
Submitted:
12 March 2024
Posted:
13 March 2024
You are already at the latest version
Abstract
Autoimmune hemolytic anemias (AIHAs) are conditions whereby a person produces an antibody to their own red blood cells (RBCs). These conditions can be primary with unknown cause or secondary, associated with diseases or infections. There are seven different categories of AIHA, warm-antibody AIHA (wAIHA); cold-antibody AIHA that includes cold agglutinin syndrome (CAS) and cold agglutin disease (CAD); mixed AIHA, also called combined cold and warm AIHA; paroxysmal cold hemoglobinuria (PCH) also termed Donath-Lansteiner test-positive AIHA; direct antiglobulin test-negative AIHA (DAT-neg AIHA); drug-induced immune hemolytic anemia (DIIHA); and passenger lymphocyte syndrome. This review will discuss each of these AIHAs and provide information on diagnosis and treatment.
Keywords:
autoimmune hemolytic anemia
; AIHA
; categories
; diagnosis
; CAD
; PCH
1. Introduction
Autoimmune hemolytic anemia (AIHA) is a hematological illness defined by the destruction of an individual’s own red blood cells (RBCs) caused by the existence of autoantibodies that target them [1–8]. This process leads to a decreased number of RBCs in circulation. In extreme instances, the lifespan of red blood cells (RBCs) is greatly reduced, dropping from the usual range of 100-120 days to only a few days [1–4]. The internal components of red blood cells are released into the circulation and surrounding tissues, producing specific symptoms related to the condition [3]. Autoantibodies usually target antigens found on virtually all RBCs [3]. Because patients with AIHA often require transfusion to address their anemia, finding compatible blood is challenging [3,9,10]. Fortunately, transfused blood survives as well as does the patient’s own, so transfusion of antigen-selected units despite crossmatch-incompatibity should not be withheld [3]. In so doing, it is imperative to rule out underlying alloantibodies that could differentially interact with the transfused donor blood [8–10].
1.1. Classification and Categories of AIHA
AIHA is classified as either primary or secondary. The disease may manifest either as a primary ailment or as a secondary condition stemming from an underlying sickness. Primary AIHA accounts for more than 60% of patients [3]. Secondary AIHA may occur due to many underlying medical disorders, including autoimmune illnesses such as Lupus, Chronic Lymphocytic Leukemia (CLL), Non-Hodgkin’s Lymphoma (NHL), and other blood malignancies, as well as infections caused by Epstein-Barr virus, Cytomegalovirus, Mycoplasma pneumonia, Hepatitis, and HIV [3,8,11,12]. Recently, AIHA has been associated with COVID-19 (13). AIHA is further classified according to the temperature at which red blood cells experience opsonization and destruction.
AIHA comprises seven different types that have been categorized mainly based on serological findings [3,14].
The seven types of AIHA are as follows: warm-antibody autoimmune hemolytic anemia (wAIHA); cold-antibody AIHA (includes cold agglutinin syndrome (CAS) and cold agglutinin disease (CAD)); mixed AIHA (also called combined cold and warm AIHA); paroxysmal cold hemoglobinuria (PCH), also called Donath-Landsteiner antibody-test positive AIHA; drug-induced immune hemolytic anemia (DIIHA); direct antiglobulin test-negative AIHA (DAT-negative AIHA); and passenger lymphocyte syndrome (related to transplantation which can appear as AIHA) [2,3,5,14–24] (see Table 1).
 |
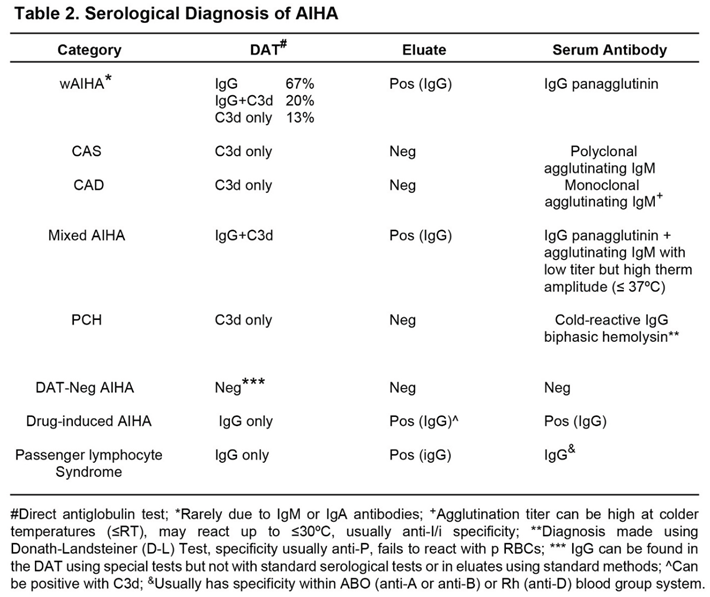
AIHA should be suspected if the patient shows signs and symptoms of anemia and other causes of the anemia, such as underproduction or bleeding, have been ruled out [3]. The most important serological test for the diagnosis of AIHAs is the direct antiglobulin test (DAT) using polyspecific and monospecific anti-human globulins (AHG) [3,14,22]. The initial testing should be performed using a polyspecific AHG that contains anti-IgG and anti-C3d. If positive, then the DAT should be performed with monospecific AHG, one containing anti-IgG and one containing anti-C3d. Table 2 shows the expected results for the DAT depending on the particular type of AIHA. The antibody may only react by indirect antiglobulin test (IAT) in wAIHA and mixed AIHA. In cold-antibody AIHA the antibody is an IgM cold agglutinin that reacts better in colder temperatures (ie- ≤ room temperature), and involves (recruits and activates) complement so that the DAT will be positive only with polyspecific and monospecific anti-C3d AHG. In mixed AIHA, the IgG antibody reacts in the IAT while the IgM antibody is an agglutinating antibody having low titer but high thermal activity (≤37oC). PCH will present with a positive DAT due to anti-C3d only; however, the serum autoantibody is an IgG bi-phasic hemolysin that is detected using the Donath-Landsteiner (D-L) test and recognizes the P-blood group system, thus, non-reactive with p RBCs. Using standard “blood bank techniques for DAT and elution”, the DAT, eluate and serum antibody are negative in DAT-neg AIHA; however, the DAT can be shown to be positive with IgG using specialized tests such as Western immunoblotting [23]. Drug-induced AIHA serologically presents as indistinguishable from wAIHA. Diagnosis in drug-dependent forms is established when stopping the suspected drug resolves the destruction, though some drugs may trigger true wAIHA by iatrogenic immune dysregulation [3]. Passenger lymphocyte syndrome is diagnosed when following a solid organ or bone marrow transplant the patient develops and antibody to the recipient’s RBCs. While presenting serologically as a wAIHA with or without a specificity, it is owing to the donor’s B-cells producing an antibody to the recipient’s residual RBCs [16,24].
 |
1.2. Incidence
Based on current calculations, the yearly occurrence of AIHA is 1-2 cases per 100,000 persons [17]. Out of all the instances, wAIHA is the most common type, accounting for two-thirds of all cases [3,17,18]. CAS/CAD is the second most common AIHA, accounting for 15-20% of cases [3,19]. Mixed AIHA is the third most common AIHA as a co-presentation of both wAIHA and cold-antibody AIHA [2,3]. PCH is a rare illness disproportionately seen in children, and associated with infections [3,20]. DAT-negative AIHA is rare [3,21] and difficult to diagnose [23]. Passenger lymphocyte syndrome occurs after transplantation and is related to the immune cells from the donor, transferred via the graft, subsequently producing host-specific anti-RBC antibodies, thereby mimicking AIHA [16,24]. In the future, the incidence of AIHA may increase due to the use of stem cell transplantation and check-point inhibitors in the treatment of cancers [25,26].
