals may experience significant lung irritation and possible damage to the lungs, eyes, and skin. Exposure to acetic acid can also lead to pharyngeal edema and chronic bronchitis.
Therefore, wearable sensors for acetic acid gas could play a vital role in protecting the health and safety of workers, ensuring regulatory compliance, and enhancing process efficiency and safety in industries where acetic acid is used.
The electrospinning technology is a versatile and scalable technique for producing nanofibers with controlled morphology and composition, offering a versatile and effective approach for designing wearable sensors with enhanced sensitivity, selectivity, flexibility, and biocompatibility, making it a promising strategy for various applications in healthcare, environmental monitoring, and beyond [29,30,31,32].
The versatility of electrospinning technology allows for the creation of advanced and sophisticated sensing layers that are compatible with electronics and electronic nanodevices [33]. Electrospun nanofibers have an exceptionally high surface area-to-volume ratio, which provides a large interface for interactions with target analytes. This increased surface area is expected to enhance the sensitivity of sensors by maximising the number of active sites available for molecular adsorption and detection. The deposition process allows for precise control over the morphology, structure, and composition of nanofibers, enabling the design of sensing layers with specific properties tailored to the requirements of the target analyte and sensing application. The resulting nanofibrous sensing layers are inherently flexible and can conform to a plethora of surfaces, making them suitable for integration into wearable and conformable sensor devices. Further, electrospinning is a scalable manufacturing technique that can produce nanofibrous sensing layers over large areas and volumes. This scalability is essential for the commercialization and widespread adoption of sensing technologies in various applications and industries.
The electrospinning technique can involve applying a high voltage, in the range of several kilovolts, between the spinneret of a spinnable polymer solution, and a collector equipped with the transducer. The collected nanofibers may undergo additional processing steps, such as drying or crosslinking, to improve their mechanical properties and stability. The choice of polymer solution (i.e. based on homopolymers, blends, nanocomposites, metal oxide precursors, etc.) depends on the desired properties of the nanofibers and the intended application of the sensing electrospun materials [34]. In the present study, the electrospun solution was composed of solubilized PVP to which a well-dispersed amount of MGC nanopowder was added. PVP looks a popular choice for electrospinning technology [35] due to the combination of a series of features like excellent solubility in a wide range of solvents including aqueous eco-friendly solvents, good electro-spinnability due to the formation of a stable jet under the influence of an electric field, blend compatibility, biocompatibility and non-toxicity, allowing for the development of diversified nanofibers with enhanced mechanical and electrical properties, and cost-effectiveness. On the other hand, PVP solubility in water renders it also a delicate material for sensing layers. Exposing it to UV-light irradiation for a brief period generates radicals, leading to the formation of new bonds between the chains within the individual fibres. This process makes the fibre insoluble or alters their solubility in various solvents, imparting new chemical and physical properties, along with enhanced stability, contingent upon the duration of UV-light exposure [36]. The use of mesoporous graphene as a nanofiller is of significant interest in sensor applications [37], due to the combination of the excellent conductivity of graphene with a network of periodic mesopores, increasing the available surface area for interactions with surrounding molecules and electrons. More specifically, MGC consists of a single layer of sp2 carbon atoms bonded in a hexagonal honeycomb crystalline arrangement with exceptional physical properties, including high carrier mobility (up to 350·103 cm2/(Vs)), thermal stability (as demonstrated by Bolotin et al., 2008 [38], and high mechanical strength (with a Young’s modulus of 1 TPa and fracture strength of 130 GPa), then able to impart both conductivity and increased mechanical strength to the nanofibers. Additionally the mesoporous structure, providing a larger available surface area (50–100 m2/g) to the nanopowder, offers the potential for enhanced selectivity through molecular size exclusion effects (with an average pore diameter of 137 Å). This material, pristine or oxidised and in combination with other compounds, has primarily found applications in the energy sector, catalysis, selective gas adsorption [39] and in bio- and chemical sensing [40,41,42,43].
It has been proven that the distribution of mesoporous graphene (MGC) within the electrospun polymer nanofibers [44,45,46,47,48] depends on factors such as the graphene/polymers mass ratio, the affinity to the hosting polymers, and the parameters of the electrospinning process. Storti et al. (2023) [49] successfully embedded graphene nanoplatelets into PVP nanofibers without further additives getting homogeneous distributions through pulsed ultrasonication and tested as an antimicrobial tool. Similarly, Del Sorbo et al. (2019) [50] developed nanocomposite fibres of PVP and non-covalently functionalized graphene through tip sonication of graphene alcoholic suspensions in the presence of PVP and used as sound absorbing material.
In this study, a unique architecture of PVP-MGC nanofibers was developed over a commercial interdigitated electrode (IDE) to function as a sensor for the rapid and selective detection of acetic acid vapours. To enhance both electrical conductivity and sensing features, the outer layer of the nanocomposite nanofibers was additionally decorated with MGC nanopowder, serving the dual roles of filler and surface binding agent.
Preliminary measurements were carried out by connecting the sensor to an electrometer to detect changes in current.
Subsequently, the sensor was incorporated into a portable sensing device using the Arduino architecture system. It was then subjected to analyte detection tests for a few tens of seconds, covering the concentration ranges of interest. This approach is in line with the objective of developing a portable system capable of providing a cost-effective and transportable solution with advanced sensing capabilities for use in workplace risk scenarios.
2. Materials and Methods
2.1. Materials
All the materials and chemicals used in this work were of analytical grade and used as received. Mesoporous graphitized carbon nanopowder (MGC, >99.95%, <500 nm DLS, 50-100 m2/g, <200 nm particle size); polyvinylpyrrolidone (PVP, Mw~1,300,000), ethanol anhydrous (EtOH 99,8%), polyethylenimine (PEI, Mw~423), acetic acid (HAc, ≥99%), formic acid (FA, ≥98%) methanol (MeOH, ≥99.8%), acetone (Ac, ≥99.5%), triethylamine (TEA, ≥99.5%) and butylamine (ButA, ≥99.5%) were purchased from Sigma Aldrich.
Interdigitated Electrodes (IDEs), supplied by Micrux Technologies (Spain), were fabricated on glass substrates (IDE dimensions: 10 x 6 x 0.75 mm, Pt/Ti electrodes, 120 pairs, 10 μm wide × 5 mm long × 150 nm thick, with a 10 μm gap). Prior to use, the electrodes were cleaned with a soap solution and a "base piranha" mixture at 60°C for approximately 30 minutes (3:1, v:v, ammonia water and hydrogen peroxide water solution), followed by rinsing with Milli-Q water (~18 MΩ cm).
2.2. Sensor Material Growth
The MGC suspension, prepared by subjecting it to pulsed ultrasonication (10 min, Branson 1800) followed by alternating vortexing and magnetic stirring until a dark ink-like liquid was achieved, was combined with the PVP/EtOH solution (C: 58.2 mg/mL). The mixture was blended until a PVP:MGC solution ratio of 1:0.0045 (w:w) was obtained for electrospinning and loaded into a syringe placed inside the electrospinning deposition chamber.
The fibre deposition process was carried out using a Fluidnatek® LE-50 electrospinning machine (Bioinicia, Spain). To ensure the production of uniform and dry fibres, the distance between the needle and the collector was set at 9 cm, with a solution flow rate of 400 µL/h. The setup comprised two high-voltage sources: one at the needle with a voltage of +4,6 kV and the other at the collector with a voltage of -2 kV. A rotating drum collector (500 rpm) was applied to promote a more organised alignment of the fibres during deposition. Once the electric potential was applied, the polymeric dispersion jet coated the interdigitated electrodes (IDEs) secured to the collector using conductive tape and positioned inside the deposition cone Figure 1 (a).
The UV-light photocrosslinking of the polymer composite nanofibers occurred using a 500W UV lamp (Polimer Helios Italquartz,Italy) emitting in a spectrum range from 180 nm to visible light for 10 min. Samples were placed 10 cm away from the light source, with the film temperature set to 28°C by a Peltier cell, Figure 1 (b).
The dipping suspension contained polyethylenimine (PEI, C: 10,7 mg/mL) and MGC (C: 0,4 mg/mL) in EtOH, previously subjected to pulsed ultrasonication, vortexing and magnetic stirring.
The deposition by dipping occurred using a custom-built system, enabling precise control and regulation of both immersion and withdrawal speeds set at 1 mm per second, Figure 1 (c).
A scheme of the entire deposition process and sensor development is represented in Figure 1.
2.3. Material Characterization
The Scanning Electron Microscopy (SEM) technique was employed to characterise the size, shape, architecture, and surface properties of the nanofibers. Specifically, nanofibrous fabrics produced via electrospinning were deposited onto thin SiO2 wafers and gold sputter-coated using a Balzers MED 010 unit. These samples were then analysed using a JEOL JSM 6010LA electron microscope at the High Equipment Centre, University of Tuscia, Viterbo, Italy.
The average diameter of the fibres was determined using Gwyddion© 2.64 software (GNU General Public License), with measurements conducted on a total of 150 fibres per sample. The normal probability distribution was calculated based on the respective means and standard deviations using Microsoft Excel©.
Quality assessment of fibres on the IDE surface was conducted utilising an optical microscope, specifically the Leica DM2700M (Leica Microsystems CMS GmbH), with observations made at magnifications of 20X, 50X, and 100X. The images were captured by Leica DMC4500 camera under incident light in brightfield using the LED lamp LH113, as well as in fluorescence using the Lamp EL6000 with FITC-Texas filters for green and blue excitation, respectively.
2.4. Sensor Measurement Systems Setup
The measurements presented in this study were initially conducted on a laboratory bench system and later replicated (in the same measurement conditions) using a low-cost portable measurement device. The two system setup are depicted in Figure 2 (a) and (b).
As previously mentioned, the vapour tests were performed with the stationary measurement system, Figure 2 (a). In this way, the resulting chemiresistor (consisting of IDEs + NFs, where NFs refers to nanofibers) was enclosed in a measuring chamber with a volume of approximately 3 mL, and then linked to an electrometer (Keithley 6517, Solon, Ohio, USA) capable of both powering and measuring its electrical outputs, with data transmission to a PC facilitated by LabVIEW Software from National Instruments (Austin, TX, USA). The current, recorded under clean air conditions, was monitored by applying potential values ranging from 0 to 2.0 V in increments of 0.2 V, with humidity percentages and temperature values strictly controlled. The resistance (R) of the fibrous coating and its relationship to humidity at 25°C (45% RH) were determined using Ohm’s Law, which states that the resistance of a circuit equals the voltage across it divided by the current flowing through it. VOCs measurements were carried out by applying 1 Volt of potential. The measurement chamber was conditioned by a customised pneumatic system necessary to generate the desired vapour concentrations. It comprised a mass flow control (MFC) with a range capacity of 0-200 standard cubic centimetres per minute (sccm) managed by a 4-channel readout controller (MKS Type 247), an electrovalve to switch the fluxes (S070C-RAG-32 from SMC Corporation) and an air pump (NMP015, KNF) to generate a carrier ambient air flux, which was previously purified passing through a carbon activated cartridge. All the connections between Teflon tubes were guaranteed by Festo (Festo AG & Co. KG, Esslingen) push-in connectors. Air cylinder (Nippon Gas, 5.0) was used as carrier gas in the circuit dedicated to the bubbling and collection of VOC vapours. This configuration facilitated the generation of precisely controlled concentrations of volatile organic compounds (VOCs). The concentration range for acetic acid was determined based on its application range (0.95-30 ppm), while for all other VOCs, the concentration range was selected to ensure it reached at least a value detectable by the sensor (FA: 5-440 ppm, NH3: 7-755 ppm, MeOH: 14-2245 ppm, EtOH: 7-930 ppm, Ac: 25-4125 ppm, TEA: 17-1350 ppm and ButA: 16-1690 ppm.
