
Submitted:
01 March 2024
Posted:
13 March 2024
You are already at the latest version
Abstract
Lumpy skin disease (LSD), caused by a virus within the Poxviridae family and Capripoxvirus genus, induces nodular skin lesions in cattle, spreading through direct contact and insect vectors, significantly affecting global cattle farming. Despite available vaccines, their effectiveness is limited by poor bioavailability and adverse effects. Our study aimed to identify potential inhibitors targeting LSD-associated DNA polymerase protein, selecting LSDV039 for further investigation through comprehensive analysis and computational methods. Virtual screening revealed rhein and taxifolin as potent binders among 380 phytocompounds, with respective affinities of -8.965 and -7.195 kcal/mol. Canagliflozin and tepotinib exhibited strong affinities (-9.858 and -8.856 kcal/mol) among 718 FDA-approved antiviral drugs. Molecular dynamics simulations of canagliflozin, tepotinib, rhein, and taxifolin highlighted taxifolin's superior stability and binding energy. Rhein displayed compactness in RMSD and RMSF but fluctuations in Rg and SASA, while canagliflozin demonstrated stability compared to tepotinib. The study highlights the promising potential of repurposed drugs and phytocompounds as LSD therapeutics. However, extensive validation through in-vitro, in-vivo, and clinical trials is crucial for their practical application.
Keywords:
Lumpy skin disease
; DNA polymerase
; potential inhibitors
; phytocompounds
; antiviral drug compounds
; molecular docking
1. Introduction
The Lumpy skin disease (LSD) is a contagious viral disease that affects ruminant animals such as cattle, water buffaloes, and giraffes, posing a significant threat across borders [1,2,3,4]. This disease is attributed to the Lumpy skin disease virus (LSDV), a member of the Capripoxvirus genus within the Poxviridae family that exhibits close kinship with the sheep pox virus (SPPV) and goat pox viruses (GTPV) [5]. It has a genome spanning 151 kilobase pairs (kb) and 156 putative genes [6]. Similar to other poxviruses, LSDV possesses several conserved genes involved in its fundamental replicative mechanisms. Seven homologues of genes found in chordopoxviruses are associated with DNA replication. These genes, namely, LSDV039, LSDV077, LSDV082, LSDV083, LSDV112, LSDV133, and LSDV139, are either known or potentially involved in DNA replication processes [6]. However, the virus is most likely spread mechanically by blood-sucking arthropods such as flies, mosquitoes, and ticks. Direct contact between animals can also transmit the virus to a lesser extent [7,8]. Additionally, infected animals can spread the virus through milk, blood, nasal secretions, and saliva, which serve as alternative transmission routes through feeding or drinking [9]. Affected animals primarily exhibit symptoms such as fever, nodular skin lesions, dramatic decrease in milk production, and weight loss [10]. The introduction of LSDV into a herd can result in a high incidence rate, ranging from 5 to 45%, and a case death rate from 0.5% to 7.0% [11,12]. Consequently, LSD poses a significant economic threat to the livestock industry worldwide because it causes vast economic losses, including abortions in females and sterility in males [13].
It was first reported in Africa (Zambia) in 1929 and was identified as a communicable disease in the 1940s [14]. It has also spread to countries in Central and Eastern Africa, the Middle East, Asia, and Eastern Europe [15]. LSD outbreak data were accessed from the World Organization for Animal Health (WOAH) World Animal Health Information System (WAHIS) database in Southeast Asian countries during the study period between October 2020 and October 2021. During the epidemic period, 866 LSD outbreaks were reported in six Southeast Asian countries, including 1,758,923 susceptible cattle, 93,465 cases, 5,936 deaths, and 1,117 culled cattle [16]. In Bangladesh, the outbreak first emerged in the Chattogram region in July 2019 and then quickly spread throughout the entire country [17].
To date, the scientific community has preferred some homologous and heterologous vaccines to develop immunity owing to their cross-protection against lumpy skin diseases, such as Kenyan sheep and goat pox (KSGP) O-180 strain vaccines and Gorgan goat pox (GTP) vaccines. While the KSGPO-180 vaccine failed to protect cattle against LSDV, the Gorgan GTP vaccine successfully prevented the clinical symptoms of LSD in all vaccinated calves [18]. Certain SPPV vaccines provide only partial or incomplete protection against LSDV. Despite the use of goat pox vaccine virus KSGPO-240 and Romanian SPPV strains, their attenuation levels were insufficient to ensure the safety of cattle. Consequently, vaccinated animals remain ill [19]. The Neethling vaccine was developed in 2016 and tested in six Balkan countries. Its effectiveness was found to be average at 79.8% (95% CI: 73.2–84.7) in the Balkan region, with varying rates in different countries. Albania had a 62.5% effectiveness rate, whereas Bulgaria and Serbia had effectiveness rates of 97% [20].
Over the past three decades, many diseases have been treated using repurposed drugs. Zidovudine (AZT), the first successful repurposed drug used to treat Human Immunodeficiency Virus (HIV) in 1987, was originally developed to treat cancer [21,22]. Recently, a drug named Remdezivir (RDV), a repurposed drug approved by Food and Drug Administration (FDA) in 2020, was used to treat Covid-19 patients [23]. Additionally, a recent in vitro study showed that ivermectin, an antiparasitic drug, is effective against capripoxviruses [24]. This study found that ivermectin demonstrated significant inhibitory effects on viral replication and the attachment and penetration stages of the LSDV virus. Specifically, the results showed that ivermectin reduced viral replication by 99.82 and 99.87% at the replication stage, respectively. It also exhibited inhibitory effects of 68.38 and 25.01% at the attachment stage and 57.83 and 0.0% at the penetration stage [24]. Nevertheless, plant-based phytocompounds with antiviral activity are suitable natural solutions and show high efficacy in inhibiting several viral diseases, such as HIV and Covid-19 [25]. For the alternative treatment approach, propolis-alginate nanoparticles (Propolis-ALg NPs) have a potential therapeutic approach through different routes, including eye drops, oral routes, and topical spray. Transmission electron microscopy was used to characterize the propolis-ALg NPs. Propolis-ALg NPs effectively treat infected animals by reducing fever and boosting overall health [13]. Furthermore, colchicine, a potent natural alkaloid, has the potential to effectively treat a range of skin diseases, either as a standalone therapy or in combination with other drugs [26]. In this study, ivermectin was used as a control drug to serve as a benchmark for comparison with repurposed drugs and plant-based compounds investigated for their antiviral properties against LSDV. Although these have demonstrated some level of effectiveness, the efficacy and safety of these treatments for LSDV remain unclear. Considering these constraints, it is crucial to identify a specific drug that can effectively inhibit or reduce LSDV infection [27,28].
Currently, there is no cure for LSD, and available treatments mainly focus on relieving the symptoms of the disease [13]. Researchers are exploring new drug targets for LSD to develop more effective treatments. Computer-aided drug design is a valuable tool in this process because it allows for the design of new drugs based on the structure of protein targets within the virus [29]. Computational methods have been employed to analyze the protein targets of the LSD virus and design novel lead molecules or repurposed drugs for drug development. Target selection is a critical step in drug discovery, particularly for viruses with evasion mechanisms [29]. The identification of highly conserved and essential targets for viral survival and replication is crucial for successful drug development. Therefore, we targeted the DNA replication associated with DNA replication, including the DNA polymerase LSDV039, which is potentially involved in DNA replication.
Considering the morbidity and mortality, a truly effective antiviral drug against LSDV has emerged. Repurposed drugs [27,28,30], which can be rapidly used, are readily available, and show significance as antivirals, are nothing but a great choice to eradicate LSDV. Hence, this study aimed to identify LSDV inhibitors that could effectively inhibit viral replication. Virtual screening and molecular modeling techniques enable the identification of compounds (plant-based compounds and repurposed drugs) with high binding affinities for viral proteins. Further experimental validation will provide valuable insights into the effectiveness and safety of potential inhibitors and pave the way for the development of new therapeutic strategies against LSDV.
2. Materials and Methods
2.1. Target Selection and Validation
In this study, target proteins were screened after an extensive literature review (Supplementary Table S1). These proteins are essential for LSDV replication and survival of the LSDV [6]. Interestingly, most of these proteins share sequence similarities with sheeppox virus (SPPV) and goatpox (GTPV) [31]. The amino acid sequences of these proteins were obtained from UniProt KB (https://www.uniprot.org/), a widely used protein database, and their structures were predicted using AlphaFold2, an advanced protein structure prediction tool [32]. To validate the predicted structures, we used the PROCHECK [33] and ERRAT plot [34] programs available from the Structural Analysis and Verification server (SAVES) (http://nihserver.mbi.ucla.edu/SAVES), which is commonly used to assess the quality of protein structures [35]. The conserved domains and superfamilies of the selected proteins were analyzed using Chimera X software [36], InterProScan [37], and Pfam (Proteins Families Database) [38]. InterProScan provides structure-based categorization, domain and homologous superfamily predictions, and other high-level results. The Pfam database stores a curated set of sequence-aligned and curated data on protein families. Additionally, the active sites of the selected proteins were predicted using the SiteMap module of Schrödinger suite 2021-2 (Schrödinger, New York, United States), a suite of computational tools widely used in drug discovery and molecular modeling [39].
2.2. Ligand Selection
In the present study, we investigated the potential use of antiviral compounds as LSDV inhibitors. To create a dataset of active compounds, we conducted an extensive literature search of various databases and compiled a dataset comprising active phytocompounds and repurposed drug compounds. We created a library of active antiviral compounds by conducting a comprehensive search of related literature in various databases, including PubMed, Google Scholar, Web of Science, and Scopus [40]. We retrieved the 3D structures of active phytocompounds and drug compounds in SDF (Structure-Data File) format from the PubChem [41] and DrugBank [42] databases. The SDF format is a file format commonly used to represent chemical structures and associated data. Open Babel software was used to convert the SDF into the PDB format [43]. After obtaining the 3D structures of the phytocompounds, their molecular interactions, binding modes, and potential as LSDV inhibitors were investigated (Figure 1).
2.3. Ligand Preparation
LigPrep, a tool in the Schrödinger suite (Maestro 12.8) [44], was utilized for ligand preparation, which accounted for the metal-binding states, desalting, and generation of tautomers and stereoisomers. It also facilitated the generation of various possible ionization states at a targeted pH of 7.0 ± 2.0 using Epik. An OPLS4 force field was used during the preparation.
2.4. Protein Preparation
The first step in protein processing was to extract the desired protein structure from the AlphaFold2. Subsequently, the Protein Preparation Wizard (PrepWizard), part of the Schrödinger suite (Maestro 12.8) [45], was used to perform several preprocessing steps on the protein structure. These steps involve the addition of missing hydrogen atoms, side-chain optimization, correction of the ionization state of the protein, and assignment of bond orders and formal charges. In addition, advanced algorithms have been used to refine protein structures, such as the removal of water molecules and energy minimization using the OPLS4 force field.
2.5. Site map Analysis
The selected protein was deposited on a Site Map, a module of the Schrödinger suite (Maestro 12.8), for binding-site analysis [46]. This software employs advanced algorithms to identify binding sites, evaluate the location of binding sites with a high degree of confidence, and predict the druggability of these sites.
2.6. Receptor Grid Generation
The interaction region between the protein and ligand was determined by generating a receptor grid using the receptor grid generation tool in Maestro 12.8 [47,48]. This grid defined the specific area surrounding the active site of the protein, and its dimensions were established based on the x-, y-, and z-coordinates. The receptor grid box had a resolution centered at coordinates 4.2, 15.18, and 1.51 along the x-, y-, and z-axes, respectively.
2.7. Molecular Docking
2.7.1. Virtual Screening and Ligand Docking
During drug development, a computational approach known as virtual screening or virtual ligand screening is used to search small-molecule libraries and identify drug-like compounds that can bind to therapeutic targets [49]. It has been used to discover novel chemical entities in structure-based drug design. We assigned 718 compounds, including 381 phytocompounds and 337 drug compounds, to the virtual screening workflow (VSW) in Maestro 12.8 [50,51,52].
The high-throughput virtual screening (HTVS) mode of Glide is executed during the initial step. The top 50% of the resulting ligands were retained for further analysis in the subsequent stage, which involved Glide Standard Precision (SP). Once again, the top-scoring 50% of the ligands were selected to proceed with the Glide Extra Precision (XP) modes [53].
