
Submitted:
02 March 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
The retinal pigment epithelium (RPE), which ensures the normal functioning of the neural retina, is a pigmented single-cell layer that separates the retina from the Bruch's membrane and the choroid. There are three main types of pigment granules in the RPE cells of the human eye: lipofuscin granules (LG) containing the fluorescent “age pigment” lipofuscin, melanoprotein granules (melanosomes, melanolysosomes) containing the screening pigment melanin and complex melanolipofuscin granules (MLG) containing simultaneously both types of pigments - melanin and lipofuscin. This review examines the functional role of pigment granules in the aging process and in the development of oxidative stress and associated pathologies in RPE cells. The focus is on the process of light-induced oxidative degradation of pigment granules caused by reactive oxygen species. The reasons leading to increased oxidative stress in RPE cells as a result of oxidative degradation of pigment granules are considered. A mechanism has been proposed to explain the phenomenon of age-related decline in melanin content in RPE cells. The essence of the mechanism is that when the lipofuscin part of the melanolipofuscin granule is exposed to light, reactive oxygen species are formed, which destroy its melanin part. As more melanolipofuscin granules are formed with age and the development of degenerative diseases, the melanin in pigmented epithelial cells ultimately disappears.
Keywords:
Retinal Pigment Epithelium
; Reactive Oxygen Species
; Lipofuscin
; Bisretinoids
; Oxidative stress
; Melanin
; Melanolipofuscin
; Aging
1. Introduction
The retinal pigment epithelium (RPE) is a monolayer of epithelial cells closely adjacent on one side to the cells of the neural retina, and on the other side to the layer of choroidal capillaries [1,2,3]. On their apical surface, RPE cells have very long and thin microvilli, which project into the interphotoreceptor matrix, where they interact with the outer segments of photoreceptor cells, the rods and cones. On their basal surface, RPE cells are separated from the choriocapillaris layer by Bruch's membrane (Figure 1). The RPE basal membrane has numerous folds and is part of Bruch's membrane, which consists of five different layers and separates RPE cells from blood vessels. This unique position of RPE cells, which affects metabolite exchange between photoreceptor cells and blood vessels, determines the main functions of this tissue. These functions include phagocytosis of shed photoreceptor outer segments, transport and removal of metabolites from photoreceptor cells, regulation of vitamin A metabolism and control of the visual cycle, absorption of scattered light, regulation of ion flows, production of growth factors for photoreceptors, and maintenance of the blood-retinal barrier [3,4,5].
Post-mitotic RPE cells undergo significant morphological changes with age. Thus, a decrease in the density of RPE cells is observed due to their loss [6,7].
Figure 1.
Scheme of the structure of the fundus. RPE – retinal pigment epithelium; IPM – interphotoreceptor matrix [8].
Figure 1.
Scheme of the structure of the fundus. RPE – retinal pigment epithelium; IPM – interphotoreceptor matrix [8].
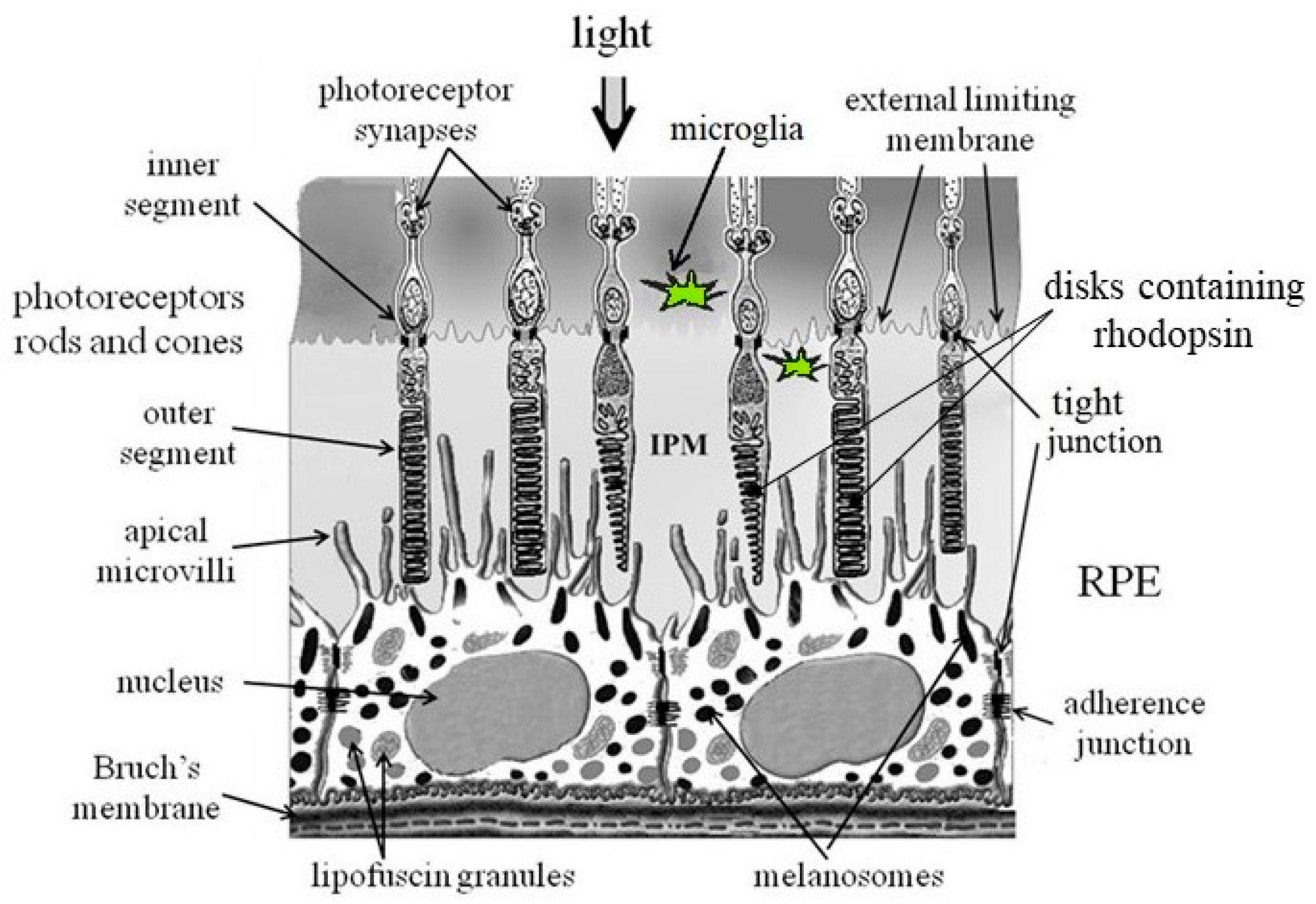
Outside the macular region, RPE cells increase in width and decrease in height, resulting in an overall thinning of the peripheral cell layer [9,10,11]. Cell cytoplasm decreases in volume, vacuolates, and the cells become pleomorphic in terms of size, volume, and content of nuclei and pigment granules [9,12]. Some RPE cells become multinucleated with age, especially outside the macular region [13]. Changes in the cytoskeleton of RPE cells are also observed. They lose their hexagonal shape and become larger and more elongated with age [14]. In addition, age-related RPE cells show an increase in the number of residual bodies (non-recyclable residues resulting from disfunction of the phagocytosis process), accumulation of basal deposits on Bruch’s membrane, thickening of Bruch’s membrane, atrophy of RPE cell microvilli, and formation of drusen between the basement membrane of the RPE and the inner collagen layer of Bruch’s membrane. [4,15]. However, despite changes in RPE cell structure and density during normal aging, the cell monolayer remains intact [16].
Changes in RPE cells become more pronounced with increased oxidative stress and the development of chronic ocular diseases such as cataracts, glaucoma, age-related macular degeneration (AMD), Stargardt disease and diabetic retinopathy [11,17,18,19]. Thus, AMD is characterized by chronic and progressive degeneration of RPE cells, photoreceptors, and retinal neurons [20]. In this case, the characteristic histological changes are expressed to a much greater extent than in aging RPE cells [21,22,23]. Characteristic histological features in RPE cells at early and intermediate stages of AMD are the presence of drusen on the basal side of the RPE along Bruch's membrane, accumulation of basal deposits within Bruch's membrane, and pigment abnormalities within the RPE cells themselves [16,24,25]. In addition, extracellular deposits between photoreceptors and the RPE, called reticular pseudodrusen, are observed [25]. Deposition of soft drusen with age in the macular region of the retina is thought to precede the development of AMD and lead to vision loss [4].
2. Cytotoxic Properties of Lipofuscin Granules and Its Role in the Development of Oxidative Stress in RPE Cells
Lipofuscin granules accumulate in RPE cells during aging and typically remain there until the end of life and can occupy up to 30% of the volume of the RPE cell cytoplasm [31,32]. The intensity of LG accumulation in RPE cells increases with the development of a number of ocular pathologies, such as Stargardt disease, Best’s macular dystrophy, and retinitis pigmentosa [19,33,34,35,36,37,38,39,40]. AMD has also been suggested to be associated with progressive LG accumulation [41,42,43]. There may also be increased accumulation of complex melanolipofuscin granules (MLG) in AMD [44,45]. LG in the RPE is a by-product of phagocytosed shed photoreceptor outer segments, which accumulates as a result of their incomplete lysosomal digestion [46]. LGs are heterogeneous and consist of a mixture of lipids, minor amounts of proteins modified and oxidized by end products of glycation and lipid peroxidation, and bisretinoid fluorophores that absorb light in the blue region of the spectrum and determine the autofluorescent properties of the granules [41,47,48,49,50,51]. Structurally, LGs are yellow-orange granules surrounded by a lipid membrane and having average diameter about 1.0 micron [52,53,54,55].