The second measuring setup (Figure 2 (b)), similar on the pneumatic side, consisted of an electronic board (Analog board) connected to an IDE that converted the electric resistance variations into voltage variations Figure 3(A). A 16-bit analog to digital convert (ADC) module (ADS1115 by Analog Devices) Figure 3 (B) converted the analog signals into digital ones. A microcontroller (Arduino Nano), Figure 2 (b), acquired and transmitted the generated data to a computer unit through a USB port. A software program (developed by Labview), elaborated, plotted and stored these data in real-time. The software executed measurement cycles and generated text files (.txt or .csv).
In this way, the system could register the presence of possible threshold levels and provided the possibility to subsequently analyse the stored data, giving a long-term average concentration to which the operator was exposed.
Finally, in order to operate, the entire portable system required a power supply (5V DC), connected to the mains power. It was located at the bottom of the system, as depicted in Figure 3 (D).
3. Results and Discussion
3.1. Sensing Material Characterization
Electrospinning technology facilitated the one-step creation of nanocomposite nanofibrous layers using a single needle. Depositions were easily performed on various substrates, including silicon dioxide thin slices for morphological and optical characterization of the fibres, and customised borosilicate interdigitated electrode (IDE) transducers for measuring the electrical and sensing properties of the thin nanofibrous coating.
Each substrate, securely fixed onto the grounded rotating cylinder and aligned with the needle tip, efficiently collected the ejected fibres. The electrospun jet streams maintained uninterrupted flow, leading to the formation of a fibrous network within a mere four minutes. The exposure of PVP nanofibers to UV light irradiation was expected to initiate the photocrosslinking of the polymer in the solid state, aided by the production of O3 radicals in the surrounding air.
PVP, being water-soluble and also soluble in ethanol and most polar solvents, is inherently fragile and not ideal for sensing applications. Previous studies have demonstrated that UV irradiation can form insoluble and photocrosslinked PVP structures [51]. Depending on the duration of exposure, these bonds made the PVP fibres insoluble or differently soluble in various solvents, endowing them with altered chemical and physical properties and increased stability. As reported in Figure 4 (A), the interdigitated electrodes with electrospun nanofibers exhibited uniform coverage, forming a network architecture characterised by consistent microporosity. These pores were supposed to serve as pathways for gas transport, thereby bolstering the material’s suitability for gas-VOC sensing applications.
Optical microscope pictures of Figure 4,(B,E) and SEM micrograph of Figure 5 (A-inset) highlight a partially aligned orientation of nanofibers as deposited and subjected to UV-irradiation, suggesting a certain degree of directional organisation of the material. The following dipping of the layer into a solution of PEI-MGC altered the shape distribution of the fibres, leading to bundling, branching, and undulations in the fibres as observable in both Figure 4, (A,D,F) and Figure 5, (A,B). This effect may happen for the interactions between EtOH and the polymer chains and be responsible for a partial swelling and then disruption of the linear arrangement. Such a fibres shape change is highlighted by fluorescence microscope pictures (Figure 4, E,F) of a looser mesh network before and after dipping. Indeed, the fibres exhibited a notable brightness, leaning towards a green hue (Figure 4,E). PVP is not typically regarded as a fluorescent polymer but it exhibits significant intrinsic fluorescence, particularly when subjected to photo-oxidation [52,53]. Moreover, when coated with PEI oligomers and MGC nanopowder, PVP nanofibers luminosity intensified further, accompanied by a shift in emission. Under the fluorescent optical microscope, the presence of a thin, radiant coating on the fibres was clearly observable (Figure 4,F) due to bright blue fluorescence of PEI [54] confirming the dipping deposition
However, following the dipping, analysis through both optical and scanning electron microscopy revealed that the fibres collected in denser networks (Figure 4, (A,D) and Figure 5 (A,B) exhibited enhanced adhesion to the substrate (IDEs and SiO2 wafer respectively) and maintained interconnectivity with one another, despite experiencing partial loss of their linear structure.
SEM images of Figure 5 (A-inset) proved that the individual nanofibers (∅: 178±40 nm) within the network exhibited intersecting trajectories, creating points of contact and potential bonding between adjacent fibres. These intersections contribute to the formation of junctions, thereby enhancing the structural integrity of the three-dimensional nanofiber network. The increased complexity of the network is evident in the emergence of features such as junctions, cross-linkages, and overlapping segments of nanofibers. These characteristics are expected to bolster the overall stability and mechanical strength of the three-dimensional structures, while also increasing the available surface area with adsorption sites. Additionally, during the dipping process, the fibres transitioned from smooth and uniform surfaces to surfaces adorned with rough sleeves. This transformation imparted a wrinkled appearance to the fibres and increased their diameter and heterogeneity in shape (∅: 281±126 nm). As a result, the nanofibers presented clusters of particulate matter distributed along their length with different surface density but firmly adhering to the fibre due to the presence of PEI Figure 5 (A,B).
3.2. Sensing Electrical Characterization
Due to PVP nanofibers intrinsic poor electrical conductivity [55] the addition of MGC as conductive filler, is expected to enhance the overall electrical conductivity of the composite material. Figure 6 (A)(B) illustrates the current-voltage (I-V) plot for the IDE coated with PVP-MGC nanofibers before and after UV photocuring. The x-axis of the plot denotes the applied voltage across the electrode, ranging from 0 to 2 V, while the y-axis represents the current passing through the electrode. As the voltage is incrementally raised in the positive direction, both IDEs exhibit a linear increase in current, characterised by comparable slopes (≈23 MOhm). A very slight increase in electrical resistance is observed when PVP-MGC nanofibers are UV irradiated (PVP-MGC)UV, Figure 6 (A). It could be attributed to the alteration of the nanofiber structure induced by photoxidation/crosslinking, leading to changes in the conductivity pathways within the nanofiber network or in chemical/physical changes of the PVP-MGC interface.
However, the linear shape observed in the current-voltage curve of the PVP-mesoporous graphene nanofibers within the range of 0V and +2V suggests that MGC may be uniformly distributed inside the fibres and that the contact between fibres and electrodes implies that there is no significant energy barrier at the interface. This uniform distribution facilitates consistent electrical conductivity across the entire length and volume of the nanofibers. Further, it presumably indicates effective integration of the conductive material within the polymer matrix, ensuring efficient electron transport pathways. The uniform integration of the nanofillers seems to be confirmed also by the SEM images, as the fibres appeared homogeneous and smooth, even on the surface (Figure 5, (A-inset). Graphene, being a highly conductive material, could introduce n-type doping characteristics to the composite nanofibers. The presence of defects or functional groups on the graphene surface may donate electrons to the PVP matrix, leading to an excess of negative charge carriers (electrons) and resulting in n-type semiconductor behaviour. Interaction between the PVP polymer and graphene mesoporous structures may facilitate charge transfer processes.
The PEI-MGC decoration of fibres through dipping significantly boosted sensor conductivity (R: ≈34 kOhm), while maintaining a linear relationship between the applied voltage and the measured current (Figure 6,B). The inset graph in Figure 6, (B) is identical to the one depicted in the same figure, except for the y-axis, which is presented in a logarithmic scale. This adjustment enables the visualisation of both IV curves simultaneously. Such an increase in current is presumably due to the outer MGC being able to provide additional conductive pathways with the nanofiber nanofillers network. PEI may further improve electrical conductivity by promoting better dispersion and adhesion of the mesoporous graphene onto the nanofiber matrix. Additionally, the decoration with mesoporous graphene and PEI could increase the surface area of the nanofibers, providing more active sites for electron transfer. This increased surface area is expected to facilitate a better interaction between the nanofibers and the surrounding environment, leading to enhanced sensing performance. Furthermore PEI, known for its ability to promote charge carrier mobility [56], could contribute to the movement of electrons through the nanofiber network.
To evaluate the sensing features, we subjected both (PVP-MGC)UV and (PVP-MGC)UV/MGC-PEI sensors to airflow under conditions of constant temperature and relative humidity. Each measure was carried out to detect various common solvents and chemical compounds that could potentially interfere with the sensors and might be commonly encountered in environments of laboratories and industries. For these measurements defined amounts of fluxes were partialized and controlled for generating the necessary concentrations of the desired target vapours.
Upon exposure to each VOC, both the sensors demonstrated an increase in current. However, the responses of the (PVP-MGC)UV/MGC-PEI sensor were notably faster, as expected, and more reproducible than (PVP-MGC)UV (data not shown), presumably attributable to an improved stability conferred by the addition of an outer skeleton of a mixture of nanopowder and oligomers, i.e. MGC and PEI, respectively.
For each of the VOCs tested, (PVP-MGC)UV/MGC-PEI sensor detected up to eight concentrations, starting from their saturated vapour pressure. From these measurements, variations in current corresponding to vapour concentrations were observed in the sensor output.
The graph in Figure 7(A) depicts a comparison of the sensor’s transient responses upon exposure to different tested VOCs. These responses are calculated as the ratio between the changes in measurement current (ΔI) and the baseline current (I0) over time. Notably, for acetic acid (HAc) (at a concentration of 30 ppm), the current exhibited a rapid increase, stabilising in t90=90 s, i.e. the time it takes for the sensor to reach 90% of its final stable response.
Conversely, the sensor didn’t reveal any signal to all the other VOCs at equivalent concentrations. However, it’s noteworthy that these VOCs generate varying concentrations in parts per million (ppm) at room temperature due to differences in their partial pressures. Thus, the transient measurements in Figure 7(A) describe a comparison of responses among different concentration levels. In the case of formic acid (FA) that has a vapour pressure of 4,66 kPa (HAc, Pvap: 1,54 kPa) calculated by Antoine Equation [57], the sensor demonstrated a distinct increase in current when exposed to approximately 440 ppm, although with slower kinetics and without reaching apparent equilibrium within the same exposure time defined vs all the VOCs. Therefore, VOC molecules adsorbed onto the nanocomposite fibres, by changing the charge distribution, led to an increase in conductivity. However, despite being measured at concentrations ranging from hundreds to thousands of ppm, all other VOCs minimally influenced the current variation, as confirmed also in Figure 7, (B). Conversely, the sensor response size and shape to HAc suggested a rapid and selective detection of the target analyte, which is essential in applications where real-time monitoring or quick identification of substances is required, such as environmental monitoring or industrial process control, overall where delays in sensor response could result in missed events or inaccurate readings. Moreover, the rapid response time correlated with the highest response signal enables the sensor to detect even low concentrations of analytes quickly. This is vital for ensuring the sensor’s effectiveness across a wide range of concentrations and for detecting trace amounts of acetic acid.
The graph depicted in Figure 7(B) showcases the correlation between normalised sensor responses and increasing concentrations of VOCs, spanning from 0 to 4125 ppm. Each concentration interval aligns with the sensor’s sensitivity, delineating clear and discernible data points across the graph. All curves exhibit linearity across the tested concentration range. Particularly noticeable is the response curve for acetic acid, which stands out by overlapping the y-axis also at lower concentrations (as depicted in the Figure 7 (B) inset), underscoring the sensor’s heightened sensitivity to acetic acid compared to the other tested VOCs. The slope of each curve acts as a sensitivity metric, further emphasising the sensor’s strong affinity for the analyte. Among the tested VOCs, FA emerges as the sole compound significantly impacting sensor responses, albeit at elevated concentrations. The bar-plot in Figure 8 depicts more in detail the sensitivity values of the sensor to the tested VOCs. In order to be able to display all values, the y-axis was fragmented. The sensor exhibits minimal sensitivity to polar and small compounds like ethanol (EtOH) and methanol (MeOH). However, its sensitivity to amines, regardless of their structure (primary, secondary, or tertiary), and to ketones is negligible. This effect could be due to the mesoporous structure of the shell enabling selective permeability. It could allow smaller molecules such as HAc and FA to diffuse through while excluding larger molecules. Additionally these mesopores may provide an extended surface area, enhancing their interaction with the functional groups and increasing adsorption. The presence of PEI in the shell, introducing amino groups able to form hydrogen bonds with the carbonyl groups of HAc and FA, could facilitate the selective adsorption of these carboxylic acids. The combined effects of the surface chemistry and pore structure result in increased sensitivity to acetic acid. Additionally, at higher concentrations, the enhanced diffusion of formic acid FA molecules through the shell leads to detectable levels of formic acid FA adsorption, expanding the detection capabilities of the sensor. Furthermore, the significant increase in current observed during the interaction between HAc molecules (acting as Lewis acids) and the MGC-PEI outer layer (with Lewis base sites) could result from the transfer of electrons from the Lewis base sites to the HAc molecules, thereby enhancing current flow. Additionally, the protonation of PEI molecules by acetic acid may modify the charge distribution within the composite, consequently increasing conductivity. An estimation of sensor selectivity [58] among the tested VOCs, calculated as:
(where Sel is the selectivity, S the sensitivity and A is the analyte) reveals that the sensor exhibits 96% sensitivity to acetic acid, 3% to formic acid, and 0.2% to ethanol. The other values are negligible.
