
Submitted:
08 March 2024
Posted:
11 March 2024
You are already at the latest version
Abstract
The influenza virus has been the primary cause of the pandemic, posing a constant threat to human society. Due to its genetic evolution and continuous outbreak, antiviral research currently focuses on exploring a novel lead agent. A comprehensive antiviral screening discovered a marine bacterium whose extract exerted excellent efficacy against influenza viruses. Parerythrobacter sp. M20A3S10, a novel strain under the family Erythrobacteraceae, could produce carotenoids exhibiting antiviral and anticancer activity by enhancing the cellular immune system. Post-treatment of M20A3S10 extract showed outstanding therapeutic indexes: against influenza virus A/PR8 (H1N1) [selectivity index (SI) = 24.0], A/Wisconsin/15/2009 (H3N2) (SI = 30.1) and B/Florida/78/2015 (SI = 38.2). Comparably, the effectiveness was demonstrated against Zika virus (ZIKV) and dengue virus type 2 (DENV2) with an SI of 22.5 and 24.1, respectively, namely broad-spectrum activity. Of note, the antiviral responses resulted from the common replication mechanism between IAV, ZIKV, and DENV2. The stimulation of apoptosis-mediated cellular immunity prevented the viral release and protected the host, suggesting that switching from necroptosis to apoptosis is a novel antiviral target. Although the specific compound affecting the antiviral activity was not identified, its promising efficacy with broad activity will contribute to developing a strategy for preventing future pandemics.
Keywords:
marine bacterium
; influenza virus
; Zika virus
; dengue virus
; apoptosis
; broad antiviral activity
1. Introduction
Infectious pathogens, comprising viruses, bacteria, and protozoa, have had a profound impact on global public health and the economy. Among these pathogens, the influenza A virus (IAV) poses a persistent threat to various hosts, highlighting the significance of its pandemic potential and prompting international surveillance networks to monitor its emergence and circulation [1]. The virus's ability to rapidly adapt to multiple hosts and evade the host immune system has resulted in significant challenges in preventing and controlling outbreaks [2]. Furthermore, its remarkable genetic diversity, such as antigenic drift and shift, can potentially lead to a pandemic, which can transmit across a broad spectrum of hosts, including humans, mammals, and birds, as evidenced by historical records [3].
On the other hand, flaviviruses represent the next significant pathogen for future pandemics and cause medically relevant illnesses in various hosts. For instance, Zika virus (ZIKV) and dengue virus (DENV) pose considerable health threats in Latin America [4]. Following the ZIKV outbreak in the Americas, which resulted in a dramatic increase in microcephaly and brain malformations in newborns, ZIKV disease became an international public health emergency. Similarly, a significant increase in DENV cases has been reported in the Americas in recent decades, with an estimated 390 million infections per year [5]. While many DENV infections resolve without complications, severe dengue remains a significant cause of illness and death in some Asian and Latin American countries [4]. Therefore, the pandemic potential of problematic RNA viruses has recently attracted considerable attention in many governments and diverse industries, necessitating the development of antiviral strategies to tackle these challenges.
Numerous attempts have been made to discover antiviral compounds in natural resources with evidence of safety and strong effectiveness, which could be applied to clinical studies [6,7,8]. These trials have led to exploring various sources, such as herbs and microorganisms from fresh or seawater. Notably, many studies in marine microbe revealed that carotenoids abundantly produced by the family Erythrobacteraceae exhibit antiviral functions by inhibiting viral genome transcription, virus-induced pro-inflammatory response, or cell death regardless of RNA or DNA viruses [9,10,11,12]. Carotenoids from Qipengyuania pacifica, another member of the Erythrobacteraceae family, have also been demonstrated to have anti-bacterial capabilities, especially against bacterial infections that are resistant to a broad array of antibiotics [13]. Collectively, compounds sourced from marine bacteria have become an active area of investigation in antiviral research.
Interestingly, infectious RNA viruses, including IAV, ZIKV, and DENV, induce necroptosis, while apoptosis is a limiting factor for producing infectious virus particles [14,15]. Host immune reaction develops a formation of apoptotic bodies containing intracellular organelles, other cytosolic components, and premature and mature viral particles [16]. Subsequently, newly expressed ligands of apoptotic bodies stimulate phagocytosis, preventing the release of infection progeny and protecting the entire organism [17]. Indeed, an extract from a marine microbe exhibited tremendous in vitro antiviral activity with guaranteed safety against IAV and influenza B virus (IBV) [18]. Therefore, apoptosis-mediated host protection has been a new antiviral target, providing important insight into developing novel therapeutic strategies against RNA viruses.
The present study carried out antiviral screening using extracts of various marine bacteria. Among them is an extract from Parerythrobacter sp. M20A3S10, belonging to the family Erythrobacteraceae, which showed remarkable antiviral efficacies in vitro against influenza virus A/Puerto Rico/8/34 (H1N1) (A/PR8) [half maximal inhibitory concentration (IC50) = 51.1 µg/mL, selectivity index (SI) = 24.0], influenza virus A/Wisconsin/15/2009 (H3N2) (A/Wisconsin) (IC50 = 40.6 µg/mL, SI = 30.1), influenza virus B/Florida/78/2015 Victoria lineage (B/Florida) (IC50 = 32.1 µg/mL, SI = 38.2), ZIKV (IC50 = 69.6 µg/mL, SI = 22.5) and Dengue virus type 2 (DENV2) (IC50 = 65.1 µg/mL, SI = 24.1), namely a broad spectrum of antiviral potential.
Notably, the Parerythrobacter extract is expected to contain large amounts of carotenoids [9,10,19], which have been shown to exhibit antiviral functions by attenuating cholesterol synthesis, affecting the general replication cycle of enveloped viruses. However, the specific compound that affects apoptosis and viral replication is yet to be identified and quantified.
Nevertheless, the M20A3S10 extract suppresses viral replication by stimulating apoptosis-mediated host immunity in IAV, ZIKV, and DENV2 infections. These findings provide important insights for developing novel therapeutic strategies against enveloped RNA viruses. With further studies to identify and quantify the specific chemical within the Parerythrobacter extract, the antiviral evaluation in this study sheds light on a promising strategy for targeting broad infection by future pandemics.
2. Materials and Methods
2.1. Bacterial Isolation and Culture Conditions
Several bacterial strains, including Parasphingopyxis, Roseibium, Parerythrobacter, Neo-rhizobium, and Qipengyuania species, were collected from seawater in Seosan (Chungnam Province, KR) on March 16, 2020. The water sample was spread on ZoBell medium (0.5 g peptone, 0.1 g yeast extract, and 0.001 g ferric phosphate [FePO4] per liter of 20% distilled water and 80% filtered seawater) using the standard dilution-plating method. The inoculated plates were incubated at 25°C for 5 days, and individual colonies were streaked on marine agar 2216 for purification. The purified strains were routinely cultured on MA at 25°C and preserved with 20% (v/v) glycerol at -80°C. The bacterial isolates were deposited under the numbers MI00005948, MI00006287, MI00006290, MI00006301, MI00007026, and MI00007049, respectively, at the Microbial Marine BioBank (MMBB) of the National Marine Biodiversity Institute of Korea (MABIK).
2.2. Phylogeny of 16S rRNA Gene Sequences
The genomic DNA of the bacterial strains was extracted using an Exgene DNA extraction kit, and the 16S rRNA gene was PCR amplified using bacteria-specific universal primers 27F and 1492R [20]. The amplified partial 16S rRNA gene sequences were sequenced and assembled by Geneious program v9.0.5 to obtain a nearly full-length 16S rRNA gene sequence. The EzBioCloud server was used to identify the phylogenetic position of strain M20A3S10, and the 16S rRNA gene sequence was compared with validly published species in the server [21]. The 16S rRNA gene sequence was deposited into GenBank under the accession OR481698. Phylogenetic trees based on 1,419 unambiguously aligned sequences were reconstructed using the neighbor-joining (NJ), maximum-likelihood (ML), and maximum-parsimony (MP) algorithms in the MEGA X [22,23,24,25]. The genetic correlation among bacterial strains used in this study and reference strains was calculated using the Kimura-2 parameter model at the nucleotide level, and phylogenetic trees were constructed using the neighbor-joining method with 1000 bootstrap replicates [26].
2.3. Preparation of the Bacterial Extracts
Bacterial extracts were prepared using a modified method from Xu et al. [9]. The bacterial including Parasphingopyxis, Roseibium, Parerythrobacter, Neorhizobium, and Qipengyuania species, were cultured in 2.5 L Erlenmeyer flasks containing 1 L of marine broth under LED light at 25°C. Subsequently, 20 L of culture using a panel or column-type photobioreactor was inoculated with the initial inoculum at a concentration of 104 CFU mL-1 in the same culture condition for 10-20 days with shaking at 150 rpm. At the end of the culture period, culture broth was extracted twice with the same volume of ethyl acetate (EtOAc). The EtOAc soluble component of bacterial culture broth was combined and dried using a vacuum evaporator. A crude total of 150 mg of bacterial extract was obtained from each species. The extracts were dissolved in dimethyl sulfoxide (DMSO) for the antiviral evaluation.
2.4. Cells and Viruses
Madin-Darby canine kidney epithelium (MDCK; ATCC CCL-34) and African green monkey kidney epithelium (Vero; ATCC CRL-1586) E6 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) form WELGENE (Seoul, KR) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin(P/S) at 37°C in a 5% CO2 atmosphere. Two strains of influenza A virus (IAV) and one strain of influenza B virus (IBV) from ATCC were used: influenza virus A/Puerto Rico/8/34 (H1N1) (A/PR8), A/Wisconsin/15/2009 (H3N2) (A/Wisconsin) strain, and B/Florida/78/2015 Victoria lineage (B/Florida). For influenza virus propagation, MDCK cells were infected with influenza viruses at a multiplicity of infection (MOI) of 1 (1 MOI), supplemented with 1 µg/mL N-tosyl-L-phenylalanine chloromethyl ketone (TPCK)-treated trypsin and incubated at 37°C in a 5% CO2 atmosphere for 48 h. Both types of flavivirus were provided from the National Culture Collection for Pathogens (Cheongju, KR): Dengue virus type 2 (DENV2) isolated from serum samples from Korean patients traveling to Singapore, India, and Thailand in 2015 and Zika virus (ZIKV) strains of Asian/American lineage, PRVABC59. For the propagation of two flaviviruses, Vero E6 cells were infected with ZIKV and DENV2 (1 MOI) and incubated at 37°C in a 5% CO2 atmosphere for 48 h.
