
Submitted:
07 March 2024
Posted:
08 March 2024
You are already at the latest version
Abstract
Ionic channels are present in eucaryotic plasmatic and intracellular membranes. They coordinate and control several functions. One of the most diverse ionic channels family are the potassium (K) channels. Within this family are the ATP-dependent potassium channels (KATP), which belong to the potassium rectifier channel family. These channels were initially described in heart muscle, yet they are also present in different types of tissues such as pancreatic, skeletal muscle, brain, and vascular and non-vascular smooth muscle. In ß-pancreatic cells, KATP are primarily responsible for maintaining membrane potential and for the depolarization mediated insulin release. KATP density/activity seems to be determinant in physiological the outcome, in pancreatic cells decrease of this density/activity may be related to Insulin resistance (IR). In extra pancreatic tissues KATP relation with IR is beginning to be explored. It is possible that in skeletal muscle, the KATP are involved in insulin dependent glucose recapture and that KATP activation leads to IR. In adipose tissue, KATP containing Kir6.2, could be related to increase in free fatty acids and insulin resistance, therefore, pathological processes leading to a prolonged adipocyte KATP inhibition might promote obesity related to insulin resistance. In central nervous system KATP activation can regulate peripheric glycemia and lead to brain IR which could be a much earlier manifestation of peripheral alterations that lead to the development of pathologies such as obesity and type 2 diabetes mellitus. In this review we aim to discuss the KATP channel characteristics, its relationship to known clinical disorders and the mechanisms and potential relations with peripheral and central insulin resistance.
Keywords:
Ionic channels
; ATP-dependent potassium channels
; Insulin resistance.
1. Introduction
Ionic channels are found in eucaryotic plasma and intracellular membranes. These channels coordinate and control several functions like neurotransmission, secretion, contraction, growth/proliferation, migration, and regulation of cell volume [1]. Ionic channels allow for the passive transport of inorganic ions, such as Na+, K+, Ca2+, and Cl−, through the cell membrane. Ionic channels can be gated and do not always allow ions to freely diffuse across the membrane. Mutations in their subunits’ structure can cause important pathologies, known as channelopathies [2].
K+ channels belong to the most diverse families of ionic channels. K+ channels are present in excitable and non-excitable cells. They control K+ efflux and influx and are highly selective due to a very conserved domain known as the selective filter (SF) [3].
Based on their structure, K+ channels can be classified according to the primary amino acid sequence of the surrounding pore [4]:
- Voltage-gated potassium (Kv) channels: channels with six transmembrane motifs and one pore;
- Potassium inward rectifier channels (KIRs): channels with two transmembrane motifs and one pore;
- Weak potassium inward rectifier (K2P) channels: channels with four transmembrane motifs and two pores.
In addition, there are other classes of K+ channels: the Slo family and the Ca2+-activated SK family (SKCa) channels [5,6].
In humans, K+ channels are encoded by more than 90 genes [7]. Mutations in these genes can produce many disorders. For example, mutations in the voltage-gated K+ channel encoded by the KVLQT1 gene and the calcium-modulated K+ channel encoded by the HERG gene are responsible for long QT syndrome (LQTS) variants [8]. LQTS is a coronary disorder where a delayed heart repolarization after the heartbeat increases the risk of irregular heartbeats which can lead to palpitations, fainting, or sudden death due to ventricular fibrillation [8]. Mutations in the KCNJ1 gene encoding the KIR1.1a channel induce neonatal Bartter syndrome which is a dominant renal pathology that causes defects in the thick portion of the ascending Henle loop, characterized by hypokalemia, hypochloremia, and metabolic alkalosis [9].
The ATP-dependent potassium (KATP) channels, which belong to the potassium rectifier channel subfamily, are an important group of K+ channels. These channels were initially described in heart muscle [10] and were later found to be also present in other types of tissues such as pancreatic beta, skeletal muscle, brain, and vascular and non-vascular smooth muscle tissues [11]. These channels have an octameric structure [12], consisting of four Kir6.1 or Kir6.2 subunits, encoded by the KCNJ8 and KCNJ11 genes, respectively, and four regulatory subunits of the SUR1 or SUR2 sulfonylurea receptor, encoded by the ABCC8 and ABCC9 genes, respectively [13,14,15]. Kir6.1 is expressed in brain and smooth vascular muscle tissues [11], while Kir6.2 is expressed in brain, cardiac muscle, skeletal muscle, vascular smooth muscle, and pancreatic beta tissues [16,17]. SUR1 is primarily expressed in the brain and pancreatic beta cells [18]. On the other hand, there are two SUR2 isoforms, SUR2A and SUR2B, which are both produced by ABCC9 splicing, and are expressed in cardiac, skeletal, and vascular smooth muscle tissues [11]. Different combinations of Kir/SUR subunits generate channels with different cellular functions, tissue locations, and pharmacological profiles [19,20].
Pancreatic KATP channels are mainly SUR1/Kir6.2 channels. Regarding glucose metabolism, more than 100 mutations in SUR1/Kir6.2 channels have been identified that are associated with channelopathies, for example, the recessive form of persistent hyperinsulinemic hypoglycemia of the child, or infantile hyperinsulinemia, and at least two other persistent types of prenatal hypoglycemia [21,22].
In addition to KATP channels, delayed rectifying K+ channels (Kv1.x), calcium-activated K+ channels (maxi-K), α-adrenoreceptor-activated K+ channels, and G protein-activated K+ channels are also present in pancreatic beta cells [23,24,25]. It has been proposed that calcium-activated K+ channels have a role in triggering the insulin-releasing signal. This process is activated in the initial phases by calcium released from inositol triphosphate (IP3)-mediated intracellular signaling. Nevertheless, channel inhibition has little effect on the total electrical activity of pancreatic beta cells [25]. It is generally accepted that KATP channels are primarily responsible for maintaining the pancreatic beta cell’s membrane potential and is involved in depolarization-mediated insulin release. However, some studies suggest that there are KATP channels that are not involved in insulin release mechanisms and mainly work on the second phase of the process [26,27,28,29]. In this review, we discuss the pathophysiological features, processes, and possible relationships between KATP channels and insulin resistance in peripheral tissues and the brain.
2. ATP-Dependent Potassium Channels (KATPs): Characteristics and Regulation
From a pharmacological perspective, KATP channels can be divided into two groups based on their sensitivity to sulfonylureas. The first group corresponds to the neuronal, neuroendocrine, and beta-cell types that are 100 to 1000 times more sensitive to sulfonylureas than the channels of the second group. The channels of this group contain the SUR2 subunit and are mainly expressed in cardiac and muscular tissues [30]. Using compounds that activate them or increase the channels’ average opening time, such as diazoxide and pinacidil (potassium channel openers: KCOs), it is also possible to distinguish between the compositions of different groups of channels. KATP channels containing SUR1 and SUR2B subunits respond better to diazoxide than those with SUR2A (which are mainly expressed in cardiac tissues) [31,32,33], and it is possible to distinguish between channels constituted by SUR2A or SUR2B due to their differential sensitivity to pinacidil [33,34].
KATP channels can be considered moderate rectifier channels, i.e., they conduct K+ more efficiently into the cell than outward [30]. The degree of rectification depends on the presence of cytoplasmic Mg2+ or polyamines, such as putrescine and spermidine, which can block the entry of K+ when the membrane is depolarized [35].
3. KATP Channel Activity and Their Functional Regulation
In pancreatic beta cells, the KATP channels consist of Kir6.2/SUR1 tetramers [36]. When extracellular glucose is increased, there is a reduction in the KATP opening paired to insulin release [37]. According to these results, KATP channels are key components that relate glucose metabolism, membrane electrical activity, and insulin release [30,37]. They also provide the dominant K+ conductance at rest and determine the beta-cell membrane potential [22]. Pancreatic KATP channels are blocked by sulfonylureas and activated by diazoxide [33,38].
When extracellular glucose levels increase, glucose is transported into the cytosol by glucose transporter 2 (GLUT-2), and ATP production increases. Kir6.2 is non-cooperatively blocked by ATP and the Kir6.2/SUR1 complex’s ATP inhibitory concentration (CI50) decreases by an order of magnitude [39]. On the other hand, SUR1 is activated by ADP/Mg2+, which binds to the nucleotide-binding domain 2 (NBD) [40]. When the extracellular glucose concentration is less than 3mM, the membrane potential remains between -60 to -70 mV. The increase in cytosolic glucose produces a gradual depolarization which causes KATP channel closure. After KATP channel closure, the membrane depolarizes to -50 mV, triggering an action potential that activates L-type voltage-gated Ca2+ channels (L-Ca2+) and probably also the type T (T-Ca2+) channels. This can cause an influx of Ca2+ that triggers insulin/Zn2+ transporter granule exocytosis [22,30] (Figure 1).