The limit of detection (LOD), often defined as the concentration at which the signal-to-noise (S/N) ratio equals a 3 (LOD = 3 * Baseline Noise), represents the concentration at which the signal becomes three times higher than the baseline noise, ensuring reliable detection above the noise level. In our measurements, conducted up to 950 ppb, the LOD was determined to be 160 ppb (Figure 7A).
The sensor was tested for one month at the same concentration of acetic acid to evaluate its stability over time (Figure 8,(B)). The response ((DI/I0)mean:2,94±0,15), averaged from five measurements per day, exhibited reproducibility ((DI/I0)mean:2,91±0,19) after 30 days of use. The sensors demonstrated a certain stability, with fluctuations remaining within the range of the measurement error.
In the last decade literature, as previously mentioned, most of the planned and investigated chemiresistors to monitor acetic acid used nanostructured metal oxide and their combinations which however required high temperatures (ranging between 150-380 °C) to achieve high sensing performances (Table 1) [13]. Their LODs varied between 10 ppb to 50 ppm, depending on the quality of doping and nanoarchitecture. Some dopants were selected for their catalytic properties, others were selected for their ability to create finer microstructures or grain boundaries, thereby increasing the surface area available for interaction with acetic acid molecules. For instance, electrospinning technology was employed to enhance the sensitivity of In2O3 for detecting HAc. The resulting highly porous and interconnected structure enabled the detection of the analyte at a concentration of 500 ppb when the sensor operated at 250 °C [21]. Conversely, ZnO was explored as a promising compound for detecting acetic acid at both high and room temperatures. The sensitivity of ZnO varied depending on whether it was in the form of hexagonal nanocrystals or foam surfactant [16], highlighting that the increase in the density of surface defects and active sites within a nanoarchitecture enhanced interactions with the analyte. By the way, the foam variant achieved a LOD of 500 ppb at a working temperature of 400°C. The integration of a porous metal-organic framework (Tb2O3@MOF) [17] with ZnO enabled the sensor to operate effectively at room temperature. However, to enhance sensor sensitivity and achieve a LOD of 500 ppb, UV light excitation was employed. Conversely, sensors based on GQDs–ZnO composites (GQDs: graphene quantum dots) could be operated at room temperature and exhibited a stronger response to acetic acid gas compared to a pure ZnO sensor, but detecting up to 1 ppm at room temperature [59]. The mesoporosity of a metal oxide (CuO) was utilised to create a sensor operating at 200°C [60], whereas the incorporation of graphene (RGO or G) in conjunction with metals [61] or ceramics [62] enabled chemiresistors to function at room temperature with exceptional sensitivity (achieving a limit of detection of up to 1 ppb [63]). Avossa et al., (2018) reported that a chemiresistor based on ES nanofibres of a blend of polystyrene and polyhydroxybutyrate (PS-PHB) hosting MGC (0.93% mass ratio) was sensitive and selective to acetic acid vapours, but only working at a temperature slightly higher than room temperature (T=40°C): the mesoporous structure, having a 137 Å average pore diameter, acted as a nucleation centre for entrapping and growing acetic acid. Since the sensor did not appear to reach a plateau quickly, a LOD was not reported [64]. The necessity of the sensor to work at more elevated temperatures appeared to be related with the polymer’s structure and the heterogeneous network architecture of MGC within the fibres. Changing the hosting polymer (PVP in the present study), the electrical and sensing features looked widely improved. Thus the achieved data in the present study suggest that the (PVP-MGC)UV/MGC-PEI sensor could operate within the permissible exposure limits (PELs) established for acetic acid (TWA: 10 ppm for an average 8-hour workday; STEL: 20 ppm during a 15-minute exposure) and serve as a promising candidate for integration into portable monitoring systems aimed at protecting workers.
3.3. Portable Sensing System
Refocusing our attention on human risks associated with exposure to hazardous concentrations of acetic acid, often challenging to measure in workplace environments, direct tests of this sensor using the previously described portable sensing system were carried out to assess its efficacy and practicality. As depicted earlier (Figure 2 (B)), unlike the stationary system where output is measured in current (A), the portable measurement system delivers output signals in voltage (V). The sensor’s interdigitated electrode (IDE) was connected to a dedicated electronic circuit board to ensure dependable measurements, and housed within a Teflon measuring chamber installed on the board. Purified ambient air at 20°C (±1°C) and 45% relative humidity (±5%RH) was employed as the gas carrier, replicating realistic ambient conditions for these assessments. Each measurement was carried out for a duration of up to 90 seconds to ascertain if this timeframe allowed the sensor adequate time to detect the presence of acetic acid in the environment.
Therefore, a sequence of progressively increasing exposure concentrations within 7 and 47 ppm, was generated and directed into the measurement chamber to evaluate the sensor’s linearity and sensitivity (refer to Figure 9).
After each exposure, the sensor was purged with purified ambient air only, facilitating the desorption of acetic acid from the sensor surface and restoring its original conductivity.
Based on the maximum signal attained, a calibration curve was derived (Figure 10).
The range encompasses all the concentrations under consideration, resulting in a linear fit curve (red) with a slope of 0.01033 ppm-1.
The integration of the sensor on a portable and low-cost system has reduced the sensitivity of the sensor, achieving a 1 ppm LOD and 3 ppm LOQ (limit of quantification). LOQ represents the lowest concentration accurately quantifiable by the sensor with an acceptable level of precision and accuracy [76].
4. Conclusions
In this study, we investigated a fibres mat of PVP-MGC decorated with PEI-MGC deposited on an IDE, exploring its sensing properties. The nanomaterial dimensions of the fibres, achieved through electrospinning technologies, provided a high surface area-to-volume ratio, enhancing the material’s sensing properties. Initial measurements revealed a notable sensing response of the nanofibers to carboxylic acids, particularly towards acetic acid vapours. The thin shell of polyethylenimine (PEI) and mesoporous carbon graphitized (MGC) on nanocomposite fibres (PVP-MGC) provides an ideal platform for the adsorption and detection of acetic acid and a minor response towards the detection of formic acid. This behaviour can be attributed to its surface chemistry, with a well-defined pore structure allowing selective permeability and increasing the sensitivity of the target analytes. The developed (PVP-MGC)UV/MGC-PEI structure is a combination of nanoarchitectures, strategies and technologies validated by literature focused on acetic acid detection. However, in this work we introduce an original composite material made of electrospun nanofibers of PVP filled with mesoporous graphene, offering distinct advantages over traditional sensing materials and facilitating detection capabilities. Finally, the synergistic effects of combining PVP nanofibers, MGC and PEI created a multifunctional sensing platform with enhanced performance characteristics. This synergism led to improved detection limits, a fast response times and a good stability compared to conventional sensing materials. We tested two measurement systems: a stationary and a portable/low-cost system. Both the systems demonstrated similar responses, with the stationary system exhibiting better accuracy offset compared to the portability of the latter. Here the sensor looks to work stably and selectively at room temperature, detecting up to 160 ppb HAc (LOD) in ambient air, with a selectivity of 96% among the tested VOCs. In contrast, the portable system can be directly used in workplaces, monitoring and storing data on workers’ exposure to acetic vapours. On the other hand, it displays a LOD of ~1 ppm and a LOQ of ~3 ppm, staying within workplace exposure limits based on TWA and STEL values. Based on this encouraging data, further developments could focus on enhancing the portable sensor unit for real-time monitoring, enabling timely alerts to workers in the event of dangerous concentrations and facilitating wearable applications. The sensor’s design not only ensures rapid and reliable results but also opens up a world of possibilities for enhanced process control and risk mitigation in various industries. With its compact and cost effective architecture, it may ensure prevention of potential health risk, promoting a safer work environment.
Author Contributions
Conceptualization, A.M.; methodology, A.M.; formal analysis: P.P.; validation, P.P., and E.Z.; investigation, P.P, E.Z., A.M.; software: A.C.; resources, G.T.; data curation, P.P., C.DN, FN.M., F.DC.; writing—original draft preparation, A.M. and P.P.; writing—review and editing, all the Authors; project administration, A.M.; funding acquisition, A.M., C.DN., and G.T. All Authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Istituto Nazionale Assicurazione contro gli Infortuni sul Lavoro (INAIL) through the italian project BRIC2019-ID 07 - Integrated system of mobile and fixed sensors for dynamic spatio-temporal mapping of volatile compounds in work environment.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
All data that support the findings of this study are available after the reasonable request to the corresponding author.
