
Submitted:
12 March 2024
Posted:
12 March 2024
You are already at the latest version
Abstract
There is little doubt that the efficacy of MnDPDP and its therapeutic improved counterpart Ca4Mn(DPDP)5 mainly depends on their MnSOD-mimetic activity when it comes to their potential use as rescue medicines, during e.g., acute myocardial infarction. However, pharmacokinetic considerations suggest that the efficacy of MnDPDP on Pt2+-associated neurotoxicity depends on another action of this drug. Electron Paramagnetic Resonance (EPR) studies have demonstrated that Pt2+ outcompetes Mn2+ and endogenous Zn2+ from binding to fodipir (DPDP), and hence suggest that the previously reported CIPN protective efficacy of MnDPDP is a result of chelation and elimination of Pt2+ by DPDP, which in turn suggests that Mn2+ is unnecessary for efficacy when it comes to oxaliplatin-associated CIPN.
Keywords:
Calmangafodipir
; mangafodipir
; manganese
; platinum
; MnSOD mimetics
; oxaliplatin-associated CIPN
; oxidative stress
; nitrosative stress
; chelation therapy
1. Background
Addition of oxaliplatin to 5-FluoroUracil (5-FU) or capecitabine chemotherapy of ColoRectal Cancer (CRC) according to FOLFOX or CAPOX regimen has increased tumoricidal efficacy, both in the adjuvant and palliative settings [1,2,3,4,5,6,7,8,9]. However, an addition that has increased unwanted toxicity, where Chemotherapy-Induced Peripheral Neuropathy (CIPN) is the most problematic dose-limiting toxicity of oxaliplatin. Neutropenia is another troublesome toxicity of oxaliplatin plus 5-FU experienced by many patients. Various approaches to prevent CIPN caused by oxaliplatin, and other platinum-containing drugs have generally failed [10]. The exact mechanism behind oxaliplatin- and cisplatin-associated CIPN is poorly understood. However, it seems reasonable to assume that it is uptake and retention of Pt2+ in the peripheral sensory nerve system that cause the toxicity. This system is lacking a blood-brain barrier, a draining lymph system, and cerebrospinal fluid [11]. This makes potentially dangerous substances, such as chemotherapy drugs, to accumulate in the peripheral nerve system and cause oxidative stress and detrimental nerve injuries.
Although there are many similarities between the pharmacodynamic, pharmacokinetic and (unwanted) toxicological properties of oxaliplatin and other platinum-containing drugs, particularly those of cisplatin, there are also major differences. In the current article we will emanate from oxaliplatin with the aim of scrutinizing the possibility of preventing oxaliplatin-associated CIPN by chelation therapy with fodipir (DPDP), similar to that lately obtained with Sodium ThioSulfate (STS) in pediatric patients (see below).
At start of oxaliplatin- or cisplatin-based treatment the terminal elimination half-life of Pt2+ is about one month. However, over time the half-life will increase and exceed one year at 10 to 20 years post chemotherapy [12], clearly illustrating the problem with platinum retention. As in other form of heavy metal toxicity, chelation therapy, i.e., administration of a suitable low weight metal-binding ligand forming a nontoxic metal complex small enough for being filtered through the kidneys (or alternatively redistributed to a less critical tissue) appears as an attractive possibility [13].
Chelation therapy to prevent Pt2+-associated CIPN has until now remained an unproven option. However, in 2022 FDA approved co-treatment with the metal chelator Sodium ThioSulfate (STS) (Pedmark®) to prevent severe ototoxicity of cisplatin in treatment of pediatric patients with localized, non-metastatic solid tumours. The decision was based on two multicentre trials, i.e., the SIOPEL 6 trial and the ACCL0431 trial [14,15]. Importantly, the ACCL0431 design included multiple cancer types at any stage and post hoc analyses showed that although survival in the STS group was equivalent to that in the control group among patients with localized disease, it was significantly lower among those with disseminated disease [15]. Results reflecting the difficulty of finding, alternatively creating selective protectants, i.e., compounds that protect against “unwanted” toxicity but not against the “wanted” tumoricidal toxicity. Although chelation therapy obviously does not provide any quick fix, it anyhow provides hope that chelation therapy may be a passable way of solving the oxaliplatin-associated CIPN problem.
Multiple lines of evidence indicate that the platinum-containing cancer drugs enter cells, are distributed to various subcellular compartments, and are exported from cells via transporters that evolved to manage copper homeostasis [16] (Figure 1). Copper (Cu2+) and Pt2+ have similar sulfur binding characteristics, and the presence of stacked rings of methionines and cysteines in the Copper TRansporter 1 (CTR1) suggest a mechanism where CTR1 selectively transports copper and platinum-containing drugs via sequential trans-chelation reactions. The similarities in binding characteristics of these metal cations also include nitrogen ligands, where binding of cisplatin- or oxaliplatin-associated Pt2+ to purine bases, preferentially guanine, which in fact reveal the tumoricidal mechanisms of both oxaliplatin and cisplatin [17] (Figure 2).
Interestingly, copper deficiency causes neurological symptoms similar to those seen in Pt2+-associated CIPN and hyper-physiological intake of Zn2+ is a common cause behind copper deficiency in humans [18]. This may in turn suggest that Pt2+ interferes with copper handling in a manner that causes a deficiency and a subsequent increase in oxidative/nitrosative stress in the Dorsal Root Ganglion (DRG) cells. Cytosolic Copper SuperOxide Dismutase (CuSOD) and cytochrome c activities depend fully on the presence of redox active copper. Copper deficiency by itself hence results in oxidative/nitrosative stress causing severe cellular injuries, including DRG cells.
2. Randomized Clinical Phase III Trials Failed to Demonstrate Positive Effects of Ca4Mn(DPDP)5
Pfeiffer et al. reported in the end of 2022 results from the POLAR A and M phase III trials in CRC patients testing the efficacy of the metal complex and MnSOD mimetic Ca4Mn(DPDP)5 (PledOx®) against oxaliplatin-associated CIPN, with the somewhat confusing aim of lowering the incidence of persistent CIPN from 40% to 20% [19]. However, in the preceding PLIANT phase II trial in CRC patients displayed an exceedingly lower frequency of adverse events, including also CIPN, than those expected from 5-FU plus oxaliplatin [20,21]. The frequency was more in line with what one expects from 5-FU alone.
The POLAR trials failed to show positive efficacy. Instead of a hypothesized 50% improvement in the incidence of persistent CIPN the real outcome was an about 50% worsening of this highly handicapping toxicity. Mechanisms that may explain the outcome, with a statistically significant number of patients being seriously injured after having received Ca4Mn(DPDP)5, indicate interactions between Pt2+-containing oxaliplatin and Mn2+-containing Ca4Mn(DPDP)5 [22] (Figure 3). The POLAR failures showed with no doubt that the positive effects of Ca4Mn(DPDP)5 on CIPN claimed by the authors of the PLIANT trial report [20] were not real, in line with Karlsson and Jynge’s criticism [21].
3. Promising Clinical Findings with MnDPDP
In 2009 Yri et al. [23] published a case report describing a young patient who received fifteen palliative cycles of oxaliplatin plus 5-FU (“Nordic FLOX” regimen), suggesting that MnDPDP may protect against oxaliplatin-associated CIPN. In fourteen of the cycles, the patient received pretreatment with MnDPDP. The patient received an accumulated dose of 1275 mg/m2 oxaliplatin, which is a dose likely to give severe neurotoxic symptoms. However, CIPN was not seen, except for the fifth cycle, when MnDPDP was left out and the patient experienced CIPN. After five cycles, the performance status for the patient was drastically improved, and the demand for analgesics was reduced. Furthermore, neutropenia did not occur during any of the chemotherapy cycles.
The case report was followed by an important paper by Coriat et al., 2014 [24] published in the Journal of Clinical Investigation. The results suggested that co-treatment with MnDPDP not only protected the patients from CIPN, but in fact also cured ongoing oxaliplatin-related CIPN. The same group headed by Batteux had previously presented results showing that MnDPDP increased the therapeutic index of both oxaliplatin and paclitaxel by simultaneously increasing the tumoricidal and the cytoprotective activity of these cytostatic drugs [25,26,27]. Karlsson et al., 2012 have reported similar results with Ca4Mn(DPDP)5 [28]. These findings clearly suggested that MnDPDP or the MnDPDP metabolite MnPyridoxyL EthylDiamine (MnPLED) might solve the oxaliplatin-associated CIPN problem.
Coriat, Batteux et al. [24] naturally assumed that the therapeutic effects of MnDPDP were due to its MnSOD-mimetic activity, i.e., a property of lowering oxidative and nitrosative cellular stress by targeting superoxide (O2•-) (Figure 4). However, this explanation is questionable from a pharmacokinetic perspective, where the MnSOD-mimetic activity last only a couple of hours [13,29]. A more plausible explanation is that DPDP or its metabolite PLED binds Pt2+ and forms an excretable metal complex or, alternatively, redistributes Pt2+ from the DRG compartment to a non-neuronal compartment and thereby lessen its negative effects on the peripheral sensory nerve system.