The screened compounds were again examined in the ligand-docking module in the extra precision (XP) mode, as it provides greater accuracy. The docking process used a flexible docking mode that automatically generated conformations for each ligand input. The G Score of the glide was examined as follows:
G Score = a × vdW+b × Coul + Lipo + H-bond + Metal + BuryP + RotB + site
Here, vdW is the Van der Waals energy, Coul is the Coulomb energy, Lipo is the lipophilic contact, HBond denotes hydrogen bonding, Metal means metal binding, BuryP indicates the penalty for buried polar groups, RotB means the penalty for freezing rotatable bonds, Site denotes polar interactions in the active site, and a=0.065 and b=0.130 are the coefficients of vdW and Coul, respectively.
Furthermore, additional settings were configured, including the incorporation of Epik state penalties into the docking score, execution of post-docking minimization, and computation of the root-mean-square deviation (RMSD) with respect to the input ligand geometries. The protocol provides the top-scoring ligands in the XP description based on the glide score and glide energy. Glide calculates the energies of a wide variety of interactions between ligands and proteins, such as hydrophobic interactions, hydrogen bonds, internal energies, pi-stacking interactions, salt bridges, desolvation, and RMSDs.
2.7.2. Free Energy Calculation by MM-GBSA
Following docking in Glide, both the receptor and ligands were considered Prime MM-GBSA. The Prime MM-GBSA module of the Schrodinger suite 2021-2 was employed to calculate the binding free energies of the protein-ligand complexes with high accuracy [54,55]. After selecting the receptor and ligand, Prime MM-GBSA uses the pose viewer file (pv.maegz) to show the appropriate descriptions [56]. The ligand-protein binding energy (G bind) was estimated using the following equation:
ΔG bind = G complex ₋ (G protein + G ligand)
In the given context, the terms G complex, G protein, and G ligand represent the minimum free energies associated with the protein-ligand complex, free protein, and free ligand, respectively.
2.7.3. ADMET Profiling of Novel Antiviral Compounds against LSDV
Ligands with the highest glide scores were carefully selected after virtual screening and ligand docking. To ensure that these selected ligands have the potential to be effective drug candidates, we performed a thorough evaluation using the Lipinski rule and ADMET analysis. We conducted this analysis using the QikProp module of the Schrödinger suite, specifically Maestro 12.8 [57].
2.7.4. Molecular Dynamics (MD) Simulation
MD simulations were performed using the Schrödinger LLC Desmond software for a duration of 100 nanoseconds [58]. Prior to MD simulations, a crucial docking step was performed to predict the static binding position of the ligand at the active site [59]. The MD simulations employed Newton's classical equation of motion to simulate atomic movements over time and predict the ligand-binding status in a physiologically relevant environment [60]. The ligand-receptor complex was prepared using Maestro's Protein Preparation Wizard, which involved the optimization, minimization, and filling of missing residues, if necessary. The system was created using a built-in tool.
The MD simulations were performed with the TIP3P (Intermolecular Interaction Potential 3 Points Transferable) solvent model in an orthorhombic box, which allowed for a 10 Å buffer region between protein atoms and box sides while maintaining a temperature of 300 K and pressure of 1 atm. Optimized Potentials for Liquid Simulation (OPLS 2005) force field were employed to describe the interactions within the system [61].
To mimic physiological conditions, counterions and 0.15 M sodium chloride were added to neutralize the total charge of the system. The models were loosened prior to the actual simulation. Trajectories were stored and inspected at regular intervals of 100 ps for further analysis. Furthermore, the trajectories obtained from the simulations were utilized in the Veusz software v. 3.6.2 (https://veusz.github.io/) to analyze structural insights and stability through root-mean-square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and solvent-accessible surface area (SASA).
3. Results
3.1. LSDV DNA Polymerase Protein Has a Groove-Like Active Site
Identifying the key proteins associated with diseases is a fundamental step in understanding the underlying mechanisms and developing targeted therapeutic interventions [62]. In this study, we focused on the ability of the protein to replicate DNA during the LSDV life cycle.
Based on their DNA replication ability, we found 12 out of 156 putative genes that could replicate the DNA of the LSDV (Table S1) [6,63]. From these genes, we selected only LSDV039 genes that code DNA polymerase enzymes, and its principal function is to faithfully duplicate the genome, ensuring that genetic information is preserved and passed on from generation to generation [6,64]. Furthermore, DNA polymerase is important for the growth and survival of all living organisms, from tiny viruses and bacteria to more complex organisms such as humans [65,66]. Unregulated DNA replication is often linked to various medical conditions such as cancer, autoimmune diseases, and viral and viral/bacterial infections [67].
Analysis using Chimera X software revealed that the DNA polymerase belongs to the DNA-directed DNA polymerase family B. It has four functional domains: DNA polymerase B exonuclease N-terminal (residues 1-22), DNA-directed DNA polymerase family B exonuclease (residues 64-349), DNA-directed DNA polymerase family B viral insert (residues 350-481) and DNA-directed DNA polymerase family B multifunctional (residues 493-989) (Figure 2). Additionally, the findings from the InterProScan and Pfam analyses supported the previously mentioned information. According to InterProScan, the conserved site within the 2–515 residues and 520-1003 residues of amino acid residues indicates that it belongs to the Ribonuclease H-like superfamily and the DNA/RNA polymerase superfamily.
DNA polymerase protein structure prediction was carried out using AlphaFold 2, and analysis of its binding site through the site map revealed that the active site structure closely resembles a groove. Additionally, a site map with a high DScore of 1.048 was selected as the active site. Proteins with Dscores between 0.7 and 0.8 are moderately druggable, while those with Dscores greater than 1.0 are considered very druggable [68]. Active-site residues were used for receptor grid generation, followed by molecular docking (Table 1, Figure 3).
3.2. Structure Validation
To assess the quality of the 3D model, a Ramachandran plot was constructed using the PROCHECK software (Figure 4). Ramachandran plot analysis of the predicted model revealed that 91% of the residues were located in the most favorable regions, whereas 8.0% were in the allowed regions. This confirmed that the predicted model was of excellent quality. ERRAT is a measure of the overall quality of non-bonded atomic interactions, where a higher score denotes superior quality. A high-quality model is generally considered to have a range of >50. According to the ERRAT server, the current 3D model's overall quality factor was 88.777 (Figure 5). The overall structural validation results are presented in Table 2.
3.3. Binding Profile Analysis of Bonded Interactions
Four potential antiviral compounds were screened from 1098 compounds using a Virtual Screening Workflow (VSW), followed by ligand docking in Glide Extra Precision (XP) modes. Among them, 380 were phytocompounds and 718 were drugs. Virtual Screening Workflow (VSW) separated 160 conformers out of 1098, and further interaction analyses of the protein-ligand complex yielded the 10 best compounds from the result of ligand docking in Glide Extra Precision (XP) modes. Compounds with a higher Glide Gscore (Gscore is a specific scoring function used to quantify the strength of the binding interaction between the ligand and protein) and binding interactions were evaluated as the best conformers.
Ligand docking performed by Maestro (Schrodinger) showed that canagliflozin had the best Glide Gscore of −9.858 kcal/mol, with a binding affinity of −45.68 kcal/mol. Compound tepotinib had the Glide Gscore −8.856 kcal/mol and its binding energy was −47.99 kcal/mol. Rhein and taxifolin got the Glide Gscore −8.965 kcal/mol and −7.195 kcal/mol, respectively, showing binding affinity −44.72 kcal/mol and −44.48 kcal/mol (Table 3). The positive control was also depicted with a Glide Gscore of −5.630 kcal/mol, which has a binding affinity of −26.28 kcal/mol.
The interaction of amino acid residues with selected ligands showed that canagliflozin establishes two non-covalent bonds with the amino acid residues Lys 383 and Asn 659 (Table 4, Figure 7 a). Canagliflozin formed two hydrogen bonds with Asn 659 with a bond distance of 1.80 and 1.73 Å, respectively. In addition, a salt bridge was formed between Lys 383 and canagliflozin, with a bond distance of 2.61 Å. Tepotinib formed two non-covalent interactions involving hydrogen bonds with Glu 339 residues with a bond distance of 1.93 Å and a salt bridge with Glu 399 with a bond distance of 2.94 Å. Moreover, it also establishes four Pi interactions in which Tyr 477, Phe 499, and Tyr 663 amino acid residues are involved in pi-pi stacking with a bond distance of 5.49, 4.89, and 5.38 Å, respectively, whereas Pi cation is only formed with amino acid residue Lys 483 having a bond distance of 4.01 Å (Figure 7 b). Rhein formed two non-covalent interactions involving two hydrogen bonds with Val 498 and Thr 664 residues having a bonding distance of 2.12 and 2.09 Å, respectively (Figure 7 c). It also established a salt bridge with Lys 337 with a bond distance of 2.71 Å. Taxifolin is involved in forming five non-covalent hydrogen bonds with amino acid residues Ser 338, Glu 339, Ser 656, Lys 483, and Thr 664 having a bond distance of 2.10, 1.78, 2.02, 2.00, and 2.05 Å, respectively, and a Pi interaction specifically Pi-cation with Lys 483 having a bond distance of 3.90 Å (Figure 7 d). In case of positive control, it formed three non-covalent hydrogen bonding with Asp 337, Ser 338, and Glu 339 residues where the bond distance between them was 1.55, 2.16, and 1.83 Å, respectively (Figure 6).
3.4. Binding Free Energy Calculations for Non-Bonded Interactions
Non-bonded interactions are more abundant than bonded interactions during protein-ligand docking. One of the significant non-bonded interactions is the Van der Waals energy. Coulomb energy is also one of the major forms of non-bonded energy. In addition, lipophilic energy, which is an important non-bonded interaction, is a ligand–receptor complex. Binding free energy calculations by Prime MMGBSA module reveals that canagliflozin formed a Van der Waals interactions having energy −42.95 kcal/mol. Tepotinib formed Van der Waals energy having energy −58.20 kcal/mol. Rhein and taxifolin are involved in interaction with Van der Waals energy having −34.55 kcal/mol and −24 kcal/mol, respectively. Positive control formed that energy with −63.90 kcal/mol. Other forms of non-bonded interactions and their energies are listed in Table 5.
To determine the non-bonded interaction energy in the protein ligand complex, the following equation was employed: MMGBSA dG Bind (NS) = MMGBSA dG Bind - Rec Strain - Lig Strain.
3.5. ADMET Profiling of Novel Antiviral Compounds against LSDV
We identified the top ten potential antiviral compounds based on their high Glide Gscore, indicating their promising binding affinity to viral targets. These compounds were subjected thorough ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) analyses using the QikProp module, which assesses crucial drug properties. The selected compounds exhibited favorable physicochemical and ADMET properties, making them excellent candidates for antiviral drug studies (Table 6).
3.6. Molecular Dynamics (MD) Simulation
MD simulations were performed to study the stability and behavior of the protein-ligand complexes over time. In this study, we examined the control complex and the four newly chosen compounds to understand their characteristics through a range of analyses, including RMSD, RMSF, Rg, and SASA. As shown in Figure 8a, the selected compound Rhein showed the lowest RMSD values compared to the positive control and the other selected compounds. A lower RMSD value indicates a higher level of system compactness [69]. The average RMSD values of ivermectin B1a, canagliflozin, tepotinib, rhein and taxifolin were 0.88 Å, 0.88 Å, 1.44 Å, 0.33 Å and 0.51 Å, respectively (Table 7). Canagliflozin exhibited a stable conformation from 15 to 63 ns. Tepotinib showed a stable conformation from 1 to 77 ns, after which fluctuations were observed from 78 to 100 ns. Rhein exhibited structural stability from 30 to 63 ns, whereas the remainder fluctuated. Taxifolin showed an initial fluctuation until 10 ns, after which a specific stabilization could be observed post-10 ns up to 89 ns; again, fluctuation occurred from 90 to 91 ns, and the rest showed structural stability. The control drug Ivermectin B1a showed stability and compactness at 16–36 ns and 44–100 ns, respectively.
Additionally, RMSF analysis was employed to examine the average atomic displacement from the mean positions in the residues during molecular dynamics simulation (Figure 8(b)). The canagliflozin showed the highest average RMSF value (1.69 Å), while the tepotinib showed the lowest RMSF value (1.26) among all systems. Higher RMSF values indicate that the system has greater flexibility during the molecular dynamics simulations [69]. The average RMSF value of Rhein (1.62 Å) was quite similar to the RMSF value of taxifolin (1.63 Å). However, Ivermectin B1a (control) showed average RMSF values of 1.58 Å (Table 7).
The radius of gyration (Rg) was used to measure the compactness of the system throughout the simulation (Figure 8(c)). Herein, the positive control ivermectin B1a showed the lowest Rg value of 32.72 Å among all the systems. In contrast, canagliflozin showed an average effect.
