
Submitted:
19 February 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
Mycobacterium tuberculosis was responsible for 1.3 million deaths worldwide in 2022. Bacillus Calmette-Guérin (BCG) is the only licensed vaccine against tuberculosis (TB); however, it has limited protective efficacy in adults. In this study, we constructed a recombinant vaccinia virus expressing Ag85B from M. tuberculosis using a novel attenuated vaccinia virus (KVAC103). We then analyzed the immunogenicity of prime-boost inoculation strategies using recombinant KVAC103 expressing Ag85B (rKVAC85B) compared to BCG. In both the rKVAC85B prime-boost and the BCG prime-rKVAC85B boost inoculation regimens, rKVAC85B induced the generation of specific IgG and the secretion of IFN-γ by immune cells. In vitro analysis of Mycobacterium growth inhibition revealed a comparable immune-mediated pattern of outcomes. Furthermore, bacterial loads in the lungs were significantly lower in mice inoculated with the BCG prime-rKVAC85B boost, than in the BCG-only group following a rechallenge infection with both H37Rv and HN878 strains of M. tuberculosis. These findings collectively suggest that KVAC103, incorporated into a viral vector, is a promising candidate for a novel TB vaccine platform that is effective against multiple TB strains, including H37Rv and HN878, and that rKVAC85B effectively stimulates immune responses against TB infection.
Keywords:
Mycobacterium tuberculosis
; tuberculosis
; vaccine
; attenuated vaccinia virus
; Antigen 85B
; immunogenicity
; bacterial loads
1. Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is the second leading cause of death from single infectious agents, with 7.5 million people newly diagnosed and 1.3 million deaths in 2022. TB also seriously threatens public health worldwide, such as HIV and malaria [1]. BCG, a live attenuated vaccine developed using Mycobacterium bovis (M. bovis), is commonly used to vaccinate newborns, effectively preventing severe incidence and transmission of the disease in childhood and adolescence [1,2,3,4]. However, the duration of protective efficacy is unstable in adults and adolescents, and the efficacy of BCG varies by age, area, and ethnicity. Additionally, the rate of infection and death due to tuberculosis rapidly increased after the COVID-19 pandemic, particularly in low-to-middle-income countries. Therefore, developing novel vaccines, either BCG replacements or BCG boosters, is urgently needed. Fourteen vaccine candidates are currently in different stages of clinical trials and are expected to prevent TB infections and disease development [5,6]. Most vaccines in clinical trials are composed of protein subunits combined with adjuvants, viral vectors based on either modified vaccinia virus, adenovirus, or influenza virus, or mRNA vaccines.
Viral vector vaccines, including MVA 85A such as Modified Vaccinia Ankara (MVA), have consistently remained in the TB vaccine pipeline. The attenuated vaccinia virus has been used as a vaccine platform as it can deliver and express foreign or large gene(s) and induce strong CD4+ and CD8+ cellular immunity. Hence, cytoplasmic gene expression could prevent the risk of integration into the host genome [6,7,8,9,10,11,12,13].
KVAC103, an innovative third-generation smallpox vaccine candidate, was developed using the Lancy-Vaxina strain [14]. This candidate has demonstrated a reduction in virulence and an augmentation in immune response, evidenced by elevated levels of neutralizing antibodies and IFN-γ production in murine and rabbit models. Genomic analysis identified significant deletions compared with the progenitor strain, indicating an enhancement in both the safety and efficacy profiles of KVAC103, making it a viable candidate for smallpox vaccination [14]. Concurrently, research efforts have focused on using attenuated vaccinia virus vectors to develop vaccines against infectious diseases and therapeutic vaccines for cancer [15,16]. Recent advancements in vaccine development using KVAC103 as a viral vector have underscored its versatility and potential for addressing various infectious diseases. Notably, a bivalent vaccine targeting anthrax and smallpox was formulated by integrating the gene encoding the protective antigen of Bacillus anthracis into the KVAC103 genome [7]. This innovative approach utilizing KVAC103 demonstrates the adaptability of the platform for developing vaccines against different pathogens, including a promising tuberculosis vaccine candidate.
The antigen 85 complex consists of the genes fbpA, fbpB, and fbpC encoding Ag85A, Ag85B, and Ag85C, respectively, which display enzymatic activity in the mycobacterial envelope. The Ag85 complex is correlated with human tuberculosis and comprises 20–30% of constitutive proteins present in the supernatant in short-term culture [8,9,10]. In particular, Ag85A (fbpA) and Ag85B (fbpB) have been selected as targets for developing TB vaccine candidates [7,9,10,11,12,13,14,15,16,17,18], and at least seven vaccines are currently in the clinical phase including Ag85A and Ag85B.
We determined a recombinant virus expressing the Ag85B protein based on attenuated vaccinia virus KVAC103 (rKVAC85B) and evaluated the vaccine efficacy for two types of vaccination, the prime-boost of rKVAC85B and BCG-prime-rKVAC85B-boost. Additionally, the mice were challenged via aerosol using 2-strains, M. tuberculosis H37Rv and the hypervirulent strain HN878, to evaluate their protective effects on the bacterial load in the lung.
2. Materials and Methods
2.1. Animals and Ethics Statement
Four to five-week-old female C57BL/6 mice were procured from DooYeol Biotech (Seoul, Korea). Upon arrival, the mice were allowed to acclimatize for a period of one week in the controlled environment of the animal care facility at the Korea Disease Control and Prevention Agency (KDCA). The subsequent experimental procedures were conducted in strict adherence to the ethical guidelines of the Institutional Animal Care and Use Committee (IACUC) of the KDCA under the approved protocol (permit number: IACUC-KCDC-124-19-2A).
2.2. Bacterial Strains
M. bovis BCG Pasteur, M. tuberculosis H37Rv, and M. tuberculosis HN878 strains were cultured in Middlebrook 7H9 broth (BD, NJ, USA) enriched with 0.02% glycerol (Sigma, MO, USA), 0.05% Tween 80 (Sigma), and 10% albumin dextrose catalase (BD) until they reached an optical density (OD) of 0.6–0.8. The cultures were then centrifuged at 4290 x g for 20 min and washed three times with PBS. To reduce clumping, the pellets were homogenized using a 26-gauge syringe. Colony-forming units (CFUs) for each strain were determined by plating on Middlebrook 7H10 agar (BD) supplemented with 0.05% glycerol and 10% oleic acid albumin dextrose catalase (BD), and incubating at 37 °C for 3–4 weeks.
2.3. Generation and Verification of Recombinant KVAC103 Expressing Ag85B
Ag85B, codon-optimized for human expression, was cloned into the vaccinia virus shuttle vector pVVT-1, which included the TK-R and TK-L regions, for homologous recombination to generate rKVAC85B (Figure 1A). Vero cells were cultured in DMEM (Gibco, NY, USA) supplemented with 10% FBS (Gibco) and antibiotics. The medium was replaced with Opti-MEM (Gibco) containing 2% FBS to the produce KVAC103 expressing GFP (rKVAC-GFP) and rKVAC85B cells. Cells adapted to 2% FBS were infected with rKVAC103 at a Multiplicity of Infection (MOI) of 0.02 for 2 hours in an environment maintained at 37 °C and 5% CO2, followed by transfection with 1.5 µg pVVT-Ag85B using Lipofectamine 2000 (ThermoFisher, CA, USA). After 5 h, the supernatants were replaced with Opti-MEM containing 2% FBS, and plaque isolation was performed after two days to select the recombinants, confirmed by using PCR and western blotting. For western blotting, polyclonal antibodies specific for M. tuberculosis Ag85B (1:1000; Abcam, Cambridge, UK) were used to detect the expressed proteins. The PCR confirmation of rKVAC85B used specific primers: forward 5′-tttgaagcattggaagcaact-3′ and reverse 5′-acgttgaaatgtcccatcgagt-3′. The attenuated vaccinia viruses, KVAC103 and rKVAC85B were grown in Opti-MEM containing 2% FBS without antibiotics for three days and titrated using the plaque-formation assay.
2.4. Animal Immunization and M. tuberculosis H37Rv and HN878 Infection via Aerosol
Mice were subjected to two distinct immunization strategies: a direct prime-boost with rKVAC85B, an administration via single inoculations at four-week intervals, and a BCG prime followed by a rKVAC85B boost, with the latter administered 10 weeks after the initial BCG administration (2 × 105 CFUs per mouse). Immunogenicity assessments were performed one week after the final immunization at 15 weeks of age. Five groups of mice were euthanized via CO2 inhalation. The immune response was evaluated by analyzing lung lymphocytes and splenocytes using an ELISpot assay, intracellular cytokine staining (ICS), quantification of IgG titers, and an MGIA assay.
To challenge with M. tuberculosis, a separate cohort of mice (n = 5) was exposed to the H37Rv and HN878 strains three weeks post-final immunization. Infection was performed using a Glas-Col aerosol generator (Glas-Col LLC., IN, USA) to ensure a consistent initial dose of 100–200 CFUs per mouse. To ascertain the initial bacterial load, lung tissues from the infected mice were homogenized in DPBS the day after exposure. Homogenates were cultured on 7H10 agar plates for 3–4 weeks at 37 °C to facilitate colony formation. Eight weeks post-exposure, the mice were sacrificed, and the bacterial burdens in the lungs and spleen were determined by culturing on 7H10 agar for an additional 3–4 weeks.
2.5. Measurement of Antigen-Specific IgG and Bead Base Analysis of Cytokines
ELISA was conducted to quantify the antibody concentrations within a 1:100 ratio diluted sera, with a focus on IgG levels assessed against plates coated with Ag85B recombinant protein antigen (100 ng/well; Abcam, Cambridge, UK) and the optical density at 450 nm was measured. To evaluate the immune response further, cytokines and chemokines were quantified in immune cells isolated from lung and spleen tissues. Cells were plated at a density of 5 × 105 cells/well and stimulated with a peptide mixture derived from Ag85B (JPT Peptide Technologies) for 36 h. The subsequent bead-based ELISA analysis, conducted in triplicate using the cell supernatant, enabled the measurement of ten-cytokine panel: interferon-gamma (IFN-γ), interleukin-2 (IL-2), tumor necrosis factor-alpha (TNF-α), interleukin-12p40 (IL-12p40), interleukin-12p70 (IL-12p70), interleukin-17A (IL-17A), interleukin-6 (IL-6), interleukin-10 (IL-10), granulocyte-macrophage colony-stimulating factor (GM-CSF), and monocyte chemoattractant protein-1 (MCP-1). Comprehensive cytokine profiling was performed using a customized Bio-Plex mouse cytokine 10-plex assay (Bio-Rad, Hercules, CA, USA).
2.6. INF-γ ELISPOT and Intracellular Cytokine Staining
Isolated cells from the lung and spleen were stimulated using a purchased peptide mixture of Ag85B, which was subsequently subjected to ELISPOT (for IFN-γ-released cells) and intracellular staining (for T-cells secreting three types of cytokines). For the IFN-γ ELISPOT assay, 5×105 cells/well were seeded in plates coated with anti-mouse IFN-γ mAb. The assay was performed following the protocol provided by the manufacturer (BD Bioscience). Each isolated cell was stimulated with an Ag85B peptide mixture (100 ng/well) for 6 h, stained with antibodies developed against surface markers, and the results were analyzed using FlowJo software (BD Biosciences, San Jose, CA, USA).
