
Submitted:
05 March 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
The spectrum, intensity and overlap of symptoms between FGIDs and other gastrointestinal disorders characterize patients with FIGDs, who are incredibly different in their backgrounds. An additional challenge with regard to the diagnosis and treatment's applicability is the ongoing expansion of the risk factors believed to be connected to these disorders. Many cytokines and inflammatory cells have been found causing the continuous existence of low-inflammation, which is thought to be a basic pathophysiological process. The idea of the gut-brain axis has been created to offer a basic framework for the complex interactions that occur between the nervous system and intestinal functions, including the involvement of the gut bacteria. In this review paper, we intend to promote a hypothesis that FGIDs, should be seen through the perspective of the network of the neuroendocrine, immunological, metabolic and microbiome pathways. Hypothesis arises from an increased understanding of chronic inflammation as a systemic disorder, being omnipresent in chronic health conditions. Better understanding of inflammation role in the pathogenesis of FGIDs can be achieved by clustering markers of inflammation with data indicating symptoms, comorbidities, and psycho-social factors. Finding subclasses among related entities of FGIDs, may reduce patient heterogeneity and help clarify pathophysiology for better treatment.
Keywords:
gastrointestinal diseases
; chronic diseases
; cytokines
; gastrointestinal microbiome
; inflammation
1. Introduction - Motivation for This Review
Up to today, inflammatory bowel diseases (IBD), ulcerative colitis (UC), and Crohn`s disease (CD), which are classified within autoinflammatory diseases, have been considered the only chronic inflammatory diseases that affect the gastrointestinal tract. The increasing evidence suggests that the development of these diseases may be triggered by a disturbance in the balance between the gut commensal microflora and the mucosal immune system [1]. There was a revolutionary discovery when, four decades ago, it was realized that colonization of gastric mucosae with the microorganism Helicobacter pillory (HP), and inflammation associated with this infection, underlie the most frequent disorders of the gastrointestinal tract, chronic gastritis, and ulcer disease. This discovery has changed our perception of these common human diseases, for which impaired secretion of gastric acid has been considered the major etiological factor [2].
A decade later, the Rome criteria were founded to enable the diagnosis of a large set of functional gastrointestinal disorders (FGIDs), for which little attention has been devoted previously [3]. Routine diagnostic examinations for these illnesses do not reveal any underlying structural defects that can explain the symptoms. The Rome classification is based on symptom clustering, taking into account the regional anatomy of the gastrointestinal tract (esophageal, gastroduodenal, bowel, biliary, and anorectal). The last update of this classification (Rome IV, 2016), recognizes 33 adult and 17 pediatric disorders. The most common adult disorders are irritable bowel syndrome (IBS) and functional dyspepsia (FD).
Although the Rome classification has led to better characterization of these disorders and improved clinical practice, it is still insufficient to enable individualized treatment, as well as accurate patient selection for clinical trials. Patients with those illnesses are exceedingly diverse, with symptoms ranging in scope, severity, frequency, and duration, as well as overlap between FGIDs and other gastrointestinal diseases. There are no suitable operational definitions or biomarkers for the accurate diagnosis of any of the aforementioned diseases [4]. Also challenging, concerning the relevance of the diagnosis and treatment, is the fact that the number of the risk and pathophysiology factors, that are thought to be associated with these disorders, constantly expanding, as new evidence becomes available. For instance, in addition to increased visceral sensitivity and altered motility, as has long been believed, decreased intestinal microbiota diversity and mucosal immune system activation have also been linked to the development of these illnesses [5].
Furthermore, experimental research has confirmed epidemiological observations that altered emotions frequently co-occur with these disorders. The concept of the gut-brain axis has been developed to provide a general framework for the intricate interactions between the nervous systems and intestinal functions, which include the gut microbiota [6,7]. A view has emerged, including a bio-psycho-social perspective, to provide a comprehensive understanding of FGIDs, by trying to explain how a large array of environmental, psychological, and biological factors contribute to the expression of symptoms of these disorders [8].
In this review paper, we intend to promote a hypothesis that FGIDs, notably referring to FD and IBS, should be seen through the perspective of the network that integrates the neuroendocrine, immunological, and metabolic pathways, including their linkages to the gut microbiome. This hypothesis arises from an increased understanding of chronic inflammation as a systemic disorder, that is omnipresent in chronic health conditions. We propose that a better understanding of the role of inflammation in the pathogenesis of FGIDs can be achieved by clustering markers of inflammation with data indicating symptoms, comorbidities, and psycho-social factors. This method, finding subclasses among one or more related entities of FGIDs, may reduce patient heterogeneity and help clarify pathophysiology pathways connected with these disorders.
2. Autoinflammatory and Autoimmune Diseases
It is commonly known that inflammation plays a significant part in the etiology of several diseases, including rheumatic and autoimmune musculoskeletal disorders [9,10]. The latter category, referred to as autoinflammatory diseases, has often been seen as existing independently of autoimmune diseases. Autoinflammatory diseases are characterized by abnormal innate immune responses but without the involvement of autoantibodies or autoreactive T lymphocytes, which are the hallmarks of autoimmune diseases [11]. As a result, our understanding of the pathophysiology of inflammation-mediated disorders has grown in complexity.
The knowledge has advanced from the view that autoimmune diseases are caused by a defective elimination of autoreactive T cell clones, as the direct effect of the polymorphism of genes involved in the immune function, to the view that the key factor in their development is the imbalance between the regulatory T cells (Treg cells) and the effector T cells (Teff cells) that can be developed under the influence of external factors or immune disturbances caused by preexisted pathologies [9,12,13]. The key element in this view is the evidence suggesting a high degree of plasticity of Treg cells under altering microenvironmental settings [14]. This feature enables Treg cells to switch from anti-inflammatory and tissue-safeguarding qualities to effector phenotypes and functions, hence assisting in the maintenance of the established type of immune response, as long as the pro-inflammatory signals disappear. While it can be a useful homeostatic mechanism in case of infection, in conditions of “sterile inflammation”, such as obesity or the presence of chronic diseases, this characteristic of Treg cells can lead to autoimmune diseases [15].
The dichotomy between autoimmune and autoinflammatory illnesses has also been questioned. Instead, an immunological continuum has been proposed, with these two types of diseases positioned on the opposing ends of the spectrum, so they still share both innate and adaptive pathways, but with varied involvement rates [16]. This viewpoint allows for the overlap of autoimmune and autoinflammatory disorders. Autoinflammatory and autoimmune disorders, like UC and CD, frequently coexist with other conditions like ankylosing spondylitis, rheumatoid arthritis, psoriasis, primary sclerosing cholangitis, uveitis, episcleritis, celiac disease, and systemic lupus erythematosus [17].
3. The Role of Inflammation in the Pathogenesis of Other Chronic Diseases
The two major findings contributed greatly to our knowledge of the function of inflammation and immunological pathways in the etiology of chronic health disorders. The first was the finding that various stimuli, not just microorganisms, can activate the innate immune receptors, increasing the production of pro-inflammatory cytokines such as interleukins, IL-1β, IL-18, and IL-6, as well as tumor necrosis factor-α (TNF-α) [18]. These “danger signals” may include components of damaged cells and tissues, metabolic intermediates, reactive oxygen species (ROS), and also conditions in the microenvironment characterized by hypoxia or nutrient deprivation. The chemical complex known as the inflammasome plays a vital function within immune cells in integrating multiple danger signals and translating them into the innate immune response [19]. Regardless of the initial stimuli that cause inflammation, conditions can be established in inflamed tissue that results in the loss of tolerance to autoantigens and the activation of Teff cells. This leads to the cooperation of the innate and adaptive (specific) immune responses in tissue damage and remodeling [9]. Which type of immune response will dominate, either type 1 (cell-mediated), which involves innate immune cells, like dendritic cells (DCs), macrophages, natural killer (NK) cells, and T helper cells type 1 (Th1 cells), or type 2 (humoral, antibody-mediated), that is characterized by activation of Th2 and B cells and antibodies production, and that involves tissue infiltration with eosinophils and mast cells, will depend on the local cytokine milieu and an individual`s genetic makeup [11,12].
Another important finding was the Th17 subtype of Teff cells, which has greatly enhanced our understanding of chronic illness etiology [20]. Cytokines that are preferentially generated by this lymphocyte subtype are cytokines of the IL-17 family, IL-21 and IL-22. These cytokines have been shown to increase inflammation through mechanisms such as the recruitment of immune cells (lymphocytes) and inflammatory effector cells, including monocytes/macrophages, neutrophils, eosinophils, mast cells, and basophils, from the circulation to the site of inflammation, which play a role in the elimination of detritus and the preparation of tissue for repair [21].
Thus, Th17 cells appear to be crucial in maintaining the inflammatory response and are responsible for chronic inflammation. The balance between tissue damage and tissue repair and fibrosis is thought to be regulated by oscillation in the predomination of either Treg or Th17 cells, based on the combined impact of several tissue- and systemically-specific pro-inflammatory signals and regulatory systems. For example, low transforming growth factor-β (TGF-β) concentrations in the microenvironment, combined with pro-inflammatory cytokines like IL-6 or IL-23, promote Th17 cell development, thus promoting inflammation. Conversely, higher TGF-β concentrations, override pro-inflammatory stimuli, shifting the Treg/Th17 balance towards Treg cell predomination and tissue repair processes, thus promoting fibrosis. This oscillating dynamic is seen as crucial in dictating the dynamic of chronic disease progression [22,23]. The plasticity of some other immune cells, in particular macrophages, which can oscillate between the pro-inflammatory M1 and reparatory M2 phenotype, depending on the conditions, may significantly contribute to the dynamic of tissue damage progression [24].
4. Inflammation Is the Vital Tissue and the Whole Body`s Reaction
Inflammation has been initially defined as an evolutionarily conserved response that protects the host from invading microorganisms and tumors while promoting tissue repair following injury [25]. Subsequently, it has been recognized that the immune system regulates a wide range of physiological processes, including neurological and gastrointestinal system function, metabolism, thermogenesis, and tissue regeneration and remodeling [25,26]. In addition, it is also involved in all types of homeostasis perturbations that may be caused by different factors, including changes in diet and environmental temperature, emotional disturbances, sleep deprivation, exposure to infections, toxins, and injuries.
Thus, inflammation can be understood as a multistep process of mobilizing defense mechanisms to eradicate the cause of homeostasis disruption. It may range from the regulatory physiological responses that occur in the absence of tissue damage to acute, time-limited reactions to infections or injury noxious, which can terminate with restitutio ad integrum or tissue remodeling, but can also lead to rapidly progressive homeostasis breakdown (in the form of sepsis). Lastly, there is the potential of chronic inflammation, which is typically brought on by non-infectious causes and is associated with the gradual deterioration of homeostasis, which eventually results in the emergence of chronic diseases [25,26,27]. In line with this view, a growing body of evidence indicates that inflammation is linked to almost all human diseases [28].
5. An Interplay between the Neuroendocrine, Immune, and Metabolic Pathways in Aging and Obesity, as a Driver of Chronic Disease Development
Unlike acute inflammation, which is often restricted to the local tissue environment and does not involve the whole body`s (systemic) reactions, chronic inflammation always involves systemic metabolic and neuroendocrine changes, that appear along with alterations in the structure and functions of a variety of tissues and organs [28]. These impacts may occasionally include just functional changes rather than structural changes [25,26]. The heterogeneity of pathology and variations in the dynamic of tissue damage may depend not only on the type and magnitude of inflammatory response but also on responsiveness/resistance of target tissues to inflammatory challenges, as well as the ability of control mechanisms to counteract the negative cost of inflammation and restore the body balance [26,29]. A period of time until which the adaptive control mechanisms can buffer the deleterious effects of inflammation-related mechanisms on tissues is determined by an individual`s genuine protective capacity and the degree to which the homeostatic mechanisms are undermined by past challenges [30].