It is well known that LGs produce reactive oxygen species (ROS), such as superoxide radicals and singlet state oxygen, when exposed to visible light, especially in the blue-green region of the spectrum [56,57,58,59,60]. ROS generated by LGs under the influence of light stimulate the oxidation of lipids and proteins and can cause the development of oxidative stress in the RPE [60,61,62,63,64,65]. For example, irradiation of a culture of RPE cells loaded with lipofuscin granules leads to an increase in oxidized lipids and proteins and consequent damage to intracellular structures [43,66,67]. Oxidative stress is a chronic cellular condition in which pro-oxidant factors, such as ROS, suppress the activity of cellular antioxidant defense systems and initiate damage to intracellular proteins and lipids, leading to cell dysfunction. The phototoxicity of LG is due to the presence in them bisretinoid fluorophores, one of which, N-retinylidene-N-retinyl ethanolamine (A2E), has been shown to be localized not only in the RPE, but also in lysosomes [68,69], and to a lesser extent in mitochondria, the Golgi apparatus and cytoplasmic membrane [70]. A2E may accumulate in RPE cells during aging [71] and act as an auto-oxidant by increasing oxidative stress [72] through photogenerating of ROS such as superoxide radicals [73,74,75] and singlet oxygen [73,76]. In addition, due to its chemical structure, A2E can act as an amphiphilic detergent that can destroy the membrane structures of intracellular RPE organelles and induce apoptosis [60,77,78,79].
In the presence of oxygen, irradiation of LG leads to the oxidation of bisretinoids and the formation of toxic products, namely epoxides, peroxides, aldehydes and ketones [80,81,82,83,84]. The formation of aldehydes and ketones in human LG irradiated with visible light in the blue-green region of the spectrum has been demonstrated by TOF-SIMS mass spectrometry and femtosecond broadband CARS [85]. Among the carbonyl products formed during the oxidation of LG, lipid peroxidation products such as reactive aldehydes and dialdehydes have also been found: 4-hydroxy-nonenal (4-NHE) and malondialdehyde (MDA) [48,86]. Active carbonyls formed by both during photoinduced oxidation of LG and oxidation of LG by superoxide radicals can remain either inside lipofuscin granules (hydrophobic oxidation products) [81,84,87] or diffuse into the cell cytoplasm (hydrophilic and amphiphilic oxidation products) [84]. It has been shown that water-soluble products of photoinduced oxidation of LG and A2E can modify water-soluble proteins as well as proteins and lipids of the outer segments of photoreceptors with the formation of fluorescent Schiff bases [84,88]. These water-soluble carbonyls are extremely toxic [89] and are believed to be precursors for the formation of advanced glycation end products (AGEs) [90,91]. AGE products are suggested to be the initiators of the development of age-related cellular dysfunction, since they cause the formation of covalent protein cross-links, leading to a decrease in protein mobility and solubility, a decrease in enzymatic activity, and loss of receptor recognition function [92,93]. It is known that AGE products such as pentosidine, carboxymethyllysine and carboxyethyllysine accumulate with age in significant quantities in Bruch's membrane [94]. Oxidatively damaged molecules such as carboxyethylpyrrole, MDA, 4-NHE and AGEs can accumulate in the macular area and are sources of oxidative stress [95,96,97,98]. Damage to proteins and lipids during oxidative stress can lead to inhibition of the process of utilization of photoreceptor outer segments phagocytosed by the RPE and an increase in protein resistance to lysosomal proteinases [97,99,100].
Modified and damaged proteins, that cannot be repaired by heat shock proteins, are utilized in proteasomes and by autophagy. Autophagy is also used by RPE cells to recycle damaged mitochondria. Autophagy is a complex lysosomal process of clearance to eliminate large damaged molecular structures such as intracellular organelles (mitochondria, endoplasmic reticulum, peroxisomes), ubiquitinated macromolecules and pathogens [16,101,102]. However, when the activity of lysosomal enzymes decreases, for example under oxidative stress, the autophagy process declines, resulting in the accumulation of endogenous pathogenicity and danger signals (DAMPs) in cells, which can activate the inflammasome [103]. Endogenous molecular patterns associated with danger typically represent damaged macromolecules that can accumulate with age as a result of increased production (oxidative stress) and/or insufficient clearance [104,105]. The NLRP3 inflammasome is an important link in the development of the inflammatory response to tissue damage, and during aging, it is an important factor in the development of so-called “inflammaging”, when the immune reactions necessary for tissue repair, as damage accumulates, turn into a chronic non-adaptive form [106].
The NLRP3 inflammasome, present in human RPE cells [107,108] consists of three components: the cytoplasmic NOD-like receptor (NLR), the adapter protein ASC, and the cysteine proteinase caspase-1 in an inactive form. When DAMP interacts with the inflammasome receptor (Figure 2), caspase is activated and converts the pro-inflammatory cytokine interleukin-1β (IL-1β) into its active form [109,110].
Activation of the inflammatory process, which in its early stages manifests as low-grade parainflammation associated with the recruitment of microglial macrophages [111]. The complement cascade is also activated by RPE cells [112,113,114], which can express various components and complement factors (C3, C5, CFF, CFH), which, in turn, trigger the secretion of proinflammatory cytokines [115,116]. Resident mononuclear phagocytes, microglia, are the dominant type of immune cells found in the nerve fiber layer, as well as in the inner and outer plexiform layers of the retina. They are thought to remove cellular debris from the subretinal space to protect photoreceptors and RPE from injury and death [117]. Activated microglial cells are recruited into the subretinal space, which promotes the removal of toxic aggregates [118].
The development of the inflammatory process in the early stages is successfully regulated by RPE cells due to their release of anti-inflammatory factors, including complement inhibitors and anti-inflammatory cytokines, such as IL-10 [119,120]. However, aging and the progression of pathologies such as AMD are accompanied by increased activation of microglia and prolongation of their presence in the subretinal space, which potentially contributes to the damage of photoreceptor and RPE cells and consequently increased inflammatory process [16,121]. When large amounts of DAMPs accumulate, the inflammatory process intensifies, the blood-retinal barrier breaks down, and myeloid cells from the peripheral circulation are recruited and infiltrate from the bloodstream into the RPE, including monocytes, tissue macrophages, and dendritic cells [122], which cause an inflammatory form of programmed cell death, pyroptosis [123].
All this indicates the importance of the balance between oxidative and antioxidant factors in RPE cells, regulating the development of oxidative stress. Melanin-containing organelles, which perform the function of protecting against light-induced damage in the retina, play a great role in the regulation of the oxidative balance in RPE cells.
3. Protective Role of Melanin-Containing Organelles in RPE Cells
The protective effect of melanin in RPE cells is associated with, firstly its screening of the photosensitive elements of the retina by absorbing excess light and dissipating its energy in the form of heat; secondly, with binding of both endogenous (photo)toxic molecules formed in the retina and RPE, and exogenous xenobiotics into inactive complexes and thirdly with antioxidant and antiradical activities [8,124,125,126]. Melanosomes contain the pigment melanin, which is black or dark brown. Melanosomes are important pigment organelles of RPE cells. They develop from premelanosomes in the early stages of ontogenesis. Premelanosomes, in turn, are formed from endosomes of the Golgi complex [127]. Spherical or elongated melanosomes are observed in the RPE. The former are found mainly in the apical part of the cell and in microvilli, while the latter are localized in the middle part of the cytoplasm. In the basal region of RPE cells, melanosomes are rare. The size of the human RPE melanosome is 2.3 ± 0.5 µm in length and 0.9 ± 0.1 µm in diameter [128]. Human RPE melanosomes unlike uveal melanocytes contain mainly eumelanin-type pigment [129,130]. Eumelanins are irregular polymers containing indole-5,6-quinone monomer units in an oxidized or reduced state and having a stable ESR signal with a high concentration of paramagnetic centers.
Melanins in the eyes of vertebrates and humans perform the function of protection against the damaging effects of scattered light. They absorb light in a wide band of visible and ultraviolet irradiation, and the degree of absorption monotonically increases with decreasing wavelength of light [124]. However, there are no absorption maxima or minima in either the visible or ultraviolet regions of the eumelanin spectrum. Most of the light energy absorbed by melanin is quickly converted into heat through the mechanism of internal conversion. Light that passes through the layers of the neural retina is absorbed by melanosomes located in the apical region of the RPE cells. It is estimated that the RPE absorbs about 34–60% of incident light in the foveal region and about 21–40% in the equatorial region [131]. As a result, the risk of potentially dangerous photochemical reactions is significantly reduced.
Absence or deficiency of melanin, albinism, is known to significantly increase the risk of light damage to the retina, primarily its photoreceptor cells and RPE cells. Albinos are extremely sensitive to the damaging effects of light [132]. Albino eyes are highly sensitive not only to light, but also to ischemia and various pro-oxidants [133,134]. All these effects appear to be associated with a deficiency or absence of melanin, a decrease or complete absence of its protective effect, including screening of photosensitive structures from excess light [8,131], antioxidant protection [127,135,136,137,138] and protection from the action of toxic molecules, including bisretinoids, by their binding into inactive complexes [139,140].
As previously noted, RPE cells undergo significant biochemical and morphologic changes during the aging process, including the accumulation of the “age pigment” lipofuscin and complex pigment granules, such as melanolipofuscin granules, while simultaneously decreasing the number of melanin-containing melanosomes. While melanosomes occupy about 8% of the RPE cell volume before the age of 20 years, this volume gradually decreases to 3.5% between the ages of 41-90 years [30,31,141]. At ages 90-101 years, melanosomes are almost completely replaced by mixed melanolipofuscin granules [142,143]. It has also been shown that melanin pigmentation in the periphery of the retina declines with age [4,9,12]. This indicates the processes of age-related biodegradation of melanosomes in RPE cells. A decrease in melanin concentration in RPE cells is an important factor leading to increased oxidative stress in the cell [29,144,145,146]. The development of AMD is also accompanied by a decrease in melanin content of RPE cells, and the melanin content is inversely proportional to the degree of AMD development [147,148]. An increase in oxidative stress with a decrease in melanin concentration may also be due to the fact that melanin degradation produces products with pro-oxidant properties [145,149,150,151]. Degradation of the melanin polymer molecule leads to a decrease in its antiradical activity and the appearance of fluorescent decomposition products exhibiting toxic and phototoxic properties [138,152,153,154,155,156].