2.7. Mycobacterium Growth Inhibition Assay (MGIA)
To generate a standard curve, a series of dilutions were prepared using M. bovis BCG 1173P2 (1 × 108 CFU/mL) and phosphate-buffered saline with Tween 80. These dilutions, spanning seven 10-fold increments, were inoculated into MGIT tubes (BD Biosciences) containing MGIT PANTA (BD’s MGIT growth supplement). Time to detection (TTD) was measured using an MGIT 960 instrument, which monitored the tubes for positivity at hourly intervals. Additionally, the dilutions were cultured on 7H10 plates to enumerate CFUs. To assess the TTD of isolated splenocytes, immunized splenocytes (1 × 106 cells/300 µl per well) were first resuspended in RPMI1640 supplemented with HEPES and L-glutamine (Hyclon, Waltham, MA). The cell suspension was then transferred to a 12-well plate, and M. bovis BCG (50 CFUs/300 µl) was added using the same medium. This co-culture was maintained for four days at 37 ℃ in a 5% CO2 incubator. Following the co-culture period, the splenocyte-BCG mixture was transferred to 1.5 mL tubes and centrifuged at 16,260 x g for 10 minutes at 4 ℃. The supernatant was carefully removed, and the pellet was resuspended in 600 µl tissue-culture-grade water. The resuspended pellet was inoculated into MGIT tubes containing MGIT PANTA. The tubes were placed in an MGIT 960 instrument and continuously monitored until they were registered as positive.
2.8. Statistical Analysis
Statistical analyses to assess the significance of the results were conducted using GraphPad Prism software. For multiple group comparisons, one-way ANOVA followed by the Tukey–Kramer multiple comparison test was used. The levels of significance are marked as follows: *p < 0.05, **p < 0.01, ***p < 0.001; ‘ns’ denotes lack of statistical significance.
3. Results
3.1. Construction and Growth Analysis of rKVAC Expressing M. tuberculosis Ag85B (rKVAC Ag85B)
Human codon-optimized Ag85B was successfully cloned into the shuttle vector pVVT1. This construct was then transfected into the attenuated vaccinia virus KVAC103 expressing GFP (rKVAC-GFP), leading to recombinant virus rKVAC85B production. rKVAC85B was verified through plaque assays on Vero cells, and viral supernatants were confirmed via PCR (data not shown). The expression of rKVAC85B was further validated via western blot analysis using monoclonal antibodies against the antigen 85B (Figure1B). A comparative analysis of plaque-forming units between rKVAC-GFP and rKVAC85B indicated a minor delay in the growth of rKVAC85B at 2–3 days post-infection. However, by day 5, the growth rate of rKVAC85B was similar to that of rKVAC-GFP (Figure 1C).
3.2. Immune Responses of rKVAC85B Prime-Boost
To explore the potential of rKVAC85B as a replacement for the traditional BCG vaccine, we evaluated the immunogenic response elicited by a prime-boost immunization regimen in C57BL/6 mice. The mice were administered two consecutive injections of rKVAC85B at four-week intervals. Subsequent ELISA employing the Ag85B recombinant protein as the coating antigen demonstrated a statistically significant elevation in antigen-specific IgG titers in the sera of rKVAC85B-immunized mice. This elevation was markedly higher than that observed in the control groups that received either PBS or traditional BCG vaccination (Figure 2A). Further investigation of cellular immunity revealed an enhanced response in rKVAC85B-immunized mice. IFN-γ ELISpot assays conducted on splenic and pulmonary lymphocytes, upon stimulation with a comprehensive mixture of Ag85B peptides, showed an increase of over 2-fold and 2.5-fold in IFN-γ-secreting cells within the spleen and lungs, respectively, compared to the BCG-immunized group (Figure 2B). A more nuanced analysis of T cell functionality was performed through intracellular cytokine staining after 36-h stimulation with the Ag85B epitope peptide mixture. Flow cytometry analysis identified a substantial fraction of CD4+ and CD8+ T lymphocytes capable of simultaneously secreting multiple cytokines: specifically, IL-2, IFN-γ, and TNF-α. These multifunctional T cells comprised approximately 10% CD4+ and 11% CD8+ T-cells in the spleen, a notable increase compared to the BCG control group (Figure 2C, D).
3.3. Antigen-Specific IgG and IFN-γ Secreting Cells via BCG-Prime-rKVAC85B-Boost
In response to the diminished efficacy of the BCG vaccine observed following infant immunization, this study was conducted to evaluate potential immunogenic enhancement through a BCG prime-boost strategy, utilizing rKVAC85B as the booster. We compared the immunological responses elicited by a BCG-prime followed by an rKVAC85B boost following the immunization regimen detailed in the Materials and Methods section. The mice were euthanized 7–10 days after the final rKVAC85B inoculation, and their sera, spleens, and lungs were collected for analysis. ELISA quantitatively demonstrated a significant increase in antigen-specific IgG levels in the rKVAC85B group serum compared to both the PBS control and BCG-only groups, as shown in Figure 3A. For an in-depth evaluation of cell-mediated immunity, we meticulously homogenized the splenic and pulmonary tissues to isolate resident immune cells. These cells were then stimulated for 36 h with a carefully selected Ag85B epitope peptide mixture to assess IFN-γ secretion. The resulting analysis revealed a substantial increase in IFN-γ secreting lymphocytes in the rKVAC85B group, with an approximately 20-fold enhancement in the spleen and a 15-fold increase in the lung tissues relative to the BCG-only group (Figure 3B). These findings underscore the synergistic effect of the BCG-prime and rKVAC85B-boost in significantly augmenting both humoral and cellular immune responses, particularly in producing antigen-specific IgG and activating T-cells.
3.4. Polyfunctional T-Cell Activation via BCG Prime-rKVAC85B Boost
To assess the polyfunctional T-cell responses induced via BCG prime-rKVAC85B boost vaccination, immune cell populations isolated from lung and spleen samples were subjected to multiparametric flow cytometry. Cells were stained with a panel of antibodies specific for key cytokines: IFN-γ, IL-2, and TNF-α. This analysis aimed to identify a subset of T-cells capable of simultaneously secreting multiple cytokines. In the cohort receiving the BCG prime-rKVAC85B boost, 13% of the CD4+ T-cell population in the spleen exhibited a tri-cytokine secretion profile (IFN-γ+/IL-2+/TNF-α+). Furthermore, 19% of these cells demonstrated bi-cytokine positivity across various combinations (IFN-γ+/IL-2+, IFN-γ+/TNF-α+, IL-2+/TNF-α+). In stark contrast, the group receiving only BCG vaccination exhibited a significantly lower frequency of polyfunctional CD4+ T-cells, with only 5% showing triple or double cytokine positivity, as shown in Figure 4Ai. Similarly, the pattern of cytokine secretion of the CD8+ T-cells was mirrored in splenic CD4 + T-cells, which did not show significant polyfunctionality in either mouse group (Figure 4Aii). In the lung tissue samples, the BCG prime-rKVAC85B boost group continued to demonstrate enhanced T-cell polyfunctionality; 10% of CD4+ T-cells were tri-cytokine-positive, and 20% displayed bi-cytokine positivity. In the BCG-only group, these figures were 7% and 3% for tri- and bi-cytokine-positive CD4 + T-cells, respectively (Figure 4Bi). Among the CD8+ T-cell populations in the lungs, the BCG prime-rKVAC85B boost group exhibited 9% tri-cytokine and 12% bi-cytokine positivity, compared to 3% and 11%, respectively, in the BCG-only group (Figure 4Bii). These findings delineate a significant enhancement in polyfunctional T cell responses, particularly in CD4+ subsets, following BCG prime-rKVAC85B boost vaccination, highlighting its potential to elicit a more robust and diverse immune response.
3.5. BCG-Prime rKVAC85B Boost Enhances Lung Cytokine Response
Cytokine secretion by T-cells in both lung and spleen tissues was quantified following a 36-h stimulation with an Ag85B peptide mixture using a bead-based ELISA approach. This analysis focused on a range of cytokines, including both pro- and anti-inflammatory markers (GM-CSF, IL-12p40, IL-12p70, IL-6, MCP-1, and IL-10) and cytokines associated with Th1 and Th17 responses (IL-17A, IL-2, TNF-α, and IFN-γ), to evaluate the antigen-specific cytokine profile induced by the BCG prime-rKVAC85B boost. The results showed that all tested cytokines were elevated by more than 2-fold in the lungs following BCG prime-rKVAC85B boost immunization compared to the BCG-only group. Notably, eight of these cytokines, including IL-17A, IL-2, TNF-α, and IFN-γ, exhibited a significant increase (p < 0.001, p < 0.01) in the lung (Figure 5).
3.6. Growth Inhibition of BCG-Prime-rKVAC85B-Boost by MGIA
The TTD values for M. bovis BCG 1173P2 based on different CFUs were measured using the MGIT method. The coefficient of determination (R²) for the standard curve was significant at 0.9638, confirming the robustness of the results, as depicted in Figure 6A. Notably, splenocytes harvested from mice immunized with BCG demonstrated a statistically significant and superior reduction in CFUs compared to that in the PBS control group (p = 0.0029, Figure 6B). Interestingly, splenocytes obtained from mice subjected to BCG prime-rKVAC85B boost immunization exhibited a substantially greater reduction in CFUs than both the PBS control group (p = 0.0006, Figure 6B) and the BCG-only immunization group (p = 0.00349, Figure 6B). This difference was approximately 0.5 CFUs log10 reduction. Consequently, the results indicated a significant decrease in M. bovis BCG growth following the BCG prime-rKVAC85B boost immunization regimen.