The electrical activity of KATP channels is complex. The increase in plasma glucose levels leads to a sustained initial depolarization that promotes a slow and sustained increase in the intracellular Ca2+ concentration (presumably due to T-Ca2+channels) leading to a period of insulin release known as phase 1. Oscillatory changes in the membrane electrical activity coincide with the oscillatory changes in the intracellular Ca2+ concentration caused by L-Ca2+ channels. These changes lead to pulses of insulin release from the pancreatic islets; this period is known as phase 2 [41,42].
Patch-clamp experiments have shown that, in the absence of ATP, KATP channels remain open and lose their activity [43]. Adenine dinucleotides can inhibit channel activity in Mg2+-free solutions [33]. The mechanism of KATP channels’ on/off process is related to the ATP/Mg-ADP ratio [44], and anionic phospholipid hydrolysis stabilizes the structure of the channel into a functional state [45,46,47,48].
The ATP IC50 for pancreatic beta cell channels is 5-10 µM [49], 8-500 µM for cardiac channels [39], and 20-100 µM for SUR2A/Kir6.2 channels [50]. The physiological range of intracellular Mg-ATP concentrations is around 1 mM, which is above the in vitro concentrations needed for the inactivation of KATP channels. Under these conditions, a 99% inhibition of the channel would be predicted, which raises the question of the mechanism that ties metabolism to the in vivo channel conditions. It has been proposed that KATP channel might be regulated by the large fluctuations in intracellular ATP concentrations or the existence of some compartmentalization that allows for local changes in the ATP concentration that are capable of inhibiting KATP channels [51]. Interestingly, pancreatic KATP channels (SUR1/Kir6.2) have a higher IC50 with Mg-ATP than with ATP, suggesting that the inhibition potential is related to the magnitude of negative ion charges [52]. However, in cardiac channels, the opposite occurs (SUR2A/Kir6.2 channels), where a greater inhibition of the channel is obtained with ATP in the presence of Mg2+ [53]. Yet, when studying SUR1 polymorphisms related to infantile hyperinsulinism, it was found that the regulation of these channels includes mechanisms other than the ATP control.
In vitro studies with reconstituted SUR1 channels containing a glycine to arginine mutation at the 1479 site (SURG1479R) showed that the channels were inhibited by ATP and Mg-ATP in the same way as wild-type channels, although these mutant channels were not activated by Mg-ADP or diazoxide [54]. This mutation is located in the NBD. Additionally, it is known that ADP and other diphosphate nucleosides inhibit KATP channels in the absence of Mg2+, so the presence of Mg2+ is critical for its activation [30,55]. KATP channel regulation relies on changes in both ATP and Mg-ADP concentrations [56]. Currently, this regulation is not well understood. Despite these channels having been known for more than three decades, there are few studies on the behavior of KATP channels in response to variations in intracellular ATP/ADP concentrations at a physiological level.
In studies with truncated carboxyl terminal Kir6.2 subunits, and in the absence of SUR1 subunits, it was found that Kir6.2 subunits are sufficient to form the pore channel structure and these subunits possess the ATP-mediated inhibitory activity of KATP channels [57,58]. However, these channels are not activated by Mg-ADP or other activators such as diazoxide, and they are not inhibited by sulfonylureas [57,58]. On the other hand, these activities were restored when expressing SUR1 along with the mutated Kir6.2 subunits [48,57]. That is, the pore-forming units of the beta-cell KATP channels are the Kir6.2 subunits, which contain the ATP regulation sites. The SUR1 units are the regulatory units that can control the channel activity. The Kir6.2 pore conformation can force the channel into an “off” state in which they are not sensitive to ATP inhibition, potentially via the action of Mg-ADP and activators such as diazoxide in one or both NBD domains of SUR1 [48,59,60].
Another important KATP channel regulator is phosphatidyl inositol 4-5-bisphosphate (PIP2), which blocks the activity of these channels in a nucleotide-independent way [44]. PIP2 stabilizes the open state of the KATP channel without affecting the individual channels’ current amplitude [46,61]. It has been demonstrated that the PIP2 and KATP channel interaction occurs in the cytoplasmic Kir6.2 region; this interaction can be blocked by neomycin or poly-L-lysine and is dependent on the phosphorylation status of the Kir6.2 amino terminal region [48,58]. Similarly, the interaction between the Kir6.2 and SUR1 subunits depends on the phosphorylation of this region and is also involved in the coupling of sulfonylureas to SUR1 for Kir6.2 pore blockage [62]. PIP2 decreases the channels’ sensitivity to ATP via intracellular mechanisms [63]. It is currently proposed that short-term physiological regulation of KATP channel is mediated by Mg-ATP while long-term regulation depends on PIP2 [64,65].
4. Genetic Variability in Human KATP Channels: Insights and Implications in Insulin Resistance
The KCNJ11 (Kir6.2) and ABCC8 (SUR1) genes encode for Betaβ-pancreatic KATP channels and are located on the same human chromosome (11p5.1) with 4.5 kb between them. KCNJ11 has a unique exon that encodes for a 390-amino acid protein consisting of two transmembrane regions and the pore-forming subunit [66]. ABCC8 only has one open reading frame (ORF) with 39 exons and more than 100 kb of genomic DNA and a median of 124bp per exon.
Polymorphisms have been described for both genes, and some of them are related to pathologies such as hypo- and hyperinsulinemia (HI) of infancy and familiar or PHHI [9,67]; these polymorphisms may also influence the susceptibility to Type 2 Diabetes Mellitus (T2DM) and the channels’ pharmacological response [68,69,70,71,72].
Mutations in ABCC8 are the most frequent cause of HI. These mutations can be divided in two functional types: class I mutations which reduce the number of channels on the membrane by interrupting channel complex biogenesis and class II mutations that reduce the channel opening rate by preventing Mg-ADP activation or by producing defective proteins incapable of forming pores [22,67,73]. Mutations in KCNJ11 are less frequent causes of HI and they also show a low or no channel activity. Sulfonylureas can act as chemical chaperones correcting traffic deficiencies in class I mutations [74], while KCOs like diazoxide are used for treating class II mutations. Nevertheless, gain of function mutations keep the channels open and beta cells hyperpolarized, thereby decreasing insulin secretion [75].
5. KATP Channels’ Role in Insulin Resistance in Peripheral Tissues
In vitro studies suggested that sulfonylureas have a mimetic insulin effect in extra-pancreatic tissues by improving glucose recapture through an insulin-independent pathway [76,77] or by potentiating insulin action [78,79]. This effect might be due to the SUR subunits’ sensitivity to sulfonylureas. For example, skeletal muscle KATP channels (mainly Kir6.2/SUR2A) are between 30 and 300 times less sensitive to glibenclamide in comparison to pancreatic beta KATP channels [80].
An increase in glucose oxidation and ATP production was observed in the hearts of rats that were perfused with tolbutamide. An increase in glucose recapture was also observed in rat skeletal muscle [81,82]. On the other hand, a concentration- and time-dependent increase in glucose recapture was observed in the L6 skeletal muscle cell line treated with glyburide and gliclazide. Additionally, glucose-independent GLUT1 translocation to the cellular membrane has also been observed, but in an insulin-dependent way, with no increase in GLUT-3 and GLUT-4 translocation [79,83]. The same effect was observed for gliclazide in rat skeletal muscle ex vivo experiments, although the increase in glucose recapture was abolished by the blockage of KATP channels with diazoxide [84].
Kir6.2-knockout mice have no KATP channel activity in pancreatic beta, skeletal muscle, and cardiac cells. In general, these mice had a closed KATP channel phenotype where the pancreatic beta cell’s membrane was constitutively depolarized. In addition, at a concentration of glucose less than 3mM, significantly high concentrations of Ca2+ and levels of insulin secretion were achieved. This phenotype could not be restored by KCOs. In general, compared to wild-type mice, these mice presented a lower glucose intolerance and higher insulin sensitivity [85].
SUR1-knockout mice’s beta cells presented a similar electrophysiological phenotype to Kir6.2-knockout beta cells, but the mice maintained normal glycemic rates and presented an increase in insulin sensitivity [86].
On the other hand, in a study that analyzed different KCNJ11 polymorphisms in a Danish population, none of the studied polymorphisms affected insulin release from beta cells after glucose or tolbutamide administration, but there was a significant increase (62%) in insulin sensitivity in comparison to subjects that did not have the KCNJ11 polymorphisms [87]. It is possible that KATP channels are involved in insulin-dependent glucose recapture in skeletal muscle. It has been shown that KATP channel inhibition in peripheral tissues, mainly in skeletal muscle, improves insulin sensitivity [88].