Understanding how inflammation plays a part in the emergence of chronic diseases from a systemic viewpoint will aid in comprehending how these conditions sometimes manifest as several disorders coexisting and how their pathology gradually spreads over time [25,26,27]. Epidemiologic research indicates that the frequency and complexity of chronic illnesses are increasing with age, potentially due to a decline in the body's ability to maintain homeostasis caused by "wear-and-tear" of cells and tissues. The immune system's prolonged exposure to harmful stimuli, which occurs concurrently, causes a permanent rise in systemic inflammation, hastening the pathophysiological alterations throughout the body [31]. Chronically active innate immunity, a shift in specific immunity toward non-specific immunity, and autoimmune reactions are characteristics of the aging immune system. These factors collectively lead to a decrease in the development of specific immune responses to foreign antigens [32]. The early emergence of comorbidities accelerates aging and the development of poor health-related outcomes, even if older individuals can follow varied aging trajectories depending on lifestyle choices and life conditions they encounter throughout their lives [33].
The "sickness phenotype," which manifests in acute inflammation when pro-inflammatory cytokines alter neural circuits, may also accompany chronic inflammatory conditions in a modified form, exhibiting symptoms like fatigue, depression, low activity, altered sleep, muscle wasting, and social withdrawal. These observations lend support to the idea that chronic inflammation is a systemic disorder [26,28]. This phenotype is thought to be the neuroendocrine system’s homeostatic reaction to increased energy demand, due to the continuous immune system activity, and it is associated with fuel allocation from energy storage units to the immune cell compartment [34]. This metabolic derangement is connected with insulin resistance (reduced efficiency of insulin in glucose utilization in insulin-sensitive tissues, such as muscle, adipose tissue, the liver, and the brain).
Insulin resistance is an adaptive homeostatic mechanism that operates at the interface of metabolic derangements and inflammation. In the long run, it is always maladaptive, posing deleterious effects on health [32]. For instance, obesity is a condition linked to insulin resistance that increases the risk of developing many chronic diseases, including adult asthma, osteoarthritis, and certain types of cancer, as well as cardio-metabolic diseases like type 2 diabetes (T2D), hypertension, cardiovascular diseases (CVD), neurodegenerative diseases, and nonalcoholic fatty liver disease [35,36]. Adipose tissue in obese individuals has a high level of macrophage infiltration. It is an adaptive mechanism designed to counteract the excessive build-up of calories (as lipids) in adipose tissue due to phagocytosis by macrophages. The majority of pro-inflammatory cytokines, however, are produced by activated macrophages. Insulin resistance is a good adaptation strategy that seems to minimize additional adipose tissue inflammation and subsequent calorie storage [35]. However as time goes on, a vicious cycle of homeostasis breakdown occurs, involving mechanisms like tissue and cell resistance to adrenergic (sympathetic) stimulation and persistent activation of the hypothalamic-pituitary-adrenal (HPA) stress axis. This, combined with a disrupted circadian rhythm, eventually exacerbates numerous pathophysiology pathways and accelerates the progression of tissue and organ pathology (Figure 1) [26,34,35].
Obesity's position as a source of inflammation and a primary driving force in the development of chronic diseases requires greater clarification. There is mounting data that suggests elderly obese people with insulin resistance and abdominal obesity are more likely to have atherosclerotic CVD. In these individuals, the visceral adipose tissue is abundantly infiltrated with immune cells associated with type 1 inflammation, such as NK and NKT (share properties of NK and T cells) cells, innate lymphoid cells type 1 (ILC1), and Th1 cells. This phenotype is characterized by pro-inflammatory macrophage polarization (M1) and increased production of pro-inflammatory cytokines, including TNF-α and IL-6 (Figure 2) [26,35,37]. A variation in the degree of adipose tissue inflammation may be utilized to distinguish metabolically unhealthy from metabolically healthy obese patients [38].
Interestingly, interventions like physical activity or a healthy diet, which are known to lessen the pathological potential of obesity, have been shown to reverse type 1 to type 2 inflammations in adipose tissue of obese individuals, the process termed as browning of the white adipose tissue [39]. Type 2 inflammation is mediated by ILC2 and Th2 cells, M2 type of macrophages, and involves eosinophils and mast cells, along with type 2 cytokines, such as IL-13, IL-4, and IL-5. (Figure 2). Many details associated with this process, which otherwise might lead to efficient interventions, are not yet clear. In particular, there is a need to clarify the role of obesity in the pathogenesis of CVD and other chronic diseases in women and men, respecting the fact that men are more prone to atherosclerotic CVD, that are associated with type 1 inflammation, while women incline to type 2 inflammation and Th2/Th17 cell-mediated immune reaction [23,40,41].
6. The Role of the Gut Microbiome and the Gut Mucosal Immune System in Chronic Disease Development
The detailed presentation of the interplay between the gut microbiome and the gut mucosal immune system in the development of chronic diseases is out of the scope of this review. Yet, it is necessary to mention it briefly; because of the growing evidence indicating dysbiosis (changes in the composition and diversity of bacterial communities in the gut) and changes in the gut mucosal immune in different pathological conditions, when compared to the healthy controls (Figure 1) [42].
The gut microbiome is a large collection of microorganisms that inhabit the intestinal luminal surface and is considered as a functioning organ that plays essential roles in human physiology [43]. It is the most significant for immune system function and determining the trade-offs between health and disease since it is the largest of the microbial ecosystems in the body that colonize the skin and other mucosal surfaces [44]. The biological potential of this organ provides evidence that the number of cells in the gut microbiome exceeds the total number of cells in the human body by a factor of more than ten.
The process of colonizing the intestine and other mucosal surfaces begins from birth and lasts until 2-3 years of age when the number and the composition of microbial families must be stabilized. The gut mucosa develops along with the systemic immune system. Namely, the exposure of the gut mucosal innate immune receptors to microbial components provides necessary signals for the postnatal immune system maturation [43]. If conditions for gut microbial colonization are not favorable, like in premature infants, when a baby is not nursed, or when antibiotics are used, it will have long-term negative impacts on health (Figure 1) [45].
The intestinal microflora, if developed under unobtrusive conditions, establishes the symbiotic or beneficial equilibrium with the host, complementing human physiology in many ways, and is usually called “the commensal microflora”. These beneficial effects are achieved through several routes: 1) by protecting against infections, while 2) ensuring tolerance to foods and microflora itself, 3) by contributing to nutrient digestion and extraction of some essential nutrients from food that otherwise cannot be extracted, and 4) by keeping the intestinal epithelial barrier intact [46]. Under the influence of unfavorable environmental factors such as an unhealthy diet, antibiotic use, psychological stress, and exposure to toxins, especially in genetically susceptible individuals, this beneficial equilibrium may turn detrimental, leading to the development or worsening of chronic inflammatory diseases (Figure 1) [47].
Specifically, in maintaining the immune system homeostasis, the commensal microflora provides the microbe-associated molecular patterns, like lipopolysaccharides (LPS) and peptidoglycans that serve as a “scaffold” for immune cell education [48]. Alternatively, by providing microbial metabolites, such as short-chain fatty acids (SCFA), branched-chain amino acids (BCFA), tryptophan metabolites, butyrate, propionate, and acetate, the commensal microflora may mediate metabolism, and thus also activity, of the immune cells [49]. One more way how gut microflora may regulate immune functions is via neuroendocrine mechanisms, which include the enteric, autonomic, and central nervous system, as well as soluble factors, like neurotransmitters, neuropeptide hormones, and interleukins (Figure 1) [50].
The mucosal surfaces of the respiratory, gastrointestinal, and urogenital tract are the areas where the body comes into contact with the external world, and where most pathogens, but also harmless antigens, such as food and airborne antigens, enter the body [46]. Under these conditions, mucosal compartments, in particular that of the intestine, have developed structurally and functionally complex immune systems, capable of mounting immune responses against pathogens, while maintaining tolerance towards non-pathogenic antigens, including also those originating from the commensal microflora. However, the gut mucosal immune system can impose selective pressure on microbial strains by varying the types and intensity of defense mechanisms used, as well as the degree to which innate and adaptive immune responses are being engaged, in the event of excessive growth of gut microbes or when more aggressive strains with higher inflammatory potentials threaten to become prevalent among microbial communities and to breach the mucosal epithelial barrier [46,48]. The spatial diversity of microbial species, that was observed to exist along both the longitudinal (the stomach, the small intestine, the cecum, and the colon) and the transversal axis (the luminal, the crypts−associated, and inner mucus layers−associated microbiota) of the gastrointestinal tract, can also contribute to understanding the flexibility of the mucosal immune system and heterogeneity of immune responses that have been observed between individuals in the population [51].
To fulfill its dual function, the gut mucosal immune system is structured in a way that inductive and effector sites are spatially separated [46,52]. Inductive sites consist of the organized lymphoid structures, lymphoid follicles, and Peyer`s patches, which are situated in the subepithelial space, and most densely populated in the ileum. The effector sites include a variety of individual immune cells diffusely distributed throughout the lamina propria of the colonic mucosae (Figure 2).
The intact epithelial and mucus layers, innate immune cells that can change their phenotypes depending on conditions in the microenvironment, and secretory immunoglobulin A (IgA) antibodies, make the basis of immune response [38,44]. The IgA antibody response is highly flexible [53]. In lymphoid follicles and Peyer's patches, antigen-producing B cells can interact with follicular dendritic cells (FDCs) and follicular Th (Thf) cells to induce hypermutation of their immunoglobulin genes and select high-affinity antibodies. IgA antibodies can be produced during the specific immune response, usually in response to pathogens [53,54]. These antibodies can also be produced as a part of the innate immune response, where Th cells are not necessary for their production [44]. In this case, IgA antibodies are of low affinity and restricted diversity, and their function is to sustain commensal microflora in a homeostatic state [53].
In a healthy condition, the immune response to intestinal microbiota is focused on the mucosal surface [38,40,44]. The mechanisms that prevent the epithelial barrier from being leaked include intact epithelium and mucus layers, as well as a range of antimicrobial peptides generated by specialized epithelial cells and an efficient IgA antibody response (Figure 2). DCs and IgA antibodies play a critical role in sampling gut microbes. Sensing gut microbiota via innate cell receptors in lamina propria mucosae results in low-grade activation of innate immune cells (priming). This is an adaptive mechanism that maintains microbial populations in equilibrium and promotes epithelial barrier integrity.
As pro-inflammatory signals from the gut microbiota become stronger in the presence of perturbations, the level of activation of innate immune cells increases, creating conditions for the establishment and maintenance of an inflammatory response, as well as potentially initiating the specific immune reaction. ILCs play an important function in controlling inflammation levels and facilitating the transition from "physiologic" to "pathologic" inflammation. These cells lack surface antigen-dependent receptors but can synthesize cytokines, which initiate the differentiation of Th cells into Th1, Th2, or Th17 subtypes (Figure 2). This way, the immune response is being directed towards domination of either Th1/Th17 or Th2/Th17 type.
7. Functional Gastrointestinal Disorders
FGIDs are the most prevalent diagnosis in gastroenterology, defined by persistent symptoms caused by disorganized brain-gut connections [55]. More than 40% of individuals suffer from FGIDs globally, which affects the quality of life in individuals and healthcare use. FGIDs are distinguished by altered gut microbiota, visceral hypersensitivity, altered mucosal and immunological function, and an impaired central nervous system [56]. They are also thought to be associated with a high incidence of mental comorbidities and chronic pain problems [57]. FD and IBS, as the most common FGIDs, are identified based on the existence of gastrointestinal symptoms, but no obvious structural abnormalities. The variability of FGIDs still makes it difficult to identify causal pathways [58].
