
Submitted:
05 March 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
The quinoline derivative, N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide, was synthesized in a conventional three-step procedure from 4,7-dichloroquinoline using N-oxidation reaction/C2-amidation reaction/C4 SNAr reaction sequence. The structure of the compound was fully characterized by FT-IR, 1H‐, 13C-NMR, DEPT-135°, and ESI-MS techniques. Its physicochemical parameters (Lipinski’s descriptors) were also calculated using the online SwissADME database. Such derivatives are relevant therapeutic agents exhibiting potent anticancer, antibacterial, antifungal, and antiparasitic properties.
Keywords:
4
; 7-dichloroquinoline
; N-oxides
; C–H bond functionalization
; C2-amidation reaction
; C4 SNAr reaction
; N-(quinolinyl)morpholines
; N-(quinolinyl)amides
; Lipinski’ descriptors
1. Introduction
Quinoline and morpholine are favored molecular frameworks for medicinal and advanced materials chemistry [1,2,3,4,5,6]. (Quinolinyl)amides and N-(quinolinyl)morpholines are especially interesting biological active molecules that can serve as useful scaffolds in pharmaceutical research. They can act as human vanilloid receptor type 1 (TRPV1) antagonists and melanin-concentrating hormone 1 receptor (MCH1R) antagonists [7,8,9], antifungal, antibacterial, and trypanocidal agents [10,11,12,13]. In particular, N-(quinolin-2-yl)benzamide derivative A (Figure 1), designed and synthesized as an anticancer sorafenib congener, presented interesting antiproliferative activity (HCT-116, MCF-7, and SK-BR3 cancer cell lines) [14]. On the other hand, the 4-(6-amidoquinolin-2-yl)morpholine B is a potent MCH1R antagonist, implicated in body weight (obesity) regulation, a major contributor to the development of diseases including type 2 diabetes mellitus, coronary heart disease, certain forms of cancer, and osteoarthritis [9], and 4-(quinolin-4-yl)morpholine C exhibited some in vitro antibacterial activity [15].
Additionally, pincer palladium complexes, based on the N-(quinolin-8-yl)amide ring, have been widely explored for diverse catalytic transformations [16], and 4,7-dichloroquinoline has been extensively used in drug and functional material research as an affordable chemical intermediate for numerous bioactive functionalized 4-amino-quinoline compounds [17,18,19,20].
Due to the paramount significance of the functionalized quinoline core in the search for drug candidates and medicinal chemistry, the development of new synthetic methodologies remains an active area. Among modern methodologies, there is regioselective functionalization of the quinoline ring through C-H activation, especially via N-oxide formation, using it as a directing group [21,22]. The latter is considered one of the most powerful and, thus, attractive approaches for synthesizing C-2 and C-8 functionalized quinoline compounds under mild reaction conditions [23,24,25,26]. In particular, metal-free deoxygenative 2-amidation of quinoline N-oxides is one of the most promising constructions of N-acylated 2-aminoquinolines, in which amides and nitriles are used as the amidation reagents in the presence of several promoters, i.e., oxalyl chloride, trifluoroacetic anhydride/2-fluoropyridine mixture, TsOH·H2O or selectfluor-methylcarbazate (NH2NHCO2Me) combination, and under Brønsted acidic ionic liquid (BAIL, [BSmim][OTf]) medium [27,28,29,30,31]. Such approaches constitute a paradigm in diversity-oriented synthesis strategies, permitting the late-stage diversification of key pharmaceutical scaffolds [32].
Considering the above-stated aspects and as a continuation of our efforts to prepare new bioactive 7-chloroquinoline-based molecules [33,34,35,36,37,38], we designed, prepared, and characterized the above-mentioned N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide. Therefore, this work describes a practical, efficient method for synthesizing the title compound through the N-oxidation reaction/C2-amidation reaction/C4 SNAr reaction sequence using commercially accessible reagents.
2. Results and Discussion
The N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide (4) was easily prepared through a conventional three-step procedure from commercially available 4,7-dichloroquinoline (1), which was oxidized with a 1.2 equivalent of m-chloroperbenzoic acid (m-CPBA) in CHCl3 at room temperature for 5 h to give N-oxide (2) [39] in 81% yield (Scheme 1).
This N-oxide was also obtained in 87% yield using acetic acid and 30% hydrogen peroxide mixture [40] as an oxidant reagent at 70 oC for 1 h. The reaction of (2) with benzonitrile proceeded in refluxing dichloromethane and conc. sulfuric acid at 70 oC for 24 h with deoxygenation and provided N-(quinolin-2-yl)benzamide (3) in 92% yield (a key C2 amidation process). Note the conditions reported by Chen et al. in 2018 (TsOH·H2O, 150 oC, 12 h) to carry out C-H functionalization via C2 amidation [29] failed in our hands. The final step of the developed method is the nucleophilic aromatic substitution (SNAr) involving the displacement of the C4-chlorine atom in quinoline compound (3) to obtain titled quinoline (4) in 92% yield. That was achieved through a base-promoted amination [41], easily realized by reacting 4-chloroquinoline derivative (3) and morpholine in the presence of K2CO3 in refluxing DMF for 24 h (Scheme 1). This protocol is valuable for industrial applications due to its simplicity of operation, transition metal-free conditions, and scalability.