Nicorandil, a KCO used as a human angina treatment, decreases glucose metabolism, causing insulin resistance without interfering with insulin secretion [88]. In other in vitro studies with human skeletal muscle cells, nicorandil and PCO-400 (another KCO) inhibited insulin-mediated glucose recapture in a dose-dependent manner. This inhibition was reverted by gliclazide or glibenclamide pre-treatment, also in a dose-dependent manner [87]. In rat aorta, phorbol 12-myristate 13-acetate (PMA), a PKC activator, was able to revert PCO-400 glucose recapture inhibition in a dose-dependent manner [33,80]; however, other works have reported no change in PKC levels after PCO-400 or PMA stimulation [89].
These results suggested that KATP channel opening decreases glucose recapture in target tissues in an insulin- and high glucose concentration-dependent manner; therefore, KATP channel opening can potentially contribute to insulin resistance in vivo (Figure 2A).
Beside KATP channels’ effects on glucose metabolism, they have shown to have effects on lipid metabolism. SUR1 is expressed in human adipocytes where it regulates intracellular Ca2+ concentrations [90]. In 3T3-Li adipocytes, diazoxide inhibits the insulin-mediated activation of the fatty acid synthase (FAS) [91]. Glibenclamide stimulates FAS activity, increases glycerol 3-phophate dehydrogenase activity, and inhibits lipolysis [92].
These works suggest that the opening of KATP channels has an inhibitory effect on fatty acid synthesis. This can be related to changes in intracellular Ca2+ concentrations which are a known regulator of lipogenesis, lipolysis, and triglyceride synthesis [92]. As previously mentioned, the increase in free fatty acids (FFAs) might cause insulin resistance; therefore, pathological processes that lead to prolonged adipocyte KATP channel inhibition might promote obesity due to insulin resistance (Figure 2B).
It has been shown that there is a strong correlation between intracellular triglyceride levels and insulin resistance outcome (i.e., in skeletal muscle and hepatic tissues) [93,94]. Some studies suggested that esterified FFAs can activate KATP channels in skeletal and cardiac muscle [95,96,97]. Esterified FFAs, such as oleoyl-CoA, can activate pancreatic beta KATP channels at low concentrations (1 µM). They can also block ATP inhibition. This activation occurs through the interaction of Kir6.2 with Mg-ADP or other KCOs, but not with SUR1 [33]. This suggests that KATP channels containing Kir6.2 in other tissues, such as smooth or skeletal muscle, can similarly react to fatty acids. An increase in the intracellular esterified fatty acid concentration due long-term exposure to FFAs could increase KATP channel activity and, as consequence, cause insulin resistance [97].
6. Beyond the Blood–Brain Barrier: Understanding the Effects of Insulin Resistance on the Brain
The brain has different mechanisms to detect and regulate glucose metabolism. It receives natural impulses from peripheral tissues, but it also has neurons that can determine glycemic levels by altering their firing pattern. When the glucose level increases, glucose-responsive (GR) neurons increase their firing pattern and glucose-sensitive (GS) neurons reduce their firing [98]. These neurons are located in the lateral hypothalamic area (LH), ventral–medial hypothalamic area (VMH), nucleus tractus solitarius, and other areas [99,100,101].
It has been proposed that GR neurons use KATP channels to regulate their firing rate [102]. Brain KATP channels contain Kir6.2 and either SUR1 or SUR2 as the regulatory subunits [11,49,102]. As in pancreatic beta cells, neural KATP channels are inhibited by ATP, causing membrane depolarization and triggering action potentials or firing [103]. Nevertheless, in basal conditions, the levels of interstitial glucose are lower than peripheral levels (<2mM) and changes in the extracellular concentrations are relatively small (<1mM). It is not clear how such small changes can alter the ATP/ADP ratio enough to regulate KATP channels. Additionally, Kir6.2 and SUR1 expression is not limited to GR neuron regions [102].
In pancreatic beta cells, glucokinase (GK) is a regulator that controls ATP levels produced by glycolysis and it is also expressed in the brain [21,104]. Levin et al. [98] have proposed that KATP channels work as glucose sensors in the neurons that contain a glucose transporter and/or GK, or any other hexokinase located sufficiently close to the plasma membrane and to KATP channels (Figure 3) [98].
Insulin is not the only glucose metabolism regulator in the brain [105]. Insulin receptors (IRs) are abundantly expressed in all areas of the brain [106,107]. Insulin’s interaction with IRs activates phosphatidylinositol-3 kinase (PI3K), which regulates the electrical activity of the target neurons through KATP channel stimulation (Figure 3) [108]. The channel activation induces hyperpolarization and a reduction in activity of the target neuron [109]. Brain IR-knockout mice are overweight, insulin resistant, and glucose intolerant [110]. It has also been observed that hypothalamic IR chronic blockade leads to pancreatic insulin resistance and to an increase in hepatic glucose production (Figure 3) [111,112].
Infusion of insulin or insulin mimetic peptides in the third ventricle suppresses liver glucose production, while central suppression of insulin signaling decreases the ability of circulating glucose to inhibit glucose production. This effect of insulin on neurons was shown to be mediated by neural KATP channel activation which can be inhibited by tolbutamide [112].
Hypothalamic KATP channels are activated by insulin and can influence hepatic glucose production by diminishing the effects of 6P-glucose and phosphoenol pyruvate carboxylase that are mediated by the autonomous nervous system [113]. It is not clear yet how environment and genetic background can affect these neuronal pathways.
Finally, another insulin resistance type has been identified at the central level in obese subjects and individuals with the G972R polymorphism in IRS-1, which is associated with T2DM and with peripheral insulin resistance [84]. It is possible that brain insulin resistance could be the initial trigger for the development of T2DM and obesity, rather than a consequence [114].
7. Conclusions
KATP channels play a very important role in energetic regulation, not only in pancreatic beta cells but also in all insulin target tissues. Alterations in the correct functioning of these channels lead to pathologies such as diabetes and obesity, both of which have a metabolic outset and can originate from an increase in peripheral and central insulin resistance.
There are diverse mechanisms that lead to insulin resistance, one of which involves KATP channels and their control of the insulin pathway in different tissues.
In peripheral tissues, the activation of extra-pancreatic KATP channels decreases membrane glucose receptor expression and glucose recapture and leads to RI. The opposite occurs when these channels are inhibited. In lipidic tissue, the inhibition of KATP channels promotes fatty acid synthesis, and their activation promotes lipolysis. High fatty acid levels lead to insulin resistance in fat and other tissues. An increase in circulating free fatty acids also causes an increase in intracellular esterified fatty acids, increasing their interaction and the activation of KATP channels leading to insulin resistance.
In central tissues, KATP channels are activated by insulin, and the disruption of this pathway (by IR or PI3K inhibition) leads to peripheral or central insulin resistance; therefore, KATP channel activation protects against insulin resistance, while KATP channel inhibition promotes it.
Gain- and loss-of-function mutations in KATP channels have been associated with pathologies like hyperinsulinemia and different types of diabetes. The first manifestations of these pathologies, such as IR, are not commonly studied. It would be interesting to investigate whether there is a genetic background in the KATP channel genes that could help us to more precisely explain these channels’ role in insulin resistance.
For example, mutations that lead to an increase in the open probability of extra-pancreatic channels at the Kir6.2 level could represent an important component for predisposition to insulin resistance. Meanwhile, mutations leading to a minor open probability can have a protector effect in peripheral tissues, with the opposite effect expected in central tissues. Thus, it is important to view KATP channel research from this perspective, such as in the case of obesity and T2DM, in order to discover better prevention tools rather than treatments for these pathologies.
Author Contributions
Conceptualization, investigation, resources, writing—original draft preparation, supervision, and funding, N.S.R-R.; writing—review and editing, visualization, and supervision, D.B.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC was funded by Universidad Nacional Autónoma de México DGAPA-PAPIIT IA207723 project.
