
Submitted:
12 March 2024
Posted:
13 March 2024
You are already at the latest version
Abstract
The method for quantitative analysis is based on the direct measurement of the analyte using differential pulse voltammetry (DPV), exploiting its oxidation peak. Calibration curves are set up to evaluate the figures of merit of the analytical method (LOD, LOQ, linear range). The application of the sensor for the dosing of GA in real matrices is carried out by analyzing non-alcoholic and alcoholic beverages and comparing the results with those reported in the literature and with the content of total polyphenols determined by the Folin-Ciocalteu method. In all cases, concentrations of GA align with those previously found in the literature for the beverages examined. Moreover, the values are always lower than the total polyphenols content; this demonstrates the good selectivity of the sensor that can discriminate the target molecule from other polyphenols present.
Keywords:
Gallic Acid
; polyphenols
; electrosynthesized Molecularly imprinted Polymer (eMIP)
; screen-printed electrodes
; Electroanalysis
; differential pulse voltammetry
; voltammetric sensors
; non-alcoholic and alcoholic beverages
1. Introduction
Polyphenols have aroused interest due to their substantial benefits to health, such as anti-inflammatory and antihistaminic effects, protection against cardiovascular diseases and would also appear antitumor activities [1].
Gallic Acid (GA, 3,4,5-trihydroxy benzoic acid) is a natural polyphenol mainly present in fruits (for example, grapes, cranberries, and bananas) and beverages such as tea and wines. GA is a joint ingredient in herbal supplements, homeopathic remedies, and pharmaceutical products. Thanks to its remarkable antioxidant activities, it inhibits the interaction of radicals with health-benefit molecules by yielding H-atoms from its phenol functionalities to free radicals [2,3,4]. Based on its outstanding electrochemical activity, it is also often used as an electrochemical standard for estimating total polyphenol content, i.e., the antioxidant capacity index of the foods [5,6]. Moreover, the quantification of GA is frequently an index of the authenticity of fruit juices or alcoholic beverages [7].
Monitoring GA concentration is crucial, and several analytical methods have been proposed, such as high-performance liquid chromatography (HPLC) [8], capillary electrophoresis [9], diffuse reflectance spectrometry [10] and chemiluminescence [11]. However, some of these techniques involve complicated and time-consuming sample pretreatment steps before analysis coupled with expensive instrumentation.
Electrochemical methods are suitable alternatives to overcome these drawbacks since they provide low-cost, selective, and sensitive detection of several analytes.
GA can be efficiently determined by electroanalytical methods since it is an electroactive compound undergoing irreversible oxidation. Moreover, thanks to the development of materials science, the modification of electrode surfaces with conductive or molecularly imprinted polymers (MIPs) or metal nanoparticles allows the selective sensing of GA [12]. Different electrochemical sensors and methods have been proposed, mainly based on modifying classical macroelectrodes, such as glassy carbon, carbon paste or gold with acrylic molecularly imprinted polymer films [13,14,15]. Despite the good analytical performances, lengthy and expensive electrode surface modifications are required, limiting their use for research purposes.
As an alternative, the present work presents the employment of screen-printed cells whose working electrode is modified by an electropolymerized molecularly imprinted polypyrrole.
In the last decades, screen-printed cells (SPCs) have grown significantly in demand since the screen-printing technology offers pros in terms of cost-effectiveness, versatility, and ease of use [16]. Besides, screen-printed electrodes (SPEs) are among the most suitable sensors for in situ analysis thanks to their rapid response, low power needs, and good sensitivity [17]. The excellent versatility of SPEs is driven mainly by the numerous ways the electrodes can be modified. For example, the inks' composition may change by adding different substances, such as complexing reagents, noble metal nanoparticles, polymers, or bioreceptors. Otherwise, modifying the electrodes by depositing nanoparticles, biological material, metal or polymeric films on their surface is feasible [18]. SPEs have been indicated as effective sensor substrates for developing disposable MIP-based devices for in situ and point-of-care testing applications.