The structural elucidation of the intermediate (2) and final compounds (3-4) was achieved based on spectroscopic data, and the results are presented in the experimental section and the electronic supporting information (ESI). Their infrared spectra record the absorption bands associated with the functional groups present in their structure. Characteristic amide -C=O strain vibration appeared in 1693 and 1674 cm−1, respectively, for (3) and (4), while the band at 1287 cm-1, corresponding to the tension of the N-O bond of precursor (2), is not present that confirms successful deoxygenated amidation procedure of (2) to form (3) (ESI, Figures S1–S3). This C-2 functionalization process is also indirectly corroborated by the absence of the signal of proton H2 in the 1H NMR spectra of quinolines (3-4); meanwhile, the doublet-shaped signal centered at 8.43 ppm with a coupling constant of 6.6 Hz corresponds to the hydrogen that is in position 2 of the quinoline ring is observed in the proton spectra of N-oxide (2) (Figure S7, S9, and S11). Each product (3-4) formation was further verified by its mass spectra. The electrospray ionization (ESI) technique in the positive mode of the obtained N-(quinolin-2-yl) benzamides (3-4) allowed us to observe the molecular ion peak [M+H+] at m/z 319.0/317.0 and 370.1/368.1 for C16H10Cl2N2O and C20H18ClN3O2 molecular formula, exhibiting the isotopic contribution of 37Cl in the ratio of ~1:2 and 1:3 (37Cl/35Cl), respectively Figure S5-S6).
The 13C-NMR and DEPT-135° spectra, shown in Figures S8, S10, and S12, were analyzed to complete the characterization. Each hydrogen and carbon atom signal is assigned by carefully analyzing the homonuclear and heteronuclear correlation spectra (COSY, HMBC, and HSQC experiments) (Figure S13-S20).
According to previous literature reports [29,42,43], a plausible reaction mechanism of the key C2 amidation process could be proposed. Nitrilium ion B, generated in situ from benzonitrile A with sulfuric acid, could react with N-oxide (2) as a dipolarophile via the [3+2]-dipolar cycloaddition to afford the five-membered oxadiazolidine intermediate D or as an electrophilic reagent through nucleophilic addition to form the Reissert-type intermediate C which is susceptible to intramolecular nucleophilic attack producing the intermediate D. The latter undergoes rearomatization with rupture of the bond (N-O) and a loss of a proton to form the stable product (3) (Scheme 2).
However, the low reactivity of such N-oxides as a 1,3-dipole in the cycloaddition reaction [43] makes the direct formation of intermediate D less possible. Thus, it is believed that its formation via the trapping of highly electrophilic intermediate B with weakly nucleophilic N-oxide (2) and subsequent intramolecular nucleophilic attack [42] is the more probable.
Due to the pharmacological importance of the obtained quinoline benzamides (3-4), we addressed the SwissADME software [44], which allowed us to rapidly estimate the potential biological activity of newly synthesized molecules. According to the predictions (Table S1), benzamides (3-4) displayed good drug-likeness scores with an acceptable hydrophilic-lipophilic balance (cLogP 3.89 and cLogP 3.16, respectively), obeying Lipinski’s rule [45] and presenting the optimal range for each six physicochemical properties (lipophilicity, size, polarity, solubility, flexibility, and saturation) in the hexagon snapshots [46]. Interestingly, introducing the benzamide fragment in the C-2 position of the quinoline ring (1) increased the lipophilicity of the comp. (3), while the second functionalization of carbon C-4 lowered it to ideal parameters (comp. (4)). However, both prepared benzamides could have a good hematoencephalic barrier permeation (the total molecular polar surface area, TPSA < 140 Å2) (Table S1, Figure S21). Their ability to permeate the blood-brain barrier (BBB) is a relevant factor in drug design. Crossing the BBB easily, they could be distributed homogeneously throughout the central nervous system (CNS). Noteworthy that according to the SwissTargetPrediction tools [47], the most achievable macromolecular targets of N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide (4) would be protein kinase inhibitors (40%) or phosphodiesterase modulator (33%) while these targets are less feasible for N-(4,7-dichloroquinolin-2-yl) benzamide (3) (33% and 13.3%, respectively) (Figure S22).
3. Materials and Methods
3.1. Chemical Analysis
The solvents and reagents employed in the synthesis of the intermediate and final compounds were of purity grade for synthesis. The composition and monitoring of the reactions, as well as the preliminary analysis of the purity of the synthesized compounds, were carried out by thin-layer chromatography (TLC) on Silufol UV254 plates of 0.25 mm thickness, revealed in a UV light chamber of 254 nm or an ethanolic solution of phosphomolybdic-sulfuric acids. The melting points of the products were determined in a Fisher–Johns melting point apparatus and the values were not corrected, reporting the average of three measurements. Infrared spectra (FT–IR) were acquired on a Thermo Scientific Nicolet iS50 FT–IR spectrophotometer with Fourier transform, with attenuated total reflectance (ATR) module, acquisition range: 4000–400 cm−1 (256 scans, resolution of 2 cm−1).