2.5. Cell Viability Assay
MDCK and Vero E6 cell viability was measured with extracts using the WST assay per the manufacturer's instructions (Abfrontier, Seoul, KR) [27]. A confluent monolayer of MDCK and Vero E6 cells was prepared in a 96-well plate at a healthy density of 5.0 x 104 cells. The extracts were prepared using a continuous dilution method with an adjusted dilution factor, resulting in the following concentrations: 1000, 500, 250, 100, 50, 25, 10, 5, 2, 1, 0.5, 0.1, 0.01 µg/mL. MDCK cells and Vero E6 cells were treated with each extract concentration using the experimental scheme outlined and incubated at 37°C in a 5% CO2 atmosphere for 48 h. The cells were then gently washed with DPBS, and 50 μL of CellVia working solution (90 μL of DMEM with 10 μL of CellVia stock solution) was added to each well. The cells were then incubated for 30 min, and absorbance was measured at 450nm with an ELISA microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) Cell viability was calculated as a percentage in comparison with DMSO-treated cells. GraphPad Prism software v9. 5. 1 (GraphPad Software, CA, USA) was used to generate nonlinear regression curves and calculate median cell cytotoxicity concentration (CC50) as described previously [28].
2.6. Antiviral Assay
To screen for antiviral activity, the above-diluted extracts (100, 50, 25, 10, 5, 2, 1, 0.5, 0.1, 0.01 µg/mL) were mixed with A/PR8 strain (0.1 MOI) supplemented with 1% P/S and 1 µg/mL of TPCK-treated trypsin at RT for 1 h. The mixtures were then absorbed in confluent MDCK cells in 96-well plates for 1 h and incubated at 37°C in a 5% CO2 atmosphere for 48 h. Cell viability was measured by the inhibition level of CPE induced by the A/PR8 strain using the MTT assay, per the manufacturer's instructions [29]. The cells were washed with DPBS and received 200 μL of MTT solution (0.4 mg/mL) in each well. After incubation at 37°C in a 5% CO2 atmosphere for 2 h, DMSO was added to dissolve formazan for 10 min at RT. Relative cell viability was then measured at 570nm.
Different treatment methods were applied to identify the mechanism of action of an extract, with inoculation with A/PR8 strain (0.1 MOI) as previously described [18]: treatment 1 h before virus infection (pre-treatment), simultaneously with virus inoculum (co-treatment), or after virus absorption (post-treatment). Infected cells were incubated for 48 h, and tissue culture supernatants were collected to measure the level of mRNA and progeny virus titers. Antiviral activity was calculated based on the cell viability of infection using an MTT assay. Nonlinear regression curves and the median inhibitory concentration (IC50) were determined as described above.
ZIKV and DENV2 were used to determine whether the extract had antiviral effects on other viruses. The serial-diluted extracts were mixed with ZIKV and DENV2 (0.1 MOI) supplemented with 1% P/S at RT for 1 h. The mixtures were then absorbed in confluent Vero E6 cells in 96-well plates for 1 h and incubated at 37°C in a 5% CO2 atmosphere for 48 h.
2.7. RT-qPCR
The quantitative RT-qPCR method was used to measure the virus in infected cells, and cell supernatants were analyzed at 48 h after treatment. Virus RNA was extracted using the QIAamp Viral RNA Mini kit (QIAGEN, Hilden, DE) per the manufacturer's guidelines [30]. PCR was performed with SensiFAST™ SYBR® Lo-ROX One-Step Kit (Bioline Meridian BioScience, Cincinnati, OH, USA). For this reaction, a mixture was assembled in a total volume of 20 μL as follows: 0.2 μL reverse transcriptase (RT), 0.4 μL Ribosafe RNase inhibitor, 0.8 μL of a primer pair (10 μM), 1X SYBR Green, 100 ng of viral RNA, and nuclease-free water. The detecting primers are as follows: forward (5’-GGCCCTTCAGTTGTTCATC-3'), and reverse primers (5’-GCAGACTTCAGGAATGTG-3') against IAV PB1, forward (5’-GGTCATGATACTGCTGATTGC-3'), and reverse primers (5’-CCACTAACGTTCTTTTGCAGAC-3') against ZIKV NS5, (5’-AGTTGTTAGTCTRYGTGGACCGAC-3'), and reverse primers (5’-TTGCACCAACAGTCAATGTCTTCAGGTTC-3') against DENV prM-. After incubation at 42°C for 30 min, PCR amplification was carried out with the following conditions: For IAV, the denaturation step (95°C, 10 s), annealing step (60°C, 10 s), and extension step (72°C, 15 s) were repeated 40 times; For ZIKV and DENV2, the denaturation step (95°C, 10s), annealing step (55°C, 15 s), and extension step (72°C, 15 s) were repeated 35 times.
2.8. Virus Titration
To confirm the progeny virus production of the supernatants, a 50% tissue culture infective dose (TCID50) was used, as described previously [16]. The supernatants were prepared in a 10-fold dilution series with DMEM and absorbed in a confluent MDCK cell for 1h at 37°C in a 5% CO2 atmosphere. At 5 days post-infection, cells were fixed with 4% neutral paraformaldehyde (4% PFA, Bio-solution, Seoul, KR), stained with 0.5% crystal violet solution, and TCID50 was determined by the Reed–Muench method.
2.9. Attachment and Penetration Assays
The attachment and penetration assays were performed as described previously [18,31]. For the attachment assay, confluent MDCK cells in a 24-well plate were treated with extracts (100 µg/mL) at 4°C before infection. After 30 minutes, the inoculum was washed thoroughly with cold DPBS. Then, the A/PR8 strain (1 MOI) was absorbed for 1h on ice. After thorough washing with cold DPBS, the cells were incubated for 20 h at 37°C in a 5% CO2 atmosphere. MDCK cells in a 24-well plate received A/PR8 strain (1 MOI) for 1 h on ice for penetration assay. After thorough washing, the cells were treated with extracts for 10 minutes and incubated for 20 hours at 37°C in a 5% CO2 atmosphere.
2.10. Cell Culture Immunofluorescence Assay
MDCK cells were prepared in an 8-well chamber to check viral protein synthesis and infected with the A/PR8 strain (0.1 MOI). Vero E6 cells were also prepared and infected with ZIKV, and DENV2 (0.1 MOI) as described elsewhere [14,32]. After fixation with 4% PFA for 10 min at RT, the chamber was washed and blocked with 5% Bovine Serum Albumin (BSA) at RT for 1 h to reduce non-specific reaction. Subsequently, a primary antibody against IAV M2, flavivirus envelope protein 4G2, was applied overnight at 4°C. The next day, a secondary antibody, goat-anti mouse Alexa Flour® 488 conjugated to a fluorescent dye, was added, and the cells were incubated for 1 h at RT. The 96-well plates were washed and analyzed using a fluorescence microscope.
2.11. Flow Cytometry
The present study utilized flow cytometry to assess the induction of viral protein and apoptosis. The methodology adopted was in accordance with previously established protocols [33]. Briefly, Madin-Darby Canine Kidney (MDCK) cells were inoculated with influenza A Virus (IAV) at 0.1 MOI and incubated at 37°C for an hour in a 5% CO2 atmosphere. Following this, a vehicle or an extract at a concentration of 50 µg/mL was added to each well, and the cells were then incubated at 37°C for 48 hours. Subsequently, the cells were washed with Dulbecco's Phosphate Buffered Saline (DPBS) and centrifuged at 3000 rpm at 4°C. This procedure resulted in the formation of a cell pellet, which was subjected to the TUNEL assay to detect apoptosis using the In Situ Cell Death Detection Kit (Roche, Basel, CH) after blocking with 5% BSA at room temperature for an hour, per the manufacturer's instructions [16]. Additionally, primary antibodies against IAV M2 and secondary antibodies conjugated with AF647 were applied to evaluate viral protein. Similarly, Vero E6 cells were inoculated with ZIKV and DENV2 at 0.1 MOI and incubated at 37°C for an hour in a 5% CO2 atmosphere. Flavivirus envelope protein 4G2 was utilized as the primary antibody against ZIKV and DENV2, and a secondary antibody conjugated with AF947 was applied. Flow cytometry was performed using the AttuneTM NxT flow cytometer (Thermo Scientific), and the AttuneTM NxT software v3.1.2 was used to digitize the data from each sample.
2.12. Statistical Analysis
Statistical analysis was carried out using GraphPad Prism software, with one-way ANOVA being employed. The data was expressed as the mean ± standard deviation (SD) of at least three independent experiments. Statistical significance was considered at the following levels: *, P < 0.05; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001. The selective index (SI) was calculated using sigmoidal dose-response curves, with the following equation: SI = mean CC50 / mean IC50. The ratio of CC50 to IC50, i.e., CC50/IC50, was employed to calculate SI.
3. Result
3.1. Phylogenetic Analyses and Antiviral Screening of Marine Bacteria
Comparative analysis of the 16S rRNA gene sequences showed that Parerythrobacter sp. M20A3S10 (accession number: OR481698) was most closely related to Pseudomonas lutipelagi GH1-16T (accession number: LT797153) with a similarity of 98.56%. Phylogenetic analysis based on 16S rRNA gene sequences using the NJ, ML, and MP algorithms showed that strain M20A3S10 formed a monophyletic clade with other members of the genus Parerythrobacter (Figure 1A). Consequently, sequence comparisons based on almost complete 16S rRNA gene sequences showed a clear affiliation of the isolate to the genus Parerythrobacter.
We further investigated the 16S rRNA gene sequences using marine bacteria which were sampled in the same batch and genetically closed to Pseudomonas sp. M20A3S10 (Figure 1B). So, antiviral screening was conducted on strain M20A3S10 and other 5 selected bacteria using extracts through pre-and post-treatment in MDCK cells. CC50 was determined by the application of the extracts in mock-infected cells, and IC50 of the extracts was measured by quantitative cytopathic effect (CPE) reduction in A/PR8 strain-infected cells (0.1 MOI) (Figure 2). As a result, the extract of Parerythrobacter sp. M20A3S10 (M20A3S10 extract) solely showed outstanding antiviral activity against A/PR8 infection (CC50 = 1223 µg/mL, IC50 = 58.4 µg/mL, SI = 20.9), which is even greater than chloroquine, an FDA-approved antiviral drug that inhibits endosomal acidification for the internalization of influenza viruses.