1.3. Pathophysiology of AIHA
AIHA occurs mainly due to IgG and IgM antibodies, although rarely IgA antibodies may be causative [1–5,11,14,27]. Furthermore, complement activation is seen in all patients with CAS/CAD and the majority of patients with PCH [3,14,18]. The pathophysiology will differ based on the individual components implicated. IgG has a limited ability to activate complement and has a high affinity for the Fcγ receptor (FcγR) found on phagocytic cells [3,28]. AIHA that includes IgG can be identified by the phagocytosis of RBCs [3,29]. IgM demonstrates robust activation via the conventional complement route [3]. Therefore, AIHA characterized by IgM usually involves the destruction of red blood cells via processes involving the complement system [3]. While monocyte-macrophages (MФ) do exhibit receptors for monomeric IgM Fc, the phagocytosis of red blood cells dominates by pentameric IgM antibodies in complement activation [3,30]. This phenomenon arises due to the presence of receptors on phagocytic cells that are specifically designed to bind to complement proteins C3b and C4b, which are detectable using a DAT with anti-complement reagents, primarily anti-C3d [3,14]. In mixed AIHA, both IgG and C3d are found on the patient’s RBCs. This is due to an agglutinating IgM cold agglutinin having a thermal amplitude to 37oC able to activate complement, and a warm-reactive IgG antibody that is reactive at 37oC causing a positive IAT [3]. In PCH, a “cold-reactive” IgG antibody binds to the patient’s RBCs at cooler temperatures (below 37oC) and can activate complement when the temperature warms to 37oC, though the patient’s RBCs show only residual complement in the DAT [3]. Removal of the opsonized RBCs by MФ are by either FcγRs if IgG, and/or C3b/C4b receptors if complement has been activated. Usually, removal of opsonized RBCs in AIHA with IgG antibodies takes place in the spleen, while IgG plus complement or complement alone on the RBCs occurs in the liver, specifically in Kupffer cells [3]. In rare cases, IgA may induce AIHA [3,27,31,32]
The symptoms of AIHA vary depending on the type. The symptoms may include dyspnea, fatigue, headache, muscular weakness, pallor, and/or jaundice [3,8]. The development of acrocyanosis and Raynaud phenomenon in CAS/CAD may culminate rarely with gangrene [3,19]. Spherocytes are often seen in cases of wAIHA when there is inadequate phagocytosis of antibody-coated RBCs by MФ [3,32]. The biochemical signs of hemolysis identified in immune-mediated hemolysis include decreased hemoglobin levels, alterations in cell markers linked to hemolysis such as higher lactate dehydrogenase (LDH), reduced haptoglobin, and increased unconjugated bilirubin [3,8,33]. In addition, a compensatory reticulocytosis may occur in those with unsuppressed, feedback-responsive marrow reserves [3]. Some cases of AIHA occur with reticulocytopenia and the synergy of underproduction with destruction may result in severe anemia with life-threatening consequences [34,35].
2. Warm-Antibody Autoimmune Hemolytic Anemia (wAIHA)
wAIHA is commonly acknowledged as the primary occurrence of autoimmune-mediated hemolytic anemia [2,3,5,14,17]. Roughly half of the instances have an unknown cause, while the other half may be linked to either an existing underlying illness or the use of certain drugs [3,12,36]. Unlike cold autoimmune hemolytic anemia conditions like CAS/CAD and PCH, which exacerbate in cold temperatures between 28°C to 31°C, wAIHA occurs at normal body temperature [3]. The primary antibody isotype involved with wAIHA is IgG, but IgA and IgM may be infrequently seen [3,14,27,30–32,37]. IgG antibodies have a strong attraction to red blood cells when the temperature is at the normal level of the human body (37°C). Cold-antibody-induced hemolytic anemia, in contrast, is distinguished by antibodies that preferentially attach to red blood cells at lower temperatures, usually between 28oC and 31°C. This category includes CAS/CAD, PCH, and mixed AIHA. However, in mixed AIHA, the IgM cold-reactive antibody can react to 37oC by agglutination [37].
The hemolysis in wAIHA is classically extravascular, mediated by MФ phagocytosis of antibody- and/or complement-opsonized autologous RBCs [3,29,33,38]. However, a role for natural killer cell-mediated antibody-dependent cellular cytotoxicity (ADCC) may also play a role in some cases of wAIHA [39].
Furthermore, MΦ have the capacity to selectively remove portions of the red cell membrane, resembling a biting action, in addition to phagocytosis [3]. This mechanism is similar to trogocytosis [40]. In wAIHA, this leads to the degradation of the RBC membrane, causing transformation into spherocytes [3,32]. Spherocytes possess less pliancy compared to regular erythrocytes and are specifically singled out for removal in the red pulp of the spleen and other constituents of the mononuclear phagocyte system. The observed expansion of the spleen (splenomegaly) relates to the trapping of spherocytes and autoantibody-opsonized RBCs in the red pulp of this organ [3].
2.1. Autoantibody Specificity
The specificity of autoantibodies in wAIHA has historically tracked with the Rh blood group system, because autoantibodies that reacted with all normal RBCs would fail to react with rare RBCs lacking certain or all Rh antigens, from Rh dash-D-dash (-D-) to Rhnull RBCs, respectively. For example, warm autoantibodies that reacted with all normal RBCs as well as with -D- and Rhnull RBCs were termed anti-nl (non-deleted), while autoantibodies that would react with all normal RBCs and -D- but not Rhnull were termed anti-pdl (partially deleted). Finally, autoantibodies that would react with normal, but not with -D- and Rhnull, were termed anti-dl (deleted) autoantibodies [3]. Subsequently, other specificities were reported to exist; for example, anti-Jka, anti-Gerbich, anti-LW and anti-Kell [3,41,42]. Other investigators found autoantibodies apparently reacting with components of band 3 on the RBCs [3,41]. As investigators dealt with the various specificities reported and were using Rhnull and -D- RBCs to determine the anti-nl, anti-pdl or anti-dl specificity of these autoantibodies, two groups, independently, found that in the majority of patients with wAHIA, autoantibodies reacted preferentially with older, reticulocyte-depleted RBCs and reacted much less or not at all with younger, reticulocyte-enriched RBCs [43,44]. Both groups found that the frequency of reactivity was 80% of patients with wAIHA for the former and 20% for the latter. Thus, warm autoantibodies in 80% of the cases showed a tropism to “old RBCs” compared to “young RBCs”. One group termed this specificity as Type I wAIHA, versus Type II wAIHA (20% of patients) with wAIHA showing no RBC age preferences [44]. These investigators suggested that Type I wAIHA could be an exacerbation of the normal clearance mechanisms in RBC senescence whereby natural autoantibodies deplete old RBCs by targeting band 3 in age-dependent conformational forms [44–47].
More recently, investigators have shown that most warm autoantibodies do, in fact, target band 3 [41,48,49]. In a recent publication, the authors confirm Type I and Type II wAIHA and suggest that Type I wAIHA autoantibodies target band 3 and that the hemolytic anemia may conversely involve patient RBCs that are also aging faster than normal [48]. A hypothesis is thus created that Type I patients’ RBCs, by aging faster, expose the immune system to more senescent RBC antigen, which in turn stimulates more autoantibody to senescent band 3, producing a functional autoimmune disease [41]. In contrast, patients with Type II wAIHA showed no excess in RBC aging, suggesting rapid and equal-opportunity opsonisation and removal of RBCs [48], consistent with previous reports of Type II wAIHA being more severe than Type I wAIHA [44]. Additional investigations show that the natural autoantibody that binds to aged RBCs, indeed, recognizes band 3 [48,49]. Taken together, these findings generate the hypothesis that most wAIHA patients, 80%, produce more of the senescent RBC autoantibody that reacts with patients’ accelerated aging RBCs, for Type I wAIHA [41,48]. Another implication from this work is that the specificity of the autoantibody in Type II wAIHA may be to other (non-band 3) RBC antigens [3,44]. These hypotheses require further work.
2.2. Biomarkers
Identifying biomarkers specific to particular disease states is an ongoing quest by biomedical researchers. Cytokines provide one area of exploration. In wAIHA some cytokines appear to be associated [50–53]. In the most comprehensive report [50] of cytokines associated with wAIHA, 54 patients with DAT+ wAIHA versus 36 normal, healthy controls were evaluated for 38 cytokines, chemokines or growth factors using a multiplex approach and Luminex technology. These investigators confirmed that TNFα and interleukin10 (IL-10) are increased in patients with wAIHA but also showed that IL-8/CXCL8, IP10/CXCL10 are also increased and suggest these as possible biomarkers too. Thus, wAIHA patients have 4 possible cytokine/chemokine biomarkers, TNFα, IL-10, IL-8/CXCL8 and IP10/CXCL10; and, perhaps IL-6 [52]. Future studies may validate this repertoire.