Mass spectra were obtained in UltraScan mode using a Hitachi LaChrom Elite HPLC liquid chromatograph coupled to a Bruker Daltonics AmaZon-X mass selective detector equipped with an Apollo-type electrospray ionization or ESI (ElectroSpray Ionization) source in positive mode, and a quadrupole ion trap or QIT (Quadrupole Ion Trap) analyzer. The acquisition of nuclear magnetic resonance spectra 1H, 13C, and 2D variants was achieved using a Bruker Avance–400 spectrometer (400 MHz for 1H and 100 MHz for 13C) using deuterated chloroform (CDCl3, 99.8% Merck®) as the solvent. Chemical shift values (δ) are expressed in ppm. In 1H-NMR spectra, the scale was adjusted from the residual chloroform signal (7.26 ppm). Similarly, the 13C-NMR spectra were scaled from the signal characteristic for the solvent (CDCl3). The coupling constants (J) are given in Hz; the multiplicity of signals is expressed by the following abbreviations: (s) singlet, (d) doublet, (t) triplet, (dd) doublet of doublets, and (m) multiplet.
3.2. Step 1: Preparation of 4,7-dichloroquinoline 1-oxide (2)
In a 10 mL flask, 4,7-dichloroquinoline (1) (197 mg, 1 mmol) was dissolved in chloroform (2.5 mL) and stirred for 5 min. Then, m-CPBA (206 mg, 1.2 mmol) was added gradually. It was allowed to stir for 5 hours at room temperature. Once the reaction (TLC monitoring) was completed, the reaction mixture was neutralized with a NaHCO3 solution, and then the organic phase was extracted with ethyl acetate (3x20 mL). The resulting extracts were dried (anhydrous Na2SO4), and the solvent was removed by distillation. The crude product was purified by column chromatography with silica gel using a mixture of petroleum ether–ethyl acetate (1:3) to obtain 172.5 mg of N-oxide (2) as a white solid (81% yield). Rf = 0.41 (petroleum ether–ethyl acetate, 1:3); Mp. 164–166 °C; IR (ATR, νmax/cm-1): 3095 and 3043 (νArC-H), 1497–1604 (νArC=C), 1287 (νN-O), 1085 (νArC-Cl) 812–886 (νArC-H). 1H-NMR (400 MHz, CDCl3), δ(ppm): 8.79 (d, J = 1.9 Hz, 1H, 8-H), 8.43 (d, J = 6.6 Hz, 1H, 2-H), 8.16 (d, J = 9.0 Hz, 1H, 5-H), 7.70 (dd, J = 9.0, 1.95 Hz, 1H, 6-H), 7.37 (d, J= 6.55 Hz, 1H, 3-H). 13C-NMR (101 MHz, CDCl3), δ(ppm): 142.4 (8a-C), 138.3 (7-C), 135.9 (2-C), 130.8 (6-C), 129.8 (4-C), 126.8 (5-C), 126.6 (4a-C), 121.2 (3-C), 120 (8-C).
3.3. Step 2: Synthesis of N-(4,7-dichloroquinolin-2-yl) benzamide (3)
In a 10 mL crimper vial equipped with a magnetic stir, benzonitrile (0.824 mL, 8 mmol) and 97% H2SO4 (0.107 mL, 2 mmol) were added. It was left stirring for 1 min. at room temperature, then N-oxide (2) (213 mg, 1 mmol) in CH2Cl2 (2 mL) was added. The reaction mixture was left stirring for 2 min. at room temperature, the reaction system was sealed and heated to 70 °C in a reflux system for 24 hours. TLC monitored the progress of the reaction. Once the reaction was finished, ethyl acetate (3x20 mL) and brine were added. The organic phase was separated and dried over anhydrous Na2SO4. The solvent was removed by distillation. The crude product was purified by column chromatography with silica gel using a mixture of petroleum ether–ethyl acetate (6:1) to obtain 290.7 mg of benzamide (3) as a white solid (92% yield). Rf = 0.60 (petroleum ether–ethyl acetate, 7:1); Mp. 133–134 °C; IR (ATR, νmax/cm-1): 3334 (νN-H), 2925 and 2856 (νArC-H), 1693 (νC=O), 1576–1393 (νArC=C), 1250 (νC-N), 863–759 (γArC-H). 1H-NMR (400 MHz, CDCl3), δ(ppm): 8.82 (s, 1H, N-H, -NH), 8.77 (s, 1H, 3-H), 8.11 (d, J = 9.0 Hz, 1H, 5-H), 8.00–7.97 (m, 2H, 2’-H/6’-H), 7.83 (d, J = 2.0 Hz, 1H, 8-H), 7.65–7.59 (m, 1H, 4’-H), 7.54–7.52 (m, 2H, 3’-H/5’-H), 7.49 (dd, J = 9.0, 2.0 Hz, 1H, 6-H). 13C-NMR (101 MHz, CDCl3), δ(ppm): 165.9 (C=O), 151.5 (2-C), 147.6 (8a-C), 144.5 (4-C), 137.1 (7-C), 133.6 (1’-C), 132.8 (4’-C), 129.8 (3’-C/5’-C), 127.4 (2’-C/6’-C), 127 (6-C), 126.7 (8-C), 125.7 (5-C), 123.1 (4a-C), 114.4 (3-C). COSY correlation [δH/δH]: 8.11/7.49 [5-H/6-H], 7.97/7.54 [(2’-H/6’-H)/(3’-H/5’-H)], 7.83/7.49 [8-H/6-H], 7.62/7.54 [4’-H/(3’-H/5’-H)]. HSQC correlation [δH/δC]: 8.77/114.4 [3-H/3-C], 8.11/125.7 [5-H/5-C], 7.97/127.4 [(2’-H/6’-H)/(2’-C/6’-C)], 7.83/126.7 [8-H/8-C], 7.62/132.8 [4’-H/4’-C], 7.54/129 [(3’-H/5’-H)/(3’-C/5’-C)], 7.49/127 [6-H/6-C]. HMBC correlation [δH/δC]: 8.77/123.1/144.5 [3-H/4a-C/4-C], 8.11/137.1/144.5/147.6 [5-H/7-C/4-C/8a-C], 7.97/127.4/132.8/165.9 [(2’-H/6’-H)/(2’-C/6’-C)/4’-C/0’-C], 7.83/123.1/127./137.1 [8-H/4a-C/6-C/7-C], 7.62/127.4 [4’-H/(2’-C/6’-C)], 7.54/129/133.6 [(3’-H/5’-H)/(3’-C/5’-C)/1’-C], 7.49/123.1/126.7/137.1 [6-H/4a-C/8-C/7-C]. ESI-MS, m/z (%): calculated mass for C16H11Cl2N2O2: 318.0 (37Cl), 316.0 (35Cl); found, [M+H]+: 319.0 (61%), 317.0 (100 %).