Acknowledgments
Many thanks to Anna Rita Taddei of Tuscia University for her SEM analysis, A. Capocecera for his technical collaboration in software programming and using, and S. Berti and T. Davanzo for their administrative support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Falahati, M.; Ahmadvand, P.; Safaee, S.; Chang, Y.C.; Lyu, Z.; Chen, R.; Li, L.; Lin, Y. Smart Polymers and Nanocomposites for 3D and 4D Printing. Materials Today 2020, 40, 215–245. [Google Scholar] [CrossRef]
- Idumah, C.I.; Obele, C.M.; Emmanuel, E.O.; Hassan, A. Recently Emerging Nanotechnological Advancements in Polymer Nanocomposite Coatings for Anti-Corrosion, Anti-Fouling and Self-Healing. Surfaces and Interfaces 2020, 21, 100734. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Fahy, W.P.; Kim, S.; Kim, H.; Zhao, N.; Pilato, L.; Kafi, A.; Bateman, S.; Koo, J.H. Recent Developments in Polymers/Polymer Nanocomposites for Additive Manufacturing. Prog Mater Sci 2020, 111. [Google Scholar] [CrossRef]
- Sarfraz, J.; Gulin-Sarfraz, T.; Nilsen-Nygaard, J.; Pettersen, M.K. Nanocomposites for Food Packaging Applications: An Overview. Nanomaterials 2021, 11, 1–27. [Google Scholar] [CrossRef]
- Gong, M.; Zhang, L.; Wan, P. Polymer Nanocomposite Meshes for Flexible Electronic Devices. Prog Polym Sci 2020, 107, 101279. [Google Scholar] [CrossRef]
- Hossain, S.K.S.; Hoque, M.E. Polymer Nanocomposite Materials in Energy Storage: Properties and Applications; Elsevier Ltd. 2018; ISBN 9780081019115. [Google Scholar]
- Zhao, L.; Liu, J. Application of Polymer Nanocomposites in Biomedicine; INC. 2022; ISBN 9780323916110. [Google Scholar]
- Panicker, S.; Mohamed, A.A. Polymer Nanocomposites for Drug Delivery Applications; INC. 2022; ISBN 9780323916110. [Google Scholar]
- Ahmadi, Y.; Moeini, N.; Yadav, M.; Ahmad, S. Antimicrobial Polymer Nanocomposite Films and Coatings; INC. 2020; ISBN 9780128214978. [Google Scholar]
- Aboulrous, A.A.; Mahmoud, T. Polymer Nanocomposites for Sensing Applications; Elsevier Ltd. 2023; ISBN 9780323884310. [Google Scholar]
- Damiri, F.; Gaiji, H.; Muhamad, I.I.; Lazim, N.A.M.; Kaur, D.; Berrada, M. Design and Fabrication of Polymer Nanocomposite Sensors; Elsevier Ltd. 2022; ISBN 9780323988308. [Google Scholar]
- Pineau, N.J.; Krumeich, F.; Güntner, A.T.; Pratsinis, S.E. Y-Doped ZnO Films for Acetic Acid Sensing down to Ppb at High Humidity. Sens Actuators B Chem 2021, 327, 128843. [Google Scholar] [CrossRef]
- Wang, C.; Ma, S.; Sun, A.; Qin, R.; Yang, F.; Li, X.; Li, F.; Yang, X. Characterization of Electrospun Pr-Doped ZnO Nanostructure for Acetic Acid Sensor. Sens Actuators B Chem 2014, 193, 326–333. [Google Scholar] [CrossRef]
- Turemis, M.; Zappi, D.; Giardi, M.T.; Basile, G.; Ramanaviciene, A.; Kapralovs, A.; Ramanavicius, A.; Viter, R. ZnO/Polyaniline Composite Based Photoluminescence Sensor for the Determination of Acetic Acid Vapor. Talanta 2020, 211, 120658. [Google Scholar] [CrossRef]
- Chu, X.; Gan, Z.; Bai, L.; Dong, Y.; Rumyantseva, M.N. The Acetic Acid Vapor Sensing Properties of BaSnO3 Microtubes Prepared by Electrospinning Method. Materials Science and Engineering: B 2020, 259, 114606. [Google Scholar] [CrossRef]
- Qin, W.F.; Zhang, H.M.; Li, X.B.; Xing, Y.W.; Guo, Y.X.; Feng, Y.Y.; Li, Y.J.; Han, S.Q.; Ma, K.F.; Cao, H.H.; et al. Surfactant Modified Hexagonal ZnO Gas Sensor for Acetic Acid. Journal of Materials Science: Materials in Electronics 2023, 34, 1–13. [Google Scholar] [CrossRef]
- Ling, W.; Zhang, S.; Cao, S.; Pu, Y.; Zhu, D. Enhanced Acetic Acid Detection for Tb2O3 @MOF-Derived ZnO at Room Temperature. Sens Actuators B Chem 2023, 377, 133057. [Google Scholar] [CrossRef]
- Zappi, D.; Varani, G.; Iatsunskyi, I.; Wallaszkovits, N.; Bailer, J.; Giardi, M.T. High-Sensitivity Metal Oxide Sensors Duplex for On-the-Field Detection of Acetic Acid Arising from the Degradation of Cellulose Acetate-Based Cinematographic and Photographic Films. Chemosensors 2022, 10. [Google Scholar] [CrossRef]
- Rizani, A.; Winingsih, S.S.; Aditya, R.; Julian, T.; Hidayat, S.N.; Kusumaatmaja, A.; Roto, R.; Triyana, K. Polyacrylamide Coated on Quartz Crystal Microbalance Electrodes for Highly Sensitive Sensor of Acetic Acid. Materials Science Forum 2019, 948 MSF, 254–259. [Google Scholar] [CrossRef]
- Cai, Jingfang; Yan, Ying; Wang, Weiwei; Ma, Yuanyuan; Cai, Lankun; Icon, L.W.; Hao, Z. Detection of Formic Acid and Acetic Acid Gases by a QCM Sensor Coated with an Acidified Multi-Walled Carbon Nanotube Membrane. Environ Technol 2021, 44.6, 751–761. [Google Scholar] [CrossRef]
- Wang, Y.C.; Sun, Z. Sen; Wang, S.Z.; Wang, S.Y.; Cai, S.X.; Huang, X.Y.; Li, K.; Chi, Z.T.; Pan, S. Di; Xie, W.F. Sub-Ppm Acetic Acid Gas Sensor Based on In2O3 Nanofibers. J Mater Sci 2019, 54, 14055–14063. [Google Scholar] [CrossRef]
- Li, G.; Su, Y.; Li, Y.Y.; Li, Y.X.; Guo, Z.; Huang, X.J.; Liu, J.H. Size-Tunable Ag Nanoparticles Sensitized Porous ZnO Nanobelts: Controllably Partial Cation-Exchange Synthesis and Selective Sensing toward Acetic Acid. Nanotechnology 2018, 29. [Google Scholar] [CrossRef] [PubMed]
- Khorramshahi, V.; Karamdel, J.; Yousefi, R. Acetic Acid Sensing of Mg-Doped ZnO Thin Films Fabricated by the Sol–Gel Method. Journal of Materials Science: Materials in Electronics 2018, 29, 14679–14688. [Google Scholar] [CrossRef]
- Jin, W.X.; Ma, S.Y.; Tie, Z.Z.; Li, W.Q.; Luo, J.; Cheng, L.; Xu, X.L.; Wang, T.T.; Jiang, X.H.; Mao, Y.Z. Synthesis of Hierarchical SnO 2 Nanoflowers with Enhanced Acetic Acid Gas Sensing Properties. Appl Surf Sci 2015, 353, 71–78. [Google Scholar] [CrossRef]
- Cheng, L.; Ma, S.Y.; Wang, T.T.; Luo, J.; Li, X.B.; Li, W.Q.; Mao, Y.Z.; Gz, D.J. Highly Sensitive Acetic Acid Gas Sensor Based on Coral-like and Y-Doped SnO2 Nanoparticles Prepared by Electrospinning. Mater Lett 2014, 137, 265–268. [Google Scholar] [CrossRef]
- Web Page Available online: https://www.gas-sensing.com/ati-acid-gases-sensor-00-1045.html.
- Chen, Y.; Yang, Y.; Liu, X.; Shi, X.; Wang, C.; Zhong, H.; Jin, F. Sustainable Production of Formic Acid and Acetic Acid from Biomass. Molecular Catalysis 2023, 545, 113199. [Google Scholar] [CrossRef]
- European Union Commission Commission Directive (EU) 2017/164 of 31 January 2017 Establishing a Fourth List of Indicative Occupational Exposure Limit Values Pursuant to Council Directive 98/24/EC, and Amending Commission Directives 91/322. 2000, EEC39.
- Angkawinitwong, U.; Williams, G.R. Electrospun Materials for Wearable Sensor Applications in Healthcare; Elsevier Ltd. 2020; ISBN 9780128196113. [Google Scholar]
- Das, R.; Zeng, W.; Asci, C.; Del-Rio-Ruiz, R.; Sonkusale, S. Recent Progress in Electrospun Nanomaterials for Wearables. APL Bioeng 2022, 6. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.; Gouma, P. Novel Electrospinning Process for Wearable Sensors. ECS Meeting Abstracts 2022, MA2022-01, 1035. [Google Scholar] [CrossRef]
- Massaglia, G.; Quaglio, M. Electrospun Nanofibers for Optimized Fiber-Shaped Wearable Sensors. In Proceedings of the IOCN 2023; MDPI: Basel, Switzerland, 5 May 2023; p. 55. [Google Scholar]
- Electrospinning for High Performance Sensors; Macagnano, A., Zampetti, E., Kny, E., Eds.; NanoScience and Technology; Springer International Publishing: Cham, 2015; ISBN 978-3-319-14405-4. [Google Scholar]
- Electrospinning, Wang, L., Qin, X., Eds.; Wiley. 2024; ISBN 9783527351978.
- Utkarsh; Hegab, H.; Tariq, M.; Syed, N.A.; Rizvi, G.; Pop-Iliev, R. Towards Analysis and Optimization of Electrospun PVP (Polyvinylpyrrolidone) Nanofibers. Advances in Polymer Technology 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Maciejewska, B.M.; Wychowaniec, J.K.; Woźniak-Budych, M.; Popenda, Ł.; Warowicka, A.; Golba, K.; Litowczenko, J.; Fojud, Z.; Wereszczyńska, B.; Jurga, S. UV Cross-Linked Polyvinylpyrrolidone Electrospun Fibres as Antibacterial Surfaces. Sci Technol Adv Mater 2019, 20, 979–991. [Google Scholar] [CrossRef]
- Wang, T.; Huang, D.; Yang, Z.; Xu, S.; He, G.; Li, X.; Hu, N.; Yin, G.; He, D.; Zhang, L. A Review on Graphene-Based Gas/Vapor Sensors with Unique Properties and Potential Applications. Nanomicro Lett 2016, 8, 95–119. [Google Scholar] [CrossRef]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh Electron Mobility in Suspended Graphene. Solid State Commun 2008, 146, 351–355. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.; Kim, S.; Hwang, J. Graphene-Based Two-Dimensional Mesoporous Materials: Synthesis and Electrochemical Energy Storage Applications. Materials 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Matatagui, D.; López-Sánchez, J.; Peña, A.; Serrano, A.; del Campo, A.; de la Fuente, O.R.; Carmona, N.; Navarro, E.; Marín, P.; del Carmen Horrillo, M. Ultrasensitive NO2 Gas Sensor with Insignificant NH3-Interference Based on a Few-Layered Mesoporous Graphene. Sens Actuators B Chem 2021, 335, 129657. [Google Scholar] [CrossRef]