3.7. BCG Prime-rKVAC85B Boost Efficacy Against M. tuberculosis Strains H37Rv and HN878
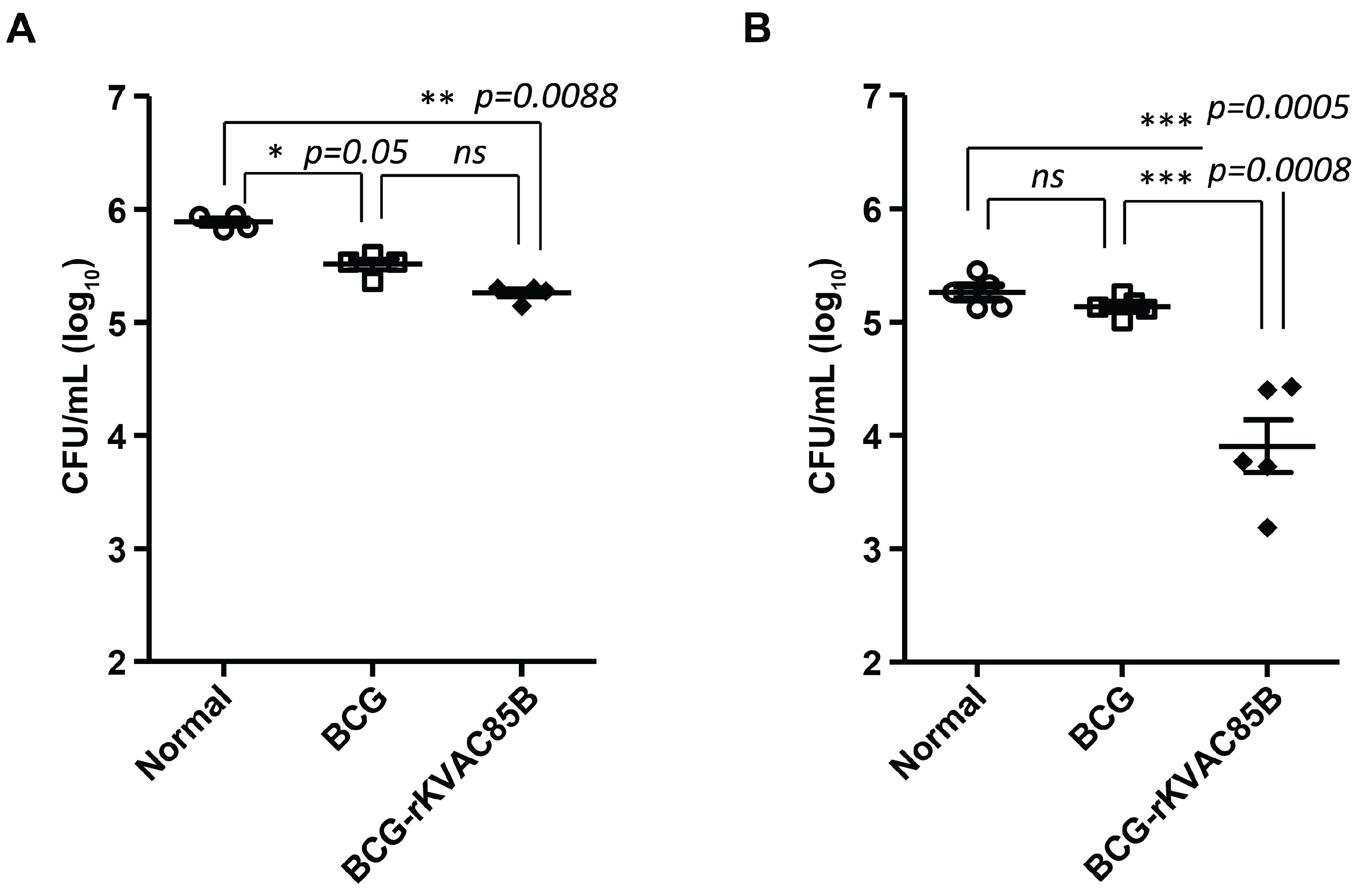
To assess the protective efficacy of rKVAC85B prime-boost and BCG prime-rKVAC85B boost, aerosol-assisted infections were conducted using the laboratory standard strain M. tuberculosis H37Rv and, hypervirulent HN878 strain three weeks after the final vaccination. In the context of the H37Rv challenge, the bacterial load in the lungs of mice receiving the BCG prime-rKVAC85B boost was significantly reduced, specifically a decrease of 0.5 log (p = 0.0088), compared to that in the unvaccinated control group (Figure 7A). In contrast, the response to the HN878 challenge showed a greater protective effect. The bacterial counts in the lungs plummeted by over 1.5 log (p = 0.0005) compared to those in the unvaccinated control group and 1 log (p = 0.0008) compared to the group receiving the BCG vaccination alone. This substantial reduction in bacterial burden against the HN878 strain, as shown in Figure 7B, along with the notable decrease observed in the H37Rv challenge, highlights the effectiveness of the BCG prime-rKVAC85B boost regimen not just against the laboratory standard strain but also against hypervirulent strains of the W-Beijing lineage, such as HN878.
4. Discussion
In the realm of TB prophylaxis, the Bacillus Calmette-Guérin (BCG) vaccine has been the predominant intervention in the last century, effectively reducing TB incidence in individuals from infancy to young adulthood. However, the efficacy of the BCG vaccine varies and tends decreases in the adult population, creating a significant gap in TB control, particularly in the absence of alternative vaccines [1,2,3]. Current research is heavily focused on developing innovative TB vaccines that either supplement or enhance the immunity provided by BCG. This research includes various advanced immunological approaches, such as heterologous prime-boost strategies that utilize protein subunits, RNA-based vaccines, and viral-vectored vaccines [9,11,19,20,21,22,23]. These methods aim to amplify the initial immune response triggered by BCG, overcoming its limitations and offering extended protection in adulthood.
According to the TB Vaccine Initiative TB vaccine development pipeline, clinical trials are currently underway to identify viral vector-based vaccine candidates, including those derived from MVA and chimpanzee adenovirus vectors [24]. These trials are exploring innovative vaccination regimens that involve mono-or heterologous prime-boost strategies, utilizing these viral vectors in various combinations to enhance their immunogenicity and efficacy against TB [6]. The use of viral vectors such as MVA, adenovirus, and parainfluenza virus 5 effectively enhances the immunity elicited by the BCG vaccine [21]. This enhancement is primarily attributed to the induction of robust CD4+ and CD8+ T-cell mediated immune responses, which include antigen-specific multifunctional T-cells and IFN-γ-secreting cells, crucial for the efficacy of TB vaccines [14,22,25,26,27].
In this study, we developed rKVAC85B, a recombinant vaccinia virus expressing the Ag85B antigen, using the attenuated vaccinia virus KVAC103 as a novel vaccine platform [6,7]. KVAC103 is a derivative of the vaccinia virus, closely related to the Lister and VACV107 strains, and demonstrates significant genomic alterations with 23 deleted and four truncated genes compared to VACV107. These alterations included the absence of virulence-related genes, such as the Bcl-2 homolog F1L, and deletions of the K1L and K3L genes, suggesting reduced virulence and cutaneous toxicity. Furthermore, KVAC103 shares 98.35% homology with MVA, indicating its potential use as a viral vector [14]. Research conducted in 2010 expanded the understanding of factors influencing MVA host range and virulence, revealing that the phenotypic traits of MVA are governed not only by known host range genes (K1L, C7L, C12L/SPI-1, E3L, and K3L) but also by additional, yet unidentified, viral genes. Among the 31 open reading frames (ORFs) in the six large deletions of MVA, it is hypothesized that one or more uncharacterized genes significantly influence the virus’s host range in vitro and reduce virulence in mammalian hosts, particularly in the parental chorioallantois vaccinia virus Ankara (CVA) background [28]. Using advanced genetic engineering with a fully sequenced bacterial artificial chromosome clone of CVA, we sequentially introduced these six major deletions into KVAC103 to avoid unwanted mutations. Interestingly, this only moderately reduced the overall virulence and significantly impaired replication in rabbit T-cells. The growth pattern of the recombinant vaccinia virus, rKVAC85B, was consistent with that of its parent strain, KVAC103, and the foreign Ag85B antigen was successfully expressed. This indicates the potential of KVAC103 as a versatile vaccine platform for TB and other infectious diseases. The insights into KVAC103’s genetic modifications, particularly its relationship with MVA and its distinct genomic traits, make it a promising candidate for vaccine development. The reduced virulence and specific host range of KVAC103, coupled with its high homology to MVA, suggest its potential advantages in safety and efficacy as a viral vector. This understanding of genetic factors, illuminated by over a decade of research, is crucial for designing and developing targeted and safe vaccines.
This study, we evaluated the immune responses elicited by two vaccination strategies in a murine model: rKVAC85B prime-boost and BCG prime-rKVAC85B boost. Administration of the rKVAC85B vaccine in both vaccination models led to a significantly increased antigen-specific IgG and IFN-γ, a cytokine essential for controlling infections caused by intracellular pathogens. The increase in IFN-γ levels following rKVAC85B vaccination highlights the vaccine’s potential to elicit a strong immune defense against intracellular infections. Our study compared two immunization strategies: rKVAC85B-prime-boost and BCG prime-rKVAC85B boost. Both strategies effectively induced IFN-γ production in response to specific antigens. However, a notable difference was observed in the magnitude of the response. The BCG-prime rKVAC85B boost strategy elicited a response that was 30-fold stronger than that achieved with BCG alone, as shown in Figure 3B. This significant enhancement in IFN-γ response with BCG prime-rKVAC85B boost underscores its superior efficacy in inducing robust and potent immune responses. Additionally, in the BCG prime-rKVAC85B boost group, there was a notable increase in the production of polyfunctional T cells. These T-cells secreted cytokines critical for Th1 immunity, specifically IL-2, IFN-γ, and TNF-α. In both vaccination models, a marked increase in the levels of IFN-γ-secreting T-cells was observed. Additionally, the BCG prime-rKVAC85B boost strategy was particularly effective in inducing potent CD4+ and CD8+ multifunctional T cell responses in the lungs, capable of secreting two or three cytokines (Figure 5). This finding is consistent with previous studies highlighting the efficacy of viral vector-assisted immunization following BCG priming in enhancing cellular immunity [9,10,11,12]. Furthermore, our analysis revealed a substantial enhancement in the levels of cytokines related to Th1 and Th17 responses (IFN-γ, TNF-α, and IL-17) in the lungs (Figure 5). Although IL-2 is generally associated with Th1 and Th2 immune responses, the presence of IL-2 alone in our study did not provide sufficient evidence of a significant Th2 response. Instead, the observed cytokine profile and T cell responses suggested that the rKVAC85B vaccine, especially in conjunction with BCG priming, primarily drives Th1 and Th17 immune responses. This comprehensive activation of the immune system underscores the potential of rKVAC85B as an effective component of tuberculosis vaccines, leveraging its ability to induce a broad and potent immune response.
Recent studies have underscored the pivotal role of IL-17 in the immune response to M. tuberculosis. This cytokine is particularly crucial in certain models of intracellular infection, where it aids in inducing the Th1 response by promoting IL-12 production. In TB, IL-17 mediates the protective effects of vaccines against M. tuberculosis. These findings suggest that the IL-17 pathway plays a significant role in the primary immune response following M. tuberculosis infection [29,30]. Choi et al. demonstrated the effectiveness of the HSP90-E6/CIA05 vaccine in enhancing BCG-induced immunity against a hypervirulent M. tuberculosis HN878 strain in mice. This effect is primarily attributed to the expansion of IFN-γ/IL-17-producing lung cells. While IFN-γ-producing T-cells are necessary for tuberculosis defense, they are insufficient on their own. Instead, the presence of cells producing both IFN-γ and IL-17 is crucial for effective protection [29]. These findings shed light on the essential role of IL-17 in TB immunity and offer new avenues for improving BCG-boosted vaccines.
In the current study, we assessed the in vitro growth inhibition ability of BCG and a BCG prime-rKVAC85B boost using MGIAs. Immunized splenocytes from the BCG prime-rKVAC85B boost group demonstrated a significantly reduced CFUs compared to those from the BCG-only group (Figure 6B). This observation is consistent with the protective efficacy observed in the BCG prime-rKVAC85B boost group.