Acknowledgments
We would like to acknowledge Dr. Tanya Plett-Torres (PECEM, Facultad de Medicina UNAM) and Dr. Barbara Nova-Franco for providing language help, writing assistance, and proofreading of this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kulbacka, J.; Choromanska, A.; Rossowska, J.; Wezgowiec, J.; Saczko, J.; Rols, M.P. Cell Membrane Transport Mechanisms: Ion Channels and Electrical Properties of Cell Membranes. Adv Anat Embryol Cell Biol 2017, 227, 39–58. [Google Scholar] [CrossRef]
- Cerrone, M.; Napolitano, C.; Priori, S.G. Genetics of Ion-Channel Disorders. Curr Opin Cardiol 2012, 27, 242–252. [Google Scholar] [CrossRef]
- Coates, L. Ion Permeation in Potassium Ion Channels. Acta Crystallogr D Struct Biol 2020, 76, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of Potassium Channels. Cell Mol Life Sci 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Salkoff, L.; Butler, A.; Ferreira, G.; Santi, C.; Wei, A. High-Conductance Potassium Channels of the SLO Family. Nat Rev Neurosci 2006, 7, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Stocker, M. Ca(2+)-Activated K+ Channels: Molecular Determinants and Function of the SK Family. Nat Rev Neurosci 2004, 5, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. 2014. [CrossRef]
- Kanters, J.K.; Fanoe, S.; Larsen, L.A.; Bloch Thomsen, P.E.; Toft, E.; Christiansen, M. T Wave Morphology Analysis Distinguishes between KvLQT1 and HERG Mutations in Long QT Syndrome. Heart Rhythm 2004, 1, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Li, D.; Edwards, N.; Hynes, A.M.; Wood, K.; Al-Hamed, M.; Wroe, A.C.; Reaich, D.; Moochhala, S.H.; Welling, P.A.; et al. Identification of Compound Heterozygous KCNJ1 Mutations (Encoding ROMK) in a Kindred with Bartter’s Syndrome and a Functional Analysis of Their Pathogenicity. Physiol Rep 2013, 1, e00160. [Google Scholar] [CrossRef]
- Sakmann, B.; Trube, G. Conductance Properties of Single Inwardly Rectifying Potassium Channels in Ventricular Cells from Guinea-Pig Heart. J Physiol 1984, 347, 641–657. [Google Scholar] [CrossRef]
- Inagaki, N.; Gonoi, T.; Iv, J.P.C.; Wang, C.-Z.; Aguilar-Bryan, L.; Bryan, J.; Seino, S. A Family of Sulfonylurea Receptors Determines the Pharmacological Properties of ATP-Sensitive K+ Channels. Neuron 1996, 16, 1011–1017. [Google Scholar] [CrossRef]
- Park, S.; Terzic, A. Quaternary Structure of KATP Channel SUR2A Nucleotide Binding Domains Resolved by Synchrotron Radiation X-Ray Scattering. J Struct Biol 2010, 169, 243–251. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P.; Boyd, A.E.; González, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; Nelson, D.A. Cloning of the Beta Cell High-Affinity Sulfonylurea Receptor: A Regulator of Insulin Secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Simon, M.C.; Le Beau, M.M.; Burant, C.F. Cloning, Tissue Expression, and Chromosomal Localization of SUR2, the Putative Drug-Binding Subunit of Cardiac, Skeletal Muscle, and Vascular KATP Channels. Diabetes 1996, 45, 1439–1445. [Google Scholar] [CrossRef]
- Hund, T.J.; Mohler, P.J. Differential Roles for SUR Subunits in KATP Channel Membrane Targeting and Regulation. Am J Physiol Heart Circ Physiol 2011, 300. [Google Scholar] [CrossRef]
- Li, L.; Shi, Y.; Wang, X.; Shi, W.; Jiang, C. Single Nucleotide Polymorphisms in K(ATP) Channels: Muscular Impact on Type 2 Diabetes. Diabetes 2005, 54, 1592–1597. [Google Scholar] [CrossRef]
- Sakura, H.; Ammala, C.; Smith, P.A.; Gribble, F.M.; Ashcroft, F.M. Cloning and Functional Expression of the CDNA Encoding a Novel ATP-Sensitive Potassium Channel Subunit Expressed in Pancreatic Beta-Cells, Brain, Heart and Skeletal Muscle. FEBS Lett 1995, 377, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Iida, A.; Sekine, A.; Miura, Y.; Ogawa, C.; Kawauchi, S.; Higuchi, S.; Nakamura, Y. Identification of 779 Genetic Variations in Eight Genes Encoding Members of the ATP-Binding Cassette, Subfamily C (ABCC/MRP/CFTR. J Hum Genet 2002, 47, 147–171. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Urciuoli, W.R.; Chatterjee, S.; Fisher, A.B.; Nehrke, K.; Brookes, P.S. KIR 6.2 Is Not the Mitochondrial KATP Channel, but Is Required for Cardioprotection by Ischemic Preconditioning. Am J Physiol Heart Circ Physiol, 2013. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol Rev 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Rodriquez de Fonseca, F.; Alvarez, E.; Chowen, J.A.; Zueco, J.A.; Gomez, R.; Eng, J.; Blázquez, E. Colocalization of Glucagon-like Peptide-1 (GLP-1) Receptors, Glucose Transporter GLUT-2, and Glucokinase MRNAs in Rat Hypothalamic Cells: Evidence for a Role of GLP-1 Receptor Agonists as an Inhibitory Signal for Food and Water Intake. J Neurochem 1996, 67, 1982–1991. [Google Scholar] [CrossRef]
- Gloyn, A.L.; Siddiqui, J.; Ellard, S. Mutations in the Genes Encoding the Pancreatic Beta-Cell KATP Channel Subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) in Diabetes Mellitus and Hyperinsulinism. Hum Mutat 2006, 27, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A.; Bokvist, K.; Arkhammar, P.; Berggren, P.O.; Rorsman, P. Delayed Rectifying and Calcium-Activated K+ Channels and Their Significance for Action Potential Repolarization in Mouse Pancreatic Beta-Cells. J Gen Physiol 1990, 95, 1041–1059. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Wheeler, M.B. Voltage-Dependent K(+) Channels in Pancreatic Beta Cells: Role, Regulation and Potential as Therapeutic Targets. Diabetologia 2003, 46, 1046–1062. [Google Scholar] [CrossRef]
- Dukes, I.D.; Philipson, L.H. K+ Channels: Generating Excitement in Pancreatic Beta-Cells. Diabetes 1996, 45, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Rosário, L.M.; Barbosa, R.M.; Antunes, C.M.; Silva, A.M.; Abrunhosa, A.J.; Santos, R.M. Bursting Electrical Activity in Pancreatic Beta-Cells: Evidence That the Channel Underlying the Burst Is Sensitive to Ca2+ Influx through L-Type Ca2+ Channels. Pflugers Arch 1993, 424, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.G.; James, R.F.; Dunne, M.J.; Sharp, G.W. Glucose Activates Both K(ATP) Channel-Dependent and K(ATP) Channel-Independent Signaling Pathways in Human Islets. Diabetes 1998, 47, 758–763. [Google Scholar] [CrossRef]
- Sato, Y.; Anello, M.; Henquin, J.C. Glucose Regulation of Insulin Secretion Independent of the Opening or Closure of Adenosine Triphosphate-Sensitive K+ Channels in Beta Cells. Endocrinology 1999, 140, 2252–2257. [Google Scholar] [CrossRef]