FD is classified as a gut-brain interaction disorder [59]. It is the most frequent FGID, with a prevalence of 20% to 40%, accounting for 3% to 5% of primary care visits. People with FD have poor quality of life due to persistent or recurring upper abdomen pain or discomfort and their quality of life is impaired in the same way that individuals with moderate heart failure are [60]. Using the Rome IV criteria it can be classified as postprandial distress syndrome (PDS) or epigastric pain syndrome (EPS), with possible overlapping. The first subcategory is defined as an uncomfortable early satiation and/or postprandial fullness that occurs at least three times weekly and the second subcategory is defined as discomfort and/or burning at least once weekly. Diagnostic criteria must be met continuously for 3 months, with symptoms appearing at least 6 months before diagnosis [59,61]. FD frequently coexists with another FGID, especially IBS [62]. The underlying pathophysiology of FD is poorly known but visceral hypersensitivity, disturbed gastric motility, post-infectious gastroenteritis, increased intestinal permeability, immune dysfunction, and gut microbiota changes, have been linked to it [59]. Knowing that more than 34% of FD patients have a psychological disorder, explains a bidirectional relationship between the gastroduodenal region and the brain (the gut-brain axis) that effectively contributes to patient symptoms [57].
IBS is a complex disorder that causes persistent stomach discomfort and irregular bowel movements. Symptoms may overlap with those of other FGIDs and up to one-third of individuals with FGIDs exhibit characteristics of many conditions, indicating a shared underlying cause [63,64]. According to Rome IV criteria, patients are classified into four categories based on their predominant bowel habit: diarrhea-predominant (IBS-D), constipation-predominant (IBS-C), mixed diarrhea/constipation (IBS-M), and unclassified (IBS-U) [65]. IBS has a predicted prevalence of 10-15 %, and is one of the leading causes for primary care visits, resulting also in increased expenditures [66]. IBS recognition has expanded in recent years but pathogenesis remains unclear, although potential causes include visceral hypersensitivity, aberrant gut motility, irregular brain-gut connection, and chronic low-grade intestinal mucosal inflammation. Studies have found differences in mast cells, T lymphocytes, B lymphocytes, and cytokine concentrations in the intestinal mucosa or systemic circulation in IBS patients compared to healthy controls [65,66]. FGIDs may be linked to atopy and microbiota imbalances. Furthermore, the research provides indirect evidence that Th17, in combination with Th2 immunological responses has an important role in the etiology of FGIDs [55,60].
8. Functional Gastrointestinal Disorders with Inflammation in the Background (PI-IBS and EoE)
Post-infection irritable bowel syndrome (PI-IBS) is an IBS condition that presents after an acute gastroenteritis episode in individuals who do not have previous IBS symptoms. It has been associated with viral, bacterial, or protozoal enterocolitis. Within one year of an acute gastrointestinal illness, there is a fourfold increase in the likelihood of developing IBS compared to non-exposed persons [67,68]. The pathophysiological process of PI-IBS involves pathogenic organisms altering the gut microbiota, leading to reduced diversity and increased Firmicutes: Bacteroides ratio (13). PI-IBS has shown severe disturbance to the core microbiome (Firmicutes, Bacteroidetes, and Actinobacteria) and a 12-fold rise in Bacteroidetes with a reduction in Firmicutes and Clostridiales when compared to healthy persons. This is also causing modifications in bile acid absorption, resulting in diarrhea [69,70,71]. Mast cells, eosinophils, T lymphocytes, and chromaffin cells release inflammatory factors (TNF-α, IFN-γ, IL-1, 6 and 8), as well as agents like histamine, leukotriene, and 5-hydroxytryptamine (5-HT), leading to increased intestinal permeability via the reorganization of proteins associated with tight junctions and visceral hypersensitivity [72,73,74].
Th1 and Th2 cells play important roles in regulating the immune response. Th2 cells, crucial for humoral immunity, secrete IL-4, IL-10, IL-13, and IL-6, whereas Th1 cells, important for cellular immunity, emit IFN-γ, IL-12, IL-2, and TNF-α. They inhibit one another to preserve a balanced immune response [75]. Studies revealed that Th1 and Th2 cytokine expression varied in the intestinal mucosa of PI-IBS patients, suggesting that the illness was caused by an immune dysregulation mechanism. Th1-derived cytokine expression (IFN-γ) increased while Th2-derived cytokine expression (IL-10) decreased in PI-IBS patients, indicating a shift towards Th1 immunodominance which may lead to chronic low-grade inflammation [75,76]. T helper 17 (Th17) polarizations are also observed in IBS and studies showed that adenosine and its receptors are involved in inflammation by promoting the Th17 polarization of CD4+ T cells [77].
Eosinophilic esophagitis (EoE) is another important FIGD, characterized by esophageal function abnormalities and extensive eosinophilic inflammation [78]. Epithelial cells and DCs release cytokines (IL-25 and IL-33), but also a thymic stromal lymphoprotein (TSLP) leading to the activation of invariant natural killer T (iNKT) cells, adaptive CD4+ effector memory T-helper 2 (Th2) cells, and innate lymphoid type 2 cells (ILC2). The predominant reaction is the Th2 immune response, secreting IL-4, IL-5, IL-13, IL-15, eotaxin-3, and periostin [79,80]. IL-5 causes eosinophils to multiply and extend from the bone marrow to all layers of the esophagus and to degranulate, triggering the release of various molecules linked to the remodeling of tissue [81]. Eotaxin-3 is overexpressed causing esophageal mucosal inflammation as a result of environmental antigens. Gene expression studies show a significant type 2 immune response in EoE, with mast cells potentially regulating the disease. Mast cells expand into the epithelium, while eosinophils enter the subepithelial tissue of the esophagus wall [82]. Individuals in clinical remission may still have a large number of mast cells. TGF-β is also important for EoE pathophysiology, causing mucosal remodeling and smooth muscle dysfunction [83].
9. Proofs That Inflammation Is Involved in the Pathogenesis of Functional Dyspepsia
FD has a complicated pathophysiology that is still unexplained. According to research, patients with FD, especially those with PDS, had increased levels of systemic cytokines and eosinophil infiltration, which is consistent with duodenal inflammation. The eosinophil-mast cell axis secretes a number of chemical mediators that may influence visceral hypersensitivity and gastrointestinal motility and may lead to neuromuscular and epithelial dysfunction. These mediators are crucial in the development of gastrointestinal symptoms of FD [84,85]. Eosinophil infiltration is estimated to cause low-grade inflammation in up to 40% of FD patients, and when these cells degranulate, symptoms occur along with impaired mucosal integrity and structural and neuronal abnormalities [86]. Mast cells are produced from bone marrow myeloid-cell progenitors (CD34+) and are important for controlling chemotaxis, nociception, innate and adaptive immunity, angiogenesis, peristalsis, fibrosis, and tissue healing in the gastrointestinal tract but also for modulating vascular and epithelial permeability. They are influenced by IL-4 and stem cell factors which control the development of different subtypes of mast cells. Unregulated or disrupted activation of mast cells can interfere with gut homeostasis, causing tissue dysfunction, and increasing inflammation [58].
The studies showed no variations in the production of IgE or the expression of the typical Th2 response cytokines (IL-4, IL-5, and IL-13), even in the presence of eosinophil involvement. So there is no firm evidence that type-2 immune responses cause duodenal eosinophilia in FD patients [64]. A literature review indicates that the Th17/Treg axis should be explored because FGID studies have shown increased levels of TNF-α, IL-1β, and IL-6, all of which are associated with the Th17 pathway. It has been demonstrated that IL-17, the main proinflammatory cytokine of the Th17 response, causes macrophages to produce IL-1β and TNF-α. The proliferation and inflammatory activity of Th17 and ILC2 populations may reduce Treg and ILC3 populations if homeostasis is disrupted in FGID patients. Given that ILC3 plays a role in maintaining homeostasis, the developing field of ILC biology in FGID patients is also worth investigating. Furthermore, eosinophils and mast cells can be recruited by Th17 and ILC2 cells [55]. Nevertheless, the observational studies confirm a close association between gastrointestinal disorders, notably including FD, and asthma, although the pathophysiology pathways are still poorly defined [87]. This association is especially reasonable when asthma is viewed as an umbrella diagnosis for several diseases with distinct and interrelating inflammatory pathways [88]. Gut microbiome dysbiosis has been proposed as the mechanism that most likely links gastrointestinal and chronic respiratory diseases.
The development and expression of FD symptoms are significantly influenced by complex brain-gut interactions. This relationship is bidirectional, with gastrointestinal symptoms and mucosal immunological activation acting as possible initiators of psychiatric comorbidities and stress, anxiety, or depression. Patients experience brain symptoms of dyspepsia and are susceptible to compounds found in the duodenal lumen such as food-derived substances, bile acids, and microbiota. Because of enhanced duodenal mucosal permeability, contents can pass through the mucosa and be identified by immune cells, which causes low-grade mucosal inflammation. Histamine, tryptase, and cytokines are among the mediators released by inflammatory cells that impair stomach motility and alter submucosal afferent neurons. The brain is the final organ to detect these alterations. These mediators enhance the permeability of the duodenal mucosa and weaken the epithelial barrier even more. And vice versa, psychological stress as an element in the pathophysiology of FD, raises duodenal permeability by stimulating mast cells via the release of corticotrophin-releasing hormone (CRH) [60,89].
Changes in the SCFA profile and improper activation of mucosal cellular stress response mechanisms, such as hypoxia-inducible factor (HIF), might also result from dysbiotic gut microbiota. The expression of HIF transcriptional targets that influence eosinophil recruitment and barrier structure in FD may be impacted downstream. For instance, duodenal eosinophilia may be facilitated by HIF-mediated elevation of TNF-α and IL-1β, which have been demonstrated to be elevated in FD inflammation [57].
10. Proofs That Inflammation Is Involved in the Pathogenesis of Irritable Bowel Syndrome
Some clinical characteristics shared by individuals with IBD may be explained by the presence of modest systemic immune activation and/or low-grade mucosal inflammation in IBS patients. Higher serum levels of TNF-α and IL-17 were found to be negatively connected with quality-of-life scores and to be correlated with IBS patients' discomfort and severity of symptoms [66]. Some other studies that measured the levels of cytokines in the serum and intestinal mucosa found no relationship between the severity of the overall symptoms and the expression of the cytokines, even though IBS patients had higher serum levels of the pro-inflammatory cytokines IL-6 and IL-8 and lower serum levels of anti-inflammatory IL-10. However, the correlation between certain clinical symptoms and inflammatory cytokines implies that immunological activation might be significant for a portion of IBS patients, such as diarrhea-predominant [90,91,92].
Because mast cells' mediators have the ability to modify enteric nerve and motor function, they are also thought to have a role in the pathophysiology of IBS. Though the majority of studies reveal higher numbers and volume of mast cells in IBS patients compared with healthy controls, mast cells counts and density vary among studies and within different segments of the intestine. Researches have demonstrated that DCs also contribute to the pathophysiology of IBS by inducing visceral hypersensitivity, activating the microcirculation, and prolonging intestinal activity. Furthermore, they have the potential to release corticotropin-releasing factor (CRF), which has the ability to cause changes in visceral hypersensitivity and intestinal motility. Even though mast cells and DCs play a role in the inflammatory state that is crucial to the pathophysiology of IBS, not all illness phenotypes showed elevated levels of these cells, which further supports the idea that IBS patient groups differ from one another. Naturally, the aforementioned can and ought to guide the creation of a customized course of care [66].