- Han, T.H.; Huang, Y.-K.; Tan, A.T.L.; Dravid, V.P.; Huang, J. Steam Etched Porous Graphene Oxide Network for Chemical Sensing. J Am Chem Soc 2011, 133, 15264–15267. [Google Scholar] [CrossRef]
- Kanjwal, M.A.; Ghaferi, A. Al Graphene Incorporated Electrospun Nanofiber for Electrochemical Sensing and Biomedical Applications: A Critical Review. Sensors 2022, 22, 8661. [Google Scholar] [CrossRef]
- Avossa, J.; Paolesse, R.; Di Natale, C.; Zampetti, E.; Bertoni, G.; De Cesare, F.; Scarascia-Mugnozza, G.; Macagnano, A. Electrospinning of Polystyrene/Polyhydroxybutyrate Nanofibers Doped with Porphyrin and Graphene for Chemiresistor Gas Sensors. Nanomaterials 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Kausar, A.; Ahmad, I.; Zhao, T.; Aldaghri, O.; Ibnaouf, K.H.; Eisa, M.H. Nanocomposite Nanofibers of Graphene—Fundamentals and Systematic Developments. Journal of Composites Science 2023, 7, 323. [Google Scholar] [CrossRef]
- De, S.; Sahoo, S.; Das, A.K.; Nayak, G.C. Recent Progress in Electrospinning Technologies for Graphene-Based Materials. 2021; pp. 1–34. [Google Scholar]
- Al-Dhahebi, A.M.; Gopinath, S.C.B.; Saheed, M.S.M. Graphene Impregnated Electrospun Nanofiber Sensing Materials: A Comprehensive Overview on Bridging Laboratory Set-up to Industry. Nano Converg 2020, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yokota, T.; Someya, T. Electrospun Nanofiber-Based Soft Electronics. NPG Asia Mater 2021, 13, 22. [Google Scholar] [CrossRef]
- Guo, Z.; Huang, J.; Xue, Z.; Wang, X. Electrospun Graphene Oxide/Carbon Composite Nanofibers with Well-Developed Mesoporous Structure and Their Adsorption Performance for Benzene and Butanone. Chemical Engineering Journal 2016, 306, 99–106. [Google Scholar] [CrossRef]
- Storti, E.; Lojka, M.; Lencová, S.; Hubálková, J.; Jankovský, O.; Aneziris, C.G. Synthesis and Characterization of Graphene Nanoplatelets-Containing Fibers by Electrospinning. Open Ceramics 2023, 15. [Google Scholar] [CrossRef]
- Del Sorbo, G.; Truda, G.; Bifulco, A.; Passaro, J.; Petrone, G.; Vitolo, B.; Ausanio, G.; Vergara, A.; Marulo, F.; Branda, F. Non Monotonous Effects of Noncovalently Functionalized Graphene Addition on the Structure and Sound Absorption Properties of Polyvinylpyrrolidone (1300 KDa) Electrospun Mats. Materials 2018, 12, 108. [Google Scholar] [CrossRef]
- Kaczmarek, H.; Szalla, A.; Kamińska, A. Study of Poly(Acrylic Acid)–Poly(Vinylpyrrolidone) Complexes and Their Photostability. Polymer (Guildf) 2001, 42, 6057–6069. [Google Scholar] [CrossRef]
- Song, G.; Lin, Y.; Zhu, Z.; Zheng, H.; Qiao, J.; He, C.; Wang, H. Strong Fluorescence of Poly( N -Vinylpyrrolidone) and Its Oxidized Hydrolyzate. Macromol Rapid Commun 2015, 36, 278–285. [Google Scholar] [CrossRef]
- Louie, S.M.; Gorham, J.M.; Tan, J.; Hackley, V.A. Ultraviolet Photo-Oxidation of Polyvinylpyrrolidone (PVP) Coatings on Gold Nanoparticles. Environ Sci Nano 2017, 4, 1866–1875. [Google Scholar] [CrossRef]
- Chen, L.; Raohao, F.; Changchang, Z.; Xiang, L.; Changhua, Z. Fluorescent Enhancement of Polyethyleneimine Nano-Polymers and the Application in Cellar Imaging. Polym Degrad Stab 2019, 163, 7–14. [Google Scholar] [CrossRef]
- Khan, W.S.; Asmatulu, R.; Eltabey, M.M. Electrical and Thermal Characterization of Electrospun PVP Nanocomposite Fibers. J Nanomater 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Gillan, L. Inkjet Printed Metal Oxide Thin Film Transistors Incorporating Polyethyleneimine. 2017. [Google Scholar]
- Roizard, D. Antoine Equation. In Encyclopedia of Membranes; Springer: Berlin, Heidelberg, 2014; pp. 1–3. [Google Scholar]
- D’Amico, A.; Di Natale, C. A Contribution on Some Basic Definitions of Sensors Properties. IEEE Sens J 2001, 1, 183–190. [Google Scholar] [CrossRef]
- Chu, X.; Dai, P.; Dong, Y.; Sun, W.; Bai, L.; Zhang, W. The Acetic Acid Gas Sensing Properties of Graphene Quantum Dots (GQDs)–ZnO Nanocomposites Prepared by Hydrothermal Method. Journal of Materials Science: Materials in Electronics 2017, 28, 19164–19173. [Google Scholar] [CrossRef]
- Geng, W.; Ma, Z.; Yang, J.; Duan, L.; Li, F.; Zhang, Q. Pore Size Dependent Acetic Acid Gas Sensing Performance of Mesoporous CuO. Sens Actuators B Chem 2021, 334, 129639. [Google Scholar] [CrossRef]
- Aziz, N.A.; Abdullah, M.F.; Badaruddin, S.A.M.; Hussin, M.R.M.; Hashim, A.M. Highly Sensitive Sub-Ppm CH3COOH Detection by Improved Assembly of Sn3O4-RGO Nanocomposite. Molecules 2022, 27. [Google Scholar] [CrossRef] [PubMed]
- Khorramshahi, V.; Karamdel, J.; Yousefi, R. High Acetic Acid Sensing Performance of Mg-Doped ZnO/RGO Nanocomposites. Ceram Int 2019, 45, 7034–7043. [Google Scholar] [CrossRef]
- He, L.; Gao, C.; Yang, L.; Zhang, K.; Chu, X.; Liang, S.; Zeng, D. Facile Synthesis of MgGa2O4/Graphene Composites for Room Temperature Acetic Acid Gas Sensing. Sens Actuators B Chem 2020, 306, 127453. [Google Scholar] [CrossRef]
- Avossa, J.; Zampetti, E.; De Cesare, F.; Bearzotti, A.; Scarascia-Mugnozza, G.; Vitiello, G.; Zussman, E.; Macagnano, A. Thermally Driven Selective Nanocomposite PS-PHB/MGC Nanofibrous Conductive Sensor for Air Pollutant Detection. Front Chem 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Cheng, L.; Ma, S.Y.; Wang, T.T.; Luo, J.; Li, X.B.; Li, W.Q.; Mao, Y.Z.; Gz, D.J. Highly Sensitive Acetic Acid Gas Sensor Based on Coral-like and Y-Doped SnO2 Nanoparticles Prepared by Electrospinning. Mater Lett 2014, 137, 265–268. [Google Scholar] [CrossRef]
- Wu, J.; Wan, Y.; Wang, Z.; Wang, Y.; Luo, Q.; Feng, C.; Yoshinobu, T. Loose Ag-Doped LaFeO 3 Nanotubes-Based Gas Sensor for Excellent Acetic Acid Sensing. IEEE Sens J 2024, 1. [Google Scholar] [CrossRef]
- Jin, W.X.; Ma, S.Y.; Tie, Z.Z.; Li, W.Q.; Luo, J.; Cheng, L.; Xu, X.L.; Wang, T.T.; Jiang, X.H.; Mao, Y.Z. Synthesis of Hierarchical SnO2 Nanoflowers with Enhanced Acetic Acid Gas Sensing Properties. Appl Surf Sci 2015, 353, 71–78. [Google Scholar] [CrossRef]
- Zhang, J.; Liao, F.; Zhu, Y.; Sun, J.; Shao, M. Visible-Light-Enhanced Gas Sensing of CdSxSe1-x Nanoribbons for Acetic Acid at Room Temperature. Sens Actuators B Chem 2015, 215, 497–503. [Google Scholar] [CrossRef]
- Gautam, M.; Jayatissa, A.H. Detection of Organic Vapors by Graphene Films Functionalized with Metallic Nanoparticles. J Appl Phys 2012, 112. [Google Scholar] [CrossRef]
- Huang, X.Y.; Chen, K.; Xie, W.; Li, Y.; Yang, F.; Deng, Y.; Li, J.; Jiang, F.; Shu, Y.; Wu, L.; et al. Chemiresistive Gas Sensors Based on Highly Permeable Sn-Doped Bismuth Subcarbonate Microspheres: Facile Synthesis, Sensing Performance, and Mechanism Study. Adv Funct Mater 2023, 33, 1–12. [Google Scholar] [CrossRef]
- Bi, W.; Liu, S. Preparation of a Hierarchical 3D Structure Composed of Co-Doped SnO2 Nanosheets with Excellent Gas Sensitivity to Acetic Acid. Materials Science and Engineering: B 2022, 286, 116006. [Google Scholar] [CrossRef]
- Zappi, D.; Varani, G.; Iatsunskyi, I.; Wallaszkovits, N.; Bailer, J.; Giardi, M.T. High-Sensitivity Metal Oxide Sensors Duplex for On-the-Field Detection of Acetic Acid Arising from the Degradation of Cellulose Acetate-Based Cinematographic and Photographic Films. Chemosensors 2022, 10, 60. [Google Scholar] [CrossRef]
- Cao, P.F.; Ma, S.Y.; Fan, R.J. Carbon-Doped Porous Hollow Alpha-Fe2O3 Microtubules Controlled by Absorbent Cotton Bio-Template to Detect Acetic Acid Vapor. Ceram Int 2022, 48, 12729–12741. [Google Scholar] [CrossRef]
- Han, D. Sol-Gel Autocombustion Synthesis of Zinc Oxide Foam Decorated with Holes and Its Use as Acetic Acid Gas Sensor at Sub-Ppm Level. Ceram Int 2020, 46, 3304–3310. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Chu, X.; Liang, S.; Kong, L. Preparation of g–C3N4–SnO2 Composites for Application as Acetic Acid Sensor. J Alloys Compd 2020, 832, 153355. [Google Scholar] [CrossRef]
- Gauglitz, G. Analytical Evaluation of Sensor Measurements. Anal Bioanal Chem 2018, 410, 5–13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme of the electrospinning deposition process (a), UV-light curing (b) and MGC-PEI coating of NFs (c).
Figure 1.
Scheme of the electrospinning deposition process (a), UV-light curing (b) and MGC-PEI coating of NFs (c).
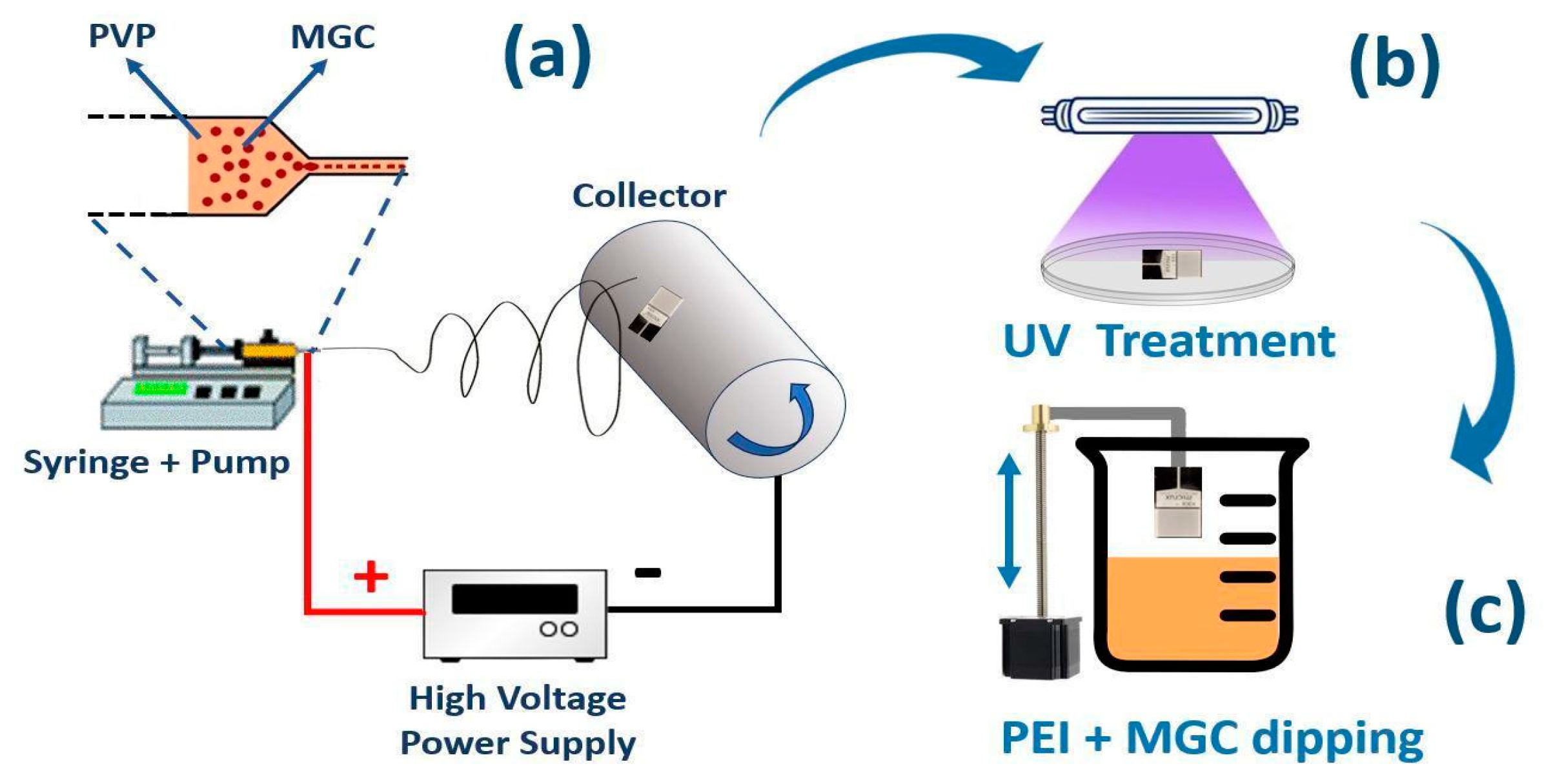
Figure 2.