The Rg value (32.97 Å) that was quite similar to tepotinib (32.99 Å) and taxifolin (32.89 Å). However, rhein showed a slightly higher Rg value of 33.18 Å (Table 7). Figure 8d illustrates the SASA analysis for all systems. The SASA value provides insights into the compactness of the systems, with lower values indicating higher compactness, and higher values suggesting openness [69]. The selected complexes had average SASA values of 47186.72, 47677.70, 47724.08, 48127.33, and 47439.90 Å for ivermectin B1a, canagliflozin, tepotinib, rhein, and taxifolin, respectively, as indicated in Figure 8. Among all systems, the positive control, ivermectin B1a, displayed the lowest average SASA value of 9829.01 Å. The average SASA values of canagliflozin (47677.70 Å), tepotinib (47724.08 Å), and taxifolin (47439.90 Å) were quite similar, while rhein showed slightly higher SASA values of 48127.33 Å among all the systems.
4. Discussion
LSD is a viral disease that affects livestock and is considered a transboundary animal disease because of its ability to cross national borders, affecting multiple countries and regions. LSDV has significantly expansion of LSDV beyond its original endemic regions in Africa. LSDV have been reported in various geographical locations worldwide, including Asian, European, and Middle Eastern countries [70]. This disease was first reported in Bangladesh in July 2019 [71], China in August 2019 [72], India in November 2019 [73], Nepal in June 2020 [74], and other Asian countries [75]. In recent years, multiple cluster outbreaks of LSD have occurred on the Asian subcontinent, leading to substantial cattle mortality and posing a serious concern for the agricultural and livestock sectors [76]. For instance, in India alone, over 155,000 cattle deaths will be reported by 2022, underscoring the urgent need for disease control and management [77].
According to a report by the Department of Livestock Services (DLS) in 2019, 553,528 cattle were affected by LSD in all eight divisions of Bangladesh. The highest incidences were observed in Chattogram (8.26%) and Khulna (6.52%), whereas the lowest was observed in Sylhet (0.01%) (DLS, 2019) [78]. In one study, the LSDV collected from two Bangladeshi strains, BD-V392.1 and BD-V395.1, were distinct from contemporary field strains found in other Asian countries such as Hong Kong, China, Taiwan, and Vietnam. This suggests a unique genetic lineage or origin for Bangladeshi LSDV strains, which differs from strains circulating in neighboring Asian countries [79]. The Livestock Research Institute (LRI) in Bangladesh provides vaccines that are used as facultative measures, meaning that it is not mandatory but optional for farmers to vaccinate their livestock against LSDV. However, vaccination coverage is limited, resulting in many small holdings remaining unvaccinated [79]. Furthermore, the genetic variation of LSDV is one of the reasons why vaccines are not effective in a specific geographical region. Currently, no subunit vaccinations or chemotherapeutic medications are available for the treatment of LSD. It is currently feasible to prevent or treat this disease using other therapies such as phytochemical-based medicines or repurposed medications.
In this study, we used computer-aided methods to identify potential DNA polymerase inhibitors. The top four compounds, two drug compounds (canagliflozin and tepotinib) and two phytocompounds (rhein and taxifolin), were identified as highly promising LSDV DNA polymerase protein inhibitors. Molecular docking results reveal that canagliflozin poses the highest Glide Gscore (−9.858 kcal/mol) and the binding affinity (−45.68 kcal/mol) among the derived compounds. tepotinib, rhein, and taxifolin exhibited higher Glide Gscores and binding affinities than those of the control, Ivermectin B1a (−5.630 kcal/mol) (Table 3). These compounds form hydrogen bonds, pi-pi interactions, salt bridges, and cation-pi bonds with the LSDV DNA polymerase protein.
The formation of hydrogen bonds between the ligand and protein is highly selective and specific [80]. This depends on the spatial arrangement of the atoms in both the binding site of the protein and the ligand. The complementarity of these structures allows precise interactions to occur. The presence of hydrogen bonds stabilizes the ligand-protein complex, enhancing binding specificity [81]. Our results showed that each of the selected compounds formed hydrogen bonds with specific active-site residues of the LSDV DNA polymerase protein (Table 4), maintaining a specific distance. Taxifolin formed the highest number of hydrogen bonds. The positive control, ivermectin B1a, formed three hydrogen bonds. The hydrogen bond distance between Asn 659 and canagliflozin is 1.80 Å and 1.73 Å, respectively. The difference in the bond distance was due to the differences in the molecules within the same amino acid residue to which they bonded. The minimum bond distance between the ligand and specific amino acid residues indicates the maximum hydrogen bond energy.
Pi-Pi interactions are frequent in protein crystal structures, which help in the interaction of proteins and small molecules. The geometry of aromatic compounds and electrostatic interactions play a role in Pi-Pi interactions [82]. Besides, Pi-Pi stacking is also involved in the binding energy of receptor ligands [83]. From our findings, only Tepotinib formed three Pi-Pi interactions with the Tyr 477, Phe 499, and Tyr 663 amino acid residues, maintaining bond distances of 5.49, 4.89, and 5.38 Å, respectively.
Salt bridges are one of the strongest non-covalent interactions in nature. It plays crucial roles in protein folding, mediates protein-protein interactions, and facilitates molecular recognition [84]. Our results suggest that canagliflozin, tepotinib, and rhein, but not taxifolin, form salt bridges. Canagliflozin forms a salt bridge with Lys 483 with a bond distance of 2.61 Å. Tepotinib forms a salt bridge with Glu 339 with a bond distance of 1.93 Å. Moreover, rhein formed a salt bridge with Lys 337 by maintaining a bond distance of 2.71 Å. The Positive control ivermectin B1a did not exhibit such bonding interactions.
Cation-pi bond interactions play a crucial role in protein stability and structure. It involves attractive forces between the aromatic ring of a small molecule and a positively charged cation in a protein [85,86,87,88]. In terms of energy, this interaction is comparable to or sometimes stronger than a hydrogen bond [89]. Research suggests that cation-pi interactions are essential for protein-ligand recognition and have valuable implications for predicting drug-receptor interactions [90]. Our study reveals that tepotinib and taxifolin from our selected compounds only formed cation-pi bonded interactions with Lys 483 having bond distances of 4.01 and 3.90 Å, respectively. In contrast, positive control did not form these bonds.
A similar binding energy pattern was observed for the non-bonded interactions (Table 5). Coulomb energy is a form of electrostatic energy and a significant non-bonded energy found in protein-ligand complexes. Rhein has the highest coulomb energy (−77.35 kcal/mol), and ivermectin B1a possesses the lowest coulomb energy without any protein-ligand strains. Lipophilic energy is important for the evaluation of drug uptake and metabolism. Canagliflozin possesses the highest lipophilic energy (−25.21 kcal/mol) without strain. The Van der Waals interaction energies showed the opposite results. The Van der Waals interaction energy without receptor and ligand strains for the positive control was the highest (−63.90 kcal/mol) compared to the selected compounds.
The drug-like properties of the compounds satisfied Lipinski's Rule of Five, indicating their potential as drug candidates. Pharmacokinetics and toxicology offer important insights into the interactions between drug molecules in the human body [91]. The properties of these compounds were calculated using ADMET analysis tools such as QikProp. These tools predict the results of various chemical and physical properties of drug candidates, including molecular weight (MW), surface area, hydrogen bond (HB) interactions, lipophilicity, and oral absorption rates in humans [92]. Similarly, Qplog Po/w, QPlogS, Qplog HERG, and Human oral absorption were the major parameters used for the assessment of chemical compounds and for identifying the pharmacokinetic features of drugs (Table 4).
In addition, most drugs on the market have a molecular mass between 200 and 600 Da, with the majority being less than 500 Daltons [93]. To determine whether Canagliflozin, Tepotinib, rhein, and taxifolin are viable options, Lipinski's Rule of Five was considered. Lipinski's Rule of Five is a set of criteria used to evaluate a compound's drug-like behavior, including assessments such as the AMES test, Veber rule, and bioavailability [94]. Canagliflozin, tepotinib, rhein, and taxifolin satisfied most of the requirements, specifically Lipinski's Rule of Five, ensuring their drug-likeliness behavior (i.e., AMES test, Veber rule with no violations, better bioavailability value, and other parameters), which could be used as drug candidates (Table 1 and Supplementary Table S1).
Molecular simulations are a powerful approach for understanding the stability and dynamics of protein-ligand complexes [95,96]. Where the Mean Square Deviation indicates the average atomic displacement, with higher RMSD values indicating structural deviation or flexibility and a lower value indicating structural stability. The Square Fluctuation quantifies the average atomic positional variance from their mean positions within a molecule, with higher RMSF values suggesting greater atomic fluctuation or flexibility, and lower RMSF values indicating less atomic fluctuation and greater stability. The radius of gyration in molecular dynamics is a measure of the compactness or spread of a molecule's structure, where a higher Rg indicates a more extended or less compact structure, and a lower Rg signifies a more compact or folded structure. The solvent-accessible surface area quantifies the surface area of a molecule that is accessible to solvent molecules, where a higher SASA value suggests a greater exposed surface area, and a lower SASA value indicates a reduced exposed surface area. These parameters collectively provide essential insights into the molecular behavior in simulations [63,97,98].
The stability and dynamics of the compounds in protein-ligand complexes were studied. From the average mean value of the molecular dynamic simulation trajectory and analysis of the whole simulation data within 100 ns, it can be determined that the phytocompound rhein showed the highest stability in RMSD and RMSF. This fluctuated slightly in comparison with that of the positive control. The Rg and SASA values indicated less compactness and less exposure to the surface area compared to the positive control. It exhibited structural stability from 30 to 63 ns, whereas the rest of the simulation fluctuated. Taxifolin maintained its compactness in the RMSD. It showed stability at 10–89 ns; after that, it fluctuated from 1 to 10 ns and 91 to 100 ns, which is comparatively less than that of the positive control and gives a slightly higher RMSF value. The Rg and SASA values provided more compactness and a greater exposed surface area, respectively, than the other three compounds, but were marginally higher than those of the positive control. Tepotinib had the highest RMSD value among all compounds, indicating a lower level of compactness and even much higher compactness than the positive control, although this drug gave a minimum value in RMSF compared to the positive control and the rest of the compounds. It exhibited compactness in the range of 1–77 ns, and instability in the range of 78–100 ns. Canagliflozin showed the same RMSD score as the positive control. The RMSF of this compound was the highest among all the other compounds, including the positive control. Rg and SASA values followed the same trends. Canagliflozin exhibited compactness in 15 to 63 ns.
Canagliflozin and tepotinib are repurposed drugs, which means they have already been approved for use in treating specific diseases, and currently, there is interest in exploring their potential use for different diseases, including LSD. Canagliflozin (Invokana) is primarily used to the treatment of type 2 diabetes. It belongs to a class of drugs known as sodium-glucose co-transporter 2 (SGLT2) inhibitors. Canagliflozin is typically prescribed as a third-line treatment option after metformin (first-line medication) and other second-line alternatives in cases where these options are insufficient to effectively manage blood sugar levels [99,100,101]. Tepotinib belongs to a class of medications known as kinase inhibitors. It works by blocking the action of an abnormal protein that signals cancer cells to multiply [102,103,104].
Similarly, in this study, two specific phytochemicals, rhein and taxifolin, were identified as top-screened compounds with drug-like properties in inhibiting the macro domain of the chikungunya virus [105], with a focus on finding phytochemical inhibitors (drug-like properties of the top-screened phytochemicals rhein and taxifolin) against a unique macro domain found in the conserved N-terminal region of the non-structural protein nsP3 of the chikungunya virus. Medicinal plants have long been used as a source of potential therapeutic compounds and may offer a promising approach to combat LSD. This study investigated the binding affinities of two phytocompounds (rhein and taxifolin) and two repurposed drugs (canagliflozin and tepotinib) to the active sites of the DNA polymerase protein LSDV039, which are important targets for antiviral drug development. Post-MM/GBSA analysis, which calculated the binding free energy, indicated that the complexes exhibited a better binding free energy at 100 ns than the pre-MM/GBSA binding free energy. This indicates that the binding of phytocompounds to target proteins became more favorable over time. This suggests that they have the potential to interact effectively with these proteins, possibly by inhibiting their activity and interfering with viral replication.
However, to develop these selected compounds as LSD-specific antivirals, it is crucial to understand their behavior in their natural hosts, confirm their ability to inhibit replication, and provide antiviral prophylaxis. Notably, the identified compounds (canagliflozin, tepotinib, rhein, and taxifolin) are publicly available, which facilitates their rapid utilization and further research. Further experimental studies are necessary to validate their efficacy as novel compounds against LSD.