MGIAs, regarded as surrogate markers for protection, have been developed to establish in vitro functional assays that correlate with protection. In this context, splenocytes immunized with BCG were subjected to MGIA, and the isolated RNA was subsequently analyzed via microarray analysis. Notably, the gene expressions associated with Th1 responses, including IFN-γ and IL-17, exhibited enhancement [30]. Furthermore, many reports have highlighted the advantages of this analytical method in the context of vaccine testing, allowing longitudinal monitoring of immunological development in the same animal subjects [31,32].
To assess the protective effect of rKVAC85B against bacterial infection, immunized mice were exposed to both H37Rv and hypervirulent HN878 strains, and the bacterial load in the lungs was subsequently measured. In our study, immunization with rKVAC85B alone resulted in some reduction in the bacterial burden in H37Rv infection via aerosols; this decrease was not significantly greater than that in the BCG-immunized group (data not shown). However, notably, the BCG prime-rKVAC85B boost markedly inhibited the growth of the hypervirulent HN878 strain more effectively than the laboratory strain H37Rv (Figure 7). This suggests that rKVAC85B can potentially enhance the vaccine efficacy of BCG, particularly against the more virulent M. tuberculosis strains. The W-Beijing family of M. tuberculosis, the predominant genetic lineage of M. tuberculosis strains, is widespread globally and especially prevalent in East Asia [33]. Epidemiological studies have revealed that W-Beijing strains are isolated more frequently from BCG-vaccinated TB patients than from non-vaccinated patients, suggesting a selective advantage of this genotype in the context of BCG vaccination [21]. Additionally, the BCG vaccination in mice offers less protection against isolates from the W-Beijing family than the standard laboratory M. tuberculosis H37Rv strain [34,35]. Notably, clinical isolate HN878, a hypervirulent strain associated with increased mortality rates, exemplifies the challenges posed by virulent strains [36,37]. These observations emphasize the critical need for vaccine development strategies effect against the most prevalent and virulent M. tuberculosis genotypes, such as the W-Beijing family.
According to the Global TB Report 2021, TB cases in individuals older than 15 years accounted for 89% of the total TB cases reported in 2020 [1], which poses the demand for a novel TB vaccine acting as a BCG booster in adults. In the present study, we used KVAC103, a new attenuated vaccinia virus vector, to develop a novel TB vaccine candidate. Using KVAC103 to express Ag85B represents a promising development in the field of TB vaccine research, particularly in adult populations. rKVAC85B, formulated using this innovative vector, has shown encouraging results by eliciting enhanced immune responses and notable protective effects in a BCG prime-boost regimen. Although these initial findings are promising, they suggest the potential of KVAC103 as an effective BCG booster and a possible frontrunner in the search for advanced TB vaccine solutions. This approach underscores the importance of exploring new avenues of virology for TB vaccine development, with KVAC103 showing considerable promise.
5. Conclusions
In this study, we developed a recombinant vaccinia virus based on the attenuated KVAC103 strain, engineered it to express the 85 B antigen, and assessed its immunogenicity using two vaccination models, rKVAC85B prime-boost and BCG prime-rKVAC85B boost. Compared to BCG alone, both vaccination strategies significantly enhanced antigen-specific IgG, IFN-γ secreting cells, and various cytokines. In addition, in vitro analysis of Mycobacterium growth inhibition demonstrated a consistent immune-mediated pattern. We also, evaluated the protective efficacy of the BCG prime-rKVAC85B boost vaccination against aerosol infections using two M. tuberculosis strains: H37Rv and hypervirulent HN878. Our results revealed that both strains had a lower bacterial burden in the rKVAC85B group than in the BCG-only immunization group. Notably, the reduced the bacterial load, particularly against the hypervirulent HN878 strain, suggests that rKVAC85B is a promising candidate for advancing TB vaccination strategies.
Author Contributions
E.S. contributed to the manuscript writing and data analysis. E.S, J.-S.Y, Y.-R.L., H.-R.C., and S-M.K. were involved in the construction of vaccine candidates, immunological analysis, and TB infection experiments and contributed to the result analysis. S.-J.S., S.-W.L., G.T.C., D.K., and J.S.Y. interpreted the data. H. S. J. and J. S. K. conceived the study design and revised the manuscript. All authors made significant contributions to the article and approved the final submitted version.
Funding
This research received financial support through grants from the Korea National Institute of Health (No. 6637-300), encompassing both intramural and external research (Nos. KNIH 2016-NG48002-00, 2019-NI085-00, and 2016-ER4202-00).
Institutional Review Board Statement
This study was ethically approved by the Institutional Animal Care and Use Committee of the Korea Centers for Disease Control and Prevention (permit number: IACUC-KCDC-124-19-2A).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We extend our gratitude to Professor Hyejon Lee from Yonsei University College of Medicine for their valuable participation in the MGIA experiment.
Conflicts of Interest
The authors declare no conflict of interest. The sponsors had no role in the design, execution, interpretation, or writing of the study.
References
- Global Tuberculosis Report 2023 (2023). Licence: CC BY-NC-SA 3.0 IGO; World Health Organization; ISBN 978-92-4-008385-1. (electronic version).
- Colditz, G. A.; Brewer, T. F.; Berkey, C. S.; Wilson, M. E.; Burdick, E.; Fineberg, H. V.; Mosteller, F. Efficacy of BCG Vaccine in the Prevention of Tuberculosis. Meta-analysis of the Published Literature. JAMA 1994, 271, 698–702. [Google Scholar] [CrossRef]
- Andersen, P.; Doherty, T. M. The Success and Failure of BCG—Implications for a Novel Tuberculosis Vaccine. Nat. Rev. Microbiol. 2005, 3, 656–662. [Google Scholar] [CrossRef]
- Elizabeth, A.; Talbot, M. D. P.; Silva, S. F. M.; Frothingham, R. Disseminated Bacille Calmette-Gue’Rin Disease After Vaccination: Case Report and Review. Clin. Infect. Dis. 1997, 24. [Google Scholar] [CrossRef]
- Ahsan, M. J. Recent Advances in the Development of Vaccines for Tuberculosis. Ther. Adv. Vaccines 2015, 3, 66–75. [Google Scholar] [CrossRef]
- Du, S.; Li, C.; Wang, Y.; Liu, C.; Ren, D.; Li, Y.; Qin, Y.; Wang, M.; Sun, D.; Zhu, N.; Jin, N. Construction and Evaluation of a New Triple-Gene Expression Cassette Vaccinia Virus Shuttle Vector. J. Virol. Methods 2012, 185, 175–183. [Google Scholar] [CrossRef]
- Hruby, D. E. Vaccinia Virus Vectors: New Strategies for Producing Recombinant Vaccines. Clin. Microbiol. Rev. 1990, 3, 153–170. [Google Scholar] [CrossRef]
- Leung-Theung-Long, S.; Gouanvic, M.; Coupet, C. A.; Ray, A.; Tupin, E.; Silvestre, N.; Marchand, J. B.; Schmitt, D.; Hoffmann, C.; Klein, M.; Seegren, P.; Huaman, M. C.; Cristillo, A. D.; Inchauspé, G. A Novel MVA-Based Multiphasic Vaccine for Prevention or Treatment of Tuberculosis Induces Broad and Multifunctional Cell-Mediated Immunity in Mice and Primates. PLOS ONE 2015, 10, e0143552. [Google Scholar] [CrossRef]
- McShane, H.; Pathan, A. A.; Sander, C. R.; Goonetilleke, N. P.; Fletcher, H. A.; Hill, A. V. Boosting BCG with MVA85A: The First Candidate Subunit Vaccine for Tuberculosis in Clinical Trials. Tuberculosis (Edinb) 2005, 85, 47–52. [Google Scholar] [CrossRef]
- Smith, G. L.; Murphy, B. J.; Law, M. Vaccinia Virus Motility. Annu. Rev. Microbiol. 2003, 57, 323–342. [Google Scholar] [CrossRef]
- Vordermeier, H. M.; Villarreal-Ramos, B.; Cockle, P. J.; McAulay, M.; Rhodes, S. G.; Thacker, T.; Gilbert, S. C.; McShane, H.; Hill, A. V.; Xing, Z.; Hewinson, R. G. Viral Booster Vaccines Improve Mycobacterium bovis BCG-Induced Protection Against Bovine Tuberculosis. Infect. Immun. 2009, 77, 3364–3373. [Google Scholar] [CrossRef]
- You, Q.; Jiang, C.; Wu, Y.; Yu, X.; Chen, Y.; Zhang, X.; Wei, W.; Wang, Y.; Tang, Z.; Jiang, D.; Wu, Y.; Wang, C.; Meng, X.; Zhao, X.; Kong, W. Subcutaneous Administration of Modified Vaccinia Virus Ankara Expressing an Ag85B-ESAT6 Fusion Protein, but Not an Adenovirus-Based Vaccine, Protects Mice Against Intravenous Challenge with Mycobacterium tuberculosis. Scand. J. Immunol. 2012, 75, 77–84. [Google Scholar] [CrossRef]
- You, Q.; Wu, Y.; Wu, Y.; Wei, W.; Wang, C.; Jiang, D.; Yu, X.; Zhang, X.; Wang, Y.; Tang, Z.; Jiang, C.; Kong, W. Immunogenicity and Protective Efficacy of Heterologous Prime-Boost Regimens with Mycobacterial Vaccines and Recombinant Adenovirus- and Poxvirus-Vectored Vaccines Against Murine Tuberculosis. Int. J. Infect. Dis. 2012, 16, e816–e825. [Google Scholar] [CrossRef]
- Lim, H.; In, H. J.; Kim, Y. J.; Jang, S.; Lee, Y. H.; Kim, S. H.; Lee, S. H.; Park, J. H.; Yang, H. J.; Yoo, J. S.; Lee, S. W.; Kim, M. Y.; Chung, G. T.; Yeo, S. G. Development of an Attenuated Smallpox Vaccine Candidate: The KVAC103 Strain. Vaccine 2021, 39, 5214–5223. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, L.; Zhao, C.; Zheng, P.; Zhang, X.; Xu, J. Vaccinia Virus-Based Vector Against Infectious Diseases and Tumors. Hum. Vaccin. Immunother. 2021, 17, 1578–1585. [Google Scholar] [CrossRef]
- Perdiguero, B.; Pérez, P.; Marcos-Villar, L.; Albericio, G.; Astorgano, D.; Álvarez, E.; Sin, L.; Gómez, C. E.; García-Arriaza, J.; Esteban, M. Highly Attenuated Poxvirus-Based Vaccines Against Emerging Viral Diseases. J. Mol. Biol. 2023, 435, 168173. [Google Scholar] [CrossRef]
- Park, D. B.; Ahn, B. E.; Son, H.; Lee, Y. R.; Kim, Y. R.; Jo, S. K.; Chun, J. H.; Yu, J. Y.; Choi, M. M.; Rhie, G. E. Construction of a Bivalent Vaccine Against Anthrax and Smallpox Using the Attenuated Vaccinia Virus KVAC103. B.M.C. Microbiol. 2021, 21, 76. [Google Scholar] [CrossRef]
- Sieling, P. A.; Torrelles, J. B.; Stenger, S.; Chung, W.; Burdick, A. E.; Rea, T. H.; Brennan, P. J.; Belisle, J. T.; Porcelli, S. A.; Modlin, R. L. The Human CD1-Restricted T-cell Repertoire Is Limited to Cross-Reactive Antigens: Implications for Host Responses Against Immunologically Related Pathogens. J. Immunol. 2005, 174, 2637–2644. [Google Scholar] [CrossRef]
- Teixeira, F. M.; Teixeira, H. C.; Ferreira, A. P.; Rodrigues, M. F.; Azevedo, V.; Oliveira, G.C. DNA Vaccine Using Mycobacterium Bovis Ag85B Antigen Induces Partial Protection against Experimental Infection in BALB/c Mice. Clin. Vaccin. Immunol. 2006, 13, 930–935. [Google Scholar] [CrossRef]
- Ulrichs, T.; Moody, D. B.; Grant, E.; Kaufmann, S. H.; Porcelli, S. A. T-Cell Responses to CD1-Presented Lipid Antigens in Humans with Mycobacterium tuberculosis Infection. Infect. Immun. 2003, 71, 3076–3087. [Google Scholar] [CrossRef]
- Billeskov, R.; Christensen, J. P.; Aagaard, C.; Andersen, P.; Dietrich, J. Comparing Adjuvanted H28 and Modified Vaccinia Virus Ankara expressing H28 in a Mouse and a Non-human Primate Tuberculosis Model. PLOS ONE 2013, 8, e72185. [Google Scholar] [CrossRef]
- Chen, Z.; Gupta, T.; Xu, P.; Phan, S.; Pickar, A.; Yau, W.; Karls, R. K.; Quinn, F. D.; Sakamoto, K.; He, B. Efficacy of Parainfluenza Virus 5 (PIV5)-Based Tuberculosis Vaccines in Mice. Vaccine 2015, 33, 7217–7224. [Google Scholar] [CrossRef] [PubMed]
- Husain, A. A.; Daginawla, H. F.; Singh, L.; Kashyap, R. S. Assessment of Immunological Markers and Booster Effects of Ag85B Peptides, Ag85B, and BCG in Blood of BCG Vaccinated Children: A Preliminary Report. Clin. Exp. Vaccin. Res. 2016, 5, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Holleman, M. The Pipeline of Tuberculosis Vaccines (2023). Available online: https://www.tbvi.eu/what-we-do/pipeline-of-vaccines/ (accessed on 31 October 2023). updated: 2022.