2.3. Treatment
wAIHA is usually initially treated with glucocorticoids, prednisone/prednisolone being most commonly used [3,55–58]. Glucocorticoids often cause some or complete remission but in difficult cases other treatments will be employed, such as off-label rituximab (anti-CD20), IVIG, or daratumumab (anti-CD38); other investigational agents such as FcγRn inhibitors (nipocalimab), Syk kinase inhibitor (fostamatinib), BTK kinase inhibitor (rilzabrutinib), or PI3 kinase, may eventually be shown to be useful [56–62]. If complement mediated hemolysis is suspected, treatment may include inhibitors of the complement pathway such as sutimlimab (anti-C1s) or eculizumab (anti-C5) [57,59,62,63]. There continues to be active research to find new therapies for wAIHA [57,63–66]. Although splenectomy was used for years with great success, it is rarely used today for resolution of wAIHA [3,59]. However, a recent study suggests that, in a cohort of 1824 patients, splenectomy was found to be an effective treatment; however, with a surgical complication rate of 12% [67].
3. Cold-Antibody Autoimmune Hemolytic Anemia
Cold-antibody autoimmune hemolytic anemia, referred to as cold agglutinin syndrome (CAS) or cold agglutinin disease (CAD), is a rare autoimmune condition characterized by the presence of high levels of cold-sensitive autoantibodies in the blood, primarily IgM, as well as autoantibodies that remain active at temperatures below 30°C (86 °F) [3,14,19,68–74]. The antibodies have a particular affinity for red blood cells, often the Ii RBC antigens [3,14,75,76], causing them to aggregate (agglutination), activate the complement system, and undergo intra- and/or extravascular hemolysis [3,14,19,65–75]. Historically, CAD included both CAD and CAS; however, these two conditions are now recognized as different and have been separated into distinct entities.
3.1. Cold Agglutinin Syndrome
Cold agglutinin syndrome (CAS) is a transient condition that is secondary to a bacterial or viral infection, such as Mycoplasma pneumoniae or Epstein-Barr virus causing mononucleosis [2,3,68,75]. CAS can also occur following certain malignancies and other autoimmune disorders [68,71–73]. CAS is also known as secondary cold agglutinin disease. CAS is self-remitting, and the condition usually is resolved when the underlying infection is resolved [3].
3.2. Cold Agglutinin Disease
Cold agglutinin disease (CAD) or primary CAD is a clonal disease whereby lymphoproliferation of a B-cell clone produces a cold-reactive (≤ 30oC) monoclonal IgM autoantibody having specificity for the I or I RBC antigens [76]. The monoclonal IgM can activate complement and cause intravascular hemolysis. CAD is a chronic condition [71–74,76].
Both CAS and CAD have similar findings for diagnosis. Both are caused by an IgM autoantibody, that targets either I or i RBC antigens. The IgM autoantibodies are naturally occurring, polyclonal anti-I or anti-i with CAS; and, in CAD there is an abnormal production of a monoclonal autoantibody, associated with a B-cell clone. The IgM autoantibody in both conditions activates complement and can cause severe intravascular hemolysis [77]. Thus, both will have a positive DAT due to only complement (C3d) on their RBCs and both will have a cold agglutinin titer at 4oC ≥64, with a thermal range ≤30oC [3].
3.3. Treatment
Treatment for CAS usually involves keeping the patient warm until the condition resolves with convalescence from the underlying disease. Manouveurs include blanketing or maintaining higher ambient room temperatures at 37oC-40oC, and if transfusion is required, using a blood warmer. In severe cases of CAS, one may use sutimlimab (anti-C1s) that inhibits the classical complement activation pathway [77].
CAD is a more difficult to treat AIHA as the antibody is usually associated with a chronic disease and the monoclonal antibody produced can cause more severe anemia than in CAS. Therefore, treatment can consist of rituximab or anti-complement therapies such as sutimlimab [57,62,83,84], or eculizumab to block the membrane attack complex of complement activation pathway [57,70,83]. To limit production of the causal antibody and to diminish the cellular biomass of the driving condition, rituximab plus strong immuosuppressive drugs such as fludarabine or bendamustine may be prescribed [57,78,83,84]. Recently, use of daratumumab (anti-CD38) has been shown to be effective in the treatment of relapsing CAD [81,82].
4. Mixed AIHA (Combined Cold and Warm AIHA)
Mixed AHIA or combined cold and warm AIHA is characterized by the simultaneous presence of an IgG warm autoantibody and a cold-reactive IgM antibody with a low titer but a broad temperature range in the blood circulation, reacting up to 37oC [2,3,37,85–91]. The syndrome is marked by significant hemolysis, resulting from both intravascular and extravascular hemolysis. The illness may be severe but shows a favourable response to steroid therapy [37]. However, mixed AIHA often follows a chronic trajectory with intermittent periods of deterioration [2,3,87,89]. The condition accounts for 5%-8% of all AIHA; of these, 50% of cases are idiopathic while 25%-42% of cases are associated with systemic lupus erythematosus (SLE) [3,4].
Treatment
Treatment usually involves glucocorticoids with patients having a good and rapid response [37]. However, again, rituximab and/or complement inhibitors may be considered ([59,89]; see treatments under wAIHA and CAD, above).
5. Paroxysmal Cold Hemoglobinuria or Donath-Landsteiner Test-Positive Hemolytic Anemia
Paroxysmal cold hemoglobinuria (PCH) is a rare autoimmune hemolytic anemia that is defined by the destruction of red blood cells in the blood vessels due to the activation of the complement system [2,3,92–102]. This destruction happens when the body is exposed to cold temperatures [3,14,98]. PCH is characterized by the presence of a cold-reactive IgG autoantibody. This antibody attaches to RBCs at temperatures below 37°C. When the RBCs are warmed to body temperature, the antibody activates complement, and the IgG antibody subsequently dissociates from the RBCs [3,14,98]. As a result, only complement is detected on the cells.
PCH was first documented in 1904 by Julius Donath and Karl Landsteiner, establishing it as one of the earliest recognized clinical conditions associated with AIHA [3]. The illness may present either as a sudden, non-recurring post-infectious event in children [3,14,20,92,95–100] or as recurring occurrences in adults with blood cancers or advanced syphilis [3,20,93,94]. Historically, PCH manifested primarily in patients having syphilis and exposure to cold resulted in paroxysms of hemoglobinuria. Today, PCH is almost always encountered as an acute transient syndrome in young children with a recent history of a viral illness [92–94], so that paroxysms resulting from cold exposure are rarely seen. Thus, it has been suggested that a better term for this condition would be Donath-Landsteiner test-positive hemolytic anemia.
5.1. Donath-Landsteiner Test
The diagnosis is made in the laboratory by the use of the Donath-Landsteiner test (D-L Test) [3,14,98]. This test utilizes the findings of anti-P as being the biphasic IgG autoantibody and RBCs positive for the P antigen and RBCs lacking the P antigen, p RBCs. RBCs that are P+ (random RBCs) and P- (p RBCs) incubate with the patient’s plasma, with a source of fresh complement or not, as a control, at room temperature. The test system then moves from room temperature to 37oC. If the P+ RBCs are hemolyzed and the p RBCs are not, then the D-LTest is positive and the diagnosis of PCH is confirmed.
5.2. Treatment
PCH in children is usually self-remitting; however, in severe cases requiring transfusion, case reports or series have described that have been successfully treated with plasma exchange, rituximab, and complement inhibitors such as eculizumab [3,95,99–102].