Aside from inflammation of the mucosa, neuroinflammation likely plays a role in the pathogenesis of IBS through the "gut–brain" axis, altering neuroendocrine pathways and glucocorticoid receptor genes. This results in a generalized pro-inflammatory phenotype as well as dysregulated serotonergic and hypothalamic-pituitary-adrenal axis functioning, which may partially explain the symptoms of IBS [65]. Recently, the importance of the gut-lung axis which can bi-directionally influence each other in health and disease has been revealed [87]. The gut and lungs are thought to communicate via various mechanisms, but gut dysbiosis-triggered GLA has been most widely studied. The opposite direction of interactions, linking inflammation of the remote sites to FGIDs, has been poorly studied so far.
Regarding the role of gut dysbiosis in pathogenesis of IBS, the fecal microbiota of individuals with IBS was found to exhibit notable dissimilarities from that of healthy participants, which may have an impact on changed bowel habits. Numerous investigations have revealed that pro-inflammatory bacterial species such Enterobacteriaceae are more prevalent than Bifidobacterium and Lactobacillus. There have been reports of both higher and lower ratios of Firmicutes/Bacteroidetes in IBS participants, which is a crude predictor of changed microbial population [93]. Established inflammatory changes in the intestine, in turn, contribute to an increase in the number of Bacteroidetes in the intestinal microbiota composition. Bacteroides, in turn, can dissolve mucosal glycoproteins, and aggravate the intestinal barrier permeability due to the splitting of dense contact proteins. Firmicutes are the main type of bacteria in the human microbiota that produce SCFAs and it has previously been demonstrated that SCFAs alter the expression of occludin, claudines 3 and 4, all of which can strengthen the epithelium's barrier qualities, thus also creating favorable conditions for prolonging the inflammatory state [94].
11. Discussion
Moving forward with the classification and individualized treatment of FGIDs will require a more comprehensive understanding of these disorders than is currently the case, according to mounting data. Therefore, the Rome Foundation Global Epidemiologic Survey results demonstrated that patients with FGIDs have a worse quality of life as the number of overlapping FGIDs increases. Additionally, the presence of mental disorders such as somatization, anxiety, and depression is associated with higher activation of neural mechanisms in patients with more symptoms [95]. According to the same line of thought, IBS and other FGID entities have been grouped with chronic fatigue and pain syndromes under the umbrella term "functional somatic disorders" [96]. Clinical practice, particularly primary care, commonly encounters somatic symptoms that are unaccounted for by the underlying physical disorders. These symptoms are typically linked to mental discomfort, and compromised well-being and low-stress coping or chronic psychological stress have been suggested as the main etiologic factors [96,97].
The fact that not all of these individuals exhibit signs of chronic stress, such as low cortisol levels in the blood or hair, contradicts this theory [97]. There are two possible explanations: 1) one biomarker is insufficient to capture the variety of pathophysiology pathways that may be present in these patients; or 2) the low sensitivity of conventional biomarkers to indicate variations in the levels of the HPA axis arousal or suppression, implying the need for new biomarkers. Whether subclinical pathology changes underlie symptoms of functional somatic disorders, cannot be said for sure, as evidence is insufficient in these terms. Growing evidence, however, coming from different research areas, indicates that these symptoms are likely to be a part of the spectrum of inflammation-mediated chronic disorders (Figure 3). This theory can be supported by several lines of evidence.
The first line of evidence is based on the finding that systemic inflammation has an essential role in generating symptoms of functional somatic disorders such as chronic fatigue syndrome. However, the results of the studies are not consistent, which means that the level of inflammation may vary among these patients [98]. Similarly, serum levels of IL-6, a traditional marker of systemic inflammation, were found to be higher in IBS patients, compared to controls, but only in one patient subtype (diarrhea predominant), which indicates that variations in the levels of inflammation are associated with different phenotypic expression [99]. Variations in the level of systemic inflammation are likely to be influenced by environmental and acquired factors, rather than they are genetically determined [99,100].
The second line of evidence indicates that mental disorders, even those with high hereditary influence, are inflammation-mediated [101]. If mental disorders develop early in life, they often result in the development of somatic comorbidities, especially cardio-metabolic disorders, which are thought to be the outcome of an allostatic load on multiple biological systems due to the chronic stress that mental illness causes. And vice versa, age-associated somatic disorders, which are inflammation-mediated, are often accompanied by mental disorders such as anxiety and depression. With everything considered, a shared framework is required to comprehend the intricate phenomena involving both bodily and brain processes. New research techniques that go beyond conventional study designs and data evaluation techniques will be needed for the initiatives focused on this area [102]. One of the recent studies, e.g., is focused on the parallel quantification of markers of systemic inflammation, IL-6 and C-reactive protein (CRP), and markers of the impaired intestinal epithelial barrier, including e.g., LPS and lipopolysaccharide-binding protein (LBP), intestinal fatty acid binding protein (IFABP), and calprotectin, in patients with mental disorders, to identify patterns of biomarkers that correspond to the types and severity of mental symptoms [103].
The third line of evidence consists of the studies that, when considered collectively, support the idea that patients with FGIDs may experience inflammatory signals from two different sources: increased mucosal inflammation on the inside and increased systemic inflammation on the outside [104]. The variations in the composition of markers of the systemic vs. the gut mucosae-associated inflammation may have the potential to create a new, more diverse classification of these disorders, which in turn can lead to etiologically-driven treatment. For example, it was shown that psychological stress in younger individuals with FD, who are still free of overt somatic comorbidities, can induce increased HP infection activity in gastric mucosae, but without an increase in the mucosal inflammatory markers like IL-6 and IL-8 [105]. Rather than increasing mucosal-related inflammation, mood disorders that predominate in the comorbidity pattern cause greater systemic inflammation, albeit the quantity, type, and severity of symptoms may be correlated with the level of systemic inflammation [103].
We supported the hypothesis that the duration and/or intensity of prior stress reactions, the state of the gut microbiome, and current comorbidities all influence the pathophysiological alterations in the guts of people with FGIDs. Only functional abnormalities such as decreased motility, visceral sensitivity, or mucosal blood flow, resulting from activated central neuroendocrine stress systems, are expected to contribute to gastrointestinal dysfunction in young individuals who do not experience stress for an extended period. Long-term stress can cause somatic mechanisms of allostatic burden to start, gut dysbiosis can occur, and eosinophil/mastocyte infiltrations can appear as indicators of an inflammatory response in the gut mucosae. This hypothesis is supported by data showing that there are a variety of effects of chronic stress on gastrointestinal function, such as altered gut wall structure and motility, and that histological immune changes, tissue changes, and changes in serum cytokines and immune cell populations vary amongst FGID studies (for diseases such as FD and IBS) [64,106]. Increased intestinal wall fibrosis, which lowers wall flexibility and the number of sensory receptors, may emerge as the main mechanism of gastrointestinal dysfunction as mucosal inflammation progresses.
As elaborated in the above sections, the gastrointestinal tract mucosae is colonized by eosinophils, which often co-localize with mast cells, in both homeostatic and inflammatory conditions [107,108,109]. The physiologic role of these innate immune cells is to maintain intestinal epithelial cell homeostasis against the overgrowth of the luminal microflora. Under stress or disease conditions, the burden of cell infiltrates and the level of their degranulation increase, which is associated with the release of plenty of inflammatory mediators, that can turn into tissue damage and fibrosis, instead of homeostatic regulation. In atopic and allergic conditions like atopic dermatitis, food allergies, asthma, and allergic rhinitis, which are mediated by a Th2-type immune reaction, involving the antibody immune response through a mediating role of IgE and IL-4 and IL-5 cytokines, eosinophils and mast cells are recognized as important players. [110]. Therefore, it is not surprising that these cells participate in gastrointestinal tract pathological conditions, considering that IgA response mediates the intrinsic immune response of gastrointestinal tract mucosae. Specifically, it has long been known that abnormal IgA antibody production plays a role in the development of clinical manifestations such as allergies, autoimmune diseases, and a higher risk of gastrointestinal and respiratory tract infections. [111,112]. Our knowledge of eosinophils' possible pathogenic function in gastrointestinal disorders is still developing.
The hypothesis that FD, a form distinguished by duodenal eosinophilic infiltration, is connected to atopic diathesis emerges but has not yet been assessed. In this case, pharmacological treatments with proven efficacy in allergic diseases such as asthma are likely to have the potential also in curing these patients [113]. Moreover, these FD patients' serum cytokine profiles are probably compatible with the Th2-mediated immune response, which may be helpful for diagnosis. The findings of a previous study, which showed that atopic patients with chronic gastritis had considerably greater serum concentrations of Th2 cytokines, IL-4 and IL-5, than did non-atopic patients with the same condition, support this theory [114]. In addition, obese middle-aged and elderly asthma, which are regarded as T2-low, and mostly mediated by neutrophils, can share with FD cytokines of Th1/Th17-type immune response [88].
Based on current research, it has been determined that the composition and concentration of serum cytokines and other inflammatory markers are influenced by various factors that characterize patients with gastric symptoms. Therefore, a thorough patient profile is necessary, encompassing variables like HP infection, the degree of mucosal inflammation (gastritis grading), and related gastrointestinal and extra-gastrointestinal comorbidities [116,117]. The evidence also suggests that major changes in the gut microbiome composition are associated with the genetic susceptibility for bowel inflammation, as in patients with IBD, and less likely to depend on environmental and lifestyle factors, as in patients with IBS or those with obesity-related metabolic disorders [116,117,118]. Rather, discrete modifications in the composition of the gut microbiome and variances in metabolite activity are driven by behavioral factors like diet and sleep quality. [119,120]. Interestingly, the studies showed that gut microbial metabolites may regulate sleep duration through the influence on circadian gene expression, which in the longer run may lead to dysbiosis and metabolic disturbances. In a bidirectional relationship, low sleep quality results in gut dysbiosis and metabolic disturbances due to activation of the HPA-axis [120].
Therefore, we hypothesize that the range of phenotypes observed in patients with FGIDs may be reflected in differences in serum pro-inflammatory, Th2- and Th17-type cytokines, as well as certain gut microbial metabolites and markers of epithelial barrier degradation. For phenotyping, a comprehensive patient description would be required, including variables such as age, sex, gastrointestinal symptoms, behavioral habits, and comorbidities. To deal with patient subgroup identification, modeling techniques based on data integration or clustering methods will be required. At one end of the continuum, as we showed in Figure 3, the location may take isolated or common gastrointestinal illnesses, such as those found in young adults who maintain unhealthy lifestyles but do not have any additional comorbidities. In this instance, it is anticipated that gut microbial metabolites and markers of damage to the gut epithelium barrier will be more prevalent in serum than indicators of systemic inflammation, such as Th1- and Th17-type cytokines. Discrete changes in the gut microbiome and alterations in microbial metabolites could be responsible for gut motility dysfunction and visceral hypersensitivity [121]. When there is more intense inflammation of the gut mucosa, the gut nerve ends nociceptive receptors become more sensitive, and the composition of serum soluble factors changes. These mediators of inflammation and activated immune cells, particularly activated eosinophils and mast cells, take over this role. On the opposite end of the spectrum, there may be older individuals who are obese and have cardio-metabolic disorders. These disorders are linked to changes in the gut microbiome and systemic inflammation, but they still fall within the category of people with low whole body entropy and allostatic load mechanisms. In this instance, central neuroendocrine and inflammatory signals, or signals from the outside, have the potential to widely sensitize the gut nerves. Lastly, there may be circumstances involving severe local and systemic inflammatory responses, such as in individuals with renal failure and CV comorbidities [122].