Layout of the measurement tests performed with the stationary (a) and the portable (b) system.
Figure 2.
Layout of the measurement tests performed with the stationary (a) and the portable (b) system.
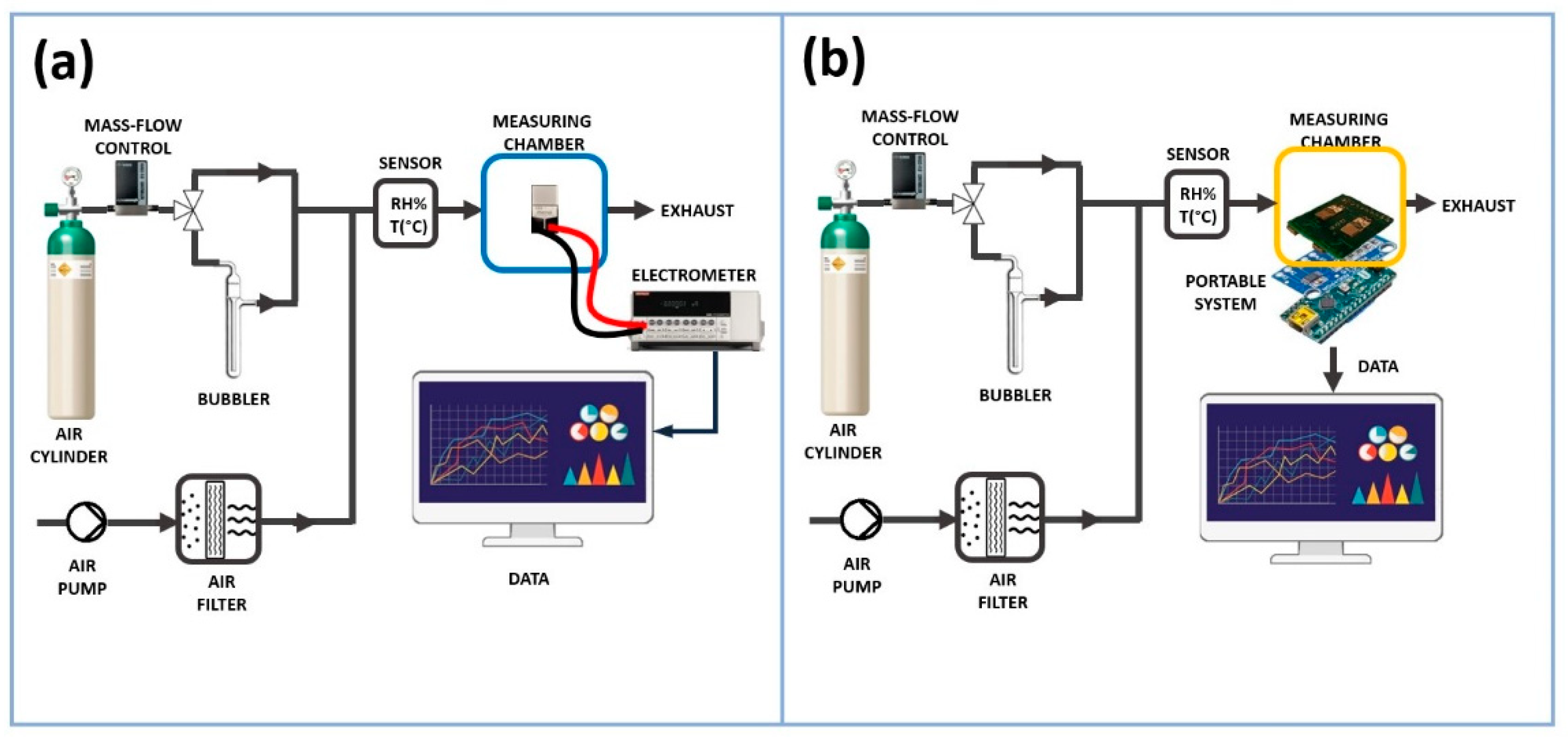
Figure 3.
Representation of the portable measurement system with a layered structure in analogue board (A), ADC module (B), microcontroller (C) and the power supply (D).
Figure 3.
Representation of the portable measurement system with a layered structure in analogue board (A), ADC module (B), microcontroller (C) and the power supply (D).
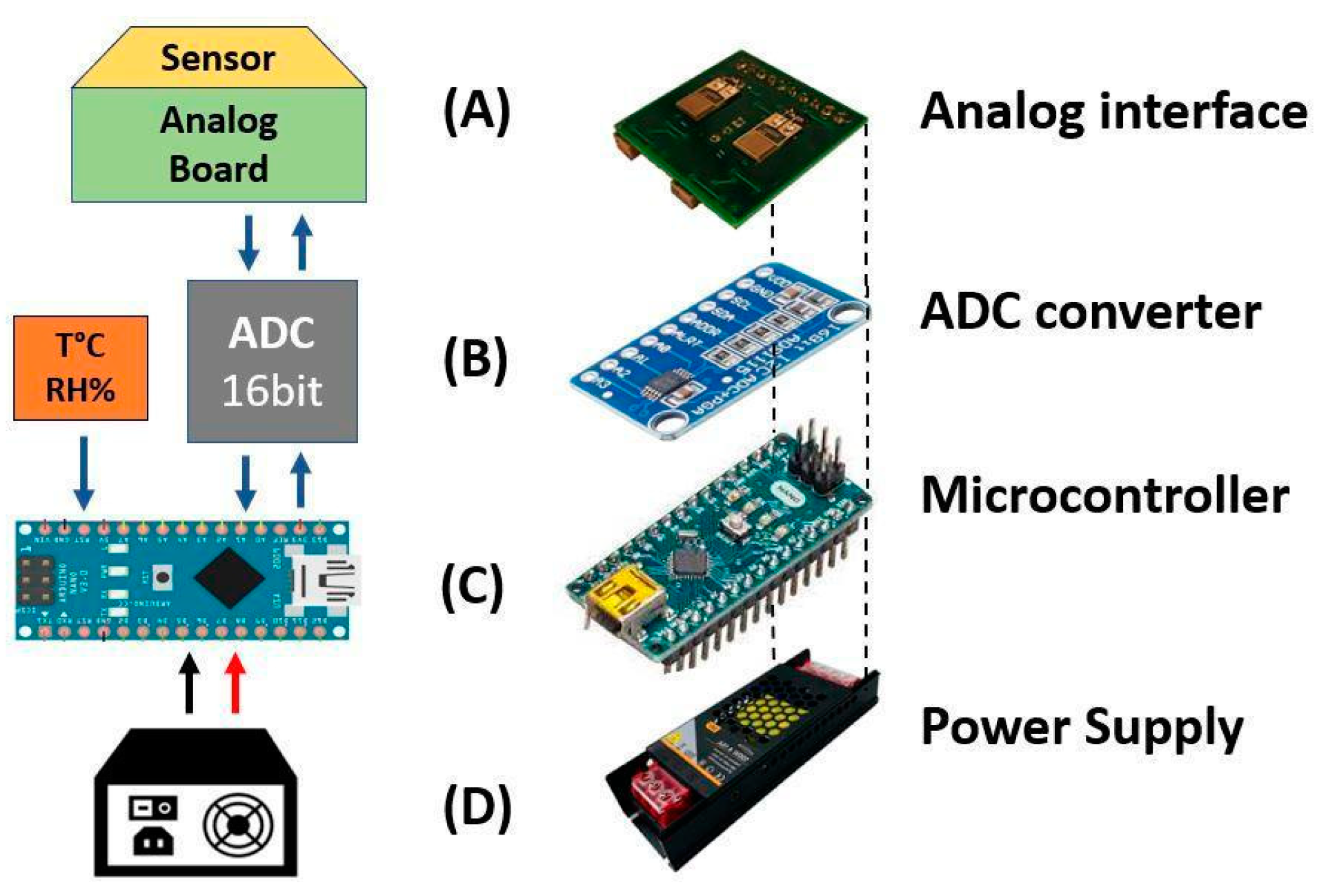
Figure 4.
– Optical microscope images depict the distribution of fibres on the IDE surface in brightfield (A, B, D) and SiO2 wafer slices under fluorescence (E, D). Specifically: (A, D) show the (PVP-MGC)UV/PEI-MGC nanofibers coating the electrodes following a 4-minute electrospinning deposition; (B) shows the (PVP-MGC)UV electrode nanofibrous coating before dipping; (E) displays the (PVP-MGC)UV nanofibrous layer from a 30-second deposition; and (F) illustrates the layer following the decoration with PEI-MGC.
Figure 4.
– Optical microscope images depict the distribution of fibres on the IDE surface in brightfield (A, B, D) and SiO2 wafer slices under fluorescence (E, D). Specifically: (A, D) show the (PVP-MGC)UV/PEI-MGC nanofibers coating the electrodes following a 4-minute electrospinning deposition; (B) shows the (PVP-MGC)UV electrode nanofibrous coating before dipping; (E) displays the (PVP-MGC)UV nanofibrous layer from a 30-second deposition; and (F) illustrates the layer following the decoration with PEI-MGC.
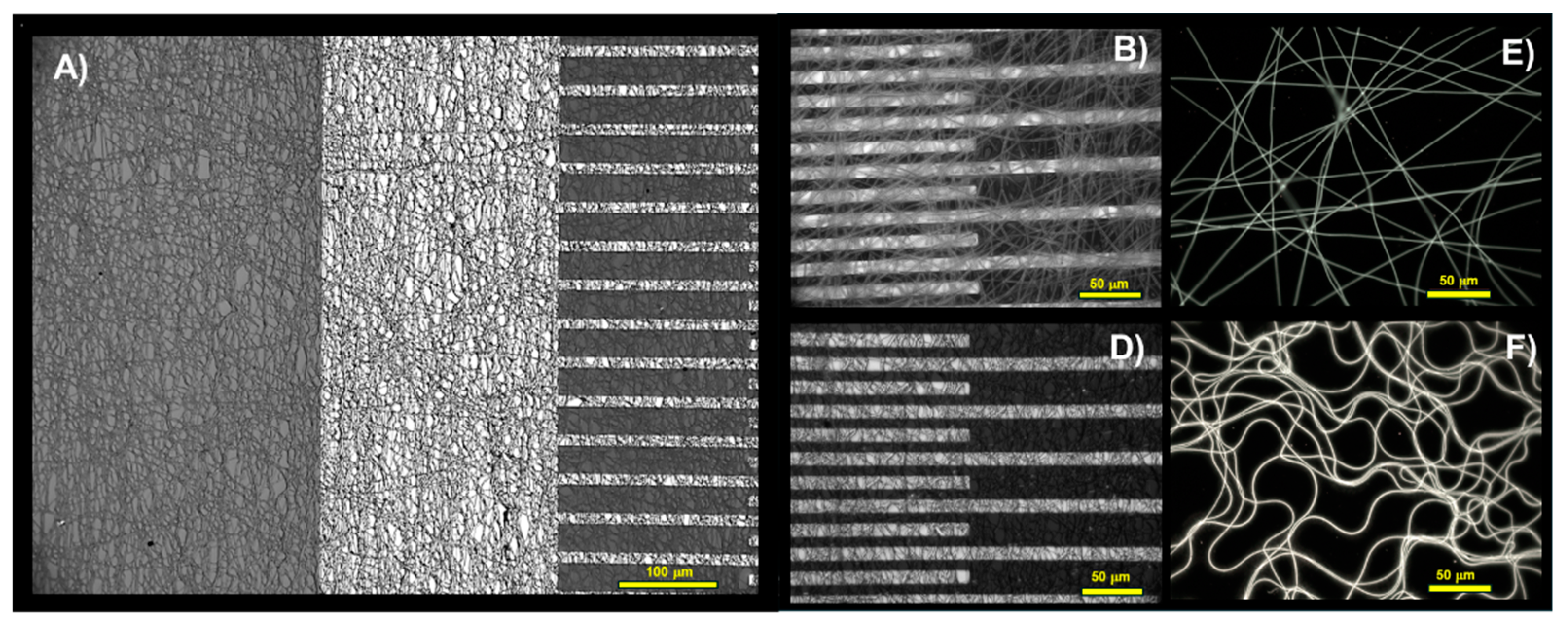
Figure 5.