4. About the Mechanisms of Age-Related Decrease in Melanin Concentration
Based on the known physicochemical properties of melanin, it can be assumed that it is resistant to degradation caused by enzymatic reactions. Most likely, melanin degradation can be caused by exposure to light quanta or chemical oxidants such as ROS. Indeed, oxidative destruction of melanin in melanosomes has been shown to occur with age. Thus, it has been shown that in elderly and old people there is an increase in fluorescence and an increase in the absorption of oxygen by RPE melanosomes [29,157]. The degradation of melanin by ultraviolet irradiation and/or hydrogen peroxide is known to produce fluorescent decomposition products [158,159,160]. It has been shown that the melanin irradiation with intense visible light and ultraviolet leads to pigment degradation [161,162]. However, to achieve this effect, high irradiation energy and long exposures are necessary. Melanin irradiation with low-intensity light does not lead to its degradation [162]. It should be noted that the structures of the eye – the cornea, lens and vitreous body – virtually do not transmit short-wave ultraviolet radiation to RPE cells containing melanin. It is also unlikely that visible light of such high intensity and duration, which is used to destroy melanin in the experiment, would ever affect the retina in vivo. Therefore, degradation of melanin in RPE melanosomes for these reasons is usually excluded for the eye, but may occur for melanin in hair and skin exposed to direct sunlight.
On the contrary, oxidizing agents such as superoxide and hydrogen peroxide can relatively easily cause melanin degradation, which is accompanied by a loss of its antioxidant activity [145,155,162]. Thus, it has been shown that superoxide radicals cause gradual destruction of melanosomes, accompanied by a drop in the ESR signal. When melanosomes are completely destroyed, a transparent solution is formed, paramagnetic centers in melanosomes disappear, and they lose their antioxidant properties [145]. As has been repeatedly mentioned, superoxide and other reactive oxygen species in large amounts can be formed in RPE cells by lipofuscin granules and bisretinoids under the influence of light, and are apparently the key molecules initiating the entire process of melanin degradation. It is highly likely that such a process may occur in mixed granules containing both lipofuscin and melanin simultaneously. Such complex granules, melanolipofuscin granules, indeed contain on average 45% less melanin than melanosomes, as shown by ESR studies for pigment granules obtained from two age groups (Figure 3, Table 1) [145,163].
Since it is generally believed that MLG is formed by the fusion of a melanosome with a lipofuscin granule [31,142], this drop in melanin concentration in the MLG granule can be explained by the degradation of melanin [145]. Indeed, we have recently shown by mass spectrometry (ToF-SIMS) using principal component analysis (PCA) that the products of oxidative degradation of RPE melanosome caused by superoxide radicals in the dark are also present in the water-soluble fraction of blue-irradiated RPE melanolipofuscin granules [156]. It is logical to assume that this decrease is associated with the degradation of melanin in the MLG through interaction with ROS generated by the lipofuscin part of the granule (Figure 4, 1).
A similar process of melanin degradation can occur in other melanin-containing granules if ROS generators are present in them simultaneously. Such generators can be either bisretinoids or photosensitive products of melanin degradation (PMD), which have recently been shown to be capable of photo-induced generation of superoxide [156]. It is known that melanosomes in RPE cells can fuse with almost any phagocytosed material [166,167,168] including, as might be expected, bisretinoids formed during the visual cycle [169]. Due to the photooxidative properties of bisretinoids, they may also be involved in the degradation of melanin [168]. PMDs, which are formed during partial degradation of melanin and are capable of photoinduced generation of superoxide [156], can participate in further degradation of the intact part of the melanin polymer and in the process of oxidation of lipofuscin (bisretinoids) (Figure 4, 2).
It has recently been shown that when melanin is exposed to exogenous free radicals (superoxide, nitric oxide), melanin enters to a high-energy state (“chemiexcitation”), in which the pigment causes degradation of lipofuscin [164,165]. Such mechanisms for removing lipofuscin using activated melanin can lead to degradation of the latter [165]. It can be speculated that the melanin polymer, contained in the granule simultaneously with bisretinoids and PMDs, changes into an excited state when exposed to light, which can lead to autodestruction of the granule (Figure 4, 3) and a decrease in the concentration of melanin in the RPE cell.
5. Conclusion
The role of melanosomes in the cells of the retinal pigment epithelium of the human eye cannot be overestimated. The disappearance of melanosomes (decrease in antioxidant protection) and the accumulation of lipofuscin granules (strengthening of ROS production) in RPE cells during aging and pathologies can lead to increased oxidative stress. Therefore, excessive accumulation of lipofuscin, as well as mixed melanolipofuscin granules in RPE cells is an important pathogenetic factor. To explain the phenomenon of age-related decline in melanin content in RPE cells, we propose a mechanism in accordance of that when light acts on the lipofuscin part of the melanolipofuscin granule, reactive oxygen species are formed, which destroy its melanin part. Since more and more melanolipofuscin granules are formed with age and with the development of degenerative diseases, melanin in pigment epithelial cells ultimately disappear. The disappearance of melanin, as well as melanosomes themselves as screening light filters and antioxidants, significantly increases the risk of developing oxidative and especially photo-oxidative stress in the structures of the eye.
Author Contributions
AD, MO—conceptualization, acquiring, analyzing data from the reviewed literature, writing and editing original draft preparation; Both authors approved the final version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation, grant agreement no. 075-15-2020-773 of 30.09.2020
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Declarations
The authors declare no competing interests.
Abbreviations
Retinal Pigment Epithelium (RPE), Age-related macular degeneration (AMD), Lipofuscin granules (LG), Melanolipofuscin granules (MLG), Reactive Oxygen Species (ROS), N-Retinyl-N- Retinyliden ethanolamine (A2E), Time-of-Flight Secondary Ion Mass Spectrometry (ToF SIMS), Coherent Anti-Stokes Raman Scattering (CARS), Hydroxynonenal (HNE), Malondialdehyde (MDA), Advanced Glycation End products (AGEs), Danger Associated Molecular Patterns (DAMP), NOD-Like Receptor (NLR), Interleukin (IL), Electron Spin Resonance (ESR), Principal Component Analysis (PCA), Products of Melanin Degradation (PMD).
References
- Bok, D.; Young, R. Phagocytic properties of the retinal pigment epithelium. In The Retinal Pigment Epithelium; Zinn, K.M., Marmor, M.F., Eds.; Harvard University Press: Cambridge, MA, USA, 1979; pp. 148–174. [Google Scholar]
- Steinberg, R.H.; Wood, I. The relationship of the RPE to photoreceptor outer segments in human retina. In The Retinal Pigment Epithelium; Zinn, K.M., Marmor, M.F., Eds.; Harvard University Press: Cambridge, MA, USA, 1979; pp. 32–42. [Google Scholar]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef]
- Bonilha, V.L. Age and disease-related structural changes in the retinal pigment epithelium. Clin. Ophthalmol. 2008, 2, 413–424. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Hyttinen, J.; Ryhanen, T.; Viiri, J.; Paimela, T.; Toropainen, E.; Sorri, I.; Salminen, A. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front Biosci-Landmark (Elite Ed). 2010, 2, 1374–1384. [Google Scholar] [CrossRef]
- Gao, H.; Hollyfield, J.G. Aging of the human retina. Differential loss of neurons and retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1–17. [Google Scholar]
- Bhatia, S.K.; Rashid, A.; Chrenek, M.A.; Zhang, Q.; Bruce, B.B.; Klein, M.; Boatright, J.H.; Jiang, Y.; Grossniklaus, H.E.; Nickerson, J.M. Analysis of RPE morphometry in human eyes. Mol. Vis. 2016, 22, 898–916. [Google Scholar]
- Ostrovsky, M.A.; Zak, P.P.; Dontsov, A.E. Vertebrate Eye Melanosomes and Invertebrate Eye Ommochromes as Screening Cell Organelles. Biol. Bull. Russ. Acad. Sci. 2018, 45, 570–579. [Google Scholar] [CrossRef]