3.2. Antiviral Evaluation by Pre- or Co-treatment of M20A3S10 Extract
To understand the antiviral mechanism of the M20A3S10 extract, MDCK cells were treated with the extract before (pre-treatment) or with viral infection (co-treatment) (Figure S1). Antiviral activity against the A/PR8 strain was measured by the inhibition effect on viral CPE using the MTT assay. However, neither treatment detected antiviral effects at any concentrations. Accordingly, viral genome copies in the infected cells acquired at 48 h post-infection were not attenuated by the treatments.
3.3. Antiviral Evaluation by Post-Treatment of M20A3S10 Extract
Therefore, the antiviral mechanism was further studied using post-treatment of the extract (Figure 3A). As shown in Figure 2, influenza virus-induced CPE was dramatically decreased by M20A3S10 extract within no observed adverse effect levels, exhibiting a great therapeutic index (CC50 = 1223 µg/mL, IC50 = 51.1 µg/mL, SI = 24.0) (Figure 3B). In addition, IFA revealed that the treatment suppressed virus protein synthesis in a dose-dependent manner (Figure 3C). According to RT-qPCR and TCID50, a gradual antiviral activity was found toward higher concentrations, resulting in significant reductions in viral genome synthesis as well as infectious virus particles (Figure 3DE).
3.4. Apoptosis-Mediated Antiviral Response through M20A3S10 Extract
In association with the replication cycle of the influenza virus, we further investigated the mechanism of antiviral response using attachment and penetration assays (Figure 2S). As a result, MDCK cells pre-treated with M20A3S10 extract, followed by A/PR8 infection, did not exhibit any reduction in viral genome copies, and while viral inoculum pre-incubated with antibody against IAV HA significantly suppressed genome synthesis (Figure S2A). Interestingly, according to the penetration assay, virus genomes were significantly reduced by post-treatment of the extract after viral binding to MDCK cells, which was cross-checked by chloroquine treatment (Figure S2B). However, this antiviral activity is expected to continue only during early infection. It is not enough to suppress persistent infection, given that the pre-and co-treatments are not enough to curb viral infection during a long incubation period, such as 48 hours of infection. In other words, the potent antiviral activity shown in Figure 3 was highly linked to the late stage of viral replication or post-entry stages.
Apoptosis is an innate cellular defense mechanism that has a critical role in preventing the growth of intracellular microbes, commonly found in viral and bacterial infections [34]. The apoptotic signal pathway requires the activation of effector proteins such as caspases, resulting in DNA fragmentation as a hallmark of the reaction [14]. To evaluate apoptosis-mediated antiviral action by the extract, the cleavage ends of DNA fragments in the infected cells with A/PR8 strain were detected by flow cytometry using an antibody against IAV nucleoprotein (NP) and TUNEL assay (Figure 4). As a result, the post-treatment of M20A3S10 extract successfully protected the host cell by activating the cellular apoptotic pathway, compared to the A/PR8-inoculated, vehicle-treated group. Moreover, the post-treatment remarkably decreased viral replication by 97% (from 23.742% of IAV-positive cells in vehicle-treated cells to 0.810% in the extract-treated cells). This data indicated that apoptosis-mediated innate immunity is a critical antiviral mechanism for this infection.
3.5. Antiviral Activity of the M20A3S10 Extract against ZIKV and DENV2
As an enveloped virus, flavivirus shares replication mechanisms with the influenza virus, such as viral binding, entry, and fusion processes. The antiviral screening was conducted to evaluate the therapeutic efficacy of the M20A3S10 extract against ZIKV and DENV2. The results revealed a significant therapeutic index against ZIKV and DENV2 infections, with the extract exhibiting greater efficacy than chloroquine. The CC50 and IC50 values for the extract were found to be 1566 µg/mL and 696M µg/mL, respectively, with an SI of 22.5 for ZIKV and 1566 µg/mL and 65.1 µg/mL, with an SI of 24.1 for DENV2 (Figure 5).
Pre- or co-treatment of the extract did not exhibit sufficient inhibition of those viruses (data not shown). In contrast, post-treatment efficiently blocked the propagation of ZIKV and DENV2 in vitro (Figure 6). Dose-dependent inhibition was demonstrated in viral genome replication, progeny production, and protein synthesis. These findings suggest that the Parerythrobacter extract could effectively control IAV, ZIKA, and DENV2 infections.
3.6. Apoptosis-Mediated Antiviral Response in ZIKV and DENV2 Infections
Regarding the common replication mechanism, IAV, ZIKV, and DENV viruses prefer necroptosis to apoptosis to produce more progeny, severely damaging respiratory organs, neurons, and immune cells. Therefore, switching from necroptosis to apoptosis could be promising antiviral targets for broad infections. The post-treatment of the M20A3S10 extract increased apoptotic reactions and reduced viral replication (Figure 7), suggesting that apoptosis is an antiviral process limiting the replication of influenza and flavivirus.
3.7. A Broad-Spectrum Antiviral Activity against Multiple Influenza Viruses
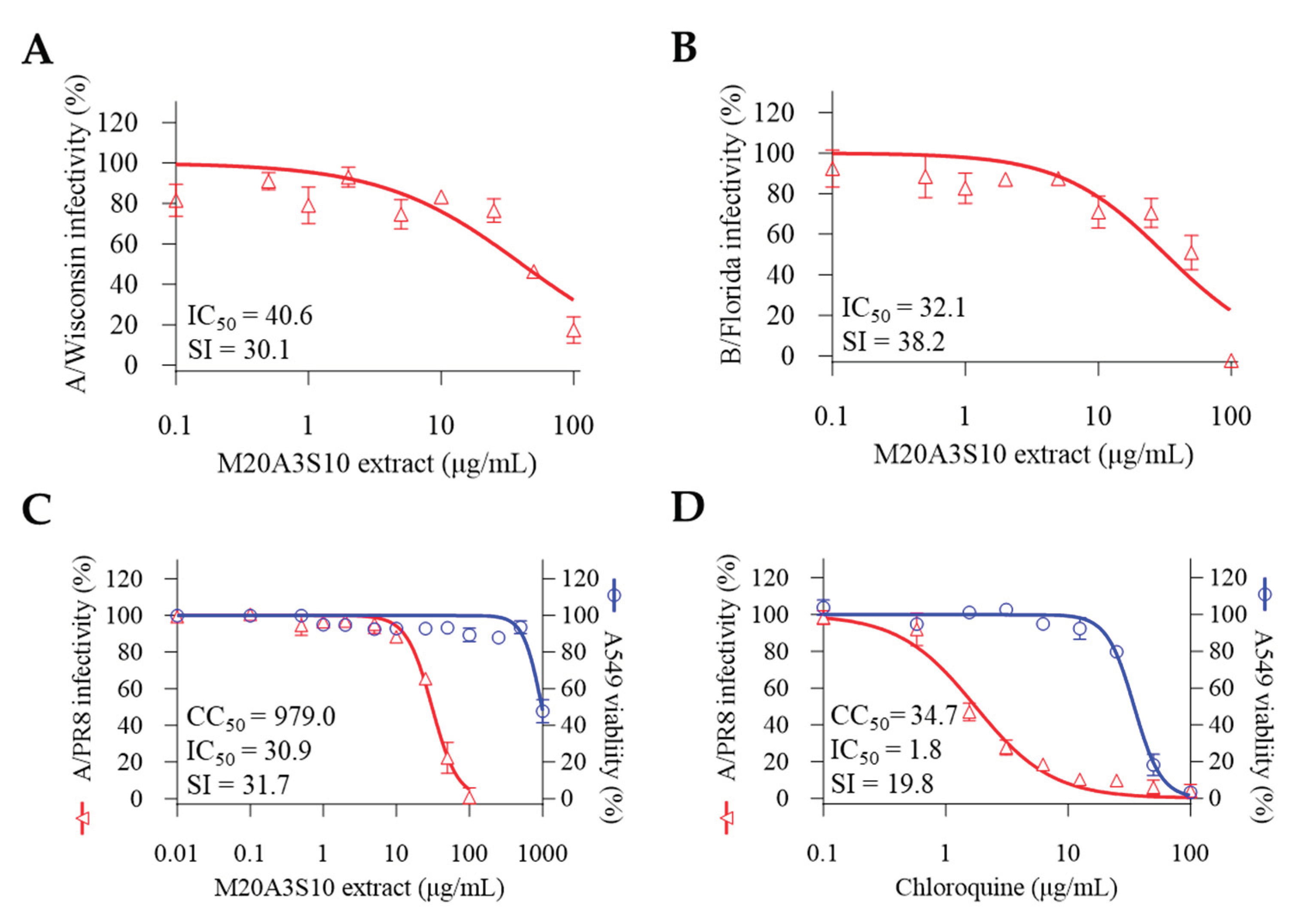
Next, antiviral evaluation was performed targeting multiple influenza virus strains aside from A/PR8 and different cell lines (Figure 8). As expected, the post-treatment of M20A3S10 extract dramatically exerted antiviral efficacies against influenza virus A/H3N2 (CC50 = 1223 µg/mL, IC50 = 40.6 µg/mL, SI = 30.1) and B/Florida (CC50 = 1223 µg/mL, IC50 = 32.1 µg/mL, SI = 38.2). Moreover, the extract more greatly protected A549 cells from influenza virus A/PR8 (CC50 = 979 µg/mL, IC50 = 30.9 µg/mL, SI = 31.7) than chloroquine. These data suggested that M20A3S10 extract has in vitro broad-spectrum antiviral potential against multiple influenza viruses, which can be applied to various cell lines originating from different organs.
4. Discussion
The COVID-19 pandemic has brought to light the importance of pandemic preparedness. The next pandemic is expected to attract even more historical attention, particularly with respect to the influenza virus and flavivirus, which have continuously posed a threat to human society due to their high contagiousness and potential to undermine public health and economies worldwide [35]. Those viruses have high mutation rates and lack proofreading mechanisms, leading to genetic diversity [36]. This diversity can give rise to mutant or variant viruses that can infect humans via zoonotic transmission, thereby raising serious public health concerns regarding climate change and newly emerging reservoirs such as bats, mosquitos, and birds [35]. Consequently, the pandemic potential of these pathogens highlights the need to develop new antiviral strategies and drugs. In this regard, marine microbes have gained significant attention, particularly as a source of natural compounds with antiviral, anti-inflammatory, anti-microbial, and anti-malarial properties [37,38,39], some of which have entered clinical studies or advanced stages for commercial uses.