Overall, LSD continues to pose significant challenges to the livestock industry, and more comprehensive control measures, including improved vaccination coverage and alternative treatment options, are required to mitigate its impact. This study emphasized the importance of pharmacokinetic properties, efficacy, and safety levels in determining therapeutic efficacy and releasing new drugs. Based on in-silico screening of natural compounds, that is, rhein and taxifolin, and repurposed drugs, that is, canagliflozin and tepotinib, combinatorial docking, molecular dynamic simulation, drug-likeliness analysis, and all other experimental data were used. Taxifolin and canagliflozin have been suggested as drug candidates for the treatment of LSD.
5. Conclusions
This study provides valuable insights into the potential of specific phytocompounds and repurposed drugs against LSD. These findings suggest that canagliflozin, tepotinib, rhein and taxifolin have strong binding affinities for important viral proteins, exhibit favorable dynamics and hydrogen-bonding patterns, and possess favorable drug-like properties. This study serves as a foundation for future research and for the design of specific drugs targeting LSD. Experimental studies are required to confirm the efficacy of these phytocompounds as potential therapeutic agents against viruses.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: LSDV DNA replicating genes and their proteins.
Author Contributions
Conceptualization, M.M.H.S., A.B., and S.A.Z.; methodology, S.A.Z., M.M.H.S., and A.R.A.; writing-original draft preparation, A.R.A., S.A.Z., M.M.H.S.; writing-review and editing, M.M.R; investigation, S.A.Z., A.R.S. and M.M.H.S.; analysis, S.A.Z., A.R.S. and M.M.H.S.; software, S.A.Z., M.M.H.S., validation, S.A.Z., M.M.H.S, figures, A.B., M.M.R. All authors revised and agreed to publish the manuscript for final submission.
Funding Statement
This work was partially supported by University Grant Commission (UGC), Bangladesh (Ref: 37.01.0000.073.03.007.20.133) and Cooperative Research Program for Agricultural Science & Technology Development (grant number PJ010859012016).
Data Availability
The data supporting the findings of the research are included in this article, and additional data are available in the supplementary file. Furthermore, the corresponding authors are ready to provide additional details upon request.
Acknowledgments
We express gratitude to the students of both the Molecular Microbiology & Genomics Laboratory, Dept. of Biotechnology and Genetic engineering, Islamic University, Bangladesh, and the Korean Wildlife Genomics Laboratory. A special thanks are extended to Professor Dr. Md. Rezuanul Islam, Professor Dr. Md. Mizanur Rahman, Professor Dr. Mohammad Minnatul Karim, Professor Dr. Md. Abu Hena Mostofa Jamal, and Professor Dr. Sudhangshu Kumar Biswas for their assistance with ethical matters and technical support related to the project.
Conflicts of Interest
The author(s) declare(s) that there is no conflict of interest regarding the publication of this paper.
References
- Das, M.; Chowdhury, M.S.R.; Akter, S.; Mondal, A.K.; Uddin, M.J.; Rahman, M.M.; Rahman, M.M. An updated review on lumpy skin disease: perspective of Southeast Asian countries. J. adv. biotechnol. exp. ther 2021, 4(3), 322–333. [Google Scholar] [CrossRef]
- Molini, U.; Boshoff, E.; Niel, A.P.; Phillips, J.; Khaiseb, S.; Settypalli, T.B.; Dundon, W.G.; Cattoli, G.; Lamien, C.E. Detection of lumpy skin disease virus in an asymptomatic eland (Taurotragus oryx) in Namibia. The Journal of Wildlife Diseases 2021, 57(3), 708–711. [Google Scholar] [CrossRef]
- Ko, Y.S.; Oh, Y.; Lee, T.G.; Bae, D.Y.; Tark, D.; Cho, H.S. Serological and molecular prevalence of lumpy skin disease virus in Korean water deer, native and dairy cattle in Korea. Korean Journal of Veterinary Service 2022, 45(2), 133–137. [Google Scholar] [CrossRef]
- Dao, T.D.; Tran, L.H.; Nguyen, H.D.; Hoang, T.T.; Nguyen, G.H.; Tran KV, D.; Nguyen, H.X.; Van Dong, H.; Bui, A.N.; Bui, V.N. Characterization of Lumpy skin disease virus isolated from a giraffe in Vietnam. Transboundary and Emerging Diseases 2022, 69(5), e3268–e3272. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.; Arif, B.; Black, D.; Dumbell, K.; Esposito, J.; Lefkowitz, E.; McFadden, G.; Moss, B.; Mercer, A.; Moyer, R. Family poxviridae. In Virus taxonomy: Classification and nomenclature of viruses. Eighth Report of the International Committee on Taxonomy of Viruses; 2005; pp. 117–133. [Google Scholar]
- Tulman, E.; Afonso, C.; Lu, Z.; Zsak, L.; Kutish, G.; Rock, D. Genome of lumpy skin disease virus. Journal of virology 2001, 75(15), 7122–7130. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Venter, E.H.; Coetzer, J.A.; Bell-Sakyi, L. Lumpy skin disease: Attempted propagation in tick cell lines and presence of viral DNA in field ticks collected from naturally-infected cattle. Ticks and tick-borne diseases 2015, 6(2), 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Bernardo, B.; Haga, I.R.; Wijesiriwardana, N.; Basu, S.; Larner, W.; Diaz, A.V.; Langlands, Z.; Denison, E.; Stoner, J.; White, M. Quantifying and modeling the acquisition and retention of lumpy skin disease virus by hematophagus insects reveals clinically but not subclinically affected cattle are promoters of viral transmission and key targets for control of disease outbreaks. Journal of virology 2021, 95(9). [Google Scholar] [CrossRef] [PubMed]
- Sprygin, A.; Pestova, Y.; Wallace, D.; Tuppurainen, E.; Kononov, A. Transmission of lumpy skin disease virus: A short review. Virus research 2019, 269, 197637. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Patial, V.; Bali, D.; Angaria, S.; Sharma, M.; Chahota, R. A review: Lumpy skin disease and its emergence in India. Veterinary research communications 2020, 44, 111–118. [Google Scholar] [CrossRef]
- Liu, P.; Li, J.; Chen, R.; Cheng, Z.; Shi, Y. The first outbreak investigation of lumpy skin disease in China. China Animal Health Inspection 2020, 37(1), 1–5. [Google Scholar]
- Wang, Y.; Zhao, L.; Yang, J.; Shi, M.; Nie, F.; Liu, S.; Wang, Z.; Huang, D.; Wu, H.; Li, D. Analysis of vaccine-like lumpy skin disease virus from flies near the western border of China. Transboundary and Emerging Diseases 2022, 69(4), 1813–1823. [Google Scholar] [CrossRef]
- Farag, T.; El-Houssiny, A.; Abdel-Rahman, E.; Hegazi, A. A new approach to the treatment of lumpy skin disease infection in cattle by using propolis encapsulated within alg nps. Adv. Anim. Vet. Sci 2020, 8(12), 1346–1355. [Google Scholar] [CrossRef]
- Sprygin, A.; Artyuchova, E.; Babin, Y.; Prutnikov, P.; Kostrova, E.; Byadovskaya, O.; Kononov, A. Epidemiological characterization of lumpy skin disease outbreaks in Russia in 2016. Transboundary and Emerging Diseases 2018, 65(6), 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Ratyotha, K.; Prakobwong, S.; Piratae, S. Lumpy skin disease: A newly emerging disease in Southeast Asia. Veterinary World 2022, 15(12), 2764. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, L.; Ward, M.P. The Spread of Lumpy Skin Disease Virus across Southeast Asia: Insights from Surveillance. Transboundary and Emerging Diseases 2023. [Google Scholar] [CrossRef]
- Hasib FM, Y.; Islam, M.S.; Das, T.; Rana, E.A.; Uddin, M.H.; Bayzid, M.; Nath, C.; Hossain, M.A.; Masuduzzaman, M.; Das, S. Lumpy skin disease outbreak in cattle population of Chattogram, Bangladesh. Veterinary Medicine and Science 2021, 7(5), 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Gari, G.; Abie, G.; Gizaw, D.; Wubete, A.; Kidane, M.; Asgedom, H.; Bayissa, B.; Ayelet, G.; Oura, C.A.; Roger, F. Evaluation of the safety, immunogenicity and efficacy of three capripoxvirus vaccine strains against lumpy skin disease virus. Vaccine 2015, 33(28), 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Pearson, C.R.; Bachanek-Bankowska, K.; Knowles, N.J.; Amareen, S.; Frost, L.; Henstock, M.R.; Lamien, C.E.; Diallo, A.; Mertens, P.P. Characterization of sheep pox virus vaccine for cattle against lumpy skin disease virus. Antiviral research 2014, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Broglia, A.; Antoniou, S.-E.; Tsiamadis, V.; Plevraki, E.; Petrović, T.; Polaček, V.; Debeljak, Z.; Miteva, A.; Alexandrov, T. Neethling vaccine proved highly effective in controlling lumpy skin disease epidemics in the Balkans. Preventive veterinary medicine 2020, 181, 104595. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, J.; Mohan, M.; Byrareddy, S.N. Drug repurposing approaches to combating viral infections. Journal of clinical medicine 2020, 9(11), 3777. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palù, G.; Legian, A. Drug repurposing for viral infectious diseases: how far are we? Trends in microbiology 2018, 26(10), 865–876. [Google Scholar] [CrossRef]
- Punekar, M.; Kshirsagar, M.; Tellapragada, C.; Patil, K. Repurposing of antiviral drugs for COVID-19 and impact of repurposed drugs on the nervous system. Microbial Pathogenesis 2022, 168, 105608. [Google Scholar] [CrossRef]
- Toker, E.B.; Ates, O.; Yeşilbağ, K. Inhibition of bovine and ovine capripoxviruses (Lumpy skin disease virus and Sheeppox virus) by ivermectin occurs at different stages of propagation in vitro. Virus research 2022, 310, 198671. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Bhattu, M.; Tripathi, A.; Verma, M.; Acevedo, R.; Kumar, P.; …; Singh, J. Potential medicinal plants to combat viral infections: A way forward to environmental biotechnology. Environmental Research 2023, 115725. [Google Scholar] [CrossRef]
- Dastoli, S.; Nisticò, S.P.; Morrone, P.; Patruno, C.; Leo, A.; Citraro, R.; Gallelli, L.; Russo, E.; De Sarro, G.; Bennardo, L. Colchicine in managing skin conditions: A systematic review. Pharmaceutics 2022, 14(2), 294. [Google Scholar] [CrossRef]
- Malabadi, R.B.; Kolkar, P.; Chalannavar, K. Outbreak of lumpy skin viral disease of cattle and buffalo in india in 2022: Ethnoveterinary medicine approach. International Journal of Innovation Scientific Research and Review 2022, 4(11), 3562–3574. [Google Scholar]
- Yadav, J.V.; Lakshman, M.; Madhuri, D. A combination of conventional and alternative ethnoveterinary medicine for the treatment of lumpy skin disease in a she-buffalo: A case report. The Pharma Innovation 2021, SP-10(1), 83–84. [Google Scholar]
- Vijayasri, S.; Hopper, W. Towards the identification of novel phytochemical leads as macrodomain inhibitors of chikungunya virus using molecular docking approach. Journal of Applied Pharmaceutical Science 2017, 2017 7, 074–082. [Google Scholar]
- Jayant, V.; Ali, R. Potential Applications of Ivermectin (IVM) in Dermatology. In Chemistry and Biological Activities of Ivermectin; 2023; pp. 199–229. [Google Scholar]
- Badhy, S.C.; Chowdhury, M.G.A.; Settypalli TB, K.; Cattoli, G.; Lamien, C.E.; Fakir MA, U.; Akter, S.; Osmani, M.G.; Talukdar, F.; Begum, N. Molecular characterization of lumpy skin disease virus (LSDV) emerged in Bangladesh reveals unique genetic features compared to contemporary field strains. BMC veterinary research 2021, 17(1), 1–11. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nature structural & molecular biology 2021, 28(9), 704–705. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of applied crystallography 1993, 26(2), 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein science 1993, 2(9), 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Banerjee, A.; Chakraborty, N.; Soren, K.; Chakraborty, P.; Bandopadhyay, R. Structural-functional analyses of textile dye degrading azoreductase, laccase and peroxidase: A comparative in silico study. Electronic Journal of Biotechnology 2020, 43, 48–54. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein science 2021, 30(1), 70–82. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: the integrative protein signature database. Nucleic acids research 2009, 37 (suppl_1), D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: the protein families database. Nucleic acids research 2014, 42(D1), D222–D230. [Google Scholar] [CrossRef]
- Schrödinger. 2021. Schrödinger Release 2021-2: SiteMap. Schrödinger, LLC. New York, NY.