12. Future Perspectives
FGIDs were once thought to be psychosomatic illnesses or motility abnormalities. The term "disorders of the gut-brain interactions" was introduced to reflect the current knowledge of these conditions, which is based on the bio-psycho-social model of chronic diseases. We went a step further in this narrative review, presenting these disorders as a spectrum of phenotypes in which symptoms are attributed to varying amounts of systemic and local gut neuro-inflammatory signals, dependent on the social, behavioral, and clinical context of an FGID patient at the time of observation. Drawing on our prior research experience, we suggest that we can gain a new understanding of these disorders with significant therapeutic implications by clustering these patients into distinct phenotypic subgroups and integrating clinical and psychosocial variables with markers of altered gut microbial metabolite production, epithelial barrier dysfunction, and inflammation. This method may lead to a new classification of FGIDs, which will likely inform individualized care and generate creative, original hypotheses that will support additional studies in this field.
13. Conclusions
The two most prevalent FIGDs that negatively impact patients' quality of life and place significant pressure on the healthcare system—particularly family physicians, who treat patients with the symptoms above most frequently—are FD and IBS. The pathophysiology of these disorders is still unclear even though the body of information supporting it is expanding daily. Moreover, the pathophysiology differs throughout patients. It is established that individuals with the mentioned disorders differ from one another, and the inability to precisely identify the mechanism underlying the emergence of symptoms in each of the groups makes therapy even more challenging today. The persistent presence of low inflammation has been linked to several cytokines and inflammatory cells, and this is believed to be a fundamental pathophysiological mechanism. The gut-brain axis concept was developed to provide a fundamental framework for the intricate relationships that exist between the neurological system and intestinal processes, including the important role of gut microbes. Systemic chronic inflammation, neuroendocrine disorders, metabolic changes, and gut microbiota dysbiosis are the basis for the emergence of many chronic diseases, the prevalence of which increases with aging and greater exposure to stress. By grouping patients with FIGDs into distinct phenotypic subgroups and combining clinical and psychosocial variables with markers of altered gut microbial metabolite production, epithelial barrier dysfunction, and inflammation, we can obtain a new understanding of these disorders that has crucial therapeutic implications. This approach might result in a new classification of FGIDs, which would probably guide tailored treatment and inspire innovative, creative theories.
Author Contributions
Conceptualization, D.Š. and L.T.M.; methodology, L.T.M. and N.V.; software, M.V. and D.Š.; validation, M.V., D.Š. and L.T.M.; formal analysis, D.Š.; investigation, T.K., V.C. and M.V.; resources, L.T.M.; data curation, L.T.M.; writing—original draft preparation, L.T.M., D.Š., M.V. and T.K.; writing—review and editing, D.Š., M.V., N.V., V.C. and L.T.M.; visualization, T.K. and V.C.; supervision, L.T.M.; project administration, L.T.M.; funding acquisition, L.T.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from the Josip Juraj Strossmayer University of Osijek, Faculty of Medicine, within the project IP-24 “Clinical inertia in prescribing medications for chronic diseases in family medicine”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| IBD | inflammatory bowel diseases |
| UC | ulcerative colitis |
| CD | Chron's disease |
| HP | Helicobacter pylori |
| FGID's | functional gastrointestinal disorders |
| IBS | irritable bowel syndrome |
| FD | functional dyspepsia |
| Treg cells | regulatory T cells |
| Teff cells | effector T cells |
| TNF-α | tumor necrosis factor-α |
| ROS | reactive oxygen species |
| DCs | dendritic cells |
| NK cells | natural killer cells |
| Th1 cells | T helper cells type 1 |
| TGF-β | transforming growth factor-β |
| T2D | type 2 diabetes |
| CVD | cardiovascular diseases |
| HPA | hypothalamic-pituitary-adrenal |
| ILC1 | innate lymphoid cells type 1 |
| LPS | lipopolysaccharides |
| SCFA | short-chain fatty acids |
| BCFA | branched-chain amino acids |
| IgA | immunolobulin A |
| FDCs | follicular dendritic cells |
| Thf cells | follicular Th cells |
| PDS | postprandial distress syndrome |
| EPS | epigastric pain syndrome |
| PI-IBS | post-infection irritable bowel syndrome |
| 5-HT | 5-hydroxytryptamine |
| Th17 | T-helper 17 |
| EoE | eosinophilic esophagitis |
| ILC3 | innate lymphoid type 3 cells |
| CRH | corticotropin-releasing hormone |
| HIF | hypoxia-inducible factor |
| CRF | corticotropin-releasing factor |
| TSLP | thymic stromal lymphoprotein |
| iNKT cells | invariant natural killer T cells |
| Th2 | T-helper 2 |
| ILC2 | innate lymphoid type 2 cells |
| CRP | C-reactive protein |
| LBP | lipopolysaccharide binding protein |
| IFABP | intestinal fatty acid binding protein |
References
- Lu, Q.; Yang, M.F.; Liang, Y.J.; Xu, J.; Xu, H.M.; Nie, Y.Q.; Wang, L.S.; Yao, J.; Li, D.F. Immunology of Inflammatory Bowel Disease: Molecular Mechanisms and Therapeutics. J Inflamm Res. 2022, 15, 1825–44. [Google Scholar] [CrossRef]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006, 19(3), 449–90. [Google Scholar] [CrossRef]
- Drossman, D.A. Functional gastrointestinal disorders: history, pathophysiology, clinical features and Rome IV. Gastroenterol. 2016, 150, 1262–79. [Google Scholar] [CrossRef] [PubMed]
- Drossman, D.A.; Tack, J. Rome Foundation Clinical Diagnostic Criteria for Disorders of Gut-Brain Interaction. Gastroenterol. 2022, 162(3), 675–9. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Singh, R.; Ro, S.; Ghoshal, U.C. Gut microbiota dysbiosis in functional gastrointestinal disorders: Underpinning the symptoms and pathophysiology. JGH Open. 2021, 5, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015, 28(2), 203–9. [Google Scholar] [PubMed]
- Foster, J.A.; McVey Neufeld, K.A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–12. [Google Scholar] [CrossRef]
- Van Oudenhove, L.; Crowell, M.D.; Drossman, D.A.; Halpert, A.D.; Keefer, L.; Lackner, J.M.; Murphy, T.B.; Naliboff, B.D.; Levy, RL. Biopsychosocial Aspects of Functional Gastrointestinal Disorders. Gastroenterol. 2016:S0016-5085(16)00218-3. [CrossRef]
- Xiang, Y.; Zhang, M.; Jiang, D.; Su, Q.; Shi, J. The role of inflammation in autoimmune disease: a therapeutic target. Front Immunol. 2023, 14, 1267091. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benkő, S.; Szűcs, G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. 2021, 17(10), 585–95. [Google Scholar] [CrossRef]
- Park, H.; Bourla, A.B.; Kastner, D.L.; Colbert, R.A.; Siegel, R.M. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol. 2012, 12(8), 570–80. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J Clin Invest 2015, 125(6), 2228–33. [Google Scholar] [CrossRef]
- Jörg, S.; Grohme, D.A.; Erzler, M.; Binsfeld, M.; Haghikia, A.; Müller, D.N.; Linker, R.A.; Kleinewietfeld, M. Environmental factors in autoimmune diseases and their role in multiple sclerosis. Cell Mol Life Sci. 2016, 73, 4611–22. [Google Scholar] [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat Immunol. 2018, 19(7), 665–73. [Google Scholar] [CrossRef]
- Medina, G.; Vera-Lastra, O.; Peralta-Amaro, A.L.; Jiménez-Arellano, M.P.; Saavedra, M.A.; Cruz-Domínguez, M.P.; Jara, LJ. Metabolic syndrome, autoimmunity and rheumatic diseases. Pharmacol Res. 2018, 133, 277–288. [Google Scholar] [CrossRef]
- Hedrich, C.M. Shaping the spectrum - From autoinflammation to autoimmunity. Clin Immunol. 2016, 165, 21–8. [Google Scholar] [CrossRef] [PubMed]
- 17. Greuter, T, Vavricka, S.R. Extraintestinal manifestations in inflammatory bowel disease – epidemiology, genetics, and pathogenesis. Exp Rev of Gastroenterol & Hepatol 2019, 13(4), 307–17. [CrossRef]
- Wang, X.; Antony, V.; Wang, Y.; Wu, G.; Liang, G. Pattern recognition receptor-mediated inflammation in diabetic vascular complications. Med Res Rev. 2020, 40(6), 2466–84. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, N.; Ouyang, W. Targeting the development and effector functions of TH17 cells. Semin Immunol. 2007, 19(6), 383–93. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371(3), 531–9. [Google Scholar] [CrossRef]
- Trtica Majnarić, L.; Guljaš, S.; Bosnić, Z.; Šerić, V.; Wittlinger, T. Neutrophil-to-Lymphocyte Ratio as a Cardiovascular Risk Marker May Be Less Efficient in Women Than in Men. Biomol. 2021, 11(4), 528. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol. 2013, 25(4), 305–12. [Google Scholar] [CrossRef]
- Mills, C.D. Anatomy of a discovery: M1 and M2 macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef]
- Medzhitov, R. The spectrum of inflammatory responses. Science. 2021, 374(6571), 1070–5. [Google Scholar] [CrossRef]
- Rankin, L.C.; Artis, D. Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell. 2018, 173(3), 554–567. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature's Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing "the Epidemic" of Chronic Diseases. Front Med (Lausanne). 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019, 25(12), 1822–32. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science. 2012, 335(6071), 936–41. [Google Scholar] [CrossRef] [PubMed]
- Fava, G.A.; McEwen, B.S.; Guidi, J.; Gostoli, S.; Offidani, E.; Sonino, N. Clinical characterization of allostatic overload. Psychoneuroendocrinol. 2019, 108, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–54. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Salvioli, S.; Garagnani, P.; de Eguileor, M.; Monti, D.; Capri, M. Immunobiography and the Heterogeneity of Immune Responses in the Elderly: A Focus on Inflammaging and Trained Immunity. Front Immunol. 2017, 8, 982. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front Med (Lausanne). 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Cutolo, M.; Buttgereit, F.; Pongratz, G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. 2010, 267(6), 543–60. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. 2017, 13(11), 633–43. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, C.; Fisichella, P.M. Epidemiology of obesity and associated comorbidities. J Laparoendoscopic Adv Surg Techniq A. 2018, 28, 919–24. [Google Scholar] [CrossRef]