4. Confusing Pharmacodynamic Difference between Ca4Mn(DPDP)5 and MnDPDP
There are few reasons to believe that the confusing difference between Ca4Mn(DPDP)5 and MnDPDP can be explained by fundamental pharmacodynamic differences between them. However, one cannot fully exclude such differences, taken in consideration the complex in vivo metabolism of MnDPDP [29] and the fact that the ready-to-use MnDPDP (Teslascan™) but not Ca4Mn(DPDP)5 contains ascorbic acid. To fully exclude such a difference, preclinical studies similar to those presented by Coriat et al. [24] should be done in order to compare the therapeutic efficacy of these two compounds, with regard of oxaliplatin-associated CIPN.
A closer look into the published report of the PLIANT trial [20] and the POLAR A and M [19] suggest that the confusing difference seems related to an until now overlooked difference in study design between the early MnDPDP trial by Coriat et al. 2014 [24]. Whereas MnDPDP displayed impressive efficacy, Ca4Mn(DPDP)5 displayed no efficacy (with regard to CIPN) at any timepoint during ongoing chemotherapy or at follow up at nine months after start of chemotherapy (See Figure 3 in the Pfeiffer et al. report [19]). Ca4Mn(DPDP)5 only showed highly devastating and amplifying efficacy. However, there was an important difference in design between the MnDPDP trial and the Ca4Mn(DPDP)5 trials. In the case of MnDPDP it was administered directly after the 2-hour infusion of oxaliplatin as a 30-minute infusion [24]. In case of Ca4Mn(DPDP)5 it was on other hand administered as a 5-minute infusion starting 10 minutes before the start of oxaliplatin infusion [19,20]. Taken in consideration the complex pharmacokinetics of oxaliplatin as well as that of MnDPDP/ Ca4Mn(DPDP)5, the difference in design may of course have had dramatic effects on the outcomes of these trials. Where addition of Ca4Mn(DPDP)5 or MnDPDP ahead of oxaliplatin may be like pouring gasoline to the fire. Why not the POLAR trials and the preceding PLIANT phase II trial used an identical administration regimen as used in the successful preceding MnDPDP trial [24] is difficult to grasp. Notable, a reference to the important and published MnDPDP report [24] is lacking in the PLIANT report, as already noted by Karlsson & Jynge 2014 [21].
Unfortunately, Karlsson et al., 2023 [22] misinterpreted the published POLAR A and M report by Pfeiffer et al., 2023 [19] in relation to the published MnDPDP trial by Coriat et al. 2014 [24], when it came to the drug administration procedures and consequences thereof. Karlsson et al. [22] concluded that “the POLAR and PLIANT trials show that PledOx and related manganese-containing compounds are unsuited for co-treatment with platinum-containing compounds”. However, as evident from the above discussion there are no data available suggesting that the drug administration procedures used by Coriat et al. [24] should cause devastating worsening like that described in the Pfeiffer et al., report [19].
Nevertheless, the above described difference in drug administration between the MnDPDP trial and that of the Ca4Mn(DPDP)5 further suggests that the negative outcome of the POLAR trials was caused by highly negative interaction between Mn2+ [associated to Ca4Mn(DPDP)5] and Pt2+ (associated to oxaliplatin) resulting in a devastating increase in cellular oxidative stress (Figure 3). Taken in consideration that oxidative and nitrosative stress is an unmistakable part of CIPN, including that of oxaliplatin [30,31], points on the interplay between •NO and O2•- and generation of strongly oxidizing and nitrating species, connected through the formation of peroxynitrite [32,33] (Figure 4), as the explanation behind the POLAR failure.
A multitude of chemical reactions may lead to formation of peroxynitrite and tyrosine (Tyr) nitration [32,33], but where transition metal-driven nitration, particularly that of Mn4+ is of a particular relevance when it comes to platinum-associated CIPN. Furthermore, nitration of Tyr34 in the mitochondrial MnSOD enzyme and Tyr74 in the cytochrome c (Figure 3), leading to irreversible inactivation of these essential mitochondrial constituents in the DRG will of course lead to highly devastating effects on the sensory nerve system.
As noted in the Background section, oxaliplatin and cisplatin might interfere negatively with the cellular handling of copper and further exacerbate the viscous circle of oxidative and nitrosative stress. Interestingly indeed, Coriat, Batteux et al. [24] analysed the activity of the SOD enzymes in erythrocyte lysates, as described in the material section of [24], from the participating patients and reported that SOD-activity was statistically significant lower in the non-responder patients than in the responders. Since mammal erythrocytes do not contain mitochondria [34] the reported SOD activity corresponds mainly to the cytosolic CuSOD. The results may indicate a connection between copper status and the severity of Pt2+-associated CIPN.
Intriguingly, nitration of Tyr34 in the mitochondrial MnSOD can be catalyzed by intra-enzymatic manganese after being oxidized into Mn4+ [32] (Figure 3). The irreversible inactivation of MnSOD and the disrupted electron transport will in turn severely amplify the initial oxidative insult. Interestingly, CIPN, associated to mitochondrial Tyr34 and Tyr74 nitration is prevented by the ONOO− decomposition catalyst, MnTE-2-PyP(5+) [30].
5. DPDP as a Presumptive Chelation Drug
Both theoretically and experimentally based considerations speak in the direction of another mechanism behind the preventive efficacy of MnDPDP [13,29] than that of an MnSOD-mimetic based mechanism as suggested by Coriat [24]. An alternative explanation is that DPDP or metabolites thereof binds Pt2+ and acts as a chelation drug, i.e., a drug that binds the metal cation in question and facilitates its mobilization and renal excretion (or its redistribution to a non-neuronal compartment) and thereby “grasps the evil by the root” [13]. Endogenous zinc (Zn2+) is the most important cation when it comes to in vivo competing binding to DPDP or its metabolite PLED. Stehr et al., 2019 [13] used the difference in Electron Paramagnetic Resonance (EPR) spectra of MnDPDP and hexaqua-Mn2+ to measure release of Mn2+ from DPDP in exchange for Pt2+ and Zn2+, as described by Schmidt et al., 2002 [35], to obtain an estimate of the formation constant (10logKML) of Pt(DPDP (Figure 5).
EPR-guided competition experiments between MnDPDP with a formation constant (10 logKML) of about 15 [36] and ZnCl2 with a corresponding constant of about 19 [36] or K2PtCl4 are presented in Figure 5. The resulting competition curve for 100 µM MnDPDP and 10–1000 µM ZnCl2 were more or less identical to that presented by Schmidt and co-workers [35]. The corresponding competition curve for 100 µM MnDPDP and 10–1000 µM K2PtCl4 lied to the left of the ZnCl2-curve. The pD2 [−10log of the concentrations (M) of a drug causing half maximal responses; EC50] (together with 95% confidence interval) values for K2PtCl4 and ZnCl2 were 4.280 (4.227–4.332) and 4.173 (4.127–4.218), respectively, i.e., there was a statistically significant difference between these two pD2 values. This suggested that Pt2+ in fact has a higher affinity than Zn2+ for DPDP. The present curve for Zn2+ and that of Schmidt et al. [35] were close to 100% exchange of Mn2+ for all concentrations of ZnCl2. Importantly, that means the formation constant for PtDPDP may be substantially higher than that for ZnDPDP, but how much higher is not possible to read out from the current experiments. The Emax (maximal response) for the K2PtCl4 and ZnCl2 were 95.62 µM (89.54–101.7) and 101.2 µM (95.18–111.2), respectively. There was a clear tendency, although not statistically significant, that the K2PtCl4 curve did not reach full metal exchange (100 µM). The K2PtCl4 + MnDPDP samples got a weak yellow-brownish colour during incubation. This was not seen in the MnDPDP control sample or in the ZnCl2 + MnDPDP samples, which may suggest that Pt2+-driven oxidation of Mn2+ had occurred to some extent in the K2PtCl4 + MnDPDP samples, which in turn may explain the somewhat lower Emax than the expected.
Whereas Mn2+ binds with six coordinates to two phenolates, two amines and two carboxylates of DPDP or its metabolite PLED, Cu2+ binds with 4 coordinates to the amines and phenolates, in a square geometry with a formation constant of about 22 [36,37]. Because of the smaller ionic radius of Cu2+ this metal ion forms a much more stable complex, with shorter bond distance [38], than that found for the Mn2+ ion [36,37]. Similarities between Cu2+ and Pt2+, hypothetically, suggest that Pt2+ may bind to DPDP and PLED in an identical manner to that of Cu2+, as illustrated in Figure 6.
6. Do DPDP and PLED Fulfil Necessary Properties of a Platinum Chelation Drug?
A platinum chelation drug, such as DPDP, must fulfil the properties of a selective cytoprotectant, i.e., a drug that protect normal cells but not cancer cells or, in one way or the other, lower the unwanted toxicity without lowering the tumoricidal efficacy.