- Kashangura, R.; Sena, E. S.; Young, T.; Garner, P. Effects of MVA85A Vaccine on Tuberculosis Challenge in Animals: Systematic Review. Int. J. Epidemiol. 2015, 44, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Sayedahmed, E. E.; Singh, V. K.; Mishra, A.; Dorta-Estremera, S.; Nookala, S.; Canaday, D. H.; Chen, M.; Wang, J.; Sastry, K. J.; Mittal, S. K.; Jagannath, C. A Recombinant Bovine Adenoviral Mucosal Vaccine Expressing Mycobacterial antigen-85B Generates Robust Protection Against Tuberculosis in Mice. Cell Rep. Med. 2021, 2, 100372. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, G.; Li, M.; Zeng, J.; Zhao, L.; Liu, X.; Wang, Y. A Recombinant Adenovirus Expressing CFP10, ESAT6, Ag85A and Ag85B of Mycobacterium tuberculosis Elicits Strong Antigen-Specific Immune Responses in Mice. Mol. Immunol. 2014, 62, 86–95. [Google Scholar] [CrossRef]
- Meisinger-Henschel, C.; Späth, M.; Lukassen, S.; Wolferstätter, M.; Kachelriess, H.; Baur, K.; Dirmeier, U.; Wagner, M.; Chaplin, P.; Suter, M.; Hausmann, J. Introduction of the Six Major Genomic Deletions of Modified Vaccinia Virus Ankara (MVA) into the Parental Vaccinia Virus Is Not Sufficient to Reproduce an MVA-Like Phenotype in Cell Culture and in Mice. J. Virol. 2010, 84, 9907–9919. [Google Scholar] [CrossRef]
- Choi, H. G.; Kwon, K. W.; Choi, S.; Back, Y. W.; Park, H. S.; Kang, S. M.; Choi, E.; Shin, S. J.; Kim, H. J. Antigen-Specific IFN-γ/IL-17-Co-producing CD4+ T-Cells Are the Determinants for Protective Efficacy of Tuberculosis Subunit Vaccine. Vaccines 2020, 8. [Google Scholar] [CrossRef]
- Marsay, L.; Matsumiya, M.; Tanner, R.; Poyntz, H.; Griffiths, K. L.; Stylianou, E.; Marsh, P. D.; Williams, A.; Sharpe, S.; Fletcher, H.; McShane, H. Mycobacterial Growth Inhibition in Murine Splenocytes as a Surrogate for Protection Against Mycobacterium tuberculosis (M. tb). Tuberculosis (Edinb) 2013, 93, 551–557. [Google Scholar] [CrossRef]
- Brennan, M. J.; Tanner, R.; Morris, S.; Scriba, T. J.; Achkar, J. M.; Zelmer, A.; Hokey, D. A.; Izzo, A.; Sharpe, S.; Williams, A.; Penn-Nicholson, A.; Erasmus, M.; Stylianou, E.; Hoft, D. F.; McShane, H.; Fletcher, H. A. The Cross-Species Mycobacterial Growth Inhibition Assay (MGIA) Project, 2010–2014. Clin. Vaccine Immunol. 2017, 24. [Google Scholar] [CrossRef]
- Zelmer, A.; Tanner, R.; Stylianou, E.; Damelang, T.; Morris, S.; Izzo, A.; Williams, A.; Sharpe, S.; Pepponi, I.; Walker, B.; Hokey, D. A.; McShane, H.; Brennan, M.; Fletcher, H. A New Tool for Tuberculosis Vaccine Screening: Ex Vivo Mycobacterial Growth Inhibition Assay Indicates BCG-Mediated Protection in a Murine Model of Tuberculosis. B.M.C. Infect. Dis. 2016, 16, 412. [Google Scholar] [CrossRef]
- Glynn, J. R.; Whiteley, J.; Bifani, P. J.; Kremer, K.; van Soolingen, D. Worldwide Occurrence of Beijing/W Strains of Mycobacterium tuberculosis: A Systematic Review. Emerg. Infect. Dis. 2002, 8, 843–849. [Google Scholar] [CrossRef]
- Gopal, R.; Monin, L.; Slight, S.; Uche, U.; Blanchard, E.; Fallert Junecko, B. A.; Ramos-Payan, R.; Stallings, C. L.; Reinhart, T. A.; Kolls, J. K.; Kaushal, D.; Nagarajan, U.; Rangel-Moreno, J.; Khader, S. A. Unexpected Role for IL-17 in Protective Immunity Against Hypervirulent Mycobacterium tuberculosis HN878 Infection. PLOS Pathog. 2014, 10, e1004099. [Google Scholar] [CrossRef] [PubMed]
- van Soolingen, D.; Qian, L.; de Haas, P. E.; Douglas, J. T.; Traore, H.; Portaels, F.; Qing, H. Z.; Enkhsaikan, D.; Nymadawa, P.; van Embden, J. D. Predominance of a Single Genotype of Mycobacterium tuberculosis in Countries of East Asia. J. Clin. Microbiol. 1995, 33, 3234–3238. [Google Scholar] [CrossRef] [PubMed]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J. M.; Barry, C. E.; Freedman, V. H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis Clinical Isolate in Mice Is Determined by Failure to Induce Th1 Type Immunity and Is Associated with Induction of IFN-alpha /Beta. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 5752–5757. [Google Scholar] [CrossRef]
- Cha, S. B.; Kim, W. S.; Kim, J. S.; Kim, H.; Kwon, K. W.; Han, S. J.; Eum, S. Y.; Cho, S. N.; Shin, S. J. Repeated Aerosolized-Boosting with Gamma-Irradiated Mycobacterium bovis BCG Confers Improved Pulmonary Protection Against the Hypervirulent Mycobacterium tuberculosis Strain HN878 in Mice. PLOS ONE 2015, 10, e0141577. [Google Scholar] [CrossRef]
Figure 1.
Construction and expression of rKVAC85B in Vero cells. (A) Schematic structure illustrating the generation of recombinant KVAC virus expressing the M. tuberculosis Ag85B protein. To create rKVACAg85B, a codon-optimized Rv1886c was inserted into the KVAC genome, containing TK-L and TK-R regions, through homologous recombination. TK: thymidine kinase. (B) Confirmation of Ag85B protein expression was conducted through western blot analysis using a rabbit polyclonal antibody against Ag85B. Purchased Ag85B protein served as a positive control, while the lysate obtained from the rKVAC-GFP infected group served as a negative control. M: protein marker; Line 1: positive control (purified Ag85B protein), Line 2: Negative control (Vero cell lysate), Line 3: rKVAC85B (Vero cell-infected lysate). (C) Comparative growth analysis was performed for the rKVAC85B and rKVAC-GFP groups. The growth titration of these two viruses was assessed in Vero cells at a multiplicity of infection (MOI) of 0.1, and the supernatants from cultured viruses were collected and harvested at 24-hour intervals. Virus titrations were determined using a plaque assay.
Figure 1.
Construction and expression of rKVAC85B in Vero cells. (A) Schematic structure illustrating the generation of recombinant KVAC virus expressing the M. tuberculosis Ag85B protein. To create rKVACAg85B, a codon-optimized Rv1886c was inserted into the KVAC genome, containing TK-L and TK-R regions, through homologous recombination. TK: thymidine kinase. (B) Confirmation of Ag85B protein expression was conducted through western blot analysis using a rabbit polyclonal antibody against Ag85B. Purchased Ag85B protein served as a positive control, while the lysate obtained from the rKVAC-GFP infected group served as a negative control. M: protein marker; Line 1: positive control (purified Ag85B protein), Line 2: Negative control (Vero cell lysate), Line 3: rKVAC85B (Vero cell-infected lysate). (C) Comparative growth analysis was performed for the rKVAC85B and rKVAC-GFP groups. The growth titration of these two viruses was assessed in Vero cells at a multiplicity of infection (MOI) of 0.1, and the supernatants from cultured viruses were collected and harvested at 24-hour intervals. Virus titrations were determined using a plaque assay.