- Straub, S.G.; Sharp, G.W. Glucose-Stimulated Signaling Pathways in Biphasic Insulin Secretion. Diabetes Metab Res Rev 2002, 18, 451–463. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Bryan, J. Molecular Biology of Adenosine Triphosphate-Sensitive Potassium Channels. Endocr Rev 1999, 20, 101–135. [Google Scholar] [CrossRef]
- D’hahan, N.; Moreau, C.; Prost, A.L.; Jacquet, H.; Alekseev, A.E.; Terzic, A.; Vivaudou, M. Pharmacological Plasticity of Cardiac ATP-Sensitive Potassium Channels toward Diazoxide Revealed by ADP. Proc Natl Acad Sci U S A 1999, 96, 12162–12167. [Google Scholar] [CrossRef]
- Gribble, F.M.; Tucker, S.J.; Seino, S.; Ashcroft, F.M. Tissue Specificity of Sulfonylureas: Studies on Cloned Cardiac and Beta-Cell K(ATP) Channels. Diabetes 1998, 47, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.; Aziz, Q.; Li, Y.; Specterman, M. ATP-Sensitive Potassium Channels and Their Physiological and Pathophysiological Roles. Compr Physiol 2018, 8, 1463–1511. [Google Scholar] [CrossRef]
- Veeraraghavan, R.; Larsen, A.P.; Torres, N.S.; Grunnet, M.; Poelzing, S. Potassium Channel Activators Differentially Modulate the Effect of Sodium Channel Blockade on Cardiac Conduction. Acta Physiol (Oxf) 2013, 207, 280–289. [Google Scholar] [CrossRef]
- Nichols, C.G.; Makhina, E.N.; Pearson, W.L.; Sha, Q.; Lopatin, A.N. Inward Rectification and Implications for Cardiac Excitability. Circ Res 1996, 78, 1–7. [Google Scholar] [CrossRef]
- Ashford, M.L.; Bond, C.T.; Blair, T.A.; Adelman, J.P. Cloning and Functional Expression of a Rat Heart KATP Channel. Nature 1994, 370, 456–459. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose Induces Closure of Single Potassium Channels in Isolated Rat Pancreatic Beta-Cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef]
- Gribble, F.M.; Proks, P.; Corkey, B.E.; Ashcroft, F.M. Mechanism of Cloned ATP-Sensitive Potassium Channel Activation by Oleoyl-CoA. J Biol Chem 1998, 273, 26383–26387. [Google Scholar] [CrossRef]
- Dabrowski, M.; Tarasov, A.; Ashcroft, F.M. Mapping the Architecture of the ATP-Binding Site of the KATP Channel Subunit Kir6.2. J Physiol 2004, 557, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Inagaki, N.; Seino, S. MgADP Antagonism to Mg2+-Independent ATP Binding of the Sulfonylurea Receptor SUR1. J Biol Chem 1997, 272, 22983–22986. [Google Scholar] [CrossRef] [PubMed]
- Kozak, J.A.; Logothetis, D.E. A Calcium-Dependent Chloride Current in Insulin-Secreting Beta TC-3 Cells. Pflugers Arch 1997, 433, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, W.F.; Satin, L.S.; Cook, D.L. Inactivation Kinetics and Pharmacology Distinguish Two Calcium Currents in Mouse Pancreatic B-Cells. J Membr Biol 1991, 119, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.L.; Hales, C.N. Intracellular ATP Directly Blocks K+ Channels in Pancreatic B-Cells. Nature 1984, 311, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Findlay, I. Effects of ADP upon the ATP-Sensitive K+ Channel in Rat Ventricular Myocytes. J Membr Biol 1988, 101, 83–92. [Google Scholar] [CrossRef]
- Furukawa, T.; Yamane, T.; Terai, T.; Katayama, Y.; Hiraoka, M. Functional Linkage of the Cardiac ATP-Sensitive K+ Channel to the Actin Cytoskeleton. Pflugers Arch 1996, 431, 504–512. [Google Scholar] [CrossRef]
- Hilgemann, D.W.; Ball, R. Regulation of Cardiac Na+,Ca2+ Exchange and KATP Potassium Channels by PIP2. Science 1996, 273, 956–959. [Google Scholar] [CrossRef]
- Fan, Z.; Makielski, J.C. Anionic Phospholipids Activate ATP-Sensitive Potassium Channels. J Biol Chem 1997, 272, 5388–5395. [Google Scholar] [CrossRef]
- Ribalet, B.; John, S.A.; Weiss, J.N. Regulation of Cloned ATP-Sensitive K Channels by Phosphorylation, MgADP, and Phosphatidylinositol Bisphosphate (PIP(2)): A Study of Channel Rundown and Reactivation. J Gen Physiol 2000, 116, 391–410. [Google Scholar] [CrossRef]
- Inagaki, N.; Gonoi, T.; Clement, J.P.; Namba, N.; Inazawa, J.; Gonzalez, G.; Aguilar-Bryan, L.; Seino, S.; Bryan, J. Reconstitution of IKATP: An Inward Rectifier Subunit plus the Sulfonylurea Receptor. Science 1995, 270, 1166–1170. [Google Scholar] [CrossRef]
- Okuyama, Y.; Yamada, M.; Kondo, C.; Satoh, E.; Isomoto, S.; Shindo, T.; Horio, Y.; Kitakaze, M.; Hori, M.; Kurachi, Y. The Effects of Nucleotides and Potassium Channel Openers on the SUR2A/Kir6.2 Complex K+ Channel Expressed in a Mammalian Cell Line, HEK293T Cells. Pflugers Arch 1998, 435, 595–603. [Google Scholar] [CrossRef]
- Gerbitz, K.D.; Gempel, K.; Brdiczka, D. Mitochondria and Diabetes. Genetic, Biochemical, and Clinical Implications of the Cellular Energy Circuit. Diabetes 1996, 45, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Kakei, M. ATP-Sensitive K+ Channels in Rat Pancreatic Beta-Cells: Modulation by ATP and Mg2+ Ions. J Physiol 1989, 416, 349–367. [Google Scholar] [CrossRef]
- Findlay, I. ATP4- and ATP.Mg Inhibit the ATP-Sensitive K+ Channel of Rat Ventricular Myocytes. Pflugers Arch 1988, 412, 37–41. [Google Scholar] [CrossRef]
- Nichols, C.G.; Shyng, S.L.; Nestorowicz, A.; Glaser, B.; Clement, J.P.; Gonzalez, G.; Aguilar-Bryan, L.; Permutt, M.A.; Bryan, J. Adenosine Diphosphate as an Intracellular Regulator of Insulin Secretion. Science 1996, 272, 1785–1787. [Google Scholar] [CrossRef]
- Nichols, C.G. KATP Channels as Molecular Sensors of Cellular Metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, WilliamF; Fatherazi, S.; Peter-Riesch, B.; Corkey, BarbaraE.; Cook, DanielL. Two Sites for Adenine-Nucleotide Regulation of ATP-Sensitive Potassium Channels in Mouse Pancreatic ?-Cells and HIT Cells. J Membr Biol 1992, 129, 287–295. [CrossRef]
- Tucker, S.J.; Gribble, F.M.; Zhao, C.; Trapp, S.; Ashcroft, F.M. Truncation of Kir6.2 Produces ATP-Sensitive K+ Channels in the Absence of the Sulphonylurea Receptor. Nature 1997, 387, 179–183. [Google Scholar] [CrossRef]
- Bushman, J.D.; Zhou, Q.; Shyng, S.L. A Kir6.2 Pore Mutation Causes Inactivation of ATP-Sensitive Potassium Channels by Disrupting PIP2-Dependent Gating. PLoS One 2013, 8, e63733. [Google Scholar] [CrossRef] [PubMed]
- Ribalet, B.; John, S.A.; Xie, L.H.; Weiss, J.N. ATP-Sensitive K+ Channels: Regulation of Bursting by the Sulphonylurea Receptor, PIP2 and Regions of Kir6.2. J Physiol 2006, 571, 303–317. [Google Scholar] [CrossRef]
- Shyng, S.; Ferrigni, T.; Nichols, C.G. Regulation of KATP Channel Activity by Diazoxide and MgADP. Distinct Functions of the Two Nucleotide Binding Folds of the Sulfonylurea Receptor. J Gen Physiol 1997, 110, 643–654. [Google Scholar] [CrossRef]
- Shyng, S.L.; Cukras, C.A.; Harwood, J.; Nichols, C.G. Structural Determinants of Pip2 Regulation of Inward Rectifier KATP Channels. J Gen Physiol 2000, 116, 599. [Google Scholar] [CrossRef]