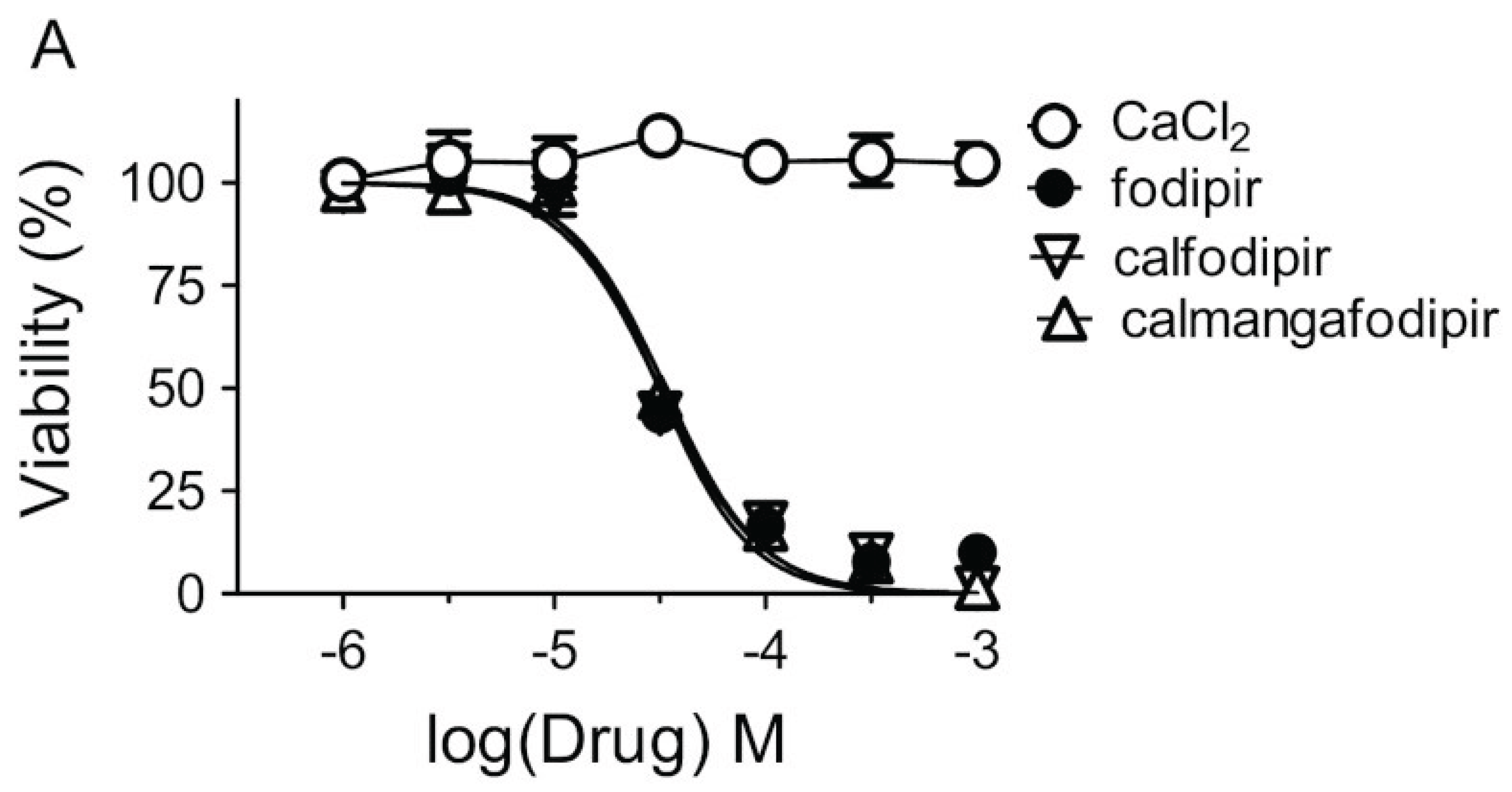
MnDPDP has preclinically been shown to protect against unwanted toxicity of oxaliplatin and simultaneously strengthen the tumoricidal efficacy of oxaliplatin in immune competent balb c mice, implanted with CT26 tumour cells [25,26]. Results noticed by the Journal of the National Cancer Institute [27]. Similar results have also been reported for Ca4Mn(DPDP)5 [28,39] (Figure 7). In vitro experiments suggest that the tumoricidal efficacy is mediated by DPDP or PLED and not by their corresponding manganese complexes (Figure 8), hypothetically by an “iron starvation” mechanism [39,40]. These properties of DPDP and PLED is of course promising when it comes to fulfil the necessary requirements of a selective protectant. The anticancer effects of DPDP and PLED are in itself striking and appear worth to follow up.
Furthermore, copper deficiency induced, for example, by the copper chelator tetrathiomolybdate suppresses tumour growth and angiogenesis in an inflammatory breast cancer xenograft in nude mice and Her2/neu cancer-prone transgenic mice [41]. The high affinity of Cu2+ for DPDP or PLED may at least theoretically induce copper deficiency and thereby suppress tumour growth, but at same time it may induce peripheral neuropathy and hence increase the neuronal insult by adding to the effect of oxaliplatin. The finding that the platinum- and copper-binding chelator STS significantly impairs the tumoricidal efficacy of cisplatin in pediatric patients with disseminated solid tumours but not in patients with localized, non-metastatic solid tumours [14,15], apparently illustrates the difficulties to predict the tumoricidal effect of copper deficiency. Whereas the signalling networks that integrate fluctuations in the abundance of growth factors, nutrients and metabolites are well established, the discovery of signalling molecules capable of mediating similar functions depending on copper availability is rudimentary [42].
Crucial for achieving the goal of a selective platinum chelation drug, is the pharmacokinetic and pharmacodynamic properties of the actual platinum drug, in this case oxaliplatin. The very high distribution volume of Pt2+ after intravenous administration of oxaliplatin, exceeding 300 litres [43], is of an immense importance. That is a property promoting rapid intracellular uptake and cellular retention. The high distribution volume of Pt2+ is apparently due to the lipophilic character of the Pt-DACH-Cl2-complex and the subsequent binding of platinum to proteins, DNA, and other cellular constituents [43].
Oxaliplatin undergoes extensive non-enzymatic biotransformation in plasma ultrafiltrate and urine and at least seventeen Pt2+-containing metabolites are observed after 24 hours [43]. A crucial point is of course whether these metabolites contribute to the tumoricidal efficacy of oxaliplatin. A retrospective comparison by McWhinney et al. 2009 [44] of the neurotoxicity versus the response rate for platinum drug treatment from fourteen Pt2+-containing trials, including oxaliplatin, cisplatin and carboplatin, did not indicate any association between neurotoxicity and tumoricidal efficacy. McWhinney et al. concluded that neurotoxicity is not merely a ‘‘necessary evil’’ but can be approached as an avoidable side effect of a platinum agent.
The Pt(DACH)Cl2 in the plasma ultrafiltrate from 10 patients receiving oxaliplatin was determined using high performance liquid chromatography and atomic absorption spectrometry [45]. Less than 3% was found undergoing biotransformation into the cell permeable and active Pt(DACH)-Cl2 metabolite (Figure 2). After uptake into the tumour cell (or the normal cell), either by diffusion or active transport and due to the low intracellular concentration of chloride, PtDACH-Cl2 will undergo sequentially hydrolyses of the chlorides and enable crosslinking at guanine residues at N7 (Figure 2) [17]. Key elements in the tumoricidal activity of oxaliplatin or cisplatin are hence (i) intracellular hydrolysis, (ii) binding to guanine bases, (iii) distortion of DNA, and (iv) changing its interactions with proteins, leading to cell killing by apoptosis.
Most platinum anticancer complexes have the general formula cis-[PtX2(NHR2)2], in which R is an organic fragment and X is a leaving group, such as chloride in cisplatin or a chelating carboxylate in oxaliplatin. Typically, platinum coordination compounds have thermodynamic strength of 100 kJ/mol or below, much weaker than covalent, C—C and C—N or C—O single and double bonds, which are between 250 and 500 kJ/mol. However, the ligand-exchange behaviour of Pt compounds is quite slow, which gives them a high kinetic stability and results in ligand-exchange reactions of minutes to days, rather than microseconds to seconds for many other coordination compounds. Furthermore, Pt2+has a strong thermodynamic preference for binding to S-donor ligands, and with so many cellular platinophiles, i.e., S-donor ligands, such as glutathione, methionine, as competing ligands in the cytosol it appears highly relevant to ask whether these few percent of PtDACH-Cl2 formed will ever reach DNA [17].
The answer to the question seems to be migration of Pt2+ from an S-ligand to purine N7, according to Reedijk 2003 [17]. The same arguments may also be valid for the two amines in DPDP and PLED, involved in binding of Cu2+ [13], and hypothetically in binding of Pt2+ in competition with the dominating non-tumoricidal Pt2+ ligands (Figure 6). Stoichiometrically, the 5 µmol/kg MnDPDP dose used in the Coriat study [24] is equivalent to that of the standard dose of 85 mg/m2 oxaliplatin in a 90 kg patient with a body area of 2 m2. As the dose limiting factor in the use of MnDPDP is neuronal retention of manganese and in case DPDP chelation therapy shows to be clinically feasible, there is apparently room for increasing the DPDP dose.