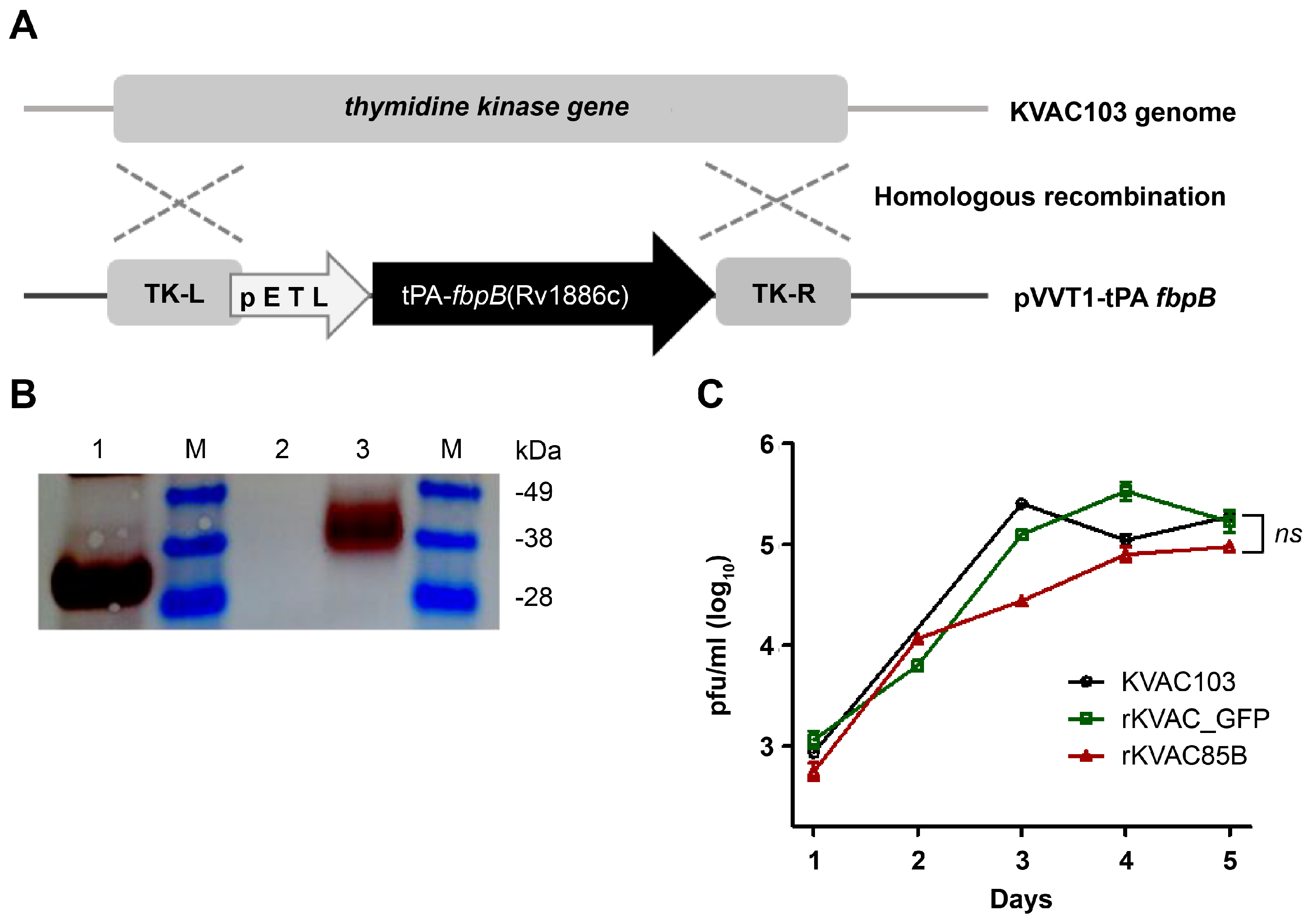
Figure 2.
Immune responses of rKVAC85B prime-boost-immunized mice. (A) IgG Antibody Response to M. tuberculosis Antigen Ag85B. Serum levels of antigen-specific IgG were quantified using ELISA, with microtiter plates coated with recombinant Ag85B protein to assess humoral immunity post-vaccination. (B) Frequency of IFN-γ-secreting T-cells in the lungs and spleens as measured via ELISPOT. Lymphocytes isolated from the lungs and spleens of vaccinated mice were stimulated ex vivo with M. tuberculosis Ag85B antigen. Spot-forming units (SFUs) per 1 × 106 cells were enumerated to determine the Ag-specific T-cell responses. (C) Polyfunctionality of CD4+ T-cells. The chart represents the percentage of CD4+ T-cells secreting different combinations of IFN-γ, TNF-α, and IL-2 upon stimulation with Ag85B protein. Pie charts illustrate the distribution of T-cells based on cytokine secretion profiles: single (gray), double (orange), and triple (blue) cytokine producers. (D) Polyfunctionality of CD8+ T-cells. Similar to panel (C), this chart depicts the percentage of CD8+ T-cells secreting cytokines IFN-γ, TNF-α, and IL-2, with accompanying pie charts showing the proportions of mono-, bi-, and tri-cytokine-secreting cells.
Figure 2.
Immune responses of rKVAC85B prime-boost-immunized mice. (A) IgG Antibody Response to M. tuberculosis Antigen Ag85B. Serum levels of antigen-specific IgG were quantified using ELISA, with microtiter plates coated with recombinant Ag85B protein to assess humoral immunity post-vaccination. (B) Frequency of IFN-γ-secreting T-cells in the lungs and spleens as measured via ELISPOT. Lymphocytes isolated from the lungs and spleens of vaccinated mice were stimulated ex vivo with M. tuberculosis Ag85B antigen. Spot-forming units (SFUs) per 1 × 106 cells were enumerated to determine the Ag-specific T-cell responses. (C) Polyfunctionality of CD4+ T-cells. The chart represents the percentage of CD4+ T-cells secreting different combinations of IFN-γ, TNF-α, and IL-2 upon stimulation with Ag85B protein. Pie charts illustrate the distribution of T-cells based on cytokine secretion profiles: single (gray), double (orange), and triple (blue) cytokine producers. (D) Polyfunctionality of CD8+ T-cells. Similar to panel (C), this chart depicts the percentage of CD8+ T-cells secreting cytokines IFN-γ, TNF-α, and IL-2, with accompanying pie charts showing the proportions of mono-, bi-, and tri-cytokine-secreting cells.
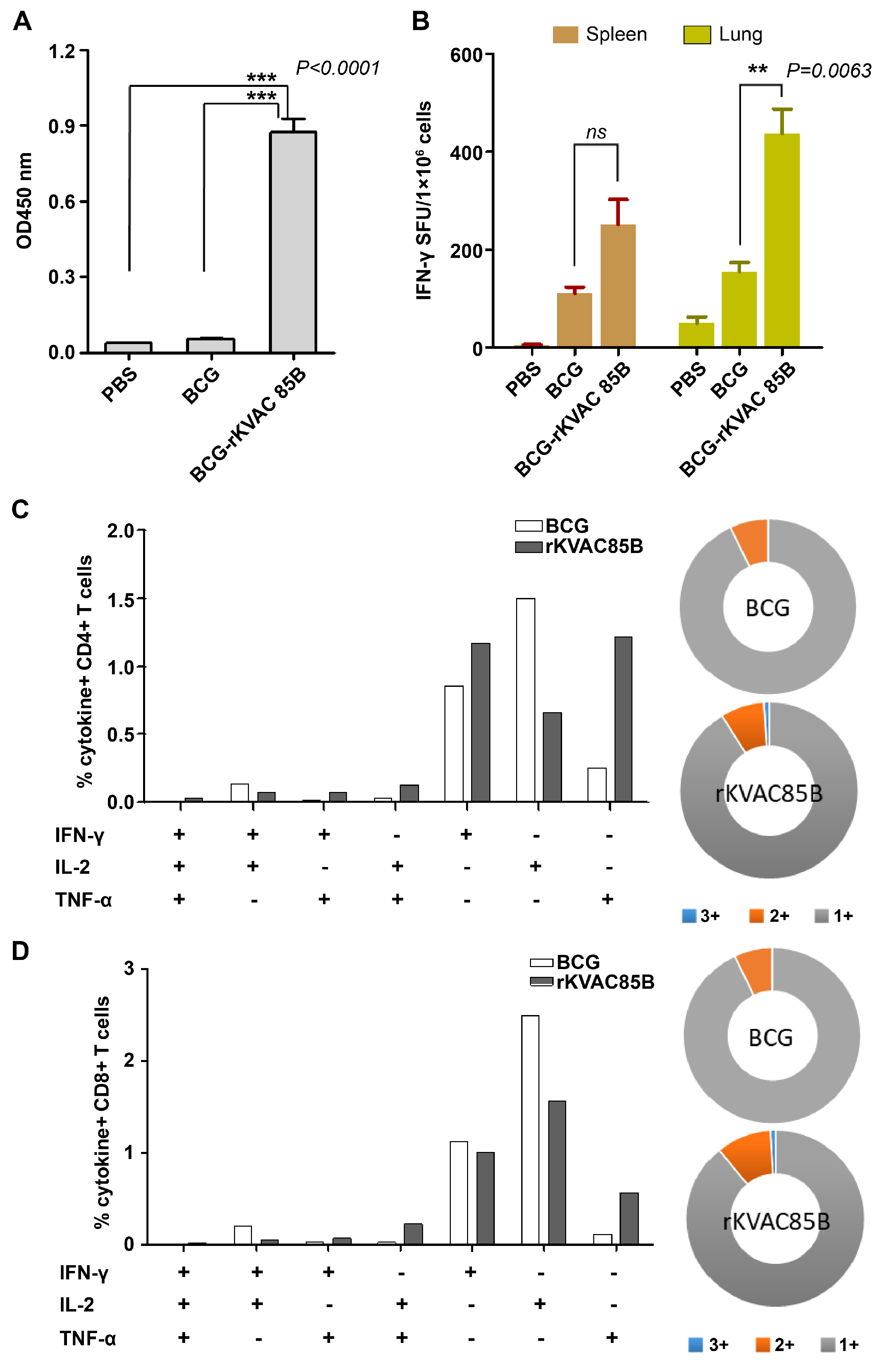
Figure 3.
Assessment of IgG titers and IFN-γ ELISPOT responses via BCG-prime rKVAC85B-boost immunization in mice. (A) Determination of Ag85B-specific IgG Titers. Sera from the immunized mice were diluted at a 1:200 ratio and applied to ELISA plates coated with the Ag85B antigen to measure specific IgG titers. The optical density was recorded at 450 nm (OD_450 nm). (B) IFN-γ ELISPOT assay to identify activated T-cells. The ELISPOT method was used to quantify IFN-γ-secreting T-cells in the spleen and lung tissues. Cells were stimulated with Ag85B peptides for 36 hours, and the frequency of IFN-γ-producing cells was measured. Notably, the lung tissue from the rKVAC85B-boosted mice showed a significant elevation in IFN-γ-secreting cells (***p < 0.0001) compared to the spleen, which displayed a less marked increase (***p = 0.0009).
Figure 3.
Assessment of IgG titers and IFN-γ ELISPOT responses via BCG-prime rKVAC85B-boost immunization in mice. (A) Determination of Ag85B-specific IgG Titers. Sera from the immunized mice were diluted at a 1:200 ratio and applied to ELISA plates coated with the Ag85B antigen to measure specific IgG titers. The optical density was recorded at 450 nm (OD_450 nm). (B) IFN-γ ELISPOT assay to identify activated T-cells. The ELISPOT method was used to quantify IFN-γ-secreting T-cells in the spleen and lung tissues. Cells were stimulated with Ag85B peptides for 36 hours, and the frequency of IFN-γ-producing cells was measured. Notably, the lung tissue from the rKVAC85B-boosted mice showed a significant elevation in IFN-γ-secreting cells (***p < 0.0001) compared to the spleen, which displayed a less marked increase (***p = 0.0009).