3.4. Step 3: Synthesis of N-(7-chloro-4-morpholinoquinolin-2-yl) benzamide (4)
In a 10 mL crimper vial equipped with a magnetic stir, benzamide (3) (316 mg, 1 mmol), morpholine (0.346 mL, 4 mmol), and K2CO3 (413 mg, 3 mmol) in DMF (2.5 mL) were mixed. The reaction system was sealed and heated at 120 °C for 24 hours. TLC monitored the progress of the reaction. Once the reaction was finished, the reaction mixture was treated with brine. The precipitate was obtained from the mother liquor. It was filtered and dried in air to give 337.7 mg of the final product (4) as a very light orange solid (92% yield). Rf = 0.79 (petroleum ether–ethyl acetate, 5:1); Mp. 223–225°C. IR (ATR, νmax/cm-1): 3279 (νN-H), 2858 and 2824 (νArC-H), 1674 (νC=O), 1577–1398 (νArC=C), 1254 (νC-N). 1H-NMR (400 MHz, CDCl3), δ(ppm): 8.76 (s, 1H, N-H), 8.17 (s, 1H, 3-H), 7.88 (d, J = 8.9 Hz, 1H, 5-H), 7.99–7.96 (m, 2H, 2’-H/6’-H), 7.78 (d, J = 2.0 Hz, 1H, 8-H), 7.62–7.57 (m, 1H, 4’-H), 7.55–7.52 (m, 2H, 3’-H/5’-H), 7.34 (dd, J = 9.0, 2.1 Hz, 1H, 6-H), 3.99 (t, J = 4.6 Hz, 4H, 2’’-H/6’’-H), 3.31 (t, J = 4.6 Hz, 4H, 3’’-H/5’’-H). 13C-NMR (101 MHz, CDCl3), δ(ppm): 166.1 (C=O), 159 (4-C), 152.8 (2-C), 148.6 (8a-C), 135.6 (7-C), 134.1 (1’-C), 132.5 (4’-C), 129 (3’-C/5’-C), 127.3 (2’-C/6’-C), 127.1 (8-C), 125.1 (5-C), 124.9 (6-C), 120 (4a-C), 101.9 (3-C), 66.8 (3’’-C/5’’-C), 52.7 (2’’-C/6’’-C). COSY correlation [δH/δH]: 7.99–7.96/7.55–7.52 [(2’-H/6’-H)/(3’-H/5’-H)], 7.88/7.34 [5-H/6-H], 7.78/7.34 [8-H/6-H], 7.62–7.57/7.55–7.52 [4’-H/(3’-H/5’-H)], 3.99/3.31 [(3’’-H/5’’-H)/(2’’-H/6’’-H)]. HSQC correlation [δH/δC]: 8.17/101.9 [3-H/3-C], 7.99–7.96/127.3 [(2’-H/6’-H)/(2’-C/6’-C)], 7.88/125.1 [5-H/5-C], 7.78/127.1 [8-H/8-C], 7.62–7.57/132.5 [4’-H/4’-C], 7.55–7.52/127.3 [(3’-H/5’-H)/(3’-C/5’-C)], 7.34/124.9 [6-H/6-C], 3.99/66.8 [(3’’-H/5’’-H)/(3’’-C/5’’-C)], 3.31/52.7 [(2’’-H/6’’-H)/(2’’-C/6’’-C)]. HMBC correlation [δH/δC]: 8.17/120 [3-H/4a-C], 7.99–7.96/127.3/132.5/166.1 [(2’-H/6’-H)/(2’-C/6’-C)/4’-C/0’-C], 7.88/135.6/148.6/159 [5-H/7-C/8a-C/4-C], 7.78/120/124.9/135.6 [8-H/4a-C/6-C/7-C], 7.62–7.57/127.3 [4’-H/(2’-C/6’-C)], 7.55–7.52/127.3/129/134.1 [(3’-H/5’-H)/(2’-C/6’-C)/(3’-C/5’-C)/1’-C], 7.49/120/127.1/135.6 [6-H/4a-C/8-C/7-C], 3.99/66.8/52.7 [(3’’-H/5’’-H)/(3’’-C/5’’-C)/(2’’-C/6’’-C)], 3.31/52.7/66.8 [(2’’-H/6’’-H)/(2’’-C/6’’-C)/(3’’-C/5’’-C)]. ESI-MS, m/z (%): calculated mass for C20H19ClN3O2: 369.1 (37Cl), 367.1 (35Cl); found, (M+H)+: 370.1 (32.6%), 368.1 (100%); 263.1 [(M+H)+-105, 94.7%].