- Ali, M.C.; Munni, Y.A.; Das, R.; Akter, N.; Das, K.; Mitra, S.; Hannan, M.A.; Dash, R. In silico chemical profiling and identification of neuromodulators from Curcuma amada targeting Acetylcholinesterase. Network Modeling Analysis in Health Informatics and Bioinformatics 2021, 10, 1–16. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2023 update. Nucleic acids research 2023, 51(D1), D1373–D1380. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic acids research 2006, 34 (suppl_1), D668–D672. [Google Scholar] [CrossRef]
- O'Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. Journal of cheminformatics 2011, 3(1), 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger 2023 Schrödinger Release 2023-2: Maestro Schrödinger, L.L.C. New York, NY.
- Schrödinger. 2023. Schrödinger Release 2023-2: Protein Preparation Wizard. Epik, Schrödinger, LLC. New York, NY.
- Schrödinger 2023 Schrödinger Release 2023-3: SiteMap Schrödinger, L.L.C. New York, NY.
- Tahlan, S.; Kumar, S.; Ramasamy, K.; Lim, S.M.; Shah SA, A.; Mani, V.; Narasimhan, B. In-silico molecular design of heterocyclic benzimidazole scaffolds as prospective anticancer agents. BMC chemistry 2019, 13(1), 1–22. [Google Scholar] [CrossRef]
- David, T.I.; Adelakun, N.S.; Omotuyi, O.I.; Metibemu, D.S.; Ekun, O.E.; Inyang, O.K.; Adewumi, B.; Enejoh, O.A.; Owolabi, R.T.; Oribamise, E.I. Molecular docking analysis of phyto-constituents from Cannabis sativa with pfDHFR. Bioinformation 2018, 14(9), 574. [Google Scholar] [CrossRef]
- Singh, N.; Chaput, L.; Villoutreix, B.O. Virtual screening web servers: designing chemical probes and drug candidates in the cyberspace. Briefings in bioinformatics 2021, 22(2), 1790–1818. [Google Scholar] [CrossRef]
- Schrödinger 2019 Schrödinger Release 2019-3: Glide Schrödinger, L.L.C. New York, NY, USA.
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins: Structure, Function, and Bioinformatics 2011, 79(10), 2794–2812. [Google Scholar] [CrossRef]
- Kadioglu, O.; Saeed, M.; Greten, H.J.; Efferth, T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Computers in biology and medicine 2021, 133, 104359. [Google Scholar] [CrossRef]
- Ghosh, S.; Nie, A.; An, J.; Huang, Z. Structure-based virtual screening of chemical libraries for drug discovery. Current opinion in chemical biology 2006, 10(3), 194–202. [Google Scholar] [CrossRef]
- Pattar, S.V.; Adhoni, S.A.; Kamanavalli, C.M.; Kumbar, S.S. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef University journal of basic and applied sciences 2020, 9(1), 1–10. [Google Scholar] [CrossRef]
- Sankar, M.; Ramachandran, B.; Pandi, B.; Mutharasappan, N.; Ramasamy, V.; Prabu, P.G.; Shanmugaraj, G.; Wang, Y.; Muniyandai, B.; Rathinasamy, S. In silico screening of natural phytocompounds towards identification of potential lead compounds to treat COVID-19. Frontiers in molecular biosciences 2021, 8, 637122. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.-Y.; Wang, Q.; Zhang, J.; Wu, T.; Zhang, F. Trastuzumab-Peptide interactions: mechanism and application in structure-based ligand design. International Journal of Molecular Sciences 2013, 14(8), 16836–16850. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger 2021 QikProp In Schrödinger Release 2021-2 Schrödinger, L.L.C. New York, NY.
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20(7), 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K. Bringing molecular dynamics simulation data into view. Trends in Biochemical Sciences 2019, 44(11), 902–913. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of lead compounds against Scm (fms10) in Enterococcus faecium using computer aided drug designing. Life 2021, 11(2), 77. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. Journal of chemical theory and computation 2010, 6(5), 1509–1519. [Google Scholar] [CrossRef]
- Espinoza-Chávez, R.M.; Salerno, A.; Liuzzi, A.; Ilari, A.; Milelli, A.; Uliassi, E.; Bolognesi, M.L. Targeted Protein Degradation for Infectious Diseases: from Basic Biology to Drug Discovery. ACS bio & med Chem Au 2022, 3(1), 32–45. [Google Scholar]
- Kara, P.; Afonso, C.; Wallace, D.; Kutish, G.; Abolnik, C.; Lu, Z.; Vreede, F.; Taljaard, L.; Zsak, A.; Viljoen, G.J. Comparative sequence analysis of the South African vaccine strain and two virulent field isolates of lumpy skin disease virus. Archives of virology 2003, 148, 1335–1356. [Google Scholar] [CrossRef]
- Garcia-Diaz, M.; Bebenek, K. Multiple functions of DNA polymerases. Critical reviews in plant sciences 2007, 26(2), 105–122. [Google Scholar] [CrossRef]
- Berdis, A.J. DNA polymerases as therapeutic targets. Biochemistry 2008, 47(32), 8253–8260. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Temporal order of evolution of DNA replication systems inferred by comparison of cellular and viral DNA polymerases. Biology direct 2006, 1(1), 1–18. [Google Scholar] [CrossRef]
- Michel, M.; Visnes, T.; Homan, E.J.; Seashore-Ludlow, B.; Hedenström, M.; Wiita E e Vallin, K.; Paulin, C.B.; Zhang, J.; Wallner, O. Computational and experimental druggability assessment of human DNA glycosylases. ACS omega 2019, 4(7), 11642–11656. [Google Scholar] [CrossRef]
- Alzyoud, L.; Bryce, R.A.; Al Sorkhy, M.; Atatreh, N.; Ghattas, M.A. Structure-based assessment and druggability classification of protein–protein interaction sites. Scientific Reports 2022, 12(1), 7975. [Google Scholar] [CrossRef]
- Ali, M.C.; Nur, A.J.; Khatun, M.S.; Dash, R.; Rahman, M.M.; Karim, M.M. Identification of potential SARS-CoV-2 main protease inhibitors from Ficus Carica Latex: An in-silico approach. Journal of Advanced Biotechnology and Experimental Therapeutics 2020, 3(4), 57–67. [Google Scholar] [CrossRef]
- Whittle, L.; Chapman, R.; Williamson, A.-L. Lumpy Skin Disease—An Emerging Cattle Disease in Europe and Asia. Vaccines 2023, 11(3), 578. [Google Scholar] [CrossRef]
- Chouhan, C.S.; Parvin, M.S.; Ali, M.Y.; Sadekuzzaman, M.; Chowdhury MG, A.; Ehsan, M.A.; Islam, M.T. Epidemiology and economic impact of lumpy skin disease of cattle in Mymensingh and Gaibandha districts of Bangladesh. Transboundary and Emerging Diseases 2022, 69(6), 3405–3418. [Google Scholar] [CrossRef]
- Lu, G.; Xie, J.; Luo, J.; Shao, R.; Jia, K.; Li, S. Lumpy skin disease outbreaks in China, since 3 August 2019. Transboundary and Emerging Diseases 2021, 68(2), 216–219. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, S.B.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Hemadri, D.; Sood, R.; Bal, G.C.; Nayak, M.K.; Pradhan, S.K.; Singh, V.P. Lumpy skin disease (LSD) outbreaks in cattle in Odisha state, India in August 2019: Epidemiological features and molecular studies. Transboundary and Emerging Diseases 2020, 67(6), 2408–2422. [Google Scholar] [CrossRef]
- Acharya, K.P.; Subedi, D. First outbreak of lumpy skin disease in Nepal. Preventive veterinary medicine 2020, 102(4), 274–283. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Chowdhury MS, R.; Akter, S.; Mondal, A.K.; Uddin, M.J.; Rahman, M.M.; Rahman, M.M. An updated review on lumpy skin disease: perspective of Southeast Asian countries. J. adv. biotechnol. exp. Ther 2021, 4(3), 322–333. [Google Scholar] [CrossRef]
- Wilhelm, L.; Ward, M.P. The Spread of Lumpy Skin Disease Virus across Southeast Asia: Insights from Surveillance. Transboundary and Emerging Diseases 2023. [CrossRef]
- Moudgil, G.; Chadha, J.; Khullar, L.; Chhibber, S.; Harjai, K. Lumpy skin disease: A comprehensive review on virus biology, pathogenesis, and sudden global emergence. 2023. [Google Scholar]
- Kayesh ME, H.; Hussan, M.T.; Hashem, M.A.; Eliyas, M.; Anower, A.M. Lumpy skin disease virus infection: An emerging threat to cattle health in Bangladesh. Hosts and Viruses 2020, 7(4), 97. [Google Scholar] [CrossRef]
- Parvin, R.; Chowdhury, E.H.; Islam, M.T.; Begum, J.A.; Nooruzzaman, M.; Globig, A.; Dietze, K.; Hoffmann, B.; Tuppurainen, E. Clinical Epidemiology, Pathology, and Molecular Investigation of Lumpy Skin Disease Outbreaks in Bangladesh during 2020–2021 Indicate the Re-Emergence of an Old African Strain. Viruses 2022, 14(11), 2529. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.E.; Haider, M.K. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Science 2010; John Wiley & Sons, Ltd.
- Wade, R.C.; Goodford, P.J. The role of hydrogen-bonds in drug binding. Progress in clinical and biological research 1989, 289, 433–444. [Google Scholar] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. Journal of medicinal chemistry 2010, 53(14), 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Farley, A.R.; Wright, G.D.; Cox, J.R. Analysis of the π-π stacking interactions between the aminoglycoside antibiotic kinase APH (3′)-IIIa and its nucleotide ligands. Chemistry & biology 2002, 9(11), 1209–1217. [Google Scholar]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. A role of salt bridges in mediating drug potency: A lesson from the N-myristoyltransferase inhibitors. Frontiers in molecular biosciences 2023, 9, 1066029. [Google Scholar] [CrossRef]
- Dougherty, D.A. Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 1996, 271(5246), 163–168. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The cation− π interaction. Chemical reviews 1997, 97(5), 1303–1324. [Google Scholar] [CrossRef]
- Scrutton, N.S.; Raine, A.R. Cation-π bonding and amino-aromatic interactions in the biomolecular recognition of substituted ammonium ligands. Biochemical Journal 1996, 319(1), 1–8. [Google Scholar] [CrossRef]
- Wouters, J. Cation-π (Na+-Trp) interactions in the crystal structure of tetragonal lysozyme. Protein science 1998, 7(11), 2472–2475. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-π interactions in structural biology. Proceedings of the National Academy of Sciences 1999, 96(17), 9459–9464. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, N.; Dougherty, D.A. Cation–π interactions in ligand recognition and catalysis. Trends in pharmacological sciences 2002, 23(6), 281–287. [Google Scholar] [CrossRef]
- Wang, C.; Dai, S.; Gong, L.; Fu, K.; Ma, C.; Liu, Y.; Zhou, H.; Li, Y. A review of pharmacology, toxicity and pharmacokinetics of 2, 3, 5, 4′-tetrahydroxystilbene-2-O-β-D-glucoside. Frontiers in Pharmacology 2022, 12, 791214. [Google Scholar] [CrossRef]
- Zhang, S.-Q.; Chen, F. Factors Influencing ADME Properties of Drugs: Advances and Applications (Part I). Current Drug Metabolism 2023, 24(1), 3–4. [Google Scholar] [CrossRef]
- Glassman, P.M.; Muzykantov, V.R. Pharmacokinetic and pharmacodynamic properties of drug delivery systems. Journal of Pharmacology and Experimental Therapeutics 2019, 370(3), 570–580. [Google Scholar] [CrossRef]
- Ahmed, S.; Ali, M.C.; Ruma, R.A.; Mahmud, S.; Paul, G.K.; Saleh, M.A.; Alshahrani, M.M.; Obaidullah, A.J.; Biswas, S.K.; Rahman, M.M. Molecular docking and dynamics simulation of natural compounds from betel leaves (Piper betle L.) for investigating the potential inhibition of alpha-amylase and alpha-glucosidase of type 2 diabetes. Molecules 2022, 27(14), 4526. [Google Scholar] [CrossRef]
- Peele, K.A.; Durthi, C.P.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Babu, D.J.; Indira, M.; Reddy, A.R.; Venkateswarulu, T. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Informatics in Medicine Unlocked 2020, 19, 100345. [Google Scholar] [CrossRef]
- Munni, Y.A.; Ali, M.C.; Selsi, N.J.; Sultana, M.; Hossen, M.; Bipasha, T.H.; Rahman, M.; Uddin, M.N.; Hosen, S.Z.; Dash, R. Molecular simulation studies to reveal the binding mechanisms of shikonin derivatives inhibiting VEGFR-2 kinase. Computational Biology and Chemistry 2021, 90, 107414. [Google Scholar] [CrossRef]
- Dash, R.; Choi, H.J.; Moon, I.S. Mechanistic insights into the deleterious roles of Nasu-Hakola disease associated TREM2 variants. Scientific Reports 2020, 10(1), 3663. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Ali, M.C.; Dash, N.; Azad MA, K.; Hosen, S.Z.; Hannan, M.A.; Moon, I.S. Structural and dynamic characterizations highlight the deleterious role of SULT1A1 R213H polymorphism in substrate binding. International Journal of Molecular Sciences 2019, 20(24), 6256. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk Gabr, N.; Hassan Mohammed, I. A comparative study of canagliflozin (INVOKANA) on type-I and type-II diabetes mellitus on adult male albino rat. Al-Azhar Medical Journal 2020, 49(1), 15–32. [Google Scholar] [CrossRef]
- Deeks, E.D.; Scheen, A.J. Canagliflozin: a review in type 2 diabetes. Drugs 2017, 77, 1577–1592. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Cersosimo, E. Use of canagliflozin in combination with and compared to incretin-based therapies in type 2 diabetes. Clinical Diabetes 2017, 35(3), 141–153. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; …; Le, X. Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. New England Journal of Medicine 2020, 383(10), 931–943. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacological research 2022, 175, 106037. [Google Scholar] [CrossRef]
- Amir, M.; Javed, S. Elucidation of binding dynamics of tyrosine kinase inhibitor tepotinib, to human serum albumin, using spectroscopic and computational approach. International Journal of Biological Macromolecules 2023, 241, 124656. [Google Scholar] [CrossRef]
- Vijayasri, S.; Hopper, W. Towards the identification of novel phytochemical leads as macrodomain inhibitors of chikungunya virus using molecular docking approach. Journal of Applied Pharmaceutical Science 2017, 7, 074–082. [Google Scholar]
Figure 1.