As well-known, MIPs are synthetic receptors that mimic the specific recognition mechanism of antibodies for antigens, receptors for hormones and enzymes for substrates [19]. The molecular imprinting process involves the polymerization of functional monomers and a crosslinker in the presence of a target analyte that acts as a template. After the polymerization, the subsequent template removal leaves specific cavities in the polymer, complementary in size and shape to the target molecule. Therefore, when contacted with a sample solution, the empty MIP can rebind the analyte or closely related molecules specifically [20,21,22,23]. MIPs are appealing for their recognition ability, like natural receptors, but also because they are available for several target analytes and possess higher physical and chemical stability than bioreceptors [24,25].
The surface imprinting method has recently been reported as the most convenient way for synthesizing the MIP directly on SPEs [26,27,28,29,30,31,32,33,34]. In particular, electropolymerization is an efficient strategy since it allows for highly controlled polymer growth on surfaces with a fine-tuning of the polymeric film thickness simply by controlling the experimental conditions [35,36,37].
The most frequently electropolymerized monomers are pyrrole, aniline and thiophene derivatives [38,39]. The attention on polypyrrole is due to its water solubility, good environmental stability, conductivity, and redox properties [40].
An electrochemical procedure that can be applied to improve the electrosynthesized molecularly imprinted polypyrrole is overoxidation, obtained by applying to the MIP film a positive electrode potential much higher than this required for the polymerization reaction. The overoxidation is helpful since it allows the formation of oxygen-containing groups such as hydroxyl, carbonyl, and carboxyl, which can form hydrogen bonds and be involved in various electrostatic interactions with the template molecules, endorsing the formation of more selective cavities [39]. Sometimes, overoxidation can be used to improve the template removal or the restoration of the sensing structure after the measurements [35].
In this scenario, a voltammetric sensor for Gallic Acid detection is developed and applied to the analyte quantification in non-alcoholic and alcoholic beverages. The sensor is obtained by modifying the graphite-ink working electrode of screen-printed cells with an electrosynthesized molecularly imprinted overoxidized polypyrrole film and using GA as the template. Differential pulse voltammetry (DPV) is selected for the analyte detection. Interference tests and analysis of tea and wine samples were performed to evaluate the selectivity and applicability of the proposed sensor.
Differently from already presented MIP-modified electrodes for GA detection [13,14,15], the emphasized pros are the cheaper electrodes and apparatus, reduced quantity of reagents and solvents, unneeded sample pretreatment step, and the possibility of in situ or online analysis thanks to portable instrumentation.
2. Materials and Methods
2.1. Reagents and Instruments
Pyrrole solution (98%, Merk Life Science S.r.l., Milan - Italy) was distilled using a Hickman distillation head until a colorless liquid was achieved and kept in darkness in a fridge at 4 °C. Gallic Acid (97.5-102.5% titration, Merk Life Science S.r.l., Milan, Italy) lithium perchlorate (LiClO4, purum p.a., ≥98.0%, Merk Life Science S.r.l., Milan, Italy), sodium chloride (NaCl, ACS reagent, 99.5%, Merk Life Science S.r.l., Milan, Italy), catechin (ACS reagent, ≥98.0%, Merk Life Science S.r.l., Milan, Italy), L-ascorbic acid (analytical standard, Merk Life Science S.r.l., Milan, Italy), 2-furaldheyde (ACS reagent, ≥98.0%, Merk Life Science S.r.l., Milan, Italy), L-tartaric acid (ACS reagent, ≥99.0%, Merk Life Science S.r.l., Milan, Italy), Folin-Cicolteau reagent (Merk Life Science S.r.l., Milan, Italy), and ethanol (for analysis, ACS reagent Merk Life Science S.r.l., Milan, Italy) were used as well as received.
For electrode surface characterization, solutions of potassium hexacyanoferrate(III), potassium chloride, and sodium chloride (Merk Life Science S.r.l., Milan, Italy) were used.
"Tavernello, cantine Caviro 2020", 10.5 %, white wine; "Tavernello cantine Caviro 2020", 11.5%, red wine; "Belvento Velarosa 2022", 13.5 %, rosè wine, "Giacomo Sperone", 17 %, marsala wine and "Lipton" green tea, were acquired in a local supermarket (Pavia, Italy).
Three-electrode screen-printed cells with graphite-ink working and counter electrodes and Ag/AgCl-ink pseudo-reference electrodes were obtained from Topflight Italia S.P.A. (Vidigulfo, Pavia, Italy).