4. Conclusions
In summary, this communication described a three-step approach for the synthesis of N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide from commercially available 4,7-dichloroquinoline through C-H amidation of 4,7-dichloroquinoline-N-oxide. The synthetic method is simple and efficient, presenting excellent yields (77–92%). The synthesized N-(7-chloro-quinolin-2-yl)benzamides are an interesting biological model for pharmacological agent research, especially regarding antineoplastic drug design or treatment of central and peripheral nervous system disorders.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org FT-IR, 1H-, 13C-NMR, and 2D variants (COSY, HSQC, and HMBC) for compounds (3) and (4).
Author Contributions
D.F.A.A and M.C.O.V. conceived the experiments; V.V.K. designed and wrote the paper. All three authors analyzed and discussed the results and data. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Universidad Industrial de Santander. M.C.O.V. thanks to the National Program 727 for her doctoral fellowship.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the study's design, collection, analysis, or interpretation of data, the writing of the manuscript, or the decision to publish the results.
References
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Iyaye, K.T.; Ademosun, O.T. Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs–a review. RSC Adv. 2022, 12, 18594–18614. [Google Scholar] [CrossRef]
- Basavarajaiah, S.M. The Versatile Quinoline and Its Derivatives as anti-Cancer Agents: An Overview. Polycycl. Aromat. Compd. 2022, 43, 4333–4345. [Google Scholar]
- Kumari, A.; Singh, R.K. Morpholine as Ubiquitous Pharmacophore in Medicinal Chemistry: Deep Insight into the Structure-Activity Relationship (SAR). Bioorg. Chem. 2020, 96, 103578. [Google Scholar] [CrossRef]
- Kourounakis, A.P.; Xanthopoulos, D.; Tzara, A. Morpholine as a Privileged Structure: A Review on the Medicinal Chemistry and Pharmacological Activity of Morpholine Containing Bioactive Molecules. Med. Res. Rev. 2020, 40, 709–752. [Google Scholar] [CrossRef]
- Tzara, A.; Xanthopoulos, D.; Kourounakis, A.P. Morpholine as a Scaffold in Medicinal Chemistry: An Update on Synthetic Strategies. Chem. Med. Chem. 2020, 15, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Daddario, N.; Minassi, A.; Moriello, A.S.; De Petrocellis, L.; Di Marzo, V. The taming of capsaicin. Reversal of the vanilloid activity of N-acylvanillamines by aromatic iodination. J. Med. Chem. 2005, 48, 4663–4669. [Google Scholar] [CrossRef]
- Jetter, M.C.; McNally, J.J.; Youngman, M.A.; McDonnell, M.E.; Dubin, A.E.; Nasser, N.; et al. N-pyridin-3-yl-and N-quinolin-3-yl-benzamides: modulators of human vanilloid receptor 1 (TRPV1). Bioorg. Med. Chem. Lett. 2008, 18, 2730–2734. [Google Scholar] [CrossRef]
- Ulven, T.; Frimurer, T.M.; Receveur, J.M.; Little, P.B.; Rist, Ø.; Nørregaard, P.K.; Högberg, T. 6-Acylamino-2-aminoquinolines as potent melanin-concentrating hormone 1 receptor antagonists. Identification, structure− activity relationship, and investigation of binding mode. J. Med. Chem. 2005, 48, 5684–5697. [Google Scholar] [CrossRef]
- Delattin, N.; Bardiot, D.; Marchand, A.; Chaltin, P.; De Brucker, K.; Cammue, B.P.; Thevissen, K. Identification of fungicidal 2,6-disubstituted quinolines with activity against Candida biofilms. Molecules 2012, 17, 12243–12251. [Google Scholar] [CrossRef]
- Basak, A.; Abouelhassan, Y.; Kim, Y.S.; Norwood, V.M.; Jin, S.; Huigens, R.W. 3rd Halogenated quinolines bearing polar functionality at the 2-position: identification of new antibacterial agents with enhanced activity against Staphylococcus epidermidis. Eur. J. Med. Chem. 2018, 155, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Collins, J.; Nyamwihura, R.; Ware, S.; Kaiser, M.; Ogungbe, I.V. Discovery of a quinoline-based phenyl sulfone derivative as an antitrypanosomal agent. Bioorg. Med. Chem. Lett. 2018, 28, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Nefertiti, A.S.G.; Batista, M.M.; Da Silva, P.B.; Batista, D.G.J.; Da Silva, C.F.; Peres, R.B.; et al. In vitro and in vivo studies of the trypanocidal effect of novel quinolines. Antimicrob. Agents Chemother. 