A summery or schematic representation of virtual screening and molecular docking using repurposed drugs and phytocompounds.
Figure 1.
A summery or schematic representation of virtual screening and molecular docking using repurposed drugs and phytocompounds.
Figure 2.
Conserved domains in DNA polymerase uncovered using Chimera X.
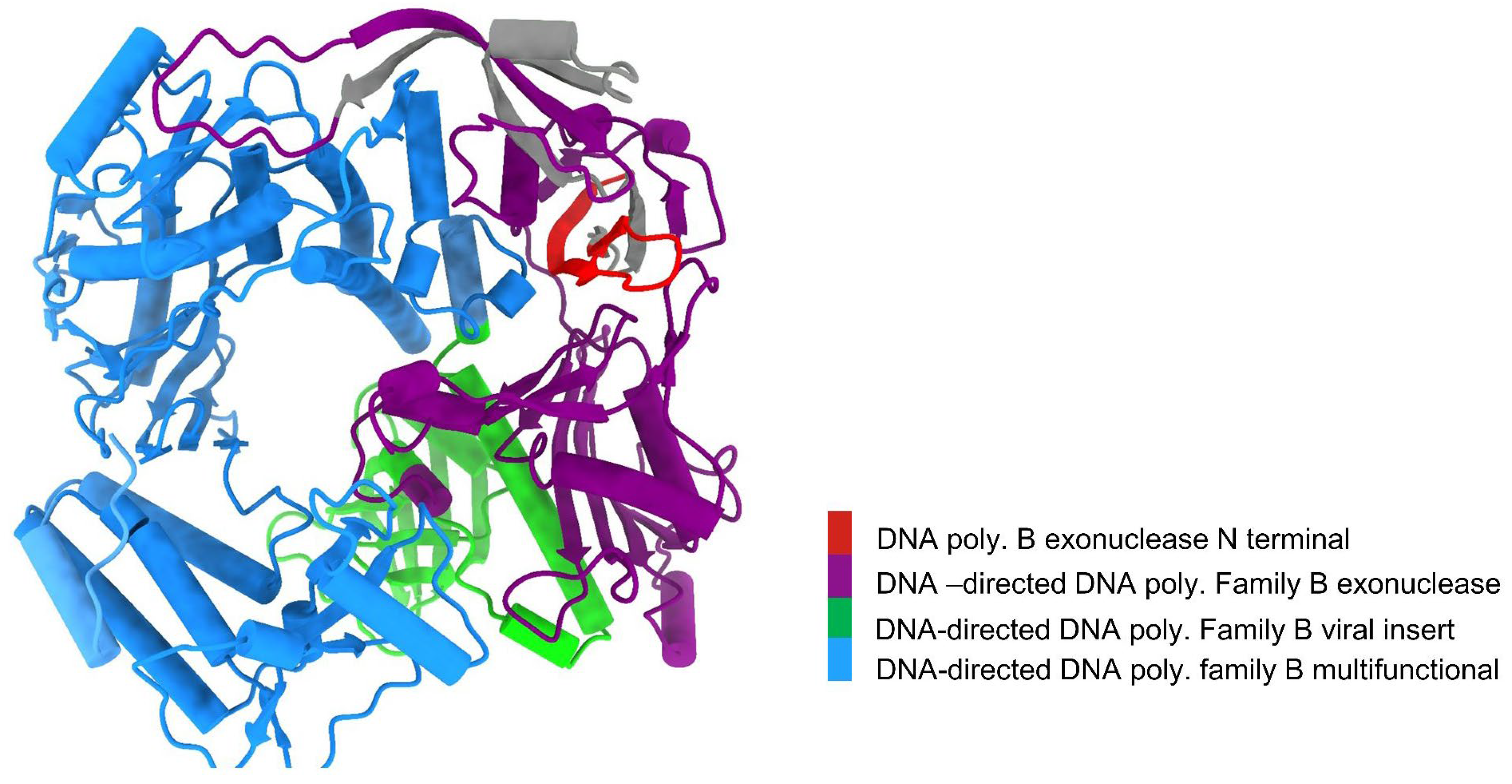
Figure 3.
DNA polymerase protein binding site residues. The red color denotes the binding site residue and the light sea green color represents the non-binding residues.
Figure 3.
DNA polymerase protein binding site residues. The red color denotes the binding site residue and the light sea green color represents the non-binding residues.
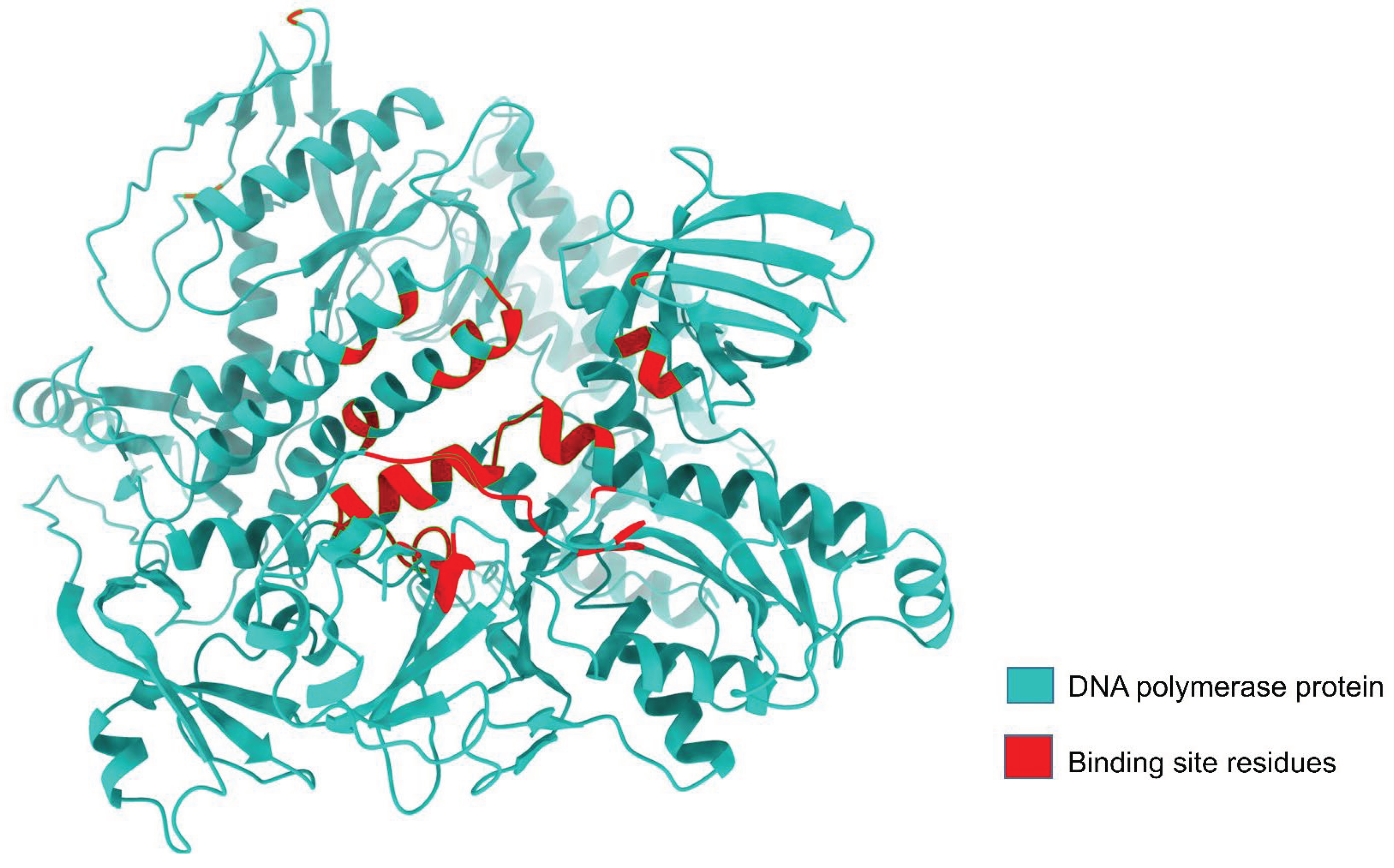
Figure 4.
Validation of DNA polymerase protein using the Ramachandran plot.
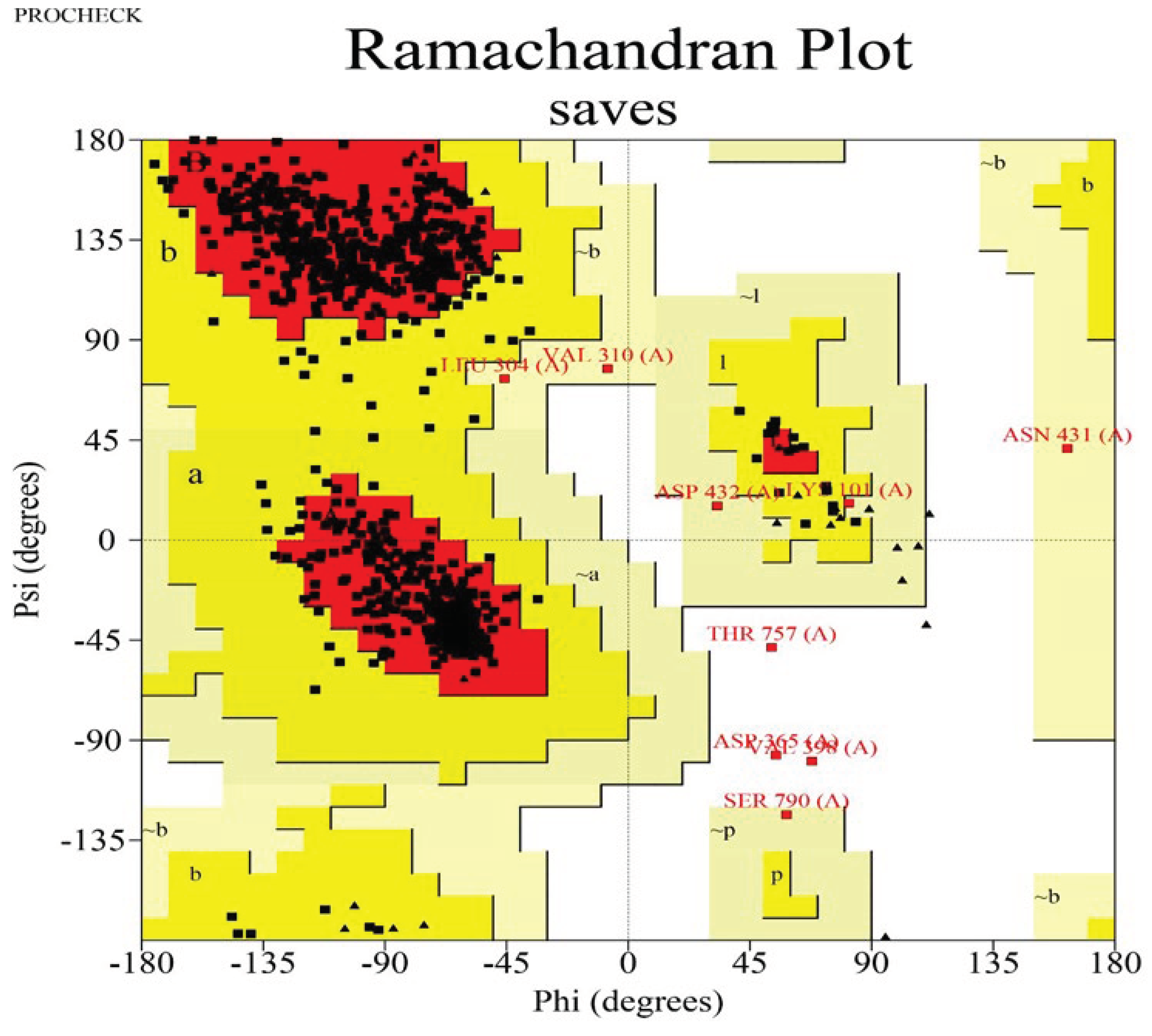
Figure 5.