We conclude that the devastating worthening of oxaliplatin-associated CIPN in the POLAR trials, as presented by Pfeiffer et al., 2022 [19], presumably was due to co-retention of Ca4Mn(DPDP)-associated Mn2+ and oxaliplatin-associated Pt2+ in DRG neurons, causing oxidative and nitrosative stress and site specific Tyr nitration and irreversible inactivation of the mitochondrial MnSOD enzyme and cytochrome c. Two extremely devastating events that lead to serious injuries of the peripheral sensory nerve system. If Pt2+ binds to DPDP with high enough affinity in vivo as previously demonstrated under in vitro condition by Stehr et al. [13], use of manganese-free DPDP may in fact solve the problem with oxaliplatin-associated CIPN. Furthermore, using a drug administration design identical to that used by Coriat et al. 2014 might potentially enabling a safe use of MnDPDP [and maybe also Ca4Mn(DPDP)5] to prevent oxaliplatin-associated CIPN, in case DPDP fails. The most important lesson to be learn from the POLAR trials is the danger to base and proceed from phase II into extensive phase III trials in an ad hoc manner, without supportive phase II data.
Author Contributions
Conceptualization: J.O.G.K. and P.J. Methodology: Not applicable for a review.; Software: not applicable for a review.; Validation: not applicable for a review.; Formal analysis: not applicable.; Investigation: not applicable.; Resources: J.O.G.K.; Writing original draft preparation: J.O.G.K.; Writing: review and editing: J.O.G.K and P.J.; Visualization: not applicable.; Supervision: not applicable.; Project administration: not applicable.; Funding acquisition: J.O.G.K. All authors have read and agreed to the published version of the manuscript.
Funding
This review was funded by Karlsson-Tuner Invest AS.
Conflicts of Interest
J.O.G.K. and P.J. are founders of PledPharma AB.; J.O.G.K. is a former employee of PledPharma; P.J. is a former scientific advisors to PledPharma; J.O.G.K. is the first inventor on three granted patent families (e.g., US8377969, US8633174, US9187509 and US10688087) covering the therapeutic use of calmangafodipir [Ca4Mn(DPDP)5] , which are owned by PledPharma/Egetis; J.O.G.K. and P.J. are, furthermore, inventors on a granted patent family (e.g., US10888553) covering the therapeutic use of fodipir (DPDP) to treat platinum-induced peripheral sensory neuropathy, which is owned by Karlsson-Tuner Invest AS; P.J. is scientific advisor to and owns shares in IC Targets AS, a company developing mangafodipir (MnDPDP) as a contrast agent for MRI.
References
- Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Tangen CM, Ungerleider JS, Emerson WA, Tormey DC, Glick JH, Veeder MH, Mailliard JA. Fluorouracil plus levamisole as effective adjuvant therapy after resection of stage III colon carcinoma: a final report. Ann Intern Med. 1995 Mar 1;122(5):321-6. [CrossRef]
- O'Connell MJ, Laurie JA, Kahn M, Fitzgibbons RJ Jr, Erlichman C, Shepherd L, Moertel CG, Kocha WI, Pazdur R, Wieand HS, Rubin J, Vukov AM, Donohue JH, Krook JE, Figueredo A. Prospectively randomized trial of postoperative adjuvant chemotherapy in patients with high-risk colon cancer. J Clin Oncol. 1998 Jan;16(1):295-300. [CrossRef]
- de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, Hendler D, de Braud F, Wilson C, Morvan F, Bonetti A. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000 Aug;18(16):2938-47. Corrected and republished in: J Clin Oncol. 2023 Nov 20;41(33):5080-5089. [CrossRef]
- André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, Topham C, Zaninelli M, Clingan P, Bridgewater J, Tabah-Fisch I, de Gramont A; Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) Investigators. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004 Jun 3;350(23):2343-51. [CrossRef]
- O'Neil BH, Goldberg RM. Innovations in chemotherapy for metastatic colorectal cancer: an update of recent clinical trials. Oncologist. 2008 Oct;13(10):1074-83. Epub 2008 Oct 15. [CrossRef]
- Sørbye H, Glimelius B, Berglund A, Fokstuen T, Tveit KM, Braendengen M, Øgreid D, and Dahl O (2004). Multicenter phase II study of Nordic fluorouracil and folinic acid bolus schedule combined with oxaliplatin as first-line treatment of metastatic colorectal cancer. J Clin Oncol 22, 31–38.
- Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004 Jan 1;22(1):23-30. Epub 2003 Dec 9. Corrected and republished in: J Clin Oncol. 2023 Jul 1;41(19):3461-3468. [CrossRef]
- Ducreux M, Bennouna J, Hebbar M, Ychou M, Lledo G, Conroy T, Adenis A, Faroux R, Rebischung C, Bergougnoux L, Kockler L, Douillard JY; GI Group of the French Anti-Cancer Centers. Capecitabine plus oxaliplatin (XELOX) versus 5-fluorouracil/leucovorin plus oxaliplatin (FOLFOX-6) as first-line treatment for metastatic colorectal cancer. Int J Cancer. 2011 Feb 1;128(3):682-90. [CrossRef]
- André T, Iveson T, Labianca R, Meyerhardt JA, Souglakos I, Yoshino T, Paul J, Sobrero A, Taieb J, Shields AF, Ohtsu A, Grothey A, Sargent DJ; for the IDEA Steering Committee. The IDEA (International Duration Evaluation of Adjuvant Chemotherapy) Collaboration: Prospective Combined Analysis of Phase III Trials Investigating Duration of Adjuvant Therapy with the FOLFOX (FOLFOX4 or Modified FOLFOX6) or XELOX (3 versus 6 months) Regimen for Patients with Stage III Colon Cancer: Trial Design and Current Status. Curr Colorectal Cancer Rep. 2013;9(3):261-269. [CrossRef]
- Albers JW, Chaudhry V, Cavaletti G, Donehower RC. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database of Systematic Reviews 2014, Issue 3. Art. No.: CD005228. [CrossRef]
- Areti A, Yerra VG, Naidu V, Kumar A. Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014 Jan 18;2:289-95. [CrossRef]
- Brouwers EE, Huitema AD, Beijnen JH, Schellens JH. Long-term platinum retention after treatment with cisplatin and oxaliplatin. BMC Clin Pharmacol. 2008 Sep 17;8:7. [CrossRef]
- Stehr JE, Lundström I, Karlsson JOG. Evidence that fodipir (DPDP) binds neurotoxic Pt2+ with a high affinity: An electron paramagnetic resonance study. Sci Rep. 2019 Nov 1;9(1):15813. [CrossRef]
- Brock PR, Maibach R, Childs M, Rajput K, Roebuck D, Sullivan MJ, Laithier V, Ronghe M, Dall'Igna P, Hiyama E, Brichard B, Skeen J, Mateos ME, Capra M, Rangaswami AA, Ansari M, Rechnitzer C, Veal GJ, Covezzoli A, Brugières L, Perilongo G, Czauderna P, Morland B, Neuwelt EA. Sodium Thiosulfate for Protection from Cisplatin-Induced Hearing Loss. N Engl J Med. 2018 Jun 21;378(25):2376-2385. [CrossRef]
- Freyer DR, Chen L, Krailo MD, Knight K, Villaluna D, Bliss B, Pollock BH, Ramdas J, Lange B, Van Hoff D, VanSoelen ML, Wiernikowski J, Neuwelt EA, Sung L. Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017 Jan;18(1):63-74. Epub 2016 Dec 1. Erratum in: Lancet Oncol. 2017 Jun;18(6):e301. PMID: 27914822; PMCID: PMC5520988. [CrossRef]
- Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol. 2010 Jun;77(6):887-94. . Epub 2010 Feb 16. [CrossRef]
- Reedijk J. New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc Natl Acad Sci U S A. 2003 Apr 1;100(7):3611-6. Epub 2003 Mar 24. [CrossRef]
- Taylor SW, Laughlin RS, Kumar N, Goodman B, Klein CJ, Dyck PJ, Dyck PJB. Clinical, physiological and pathological characterisation of the sensory predominant peripheral neuropathy in copper deficiency. J Neurol Neurosurg Psychiatry. 2017 Oct;88(10):839-845. Epub 2017 Aug 5. [CrossRef]
- Pfeiffer P, Lustberg M, Näsström J, Carlsson S, Persson A, Nagahama F, Cavaletti G, Glimelius B, Muro K. Calmangafodipir for Prevention of Oxaliplatin-Induced Peripheral Neuropathy: Two Placebo-Controlled, Randomized Phase 3 Studies (POLAR-A/POLAR-M). JNCI Cancer Spectr. 2022 Nov 1;6(6):pkac075. [CrossRef]
- Glimelius B, Manojlovic N, Pfeiffer P, Mosidze B, Kurteva G, Karlberg M, Mahalingam D, Buhl Jensen P, Kowalski J, Bengtson M, Nittve M, Näsström J. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): a placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018 Mar;57(3):393-402. Epub 2017 Nov 15. [CrossRef] [PubMed]
- Karlsson JOG, Jynge P. Is it possible to draw firm conclusions from the PLIANT trial? Acta Oncol. 2018 Jun;57(6):862-864. Epub 2017 Dec 15. [CrossRef]
- Karlsson JOG, Jynge P, Ignarro LJ. Exacerbated Neuropathy in POLAR A and M Trials Due to Redox Interaction of PledOx-Associated Mn2+ and Oxaliplatin-Associated Pt2+. Antioxidants (Basel). 2023 Mar 1;12(3):608. [CrossRef]
- Yri OE, Vig J, Hegstad E, Hovde O, Pignon I, Jynge P. Mangafodipir as a cytoprotective adjunct to chemotherapy--a case report. Acta Oncol. 2009;48(4):633-5. [CrossRef]
- Coriat R, Alexandre J, Nicco C, Quinquis L, Benoit E, Chéreau C, Lemaréchal H, Mir O, Borderie D, Tréluyer JM, Weill B, Coste J, Goldwasser F, Batteux F. Treatment of oxaliplatin-induced peripheral neuropathy by intravenous mangafodipir. J Clin Invest. 2014 Jan;124(1):262-72. Epub 2013 Dec 20. [CrossRef]
- Laurent A, Nicco C, Chéreau C, Goulvestre C, Alexandre J, Alves A, Lévy E, Goldwasser F, Panis Y, Soubrane O, Weill B, Batteux F. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005 Feb 1;65(3):948-56.