Voltammetric and EIS (Electrochemical Impedance Spectroscopy) measurements were performed using the potentiostat/galvanostat EmStat4s (PalmSens BV, Houten, The Netherlands).
2.2. Preparation of the eMIP and eNIP Sensors
Before modification, each screen-printed cell (SPC) was cleaned with ethanol and left to dry at room temperature under a hood.
The eMIP-based electrode was obtained by electrodeposition on the surface of the clean working electrode of the SPC by five cycles of cyclic voltammetry (CV) in the potential range −0.6 ÷ 0.8 V at the scan rate 0.1 V/s, in an aqueous solution of 0.1 M LiClO4, 15 mM pyrrole, and 0.1 M Gallic Acid. The overoxidized polypyrrole-imprinted film was obtained by applying a fixed potential of +1.2 V for 12 s in 10 mL of 0.1 M LiClO4 solution.
The template removal from the eMIP was performed in two steps: 1) a washing in ethanol for 20 min, followed by 15-20 CV scans from -1 to +1 V (scan rate 0.1 V/s) in 0.1 M LiClO4 at pH 3 to remove the entrapped template completely, i.e., until the disappearance of the oxidation peak corresponding to Gallic Acid.
Electropolymerized, not-imprinted polymer films (e-NIPs) on the working electrode of the SPC were prepared using the same conditions but without adding the template, i.e., GA, in the polymerization mixture.
2.3. Voltammetric and Amperometric Measurements
The electrochemically active surface was determined before and after the working electrode modification with the eMIP or eNIP. It was obtained by performing cyclic voltammetry measurements in 5 mM K3Fe(CN)6/0.1 M KCl solution at pH 7 (the electrochemical probe solution), scanning the potential from −1 to +1 V, and varying the scan rate from 0.025 to 0.5 V/s.
The intensity of the cathodic or anodic peak was registered and plotted vs. the square root of the scan rate; from the slope (K) of the obtained straight line, the effective area was computed by applying the following modified Randles–Sevick's equation [41,42]:
n is the number of electrons acquired for the reduction of the electrochemical probe (for K3Fe(CN)6 n = 1), D is the diffusion coefficient (for K3Fe(CN)6, D=3.0910−6 cm2/s), and C is the concentration (5 mM) of the electrochemical probe.
The double-layer capacitance of the working electrodes [42,43,44] before and after the electrode modification was measured by performing cyclic voltammetry in 0.1 M NaCl solution at different scan rates (from 0.025 to 0.5 V/s.), scanning the potential in an interval by which non-faradic current is expected, i.e., from +0.05 to −0.05 V. The difference between the cathodic and anodic current values read at 0.02 V was plotted vs. the scan rate, and the slope of the straight line obtained is the capacitance. The capacitance of the double layer can be obtained by dividing this value by two.
Before and after modification of the electrode surface, Electrochemical Impedance Spectroscopy (EIS) measurements were also undertaken to characterize the electrode surface further and describe the electrochemical processes at the electrolyte–electrode interface [45,46]. The EIS measurements were performed in 10 mL of K3Fe(CN)6 5 mM and KCl 0.1 M at pH 7.5 as the probe solution. The impedance was registered in the 100 kHz–10 mHz frequency range with a sinusoidal potential modulation of 0.05 V superimposed on a dc potential of 0.2 V.
The electrochemical oxidation of GA was studied using cyclic voltammetry and exhaustive coulometry. In particular, the number of electrons involved in the electrochemical process was obtained by exhaustive coulometry (EC) [47,48,49] by electrolyzing the analyte at a potential slightly higher (50 or 100 mV) than the oxidation peak in a three-electrode cell under a static nitrogen atmosphere. A Pt gauze was used as the working electrode, a Pt wire was used as the counter electrode, and Ag/AgCl/3 M KCl was used as the reference electrode. The number of electrons was computed from the calculated charge, Q, required to electrolyze the analyte exhaustively (i.e., when the current was reduced to 5 % of the initial value).
The other parameters for the electrochemical oxidation characterization, i.e., the diffusion coefficient D, the standard potential E°, the charge transfer coefficient α, the electron transfer kinetic constant k0, and the reaction order, were derived from the cyclic voltammograms registered in 10 mL LiClO4 0.1 M/GA 2.5 mM at pH 3 solution, from -1 V to +1 V at different scan rate from 0.01 to 2 V/s.