SEM micrographs depict fibres of PVP-MGC after UV-light irradiation (A-inset) and subsequent PEI-MGC decoration through dipping (A), along with a magnified view of a section (B).
Figure 5.
SEM micrographs depict fibres of PVP-MGC after UV-light irradiation (A-inset) and subsequent PEI-MGC decoration through dipping (A), along with a magnified view of a section (B).

Figure 6.
- I/V measurement of the nanocomposite fibres before (PVP-MGC) and after the UV treatment (PVP-MGC)UV) (A); comparison between the I-V curves of (PVP-MGC)UV and (PVP-MGC)UV/PEI-MGC (B).
Figure 6.
- I/V measurement of the nanocomposite fibres before (PVP-MGC) and after the UV treatment (PVP-MGC)UV) (A); comparison between the I-V curves of (PVP-MGC)UV and (PVP-MGC)UV/PEI-MGC (B).
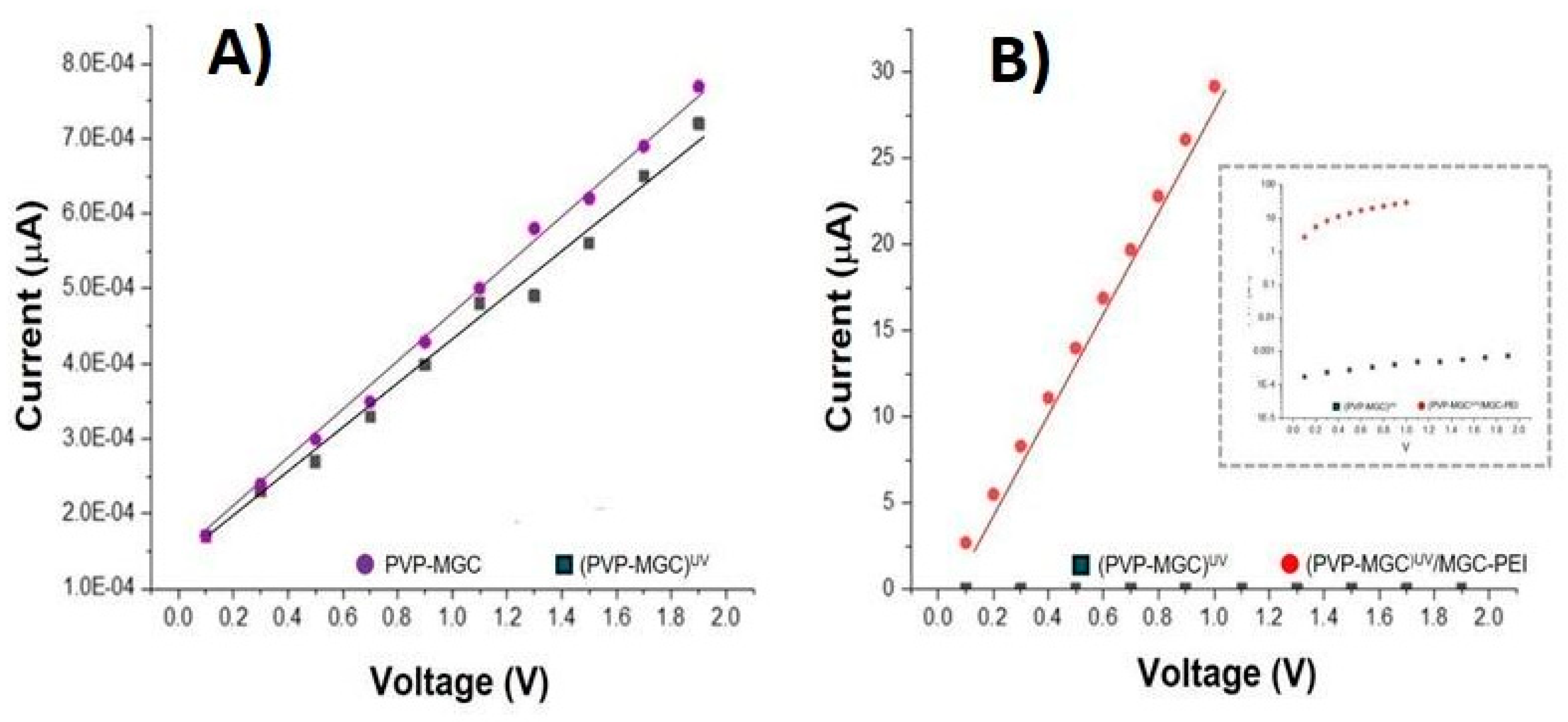
Figure 7.
Normalised transient responses (ΔI/I0) towards the highest concentration of VOCs measured (HAc: 30 ppm, FA: 440 ppm, NH3: 754 ppm, MeOH: 2245, EtOH: 930 ppm, Ac: 4126 ppm, TEA: 1349 ppm, ButA: 1691 ppm) (A) and normalised response curves plotted against increasing concentrations (ppm) of VOCs (B).
Figure 7.
Normalised transient responses (ΔI/I0) towards the highest concentration of VOCs measured (HAc: 30 ppm, FA: 440 ppm, NH3: 754 ppm, MeOH: 2245, EtOH: 930 ppm, Ac: 4126 ppm, TEA: 1349 ppm, ButA: 1691 ppm) (A) and normalised response curves plotted against increasing concentrations (ppm) of VOCs (B).
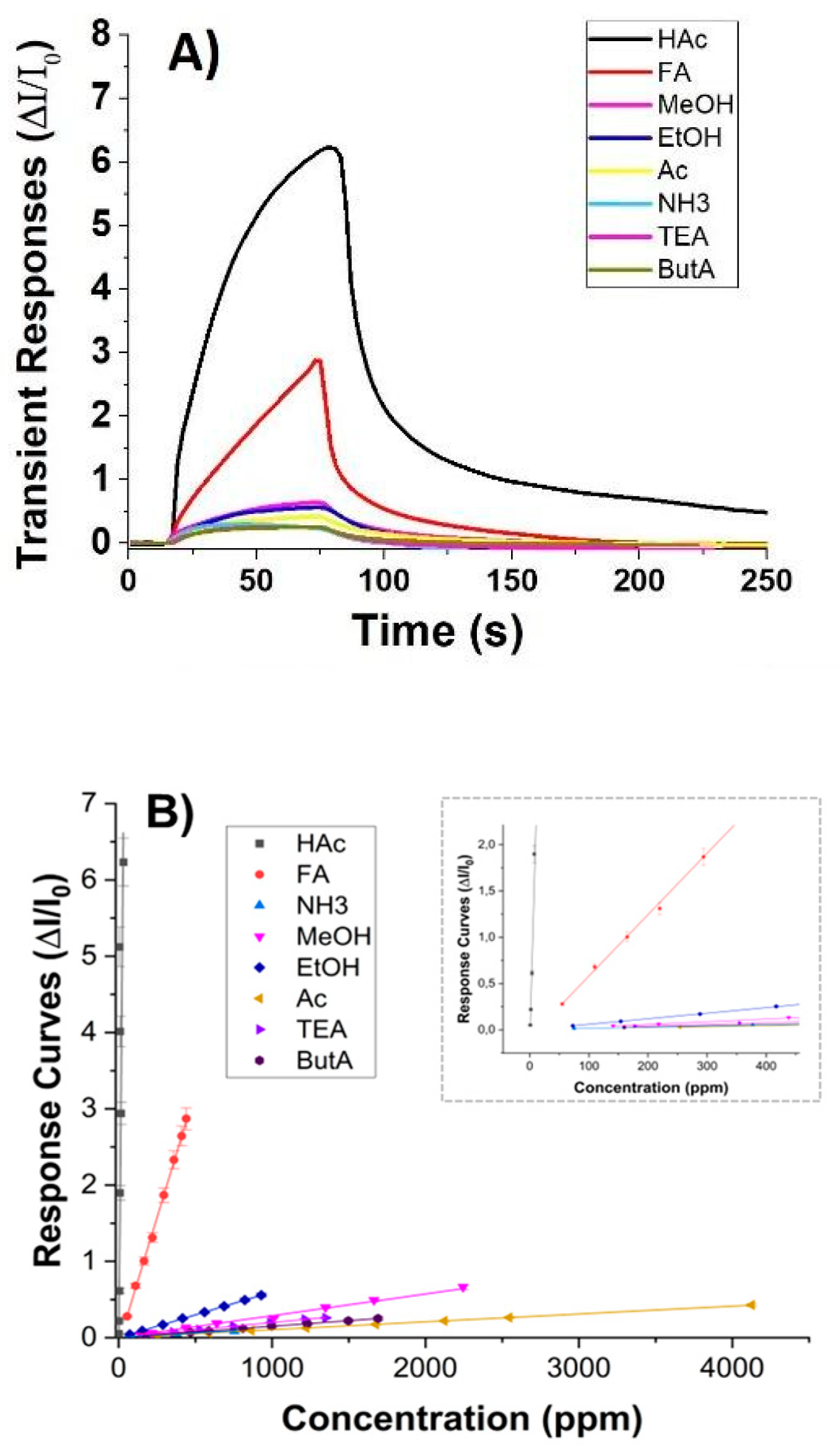
Figure 8.
Sensitivity of the sensor towards each vapour solvent (A); sensor responses to 15 ppm HAc in 30 days (B).
Figure 8.
Sensitivity of the sensor towards each vapour solvent (A); sensor responses to 15 ppm HAc in 30 days (B).
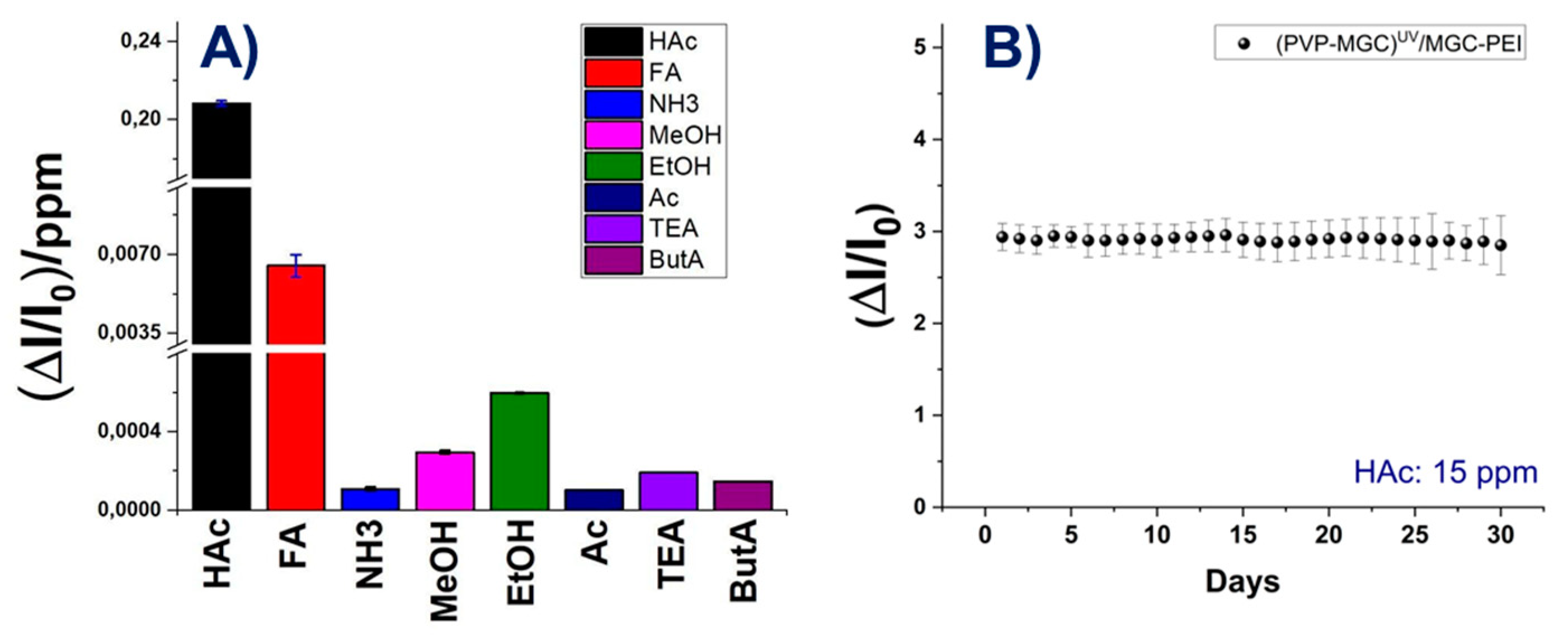
Figure 9.