- Alexandre J, Nicco C, Chéreau C, Laurent A, Weill B, Goldwasser F, Batteux F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006 Feb 15;98(4):236-44. [CrossRef]
- Doroshow, JH. Redox modulation of chemotherapy-induced tumor cell killing and normal tissue toxicity. J Natl Cancer Inst. 2006 Feb 15;98(4):223-5. [CrossRef]
- Karlsson JO, Kurz T, Flechsig S, Näsström J, Andersson RG. Superior therapeutic index of calmangafodipir in comparison to mangafodipir as a chemotherapy adjunct. Transl Oncol. 2012 Dec;5(6):492-502. Epub 2012 Dec 1. [CrossRef]
- Toft KG, Hustvedt SO, Grant D, Martinsen I, Gordon PB, Friisk GA, Korsmo AJ, Skotland T. Metabolism and pharmacokinetics of MnDPDP in man. Acta Radiol. 1997 Jul;38(4 Pt 2):677-89. [CrossRef]
- Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, Dagostino C, Ryerse J, Rausaria S, Kamadulski A, Neumann WL, Salvemini D. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci. 2012 May 2;32(18):6149-60. [CrossRef]
- Janes K, Doyle T, Bryant L, Esposito E, Cuzzocrea S, Ryerse J, Bennett GJ, Salvemini D. Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain. 2013 Nov;154(11):2432-2440. Epub 2013 Jul 25. [CrossRef]
- Radi, R. Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res. 2013 Feb 19;46(2):550-9. Epub 2012 Nov 16. [CrossRef]
- Bartesaghi S, Radi R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018 Apr;14:618-625. Epub 2017 Sep 19. [CrossRef]
- Bratosin D, Estaquier J, Petit F, Arnoult D, Quatannens B, Tissier JP, Slomianny C, Sartiaux C, Alonso C, Huart JJ, Montreuil J, Ameisen JC. Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 2001 Dec;8(12):1143-56. [CrossRef]
- Schmidt PP, Toft KG, Skotland T, Andersson K. Stability and transmetallation of the magnetic resonance contrast agent MnDPDP measured by EPR. J Biol Inorg Chem. 2002 Mar;7(3):241-8. Epub 2001 Oct 19. [CrossRef]
- Rocklage SM, Cacheris WP, Quay SC, Hahn FE, Raymond KN. Manganese(II) N,N′-dipyridoxylethylenediamine-N,N′diacetate 5, 5-bis(phosphate). Synthesis and characterization of a paramagnetic chelate for magnetic resonance imaging enhancement. Inorg Chem. 1989;28:477–485. [CrossRef]
- Rocklage SM, et al. Structural and Thermodynamic Characterization of Manganese (II) N,N′.
- -Dipyridoxylethylenediamine-N, N′-diacetate. A Novel Manganese (II) Chelate. Inorg. Chem. 1988;27:3530–3534. [CrossRef]
- Shannon, RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. 1976;A32:751–767. [CrossRef]
- Karlsson JO, Ignarro LJ, Lundström I, Jynge P, Almén T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov Today. 2015 Apr;20(4):411-21. Epub 2014 Nov 20. [CrossRef]
- Kurz T, Grant D, Andersson RG, Towart R, De Cesare M, Karlsson JO. Effects of MnDPDP and ICRF-187 on Doxorubicin-Induced Cardiotoxicity and Anticancer Activity. Transl Oncol. 2012 Aug;5(4):252-9. Epub 2012 Aug 1. PMID: 22937177; PMCID: PMC3431035. [CrossRef]
- Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C, De Carvalho M, Mesri EA, Robins DM, Dick RD, Brewer GJ, Merajver SD. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002 Sep 1;62(17):4854-9.
- Ge EJ, Bush AI, Casini A, Cobine PA, Cross JR, DeNicola GM, Dou QP, Franz KJ, Gohil VM, Gupta S, Kaler SG, Lutsenko S, Mittal V, Petris MJ, Polishchuk R, Ralle M, Schilsky ML, Tonks NK, Vahdat LT, Van Aelst L, Xi D, Yuan P, Brady DC, Chang CJ. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022 Feb;22(2):102-113. Epub 2021 Nov 11. [CrossRef]
- Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E. Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res. 2000 Apr;6(4):1205-18.
- McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009 Jan;8(1):10-6. PMID: 19139108; PMCID: PMC2651829. [CrossRef]
- Shord SS, Bernard SA, Lindley C, Blodgett A, Mehta V, Churchel MA, Poole M, Pescatore SL, Luo FR, Chaney SG. Oxaliplatin biotransformation and pharmacokinetics: a pilot study to determine the possible relationship to neurotoxicity. Anticancer Res. 2002 Jul-Aug;22(4):2301-9.
Figure 1.
Schematic Illustration of the tightly regulated cellular handling of copper, where CuSOD (SOD) and cytochrome c (CytC) are of particular importance for cells at high energy demand, such as Dorsal Root Ganglion (DRG) cells. The Copper Transporter 1 (CTR 1) is implicated in the uptake of oxaliplatin and other Pt2+-containing drugs.
Figure 1.
Schematic Illustration of the tightly regulated cellular handling of copper, where CuSOD (SOD) and cytochrome c (CytC) are of particular importance for cells at high energy demand, such as Dorsal Root Ganglion (DRG) cells. The Copper Transporter 1 (CTR 1) is implicated in the uptake of oxaliplatin and other Pt2+-containing drugs.
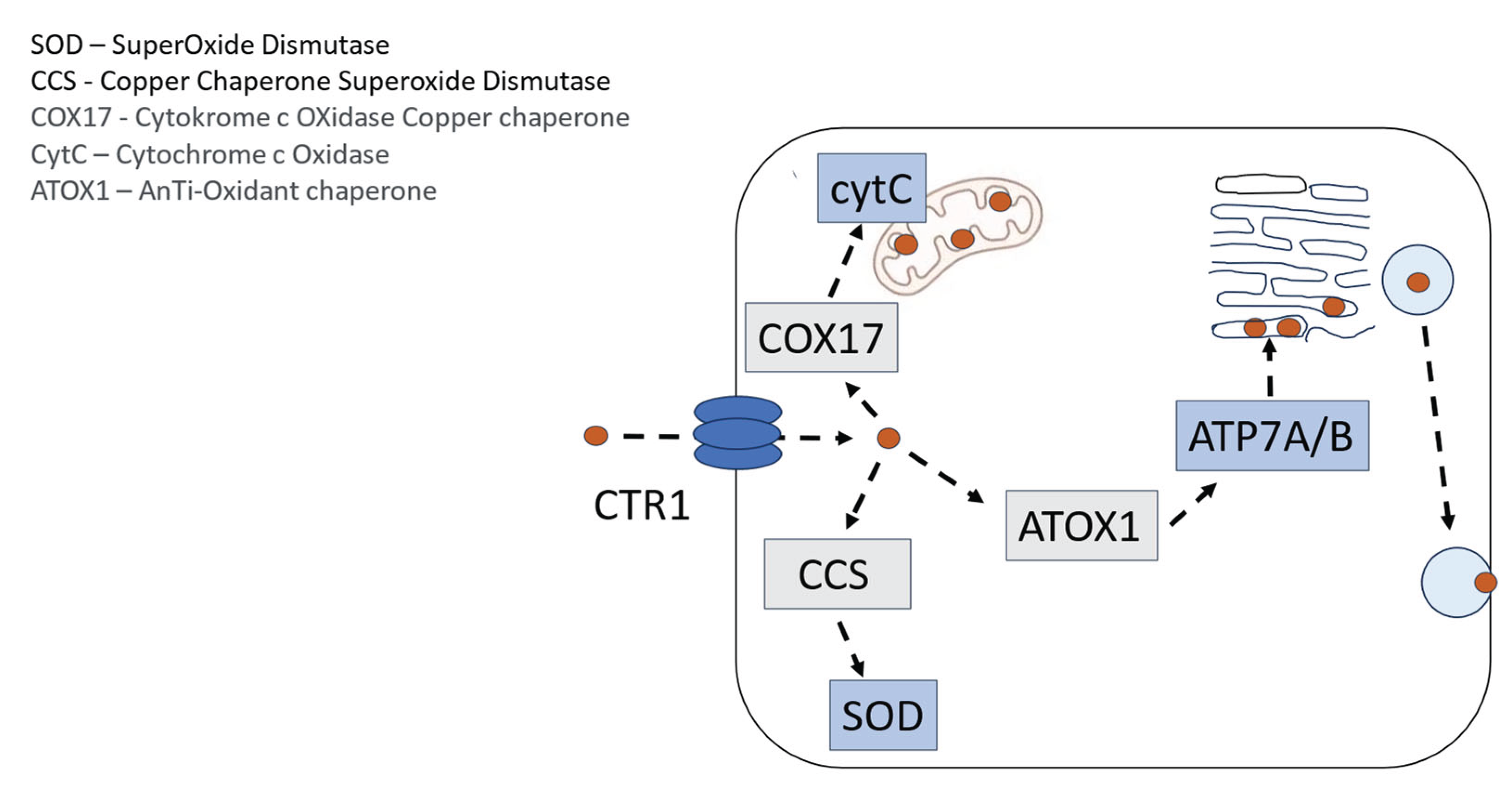
Figure 2.