An extract of Mameliella sp., a Gram-negative, non-motile, rod-shaped marine bacterium, recently exhibited significant antiviral activity against IAV and IBV [18]. In parallel, we conducted preliminary antiviral screening with 200 marine bacteria isolated from the coastal seawater. We identified Parerythrobacter sp. M20A3S10 was the bacterium whose extract exhibited the most significant safety and effectiveness as an antiviral against A/PR8 infection. This novel strain of the rare genus Parerythrobacter was isolated from the seawater closest to Parerythrobacter lutipelagi (98.11%). Notably, the Erythrobacteraceae family, including Parerythrobacter, is known to produce carotenoids [9]. Carotenoids are organic pigments studied for their antioxidant properties, which could interfere with viral replication mechanisms [12,40]. Carotenoids from Qipengyuania pacifica, another member of the Erythrobacteraceae family, have also been demonstrated to have anti-bacterial capabilities, especially against bacterial infections that are resistant to a broad array of antibiotics [13].
In other words, influenza viruses have lipid envelopes that are susceptible to disruption [40], so it is plausible that carotenoid-rich extracts could destabilize the viral membrane. Additionally, some members of the Erythrobacteraceae family have been found in marine environments rich in bioactive compounds that could have broader antiviral effects [13]. While empirical studies are needed to substantiate these claims, the unique biochemical profile of carotenoid-producing bacteria within the Erythrobacteraceae family offers a promising target for developing novel antiviral agents against influenza viruses [13,40].
Post-treatment of M20A3S10 extract exhibited remarkable antiviral activity against A/PR8 infection (IC50 = 51.1 µg/mL, SI = 24.0), which was more significant than chloroquine, a commercialized drug. In addition, dose-dependent antiviral activity was found in viral protein synthesis, genome synthesis, and progeny production. However, pre-and co-treatment of the extract did not suppress the influenza virus. Even if the penetration assay revealed a significant reduction of the viral genome, it is likely due to some residual components from the extract within the cell during pre-treatment. Collectively, it indicates that the antiviral action of the extract is supposedly involved in post-entry stages, suggesting post-treatment therapy is available after viral infection, which is more adaptable in clinical uses and more feasible for controlling epidemic diseases than prophylactic therapy.
Regarding compounds of marine extracts, carotenoids could exert antiviral activities in the late stage of viral replication, primarily during genome replication and caspase-mediated apoptotic clearance [41,42]. Accordingly, we found that M20A3S10 extract remarkably suppressed virus replication by enhancing apoptotic reactions. In many viral and bacterial infections, apoptosis prevents the growth of intracellular microbes as an innate defense mechanism [34]. In other words, apoptosis exhibits a significant antiviral reaction by inhibiting IAV replication and respiratory immunopathology [43].
Interestingly, flavivirus shares common characteristics with the influenza virus during its replication. Flavivirus has cholesterol as a component of the envelope, which is critical for viral binding, entry, and fusion processes. Therefore, a drug modulating cholesterol synthesis, such as statins, could efficiently control the influenza virus and flavivirus [4]. Indeed, M20A3S10 extract significantly inhibits viral protein synthesis, replication, and progeny production against either ZIKV or DENV infection in vitro. The therapeutic indexes are IC50 = 69.6 µg/mL and SI = 22.5 against ZIKV, IC50 = 65.1 µg/mL, and SI = 24.1 against DENV, suggesting a cholesterol-destabilizing component within the extract should be further investigated to control influenza virus and flavivirus.
Regarding the replication mechanism, IAV, ZIKV, and DENV induce necroptosis through the NLRP3 inflammasome while evading apoptosis, thereby allowing more reproduction of progeny. However, hosts exhibit an essential innate immune reaction, where viral RNA is detected by RIG-I-like receptors, triggering IRF3-mediated apoptosis, which limits viral propagation [44]. As expected, post-treatment of the M20A3S10 extract enhanced apoptotic reaction, reducing viral replication in ZIKV and DENV infections.
A broad antiviral mechanism was investigated using multiple strains of influenza viruses, resulting in promising therapeutic indexes against A/Wisconsin (IC50 = 40.6 µg/mL, SI = 30.1) and B/Florida (IC50 = 32.1 µg/mL, SI = 38.2). Moreover, the extract's effectiveness was guaranteed in a lung epithelial cell line against A/PR8 infection (IC50 = 30.9 µg/mL, SI = 31.7), greater than chloroquine. Given the similarity of the viral architecture and replication mechanism, it is plausible that apoptosis-mediated innate immunity is the limiting factor in controlling broad-spectrum infections by IAV, IBV, ZIKV, and DENV, which the M20A3S10 extract could efficiently induce.
Since The Great Flu of 1918, the influenza virus has been the focus of active investigation in public health, requiring significant effort for antiviral treatment and drug development. Multidisciplinary research, including microbiology, natural product chemistry, and molecular biology, has been undertaken [45,46]. Currently, antiviral studies have focused on creating a novel lead agent from natural compounds due to their notable efficacy and safety in controlling various viral diseases [18,50]. The extract of Parerythrobacter sp. M20A3S10 has been identified as a promising source of broad-spectrum antiviral agents that are both effective and safe.
Further research is required to identify antiviral components within the extract, including carotenoids, and to explore their properties and profiles using LC-MS/MS-based metabonomics. Animal experiments will also be required to evaluate the in vivo antiviral potential. Nevertheless, given its promising efficacy, possible antiviral mechanism of action, and broad-spectrum activity, antiviral research with the M20A3S10 extract could be extended to pre-clinical studies targeting current viral diseases and emerging pandemics in the future. Additionally, the antiviral mechanism of carotenoid-producing bacteria from the Erythrobacteraceae family against influenza viruses will be an active area of further investigation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Antiviral effect of M20A3S10 extract by pre-treatment (A-C) and co-treatment (D-F) against A/PR8 strain. (A) Schematic diagram of virus inoculation and extract treatment. (B) IC50 and SI of M20A3S10 extract measured by CPE-inhibition assay. (C) Viral genome copies detected by RT-qPCR. (D) Schematic diagram of virus inoculation and extract treatment. (E) IC50 and SI of M20A3S10 extract measured by CPE-inhibition assay. (F) Viral genome copies detected by RT-qPCR. All data in the graphs are presented as arithmetic means ± S.D. from 3 independent experiments; Figure S2: Virus attachment (A) and penetration assay (B). (A) Viral genome copies by pre-treatment of the extract before virus binding. (B) Viral genome copies by post-treatment after virus binding. All data in the graphs are presented as arithmetic means ± S.D. from 3 independent experiments. HA, haemagglutinin. *, P < 0.05; **, P < 0.01; ****, P < 0.0001
Author Contributions
Conceptualization, K.-S. M. and G. C.; methodology, M. Y. S., J.-Y. Y., J. A. C.; software, H.-J. K.; validation, Y. M. K. and E.-S. C.; formal analysis, J.-G. P.; investigation, S.-B. J., K.-S. M.; resources, G. C.; data curation, S.-B. J.; writing—original draft preparation, K.-S. M.; writing—review and editing, G. C.; visualization, H.-J. K.; supervision, Y.-B. B. project administration, J.-G. P.; funding acquisition, S.-I. P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (No. NRF-2022R1A2C1011742). It was also funded by an in-house grant from the National Marine Biodiversity Institute of Korea (grant number MABIK 2024M00600).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
The data supporting the conclusions of this article are included within the article. Raw data are available from the corresponding author upon reasonable request.
Acknowledgments
We express our deepest gratitude to the National Marine Biodiversity Institute of Korea (MABIK) for providing the bacterial isolate, which is deposited at the Microbial Marine BioBank (MMBB) under the number MI00006287.
Conflicts of Interest
The authors declare they have no conflict of interest.