Structure validation using ERRAT2. The red bars indicate the misfolded region, yellow bars indicate the error region, and white bars represent the region with a lower error rate.
Figure 5.
Structure validation using ERRAT2. The red bars indicate the misfolded region, yellow bars indicate the error region, and white bars represent the region with a lower error rate.
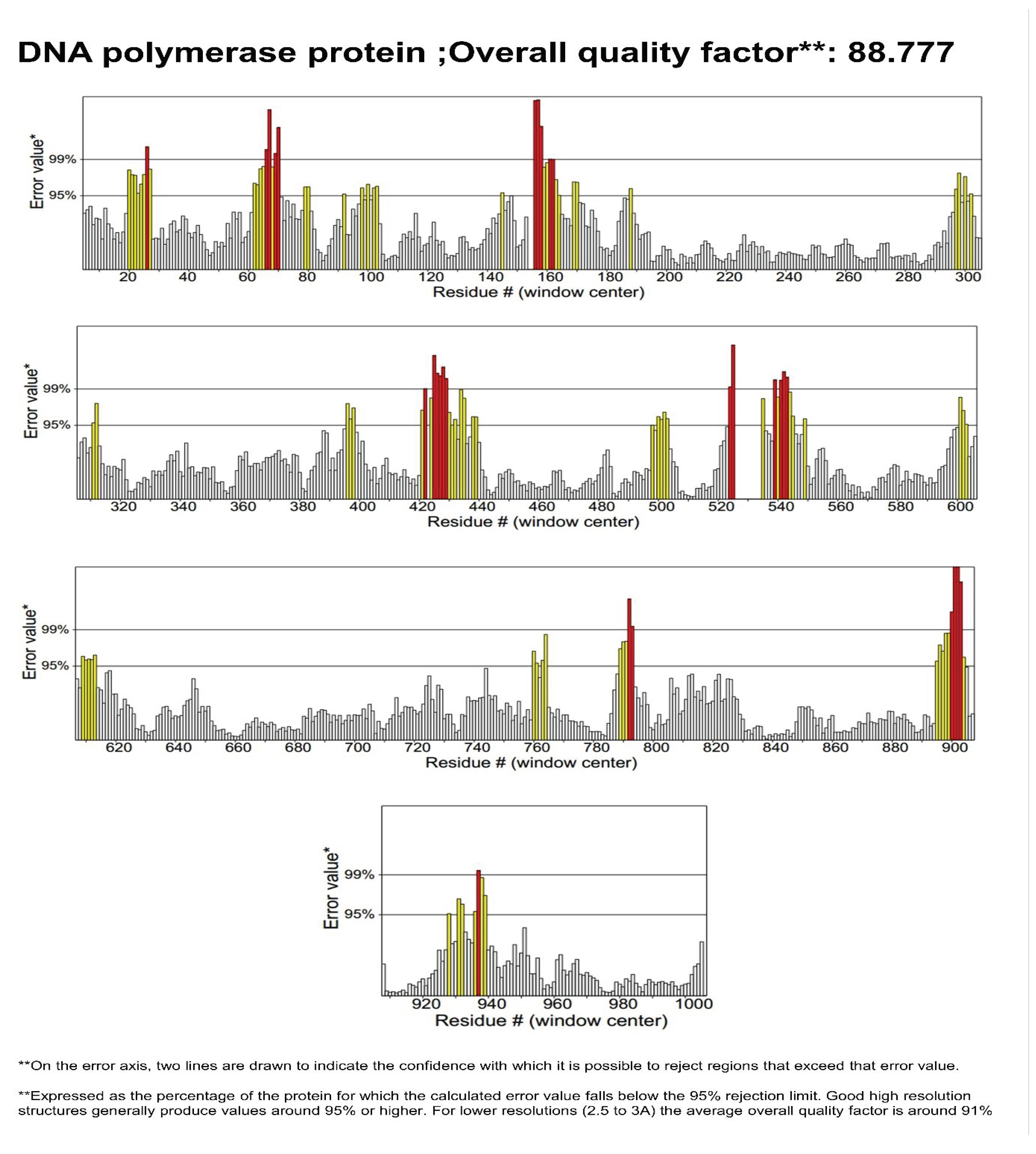
Figure 6.
(a) Docking interactions of Ivermectin B1a (positive control) with DNA Polymerase Protein (b) Docking pose Alignment of two Drugs and two phytocompounds.
Figure 6.
(a) Docking interactions of Ivermectin B1a (positive control) with DNA Polymerase Protein (b) Docking pose Alignment of two Drugs and two phytocompounds.
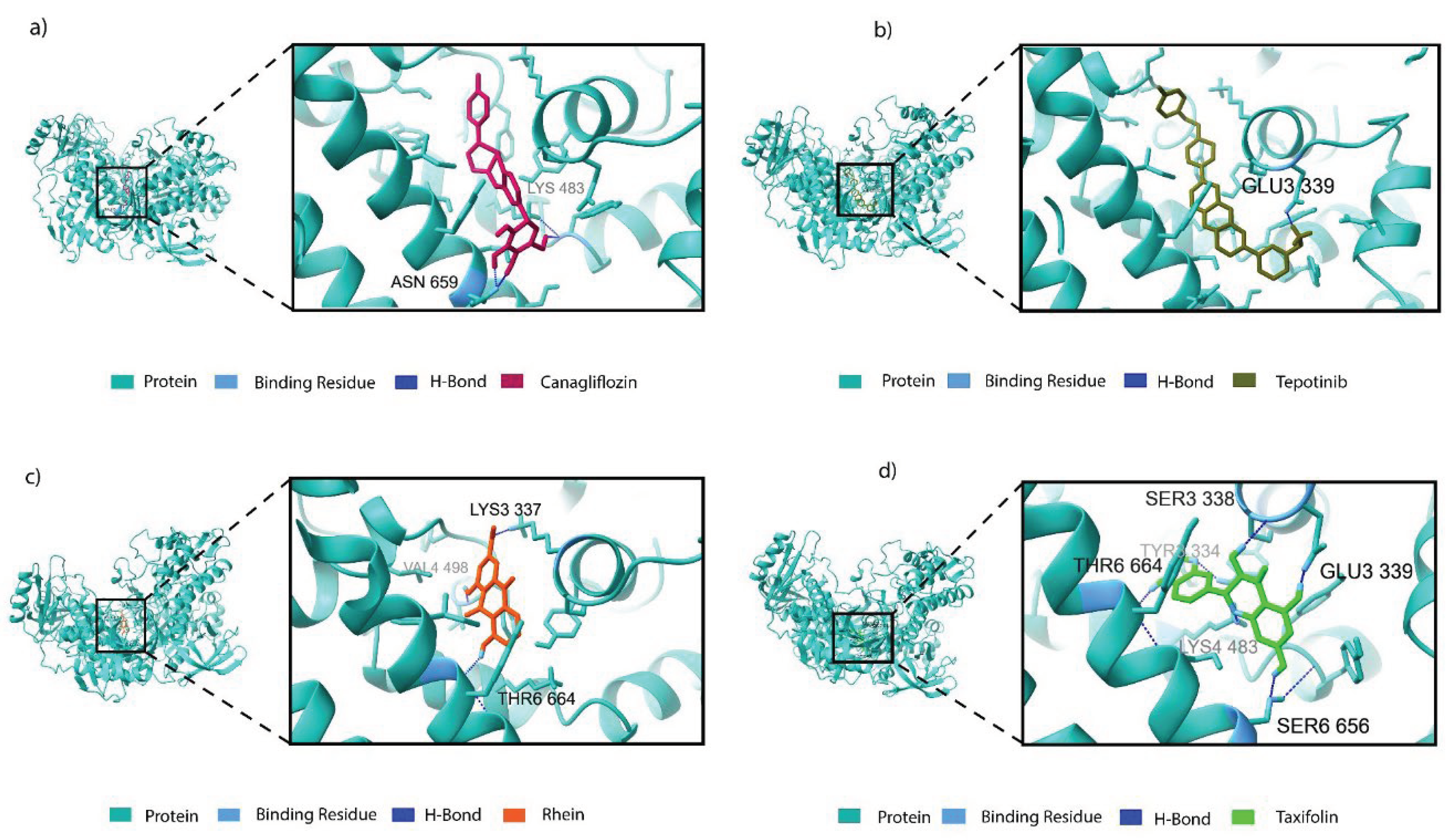
Figure 7.
Docking interactions of (a) Canagliflozin (b)Tepotinib (c) Rhein (Cassic Acid) (d) Taxifolin (Dihydroquercetin) with DNA Polymerase Protein.
Figure 7.
Docking interactions of (a) Canagliflozin (b)Tepotinib (c) Rhein (Cassic Acid) (d) Taxifolin (Dihydroquercetin) with DNA Polymerase Protein.
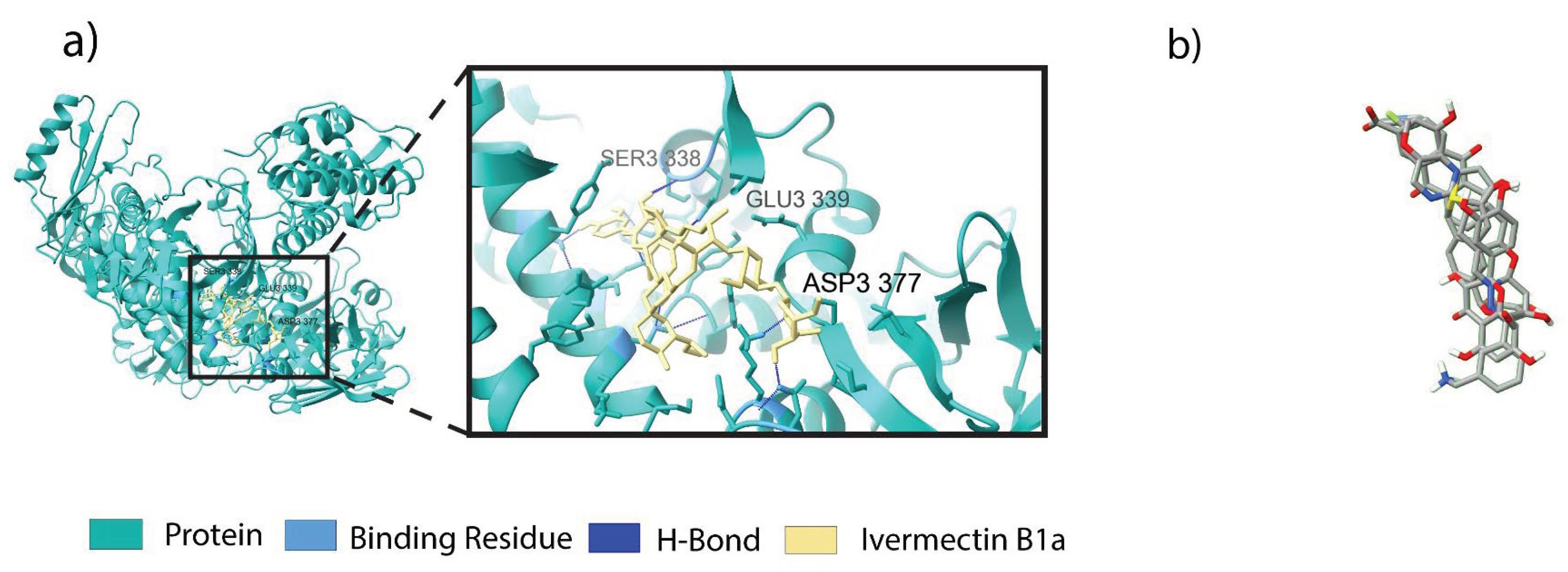
Figure 8.