Transport and non-enzymatic metabolism of oxaliplatin into PtDACH-Cl2 that after sequential intracellular exchange of the chlorides against water (aquation) crosslinks purines at N7.
Figure 2.
Transport and non-enzymatic metabolism of oxaliplatin into PtDACH-Cl2 that after sequential intracellular exchange of the chlorides against water (aquation) crosslinks purines at N7.
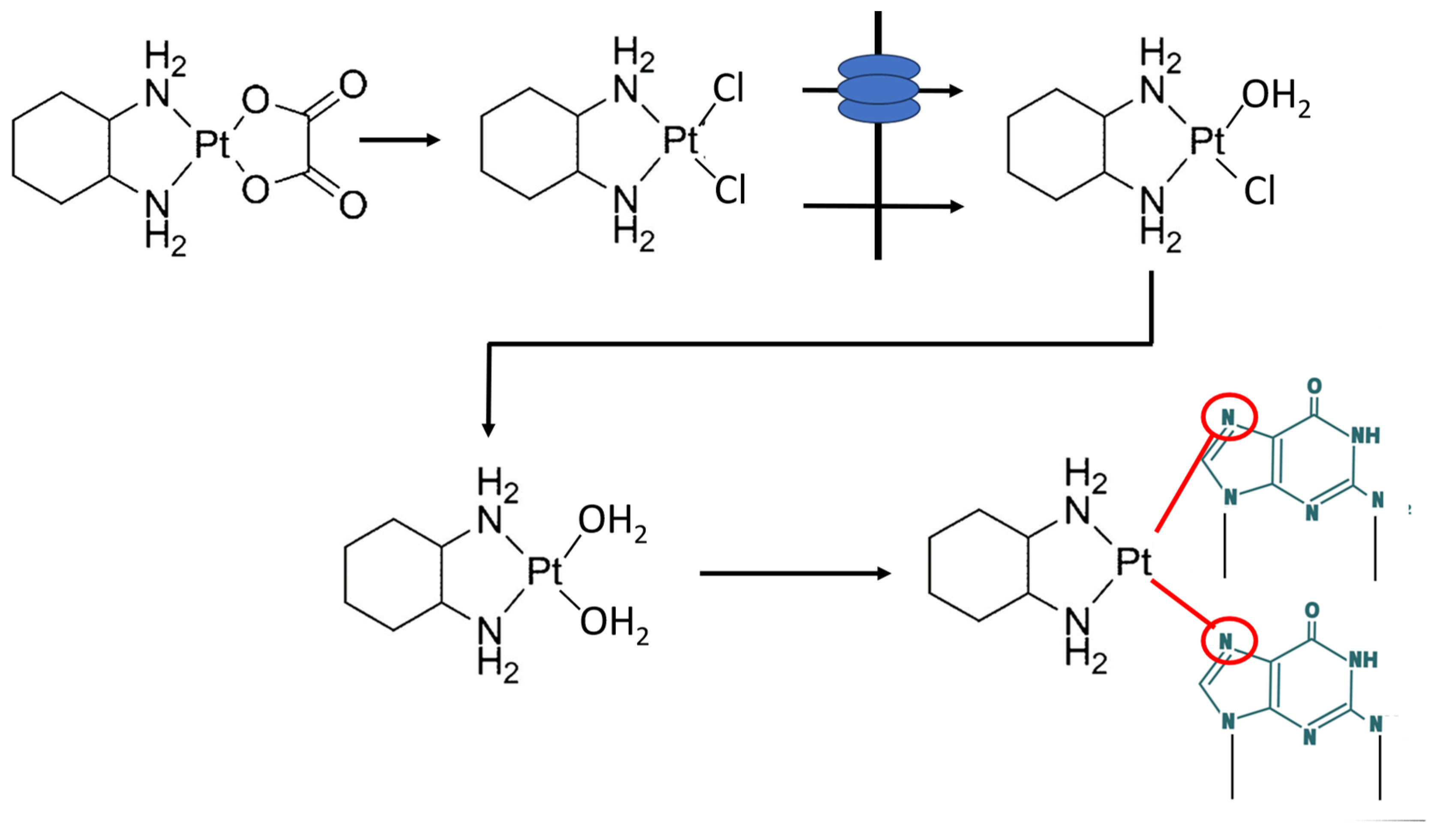
Figure 3.
Schematic illustration of a DRG and two important targets for Pt2+-associated oxidative and nitrosative stress, namely MnSOD and cytochrome c (Cyt c), and the transition metal-driven chemistry leading to cite specific tyrosine nitration (Tyr34 of the MnSOD-enzyme and Tyr74 of cytochrome c) and irreversible inactivation of MnSOD and disruption of normal electron transfer in the respiratory chain.
Figure 3.
Schematic illustration of a DRG and two important targets for Pt2+-associated oxidative and nitrosative stress, namely MnSOD and cytochrome c (Cyt c), and the transition metal-driven chemistry leading to cite specific tyrosine nitration (Tyr34 of the MnSOD-enzyme and Tyr74 of cytochrome c) and irreversible inactivation of MnSOD and disruption of normal electron transfer in the respiratory chain.
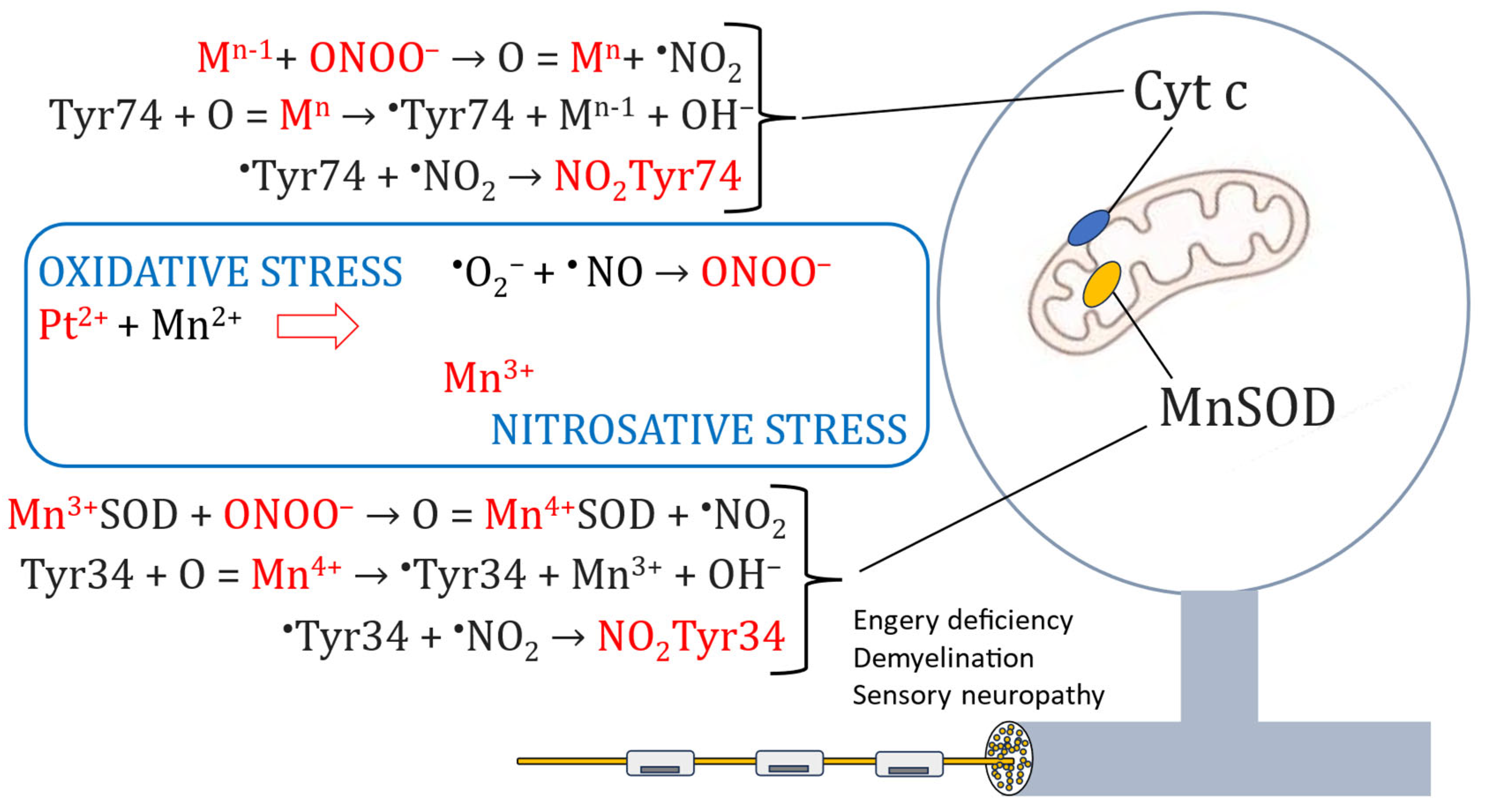
Figure 4.