References
- Harrington, W.N.; Kackos, C.M.; Webby, R.J. The evolution and future of influenza pandemic preparedness. Exp Mol Med 2021, 53, 737–749. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef]
- Wong, K.H.; Lal, S.K. Alternative antiviral approaches to combat influenza A virus. Virus Genes 2023, 59, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Farfan-Morales, C.N.; Cordero-Rivera, C.D.; Reyes-Ruiz, J.M.; Hurtado-Monzón, A.M.; Osuna-Ramos, J.F.; González-González, A.M.; De Jesús-González, L.A.; Palacios-Rápalo, S.N.; del Ángel, R.M. Anti-flavivirus Properties of Lipid-Lowering Drugs. Frontiers in Physiology 2021, 12, Review. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chisti, Y.; Banerjee, U.C. Production of shikimic acid. Biotechnol Adv 2012, 30, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Karpf, M.; Trussardi, R. Efficient access to oseltamivir phosphate (Tamiflu) via the O-trimesylate of shikimic acid ethyl ester. Angew Chem Int Ed Engl 2009, 48, 5760–5762. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.D.; Shi, X.X.; Ko, K.H.; Lu, W.D. A short and practical synthesis of oseltamivir phosphate (Tamiflu) from (-)-shikimic acid. J Org Chem 2009, 74, 3970–3973. [Google Scholar] [CrossRef]
- Xu, L.; Sun, C.; Fang, C.; Oren, A.; Xu, X.W. Genomic-based taxonomic classification of the family Erythrobacteraceae. Int J Syst Evol Microbiol 2020, 70, 4470–4495. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Liu, Y.; Wang, L.; Wang, W.; Shao, Z. Erythrobacter atlanticus sp. nov., a bacterium from ocean sediment able to degrade polycyclic aromatic hydrocarbons. Int J Syst Evol Microbiol 2015, 65, 3714–3719. [Google Scholar] [CrossRef]
- Cai, X.; Chen, Y.; Xie, X.; Yao, D.; Ding, C.; Chen, M. Astaxanthin prevents against lipopolysaccharide-induced acute lung injury and sepsis via inhibiting activation of MAPK/NF-kappaB. Am J Transl Res 2019, 11, 1884–1894. [Google Scholar]
- Khalil, A.; Tazeddinova, D.; Aljoumaa, K.; Kazhmukhanbetkyzy, Z.A.; Orazov, A.; Toshev, A.D. Carotenoids: Therapeutic Strategy in the Battle against Viral Emerging Diseases, COVID-19: An Overview. Prev Nutr Food Sci 2021, 26, 241–261. [Google Scholar] [CrossRef] [PubMed]
- Tareen, S.; Risdian, C.; Müsken, M.; Wink, J. Qipengyuania pacifica sp. nov., a novel carotenoid-producing marine bacterium of the family Erythrobacteraceae, isolated from sponge (Demospongiae), and antimicrobial potential of its crude extract. Diversity 2022, 14, 295. [Google Scholar] [CrossRef]
- Baek, Y.B.; Kwon, H.J.; Sharif, M.; Lim, J.; Lee, I.C.; Ryu, Y.B.; Lee, J.I.; Kim, J.S.; Lee, Y.S.; Kim, D.H.; et al. Therapeutic strategy targeting host lipolysis limits infection by SARS-CoV-2 and influenza A virus. Signal Transduct Target Ther 2022, 7, 367. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Okamoto, T.; Katoh, H.; Sugiyama, Y.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Fukuhara, T.; Ikawa, M.; et al. Infection with flaviviruses requires BCLXL for cell survival. PLOS Pathogens 2018, 14, e1007299. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.; Seo, J.Y.; Baek, Y.B.; Park, J.G.; Kang, M.I.; Cho, K.O.; Park, S.I. Opposite Effects of Apoptotic and Necroptotic Cellular Pathways on Rotavirus Replication. J Virol 2022, 96, e0122221. [Google Scholar] [CrossRef]
- Erwig, L.P.; Henson, P.M. Clearance of apoptotic cells by phagocytes. Cell Death Differ 2008, 15, 243–250. [Google Scholar] [CrossRef]
- Kim, H.-J.; Park, J.-G.; Moon, K.-S.; Jung, S.-B.; Kwon, Y.M.; Kang, N.S.; Kim, J.-H.; Nam, S.-J.; Choi, G.; Baek, Y.-B.; et al. Identification and characterization of a marine bacterium extract from Mameliella sp. M20D2D8 with antiviral effects against influenza A and B viruses. Archives of Virology 2024, 169, 41. [Google Scholar] [CrossRef]
- Koblizek, M.; Beja, O.; Bidigare, R.R.; Christensen, S.; Benitez-Nelson, B.; Vetriani, C.; Kolber, M.K.; Falkowski, P.G.; Kolber, Z.S. Isolation and characterization of Erythrobacter sp. strains from the upper ocean. Arch Microbiol 2003, 180, 327–338. [Google Scholar] [CrossRef]
- Lane, D. 16S/23S rRNA sequencing. Nucleic acid techniques in bacterial systematics 1991. [Google Scholar]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 1987, 4, 406–425. [Google Scholar] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J Mol Evol 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Systematic Biology 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput Biol 2007, 3, e56. [Google Scholar] [CrossRef]
- Hong, J.Y.; Ban, J.J.; Quan, Q.L.; Eom, J.E.; Shin, H.S.; Chung, J.H. Mixture of Tomato and Lemon Extracts Synergistically Prevents PC12 Cell Death from Oxidative Stress and Improves Hippocampal Neurogenesis in Aged Mice. Foods 2022, 11. [Google Scholar] [CrossRef]
- Park, J.G.; Avila-Perez, G.; Madere, F.; Hilimire, T.A.; Nogales, A.; Almazan, F.; Martinez-Sobrido, L. Potent Inhibition of Zika Virus Replication by Aurintricarboxylic Acid. Front Microbiol 2019, 10, 718. [Google Scholar] [CrossRef]
- Sharif, M.; Baek, Y.B.; Nguyen, T.H.; Soliman, M.; Cho, K.O. Porcine sapovirus-induced RIPK1-dependent necroptosis is proviral in LLC-PK cells. PLoS ONE 2023, 18, e0279843. [Google Scholar] [CrossRef]
- Alfajaro, M.M.; Kim, J.Y.; Barbe, L.; Cho, E.H.; Park, J.G.; Soliman, M.; Baek, Y.B.; Kang, M.I.; Kim, S.H.; Kim, G.J.; et al. Dual Recognition of Sialic Acid and alphaGal Epitopes by the VP8* Domains of the Bovine Rotavirus G6P[5] WC3 and of Its Mono-reassortant G4P[5] RotaTeq Vaccine Strains. J Virol 2019, 93. [Google Scholar] [CrossRef]
- Dohme, A.; Knoblauch, M.; Egorova, A.; Makarov, V.; Bogner, E. Broad-spectrum antiviral diazadispiroalkane core molecules block attachment and cell-to-cell spread of herpesviruses. Antiviral Res 2022, 206, 105402. [Google Scholar] [CrossRef]
- Weider, T.; Genoni, A.; Broccolo, F.; Paulsen, T.H.; Dahl-Jørgensen, K.; Toniolo, A.; Hammerstad, S.S. High Prevalence of Common Human Viruses in Thyroid Tissue. Frontiers in Endocrinology 2022, 13, Original. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Horsel, J.; Schulze, M.; Agalaridis, G.; Genzel, Y.; Reichl, U. Infection dynamics and virus-induced apoptosis in cell culture-based influenza vaccine production-Flow cytometry and mathematical modeling. Vaccine 2009, 27, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Arnett, E.; Weaver, A.M.; Woodyard, K.C.; Montoya, M.J.; Li, M.; Hoang, K.V.; Hayhurst, A.; Azad, A.K.; Schlesinger, L.S. PPARgamma is critical for Mycobacterium tuberculosis induction of Mcl-1 and limitation of human macrophage apoptosis. PLoS Pathog 2018, 14, e1007100. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Sobur, M.A.; Islam, M.S.; Ievy, S.; Hossain, M.J.; El Zowalaty, M.E.; Rahman, A.T.; Ashour, H.M. Zoonotic Diseases: Etiology, Impact, and Control. Microorganisms 2020, 8. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Genetic diversity and evolution of viral populations. Encyclopedia of virology 2021, 53. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat Prod Rep 2016, 33, 382–431. [Google Scholar] [CrossRef]
- Barzkar, N.; Tamadoni Jahromi, S.; Poorsaheli, H.B.; Vianello, F. Metabolites from Marine Microorganisms, Micro, and Macroalgae: Immense Scope for Pharmacology. Mar Drugs 2019, 17. [Google Scholar] [CrossRef]
- Wang, Y.P.; Lei, Q.Y. Metabolite sensing and signaling in cell metabolism. Signal Transduct Target Ther 2018, 3, 30. [Google Scholar] [CrossRef]
- Lomartire, S.; Goncalves, A.M.M. Antiviral Activity and Mechanisms of Seaweeds Bioactive Compounds on Enveloped Viruses-A Review. Mar Drugs 2022, 20. [Google Scholar] [CrossRef]
- Yim, S.K.; Kim, I.; Warren, B.; Kim, J.; Jung, K.; Ku, B. Antiviral Activity of Two Marine Carotenoids against SARS-CoV-2 Virus Entry In Silico and In Vitro. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, G.E.; Abu-Serie, M.M.; Abo-Elela, G.M.; Ghozlan, H.; Sabry, S.A.; Soliman, N.A.; Abdel-Fattah, Y.R. In vitro dual (anticancer and antiviral) activity of the carotenoids produced by haloalkaliphilic archaeon Natrialba sp. M6. Sci Rep 2020, 10, 5986. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.E.; Niles, J.A.; Fleming, E.H.; Roberts, N.J. The role of cell surface expression of influenza virus neuraminidase in induction of human lymphocyte apoptosis. Virology 2019, 534, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Sen, G.C. RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): A new antiviral pathway. Protein Cell 2017, 8, 165–168. [Google Scholar] [CrossRef]
- Fraser, C.; Donnelly, C.A.; Cauchemez, S.; Hanage, W.P.; Van Kerkhove, M.D.; Hollingsworth, T.D.; Griffin, J.; Baggaley, R.F.; Jenkins, H.E.; Lyons, E.J.; et al. Pandemic potential of a strain of influenza A (H1N1): Early findings. Science 2009, 324, 1557–1561. [Google Scholar] [CrossRef]
- Knipe, D.; Howley, P.; Griffin, D.; Lamb, R.; Martin, M.; Roizman, B.; Straus, S. Fields Virology, Volumes 1 and 2; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Cheng, A.J.T.; Macalino, S.J.Y.; Billones, J.B.; Balolong, M.P.; Murao, L.A.E.; Carrillo, M.C.O. In silico evaluation of Philippine Natural Products against SARS-CoV-2 Main Protease. J Mol Model 2022, 28, 345. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic analysis of marine bacteria either in use or for comparison with isolated strains. (A) A neighbor-joining tree based on 16S rRNA gene, showing the phylogenetic relationships of strain M20A3S10 (in bold type) and closely related taxa with validly published names. GenBank accession numbers are given in parentheses. Bootstrap values above 70 % are shown on nodes in percentages of 1,000 replicates. Bar, 0.01 changes per nucleotide position. (B) Marine bacteria whose extracts were subjected to preliminary antiviral screening. Bar, 0.04 changes per nucleotide position.
Figure 1.
Phylogenetic analysis of marine bacteria either in use or for comparison with isolated strains. (A) A neighbor-joining tree based on 16S rRNA gene, showing the phylogenetic relationships of strain M20A3S10 (in bold type) and closely related taxa with validly published names. GenBank accession numbers are given in parentheses. Bootstrap values above 70 % are shown on nodes in percentages of 1,000 replicates. Bar, 0.01 changes per nucleotide position. (B) Marine bacteria whose extracts were subjected to preliminary antiviral screening. Bar, 0.04 changes per nucleotide position.