Analysis of Molecular Dynamic Simulation 100 (ns) for DNA-polymerase where (a) RMSD (Root mean square deviation); (b) RMSF (Root mean square fluctuations); (c) Rg (Radius of gyration); (d) SASA (Solvent accessible surface area).
Figure 8.
Analysis of Molecular Dynamic Simulation 100 (ns) for DNA-polymerase where (a) RMSD (Root mean square deviation); (b) RMSF (Root mean square fluctuations); (c) Rg (Radius of gyration); (d) SASA (Solvent accessible surface area).
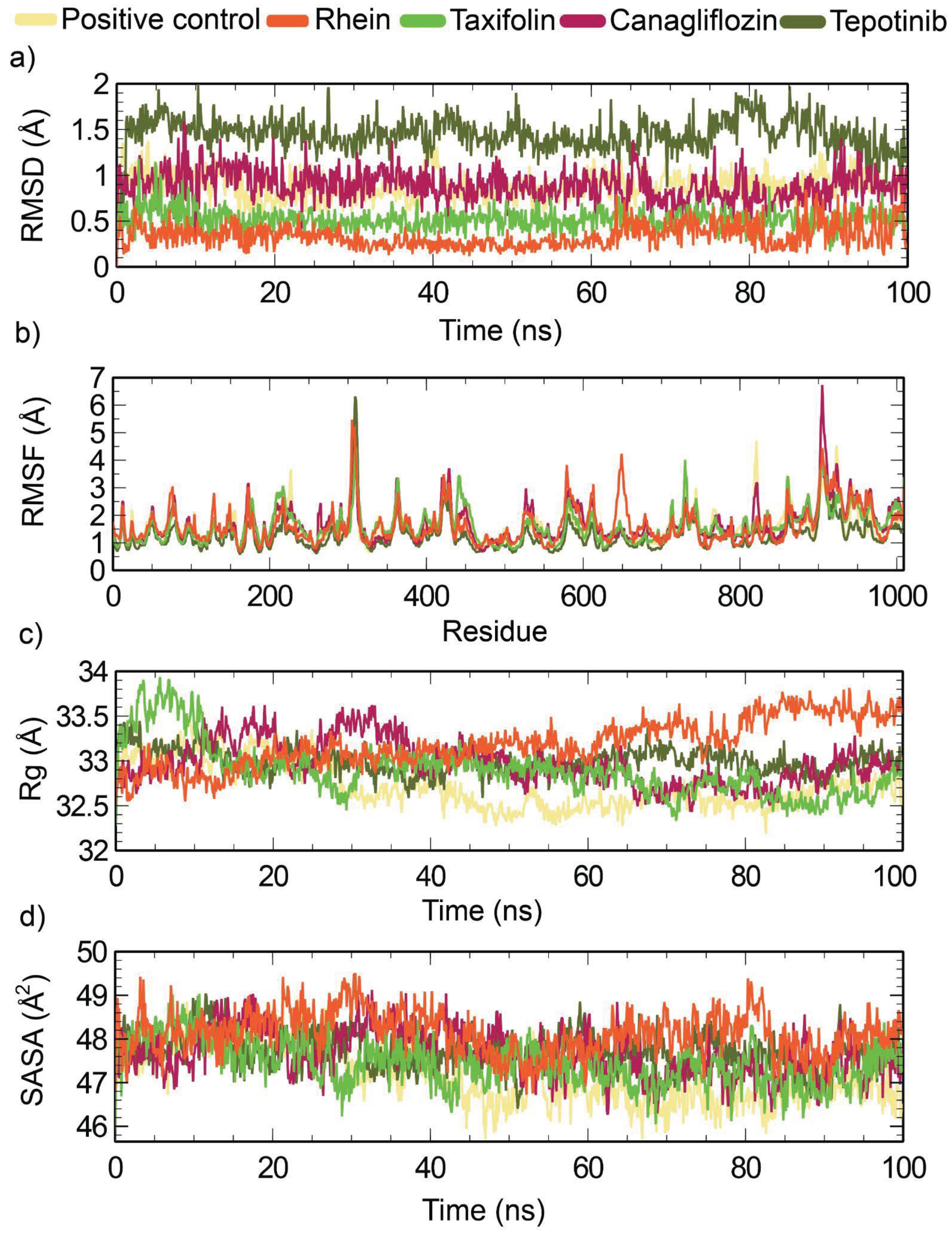

Table 1.
Binding site residues used as input for receptor grid generation during docking.
| Protein name | Binding site residues |
|---|---|
| DNA Polymerase (LSDV 039) | Lys 4, Glu 122, Gly 123, Cys 124, Arg 155, Phe156, Asn 157, Ile 158, Asn 159, Arg 160, Tyr 162, Phe 164, Ile 191, Asn 195, Leu 304, Phe 329, Thr 333, Tyr 334, Lys 337, Ser 338, Glu 339, Lys 340, Asn 352, Ala 353, Phe 354, Ser 355, Cys 356, Asn 374, Ile 379, Gly 380, Lys 381, Ile 382, Ser 383, Ser 384, Phe 385, Glu 387, Val 388, Asp 412, Tyr 473, Trp 475, Asn 476, Tyr 477, Tyr 478, Gly 479, Ile 480, Glu 481, Thr 482, Lys 483, Asp 485, Ala 486, Gly 487, Phe 489, Tyr 491, Val 498, Phe 499, Glu 500, Tyr 501, Arg 502, Ala 503, Leu 506, Tyr642, Tyr646, Leu 651, Ser 652, Thr 653, Lys 655, Ser 656, Ile 657, Tyr 658, Asn 659, Ser 660, Met 661, Glu 662, Tyr 663, Thr 664, Tyr 665, Ile 667, Ile 668, Ser 671 |
Table 2.
Protein structure validation through PROCHECK (Ramachandran plot) and Errat plot.
| Gene | Uniprot ID | Site Map Analysis | Errat value | Ramachandran plot | |||
|---|---|---|---|---|---|---|---|
| Most favored regions | Additional allowed regions | Generously allowed regions | Disallowed region | ||||
| LSDV039 | Q91MW8 | 1.048 | 88.777 | 91% | 8.0% | 0.6% | 0.3% |
Table 3.
Glide Gscore, Glide emodel, and prime MM/GBSA binding energies for hits.
| Compound type | Compound ID | Name | Glide Gscore (kcal/mol) | Glide emodel (kcal/mol) | MMGBSA dGbind (kcal/mol) |
|---|---|---|---|---|---|
| Positive control Antiviral drugs |
CID6321424 | Ivermectin B1a | −5.630 | −67.888 | −26.28 |
| DB08907 | Canagliflozin | −9.858 | −69.368 | −45.68 | |
| DB15133 | Tepotinib | −8.856 | −88.691 | −47.99 | |
| Phytocompounds | CID 10168 | Rhein | −8.965 | −43.078 | −44.72 |
| CID 439533 | Taxifolin | −7.195 | −51.868 | −44.48 |
Table 4.
Interacting residues of protein with Selected Ligands with bond type.
| Compound type | Compound ID | Name | Residues in interaction | Bond distance (Å) | Bond type |
|---|---|---|---|---|---|
| Positive control Antiviral drugs |
CID6321424 (positive control) | Ivermectin B1a | Asp 337 Ser 338 Glu 339 |
1.83 2.16 1.55 |
H-bond H-bond H-bond |
| DB08907 | Canagliflozin | Lys 483 Asn 659 Asn 659 |
2.61 1.80 1.73 |
Salt bridge H-bond H-bond |
|
| DB15133 | Tepotinib | Glu 339 Glu 339 Tyr 477 Lys 483 Phe 499 Tyr 663 |
1.93 2.94 5.49 4.01 4.89 5.38 |
H-bond Salt bridge Pi-Pi Stacking Pi-cation Pi-Pi Stacking Pi-Pi Stacking |
|
| Phytocompounds | CID 10168 | Rhein | Lys 337 Val 498 Thr 664 |
2.71 2.12 2.09 |
Salt bridge H-bond H-bond |
| CID 439533 | Taxifolin | Ser 338 Glu 339 Lys 483 Lys 483 Ser 656 Thr 664 |
2.10 1.78 2.00 3.90 2.02 2.05 |
H-bond H-bond H-bond Pi-cation H-bond H-bond |
Table 5.
Binding free energy of 4 selected ligands against LSDV DNA Polymerase.
| compound type | Compound ID | Name | ∆Gbind(NS) | ∆Gcoulomb (NS) | ∆GLipo (NS) | ∆GvdW (NS) |
|---|---|---|---|---|---|---|
| Positive control Antiviral drugs |
CID6321424 | Ivermectin B1a | −39.70 | −15.52 | −17.01 | −63.90 |
| DB08907 | Canagliflozin | −51.44 | −46.81 | −25.21 | −42.95 | |
| DB15133 | Tepotinib | −54.06 | 50.09 | −23.28 | −58.20 | |
| Phytocompounds | CID 10168 | Rhein | −38.20 | −77.35 | −13.22 | −34.55 |
| CID 439533 | Taxifolin | −47.80 | −29.02 | −13.33 | −24.43 |
*NS= No Strain, ∆Gbind (NS) = Binding/interaction energy without receptor and ligand strains, ∆Gcovalent (NS) = Covalent binding energy without stains, ∆Gcoulomb (NS) = Coulomb energy without strains. ∆GLipo (NS) = Lipophilic energy without stains, ∆GvdW (NS) = Van der Waals energy without strains.
Table 6.
ADME analysis and pharmacological parameter of hits using Qikprop.
| Compound type | Name | #Star1 | Molecular Weight2 | SASA3 | FISA4 | QPlogPo/w5 | QPlogS6 | QPlogBB7 | QPlogHERG8 | QPlogKp9 | Percent Human OralAbsorption10 | Rule Of Five11 | Rule Of Tree12 |
| (Positive control) | Ivermectin B1a | 11 | 875.104 | 1232.663 | 137.092 | 6.398 | −8.094 | −2.277 | −5.978 | −2.113 | 73.189 | 3 | 2 |
| Antiviral drugs | Canagliflozin | 4 | 438.469 | 693.643 | 174.33 | 4.006 | −6.142 | −1.354 | −6.407 | −2.970 | 92.331 | 0 | 2 |
| Tepotinib | 1 | 494.595 | 881.973 | 94.452 | 4.588 | −6.747 | −0.547 | −8.583 | −3.171 | 100.00 | 0 | 2 | |
| Phytocompounds | Rhein | 0 | 284.225 | 478.121 | 271.668 | 0.979 | −2.658 | −1.968 | −2.686 | −5.511 | 47.411 | 0 | 1 |
| Taxifolin | 0 | 304.256 | 518.521 | 276.846 | 0.107 | −2.732 | −2.271 | −4.928 | −5.382 | 52.104 | 0 | 0 |
1#Star- mentions values that fall outside the 95% range of similar values seen in other drugs. Many stars suggest that a molecule is less drug-like than molecules with few stars (the recommended range is 0–5). 2Molecular weight (range 130.0 to 725.0). 3SASA-measure the total solvent accessible surface area (recommended value 300.0 to 1000.0). 4FISA-hydrophilic component of SASA (recommended value 7.0 of 330.0). 5QPlogPo/w, predicts octanol/water partition coefficient (recommended value −2.0 to −6.5).6QPlogS Predicted aqueous solubility (recommended value –6.5 to 0.5). 7QPlogBB Predicted brain/blood partition coefficient (recommended value –3.0 1.2). 8QPlogHERG Predicted IC50 value for blockage of HERG K+ channels (recommended value, concern below –5). 9QPlogKp Predicted skin permeability, log Kp (recommended value –8.0 to −1.0). Percent Human Oral Absorption 10Predicted human oral absorption on a 0–100% scale (recommended value, >80% is high and <25% is poor). 11Rule Of Five represents Lipinski’s Rule of Five (the maximum accepted value is four). 12Rule Of Three represents Jorgensen’s rule of three (the maximum accepted value is 3).
Table 7.
The average mean value of a molecular dynamics (MD) trajectory.
| System | RMSD (Å) | RMSF (Å) | Rg (Å) | SASA (Å) |
|---|---|---|---|---|
| Ivermectin B1a | 0.88 | 1.58 | 32.72 | 47186.72 |
| Canagliflozin | 0.88 | 1.69 | 32.97 | 47677.70 |
| Tepotinib | 1.44 | 1.26 | 32.99 | 47724.08 |
| Rhein | 0.33 | 1.62 | 33.18 | 48127.33 |
| Taxifolin | 0.51 | 1.63 | 32.89 | 47439.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