The diffusion coefficient D (cm2/s) was calculated from the slope of the plot intensity of the anodic peak current (ip, A) vs. square root of the scan rate (v, V/s) and by applying the following Randles-Sevick equation 2 [48,49]:
where n is the number of exchanged electrons, α is the charge transfer coefficient (V), A is the active area (cm2), [C]∞ is the molar concentration of the analyte in the bulk solution, and D is the diffusion coefficient (cm2/s).
The standard potential (E0) was obtained from the intercept of the plot anodic peak potential (Ep, V) vs. scan rate (v, V/s) [50].
The charge transfer coefficient (α, V) was calculated from the slope of the straight line obtained by plotting the potential of the anodic peak (Ep, V) vs. the logarithm of the scan rate (v, V/s) and applying the following equation 3 [51]:
where R is the gas constant (J/molK), T is the temperature (K), F is the Faraday constant (C/mol), nα is the number of exchanged electrons, and v is the scan rate (V/s).
The charge transfer kinetic constant k0 was computed by applying the following equation 4 [52,53]:
where Ep is the potential of the anodic peak, R is the gas constant (J/molK), T is the temperature (K), α is the charge transfer coefficient (V), F is the Faraday constant (C/mol), D is the diffusion coefficient (cm2/s), n is the number of exchanged electrons, and v is the scan rate (V/s).
From the intercept of the straight line obtained by plotting vs. , k0 can be evaluated.
Gallic Acid (GA) was quantitatively determined by Differential Pulse Voltammetry (DPV) in 10 mL of LiClO4 0.1 M at pH 3 or in hydro-alcoholic solutions at different percentages of ethanol and applying the following experimental conditions: Estart = −1 V; Eend = 1 V; Estep = 0.015 V; Epulse = 0.02 V; tpulse = 0.2 s; scan rate = 0.05 V/s.
3. Results
3.1. Working Electrode Modification and Characterization
Molecularly imprinted polypyrrole was electropolymerized onto the working electrode of the screen-printed cell by cyclic voltammetry (CV) scanning the potential from -0.6 V to +0.8 V at 0.1 V/s in an aqueous solution of 0.1 M LiClO4, 15 mM pyrrole, and 0.1 M Gallic Acid. The molar ratio template/functional monomer equal to 1:15 was chosen, according to previous studies that suggested avoiding molar ratios higher than 1:5 since fewer imprinted sites should be obtained; conversely, molar ratios lower than 1:20 involve high quantity of template that, during the synthesis, can influence the micro-environment of the reaction, hindering the polymerization [54]. As a good compromise between a scarce number of the imprinted cavities (with less than 5 CV scans) and a very thick polymer film with less accessible recognition sites (with more than 7-10 CV scans) [55], five CV scans were selected.
The polypyrrole-imprinted film was then overoxidized. Overoxidation involves the formation of ketone groups in the polypyrrole backbone and disrupts conjugation but without significant material loss from the electrode [56]. Besides, improved film thickness control occurs, and the background currents are stable [36,38,39].
In the eMIP-modified electrode, the template molecules are entrapped in the polypyrrole matrix by non-covalent interactions, i.e., hydrogen bonds between the hydroxyl and carboxylic groups of the Gallic Acid and the -NH functionalities of the pyrrole units. A scheme of the possible interaction mechanism is shown in the following Figure 1.
Regarding the electrochemical characterization of the working electrode surface before and after modification with eMIP or eNIP, the electrochemically active area and the double-layer capacitance were determined. Table 1 recaps the results. Moreover, Electrochemical Impedance Spectroscopy (EIS) measurements were implemented.
The active area was determined by CV measurements in an electrochemical probe solution (K3Fe(CN)6) at different scan rates. The height of the oxidation or reduction current peak was plotted vs. the square root of the scan rate; from the slope of the straight line (K) and applying the Randles–Sevick's equation (Eq. 1), the active area was computed.
From Table 1, it can be observed that the active area decreases after the electrode is coated with the polymer, and, as expected, the electroactive surface of the eNIP-based electrode is lower than that of the eMIP. Indeed, the absence of the polymer's recognition cavities in the eNIP results in a decrease in the active area.