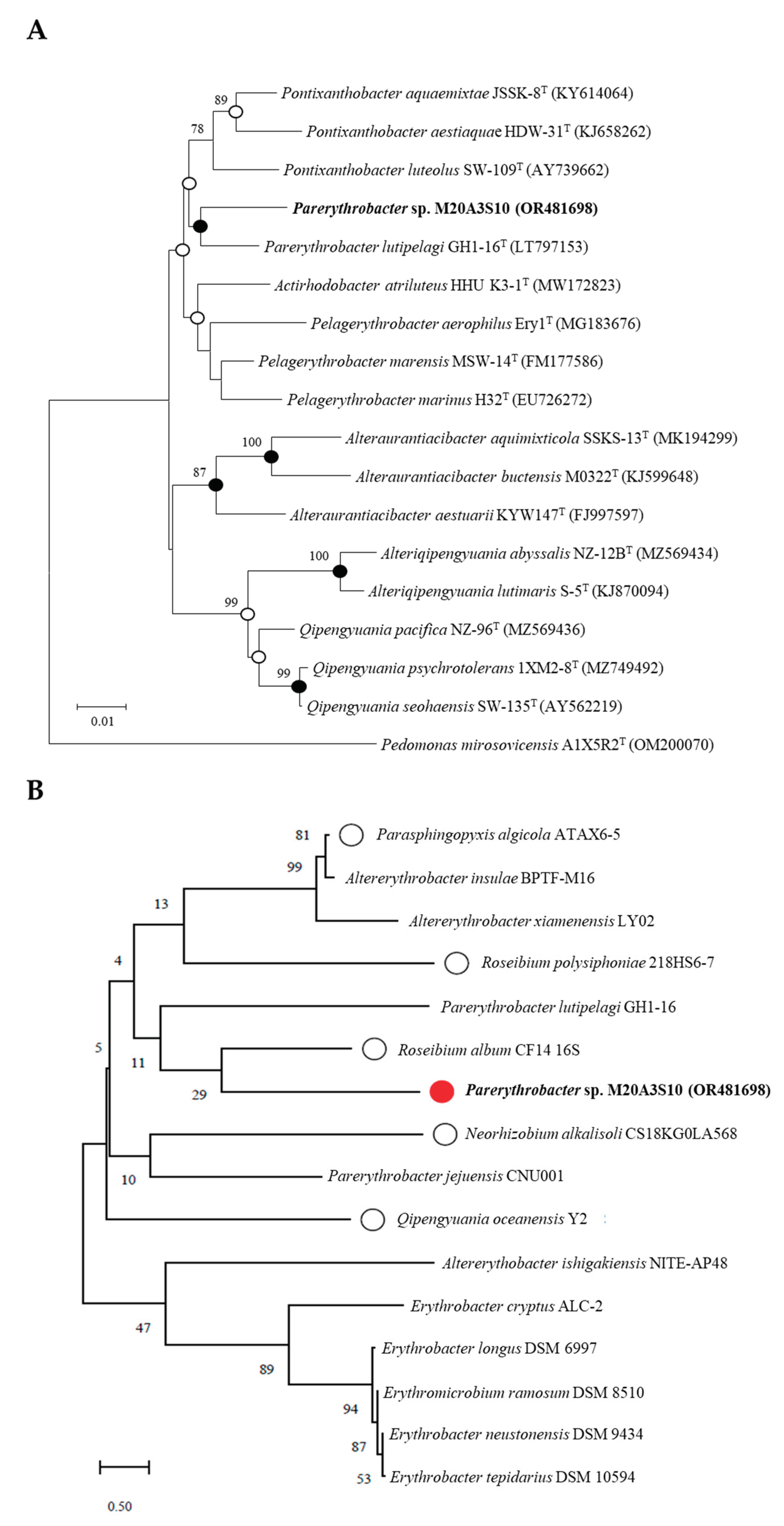
Figure 2.
Marine bacteria whose extracts were subjected to preliminary antiviral screening. (A) Parerythrobacter sp. M20A3S10, (B) Roseibium album, (C) Parasphingopyxis algicola, (D) Roseibium polysiphoniae, (E) Neorhizobium alkalisoli, (F) Qipengyuania oceanensis.
Figure 2.
Marine bacteria whose extracts were subjected to preliminary antiviral screening. (A) Parerythrobacter sp. M20A3S10, (B) Roseibium album, (C) Parasphingopyxis algicola, (D) Roseibium polysiphoniae, (E) Neorhizobium alkalisoli, (F) Qipengyuania oceanensis.
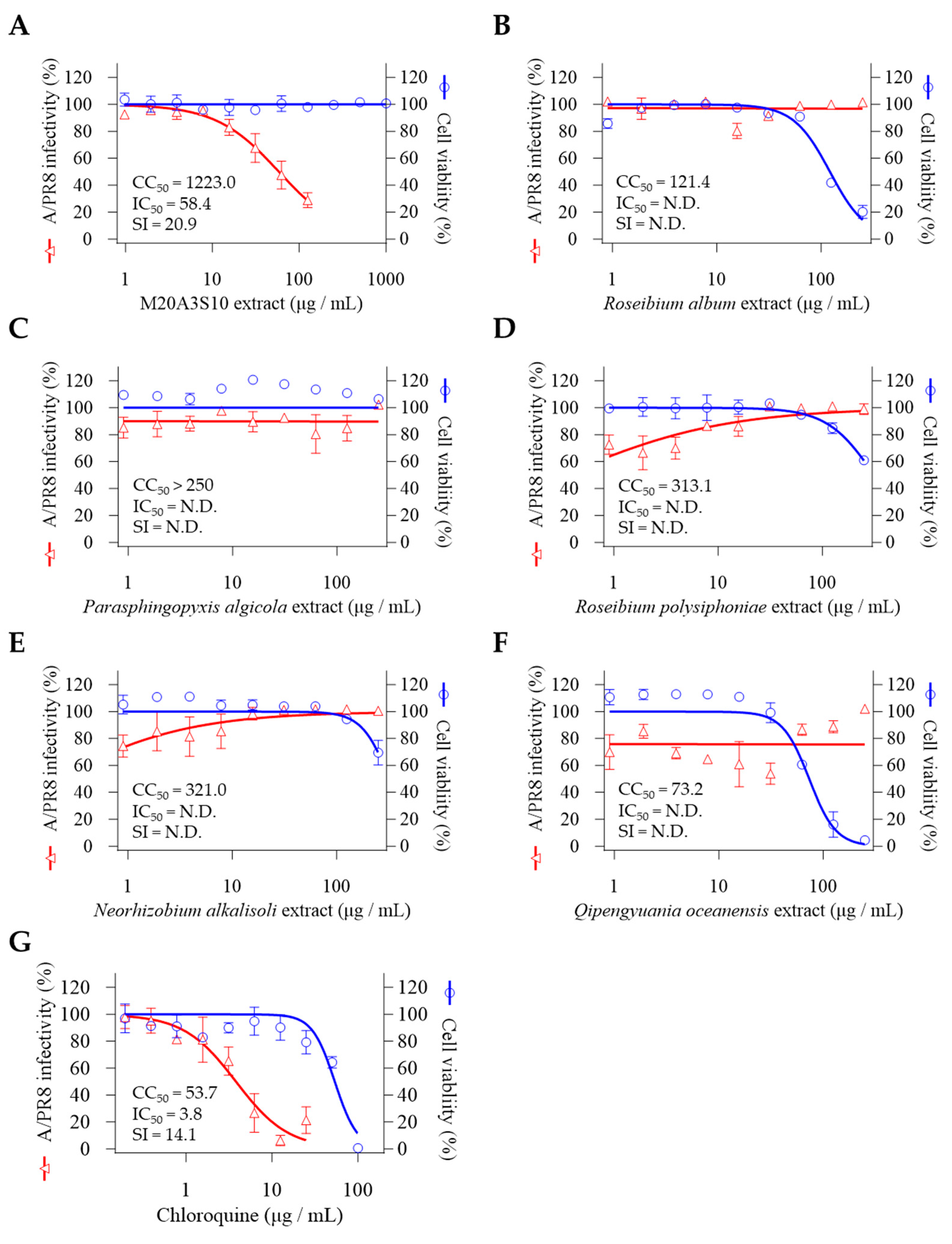
Figure 3.
Antiviral effect of M20A3S10 extract by post-treatment. (A) Schematic diagram of virus inoculation and extract treatment. (B) IC50 and SI of M20A3S10 extract measured by CPE-inhibition assay. (C) Viral protein synthesis detected by IFA. (D) Viral genome copies detected by RT-qPCR. (E) Progeny virus production measured by TCID50. All data in the graphs are presented as arithmetic means ± S.D. from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001.
Figure 3.
Antiviral effect of M20A3S10 extract by post-treatment. (A) Schematic diagram of virus inoculation and extract treatment. (B) IC50 and SI of M20A3S10 extract measured by CPE-inhibition assay. (C) Viral protein synthesis detected by IFA. (D) Viral genome copies detected by RT-qPCR. (E) Progeny virus production measured by TCID50. All data in the graphs are presented as arithmetic means ± S.D. from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001.
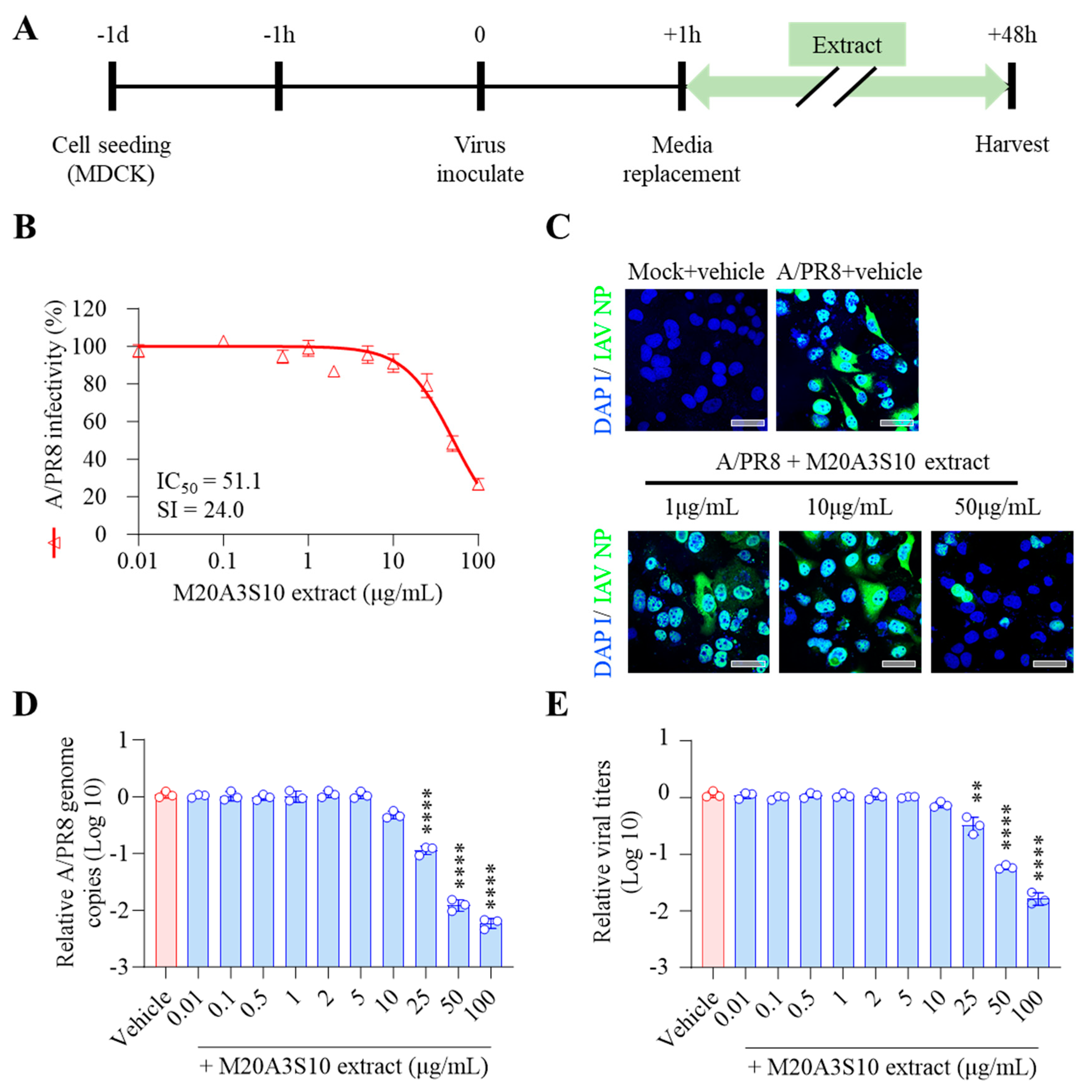
Figure 4.