- Friedman, E. The Retinal Pigment Epithelium. Arch. Ophthalmol. 1968, 79, 315–320. [Google Scholar] [CrossRef]
- Grossniklaus, H.E.; Nickerson, J.M.; Edelhauser, H.F.; Bergman, L.A.M.K.; Berglin, L. Anatomic Alterations in Aging and Age-Related Diseases of the Eye. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF23–ORSF27. [Google Scholar] [CrossRef]
- Goodman, D.; Ness, S. The role of oxidative stress in the aging eye. Life. 2023, 13, 837. [Google Scholar] [CrossRef]
- Salvi, S.M. Ageing changes in the eye. Postgrad. Med. J. 2006, 82, 581–587. [Google Scholar] [CrossRef]
- Ts’o, M.O.; Friedman, E. The retinal pigment epithelium. I. Comparative histology. Arch. Ophthalmol. 1967, 78, 641–649. [Google Scholar] [CrossRef]
- Tarau, I.S.; Berlin, A.; Curcio, C.A.; Ach, T. The Cytoskeleton of the Retinal Pigment Epithelium: From Normal Aging to Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 3578. [Google Scholar] [CrossRef]
- Curcio, C.A.; Millican, C.L. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch. Ophthalmol. 1999, 117, 329–339. [Google Scholar] [CrossRef]
- Wong, J.H.C.; Ma, J.Y.W.; Jobling, A.I.; Brandli, A.; Greferath, U.; Fletcher, E.L.; Vessey, K.A. Exploring the pathogenesis of age-related macular degeneration: A review of the interplay between retinal pigment epithelium dysfunction and the innate immune system. Frontiers in Neuroscience. 2022, 16, 1009599. [Google Scholar] [CrossRef]
- Abokyi, S.; To, C.H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef]
- Li, Q.; Wang, M.; Li, X.; Shao, Y. Aging and diabetic retinopathy: Inherently intertwined pathophysiological processes. Exp. Gerontol. 2023, 175, 112138. [Google Scholar] [CrossRef]
- Jeffery, R.C.H.; Chen, F.K. Stargardt disease: Multimodal imaging: A review. Clin. Exp. Ophthalmol. 2021, 49, 498–515. [Google Scholar] [CrossRef]
- Miller, J.W. Age-related macular degeneration revisited–piecing the puzzle: The LXIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 2013, 155, 1–35.e13. [Google Scholar] [CrossRef]
- Young, R.W. Pathophysiology of age-related macular degeneration. Surv. Ophthalmol. 1987, 31, 291–306. [Google Scholar] [CrossRef]
- Roth, F.; Bindewald, A.; Holz, F.G. Key pathophysiologic pathways in age-related macular disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 710–716. [Google Scholar] [CrossRef]
- Nowak, J.Z. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar]
- Holz, F.G.; Bellman, C.; Staudt, S.; Schutt, F.; Volcker, H.E. Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1051–1056. [Google Scholar] [CrossRef]
- Greferath, U.; Guymer, R.H.; Vessey, K.A.; Brassington, K.; Fletcher, E.L. Correlation of Histologic Features with In Vivo Imaging of Reticular Pseudodrusen. Ophthalmology. 2016, 123, 1320–1331. [Google Scholar] [CrossRef]
- Haralampus-Grynaviski, N.M.; Lamb, L.E.; Clancy, C.M.R.; Skumatz, C.; Burke, J.M.; Sarna, T.; Simon, J.D. Spectroscopic and morphological studies of human retinal lipofuscin granules. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3179–3184. [Google Scholar] [CrossRef]
- Rozanowska, M.; Pawlak, A.; Rozanowski, B.; Skumatz, C.; Zareba, M.; Boulton, M.E.; Burke, J.M.; Sarna, T.; Simon, J.D. Age-related changes in the photoreactivity of retinal lipofuscin granules: Role of chloroform-insoluble components. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1052–1060. [Google Scholar] [CrossRef]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Investig. Ophthalmol. Vis. Sci. 1986, 27, 145–152. [Google Scholar]
- Sarna, T.; Burke, J.M.; Korytowski, W.; Rozanowska, M.; Skumatz, C.M.; Zareba, A.; Zareba, M. Loss of melanin from human RPE with aging: possible role of melanin photooxidation. Exp. Eye Res. 2003, 76, 89–98. [Google Scholar] [CrossRef]
- Pollreisz, A.; Messinger, J.D.; Sloan, K.R.; Mittermueller, T.J.; Weinhandl, A.S.; Benson, E.K.; Kidd, G.J.; Schmidt-Erfurth, U.; Curcio, C.A. Visualizing melanosomes, lipofuscin, and melanolipofuscin in human retinal pigment epithelium using serial block face scanning electron microscopy. Exp. Eye Res. 2018, 166, 131–139. [Google Scholar] [CrossRef]
- Feeney-Burns, L.; Hilderbrand, E.S.; Eldridge, S. Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Investig. Ophthalmol. Vis. Sci. 1984, 25, 195–200. [Google Scholar]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: formation, distribution, and metabolic consequences. Ann. NY Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef]
- Fujinami, K.; Lois, N.; Davidson, A.E.; Mackay, D.S.; Hogg, C.R.; Stone, E.M.; Tsunoda, K.; Tsubota, K.; Bunce, C.; Robson, A.G.; Moore, A.T.; Webster, A.R.; Holder, G.E.; Michaelides, M. A longitudinal study of Stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am. J. Ophthalmol. 2013, 155, 1075–1088.e13. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Muller, P.L.; Birtel, J.; Herrmann, P.; McGuinness, M.B.; Holz, F.G.; Issa, P.C. Quantitative Fundus Autofluorescence and Genetic Associations in Macular, Cone, and Cone-Rod Dystrophies. Ophthalmol. Retina. 2020, 4, 737–749. [Google Scholar] [CrossRef]
- von Rückmann, A.; Fitzke, F.W.; Bird, A.C. In vivo fundus autofluorescence in macular dystrophies. Arch. Ophthalmol. 1997, 115, 609–615. [Google Scholar] [CrossRef]
- Bakall, B.; Radu, R.A.; Stanton, J.B.; Burke, J.M.; McKay, B.S.; Wadelius, C.; Mullins, R.F.; Stone, E.M.; Travis, G.H.; Marmorstein, A.D. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Exp. Eye Res. 2007, 85, 34–43. [Google Scholar] [CrossRef]
- Zhang, Q.; Small, K.W.; Grossniklaus, H.E. Clinicopathologic findings in Best vitelliform macular dystrophy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 745–751. [Google Scholar] [CrossRef]
- Jauregui, R.; Chan, L.; Oh, J.K.; Cho, A.; Sparrow, J.R.; Tsang, S.H. Disease asymmetry and hyperautofluorescent ring shape in retinitis pigmentosa patients. Sci. Rep. 2020, 10, 3364. [Google Scholar] [CrossRef]
- Jauregui, R.; Park, K.S.; Duong, J.K.; Sparrow, J.R.; Tsang, S.H. Quantitative Comparison of Near-infrared Versus Short-wave Autofluorescence Imaging in Monitoring Progression of Retinitis Pigmentosa. Am. J. Ophthalmol. 2018, 194, 120–125. [Google Scholar] [CrossRef]
- Meleppat, R.K.; Ronning, K.E.; Karlen, S.J.; Burns, M.E.; Pugh, E.N.Jr.; Zawadzki, R.J. In vivo multimodal retinal imaging of disease-related pigmentary changes in retinal pigment epithelium. Sci Rep. 2021, 11, 16252. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.J.; Rakoczy, P.E.; Constable, I.J. Lipofuscin of the retinal pigment epithelium: a review. Eye. 1995, 9, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Holz, F.G.; Pauleikhoff, D.; Klein, R.; Bird, A.C. Pathogenesis of lesions in late age-related macular disease. Am. J. Ophthalmol. 2004, 137, 504–510. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Boulton, M.E. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Muller, P.L.; Finger, R.P.; McGuinness, M.B.; Holz, F.G.; Issa, P.C. Quantitative Fundus Autofluorescence in Early and Intermediate Age-Related Macular Degeneration. JAMA Ophthalmol. 2016, 134, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Bermond, K.; von der Emde, L.; Tarau, I.S.; Bourauel, L.; Heintzmann, R.; Holz, F.G.; Curcio, C.A.; Sloan, K.R.; Ach, T. Autofluorescent Organelles Within the Retinal Pigment Epithelium in Human Donor Eyes with and without Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2022, 63, 23. [Google Scholar] [CrossRef] [PubMed]
- Feeney, L. The phagosomal system of the pigment epithelium: a key to retinal disease. Investig. Ophthalmol. Vis. Sci. 1973, 12, 635–638. [Google Scholar]
- Eldred, G.E.; Katz, M.L. Fluorophores of the human retinal pigment epithelium: separation and spectral characterization. Exp. Eye Res. 1988, 47, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Kopitz, J. Proteins modified by malondialdehyde,4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3663–3668. [Google Scholar] [CrossRef] [PubMed]
- Schutt, F.; Ueberle, B.; Schnolzer, M.; Holz, F.G.; Kopitz, J. Proteome analysis of lipofuscin in human retinal pigment epithelial cells. FEBS Letters. 2002, 528, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Gugiu, B.; Renganathan, K.; Davies, M.W.; Gu, X.; Crabb, J.S.; Kim, S.R.; Rózanowska, M.B.; Bonilha, V.L.; Rayborn, M.E.; Salomon, R.G.; Sparrow, J.R.; Boulton, M.E.; Hollyfield, J.G.; Crabb, J.W. Retinal pigment epithelium lipofuscin proteomics. Mol. Cell. Proteomics. 2008, 7, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Gregory-Roberts, E.; Yamamoto, K.; Blonska, A.; Ghosh, S.K.; Ueda, K.; Zhou, J. The bisretinoids of retinal pigment epithelium. Prog. Retin. Eye Res. 2012, 31, 121–135. [Google Scholar] [CrossRef]