6. DAT-Neg AIHA
Approximately 1% to 10% of persons diagnosed with AIHA have a negative DAT [3,103,104]. The DAT is a diagnostic technique that has a sensitivity range from 92% to 97% for detecting AIHA. The diagnosis of DAT-neg AIHA mostly relies on the method of excluding other possible causes [3,14,103–105]. Several key variables contribute to the development of DAT-neg AIHA. These factors consist of (a) the existence of red blood cell-bound IgG at levels that are too low to be detected using standard methods [3,20,103]; (b) the relatively weak binding strength of IgG [3]; (c) the lack of a positive eluate using standard methods and (c) the presence of RBC-bound IgA where AHG contains only antibodies to IgG [73], or in rare instances, IgA or IgM [3,21,27,30,31,106].
In the early 1970s, Gilliland and colleagues [3,103] were the first to emphasize that the amount of antibodies found on RBCs in patients with AIHA may be insufficient to detect by standard DAT reagents. They proposed that the distribution of IgG antibodies on circulating RBCs was non-uniform. RBCs accumulate IgG as they age, leading to a larger ratio of IgG to RBCs in older RBCs compared to younger ones [43,44,48,49,103].
In some instances of DAT-neg AIHA, less often encountered immunoglobulins, namely IgA and IgM, may be identified on the outside of RBCs [3,22,27,30,31,106]. IgA AIHA has a clinical picture that closely matches that of IgG wAIHA [106]. While the DAT may yield negative results when performed with traditional antiglobulin sera [3], a Western blotting method may detect low levels of IgG on RBCs in DAT-neg AIHA [23]. This technique could also be adjusted to monitor IgA and IgM levels on RBCs in DAT-neg AIHA. Most instances of IgM AIHA are caused by the presence of a pathological cold autoantibody that has a broad temperature range and may respond at temperatures ranging from 30oC to 37°C (see Sections 3 and 4, above). Diagnosing warm IgM AIHA might be difficult since there are no significant serologic findings. Antibody detection tests may exhibit little reactivity. Both pathogenic IgM cold autoantibodies and warm IgM autoantibodies exhibit the presence of complement coating on the patient’s RBCs. As a result of this feature, some persons with warm IgM AIHA may get an inaccurate diagnosis of CAD or PCH [3,30].
Treatment
Treatment for severe cases of DAT-neg AIHA would be similar as to those used for the treatment of wAIHA (see Section 2, above).
7. Drug-Induced Immune Hemolytic Anemia
Drug-induced immune hemolytic anemia (DIIHA) is a rare condition primarily caused by the existence of drug-induced antibodies, which may be classified as either drug-dependent or drug-independent [15,36,107–109]. Patients with DIIHA may have signs of rapid destruction of red blood cells inside blood vessels quickly after receiving the medicine. This may be seen, for example, in small babies who develop hemolytic anemia as a result of ceftriaxone therapy [108,109]. In contrast, some persons may have less severe signs of extravascular hemolysis, which might occur many months after therapy, as shown in instances of methyldopa-induced hemolytic anemia [31,38].
7.1. Drug-Dependent Antibodies
Drug-dependent antibodies may be detected by analyzing RBCs that have been treated with drugs, termed the “hapten-specific mechanism” or untreated RBCs in the presence of a drug solution, termed the “neoantigen-dependent mechanism” [36,108]. Both of these mechanisms require the drug to somehow interact with proteins on the RBCs, forming a hapten (drug)-carrier (RBC) complex which can elicit an antibody response to the drug (hapten). Examples include penicillin and cephalosporins [36,108]. Alternatively, a drug may complex with the RBC in vivo forming a compound antigen, a neoantigen, that requires the drug to be in the testing system in order to detect its activity [36]. Examples of this mechanism include 2nd and 3rd generation cephalosporins such as cefotetan and ceftriaxone [36,108,109].
In some instances, the antibody that is detected appears to an autoantibody resembling autoantibodies found in wAIHA [12,36,38,109]. In this case, the DAT is positive for IgG without apparent drug-dependence, and with IgG antibody detectable in plasma [12]. The prototypical drug that causes this type of DIIHA is alpha-methyldopa [38]. This phenomenon has been termed the “cross-reactive autoantibody mechanism” [36]. Finally, there is an unusual mechanism whereby following certain drug therapies, the patient’s RBCs are able to take up proteins from their surroundings, including immunoglobulins, and give a positive DAT. With IgG on the patient’s RBCs, this can result in monocyte-macrophage recognition and extravascular hemolysis. This mechanism is poorly understood but it may be dependent on an alkaline pH, so that patients in a state of metabolic alkylosis may be at risk [36,107].
7.2. Drug-Induced Autoimmune Hemolytic Anemia
The mechanism of cross-reactive autoantibody production results in an IgG warm autoantibody being made by the patient that is dependent on the drug being administered. The classic example is that of alpha-methyldopa therapy [12,38]. In these conditions, the patient presents with acquired hemolytic anemia with a DAT positive for IgG and negative for complement, and a serum antibody that reacts with all unrelated RBCs. Thus, this condition looks like a classic wAIHA. It has been postulated that the drug interacts with the RBC membrane to modify it enough that it appears as “non-self” to the patient’s immune system and this then results in the patient making an antibody to the RBCs plus the drug, but also a “crossreactive” antibody that doesn’t require the drug [36,38]. Stopping the administration of the implicated drug will resolve the anemia; but, it may take some time for the autoantibody to go away [33]. It is unknown why only a small percentage of patients will develop this condition; however, the resulting hemolytic anemia can be severe and even life-threatening. Methyldopa is rarely used anymore. Currently, drugs that can result in the production of an IgG autoantibody to the patient’s RBCs include cefotetan and ceftriaxone [36,108,109]. These drugs also act by both the hapten-specific and neoantigen-dependent mechanisms making these two drugs potentially very dangerous [108,109].
7.3. Immunoglobulin Adsorption Mechanism
The other mechanism that can present as wAIHA is when drugs cause adsorption of serum proteins onto the RBCs. This previously was termed the “nonspecific adsorption mechanism” and results in any serum protein being “adsorbed” onto the RBCs, including albumin, complement components and immunoglobulins, in particular IgG [3,107]. The patient can develop hemolytic anemia due to the IgG being on the patient’s RBCs. When the laboratory investigates the reason for the anemia, the DAT is positive for IgG. However, the eluate will be negative providing a clue that this may be drug-related. Although the implicated drug can cause adsorption of complement components, these would be C2, C3, C4 that normally circulate in the blood, but not the complement component that is tested in the DAT, which is C3d. Thus, only IgG would be detected using AHG.
Anytime a patient with acquired immune hemolytic anemia is encountered with an IgG positive DAT and a negative eluate, a drug history should be obtained as this outcome may be related to a drug-induced hemolytic anemia. The reason for this phenomenon of immunoglobulin adsorption has been postulated to be due to the chemistry of the drugs, with some drugs having chemical structures that allows these drugs to covalently bind to both RBC proteins and external proteins in the patient’s serum [36,107].
7.4. Treatment
The treatment for these drug-induced conditions often simply involves stopping the drug therapy, if possible, or switching to a different, chemically unrelated, drug. When this is done, the hemolysis may abruptly cease [12,38]. If the hemolysis is severe one can treat the patient with similar therapies as used for the treatment of wAIHA.
8. Passenger Lymphocyte Syndrome
Passenger lymphocyte syndrome is an unusual hemolytic occurrence that can resemble AIHA. Allogeneic transplantation includes bone marrow transplantation (BMT) and solid organ transplantation. In BMT, host lymphocytes are transferred into the recipient from the bone marrow or stem cell material obtained from the donor [13,93]. In solid organ transplantation, donor lymphocytes can be “passengers” in the solid organ obtained from the donor and these can be transferred into the recipient [24]. It is feasible for donor lymphocytes that have been transferred to produce antibodies that are specific to RBC antigens in the recipient’s body, but not present on the donor’s own red cells [13,24,110–113]. Indeed, sometimes these passenger lymphocytes in either BMT or solid organ transplants are obtained from donors who have been sensitized to produce alloantibodies to antigens for which they lack. Thus, passenger lymphocytes from a blood group O donor transferred into a recipient of blood group A can produce anti-A that can react with and hemolyze the residual RBCs in the recipient due to those being blood group A. Likewise, passenger lymphocytes from a donor who is Rh-negative but who has been sensitized to produce anti-D can induce a hemolytic anemia in a Rh-positive recipient, which may manifest as a delayed-type hemolytic reaction. In both instances, the hemolytic anemia can appear as an AIHA, though with an apparent specificity. Indeed, if this syndrome occurs, its presentation as immune hemolysis after a transplantation may be misinterpreted as an autoimmune hemolytic anemia. The hemolysis can be severe with outcomes such as renal failure [111,113].