Evaluation of apoptotic response by M20A3S10 extract using flow cytometry. Apoptosis was measured by TUNEL assay and virus protein by an antibody against IAV NP. (A) A/PR8-infected (0.1 MOI) vehicle-treated group. (B) A/PR8-infected (0.1 MOI), M20A3S10 extract-treated group. Quantification of TUNEL- (C) and virus-positive cells (D) treated with vehicle or M20A3S10 extract. **, P < 0.01; ****, P < 0.0001.
Figure 4.
Evaluation of apoptotic response by M20A3S10 extract using flow cytometry. Apoptosis was measured by TUNEL assay and virus protein by an antibody against IAV NP. (A) A/PR8-infected (0.1 MOI) vehicle-treated group. (B) A/PR8-infected (0.1 MOI), M20A3S10 extract-treated group. Quantification of TUNEL- (C) and virus-positive cells (D) treated with vehicle or M20A3S10 extract. **, P < 0.01; ****, P < 0.0001.
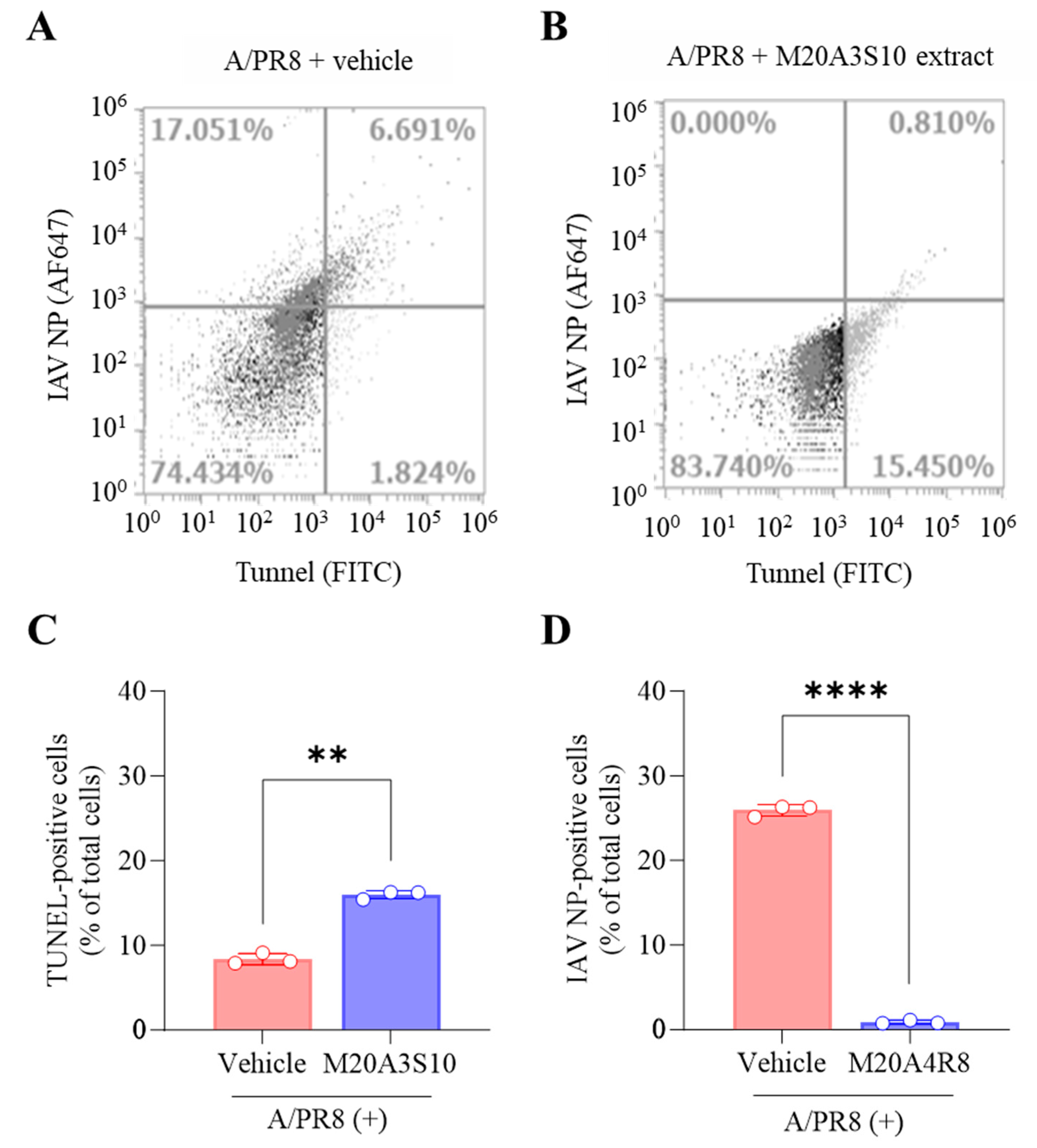
Figure 5.
Antiviral activity against flaviviruses by full-treatment of M20A3S10 extract. Antiviral actions were measured against ZIKV (A,B) and DENV (C,D) infections, treated with either the M20A3S10 extract or chloroquine.
Figure 5.
Antiviral activity against flaviviruses by full-treatment of M20A3S10 extract. Antiviral actions were measured against ZIKV (A,B) and DENV (C,D) infections, treated with either the M20A3S10 extract or chloroquine.
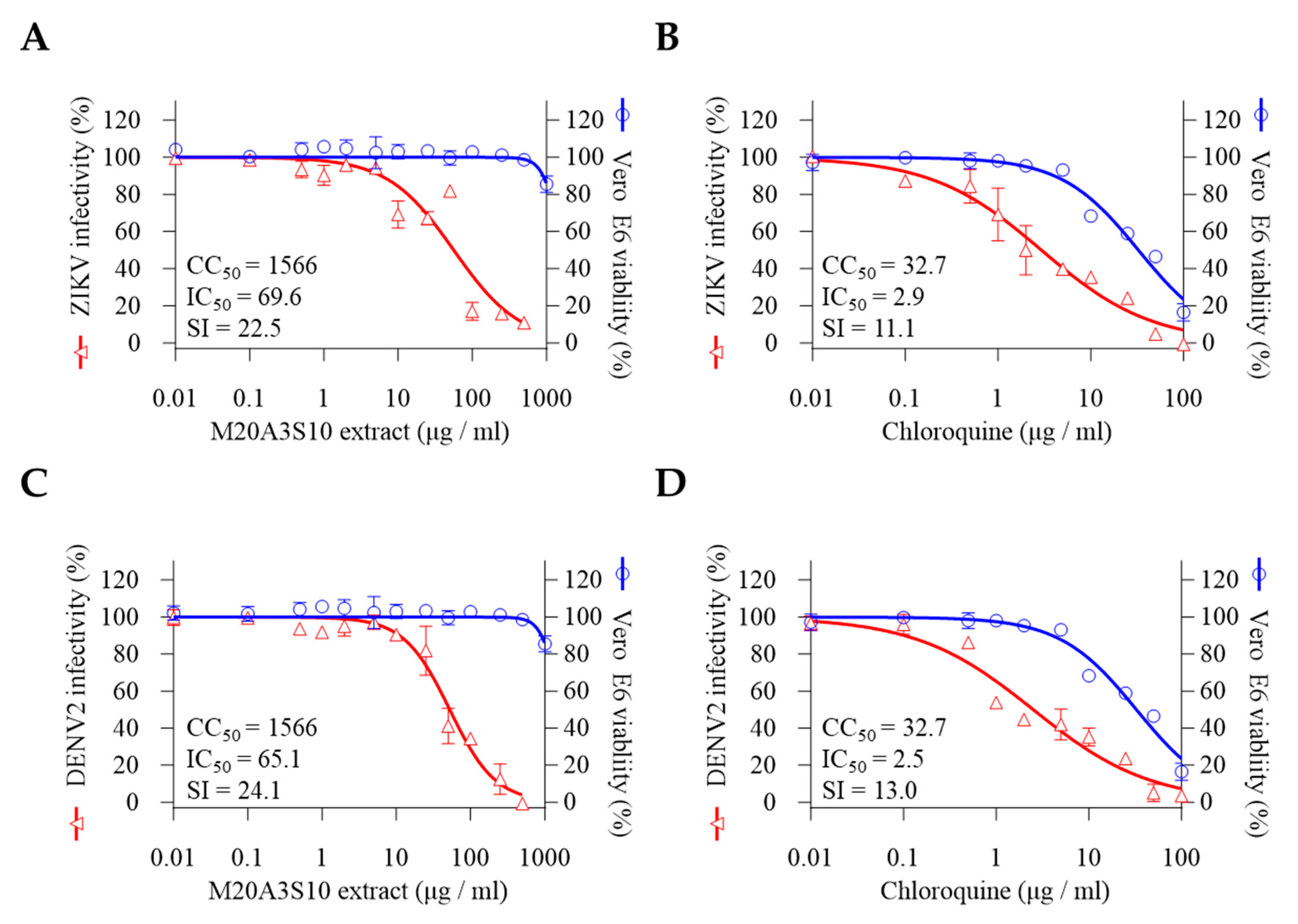
Figure 6.