- Clancy, C.M.R.; Krogmeier, J.R.; Pawlak, A.; Rozanowska, M.; Sarna, T.; Dunn, R.C.; Simon, J.D. Atomic force microscopy and nearfield scanning optical microscopy measurements of single human retinal lipofuscin granules. J. Phys. Chem. B 2000, 104, 12098–12100. [Google Scholar] [CrossRef]
- Petrukhin, A.N.; Astaf’ev, A.A.; Zolotavin, P.N.; Feldman, T.B.; Dontsov, A.E.; Sarkisov, O.M.; Ostrovsky, M.A. Heterogeneity of structure and fluorescence of single lipofuscin granule from retinal pigment epithelium of human donor eyes: study with the use of atomic force microscopy and near-field microscopy. Doklady Biochem. Biophys. 2005, 405, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, M.A.; Gulin, A.A.; Feldman, T.B.; Bel’skich, Y.C.; Arbukhanova, P.M.; Astaf’ev, A.A.; Nadtochenko, V.A.; Borzenok, S.A.; Ostrovsky, M.A. Time-of-flight secondary ion mass spectrometry to assess spatial distribution of A2E and its oxidized forms within lipofuscin granules isolated from human retinal pigment epithelium. Anal. Bioanal. Chem. 2016, 408, 7521–7528. [Google Scholar] [CrossRef] [PubMed]
- Rozanowska, M.G. Lipofuscin, its origin, properties, and contribution to retinal fluorescence as a potential biomarker of oxidative damage to the retina. Antioxidants 2023, 12, 2111. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Dontsov, A.; Jarvis-Evans, J.; Ostrovsky, M.; Svistunenko, D. Lipofuscin is a photoinducible free radical generator. J. Photochem. Photobiol. B Biol. 1993, 19, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Rozanowska, M.; Jarvis-Evans, J.; Korytowski, W.; Boulton, M.E.; Burke, J.M.; Sarna, T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem. 1995, 270, 18825–18830. [Google Scholar] [CrossRef]
- Rozanowska, M.; Wessels, J.; Boulton, M.; Burke, J.M.; Rodgers, M.A.J.; Truscott, T.G.; Sarna, T. Blue light-induced singlet oxygen generation by retinal lipofuscin in non-polar media. Free Radic. Biol. Med. 1998, 24, 1107–1112. [Google Scholar] [CrossRef]
- Boulton, M.; Rozanowska, M.; Rozanowski, B.; Wess, T. The photoreactivity of ocular lipofuscin. Photochem. Photobiol. Sci. 2004, 3, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Dontsov, A.E.; Sakina, N.L.; Ostrovsky, M.A. Comparative study of the dark and light induced toxicity of lipofuscin granules from human retinal pigment epithelium and their chromophore A2E on the cardiolipin liposome model. Rus. Chem. Bull Intl. Ed. 2012, 61, 442–448. [Google Scholar] [CrossRef]
- Wassell, J.; Davies, S.; Bardsley, W.; Boulton, M. The photoreactivity of the retinal age pigment lipofuscin. J. Biol. Chem. 1999, 274, 23828–23832. [Google Scholar] [CrossRef] [PubMed]
- Dontsov, A.E.; Glickman, R.D.; Ostrovsky, M.A. Retinal pigment epithelium pigment granules stimulate the photo-oxidation of unsaturated fatty acids. Free Radic. Biol. Med. 1999, 26, 1436–1446. [Google Scholar] [CrossRef]
- Dontsov, A.E.; Sakina, N.L.; Bilinska, B.; Krzyzanowski, L.; Feldman, T.B.; Ostrovsky, M.A. Comparison of photosensitizing effect of lipofuscin granules from retinal pigment epithelium of human donor eyes and their fluorophore A2E. Dokl. Biochem. Biophys. 2005, 405, 458–460. [Google Scholar] [CrossRef]
- Nowak, J.Z. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: focus on age-related macular degeneration. Pharmacol. Rep. 2013, 65, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Markitantova, Y.; Simirskii, V. Endogenous and exogenous regulation of redox homeostasis in retinal pigment epithelial cells: an updated antioxidant perspective. Int. J. Mol. Sci. 2023, 24, 10776. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, F.A.; Boulton, M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3041–3046. [Google Scholar]
- Nilsson, S.E.; Sundelin, S.P.; Wihlmark, U.; Brunk, U.T. Aging of cultured retinal pigment epithelial cells: oxidative reactions, lipofuscin formation and blue light damage. Doc. Ophthalmol. 2003, 106, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Parish, C.A.; Hashimoto, M.; Nakanishi, K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2988–2995. [Google Scholar]
- Holz, F.G.; Schutt, F.; Kopitz, J.; Eldred, G.E.; Kruse, F.E.; Volcker, H.E.; Cantz, M. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Investig. Ophthalmol. Vis. Sci. 1999, 40, 737–743. [Google Scholar]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Dithmar, S.; Volcker, H.E.; Kopitz, J. Accumulation of A2-E in mitochondrial membranes of cultured RPE cells. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.; Serban, B.; Bernstein, P.S. Retinal carotenoids can attenuate formation of A2E in the retinal pigment epithelium. Arch. Biochem. Biophys. 2009, 483, 175–181. [Google Scholar] [CrossRef]
- Schütt, F.; Davies, S.; Kopitz, J.; Holz, F.G.; Boulton, M.E. Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2303–2308. [Google Scholar]
- Pawlak, A.; Wrona, M.; Rozanowska, M.; Zareba, M.; Lamb, L.E.; Roberts, J.E.; Simon, J.D.; Sarna, T. Comparison of the aerobic photoreactivity of A2E with its precursor retinal. Photochem. Photobiol. 2003, 77, 253–258. [Google Scholar] [CrossRef]
- Gaillard, E.R.; Avalle, L.B.; Keller, L.M.; Wang, Z.; Reszka, K.J.; Dillon, J.P. A mechanistic study of the photooxidation of A2E, a component of human retinal lipofuscin. Exp. Eye Res. 2004, 79, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Broniec, A.; Pawlak, A.; Sarna, T.; Wielgus, A.; Roberts, J.E.; Land, E.J.; Truscott, T.G.; Edge, R.; Navaratnam, S. Spectroscopic properties and reactivity of free radical forms of A2E. Free Radic. Biol. Med. 2005, 38, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, A.; McGarvey, D.J.; Roberts, J.; Sarna, T.; Truscott, T.G. Photochemical studies of A2-E. J. Photochem. Photobiol. B: Biol. 2001, 64, 162–165. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Sakmar, T.P. Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol. 2002, 120, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.; Finnemann, S.C.; Rodriguez-Boulan, E. The lipofuscin fluorophore A2E perturbs cholesterol metabolismin retinal pigment epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 11026–11031. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, V.S.; Sokolenko, E.A.; Sokolov, A.V.; Dontsov, A.E.; Chizmadzhev, Yu.A.; Ostrovsky, M.A. Interaction of pyridinium bisretinoid (A2E) with bilayer lipid membranes. J. Photochem. Photobiol. B: Biol. 2007, 86, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Nakanishi, K.; Parish, C.A. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1981–1990. [Google Scholar]
- Wang, Z.; Keller, L.M.M.; Dillon, J.; Gaillard, E.R. Oxidation of A2E results in the formation of highly reactive aldehydes and ketones. Photochem. Photobiol. 2006, 82, 1251–1257. [Google Scholar] [CrossRef]
- Yakovleva, M.A.; Sakina, N.L.; Kononikhin, A.S.; Feldman, T.B.; Nikolaev, E.N.; Dontsov, A.E.; Ostrovsky, M.A. Detection and study of the products of photooxidation of N-Retinylidene-N-retinylethanolamine (A2E), the fluorophore of lipofuscin granules from retinal pigment epithelium of human donor eyes. Doklady Biochem. Biophys. 2006, 409, 223–225. [Google Scholar] [CrossRef]
- Yoon, K.D.; Yamamoto, K.; Ueda, K.; Zhou, J.; Sparrow, J.R. A novel source of methylglyoxal and glyoxal in retina: implications for age-related macular degeneration. PLoS ONE. 2012, 7, e41309. [Google Scholar] [CrossRef]
- Yakovleva, M.; Dontsov, A.; Trofimova, N.; Sakina, N.; Kononikhin, A.; Aybush, A.; Gulin, A.; Feldman, T.; Ostrovsky, M. Lipofuscin granule bisretinoid oxidation in the human retinal pigment epithelium forms cytotoxic carbonyls. Int. J. Mol. Sci. 2022, 23, 222. [Google Scholar] [CrossRef]
- Aybush, A.V.; Gulin, A.A.; Vasin, A.A.; Dontsov, A.E.; Nadtochenko, V.A.; Ostrovsky, M.A. Multimodal approach to reveal the effect of light irradiation on chemical composition of lipofuscin granules of human RPE tissues. J. Physics: Conference Series. 2020, 1695, 012063. [Google Scholar] [CrossRef]
- Rózanowska, M.B.; Rózanowski, B. Photodegradation of lipofuscin in suspension and in ARPE-19 cells and the similarity of fluorescence of the photodegradation product with oxidized docosahexaenoate. Int. J. Mol. Sci. 2022, 23, 922. [Google Scholar] [CrossRef]
- Wu, Y.; Yanase, E.; Feng, X.; Siegel, M.M.; Sparrow, J.R. Structural characterization of bisretinoid A2E photocleavage products and implications for age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 7275–7280. [Google Scholar] [CrossRef] [PubMed]