Normalised (ΔV/V0) response-recovery curves of the sensor when exposed to increasing concentrations of acetic acid vapours, ranging between 7 and 47 ppm.
Figure 9.
Normalised (ΔV/V0) response-recovery curves of the sensor when exposed to increasing concentrations of acetic acid vapours, ranging between 7 and 47 ppm.
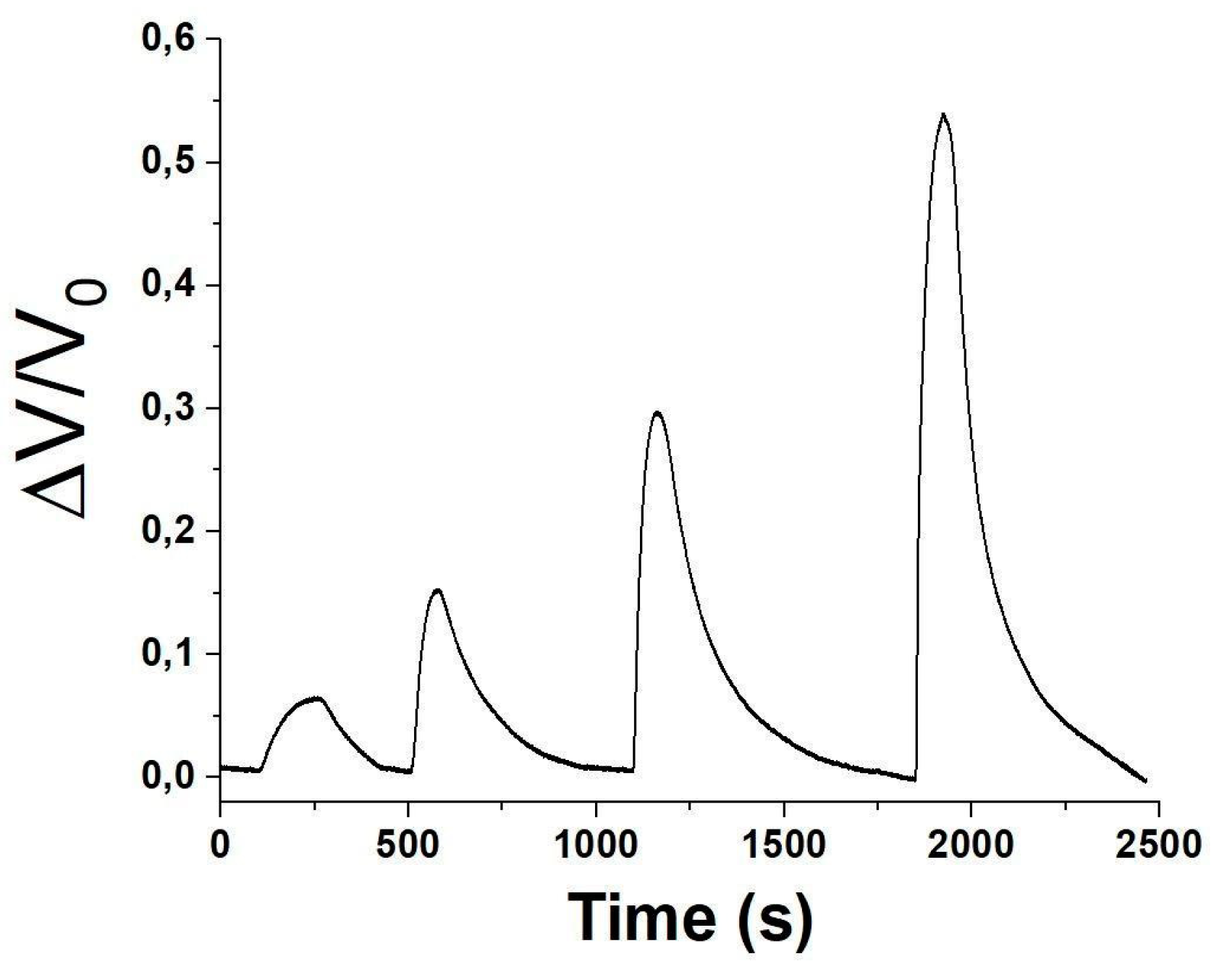
Figure 10.
Normalised voltage responses (ΔV/V0) to different acetic acid vapours concentrations (ppm).
Figure 10.
Normalised voltage responses (ΔV/V0) to different acetic acid vapours concentrations (ppm).
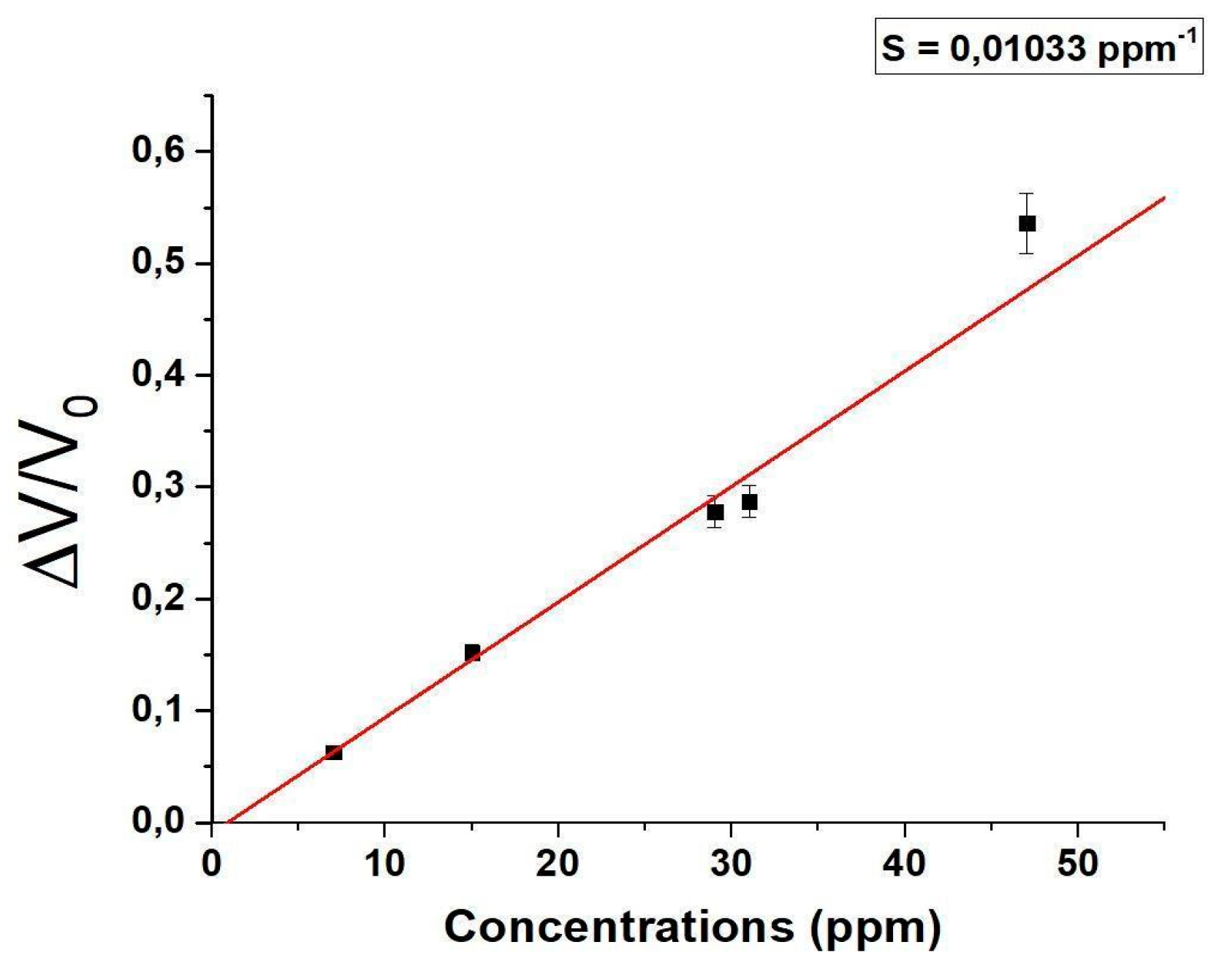

Table 1.
A summary of recent research on gas sensors for the detection of acetic acid.
| Sensing Material | Type of sensor | Temperature (°C) | LOD | Reference |
|---|---|---|---|---|
| Pr-doped ZnO | Chemiresistor | 380 | 50 ppm | [13] |
| Y-doped SnO2 | Chemiresistor | 300 | 10 ppm | [65] |
| Ag-doped LaFeO3 | Chemiresistor | 150 | 0,5 ppm | [66] |
| Flower-like SnO2 | Chemiresistor | 260 | 1 ppm | [67] |
| CdSxSe1−xnanoribbons | Chemiresistor | 100 | 0,87 ppm | [68] |
| Gr:Au and Gr:Pt | Chemiresistor | rt | 0.6%/ppm | [69] |
| Hexagonal ZnO | Chemiresistor | 230 | 10 ppm | [16] |
| Tb2O3@MOF- ZnO | Chemiresistor | 20°C | 0,5 ppm | [17] |
| Bi2O2CO3 | Chemiresistor | 150°C | 1 ppm | [70] |
| Co-doped SnO2 | Chemiresistor | 300°C | 10 ppm | [71] |
| metal oxide (WO/SnO) | Chemiresistor | rt | 30 ppb | [72] |
| Sn3O4-RGO | Chemiresistor | rt | 64%/ppm | [61] |
| C-doped α-Fe2O3 | Chemiresistor | 260°C | 1 ppm | [73] |
| Y-doped ZnO | Chemiresistor | 350°C | 10 ppb | [12] |
| mesoporous CuO | Chemiresistor | 200°C | 10 ppm | [60] |
| BaSnO3 microtubes | Chemiresistor | 245°C | 0,3 ppm | [15] |
| ZnO foam | Chemiresistor | 400°C | 0,5 ppm | [74] |
| GeC3N4eSnO2 | Chemiresistor | 185°C | 0,1 ppm | [75] |
| MgGa2O4/graphene | Chemiresistor | rt | 1 ppb | [63] |
| In2O3 nanofibers | Chemiresistor | 250°C | 500 ppb | [21] |
| Mg-doped ZnO/rGO | Chemiresistor | 250°C | 10 ppm | [62] |
| GQDs–ZnO | Chemiresistor | rt | 1 ppm | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