- Nedunchezhiyan, U.; Varughese, I.; Sun, A.R.; Wu, X.; Crawford, R.; Prasadam, I. Obesity, Inflammation, and Immune System in Osteoarthritis. Front Immunol. 2022, 13, 907750. [Google Scholar] [CrossRef] [PubMed]
- Tsatsoulis, A.; Paschou, S.A. Metabolically Healthy Obesity: Criteria, Epidemiology, Controversies, and Consequences. Curr Obes Rep. 2020, 9(2), 109–20. [Google Scholar] [CrossRef]
- Machado, S.A., Pasquarelli-do-Nascimento, G., da Silva, D. Farias, G.R.; de Oliveira Santos, I.; Baptista, L.B.; Magalhães, K.G. Browning of the white adipose tissue regulation: new insights into nutritional and metabolic relevance in health and diseases. Nutr Metab (Lond). 2022, 19(1), 61. [CrossRef]
- Razeghian-Jahromi, I.; Karimi Akhormeh, A.; Razmkhah, M.; Zibaeenezhad, M.J. Immune system and atherosclerosis: Hostile or friendly relationship. Int J Immunopathol Pharmacol. 2022, 36, 3946320221092188. [Google Scholar] [CrossRef]
- Garcia, M.; Mulvagh, S.L.; Merz, C.N.; Buring, J.E.; Manson, J.E. Cardiovascular Disease in Women: Clinical Perspectives. Circ. Res. 2016, 118, 1273–93. [Google Scholar] [CrossRef]
- Vijay, A.; Valdes, A.M. Role of the gut microbiome in chronic diseases: a narrative review. Eur J Clin Nutr. 2022, 76(4), 489–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Z.; Zhu, L.B.; Li, Z.R.; Lin, J. Bacterial colonization and intestinal mucosal barrier development. World J Clin Pediatr. 2013, 2(4), 46–53. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell 2016, 164, 337–40. [Google Scholar] [CrossRef] [PubMed]
- Conroy, M.E.; Shi, H.N.; Walker, W.A. The long-term health effects of neonatal microbial flora. Curr Opin Allergy Clin Immunol. 2009, 9, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30(6), 492–506. [Google Scholar] [CrossRef]
- Hrncir, T. Gut Microbiota Dysbiosis: Triggers, Consequences, Diagnostic and Therapeutic Options. Microorganisms. 2022, 10(3), 578. [Google Scholar] [CrossRef]
- Schlechte, J.; Skalosky, I.; Geuking, M.B.; McDonald, B. Long-distance relationships - regulation of systemic host defense against infections by the gut microbiota. Mucosal Immunol. 2022, 15, 809–18. [Google Scholar] [CrossRef]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol Immunol. 2021, 18(4), 866–77. [Google Scholar] [CrossRef]
- Kenney, M.J.; Ganta, C.K. Autonomic nervous system and immune system interactions. Compr Physiol. 2014, 4(3), 1177–200. [Google Scholar] [CrossRef] [PubMed]
- Daniel, N.; Lécuyer, E.; Chassaing, B. Host/microbiota interactions in health and diseases—Time for mucosal microbiology! Mucosal Immunol. 2021, 14, 1006–16. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.R.; Fujihashi, K. Inside the mucosal immune system. PLoS Biol. 2012, 10(9), e1001397. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, D.B.; Fagarasan, S. IgA synthesis: a form of functional immune adaptation extending beyond gut. Curr Opin Immunol. 2012, 24(3), 261–8. [Google Scholar] [CrossRef] [PubMed]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef] [PubMed]
- Burns, G.; Pryor, J.; Holtmann, G.; Walker, M.M.; Talley, N.J.; Keely, S. Immune Activation in Functional Gastrointestinal Disorders. Gastroenterol Hepatol (NY). 2019, 15(10), 539–48. [Google Scholar]
- Sperber, A.D.; Bangdiwala, S.I.; Drossman, D.A.; Ghoshal, U.C.; Simren, M.; Tack, J.; Whitehead, W.E.; Dumitrascu, D.L.; Fang, X.; Fukudo, S.; et al. Worldwide Prevalence and Burden of Functional Gastrointestinal Disorders, Results of Rome Foundation Global Study. Gastroenterology 2021, 160(1), 99–114.e3. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.; Burns, G.L.; Hoedt, E.C.; Keely, S.; Talley, N.J. Eosinophils, Hypoxia-Inducible Factors, and Barrier Dysfunction in Functional Dyspepsia. Front Allergy. 2022, 3, 851482. [Google Scholar] [CrossRef] [PubMed]
- Wouters, M.M.; Vicario, M.; Santos, J. The role of mast cells in functional GI disorders. Gut. 2016, 65(1), 155–68. [Google Scholar] [CrossRef]
- Black, C.J.; Drossman, D.A.; Talley, N.J.; Ruddy, J.; Ford, A.C. Functional gastrointestinal disorders: advances in understanding and management. Lancet. 2020, 396, 1664–74. [Google Scholar] [CrossRef]
- Oshima, T. Functional Dyspepsia: Current Understanding and Future Perspective. Digestion. 2024, 105(1), 26–33. [Google Scholar] [CrossRef]
- Stanghellini, V.; Chan, F.K.; Hasler, W.L.; Malagelada, J.R.; Suzuki, H.; Tack, J.; Talley, N.J. Gastroduodenal disorders. Gastroenterology. 2016, 150(6), 1380–92. [Google Scholar] [CrossRef]
- Van den Houte, K.; Carbone, F.; Goelen, N.; Schol, J.; Masuy, I.; Arts, J.; Caenepeel, P.; Staessen, D.; Vergauwe, P.; Van Roey, G.; et al. Effects of Rome IV Definitions of Functional Dyspepsia Subgroups in Secondary Care. Clin Gastroenterol Hepatol. 2021, 19(8), 1620–6. [Google Scholar] [CrossRef]
- Moloney, R.D.; Johnson, A.C.; O'Mahony, S.M.; Dinan, T.G.; Greenwood-Van Meerveld, B.; Cryan, J.F. Stress and the Microbiota-Gut-Brain Axis in Visceral Pain: Relevance to Irritable Bowel Syndrome. CNS Neurosci Ther. 2016, 22(2), 102–17. [Google Scholar] [CrossRef]
- Burns, G.; Carroll, G.; Mathe, A.; Horvat, J.; Foster, P.; Walker, M.M.; Talley, N.J.; Keely, S. Evidence for Local and Systemic Immune Activation in Functional Dyspepsia and the Irritable Bowel Syndrome: A Systematic Review. Am J Gastroenterol. 2019, 114(3), 429–36. [Google Scholar] [CrossRef]
- Ng, Q.X.; Soh, A.Y.S.; Loke, W.; Lim, D.Y.; Yeo, W.S. The role of inflammation in irritable bowel syndrome (IBS). J Inflamm Res. 2018, 11, 345–9. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, N.; Germanidis, G. Current insights into the innate immune system dysfunction in irritable bowel syndrome. Ann Gastroenterol. 2018, 31(2), 171–87. [Google Scholar] [CrossRef] [PubMed]
- Berumen, A.; Edwinson, A.L.; Grover, M. Post-infection Irritable Bowel Syndrome. Gastroenterol Clin North Am. 2021, 50(2), 445–61. [Google Scholar] [CrossRef] [PubMed]
- Klem, F.; Wadhwa, A.; Prokop, L.J.; Sundt, W.J.; Farrugia, G.; Camilleri, M.; Singh, S.; Grover, M. Prevalence, Risk Factors, and Outcomes of Irritable Bowel Syndrome After Infectious Enteritis: A Systematic Review and Meta-analysis. Gastroenterol 2017, 152(5), 1042–54.e1. [Google Scholar] [CrossRef] [PubMed]
- Carco, C.; Young, W.; Gearry, R.B.; Talley, N.J.; McNabb, W.C.; Roy, N.C. Increasing Evidence That Irritable Bowel Syndrome and Functional Gastrointestinal Disorders Have a Microbial Pathogenesis. Front Cell Infect Microbiol. 2020, 10, 468. [Google Scholar] [CrossRef] [PubMed]
- Barman, M.; Unold, D.; Shifley, K.; Amir, E.; Hung, K.; Bos, N.; Salzman, N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008, 76(3), 907–15. [Google Scholar] [CrossRef] [PubMed]
- Jalanka, J.; Salonen, A.; Fuentes, S.; de Vos, W.M. Microbial signatures in post-infectious irritable bowel syndrome--toward patient stratification for improved diagnostics and treatment. Gut Microbes. 2015, 6(6), 364–9. [Google Scholar] [CrossRef]
- Wang, C.; Fang, X. Inflammation and Overlap of Irritable Bowel Syndrome and Functional Dyspepsia. J Neurogastroenterol Motil. 2021, 27(2), 153–64. [Google Scholar] [CrossRef]
- Lupu, V.V.; Ghiciuc, C.M.; Stefanescu, G.; Mihai, C.M.; Popp, A.; Sasaran, M.O.; Bozomitu, L.; Starcea, I.M.; Adam Raileanu, A.; Lupu, A. Emerging role of the gut microbiome in post-infectious irritable bowel syndrome: A literature review. World J Gastroenterol. 2023, 29(21), 3241–56. [Google Scholar] [CrossRef]
- Sadeghi, A.; Biglari, M.; Nasseri Moghaddam, S. Post-infectious Irritable Bowel Syndrome: A Narrative Review. Middle East J Dig Dis. 2019, 11(2), 69–75. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Deng, Z. Imbalanced shift of cytokine expression between T helper 1 and T helper 2 (Th1/Th2) in intestinal mucosa of patients with post-infectious irritable bowel syndrome. BMC Gastroenterol. 2012, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- De Winter, B.Y.; van den Wijngaard, R.M.; de Jonge, WJ. Intestinal mast cells in gut inflammation and motility disturbances. Biochim Biophys Acta. 2012, 1822(1), 66–73. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.W.; Ma, Z.C.; Fu, J.; Huang, B.L.; Liu, F.J.; Sun, D.; Lan, C. Upregulated adenosine 2A receptor accelerates post-infectious irritable bowel syndrome by promoting CD4+ T cells' T helper 17 polarization. World J Gastroenterol. 2022, 28(25), 2955–67. [Google Scholar] [CrossRef] [PubMed]
- Dellon, E.S.; Kim, H.P.; Sperry, S.L.; Rybnicek, D.A.; Woosley, J.T.; Shaheen, N.J. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest. Endosc. 2014, 79, 577–85. [Google Scholar] [CrossRef]
- Racca, F.; Pellegatta, G.; Cataldo, G.; Vespa, E.; Carlani, E.; Pelaia, C.; Paoletti, G.; Messina, M.R.; Nappi, E.; Canonica, G.W.; et al. Type 2 Inflammation in Eosinophilic Esophagitis: From Pathophysiology to Therapeutic Targets. Front Physiol. 2022, 12, 815842. [Google Scholar] [CrossRef]
- Khokhar, D.; Marella, S.; Idelman, G.; Chang, J.W.; Chehade, M.; Hogan, S.P. Eosinophilic esophagitis: Immune mechanisms and therapeutic targets. Clin Exp Allergy. 2022, 52(10), 1142–56. [Google Scholar] [CrossRef]
- Arias, Á.; Lucendo, A.J. Molecular basis and cellular mechanisms of eosinophilic esophagitis for the clinical practice. Expert Rev Gastroenterol Hepatol. 2019, 13(2), 99–117. [Google Scholar] [CrossRef]
- Wąsik, J.; Małecka-Wojciesko, E. Eosinophilic Esophagitis—What Do We Know So Far? Journal of Clinical Medicine. 2023, 12(6), 2259. [Google Scholar] [CrossRef]