- Dontsov, A.; Yakovleva, M.; Trofimova, N.; Sakina, N.; Gulin, A.; Aybush, A.; Gostev, F.; Vasin, A.; Feldman, T.; Ostrovsky, M. Water-soluble products of photooxidative destruction of the bisretinoid A2E cause proteins modification in the dark. Int. J. Mol. Sci. 2022, 23, 1534. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.D.; Bierhaus, A.; Haring, H.U.; Nawroth, P.P.; Lehmann, R. Chemistry and pathobiology of advanced glycation end products. Contrib. Nephrol. 2001, 131, 1–9. [Google Scholar]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta – Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Montenegro, D.; Zhao, J.; Sparrow, J.R. Bisretinoids of the retina: photo-oxidation, iron-catalyzed oxidation, and disease consequences. Antioxidants. 2021, 10, 1382. [Google Scholar] [CrossRef]
- Baynes, Y.W. The role of AGEs in aging: causation or correlation. Exp. Gerontol. 2001, 36, 1527–1537. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Glenn, J.V.; Beattie, J.R.; Barret, L.; Frizzell, N.; Thorpe, S.R.; Boulton, M.E.; McGarvey, J.J.; Stitt, A.W. Confocal Raman microscopy can quantify advanced glycation end products modifications in Bruch’s membrane leading to accurate, nondestructive prediction of ocular aging. FASEB J. 2007, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; Salomon, R.G.; Hollyfield, J.G. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14682–14687. [Google Scholar] [CrossRef]
- Glenn, J.V.; Mahaffy, H.; Wu, K.; Smith, G.; Nagai, R.; Simpson, D.A.; Boulton, M.E.; Stitt, A.W. Advanced glycation end product (AGE) accumulation on Bruch's membrane: Links to age-related RPE dysfunction. Investig. Ophthalmol. Vis. Sci. 2009, 50, 441–451. [Google Scholar] [CrossRef]
- Finnemann, S.C.; Leung, L.W.; Rodriguez-Boulan, E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 3842–3847. [Google Scholar] [CrossRef]
- Klettner, A. Oxidative stress induced cellular signaling in RPE cells. Front. Biosci. -Schol. Ed. 2012, 4, 392–411. [Google Scholar] [CrossRef] [PubMed]
- Krohne, T.U.; Kaemmerer, E.; Holz, F.G.; Kopitz, J. Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp. Eye Res. 2010, 90, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell. 2012, 11, 1–13. [Google Scholar] [CrossRef]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy. 2007, 3, 433–441. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Roles for the ubiquitin-proteasome pathway in protein quality control and signaling in the retina: Implications in the pathogenesis of age-related macular degeneration. Mol. Aspects Med. 2012, 33, 446–466. [Google Scholar] [CrossRef]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cell - implications for age-related macular degeneration (AMD). Immunology Letters. 2012, 147, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Copland, D.A.; Theodoropoulou, S.; Chiu, H.A.A.; Barba, M.D.; Mak, K.W.; Mack, M.; Nicholson, L.B.; Dick, A.D. Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis. Sci. Rep. 2016, 6, 20639. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature. 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, R.T.; Cao, S.; Cui, J.Z.; Wang, A.; To, E.; Matsubara, J.A. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediators Inflamm. 2015, 2015, 690243. [Google Scholar] [CrossRef] [PubMed]
- Haiju, N. Regulation of oxidative stress and inflammatory responses in human retinal pigment epithelial cells. Acta Ophthalmol. 2022, 100, 3–59. [Google Scholar]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, M.C.; Fisher, C.R.; Kapphahn, R.J.; Stahl, M.R.; Shen, S.; Qu, J.; Montezuma, S.; Ferrington, D. Inflammasome Activation in Retinal Pigment Epithelium from Human Donors with Age-Related Macular Degeneration. Cells. 2022, 11, 2075. [Google Scholar] [CrossRef] [PubMed]
- Leveillard, T.; Philp, N.J.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. [Google Scholar] [CrossRef]
- Hollyfield, J.G. Age-related macular degeneration: the molecular link between oxidative damage, tissue specific inflammation and outer retinal disease: the Proctor lecture. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1275–1281. [Google Scholar] [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef]
- Karunadharma, P.P.; Kapphahn, R.J.; Stahl, M.R.; Olsen, T.W.; Ferrington, D.A. Dissecting Regulators of Aging and Age-Related Macular Degeneration in the Retinal Pigment Epithelium. Oxid. Med. Cell. Longev. 2022, 2022, 6009787. [Google Scholar] [CrossRef]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; Smith, R.J.H.; Silvestri, G.; Russell, S.R.; Klaver, C.C.W.; Barbazetto, I.; Chang, S.; Yannuzzi, L.A.; Barile, G.R.; Merriam, J.C.; Smith, R.T.; Olsh, A.K.; Bergeron, J.; Zernant, J.; Merriam, J.E.; Gold, B.; Dean, M.; Allikmets, R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 7227–7232. [Google Scholar] [CrossRef]
- Detrick, B.; Hooks, J.J. The RPE Cell and the Immune System. In Retinal Pigment Epithelium in Health and Disease; Klettner, A.K., Dithmar, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 101–114. [Google Scholar]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef]
- Chinnery, H.R.; McLenachan, S.; Humphries, T.; Kezic, J.M.; Chen, X.; Ruitenberg, M.J.; McMenamin, P.G. Accumulation of murine subretinal macrophages: Effects of age, pigmentation and CX3CR1. Neurobiol. Aging. 2012, 33, 1769–1776. [Google Scholar] [CrossRef]
- Luo, C.; Zhao, J.; Madden, A.; Chen, M.; Xu, H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp. Eye Res. 2013, 112, 93–101. [Google Scholar] [CrossRef]
- Lin, T.; Walker, G.B.; Kurji, K.; Fang, E.; Law, G.; Prasad, S.S.; Kojic, L.; Cao, S.; White, V.; Cui, J.Z.; Matsubara, J.A. Parainflammation associated with advanced glycation end product stimulation of RPE in vitro: Implications for age-related degenerative diseases of the eye. Cytokine. 2013, 62, 369–381. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M.; Manivannan, A.; Lois, N.; Forrester, J.V. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. 2008, 7, 58–68. [Google Scholar] [CrossRef]
- Luhmann, U.F.; Ali, R.R. Local vs. systemic mononuclear phagocytes in age-related macular degeneration and their regulation by CCL2-CCR2 and CX3CL1-CX3CR1 chemokine signaling. Adv. Exp. Med. Biol. 2012, 723, 17–22. [Google Scholar] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Sarna, T. Properties and function of the ocular melanin - a photobiophysical view. J. Photochem. Photobiol. B. Biol. 1992, 12, 215–258. [Google Scholar] [CrossRef]
- Peters, S.; Schraermeyer, U. Characteristics and functions of melanin in retinal pigment epithelium. Ophthalmologe. 2001, 98, 1181–1185. [Google Scholar]
- Ostrovsky, M.A.; Dontsov, A.E. Vertebrate Eye Melanosomes and Invertebrate Eye Ommochromes as Antioxidant Cell Organelles: Part 2. Biology Bulletin of the Russian Academy of Sciences. 2019, 46, 105–116. [Google Scholar] [CrossRef]
- Fukuda, M. Organizational cell biology. In Encyclopedia of Cell Biology; Brandshaw, R.A., Stahl, P.D., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 2, ISBN 978-0-12-394796-3. [Google Scholar]
- Pollreisz, A.; Neschi, M.; Sloan, K.R.; Pircher, M.; Mittermueller, T.; Dacey, D.M.; Schmidt-Erfurth, U.; Curcio, C.A. Atlas of Human Retinal Pigment Epithelium Organelles Significant for Clinical Imaging. Investig. Ophthalmol. Vis. Sci. 2020, 61, 13. [Google Scholar] [CrossRef]
- Hu, D.N.; Simon, J.D.; Sarna, T. Role of ocular melanin in ophthalmic physiology and pathology. Photochem. Photobiol. 2008, 84, 639–644. [Google Scholar] [CrossRef]
- Istrate, M.; Vlaicu, B.; Poenaru, M.; Hasbei-Popa, M.; Salavat, M.C.; Iliesku, D.A. Photoprotection role of melanin in the human retinal pigment epithelium. Imaging techniques for retinal melanin. Romanian J. Ophthalmol. 2020, 64, 100–104. [Google Scholar] [CrossRef]
- Różanowska, M. Properties and functions of ocular melanins and melanosomes. In Melanins and Melanosomes: Biosynthesis, Biogenesis, Physiological, and Pathological Functions; Riley, P.A., Borovansky, J., Eds.; John Wiley and Sons, Inc.: Weinheim, Germany, 2011; pp. 187–224. [Google Scholar]
- Rapp, L.M.; Williams, T.P. The role of ocular pigmentation in protecting against retinal light damage. Vis. Res. 1980, 20, 1127–1131. [Google Scholar] [CrossRef]
- Armstrong, D.; Hiramitsu, T.; Gutteridge, J.; Nilsson, S.E. Studies on experimentally induced retinal degeneration. 1. Effect of lipid peroxides on electroretinographic activity in the albino rabbit. Exp. Eye Res. 1982, 35, 157–171. [Google Scholar] [CrossRef]