ZIKV and DENV of progeny virus inhibition effect according to M20A3S10 extract treatment (A,B) ZIKV and DENV genome copies detected by RT-qPCR. (C,D) ZIKV and DENV protein synthesis detected by IFA. ***, P < 0.001; ****, P < 0.0001.
Figure 6.
ZIKV and DENV of progeny virus inhibition effect according to M20A3S10 extract treatment (A,B) ZIKV and DENV genome copies detected by RT-qPCR. (C,D) ZIKV and DENV protein synthesis detected by IFA. ***, P < 0.001; ****, P < 0.0001.
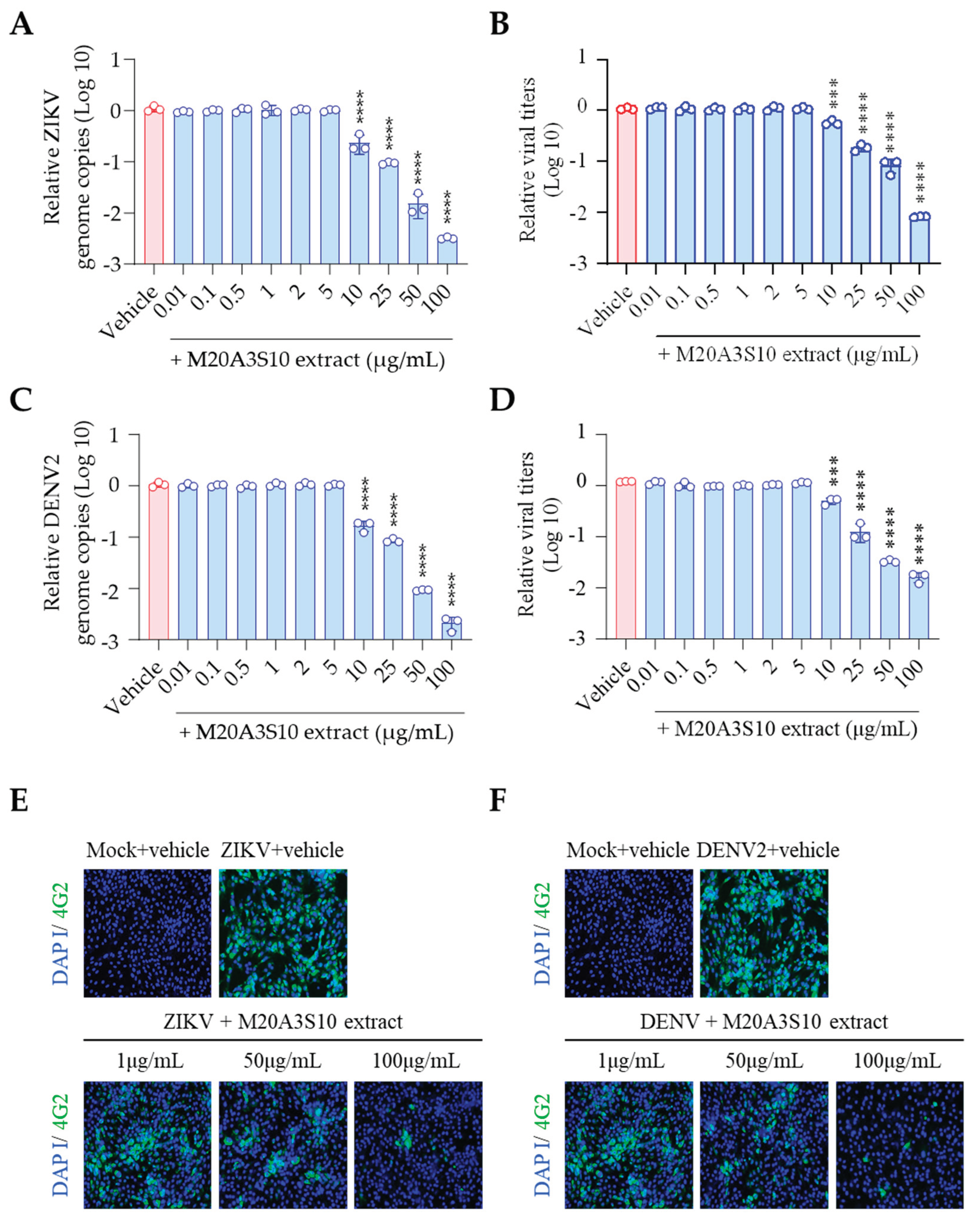
Figure 7.
Evaluation of apoptotic response by M20A3S10 extract using flow cytometry. Apoptosis was measured by TUNEL assay and virus protein by an antibody against flavivirus group envelope protein, respectively. (A-D) Summary of flow cytometry data. Quantification of TUNEL- (A,C) and virus-positive cells (B,D) treated with the vehicle or M20A3S10 extract. All data in the graphs are presented as the arithmetic mean ± S.D. from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001.
Figure 7.
Evaluation of apoptotic response by M20A3S10 extract using flow cytometry. Apoptosis was measured by TUNEL assay and virus protein by an antibody against flavivirus group envelope protein, respectively. (A-D) Summary of flow cytometry data. Quantification of TUNEL- (A,C) and virus-positive cells (B,D) treated with the vehicle or M20A3S10 extract. All data in the graphs are presented as the arithmetic mean ± S.D. from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001.
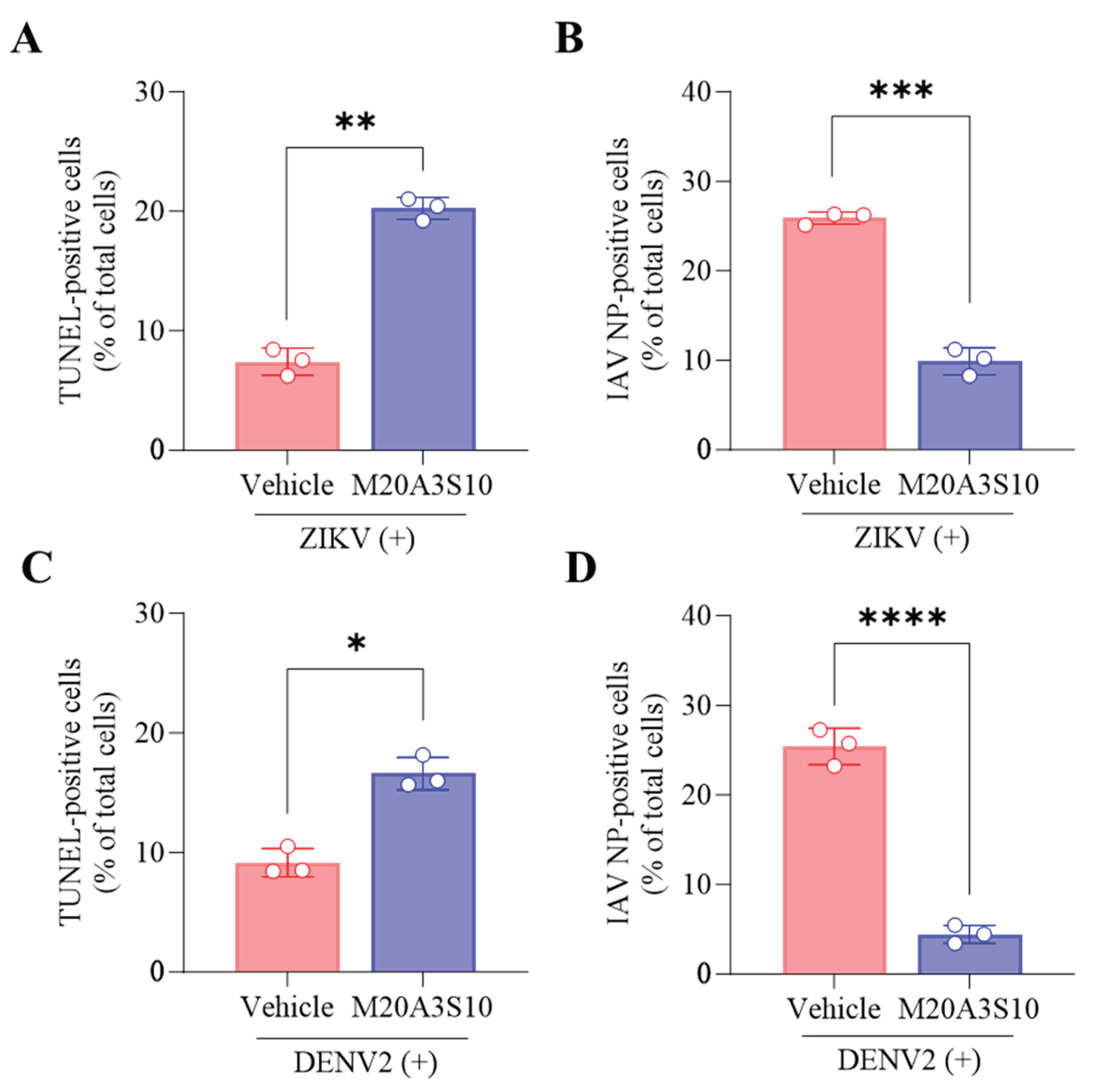
Figure 8.
The post-treatment of the M20A3S10 extract shows a broad spectrum of antiviral potential against multiple influenza viruses. Antiviral actions were measured against the A/H3N2 strain (A) and the B/Yamagata strain (B). Antiviral responses in A549 cells by the post-treatment of M20A3S10 extract (C) and chloroquine (D).
Figure 8.
The post-treatment of the M20A3S10 extract shows a broad spectrum of antiviral potential against multiple influenza viruses. Antiviral actions were measured against the A/H3N2 strain (A) and the B/Yamagata strain (B). Antiviral responses in A549 cells by the post-treatment of M20A3S10 extract (C) and chloroquine (D).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