- O'Shea, K.M.; Aceves, S.S.; Dellon, E.S.; Gupta, S.K.; Spergel, J.M.; Furuta, G.T.; Rothenberg, M.E. Pathophysiology of Eosinophilic Esophagitis. Gastroenterol. 2018, 154(2), 333–345. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.A.M.; Akhter, S.; Khan, M.R.; Saha, M.; Roy, P.K. Association of duodenal eosinophilia with Helicobacter pylori-negative functional dyspepsia. Arab J Gastroenterol. 2020, 21(1), 19–23. [Google Scholar] [CrossRef]
- Andersen, L.P.; Holck, S.; Janulaityte-Günther, D.; Kupcinskas, L.; Kiudelis, G.; Jonaitis, L.; Janciauskas, D.; Holck, P.; Bennedsen, M.; Permin, H.; et al. Gastric inflammatory markers and interleukins in patients with functional dyspepsia, with and without Helicobacter pylori infection. FEMS Immunol Med Microbiol. 2005, 44(2), 233–8. [Google Scholar] [CrossRef]
- Shah, A.; Fairlie, T.; Brown, G.; Jones, M.P.; Eslick, G.D.; Duncanson, K.; Thapar, N.; Keely, S.; Koloski, N.; Shahi, M.; et al. Duodenal Eosinophils and Mast Cells in Functional Dyspepsia: A Systematic Review and Meta-Analysis of Case-Control Studies. Clin Gastroenterol Hepatol 2022, 20(10), 2229–42.e29. [Google Scholar] [CrossRef]
- Eladham, M.W.; Selvakumar, B.; Saheb Sharif-Askari, N.; Saheb Sharif-Askari, F.; Ibrahim, S.M.; Halwani, R. Unraveling the gut-Lung axis: Exploring complex mechanisms in disease interplay. Heliyon. 2024, 10(1), e24032. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, M.E.; Lee, F.E.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin Rev Allergy Immunol. 2019, 56(2), 219–33. [Google Scholar] [CrossRef]
- Ceulemans, M.; Jacobs, I.; Wauters, L.; Vanuytsel, T. Immune Activation in Functional Dyspepsia: Bystander Becoming the Suspect. Front Neurosci. 2022, 16, 831761. [Google Scholar] [CrossRef] [PubMed]
- Bennet, S.M.; Polster, A.; Törnblom, H.; Isaksson, S.; Capronnier, S.; Tessier, A.; Le Nevé, B.; Simrén, M.; Öhman, L. Global Cytokine Profiles and Association With Clinical Characteristics in Patients With Irritable Bowel Syndrome. Am J Gastroenterol. 2016, 111(8), 1165–76. [Google Scholar] [CrossRef]
- Goral, V.; Kucukoner, M.; Buyukbayram, H. Mast cells count and serum cytokine levels in patients with irritable bowel syndrome. Hepatogastroenterol. 2010, 57(101), 751–4. [Google Scholar]
- Seyedmirzaee, S.; Hayatbakhsh, M.M.; Ahmadi, B.; Baniasadi, N.; Bagheri Rafsanjani, A.M.; Nikpoor, A.R.; Mohammadi, M. Serum immune biomarkers in irritable bowel syndrome. Clin Res Hepatol Gastroenterol. 2016, 40(5), 631–637. [Google Scholar] [CrossRef]
- Ghaffari, P.; Shoaie, S.; Nielsen, L.K. Irritable bowel syndrome and microbiome; Switching from conventional diagnosis and therapies to personalized interventions. J Transl Med. 2022, 20(1), 173. [Google Scholar] [CrossRef]
- Ivashkin, V.; Poluektov, Y.; Kogan, E.; Shifrin, O.; Sheptulin, A.; Kovaleva, A.; Kurbatova, A.; Krasnov, G.; Poluektova, E. Disruption of the pro-inflammatory, anti-inflammatory cytokines and tight junction proteins expression, associated with changes of the composition of the gut microbiota in patients with irritable bowel syndrome. PLoS One. 2021, 16(6), e0252930. [Google Scholar] [CrossRef]
- Knowles, S.R.; Skvarc, D.; Ford, A.C.; Palsson, O.S.; Bangdiwala, S.I.; Sperber, A.D.; Mikocka-Walus, A. Negative Impact of Disorders of Gut-Brain Interaction on Health-Related Quality of Life: Results From the Rome Foundation Global Epidemiology Survey. Gastroenterol. 2023, 164(4), 655–8.e10. [Google Scholar] [CrossRef]
- Tak, L.M.; Rosmalen, J.G. Dysfunction of stress responsive systems as a risk factor for functional somatic syndromes. J Psychosom Res. 2010, 68(5), 461–8. [Google Scholar] [CrossRef]
- Fischer, S.; Skoluda, N.; Ali, N.; Nater, U.M.; Mewes, R. Hair cortisol levels in women with medically unexplained symptoms. J Psychiatr Res. 2022, 146, 77–82. [Google Scholar] [CrossRef]
- Strawbridge, R.; Sartor, M.L.; Scott, F.; Cleare, A.J. Inflammatory proteins are altered in chronic fatigue syndrome-A systematic review and meta-analysis. Neurosci Biobehav Rev. 2019, 107, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Bashashati, M.; Moradi, M.; Sarosiek, I. Interleukin-6 in irritable bowel syndrome: A systematic review and meta-analysis of IL-6 (-G174C) and circulating IL-6 levels. Cytokine. 2017, 99, 132–8. [Google Scholar] [CrossRef] [PubMed]
- Kamp, K.J.; Han, C.; Shulman, R.J.; Cain, K.C.; Barney, P.; Opp, M.R.; Chang, L.; Burr, R.L.; Heitkemper, M.M. Cytokine Levels and Symptoms Among Women with Irritable Bowel Syndrome: Considering the Role of Hormonal Contraceptive Use. Biological Research For Nursing. 2021, 23(2), 171–9. [Google Scholar] [CrossRef] [PubMed]
- Finlay, S.; Rudd, D.; McDermott, B.; Sarnyai, Z. Allostatic load and systemic comorbidities in psychiatric disorders. Psychoneuroendocrinology. 2022, 140, 105726. [Google Scholar] [CrossRef] [PubMed]
- Mostafaei, S.; Kabir, K.; Kazemnejad, A.; Feizi, A.; Mansourian, M.; Hassanzadeh Keshteli, A.; Afshar, H.; Arzaghi, S.M.; Rasekhi Dehkordi, S.; Adibi, P.; et al. Explanation of somatic symptoms by mental health and personality traits: application of Bayesian regularized quantile regression in a large population study. BMC Psychiatry 2019, 19(1), 207. [Google Scholar] [CrossRef]
- Brouillet, J.Z.; Boltri, M.; Lengvenyte, A.; Lajnef, M.; Richard, J.R.; Barrau, C.; Strumila, R.; Coyac, M.; Wu, C.L.; Boukouaci, W.; et al. Association of markers of inflammation and intestinal permeability in suicidal patients with major mood disorders. Journal of Affective Disorders Reports. 2023, 14, 100624. [Google Scholar] [CrossRef]
- Volarić, M.; Šojat, D.; Majnarić, L.T.; Vučić, D. The Association between Functional Dyspepsia and Metabolic Syndrome—The State of the Art. Int. J. Environ. Res. Public Health. 2024, 21, 237. [Google Scholar] [CrossRef]
- Murni, A.W.; Darwin, E.; Zubir, N.; Nurdin, A.E. Analyzing Determinant Factors for Pathophysiology of Functional Dyspepsia Based on Plasma Cortisol Levels, IL-6 and IL-8 Expressions and H. pylori Activity. Acta Med Indones. 2018, 50(1), 38–45. [Google Scholar] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Stress and the gut: pathophysiology, clinical consequences, diagnostic approach, and treatment options. J Physiol Pharmacol. 2011, 62(6), 591–9. [Google Scholar] [PubMed]
- Jacobs, I.; Ceulemans, M.; Wauters, L.; Breynaert, C.; Vermeire, S.; Verstockt, B.; Vanuytsel, T. Role of Eosinophils in Intestinal Inflammation and Fibrosis in Inflammatory Bowel Disease: An Overlooked Villain? Front Immunol. 2021, 12, 754413. [Google Scholar] [CrossRef] [PubMed]
- Wauters, L.; Burns, G.; Ceulemans, M.; Walker, M.M.; Vanuytsel, T.; Keely, S.; Talley, NJ. Duodenal inflammation: an emerging target for functional dyspepsia? Expert Opin Ther Targets. 2020, 24(6), 511–23. [Google Scholar] [CrossRef] [PubMed]
- Robida, P.A.; Puzzovio, P.G.; Pahima, H.; Levi-Schaffer, F.; Bochner, B.S. Human eosinophils and mast cells: Birds of a feather flock together. Immunol Rev. 2018, 282(1), 151–67. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.; Walker, M.M.; Talley, N.J. Gastrointestinal eosinophils in health, disease and functional disorders. Nat Rev Gastroenterol Hepatol. 2010, 7(3), 146–56. [Google Scholar] [CrossRef] [PubMed]
- Cinicola, B.L.; Pulvirenti, F.; Capponi, M.; Bonetti, M.; Brindisi, G.; Gori, A.; De Castro, G.; Anania, C.; Duse, M.; Zicari, A.M. Selective IgA Deficiency and Allergy: A Fresh Look to an Old Story. Medicina (Kaunas). 2022, 58(1), 129. [Google Scholar] [CrossRef]
- Odineal, D.D.; Gershwin, M.E. The epidemiology and clinical manifestations of autoimmunity in selective IgA deficiency. Clin Rev Allergy Immunol. 2020, 58, 107–33. [Google Scholar] [CrossRef]
- Friesen, C.A.; Neilan, N.A.; Schurman, J.V.; Taylor, D.L.; Kearns, G.L.; Abdel-Rahman, S.M. Montelukast in the treatment of duodenal eosinophilia in children with dyspepsia: effect on eosinophil density and activation in relation to pharmacokinetics. BMC Gastroenterol. 2009, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, Z.; Zbikowska-Gotz, M.; Romański, B.; Sinkiewicz, W. Evaluating the profile of selected cytokines in patients with food allergy and chronic gastritis. Med Sci Monit. 2000, 6(6), 1128–35. [Google Scholar] [PubMed]
- Malfertheiner, P.; Camargo, M.C.; El-Omar, E.; Liou, J.M.; Peek, R.; Schulz, C.; Smith, S.I.; Suerbaum, S. Helicobacter pylori infection. Nat Rev Dis Primers. 2023, 9(1), 19. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Nishida, A- Alteration of the Gut Microbiome in Inflammatory Bowel Disease. Digestion. 2023, 104(1), 16–23. [CrossRef] [PubMed]
- Pittayanon, R.; Lau, J.T.; Yuan, Y.; Leontiadis, G.I.; Tse, F.; Surette, M.; Moayyedi, P. Gut Microbiota in Patients With Irritable Bowel Syndrome-A Systematic Review. Gastroenterol. 2019, 157(1), 97–108. [Google Scholar] [CrossRef]
- Hermes, G.D.A.; Reijnders, D.; Kootte, R.S.; Goossens, G.H.; Smidt, H.; Nieuwdorp, M.; Blaak, E.E.; Zoetendal, E.G. Individual and cohort-specific gut microbiota patterns associated with tissue-specific insulin sensitivity in overweight and obese males. Sci Rep. 2020, 10(1), 7523. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Rijnaarts, I.; Hermes, G.D.A.; de Roos, N.M.; Witteman, B.J.M.; de Wit, N.J.W.; Govers, C.; Smidt, H.; Zoetendal, E.G. Fecal Microbiota Signatures Are Not Consistently Related to Symptom Severity in Irritable Bowel Syndrome. Dig Dis Sci. 2022, 67(11), 5137–48. [Google Scholar] [CrossRef]
- Matenchuk, B.A.; Mandhane, P.J.; Kozyrskyj, A.L. Sleep, circadian rhythm, and gut microbiota. Sleep Med Rev. 2020, 53, 101340. [Google Scholar] [CrossRef]
- Vanuytsel, T.; Bercik, P.; Boeckxstaens, G. Understanding neuroimmune interactions in disorders of gut-brain interaction: from functional to immune-mediated disorders. Gut. 2023, 72(4), 787–98. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol Dial Transplant. 2015, 30(6), 924–33. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Interplay of systemic chronic inflammation, neuroendocrine disorders, metabolic changes and gut microbiota dysbiosis in chronic disease development including functional gastrointestinal disorders.
Figure 1.
Interplay of systemic chronic inflammation, neuroendocrine disorders, metabolic changes and gut microbiota dysbiosis in chronic disease development including functional gastrointestinal disorders.