- Safa, R.; Osborne, N.N. Retinas from albino rats are more susceptible to ischemic damage than age-matched pigmented animals. Brain Res. 2000, 862, 36–42. [Google Scholar] [CrossRef]
- Ostrovsky, M.A.; Sakina, N.L.; Dontsov, A.E. An antioxidative role of ocular screening pigments. Vis. Res. 1987, 27, 893–899. [Google Scholar] [CrossRef]
- Wang, Z.; Dillon, J.; Gaillard, E.R. Antioxidant properties of melanin in retinal pigment epithelial cells. Photochem. Photobiol. 2006, 82, 474–479. [Google Scholar] [CrossRef]
- Hu, D.N.; Simon, J.D.; Sarna, T. Role of ocular melanin in ophthalmic physiology and pathology. Photochem. Photobiol. 2008, 84, 639–644. [Google Scholar] [CrossRef]
- Burke, J.M.; Kaczara, P.; Skumatz, C.M.; Zareba, M.; Raciti, M.W.; Sarna, T. Dynamic analyses reveal cytoprotection by RPE melanosomes against non-photic stress. Mol. Vis. 2011, 17, 2864–2877. [Google Scholar]
- Dontsov, A.E.; Koromyslova, A.D.; Sakina, N.L. ; Lipofuscin component A2E does not reduce antioxidant activity of DOPA-melanin. Bull. Exp. Biol. Med. 2013, 154, 624–627. [Google Scholar] [CrossRef]
- Sakina, N.L.; Koromyslova, A.D.; Dontsov, A.E.; Ostrovsky, M.A. RPE melanosomes bind A2E fluorophore of lipofuscin granules and products of its photooxidation. Sechenov Ross. Fiziol. Zh. 2013, 99, 642–653. [Google Scholar]
- Davies, S.; Shamsi, F.A; Zadło, A.; Dayhaw-Barker, P.; Rozanowska, M.; Sarna, T.; Boulton, M.E. The Phototoxicity of Aged Human Retinal Melanosomes. Photochem. Photobiol. 2008, 84, 650–657. [Google Scholar]
- Feeney-Burns, L.; Berman, E.R.; Rothman, H. Lipofuscin of human retinal pigment epithelium. Am. J. Ophthalmol. 1980, 90, 783–791. [Google Scholar] [CrossRef]
- Feeney-Burns, L.; Burns, R.P.; Gao, C.-L. Age-related macular changes in humans over 90 years old. Am. J. Ophthalmol. 1990, 109, 265–278. [Google Scholar] [CrossRef]
- Schmidt, S.Y.; Peisch, R.D. Melanin concentration in normal human retinal pigment epithelium. Regional variation and age-related reduction. Investig. Ophthalmol. Vis. Sci. 1986, 27, 1063–1067. [Google Scholar]
- Dontsov, A.E.; Sakina, N.L.; Ostrovsky, M.A. Loss of melanin by eye retinal pigment epithelium cells is associated with its oxidative destruction in melanolipofuscin granules. Biochemistry (Moscow) 2017, 82, 916–924. [Google Scholar] [CrossRef]
- Yacout, S.M.; McIlwain, K.L.; Mirza, S.P.; Gaillard, E.R. Characterization of retinal pigment epithelial melanin and degraded synthetic melanin using mass spectrometry and in vitro biochemical diagnostics. Photochem. Photobiol. 2019, 95, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Hyman, L.G.; Lilienfeld, A.M.; Ferris, F.L., 3rd.; Fine, S.L. Senile macular degeneration: a case-control study. Am. J. Epidemiol 1983, 118, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Relationship of senile macular degeneration to ocular pigmentation. Am. J. Ophthalmol. 1985, 99, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Zareba, M.; Szewczyk, G.; Sarna, T.; Hong, L.; Simon, J.D.; Henry, M.M.; Burke, J.M. Effects of photodegradation on the physical and antioxidant properties of melanosomes isolated from retinal pigment epithelium. Photochem. Photobiol. 2006, 82, 1024–1029. [Google Scholar] [CrossRef]
- Mahendra, C.K.; Tan, L.T.H.; Pusparajah, P.; Htar, T.T.; Chuah, L.; Lee, V.S.; Low, L.E.; Tang, S.Y.; Chan, K.-G.; Goh, B.H. Detrimental effects of UVB on retinal pigment epithelial cells and its role in age-related macular degeneration. Oxid. Med. Cell. Longev. 2020, 2020, 1904178. [Google Scholar] [CrossRef]
- Olchawa, M.M.; Szewczyk, G.M.; Zadlo, A.C.; Krzysztynska-Kuleta, O.I.; Sarna, T.J. The effect of aging and antioxidants on photoreactivity and phototoxicity of human melanosomes: an in vitro study. Pigment Cell & Melanoma Research 2021, 34, 670–682. [Google Scholar]
- Zadlo, A.; Burke, J.M.; Sarna, T. Effect of untreated photobleached bovine RPE melanosomes an the photoinduced peroxidation of lipids. Photochem. Photobiol. Sci. 2009, 8, 830–837. [Google Scholar] [CrossRef]
- Ito, S.; Pilat, A.; Gerwat, W.; Skumatz, C.M.; Ito, M.; Kiyono, A.; Zadlo, A.; Nakanishi, Y.; Kolbe, L.; Burke, J.M.; Sarna, T.; Wakamatsu, K. Photoaging of human retinal pigment epithelium is accompanied by oxidative modifications of its eumelanin. Pigment Cell & Melanoma Research. 2013, 26, 357–366. [Google Scholar]
- Ito, S.; Wakamatsu, K.; Sarna, T. Photodegradation of eumelanin and pheomelanin and its pathophysiological implications. Photochem. Photobiol. 2018, 94, 409–420. [Google Scholar] [CrossRef]
- Gulin, A.A.; Dontsov, A.E.; Yakovleva, M.A.; Trofimova, N.N.; Aybush, A.V.; Vasin, A.A.; Ostrovsky, M.A. Oxidative destruction of human RPE melanosomes induced by superoxide radicals leads to the formation of reactive aldehydes and ketones. St. Petersburg State Polytechnical University Journal. Physics and Mathematics. 2022, 15, 311–316. [Google Scholar]
- Dontsov, A.E.; Yakovleva, M.A.; Vasin, A.A.; Gulin, A.A.; Aybush, A.V.; Nadtochenko, V.A.; Ostrovsky, M.A. Understanding the Mechanism of Light-Induced Age-Related Decrease in Melanin Concentration in Retinal Pigment Epithelium Cells. Int. J. Mol. Sci. 2023, 24, 13099. [Google Scholar] [CrossRef] [PubMed]
- Rozanowska, M.; Korytowski, W.; Rozanowski, B.; Skumatz, C.; Boulton, M.E.; Burke, J.M.; Sarna, T. Photoreactivity of aged human RPE melanosomes: a comparison with lipofuscin. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2088–2096. [Google Scholar]
- Kayatz, P.; Thumann, G.; Luther, T.T.; Jordan, J.F.; Bartz-Schmidt, K.V.; Esser, P.J.; Schraermeyer, U. Oxidation causes melanin fluorescence. Investig. Ophthalmol. Vis. Sci. 2001, 42, 241–246. [Google Scholar]
- Elleder, M.; Borovansky, J. Autofluorescence of melanin induced by ultraviolet radiation and near ultraviolet light. A histochemical and biochemical study. Histochem. J. 2001, 33, 273–281. [Google Scholar] [CrossRef]
- Borovansky, J.; Elleder, M. Melanosomes degradation: fact or fiction. Pigment Cell Res. 2003, 16, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Zadlo, A.; Rozanowska, M.B.; Burke, J.M.; Sarna, T.J. Photobleaching of retinal pigment epithelium melanosomes reduces their ability to inhibit iron-induced peroxidation of lipids. Pigment Cell Res. 2006, 20, 52–60. [Google Scholar] [CrossRef]
- Dontsov, A.E.; Sakina, N.L.; Koromyslova, A.D.; Ostrovsky, M.A. Effect of UV radiation and hydrogen peroxide on the antiradical and antioxidant activities of DOPA-melanin and melanosomes from retinal pigment epithelial cells. Russian Chem. Bull. 2015, 64, 1623–1628. [Google Scholar] [CrossRef]
- Sakina, N.L.; Dontsov, A.E.; Degtyarev, E.N.; Kovarsky, A.L.; Arbukhanova, P.M.; Borzenok, S.A.; Ostrovsky, M.A. Comparative evaluation of melanin content in melanosomes and melanolipofuscin granules from the human retinal pigment epithelium cells. Fyodorov Journal of Ophthalmic Surgery. 2018, 1, 78–82. [Google Scholar] [CrossRef]
- Lyu, Y.; Tschulakow, A.V.; Schraermeyer, U.A. Melanosomes degrade lipofuscin and precursors that are derived from photoreceptor membrane turnover in the retinal pigment epithelium—an explanation for the origin of the melanolipofuscin granule. Preprint. 2022. [Google Scholar] [CrossRef]
- Lyu, Y.; Tschulakow, A.V.; Wang, K.; Brash, D.E.; Schraermeyer, U. Chemiexcitation and melanin in photoreceptor disc turnover and prevention of macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 2023, 120, e2216935120. [Google Scholar] [CrossRef]
- Schraermeyer, U.; Heimann, K. Current understanding on the role of retinal pigment epithelium and its pigmentation. Pigment Cell Res. 1999, 12, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Schraermeyer, U.; Stieve, H. A newly discovered pathway of melanin formation in cultured retinal pigment epithelium of cattle. Cell Tissue Res. 1994, 276, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Schraermeyer, U.; Peters, S.; Thumann, G.; Kociok, N.; Heimann, K. Melanin granules of retinal pigment epithelium are connected with the lysosomal degradation pathway. Exp. Eye Res. 1999, 68, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Taubitz, T.; Fanga, Y.; Biesemeier, A.; Julien-Schraermeyer, S.; Schraermeyer, U. Age, lipofuscin and melanin oxidation affect fundus near-infrared autofluorescence. EBioMedicine. 2019, 48, 592–604. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Scheme illustrating the role of lipofuscin granules and bisretinoids in photooxidative stress in RPE cells.
Figure 2.
Scheme illustrating the role of lipofuscin granules and bisretinoids in photooxidative stress in RPE cells.
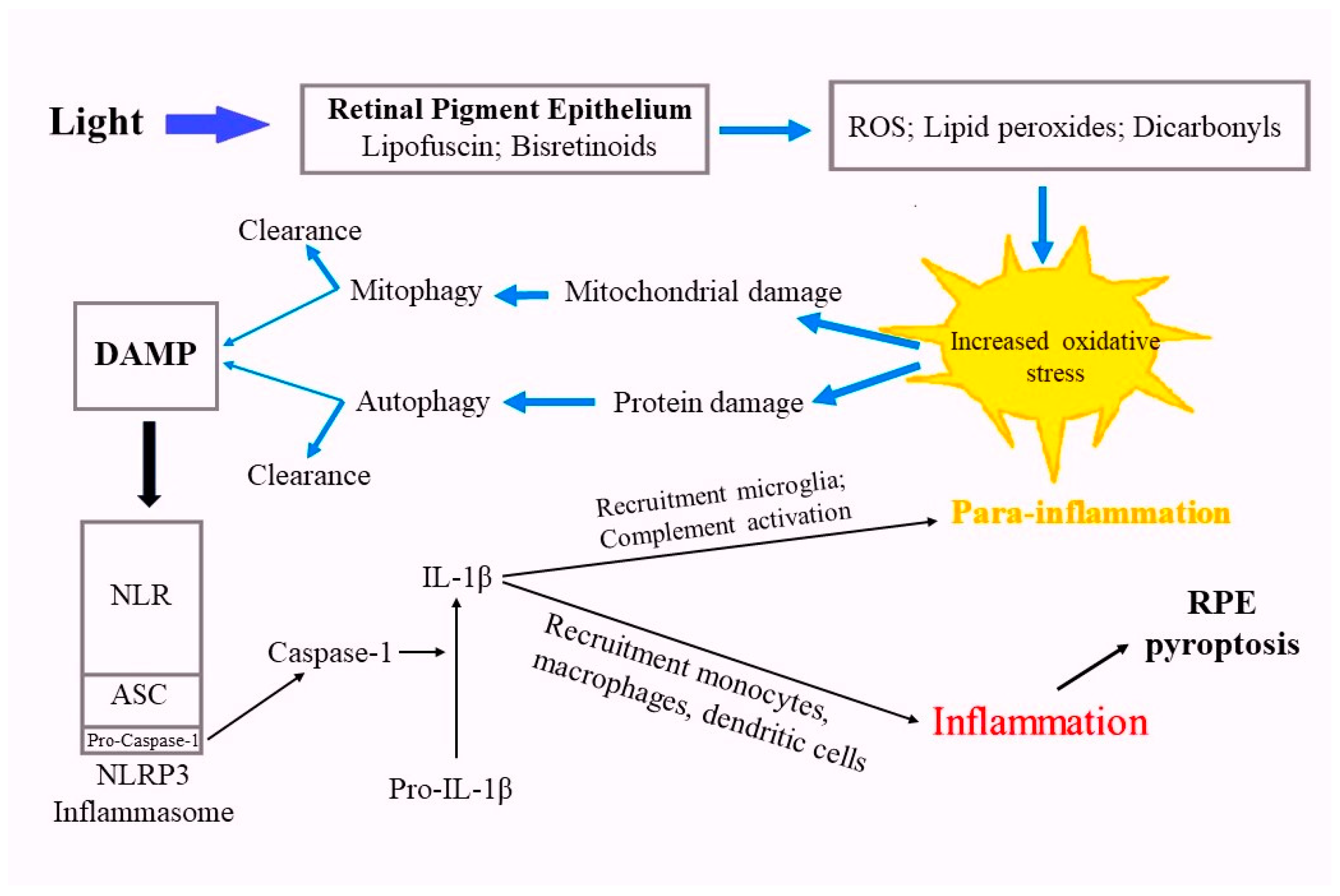
Figure 3.