2018, 62, 10.1128/aac.01936-17. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Seo, S.H.; Cho, N.C.; Kang, S.B.; Pae, A.N.; Kim, K.S.; Keum, G. Design, synthesis, in-vitro antiproliferative activity and kinase profile of new picolinamide based 2-amido and ureido quinoline derivatives. Eur. J. Med. Chem. 2015, 101, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.J.; Patel, A.P.; Chikhalia, K.H. Design and synthesis of some novel 7-substituted thiosemicarbazinyl-quinolines via Ullmann coupling reaction and examination of their antimicrobial activities. Res. Chem. Intermed. 2018, 44, 813–828. [Google Scholar] [CrossRef]
- Pandiri, H.; Soni, V.; Gonnade, R.G.; Punji, B. Development of (quinolinyl) amido-based pincer palladium complexes: a robust and phosphine-free catalyst system for C–H arylation of benzothiazoles. New J. Chem. 2017, 41, 3543–3554. [Google Scholar] [CrossRef]
- O'Neill, P.M.; Bray, P.G.; Hawley, S.R.; Ward, S.A.; Park, B.K. 4-Aminoquinolines—Past, present, and future; A chemical perspective. Pharmacol. Therapeut. 1998, 77, 29–58. [Google Scholar] [CrossRef]
- Solomon, V.R.; Pundir, S.; Lee, H. Examination of novel 4-aminoquinoline derivatives designed and synthesized by a hybrid pharmacophore approach to enhance their anticancer activities. Sc. Rep. 2019, 9, 6315. [Google Scholar] [CrossRef]
- Nandi, S.; Chauhan, B.; Tarannum, H.; Khede, M.K. Multi-target polypharmacology of 4-aminoquinoline compounds against malaria, tuberculosis and cancer. Curr. Top. Med. Chem. 2023, 23, 403–414. [Google Scholar] [CrossRef]
- Ravindar, L.; Hasbullah, S.A.; Rakesh, K.P.; Raheem, S.; Agustar, H.K.; Ismail, N.; Ling, L.Y.; Hassan, N. I. Exploring diverse frontiers: Advancements of bioactive 4-aminoquinoline-based molecular hybrids in targeted therapeutics and beyond. Eur. J. Med. Chem. 2023, 264, 116043. [Google Scholar] [CrossRef]
- Corio, A.; Gravier-Pelletier, C.; Busca, P. Regioselective Functionalization of Quinolines through CH Activation: A Comprehensive Review. Molecules 2021, 26, 5467. [Google Scholar] [CrossRef] [PubMed]
- Singha, K.; Habib, I.; Hossain, M. Quinoline N-Oxide: A Versatile Precursor in Organic Transformations. ChemistrySelect 2022, 7, e202203537. [Google Scholar] [CrossRef]
- Yan, G.; Borah, A.J.; Yang, M. Recent Advances in Catalytic Functionalization of N-Oxide Compounds via C-H Bond Activation. Adv. Synth. Catal. 2014, 356, 2375–2394. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Vargas Méndez, L.Y.; Puerto Galvis, C.E.; Ortiz Villamizar, M.C. The direct C–H alkenylation of quinoline N-oxides as a suitable strategy for the synthesis of promising antiparasitic drugs. New J. Chem. 2020, 44, 12–19. [Google Scholar] [CrossRef]
- Dong, D.; Sun, Y.; Li, G.; Yang, H.; Wang, Z.; Xu, X. Recent Progress in the Functionalization of Quinoline N-Oxide. Chin. J. Org. Chem. 2020, 40, 4071–4086. [Google Scholar] [CrossRef]
- Wang, D.; Désaubry, L.; Li, G.; Huang, M.; Zheng, S. Recent Advances in the Synthesis of C2-Functionalized Pyridines and Quinolines Using N-Oxide Chemistry. Adv. Synth. Catal. 2021, 363, 2–39. [Google Scholar] [CrossRef]
- Couturier, M.; Caron, L.; Tumidajski, S.; Jones, K.; White, T.D. Mild and direct conversion of quinoline N-oxides to 2-amidoquinolines with primary amides. Org. Lett. 2006, 8, 1929–1932. [Google Scholar] [CrossRef]
- Medley, J.W.; Movassaghi, M. Direct dehydrative N-pyridinylation of amides. J. Org. Chem. 2009, 74, 1341–1344. [Google Scholar] [CrossRef]
- Chen, X.; Peng, M.; Huang, H.; Zheng, Y.; Tao, X.; He, C.; Xiao, Y. TsOH· H2O-mediated N-amidation of quinoline N-oxides: facile and regioselective synthesis of N-(quinolin-2-yl) amides. Org. Biomol. Chem. 2018, 16, 6202–6205. [Google Scholar] [CrossRef]
- Xie, L.Y.; Peng, S.; Liu, F.; Yi, J.Y.; Wang, M.; Tang, Z.; Xu, X.; He, W.M. Metal-free deoxygenative 2-amidation of quinoline N-oxides with nitriles via a radical activation pathway. Adv. Synth. Catal. 2018, 360, 4259–4264. [Google Scholar] [CrossRef]