8.1. Role of Cyclosporine
The specific influence of cyclosporine on the promotion of antibody production from the donor is still not fully understood. Because hemolysis has been most frequently related to cyclosporine, a medication that suppresses the immune system, it is suggested that donor B lymphocytes proliferated and produced antibodies because of cyclosporine effects to selectively inhibit T-cell function [114]. Alternatively, previously sensitized lymphocytes when exposed to an antigen in the presence of cyclosporine can still respond to antigens on recipient RBCs [115].
8.2. Treatment
It is critical to recognize that the stem cell or solid organ transplant patient who is hemolyzing may be manifesting a passenger lymphocyte syndrome. Treatment may require transfusion of antibody-evading (donor blood type) RBCs, ie- blood group O cells if hemolysis is due to ABO antibodies (in an group O donor to a non-O recipient). If anti-D or other alloantibodies are identified as the cause of the hemolysis, then antigen-negative donor blood should be used for transfusion [112,116]. If hemolysis is severe, besides transfusion, if required, one can treat with immunosuppressive drugs [116,117], or a combination of immunosuppressive drugs, IVIG, and plasmapheresis [114].
9. Current Opinion
We have often wondered if hyperhemolysis syndrome (context sickle cell disease (SCD) or not) deserves its own category within AIHA, as, although it is a distinctly transfusion-triggered event, with the absence or presence of an involved alloantibody cognate to a triggering unit. Once triggered, however, it appears to be an autoimmune-like phenomenon with extreme bystander hemolysis [ref] (kinetically akin to post transfusion purpura (PTP)), and with a particular role of the complement cascade (as judged by the response of some cases to eculizumab), as well as hyperinflammation (as judged by the greater responsiveness to IVIG with high-dose steroids, +/- the utility of anti-cytokine storm interventions such as tocilizumab). In virtually every regard, this is a pathology that is not addressed by any of the conventional AIHA categories. However, because it manifests in part with an autologous response, there is an argument for its inclusion in the taxonomy of AIHA, just as there are arguments for the inclusion of passenger lymphocyte syndrome.
10. Summary and Perspectives
The review herein has scoped the range of autoimmune hemolytic anemias. There are seven categories (Table 1), including passenger lymphocyte syndrome, which though not endogenous, may appear as an AIHA. Within each category the diagnostic features and management options are compared and contrasted. The promise of novel immune modifiers (anti-CD38, complement cascade specific monoclonals, [57,81,82,118], anti-rejection medications [57,119,120]/mTOR therapies [126], kinase inhibitors [121–123], protease inhibitors [124], and co-stimulation blockade agents [125], awaits testing to broaden the range of treatment options.
Institutional Consent Statement
Not applicable.
Author Contributions
Conceptualization, M.L., C.C-G., and D.R.B.; writing - original draft preparation, M.L.; writing – review and editing, C.C-G., and D.R.B.; All authors have read and agree to the published version of the manuscript.
Funding
There was no external funding received.
Data Availability Statement
There is no data to share.
Conflict of Interest
The authors declare no conflict of interest.
References
- Dacie, J.V. Autoimmune haemolytic anaemias. Br. Med. J. 1970, 2, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Hewitt, S.; Stamps, B.K. Autoimmune haemolysis: An 18-year study of 865 cases referred to a regional transfusion centre. Br Med J (Clin Res Ed). 1981, 282, 2023–2027. [Google Scholar] [CrossRef] [PubMed]
- Petz, L.D.; Garratty, G. Immune Hemolytic Anemias. 2nd ed, Philadelphia, PA: Churchill Livingstone, 2004.
- Hashimoto, C. Autoimmune hemolytic anemia. Clin. Rev. Allergy Immunol. 1998, 16, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Barcellini, W. Autoimmune Hemolytic Anemias. N. Engl. J. Med. 2021, 385, 1407–1419. [Google Scholar] [CrossRef]
- Mulder, F.V.M.; Evers, D.; de Haas, M.; et al. Severe autoimmune hemolytic anemia; epidemiology, clinical management, outcomes and knowledge gaps. Front. Immunol. 2023, 14, 1228142. [Google Scholar] [CrossRef]
- Giannotta, J.A.; Capecchi, M.; Fattizzo, B.; Artoni, A.; Barcellini, W. Intravenous immunoglobulins in autoimmune cytopenias: An old tool with an alternative dosing schedule. Blood Transfus. 2023, 21, 557–560. [Google Scholar] [PubMed]
- Tranekær, S.; Hansen, D.L.; Frederiksen, H. Epidemiology of Secondary Warm Autoimmune Haemolytic Anaemia-A Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 1244. [Google Scholar] [CrossRef] [PubMed]
- Branch, D.R.; Petz, L.D. Detecting alloantibodies in patients with autoantibodies. Transfusion 1999, 39, 6–10. [Google Scholar]
- Petz, L.D. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: Should we eliminate this meaningless term? A commentary for clinicans and transfusion medicine professionals. Transfusion 2003, 43, 503–507. [Google Scholar] [CrossRef]
- Barros, M.M.; Blajchman, M.A.; Bordin, J.O. Warm autoimmune hemolytic anemia: Recent progress in understanding the immunobiology and the treatment. Transfus. Med. Rev. 2010, 24, 195–210. [Google Scholar] [CrossRef]
- Al Hadidi, S.; Udden, M. Autoimmune hemolytic anemia in a patient with chronic lymphocytic leukemia. Clin. Case Rep. 2020, 8, 1112–1113. [Google Scholar] [CrossRef] [PubMed]
- Zebardast, A.; Hasanzadeh, A.; Shiadeh, S.A.E.; Tourani, M.; Yahyapour, Y. COVID-19: A trigger of autoimmune diseases. Cell Biol. Int. 2023, 47, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Petz, L.D.; Branch, D.R. Serological tests for the diagnosis of immune hemolytic anemias. IN: Methods in Hematology: Immune Cytopenias, vol. 9 (R. McMillan, ed.), Churchill Livingstone, New York, NY.; 1983; 9-48.
- Petz, L.D.; Branch, D.R. Drug-induced immune hemolytic anemia. IN: Methods in Hematology: Immune Hemolytic Anemias, vol. 12 (H. Chaplin, ed.), Churchill Livingstone, New York, NY.; 1985; 47-94.