As regards the double-layer capacitance, a value of 0.7(5) μF was obtained for the bare electrode, significantly low if compared to that generally achievable with glassy carbon electrodes and this can be due to the different structures of the graphite ink of the screen-printed electrode employed here, with a predominance of basal planes compared to edge plane pyrolytic graphite electrodes that exhibit faster electrochemical kinetics, as previously suggested [44]. The double-layer capacitance increased from the bare electrode to the eNIP and eMIP-modified electrodes (see Table 1); this indicates that the presence of the polymer layer increases the possibility of accumulating electrical charges onto the electrode surface.
The characterization of the bare and modified electrodes was also deployed by EIS measurements. The data obtained from the EIS plot can be correlated to the physical and chemical properties of the electrode surface, modeling the electrochemical responses by an equivalent electrical circuit (Randles circuit). Figure 2 shows the Nyquist plot of bare and modified electrodes (imaginary impedance -Z vs. real impedance Z).
All phenomena at the bare, eMIP and eNIP-modified electrode surfaces may be schematized with the Randles circuit reported as inset in Figure 2. R represents the interface electrode/electrolyte resistance, while RCT is the charge transfer resistance (diameter of the semi-circle in the Nyquist plot). W is the Warburg element representing the analyte diffusion in the bulk of the solution, and C is the capacitor at the electrode/electrolyte interface.
The linear part of the Nyquist plot (the straight line at 45°) characteristic for a process limited by mass diffusion is evident only for the bare electrode. Moreover, it has the lowest charge transfer resistance since the graphite-ink of the electrode is a good conductor. The resistance increases if a polymeric film on the working electrode surface prevents the electrons’ transfer. Since the eMIP possesses the recognition cavities, a lower resistance is observed for the eMIP-modified electrode after template removal compared to that that has the cavities occupied by the analyte; indeed, the RCT increases (the diameter of the semicircle increases) by passing from the eMIP after template removal to the eMIP before template removal and finally to the eNIP, and consequently the diffusion becomes less evident (linear part of the plot. This behavior is explained by considering that when the analyte molecules are present within the polymer cavities and even more so when the electrode is coated with the eNIP, the access of the electrochemical probe to the electrode surface is prevented, thus increasing the resistance to electronic transfer.
3.2. Characterization of the Gallic Acid Oxidation Reaction at the Bare and eMIP-Modified Electrode Surface
The electrochemical oxidation reaction of Gallic Acid was studied using the bare and the eMIP-modified electrodes, both in LiClO4 0.1 M solution and hydro-alcoholic media containing LiClO4 0.1 M the 20 % of ethanol, to verify if the process could be affected by the polymeric film or the presence of ethanol.
Exhaustive coulometry (EC) was used to evaluate the number of electrons involved in the redox process. The electrode was immersed in 10 mL of 2.5 mM Gallic Acid solution; the potential was set to +0.7 V and kept fixed for two hours. From the obtained total charge quantity it has been obtained that the total number of electrons involved is equal to 2. It is necessary to observe that the first oxidation peak around + 0.25 V (in aqueous medium) is the most intense and appreciable one from the measurements in CV. The second, at more positive potentials (about + 0.6 V in aqueous solution), is much less intense and is confused with background noise, especially for measurements conducted with the eMIP electrode; this peak is, therefore, unusable for quantitative analysis, as also reported by other authors [57]. The following mechanism (Figure 3) can thus be assumed, according to previously published works [57]:
Table 2 summarizes the results of the characterization of the oxidation process.
The value of the charge transfer coefficient α is obtained from the graphs Ep vs. log v; in fact, from the slope, it is possible to derive α·nα, from which, knowing the number of electrons involved in the process, α is computed (see equation 3). Acceptable α values for compounds with irreversible behavior are between 0 and 1. Faster charge transfer processes are characterized by lower values of the charge transfer coefficient and vice versa for slower charge transfer processes. The results are given in Table 2, first column. Since the values of α are all in the range 0-1, irreversible behavior can be claimed. The values obtained with the bare electrode and the electrode coated with the eMIP are not significantly different, demonstrating that the polymer does not interfere with the electrochemical process. Even in the presence of ethanol, there is no significant change in the α value.