- Xie, L.Y.; Peng, S.; Lu, L.H.; Hu, J.; Bao, W.H.; Zeng, F.; Tang, Z.; He, W.M. Brønsted acidic ionic Liquid-Promoted amidation of quinoline N-Oxides with nitriles. ACS Sustain. Chem. Engin. 2018, 6, 7989–7994. [Google Scholar] [CrossRef]
- Josephitis, C.M.; Nguyen, H.M.; McNally, A. Late-Stage C–H Functionalization of Azines. Chem. Rev. 2023, 123, 7655–7691. [Google Scholar] [CrossRef]
- Rojas Ruiz, F.A.; Kouznetsov, V.V. Property-based Design and Synthesis of New Chloroquine Hybrids via Simple Incorporation of 2-Imino-thiazolidin-4-one or 1H-Pyrrol-2,5-dione Fragments on the 4-Amino-7-chloroquinolines Side Chain. J. Braz. Chem. Soc. 2011, 22, 1774–1781. [Google Scholar] [CrossRef]
- Fonseca-Berzal, C.; Rojas Ruiz, F.A.; Escario, J.A.; Kouznetsov, V.V.; Gómez-Barrio, A. In vitro phenotypic screening of 7-chloro-4-amino(oxy)quinoline derivatives as putative anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. Lett. 2014, 24, 1209–1213. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Sojo, F.; Rojas Ruiz, F.A.; Merchán-Arenas, D.R.; Arvelo, F. Synthesis and cytotoxic evaluation of 7-chloro-4-phenoxyquinolines with formyl, oxime and thiosemicarbazone scaffolds. Med. Chem. Res. 2016, 25, 2718–2727. [Google Scholar] [CrossRef]
- Luna-Parada, L.K.; Kouznetsov, V.V. 5-Chloro-8-{[1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl] methoxy}quinoline. Molbank 2019, 2019, M1038. [Google Scholar] [CrossRef]
- Rosado-Solano, D.N.; Barón-Rodríguez, M.A.; Sanabria-Florez, P.L.; Luna-Parada, L.K.; Puerto-Galvis, C.E.; Zorro-González, A.F.; Kouznetsov, V.V.; Vargas-Méndez, L.Y. Synthesis, Biological Evaluation and in silico Computational Studies of 7-Chloro-4-(1H-1,2,3-triazol-1-yl)quinoline Derivatives. Search for new controlling agents against Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae. J. Agric. Food Chem. 2019, 67, 9210–9219. [Google Scholar] [CrossRef]
- Rodríguez Enciso, D.A.; Puerto Galvis, C.E.; Kouznetsov, V.V. Microwave-assisted synthesis of pharmacologically active 4-phenoxyquinolines and their benzazole-quinoline hybrids through SNAr reaction of 4,7-dichloroquinoline and phenols using [bmim][PF6] as a green solvent. Curr. Org. Synth. 2023, 20, 546–559. [Google Scholar]
- Elslager, E.F.; Gold, E.H.; Tendick, F.H.; Werbel, L.M.; Worth, D.F. Amodiaquine N-oxides and other 7-chloro-4-aminoquinoline N-oxides. J. Heterocyclic Chem. 1964, 1, 6–12. [Google Scholar] [CrossRef]
- Solomon, V.R.; Haq, W.; Srivastava, K.; Puri, S.K.; Katti, S.B. Design and synthesis of 3-[(7-chloro-1-oxidoquinolin-4-ylamino) alkyl]-1,3-thiazolidin-4-ones as antimalarial agents. J. Enzyme Inhib. Med. Chem. 2013, 28, 1048–1053. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, F.; Deng, G.J.; Gong, H. Amination of aromatic halides and exploration of the reactivity sequence of aromatic halides. J. Org. Chem. 2018, 84, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Vamos, M.; Cosford, N.D. 2-Aminopyridines via reaction of pyridine N-oxides and activated isocyanides. J. Org. Chem. 2014, 79, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Ryzhakov, A.V.; Rodina, L.L. Aromatic N-oxides as 1,3-dipoles and π-donors in reactions with unsaturated compounds. Review. Chem. Heterocycl. Compd. 1992, 28, 483–493. [Google Scholar] [CrossRef]
- Swiss Institute of Bioinformatics. Available online: http://www.swissadme.ch/ (accessed on 26 February 2024).
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ortiz Villamizar, M.C.; Puerto Galvis, C.E.; Pedraza Rodríguez, S.A.; Zubkov, F.I.; Kouznetsov, V.V. Synthesis, In Silico and In Vivo Toxicity Assessment of Functionalized Pyridophenanthridinones via Sequential MW-Assisted Intramolecular Friedel-Crafts Alkylation and Direct C–H Arylation. Molecules 2022, 27, 8112. [Google Scholar] [CrossRef]
- Swiss Institute of Bioinformatics. Available online: http://www.swisstargetprediction.ch/ (accessed on 26 February 2024).
Figure 1.
Representative examples of pharmacological agents based on N-(quinolinyl)amide and N-(quinolinyl)morpholine scaffolds.
Figure 1.
Representative examples of pharmacological agents based on N-(quinolinyl)amide and N-(quinolinyl)morpholine scaffolds.