- Hows, J.; Beddow, K.; Gordon-Smith, E.; et al. Donor-derived red blood cell antibodies and immune hemolysis after allogeneic bone marrow transplantation. Blood. 1986, 67, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.L.; Möller, S.; Andersen, K.; Gaist, D.; Frederiksen, H. Increasing incidence and prevalence of acquired hemolytic anemias in Denmark, 1980-2016. Clin. Epidemiol. 2020, 12, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Packman, C.H. Hemolytic anemia due to warm autoantibodies. Blood Rev. 2008, 22, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Röth, A.; Randen, U.; Jilma, B.; Tjønnfjord, G.E. Cold agglutinin disease: Current challenges and future prospects. J. Blood Med. 2019, 10, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.W.; Figueroa Villalba, C.A.; Booth, G.S.; Woo, J.S.; Stephens, L.D.; Adkins, B.D. Clinical and epidemiological features of paroxysmal cold hemoglobinuria: A systematic review. Blood Adv. 2023, 7, 2520–2527. [Google Scholar] [CrossRef]
- Rodberg, K. DAT-Negative Autoimmune Hemolytic Anemia. Hematol Oncol Clin North Am. 2022, 36, 307–313. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. Stategies to overcome the diagnostic challenges of autoimmune hemolytic anemias. Expert Rev. Hematol. 2023, 16, 515–524. [Google Scholar] [CrossRef]
- Bloch, E.M.; Sakac, D.; Branch, H.A.; Cserti-Gazdewich, C.; Pendergrast, J.; Pavenski, K.; Branch, D.R. Western immunoblotting as a new tool for investigating direct antiglobulin test-negative autoimmune hemolytic anemias. Transfusion 2015, 55, 1529–1537. [Google Scholar] [CrossRef]
- ElAnsary, M.; Hanna, M.O.F.; Saadi, G.; et al. Passenger lymphocyte syndrome in ABO and Rhesus D minor mismatched liver and kidney transplantation: A prospective analysis. Hum. Immunol. 2015, 76, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Khosla, A.; Sandhu, R.S.; Singhal, S.; Koka, J.-M. Atezolizumab-induced direct antiglobulin test-negative autoimmune hemolytic anemia. Am. J. Ther. 2023; Jul 11, online ahead of print. [Google Scholar]
- Greenmyer, J.R.; Anagno, S.; Ali, A.; et al. Autoimmune cytopenias following pediatric hematopoietc cell transplant. Bone Marrow Transplant. 2024, 59, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Booker, D.J.; Stamps, R.; Booth, J.R.; Hook, V. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997, 37, 175–181. [Google Scholar] [CrossRef]
- Abramson, N.; Gelfand, E.W.; Jandl, J.H.; Rosen, F.S. The interaction between human monocytes and red cells. Specificity for IgG subclasses and IgG fragments. J. Exp. Med. 1970, 132, 1207–1215. [Google Scholar] [CrossRef]
- Gallagher, M.T.; Branch, D.R.; Mison, A.; Petz, L.D. Evaluation of reticuloendothelial function in autoimmune hemolytic anemia using an in vitro assay of monocyte-macrophage interaction with erythrocytes. Exp. Hematol. 1983, 11, 82–89. [Google Scholar]
- Arndt, P.A.; Leger, R.M.; Garratty, G. Serologic findings in autoimmune hemolytic anemia associated with immunoglobulin M warm autoantibodies. Transfusion. 2009, 49, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, N.; Dua, S.; Arora, S. IgA-mediated autoimmune hemolytic anemia in an infant. Tranfus. Apher. Sci. 2020, 59, 102695. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, X.-M. Autoimmune hemolytic anemia as an initial presentation in children with systemic lupus erythematosus: Two case reports. J. Int. Med. Res. 2022, 50, 3000605221115390. [Google Scholar] [CrossRef]
- Pendergrast, J.; Armali, C.; Callum, J.; et al. A prospective observational study of the incidence, natural history, and risk factors for intravenous immunoglobulin-mediated hemolysis. Transfusion 2021, 61, 1053–1063. [Google Scholar] [CrossRef]
- Conley, C.L.; Lippman, S.M.; Ness, P. Autoimmune hemolytic anemia with reticulocytopenia. A medical emergency. JAMA. 1980, 244, 1688–1690. [Google Scholar] [CrossRef]
- Conley, C.L.; Lippman, S.M.; Ness, P.M.; Petz, L.D.; Branch, D.R.; Gallagher, M.T. Autoimmune hemolytic anemia with reticulocytopenia and erythroid marrow. N. Engl. J. Med. 1982, 306, 281–286. [Google Scholar] [CrossRef]
- Branch, D.R. Drug-induced immune haemolytic anaemias. ISBT Science Series 2019, 14, 49–52. [Google Scholar] [CrossRef]
- Shulman, I.A.; Branch, D.R.; Nelson, J.M.; Thompson, J.C.; Petz, L.D. Autoimmune hemolytic anemia with both cold and warm autoantibodies. JAMA. 1985, 253, 1746–1748. [Google Scholar] [CrossRef]
- Branch, D.R.; Gallagher, M.T.; Shulman, I.A.; Mison, A.P.; Sy Siok Hian, A.L.; Petz, L.D. Reticuloendothelial cell function in alpha-methyldopa-induced hemolytic anemia. Vox Sang. 1983, 45, 278–287. [Google Scholar]
- Branch, D.R.; Boligan, K.; Sandhu, G.; Munn, K.; Halverson, G. Antibody-dependent cellular cytotoxicity is an important mechanism of red blood cell destruction in antibody-mediated hemolysis. Transfusion 2023, Vol. 63, No.S5, 29A. [Google Scholar] [CrossRef]
- Schriek, P.; Villadangos, J.A. Trogocytosis and cross-dressing in antigen presentation. Curr. Opin. Immunol. 2023, 83, 102331. [Google Scholar] [CrossRef]
- Branch, D.R. Warm autoimmune hemolytic anemia: New insights and hypotheses. Curr. Opin. Hematol. 2023, 30, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.W.; Abels, E.; Binns, T.C.; Tormey, C.A.; Sostin, N. Warm autoimmune hemolytic anemia with anti-Jka specificity following babesiosis masquerading as a delayed hemolytic transfusion reaction. Transfusion 2023, 63, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.S.; Kleeman, J.E.; Masouredis, S.P. Differential binding of IgG anti-D and IgG autoantibodies to reticulocytes and red blood cells. Br. J. Haematol. 1983, 55, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Branch, D.R.; Shulman, I.A.; Sy Siok Hian, A.L.; Petz, L.D. Two distinct categories of warm autoantibody reactivity with age-fractionated red cells. Blood 1984, 63, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Lutz HU, Stringaro-Wipf, G. Senescent red cell-bound IgG is attached to band 3 protein. Biomed Biochim Acta. 1983;42(11-12):S117-S121.
- Kay, M. Immunoregulation of cellular life span. Ann N Y Acad Sci. 2005;1057:85-111.
- Lutz HU, Bogdanova, A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front Physiol. 2013;4:387.
- Bloch, E.M.; Branch, H.A.; Sakac, D.; Leger, R.M.; Branch, D.R. Differential red blood cell age fractionation and Band 3 phosphorylation distinguish two different subtypes of warm autoimmune hemolytic anemia. Transfusion 2020, 60, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Badior, K.E.; Bloch, E.M.; Branch, D.R. A naturally present autoantibody to senescent red blood cells? Transfusion 2022, 62, 1311–1312. [Google Scholar] [CrossRef] [PubMed]
- Branch, D.R.; Leger, R.M.; Sakac, D.; et al. The chemokines IP-10/CXCL10 and IL-8/CXCL8 are potential novel biomarkers of warm autoimmune hemolytic anemia. Blood Adv. 2023, 7, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Zaninoni, A.; Fattizzo, B.; Pettine, L.; Vercellati, C.; Marcello, A.; Barcellini, W. Cytokine polymorphisms in patients with autoimmune hemolytic anemia. Front. Immunol. 2023, 14, 1221582. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, L.; Yang, J.; et al. Interleukin 6 exacerbates the progression of warm autoimmune hemolytic anemia by influencing the activity and function of B cells. Sci. Rep. 2023, 13, 13231. [Google Scholar] [CrossRef] [PubMed]
- Ciudad, M.; Ouandji, S.; Lamarthee, B.; et al. Regulatory T-cell dysfunctions are associated with increase in tumor necrosis factor α in autoimmune hemolytic anemia and participate in Th17 polarization. Haematologica 2024, 109, 444–457. [Google Scholar] [CrossRef]
- Murakhovskaya, I.; Crivera, C.; Leon, A.; et al. Healthcare resource utilization of patients with warm autoimmune hemolytic anemia initiating first line therapy of oral corticosteroids with or without rituximab. Ann. Hematol. 2024, Feb 1. Online ahead of print.
- Barcellini, W.; Fattizzo, B. How I treat warm autoimmune hemolytic anemia [erratum appears in Blood, 2023, 141, 438–439]. Blood 2021, 137, 1283–1294. [Google Scholar] [CrossRef]
- Michalak, S.S.; Olewicz-Gawlik, A.; Rupa-Matysek, J.; Wolny-Rokicka, E.; Nowakowska, E.; Gil, L. Autoimmune hemolytic anemia: Current knowledge and perspectives. Immun. Ageing 2020, 17, 38. [Google Scholar] [CrossRef]
- Fattizzo, B.; Berentsen, S.; Barcellini, W. Editorial: Practical recommendations and consensus for the management of immune mediated hematologic diseases. Front. Immunol. 2024, 15, 1364227. [Google Scholar] [CrossRef]
- Sakamoto, A.S.; Sequeira, F.S.; Blanco, B.P.; Garanito, M.P. Pediatric autoimmune hemolytic anemia: A single certer retrospective study. Hemtol. Transfus. Cell Ther. 2024, Feb 19, online ahead of print.