- Reimann, F.; Tucker, S.J.; Proks, P.; Ashcroft, F.M. Involvement of the N-Terminus of Kir6.2 in Coupling to the Sulphonylurea Receptor. J Physiol 1999, 518 Pt 2, 325–336. [Google Scholar] [CrossRef]
- Shyng, S.L.; Nichols, C.G. Membrane Phospholipid Control of Nucleotide Sensitivity of KATP Channels. Science 1998, 282, 1138–1141. [Google Scholar] [CrossRef]
- Shyng, S.L.; Cukras, C.A.; Harwood, J.; Nichols, C.G. Structural Determinants of PIP(2) Regulation of Inward Rectifier K(ATP) Channels. J Gen Physiol 2000, 116, 599–608. [Google Scholar] [CrossRef]
- Pipatpolkai, T.; Usher, S.G.; Vedovato, N.; Ashcroft, F.M.; Stansfeld, P.J. The Dynamic Interplay of PIP2 and ATP in the Regulation of the KATP Channel. J Physiol 2022, 600, 4503–4519. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Clauin, S.; Bellanne-Chantelot, C.; de Lonlay, P.; Harries, L.W.; Gloyn, A.L.; Ellard, S. Update of Mutations in the Genes Encoding the Pancreatic Beta-Cell K(ATP) Channel Subunits Kir6.2 (KCNJ11) and Sulfonylurea Receptor 1 (ABCC8) in Diabetes Mellitus and Hyperinsulinism. Hum Mutat 2009, 30, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G.; York, N.W.; Remedi, M.S. ATP-Sensitive Potassium Channels in Hyperinsulinism and Type 2 Diabetes: Inconvenient Paradox or New Paradigm? Diabetes 2022, 71, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Nestorowicz, A.; Wilson, B.A.; Schoor, K.P.; Inoue, H.; Glaser, B.; Landau, H.; Stanley, C.A.; Thornton, P.S.; Clement, J.P.; Bryan, J.; et al. Mutations in the Sulonylurea Receptor Gene Are Associated with Familial Hyperinsulinism in Ashkenazi Jews. Hum Mol Genet 1996, 5, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Glamoclija, U.; Jevric-Causevic, A. Genetic Polymorphisms in Diabetes: Influence on Therapy with Oral Antidiabetics. Acta Pharm 2010, 60, 387–406. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Nicolson, T.J.; Riveline, J.P.; Taneja, T.K.; Baldwin, S.A.; Baldwin, J.M.; Charpentier, G.; Gautier, J.F.; Froguel, P.; Vaxillaire, M.; et al. A Rare Mutation in ABCC8/SUR1 Leading to Altered ATP-Sensitive K+ Channel Activity and Beta-Cell Glucose Sensing Is Associated with Type 2 Diabetes in Adults. Diabetes 2008, 57, 1595–1604. [Google Scholar] [CrossRef]
- Bonfanti, D.H.; Alcazar, L.P.; Arakaki, P.A.; Martins, L.T.; Agustini, B.C.; de Moraes Rego, F.G.; Frigeri, H.R. ATP-Dependent Potassium Channels and Type 2 Diabetes Mellitus. Clin Biochem 2015, 48, 476–482. [Google Scholar] [CrossRef]
- Martin, G.M.; Sung, M.W.; Shyng, S.L. Pharmacological Chaperones of ATP-Sensitive Potassium Channels: Mechanistic Insight from CryoEM Structures. Mol Cell Endocrinol 2020, 502, 110667. [Google Scholar] [CrossRef]
- Ashcroft, F.M. ATP-Sensitive Potassium Channelopathies: Focus on Insulin Secretion. J Clin Invest 2005, 115, 2047–2058. [Google Scholar] [CrossRef]
- Sato, M.; Ozawa, T.; Yoshida, T.; Umezawa, Y. A Fluorescent Indicator for Tyrosine Phosphorylation-Based Insulin Signaling Pathways. Anal Chem 1999, 71, 3948–3954. [Google Scholar] [CrossRef]
- Gloyn, A.L.; Pearson, E.R.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.C.L.; Molnes, J.; et al. Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes. N Engl J Med 2004, 350, 1838–1849. [Google Scholar] [CrossRef]
- Cooper, D.R.; Vila, M.C.; Watson, J.E.; Nair, G.; Pollet, R.J.; Standaert, M.; Farese, R. V Sulfonylurea-Stimulated Glucose Transport Association with Diacylglycerollike Activation of Protein Kinase C in BC3H1 Myocytes. Diabetes 1990, 39, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.J.; Standaert, M.L.; Pollet, R.J. Direct Effects of Sulfonylurea Agents on Glucose Transport in the BC3H-1 Myocyte. Diabetes 1987, 36, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Maloff, B.L.; Lockwood, D.H. In Vitro Effects of a Sulfonylurea on Insulin Action in Adipocytes. Potentiation of Insulin-Stimulated Hexose Transport. J Clin Invest 1981, 68, 85–90. [Google Scholar] [CrossRef]
- Wang, P.H.; Moller, D.; Flier, J.S.; Nayak, R.C.; Smith, R.J. Coordinate Regulation of Glucose Transporter Function, Number, and Gene Expression by Insulin and Sulfonylureas in L6 Rat Skeletal Muscle Cells. J Clin Invest 1989, 84, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Barrett-Jolley, R.; Davies, N.W. Kinetic Analysis of the Inhibitory Effect of Glibenclamide on KATP Channels of Mammalian Skeletal Muscle. J Membr Biol 1997, 155, 257–262. [Google Scholar] [CrossRef]
- Kramer, J.H.; Lampson, W.G.; Schaffer, S.W. Effect of Tolbutamide on Myocardial Energy Metabolism. Am J Physiol 1983, 245, H313-9. [Google Scholar] [CrossRef]
- Daniels, E.L.; Lewis, S.B. Acute Tolbutamide Administration Alone or Combined with Insulin Enhances Glucose Uptake in the Perfused Rat Hindlimb. Endocrinology 1982, 110, 1840–1842. [Google Scholar] [CrossRef]
- Tsiani, E.; Ramlal, T.; Leiter, L.A.; Klip, A.; Fantus, I.G. Stimulation of Glucose Uptake and Increased Plasma Membrane Content of Glucose Transporters in L6 Skeletal Muscle Cells by the Sulfonylureas Gliclazide and Glyburide. Endocrinology 1995, 136, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Pulido, N.; Casla, A.; Suárez, A.; Casanova, B.; Arrieta, F.J.; Rovira, A. Sulphonylurea Stimulates Glucose Uptake in Rats through an ATP-Sensitive K+ Channel Dependent Mechanism. Diabetologia 1996, 39, 22–27. [Google Scholar] [CrossRef]
- Miki, T.; Nagashima, K.; Tashiro, F.; Kotake, K.; Yoshitomi, H.; Tamamoto, A.; Gonoi, T.; Iwanaga, T.; Miyazaki, J.; Seino, S. Defective Insulin Secretion and Enhanced Insulin Action in KATP Channel-Deficient Mice. Proc Natl Acad Sci U S A 1998, 95, 10402–10406. [Google Scholar] [CrossRef]
- Wasada, T. Adenosine Triphosphate-Sensitive Potassium (K(ATP)) Channel Activity Is Coupled with Insulin Resistance in Obesity and Type 2 Diabetes Mellitus. Intern Med 2002, 41, 84–90. [Google Scholar] [CrossRef]
- Hansen, L.; Echwald, S.M.; Hansen, T.; Urhammer, S.A.; Clausen, J.O.; Pedersen, O. Amino Acid Polymorphisms in the ATP-Regulatable Inward Rectifier Kir6.2 and Their Relationships to Glucose- and Tolbutamide-Induced Insulin Secretion, the Insulin Sensitivity Index, and NIDDM. Diabetes 1997, 46, 508–512. [Google Scholar] [CrossRef]
- Wasada, T.; Watanabe, C.; Nakagami, T.; Iwamoto, Y. Adenosine Triphosphate-Sensitive Potassium Channels Are Involved in Insulin-Mediated Glucose Transport in Humans. Metabolism 1999, 48, 432–436. [Google Scholar] [CrossRef]
- Wasada, T.; Yano, T.; Ohta, M.; Yui, N.; Iwamoto, Y. ATP-Sensitive Potassium Channels Modulate Glucose Transport in Cultured Human Skeletal Muscle Cells. Endocr J 2001, 48, 369–375. [Google Scholar] [CrossRef]