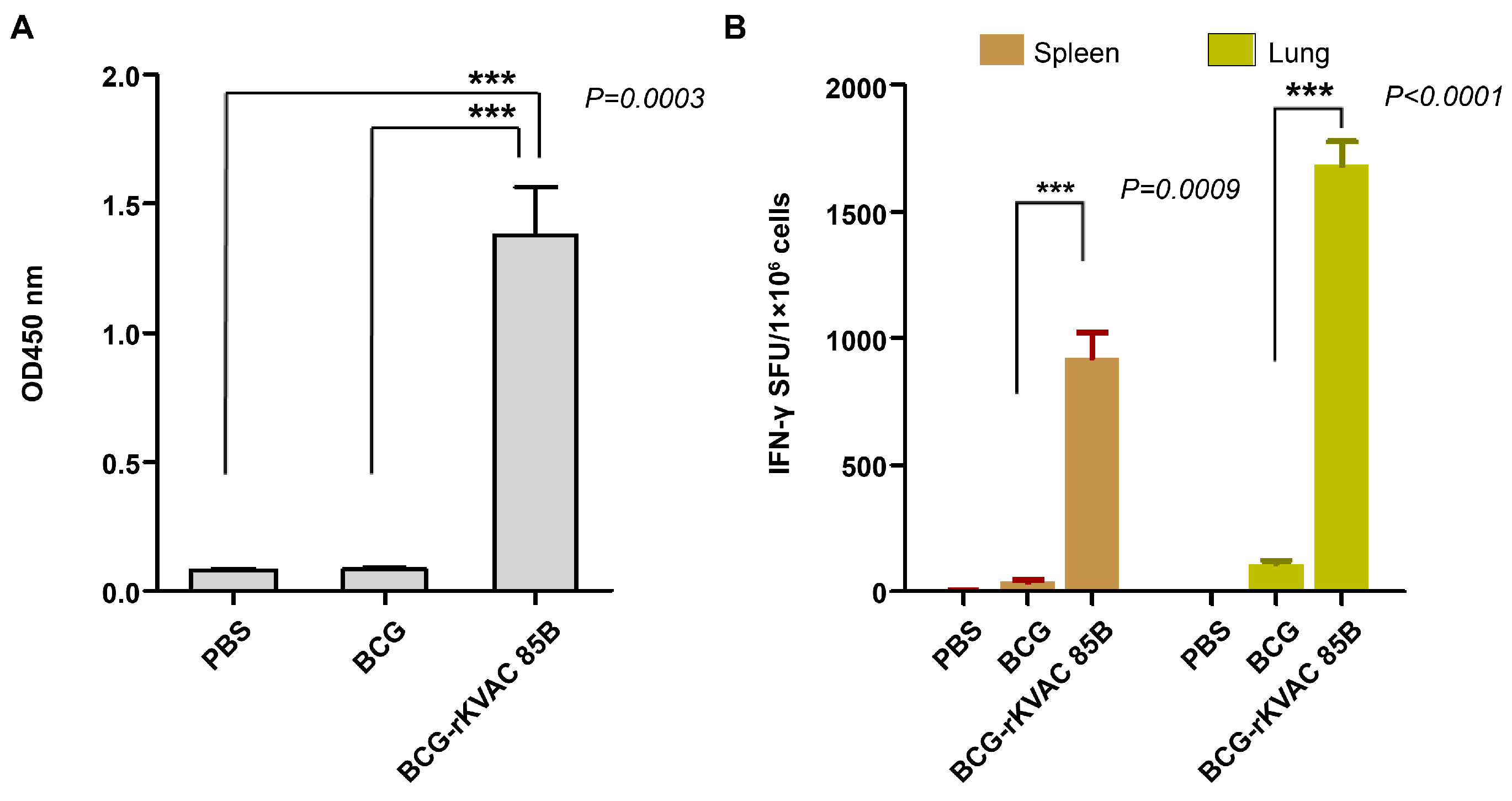
Figure 4.
Polyfunctional T-cell responses via BCG-prime-rKVAC85B-boost immunization in mice. Polyfunctional T-cell profiling was conducted to assess the capacity of CD4+ and CD8+ T-cells to secrete the cytokines IFN-γ, TNF-α, and IL-2 following antigenic stimulation. (A) Frequency of polyfunctional CD4+ (i) and CD8+ T-cells (ii) in the spleen. Data are presented as the percentage of cells secreting any combination of the three cytokines, with pie charts reflecting the cell proportion secreting mono-, bi-, or tri-cytokines. (B) Frequency of polyfunctional CD4+ T-cells (i) and CD8+ T-cells (ii) in the lungs. Pie charts show the distribution of T-cells based on their cytokine secretion profiles, categorized by their ability to secrete mono-, bi-, or tri-cytokines.
Figure 4.
Polyfunctional T-cell responses via BCG-prime-rKVAC85B-boost immunization in mice. Polyfunctional T-cell profiling was conducted to assess the capacity of CD4+ and CD8+ T-cells to secrete the cytokines IFN-γ, TNF-α, and IL-2 following antigenic stimulation. (A) Frequency of polyfunctional CD4+ (i) and CD8+ T-cells (ii) in the spleen. Data are presented as the percentage of cells secreting any combination of the three cytokines, with pie charts reflecting the cell proportion secreting mono-, bi-, or tri-cytokines. (B) Frequency of polyfunctional CD4+ T-cells (i) and CD8+ T-cells (ii) in the lungs. Pie charts show the distribution of T-cells based on their cytokine secretion profiles, categorized by their ability to secrete mono-, bi-, or tri-cytokines.
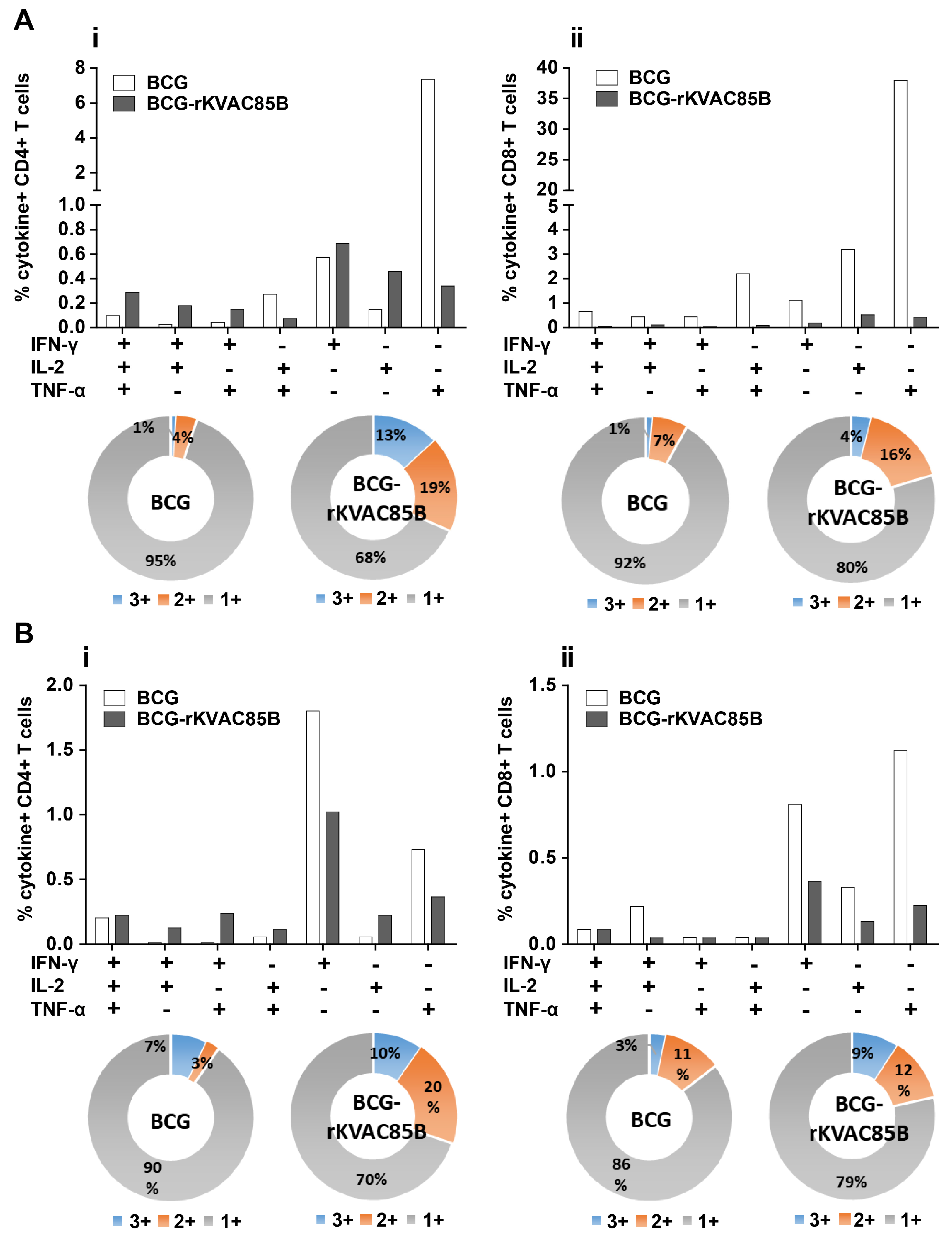
Figure 5.
Augmented pulmonary cytokine response following BCG-prime-rKVAC85B-boost. Cytokine responses in the pulmonary compartment were quantified following a prime-boost vaccination regimen using bead-based ELISA. Lung cells were harvested and stimulated with antigen Ag85B peptides for 36 h to assess post-vaccination cytokine levels. This comparative analysis delineated cytokine induction across three groups: PBS control, BCG priming alone, and BCG priming followed by rKVAC85B booster. The cytokines measured include IFN-γ, IL-2, TNF-α, IL-12p40, IL-12p70, IL-17A, IL-6, IL-10, GM-CSF, and MCP-1. Results are expressed as the mean ± standard deviation (SD), and levels of statistical significance are marked by asterisks: **p < 0.01, ***p < 0.001, indicating a statistically significant increase in cytokine levels in the BCG and rKVAC85B co-immunized group.
Figure 5.
Augmented pulmonary cytokine response following BCG-prime-rKVAC85B-boost. Cytokine responses in the pulmonary compartment were quantified following a prime-boost vaccination regimen using bead-based ELISA. Lung cells were harvested and stimulated with antigen Ag85B peptides for 36 h to assess post-vaccination cytokine levels. This comparative analysis delineated cytokine induction across three groups: PBS control, BCG priming alone, and BCG priming followed by rKVAC85B booster. The cytokines measured include IFN-γ, IL-2, TNF-α, IL-12p40, IL-12p70, IL-17A, IL-6, IL-10, GM-CSF, and MCP-1. Results are expressed as the mean ± standard deviation (SD), and levels of statistical significance are marked by asterisks: **p < 0.01, ***p < 0.001, indicating a statistically significant increase in cytokine levels in the BCG and rKVAC85B co-immunized group.