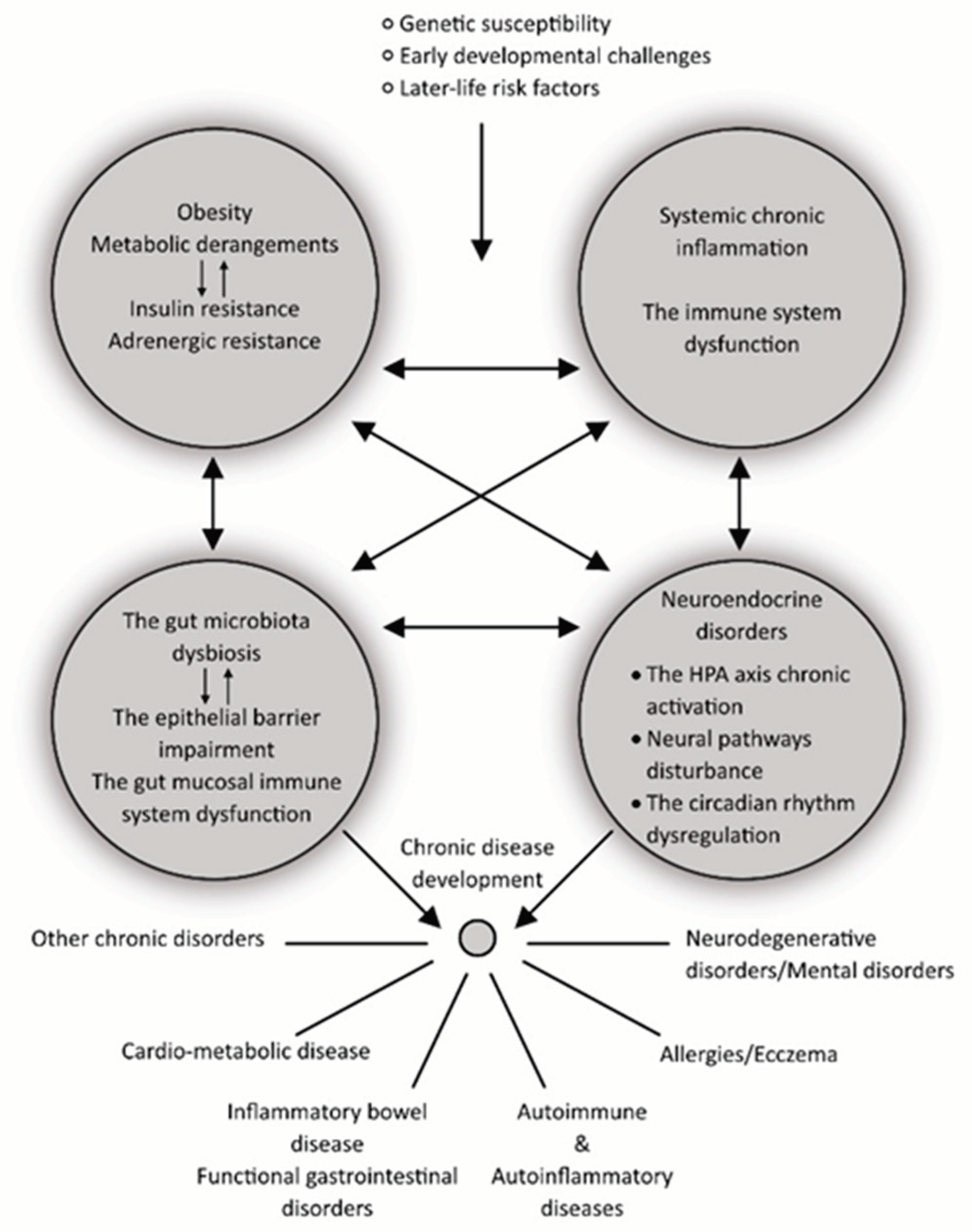
Figure 2.
Presentation of dual structure (inductive and effector sites) of the gut mucosal immune system.
Figure 2.
Presentation of dual structure (inductive and effector sites) of the gut mucosal immune system.
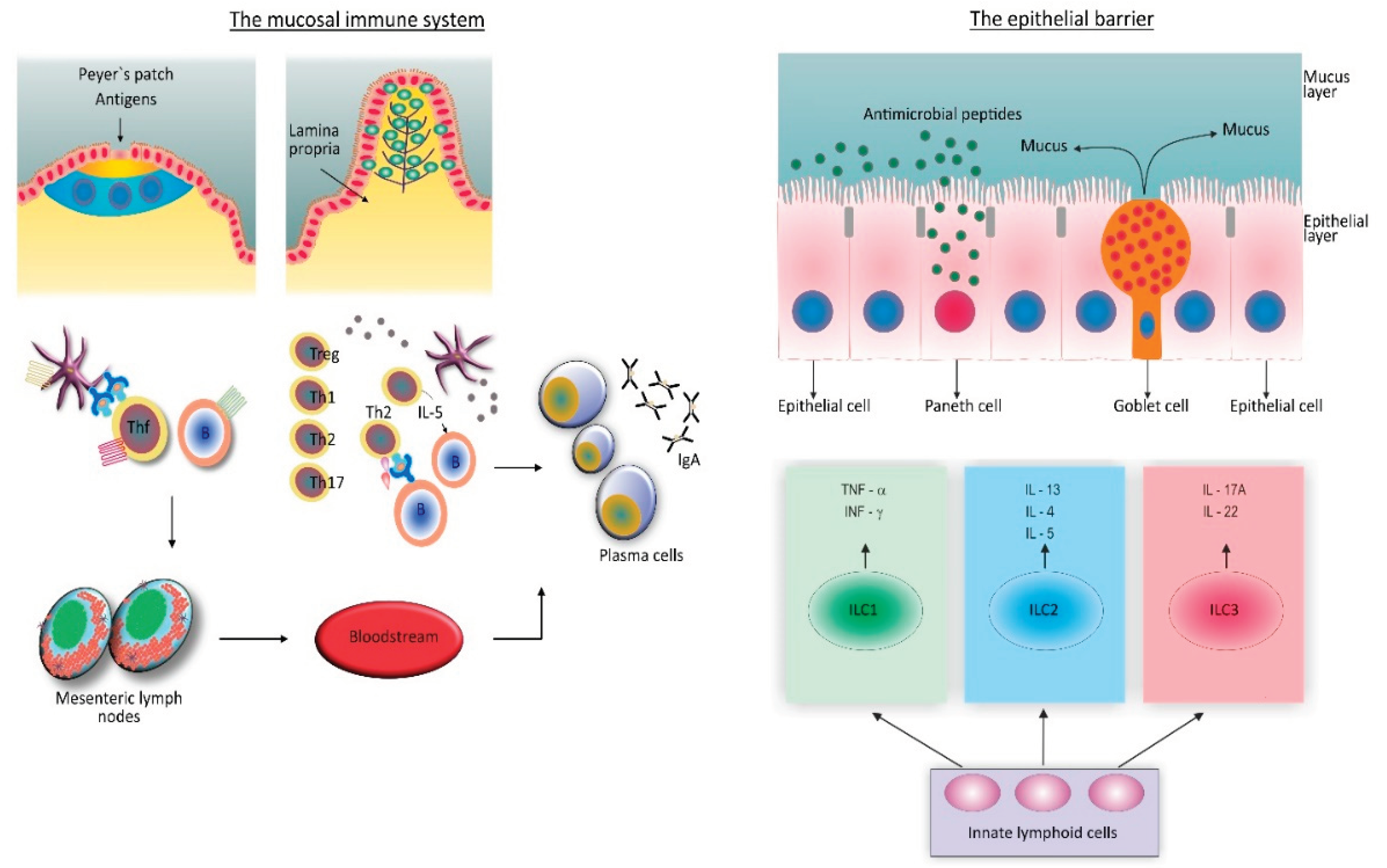
Figure 3.
The spectrum of inflammation-mediated chronic disorders communicating through systemic immunity, and mucosal immunity, and ageing connected obesity.
Figure 3.
The spectrum of inflammation-mediated chronic disorders communicating through systemic immunity, and mucosal immunity, and ageing connected obesity.
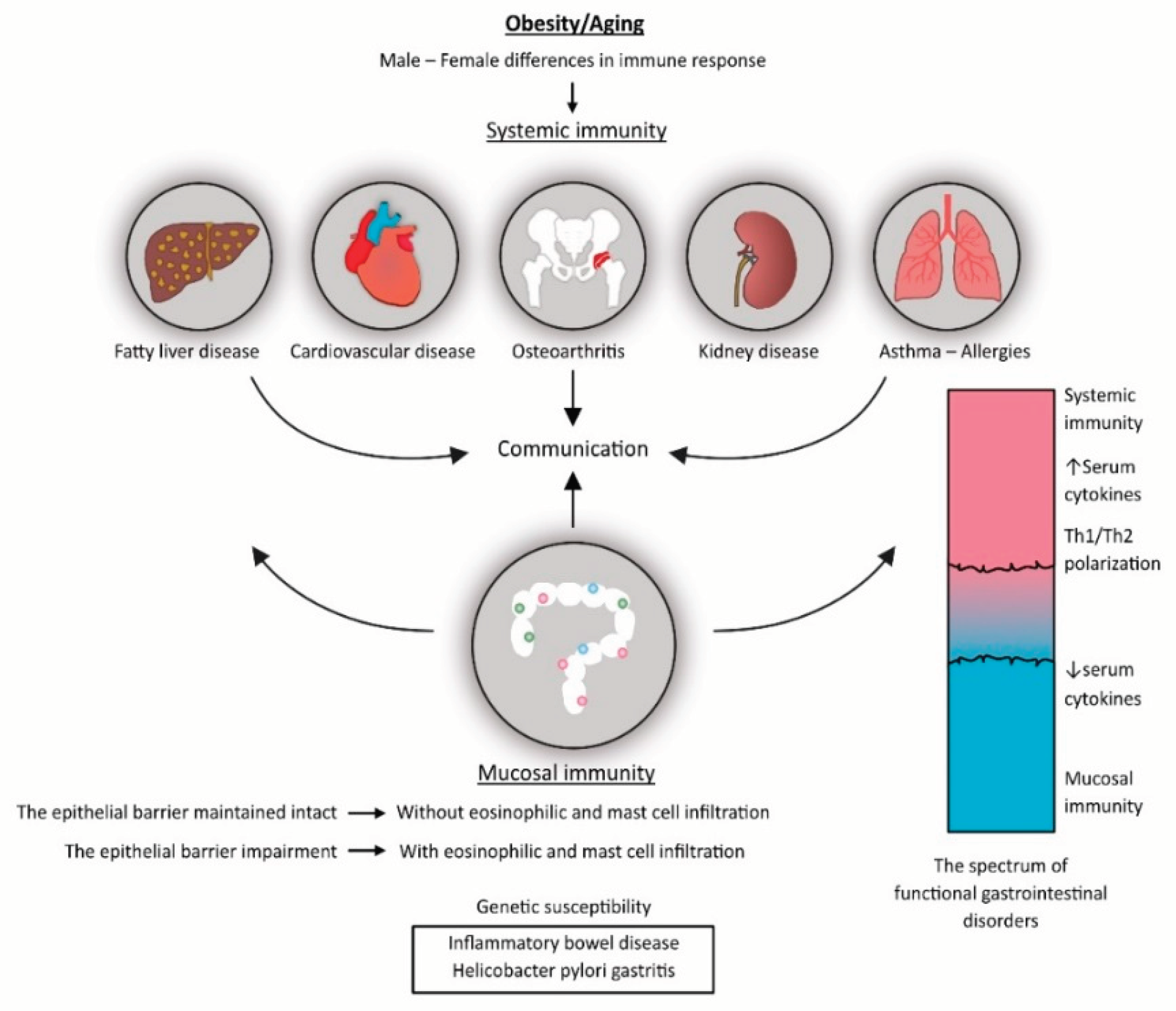
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