- Berensten, S.; Fattizzo, B.; Barcellini, W. The choice of new treatments in autoimmune hemolytic anemia: How to pick from the basket? Front. Immunol. 2023, 14, 1180509. [Google Scholar]
- Kuter, D.J. Warm autoimmune hemolytic anemia and the best treatment strategies. Hematology Am. Soc. Hematol. Educ. Program 2022, 2022, 105–113. [Google Scholar] [CrossRef]
- Cavallaro, F.; Barcellini, W.; Fattizzo, B. Antibody based therapeutics for autoimmune hemolytic anemia. Expert Opin. Biol. Ther. 2023, 23, 1227–1237. [Google Scholar] [CrossRef]
- Fattizzo, B.; Barcellini, W. Autoimmune hemolytic anemia: Causes and consequences. Expert Rev. Clin. Immunol. 2022, 18, 731–745. [Google Scholar] [CrossRef]
- Bortolotti, M.; Barcellini, W.; Fattizzo, B. Molecular pharmacology in complement-mediated hemolytic disorders. Eur. J. Haematol. 2023, 111, 326–336. [Google Scholar] [CrossRef]
- Premnath, N.; Pandey, U.; Pandey, M.; Venuprasad, K. Possible role of IL-23 inhibitor in autoimmune hemolytic anemia. Case Reports 2023, 111, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Murakhovskaya, I. Development of new drugs for autoimmune hemolytic -anemia. Pharmaceutics 2022, 14, 1035. [Google Scholar] [CrossRef]
- Loriamini, M.; Lewis-Bakker, M.M.; Frias Boligan, K.; et al. Small molecule drugs that inhibit phagocytosis. Molecules 2023, 28, 757. [Google Scholar] [CrossRef]
- Ogbue, O.D.; Bahaj, W.; Kewan, T.; et al. Splenectomy outcomes in immune cytopenias: Treatment outcomes and determinants of response. J. Intern. Med. 2024, 295, 229–241. [Google Scholar] [CrossRef]
- Malhotra, V.; Abraham, T.; Vesona, J.; Chopra, A.; Radakrishna, N. Infectious mononucleosis with secondary cold agglutinin disease causing autoimmune haemolytic anaemia. BMJ Case Rep. 2009, 2009, bcr12.2008.1390.
- Osorio-Toro, L.M.; Quintana-Ospina, J.H.; Melo-Burbano, L.A.; Ruiz-Jimenez, P.A.; Daza-Arana, J.E.; Rivas-Tafurt, G.P.; Izquierdo-Loaiza, J.H. Autoimmune hemolytic anemia caused by cold agglutinin antibodies in systemic lupus erythematosus—A rare association: Case report. J. Blood Med. 2023, 14, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Updates on the diagnosis and management of cold autoimmune hemolytic anemia. Hematol. Oncol. Clin. North Am. 2022, 36, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Swiecicki, P.L.; Hegerova, L.T.; Gertz, M.A. Cold agglutinin disease. Blood. 2013, 122, 1114–1121. [Google Scholar] [CrossRef]
- Jaffe, C.J.; Atkinson, J.P.; Frank, M.M. The role of complement in the clearance of cold agglutinin-sensitized erythrocytes in man. J Clin Invest. 1976, 58, 942–949. [Google Scholar] [CrossRef]
- Petz, L.D. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008, 22, 1–15. [Google Scholar] [CrossRef]
- Berensten, S.; Barcellini, W.; D’Sa, S.; et al. Cold agglutinin disease revisited: A multinational observational study of 232 patients. Blood 2020, 136, 480–488. [Google Scholar]
- Nunes, B.S.; Gouveia, C.; Kjollstrom, P.; Neves, J.F. Cold agglutinin syndrome and hemophagocytic lymphohistocytosis: An unusual combination caused by Epstein-Barr Virus infection. Cureus 2024, 16, e52179. [Google Scholar]
- Malecka, A.; Ostlie, I.; Troen, G.; et al. Gene expression analysis revealed downregulation of complement receptor 1 in clonal B cells in cold agglutinin disease. Clin. Exp. Immunol. 2023, Dec 22, online ahead of print.
- Broome, C.M. Complement-directed therapy for cold agglutinin diseae: Sutimlimab. Expert Rev. Hemaol. 2023, 16, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Despotovic, J.M.; Kim, T.O. Cold AIHA and the best treatment strategies. Hematology Am. Soc. Hematol. Educ. Program 2022, 2022, 90–95. [Google Scholar] [CrossRef]
- Berensten, S. Sutimlimab for the treatment of cold agglutinin disease. Hemasphere 2023, 7, e879. [Google Scholar]
- Roth, A.; Barcellini, W.; D’Sa, S.; et al. Sustained inhibition of complement C1s with sutimlimab over 2 years in patients with cold agglutinin disease. Am. J. Hematol. 2023, 98, 1246–1253. [Google Scholar] [CrossRef]
- Zaninoni, A.; Giannotta, J.A.; Galli, A. The immunomodulatory effect and clinical efficacy of daratumumab in a patient with cold agglutinin disease. Front. Immunol. 2023, 12, 649441. [Google Scholar] [CrossRef]
- Mohamed, A.; Alkhatib, M.; Alshurafa, A.; El Omri, H. Refractory cold agglutinin diseae successfully treated with daratumumab. A case report and review of literature. Hematology 2023, 28, 2252651. [Google Scholar] [CrossRef]
- Berentsen, S.; Tjønnfjord, G.E. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012, 26, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Berensten, S.; Barcellini, W.; D’Sa, S.; Jilma, B. Sutimlimab for treatment of cold agglutinin disease: Why, how and for whom. Immunotherapy 2022, 14, 1191–1204. [Google Scholar]
- Morselli, M.; Luppi, M.; Potenza, L.; et al. Mixed warm and cold autoimmune hemolytic anemia: Complete recovery after 2 courses of rituximab treatment. Blood. 2002, 99, 3478–3479. [Google Scholar] [CrossRef]
- Kajii, E.; Miura, Y.; Ikemoto, S. Characterization of autoantibodies in mixed-type autoimmune hemolytic anemia. Vox Sang. 1991, 60, 45–52. [Google Scholar]
- Mayer, B.; Yurek, S.; Kiesewetter, H.; Salama, A. Mixed-type autoimmune hemolytic anemia: Differential diagnosis and a critical review of reported cases. Transfusion 2008, 48, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.; Amardeep, K.; Yee, A.; Berson, B.; Cook, P. Mixed warm and cold autoimmune hemolytic anemia with concomitant immune thrombocytopenia following recent SARS-CoV-2 infection and ongoing rhinovirus infection. Cureus 2023, 15, e38509. [Google Scholar] [CrossRef]
- Smith, E.C.; Kahwash, N.; Piran, S. Management of mixed warm/cold autoimmune hemolytic anemia: A case report and review of the current literature. Case Rep. Hematol. 2023, 2023, 1381861. [Google Scholar] [CrossRef]
- Hanna, M.; Carcao, M. Rare case of refractory mixed autoimmune hemolytic anemia in a 6-year-old child: A case report. J. Med. Case Rep. 2023, 17, 418. [Google Scholar] [CrossRef]
- Turudic, D.; Bekic, S.D.; Mucavac, L.; Pavlovic, M.; Milosevic, D.; Billic, E. Case report: Autoimmune hemolytic anemia caused by warm and cold autoantibodies with complement activation- etiological and therapeutic issues. Front.Pediatr. 2023, 11, 1217536. [Google Scholar] [CrossRef]
- Göttsche, B.; Salama, A.; Mueller-Eckhardt, C. Donath-Landsteiner autoimmune hemolytic anemia in children. A study of 22 cases. Vox Sang. 1990, 58, 281–286. [Google Scholar] [PubMed]
- Shanbhag, S.; Spivak, J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015, 29, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Leibrandt, R.; Angelino, K.; Vizel-Schwartz, M.; Shapira, I. Paroxysmal Cold Hemoglobinuria in an Adult with Respiratory Syncytial Virus. Case Rep Hematol. 2018, 2018:7586719.