MLG from human RPE cells contain less melanin than MG. ESR spectra of MG (A) and MLG (B) from human RPE cells.
Figure 3.
MLG from human RPE cells contain less melanin than MG. ESR spectra of MG (A) and MLG (B) from human RPE cells.
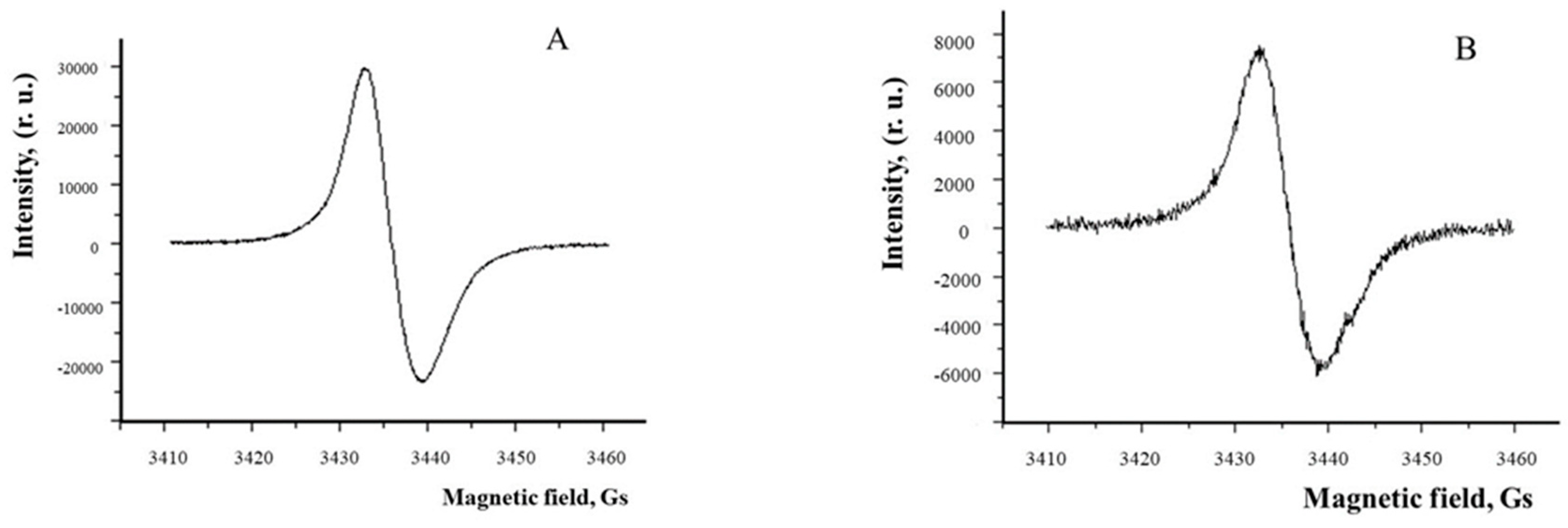
Figure 4.
Scheme of the mechanisms involved in melanin degradation in the melanolipofuscin granule. Abbreviations: PMD—melanin degradation products, ROS— reactive oxygen species, LIP-ox—oxidized lipofuscin. 1. Light in the presence of oxygen activates ROS generation mediated by lipofuscin fluorophores. The resulting ROS can oxidize both melanin, causing its degradation and the formation of PMD, and lipofuscin, causing the formation of reactive dicarbonyls. 2. The resulting photosensitive melanin degradation products (PMDs) generate ROS when exposed to light and, in turn, cause further degradation of melanin and lipofuscin. 3. In a granule containing all three components, namely melanin, lipofuscin (bisretinoids) and PMD, light and ROS activate the transition of melanin to a high-energy state in which the excited pigment causes degradation of lipofuscin [164,165].
Figure 4.
Scheme of the mechanisms involved in melanin degradation in the melanolipofuscin granule. Abbreviations: PMD—melanin degradation products, ROS— reactive oxygen species, LIP-ox—oxidized lipofuscin. 1. Light in the presence of oxygen activates ROS generation mediated by lipofuscin fluorophores. The resulting ROS can oxidize both melanin, causing its degradation and the formation of PMD, and lipofuscin, causing the formation of reactive dicarbonyls. 2. The resulting photosensitive melanin degradation products (PMDs) generate ROS when exposed to light and, in turn, cause further degradation of melanin and lipofuscin. 3. In a granule containing all three components, namely melanin, lipofuscin (bisretinoids) and PMD, light and ROS activate the transition of melanin to a high-energy state in which the excited pigment causes degradation of lipofuscin [164,165].
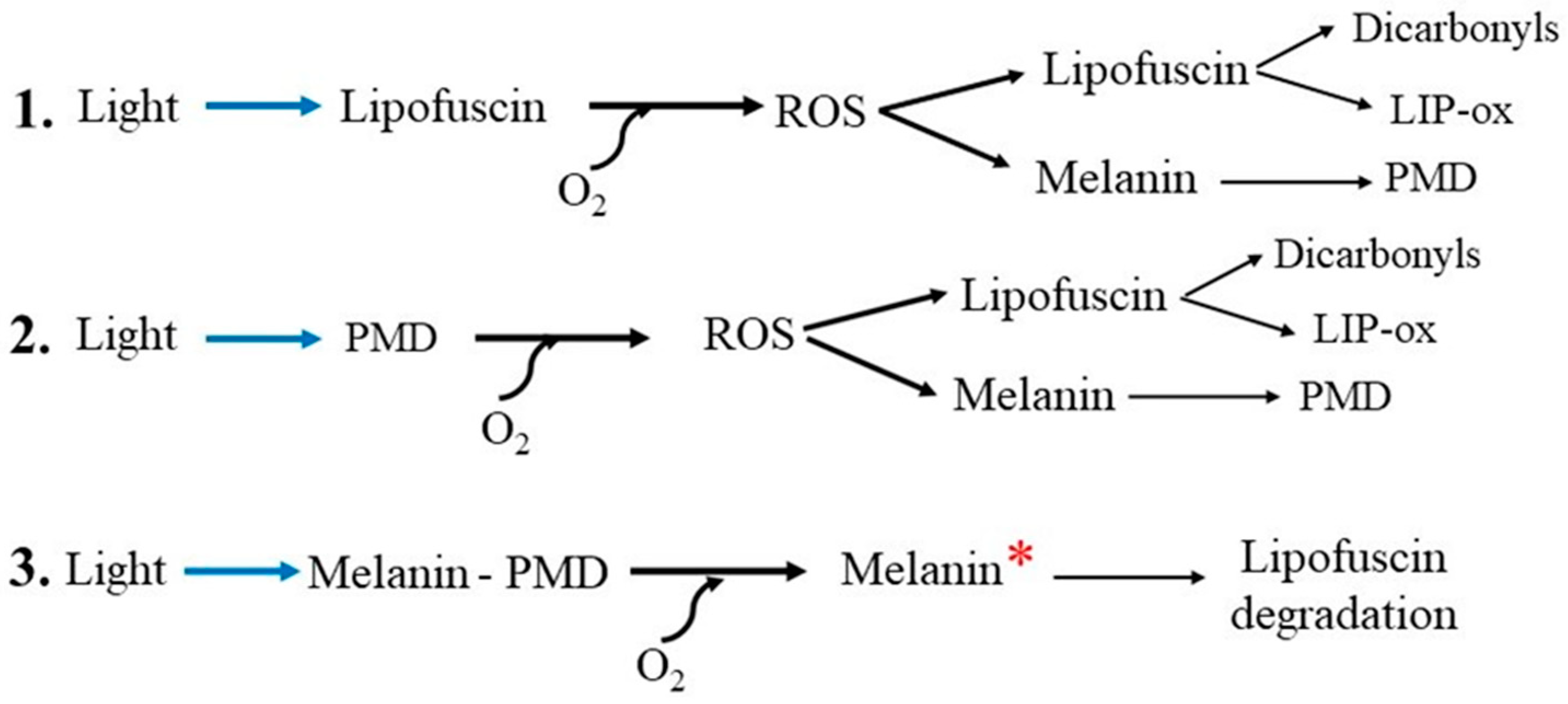

Table 1.
Comparison of melanin concentrations in MG and MLG from RPE cells for donors of two age categories.
Table 1.
Comparison of melanin concentrations in MG and MLG from RPE cells for donors of two age categories.
| Age groups | Melanin concentration, mg/per granule | |
|---|---|---|
| Melanosomes (MG) | Melanolipofuscin (MLG) | |
| 30-60 years old, (50 eyes) | (2.3 ± 0.4) x10-10 | (1.3 ± 0.5) x10-10 |
| 60-75 years old (30 eyes) | (2.2 ± 0.8) x10-10 | (1.2 ± 0.4) x10-10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