- Linde, C.; Löffler, C.; Quast, U. Inhibition by Protein Kinase C of the 86Rb+ Efflux and Vasorelaxation Induced by P1075, a K(ATP) Channel Opener, in Rat Isolated Aorta. Naunyn Schmiedebergs Arch Pharmacol 1997, 356, 425–432. [Google Scholar] [CrossRef]
- Standridge, M.; Alemzadeh, R.; Zemel, M.; Koontz, J.; Moustaid-Moussa, N. Diazoxide Down-Regulates Leptin and Lipid Metabolizing Enzymes in Adipose Tissue of Zucker Rats. FASEB J 2000, 14, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moustaid-Moussa, N.; Wilkison, W.O.; Zemel, M.B. Role of the Sulfonylurea Receptor in Regulating Human Adipocyte Metabolism. FASEB J 1999, 13, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered Fat Storage and Mobilization in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Endocr Rev 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Koyama, K.; Chen, G.; Lee, Y.; Unger, R.H. Tissue Triglycerides, Insulin Resistance, and Insulin Production: Implications for Hyperinsulinemia of Obesity. Am J Physiol 1997, 273, E708-13. [Google Scholar] [CrossRef]
- Kelley, D.E.; Mandarino, L.J. Fuel Selection in Human Skeletal Muscle in Insulin Resistance: A Reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef]
- Ordway, R.W.; Walsh, J.V.; Singer, J.J. Arachidonic Acid and Other Fatty Acids Directly Activate Potassium Channels in Smooth Muscle Cells. Science 1989, 244, 1176–1179. [Google Scholar] [CrossRef]
- Larsson, O.; Deeney, J.T.; Bränström, R.; Berggren, P.O.; Corkey, B.E. Activation of the ATP-Sensitive K+ Channel by Long Chain Acyl-CoA. A Role in Modulation of Pancreatic Beta-Cell Glucose Sensitivity. J Biol Chem 1996, 271, 10623–10626. [Google Scholar] [CrossRef]
- Levin, B.E.; Dunn-Meynell, A.A.; Routh, V.H. Brain Glucose Sensing and Body Energy Homeostasis: Role in Obesity and Diabetes. Am J Physiol 1999, 276, R1223-31. [Google Scholar] [CrossRef] [PubMed]
- Kow, L.M.; Pfaff, D.W. Actions of Feeding-Relevant Agents on Hypothalamic Glucose-Responsive Neurons in Vitro. Brain Res Bull 1985, 15, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Oomura, Y. Glucose Responding Neurons in the Nucleus Tractus Solitarius of the Rat: In Vitro Study. Brain Res 1984, 307, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Oomura, Y.; Ooyama, H.; Sugimori, M.; Nakamura, T.; Yamada, Y. Glucose Inhibition of the Glucose-Sensitive Neurone in the Rat Lateral Hypothalamus. Nature 1974, 247, 284–286. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Rawson, N.E.; Levin, B.E. Distribution and Phenotype of Neurons Containing the ATP-Sensitive K+ Channel in Rat Brain. Brain Res 1998, 814, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ashford, M.L.; Boden, P.R.; Treherne, J.M. Tolbutamide Excites Rat Glucoreceptive Ventromedial Hypothalamic Neurones by Indirect Inhibition of ATP-K+ Channels. Br J Pharmacol 1990, 101, 531–540. [Google Scholar] [CrossRef]
- Jetton, T.L.; Liang, Y.; Pettepher, C.C.; Zimmerman, E.C.; Cox, F.G.; Horvath, K.; Matschinsky, F.M.; Magnuson, M.A. Analysis of Upstream Glucokinase Promoter Activity in Transgenic Mice and Identification of Glucokinase in Rare Neuroendocrine Cells in the Brain and Gut. J Biol Chem 1994, 269, 3641–3654. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Damberg, G.S.; Tkac, I.; Gruetter, R. The Effect of Insulin on in Vivo Cerebral Glucose Concentrations and Rates of Glucose Transport/Metabolism in Humans. Diabetes 2001, 50, 2203–2209. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Figlewicz, D.P.; Baskin, D.G.; Woods, S.C.; Porte, D. Insulin in the Brain: A Hormonal Regulator of Energy Balance. Endocr Rev 1992, 13, 387–414. [Google Scholar] [CrossRef]
- Hörsch, D.; Kahn, C.R. Region-Specific MRNA Expression of Phosphatidylinositol 3-Kinase Regulatory Isoforms in the Central Nervous System of C57BL/6J Mice. J Comp Neurol 1999, 415, 105–120. [Google Scholar] [CrossRef]
- Choudhury, A.I.; Heffron, H.; Smith, M.A.; Al-Qassab, H.; Xu, A.W.; Selman, C.; Simmgen, M.; Clements, M.; Claret, M.; Maccoll, G.; et al. The Role of Insulin Receptor Substrate 2 in Hypothalamic and Beta Cell Function. J Clin Invest 2005, 115, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, U. Where Does Insulin Resistance Start? The Brain. Diabetes Care 2009, 32 Suppl 2, S174-7. [Google Scholar] [CrossRef]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing Hypothalamic Insulin Receptors Causes Hyperphagia and Insulin Resistance in Rats. Nat Neurosci 2002, 5, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic Insulin Signaling Is Required for Inhibition of Glucose Production. Nat Med 2002, 8, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) Channels Control Hepatic Glucose Production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Tschritter, O.; Preissl, H.; Hennige, A.M.; Stumvoll, M.; Porubska, K.; Frost, R.; Marx, H.; Klosel, B.; Lutzenberger, W.; Birbaumer, N.; et al. The Cerebrocortical Response to Hyperinsulinemia Is Reduced in Overweight Humans: A Magnetoencephalographic Study. Proc Natl Acad Sci U S A 2006, 103, 12103–12108. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A) Schematic representation of SURx with Kir6.x subunit showing the most studied Kir domains, the pore region (M1 and M2), the potassium sensor region or selective filter (SF), the ATP-binding site, and the carboxyl end where the membrane transport signal is located. B) Schematic representation of pancreatic beta KATP channel signaling. When glucose enters the cell (1), its metabolism increases the ATP concentration (2); ATP blocks KATP channels (3), causing membrane depolarization (4) that leads to the opening of voltage-dependent Ca2+ channels (5). The increase in the intracellular Ca2+ concentration triggers the release of insulin-containing vesicles (6). (Modified from Aguilar-Bryan and Bryan, 1999, and Flanagan et al., 2009.).
Figure 1.
A) Schematic representation of SURx with Kir6.x subunit showing the most studied Kir domains, the pore region (M1 and M2), the potassium sensor region or selective filter (SF), the ATP-binding site, and the carboxyl end where the membrane transport signal is located. B) Schematic representation of pancreatic beta KATP channel signaling. When glucose enters the cell (1), its metabolism increases the ATP concentration (2); ATP blocks KATP channels (3), causing membrane depolarization (4) that leads to the opening of voltage-dependent Ca2+ channels (5). The increase in the intracellular Ca2+ concentration triggers the release of insulin-containing vesicles (6). (Modified from Aguilar-Bryan and Bryan, 1999, and Flanagan et al., 2009.).
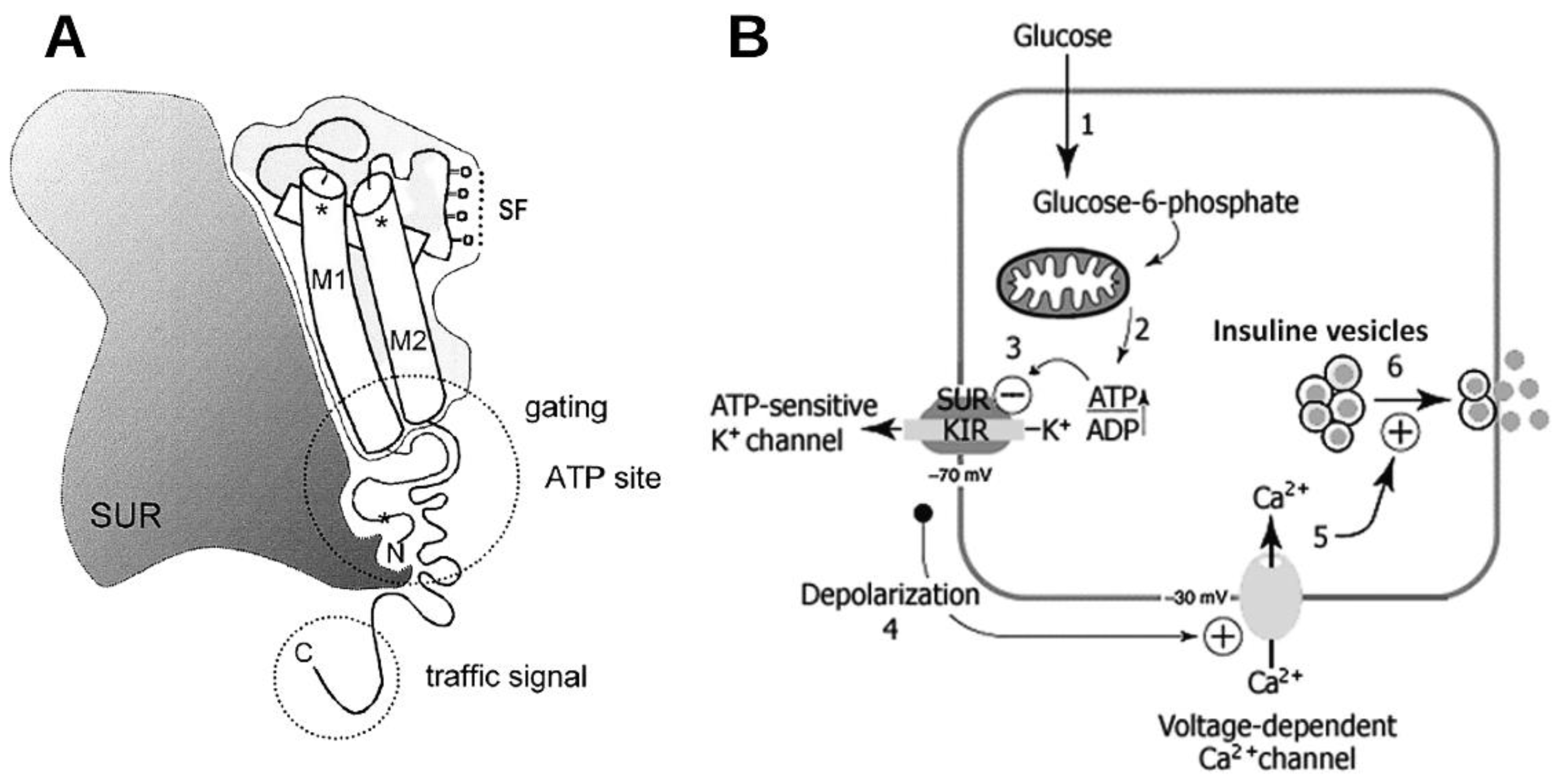
Figure 2.