Schematic illustration of the connection between oxidative and nitrosative stress. Under normal redox conditions MnSOD (together with CuSODs) will keep the production of superoxide (O2•-) in check by dismutation O2•- into hydrogen peroxide (H2O2) and water. At severe oxidative stress, however, the production of O2•- will exceed the capacity of the SOD enzymes and O2•- will react with •NO and forming highly toxic peroxynitrite (ONOO-). Driven by reduction of Mn4+ to Mn3+, ONOO- will nitrate tyrosine residues (NO2-Tyr), which will cause mitochondrial MnSOD inactivation and disruption of electron transport through the respiratory chain. Two highly devastating effects on high energy demanding tissue, such as that of the peripheral sensory nerve system.
Figure 4.
Schematic illustration of the connection between oxidative and nitrosative stress. Under normal redox conditions MnSOD (together with CuSODs) will keep the production of superoxide (O2•-) in check by dismutation O2•- into hydrogen peroxide (H2O2) and water. At severe oxidative stress, however, the production of O2•- will exceed the capacity of the SOD enzymes and O2•- will react with •NO and forming highly toxic peroxynitrite (ONOO-). Driven by reduction of Mn4+ to Mn3+, ONOO- will nitrate tyrosine residues (NO2-Tyr), which will cause mitochondrial MnSOD inactivation and disruption of electron transport through the respiratory chain. Two highly devastating effects on high energy demanding tissue, such as that of the peripheral sensory nerve system.
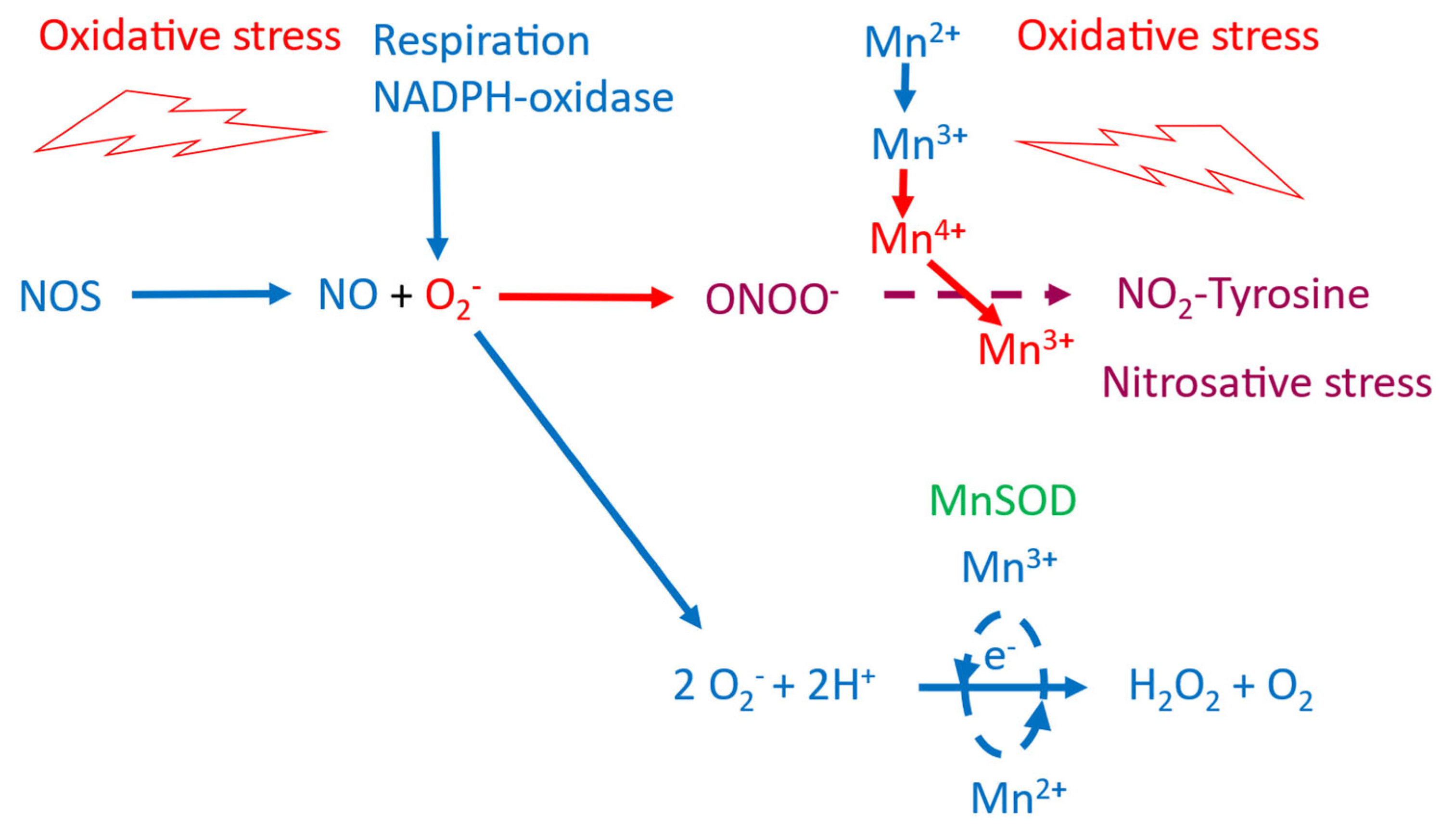
Figure 5.
The EPR standard curve for free Mn2+ is shown in the left graph. Where the double integral of the EPR signal (insert) in arbitrary units is plotted against the concentration of MnCl2. Metal exchange of MnDPDP by Zn2+ and Pt2+is shown in the right graphs. The EPR invisibility of MnDPDP was used to monitor exchange of Mn2+ for Zn2+and Pt2+ [13].
Figure 5.
The EPR standard curve for free Mn2+ is shown in the left graph. Where the double integral of the EPR signal (insert) in arbitrary units is plotted against the concentration of MnCl2. Metal exchange of MnDPDP by Zn2+ and Pt2+is shown in the right graphs. The EPR invisibility of MnDPDP was used to monitor exchange of Mn2+ for Zn2+and Pt2+ [13].
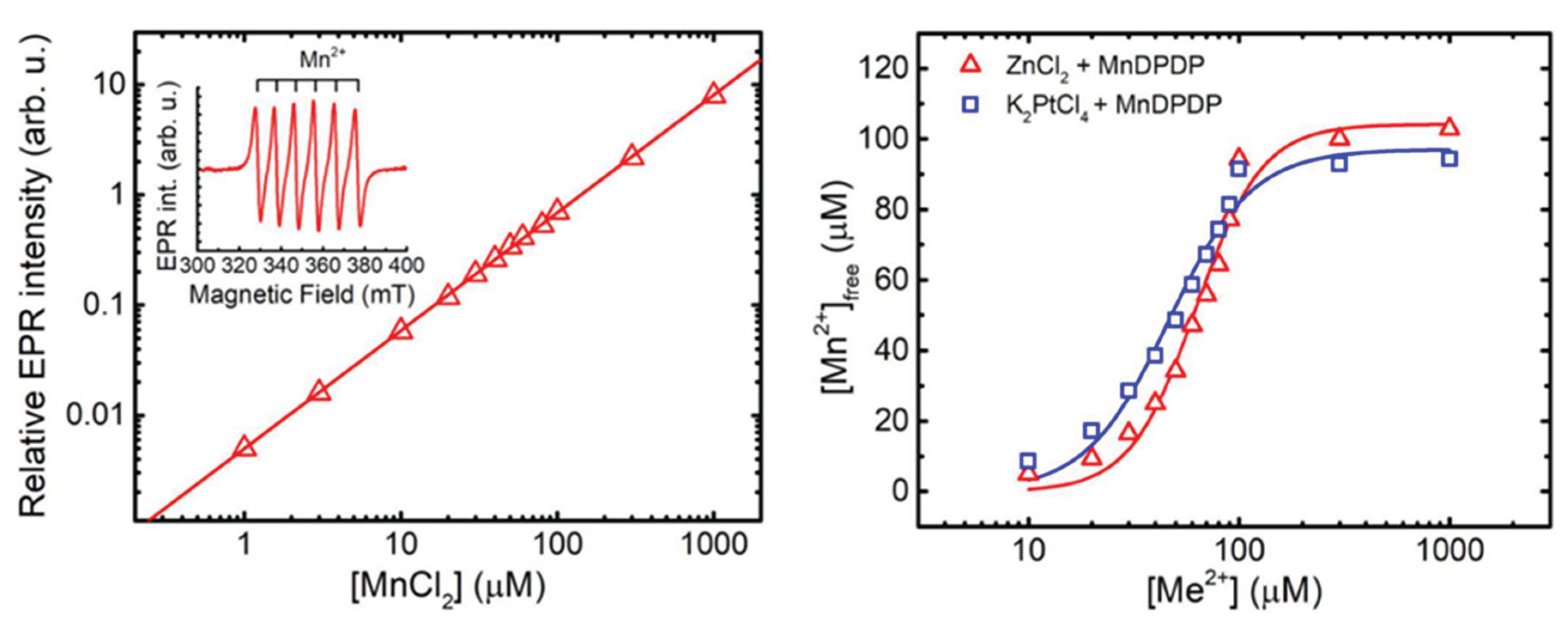
Figure 6.