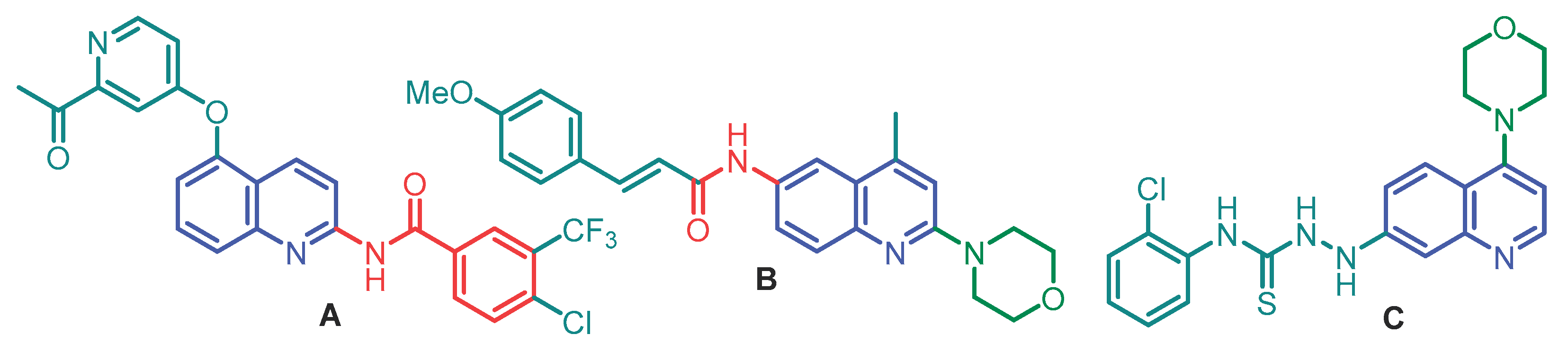
Scheme 1.
Three-step synthesis of N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide (4) from commercially available 4,7-dichloroquinoline (1): (a) Oxidation step: m-CPBA, CHCl3, rt, 5 h; (b) C2 functionalization (amidation) process: Ph-C≡N, 97% H2SO4, CH2Cl2, 70 oC, 24 h; (c) C4 functionalization (amination) process: Morpholine, K2CO3, DMF, 120 oC, 24 h.
Scheme 1.
Three-step synthesis of N-(7-chloro-4-morpholinoquinolin-2-yl)benzamide (4) from commercially available 4,7-dichloroquinoline (1): (a) Oxidation step: m-CPBA, CHCl3, rt, 5 h; (b) C2 functionalization (amidation) process: Ph-C≡N, 97% H2SO4, CH2Cl2, 70 oC, 24 h; (c) C4 functionalization (amination) process: Morpholine, K2CO3, DMF, 120 oC, 24 h.
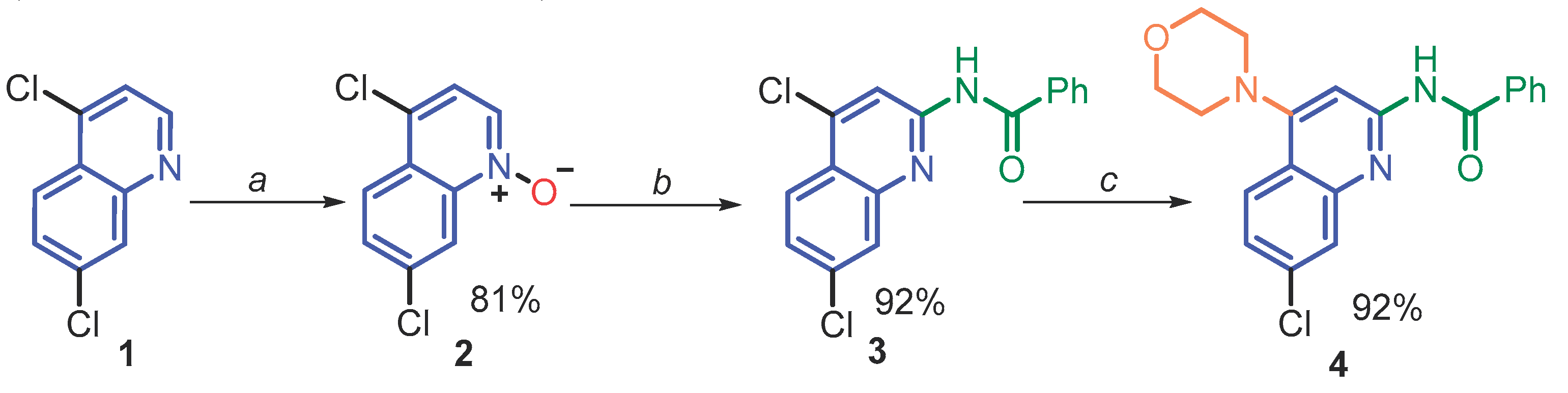
Scheme 2.
Plausible reaction mechanism of C2 amidation of N-oxide (2) with PhC≡N to form N-(quinolin-2-yl) benzamide (3).
Scheme 2.
Plausible reaction mechanism of C2 amidation of N-oxide (2) with PhC≡N to form N-(quinolin-2-yl) benzamide (3).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