- Blackall, D.; Dolatshahi, L. Autoimmune hemolytic anemia in children: Laboratory investigation, disease associations, and treatment strategies. J. Pediatr. Hematol. Oncol. 2022, 44, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Eldar, D.; Ganzel, C. Erythrophagocytosis in a young adult with mycoplasma pneumonia-induced paroxysmal cold hemoglobinuria. Blood 2021, 137, 1432. [Google Scholar] [CrossRef]
- Radonjic, Z.; Andric, B.; Serbic, O.; Micic, D.; et al. A rare case report of autoimmune haemolytic anemia in a female child due to a Donath-Landsteiner antibody. Transfus. Clin. Biol. 2023, 27, 83–86. [Google Scholar] [CrossRef]
- Kilty, M.; Ipe, T.S. Donath-Landsteiner test. Immunohematology 2019, 35, 3–6. [Google Scholar] [CrossRef]
- Hogan, K.O.; Oroszi, G. Paroxysmal cold hemoglobinuria: A diagnostic dilemma in a paediatric patient. Transfus. Med. 2023, 33, 416–419. [Google Scholar] [CrossRef]
- Pelletier, J.; Ward, C.; Borloz, M.; Ickes, A.; Guelich, S.; Edwards Jr, E. A case of childhood severe paroxysmal cold hemoglobinuria with acute renal failure successfully treated with plasma exchange and eculizumab. Case Rep. Pediatr. 2022, 2022, 3267189. [Google Scholar] [CrossRef]
- Lau-Braunhut, S.A.; Stone, H.; Collins, G.; Berensten, S.; Braun, B.S.; Zinter, M.S. Paroxysmal cold hemoglobinuria successfully treated with complement inhibition. Blood Adv. 2019, 3, 3575–3578. [Google Scholar] [CrossRef]
- Hiranuma, N.; Koba, Y.; Kawata, T.; Tamekane, A.; Watanabe, M. Successful treatment of warm autoimmune hemolytic anemia with a positive Donath-Landsteiner test using rituximab. Intern. Med. 2023, Dec 11, online ahead of print.
- Gilliland, B.C.; Baxter, E.; Evans, R.S. Red-cell antibodies in acquired hemolytic anemia with negative antiglobulin serum tests. N Engl J Med. 1971, 285, 252–256. [Google Scholar] [CrossRef]
- Karafin, M.S.; Denomme, G.A.; Schanen, M.; Gottschall, J.L. Clinical and reference lab characteristics of patients with suspected direct antiglobulin test (DAT)-negative immune hemolytic anemia. Immunohematology 2015, 31, 108–115. [Google Scholar] [CrossRef]
- Segel, G.B.; Lichtman, M.A. Direct antiglobulin (“Coombs”) test-negative autoimmune hemolytic anemia: A review. Blood Cells Mol. Dis. 2014, 52, 152–160. [Google Scholar] [CrossRef]
- Gollamudi, J.; Dasgupta, S.K.; Thiagarajan, P. Erythrophagocytosis in autoimmune immunoglobulin A-mediated hemolysis. Transfusion 2023, 63, 1978–1982. [Google Scholar] [CrossRef]
- Branch, D.R.; Sy Siok Hian, A.L.; Petz, L.D. Mechanism of nonimmunologic adsorption of proteins using cephalothin-coated red cells. Transfusion 1984, 24, 415. [Google Scholar]
- Arndt, P.A. Drug-induced immune hemolytic anemia: The last 30 years of changes. Immunohematology 2014, 30, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.M.; Liu, L.H.; Wu, Q.; Wang, H.G. Cefoperazone/sulbactam-induced hemolytic anemia. J. Postgrad. Med. 2023, 69, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Teshigawara-Tanabe, H.; Hagihara, M.; Matsumura, A.; et al. Passenger lymphocyte syndrome after ABO-incompatible allogeneic hematopoietic stem cell transplantation; dynamics of ABO allo-antibody and blood type conversion. Hematology 2021, 26, 835–839. [Google Scholar] [CrossRef]
- Zhao, H.; Ding, Z.; Luo, Z.; et al. Passenger lymphocyte syndrome in renal transplantation: A systematic review of published case reports. Transpl. Immunol. 2022, 73, 101605. [Google Scholar] [CrossRef]
- Kohl, M.M.; Schwarz, S.; Jaksch, P.; et al. High rate of passenger lymphocyte syndrome after ABO minor incompatible lung transplantation. Am. J. Respir. Crit. Med. 2023, Dec 11, online ahead of print.
- Zhao, D.; Leung, J.; Hu, Z.; Ye, S.; Ye, Q. Passenger lymphocyte syndrome after ABO-mismatched kidney transplantation: A case report and literature review. Transpl. Immunol. 2023, 76, 101725. [Google Scholar] [CrossRef]
- Debska-Slizien, A.; Chamienia, A.; Krol, E.; et al. Hemolytic anemia after renal transplantation: Analysis of case reports. Transplant. Proc. 2003, 35, 2233–2237. [Google Scholar] [CrossRef]
- Kang, H.G.; Zhang, D.; Degauque, N.; Mariat, C.; Alexopoulos, S.; Zheng, X.X. Effects of cyclosporine on transplant tolerance: The role of IL-2. Am J Transplant. 2007, 7, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, J.L.; Petz, L.D.; Calhoun, L.; et al. Hemolysis of transfused group O red blood cells in minor ABO-incompatible unrelated-donor bone marrow transplants in patients receiving cyclosporine without posttransplant methotrexate. Blood 1992, 79, 3076–3085. [Google Scholar] [CrossRef] [PubMed]
- Zarei, E.; Shafiekhani, M.; Azadeh, N.; et al. Passenger lymphocyte syndrome as a rare cause of hemolysis in a patient after small intestine transplantation, a case report and review of the literature. Asian J. Transfus. Sci. 2022, 16, 135–139. [Google Scholar]
- Gelbengger, G.; Berensten, S.; Jilma, B. Monoclonal antibodies for treatment of cold agglutinin disease. Expert Opin. Biol. Ther. 2023, 23, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Berrueco, R.; Gonzalez-Forster, E.; Deya-Martinez, A. Mycophenolate mofetil for autoimmune cytopenias in children: High rates of response in inborn errors of immunity. Front. Pediatr. 2023, 11, 1174671. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, Q.; Yang, C.; Chen, M.; Han, B. Sirolimus is effective for primary refractory/relapsed warm autoimmune haemolytic anaemia/Evans syndrome: A retrospective single-center study. Ann. Med. 2023, 55, 2282180. [Google Scholar] [CrossRef]
- Robak, E.; Robak, T. Bruton’s kinase inhibitors for the treatment of immunological diseases: Current status and perspectives. J. Clin. Med. 2022, 11, 2807. [Google Scholar] [CrossRef]
- Kuter, D.J.; Piatek, C.; Roth, A.; et al. Fostamatinib for warm antibody autoimmune hemolytic anemia: Phase 3, randomized, double-blind, placebo-controlled, global study (FORWARD). Am. J. Hematol. 2024, 99, 79–87. [Google Scholar] [CrossRef]
- Pope, V.; Hsia, C.C. Safe utilization of ruxolitinib in simultaneous primary myelofibrosis and warm autoimmune hemolytic anemia. Ann. Hematol. 2024, 103, 677–679. [Google Scholar] [CrossRef]
- McGlothlin, J.; Abeykoon, J.; Al-Hattab, E.; et al. Bortezomib and daratumumab in refractory autoimmune hemolytic anemia. Am. J. Hematol. 2023, 98, E263–E265. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Schliesser, G.; Neubauer, A. Abatacept as salvage therapy for life-threatening refractory autoimmune hemolytic anemia: A case report. Hematology 2023, 28, 2208010. [Google Scholar] [CrossRef] [PubMed]
- Sorin, B.; Fadlallah, J.; Garzaro, M.; et al. Real-life use of mTOR inhibitor-based therapy in adults with autoimmune cytopenia highlights strong efficacy in relapsing/refractory multi-lineage autoimmune cytopenias. Ann. Hematol. 2023, 102, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