Hypothetically and similar to Cu2+, Pt2+ may bind to DPDP and PLED with 4 coordinates to the amines and phenolates, in a square geometry [36,37].
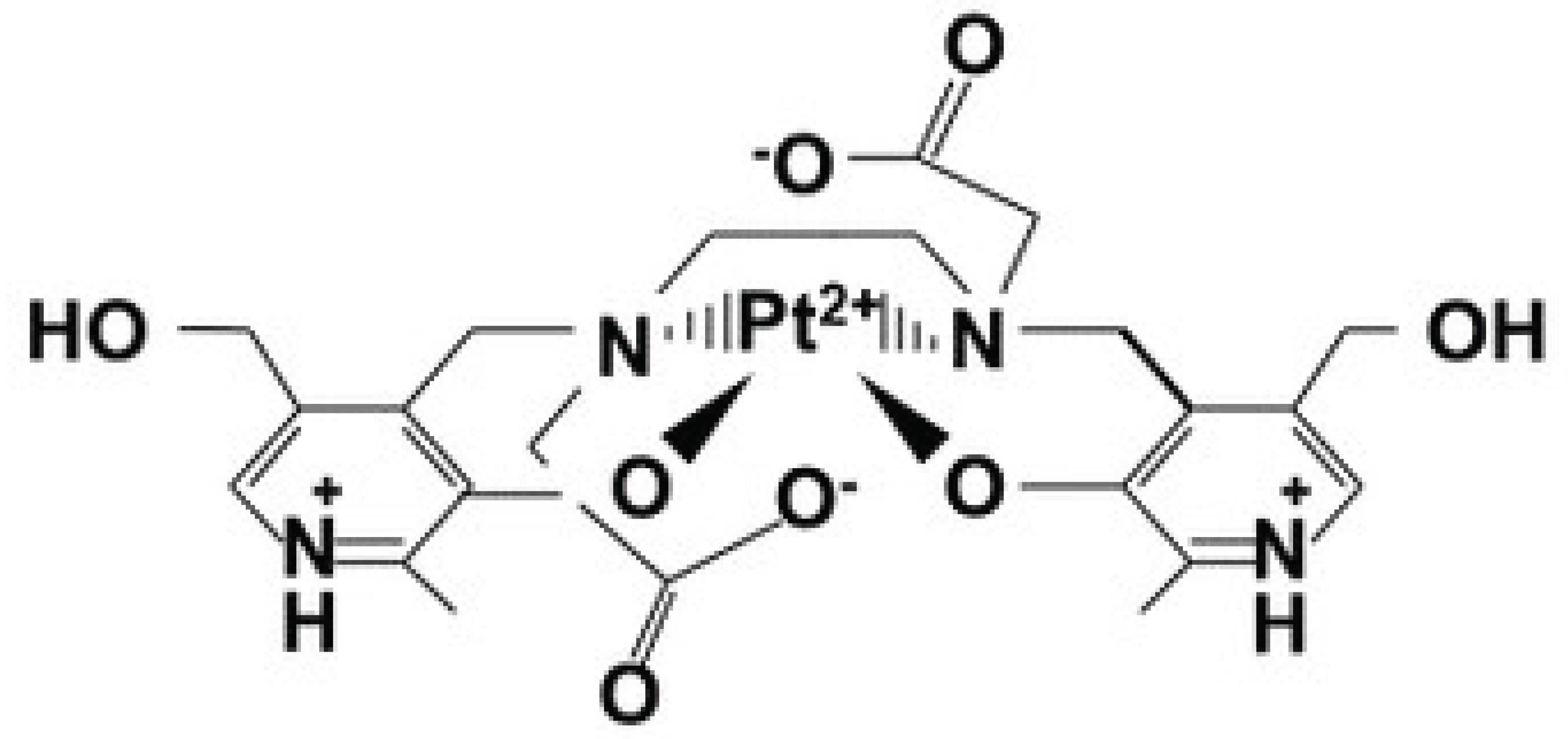
Figure 7.
Antitumor effect of oxaliplatin (10 mg/kg) in the absence and presence of 6.5 and 32.5 µmol/kg calmangafodipir [Ca4Mn(DPDP)5] in immune competent balb c mice implanted with CT26 tumours. Results are expressed as mean ± SEM (n = 5) [28].
Figure 7.
Antitumor effect of oxaliplatin (10 mg/kg) in the absence and presence of 6.5 and 32.5 µmol/kg calmangafodipir [Ca4Mn(DPDP)5] in immune competent balb c mice implanted with CT26 tumours. Results are expressed as mean ± SEM (n = 5) [28].
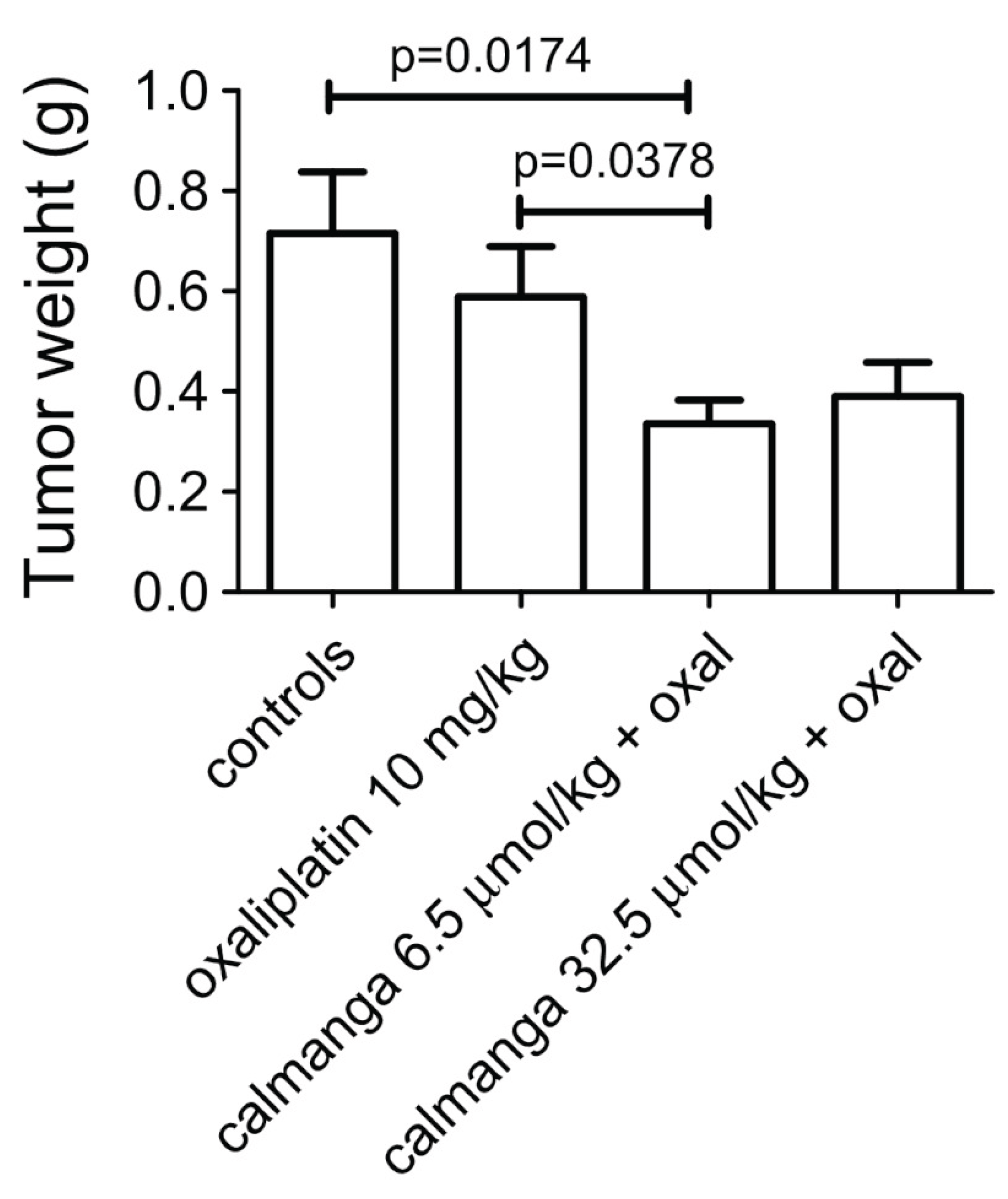
Figure 8.
Cytotoxic effects of increasing concentrations of CaCl2, fodipir (DPDP), calfodipir [CaDPDP), and calmangafodipir [Ca4Mn(DPDP)5] in CT26 tumour cells (mean ± SD; n = 3) [28].
Figure 8.
Cytotoxic effects of increasing concentrations of CaCl2, fodipir (DPDP), calfodipir [CaDPDP), and calmangafodipir [Ca4Mn(DPDP)5] in CT26 tumour cells (mean ± SD; n = 3) [28].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