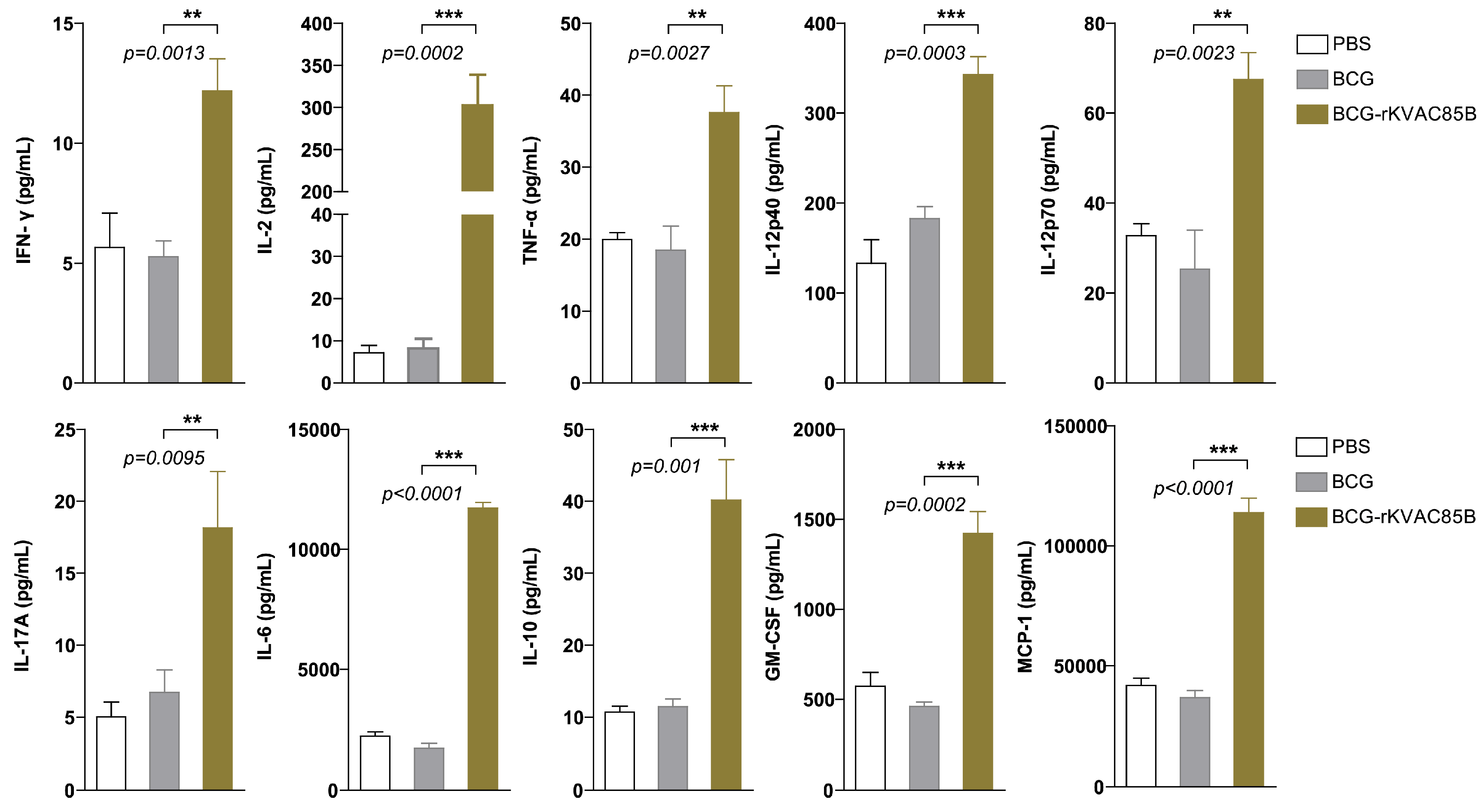
Figure 6.
In vitro growth inhibition of M. bovis BCG with splenocytes isolated from BCG-prime-rKVAC85B-boost immunized mice. (A) MGIA test standard curve, depicting the relationship between the logarithm of colony-forming units (Log_10 CFUs) and time to detection (TTD) in days. The standard curve demonstrates a high coefficient of determination (R2 = 0.9638), indicating a strong inverse correlation between Log_10 CFUs and TTD, as described by the linear regression equation y = −39.979x + 318.67. (B) At one week after final immunization, 1 × 106 splenocytes from immunized mice groups were co-cultured with 50 CFUs of M. bovis BCG. Splenocytes were obtained from six control animals in each group, represented by an individual data point. The p-values of the differences were determined via one-way ANOVA with Tukey’s multiple comparison tests.
Figure 6.
In vitro growth inhibition of M. bovis BCG with splenocytes isolated from BCG-prime-rKVAC85B-boost immunized mice. (A) MGIA test standard curve, depicting the relationship between the logarithm of colony-forming units (Log_10 CFUs) and time to detection (TTD) in days. The standard curve demonstrates a high coefficient of determination (R2 = 0.9638), indicating a strong inverse correlation between Log_10 CFUs and TTD, as described by the linear regression equation y = −39.979x + 318.67. (B) At one week after final immunization, 1 × 106 splenocytes from immunized mice groups were co-cultured with 50 CFUs of M. bovis BCG. Splenocytes were obtained from six control animals in each group, represented by an individual data point. The p-values of the differences were determined via one-way ANOVA with Tukey’s multiple comparison tests.
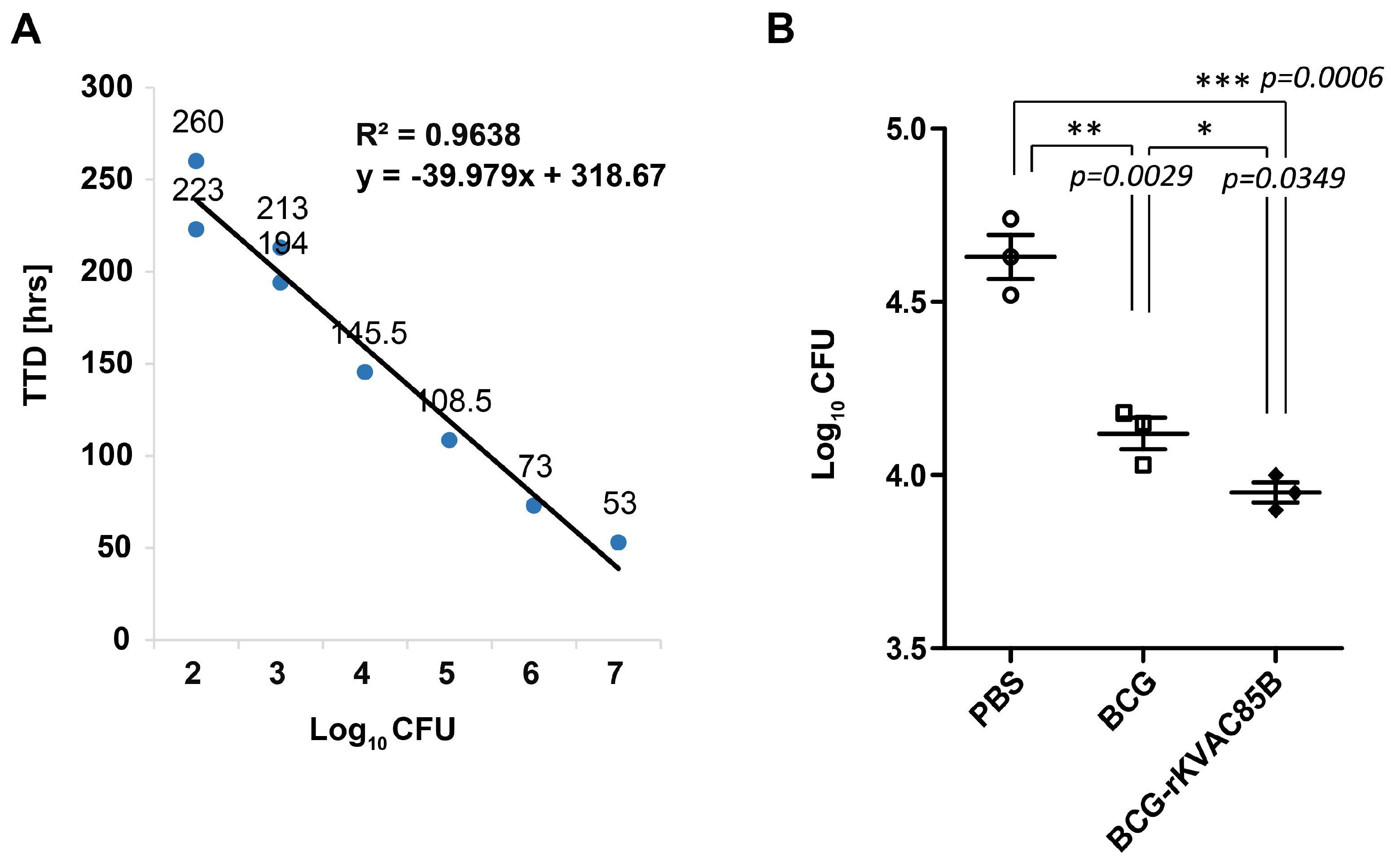
Figure 7.
Comparative efficacy of BCG-prime-rKVAC85B-boost immunization against M. tuberculosis H37Rv and HN878. Bacterial-load quantification in the lungs of mice, following an immunization schedule of BCG priming and subsequent rKVAC85B boosts, was performed to determine the immunoprotective effects against two distinct strains of M. tuberculosis. (A) H37Rv strain challenge: Mice were challenged with M. tuberculosis H37Rv three weeks after the last immunization dose. Eight weeks post-infection, CFUs in the lung tissue were enumerated to quantify the bacterial burden and infer the level of protection conferred by immunization. (B) HN878 strain challenge: In an experimental setup similar to that in (A), mice were challenged with the HN878 strain. Lung CFUs were counted after eight weeks to determine the strain-specific protective efficacy of the vaccine regimen. Statistical analyses for both (A) and (B) were performed using one-way ANOVA.
Figure 7.
Comparative efficacy of BCG-prime-rKVAC85B-boost immunization against M. tuberculosis H37Rv and HN878. Bacterial-load quantification in the lungs of mice, following an immunization schedule of BCG priming and subsequent rKVAC85B boosts, was performed to determine the immunoprotective effects against two distinct strains of M. tuberculosis. (A) H37Rv strain challenge: Mice were challenged with M. tuberculosis H37Rv three weeks after the last immunization dose. Eight weeks post-infection, CFUs in the lung tissue were enumerated to quantify the bacterial burden and infer the level of protection conferred by immunization. (B) HN878 strain challenge: In an experimental setup similar to that in (A), mice were challenged with the HN878 strain. Lung CFUs were counted after eight weeks to determine the strain-specific protective efficacy of the vaccine regimen. Statistical analyses for both (A) and (B) were performed using one-way ANOVA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