KATP channels and insulin resistance in peripheral tissues. A) In skeletal and muscle tissue KATP channel activation may be related to insulin resistance and a decrease in glucose recapture under high insulin and glucose conditions. The molecular mechanisms involved have yet to be elucidated. B) In contrast, in adipocytes, sustained KATP channel inhibition promotes fatty acid synthesis which may lead to obesity and insulin resistance.
Figure 2.
KATP channels and insulin resistance in peripheral tissues. A) In skeletal and muscle tissue KATP channel activation may be related to insulin resistance and a decrease in glucose recapture under high insulin and glucose conditions. The molecular mechanisms involved have yet to be elucidated. B) In contrast, in adipocytes, sustained KATP channel inhibition promotes fatty acid synthesis which may lead to obesity and insulin resistance.
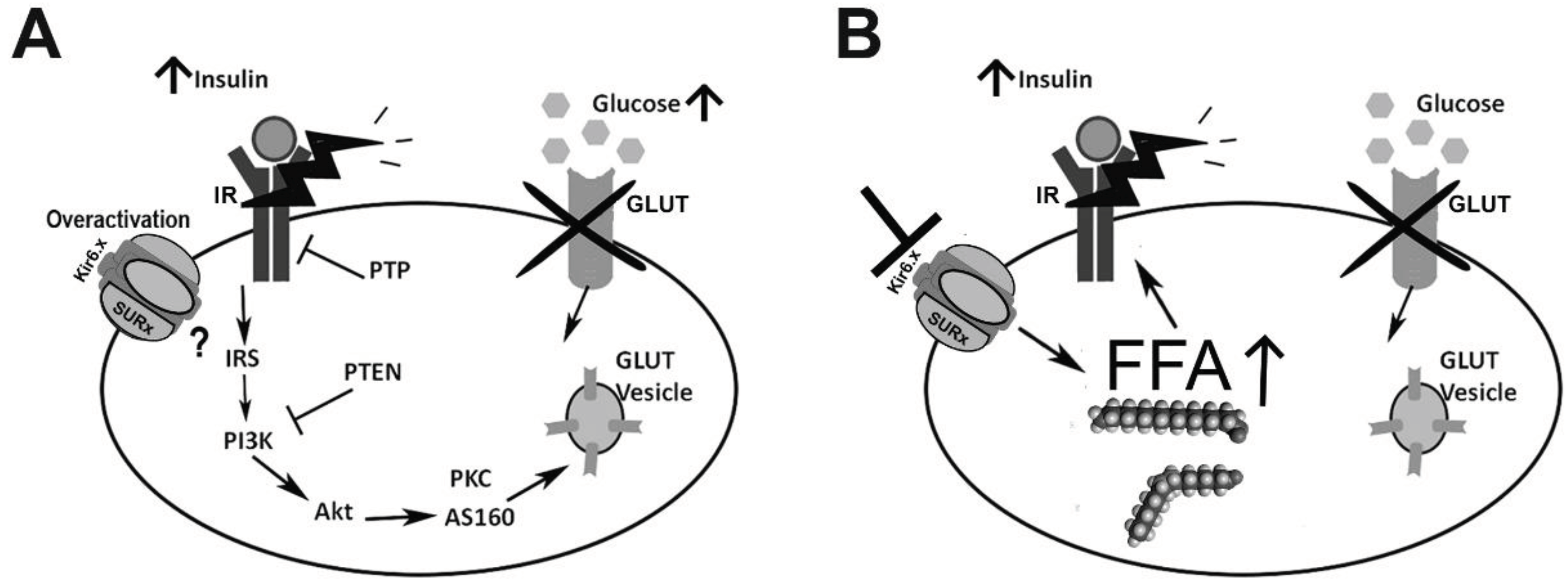
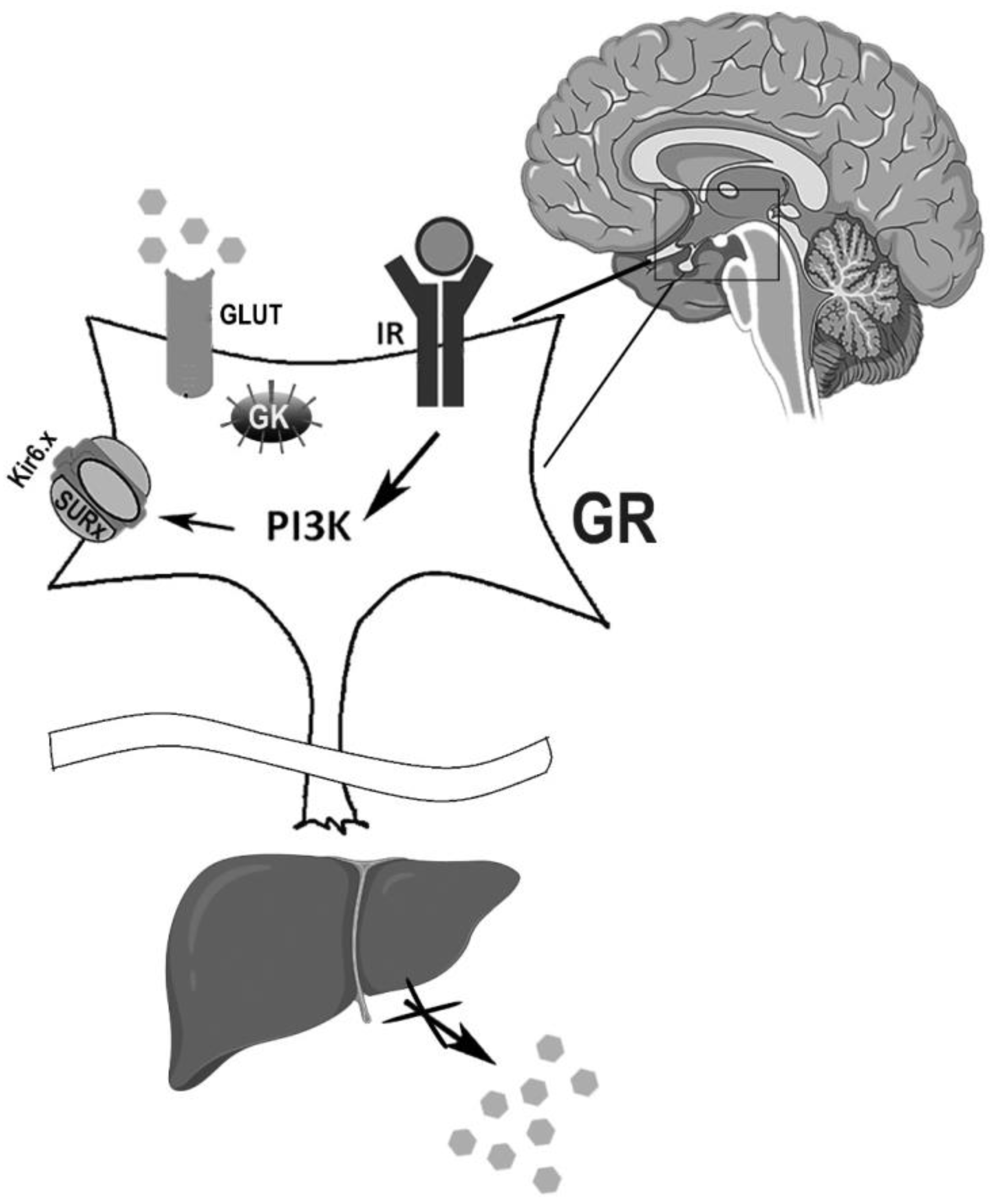
Figure 3.
Insulin-mediated KATP effects on the brain. In glucose-responsive (GR) neurons, KATP channels regulate the neurons’ firing rate and insulin regulates the KATP channel electrical activity. KATP channels act as glucose sensors in glucose transporter (GLUT)- or glucokinase (GK)-containing neurons, preventing insulin resistance and glucose intolerance. At the peripheral level, brain insulin regulation of KATP activity may prevent an increase in hepatic glucose production and protect against insulin resistance. IR: insulin receptor; PI3K: phosphatidylinositol 3-kinase. Gray hexagons represent glucose molecules.
Figure 3.
Insulin-mediated KATP effects on the brain. In glucose-responsive (GR) neurons, KATP channels regulate the neurons’ firing rate and insulin regulates the KATP channel electrical activity. KATP channels act as glucose sensors in glucose transporter (GLUT)- or glucokinase (GK)-containing neurons, preventing insulin resistance and glucose intolerance. At the peripheral level, brain insulin regulation of KATP activity may prevent an increase in hepatic glucose production and protect against insulin resistance. IR: insulin receptor; PI3K: phosphatidylinositol 3-kinase. Gray hexagons represent glucose molecules.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