The intercept of the graph Ep vs. v for points obtained at a lower scan rate (i.e., by exploiting the first linear part of the curve) corresponds to the E0. The results are shown in Table 2, in the second column. Even in this case, the values obtained with the two electrodes do not differ significantly, as expected, since the eMIP cavities do not interfere with the electrochemical process but are a "preferential way" to access the analyte to the electrode surface. However, the value of E0 is almost double in the presence of ethanol, which means that the solvent hinders the process. Therefore, it is necessary to apply more positive potentials to cause the oxidation of Gallic Acid.
From the slope of graph i vs. v1/2 and applying Eq. 2, the diffusion coefficient can be determined, D. The results are shown in Table 2, third column. D values are not significantly different for bare and eMIP electrodes; the presence of ethanol also does not substantially change the diffusion coefficient of the analyte to the electrode.
From the graphs Ep αF/RT vs. 0.5·ln(αvF/RT) and applying Eq. 4, it is possible to derive the charge transfer kinetic constant, k0: the obtained values are given in Table 2, fourth column. Since k0 depends on the potential and the presence of ethanol causes the peak potential to shift to more positive values, the values obtained in the hydroalcoholic medium are much lower than in aqueous solutions. In any case, the values are similar between the bare electrode and that modified with eMIP.
3.3. Quantitative Analysis of Gallic Acid by DPV
Preliminary tests were conducted to identify which ionic medium was most suitable for developing the voltammetric method for the quantitative determination of gallic acid. Precisely, three different ionic media were compared: acetate buffer 0.1 M at pH 4, NaCl 0.1 M and LiClO4 0.1 M. From the results, it was noted that the analysis is more sensitive when using LiClO4 0.1 M (see Figure 4); moreover, the sensitivity is not affected acidifying the solution up to pH 3.5. It was therefore chosen to work with LiClO4 0.1 M at a pH of about 3-3.5 to simulate the wines’ acidity.
To assess the effect of the presence of ethanol on the DPV measurement both in terms of the shift of the peak potential and the value of the current intensity of the anodic peak, measurements were carried out in 0.2 mM Gallic Acid solution in LiClO4 0.1 M at pH 3 and containing ethanol at a percentage ranging from 0 to 40 %. The results obtained are summarized in the histograms of Figure 5.
The graphs of Figure 5 show that the presence of ethanol in the solution up to 10% does not affect both the potential and peak current. With the increase in the alcohol content, the peak potential progressively shifts, and the current drastically reduces.
For this reason, it is crucial for the quantification of Gallic Acid in beverages, to perform calibrations in the appropriate hydroalcoholic medium or to carry out the method of standard additions. Besides, since the recorded currents of 40% or more of ethanol are of very low intensity, the voltammetric method is expected to be applied only to beverages with a lower alcohol content; otherwise, dilution of the sample is needed.
As stated before, the method for quantitative analysis is based on the direct measure of the analyte using Differential Pulse Voltammetry (DPV), exploiting the oxidation peak at about + 0.2 V.
Calibration curves were set up to evaluate the analytical method's figures of merit (sensitivity, LOD, LOQ, linear range), while the standard additions method was used to assess Gallic Acid in real samples.
Figure 6 shows the voltammograms obtained with bare, eMIP- and eNIP-modified electrodes, and Figure 7 reports the calibration curves in the linear range (three replicates per each kind of electrode).
Table 3 summarizes the analytical method's figures of merit for the three different electrodes (LOD, LOQ, linear range).
The best sensitivity and the lower LOD are obtained with the electrode modified with e-MIP. The slope of the electrode modified with e-NIP is much lower, which is desirable since, in this case, the electrode is covered by a low-porous polymer film that hinders the arrival of the analyte to the electrode surface.
The limit of quantification with the sensor modified by the e-MIP equal to about 20 μM corresponds to a content of GA ~3 mg/L. This value affirms the validity of the method for the dosage of Gallic Acid in alcoholic and non-alcoholic beverages since the amount of GA is much higher in these matrices.
To evaluate the selectivity of the method, tests were carried out considering three potential interferents: catechin as a different polyphenol, ascorbic acid, a substance used as an antioxidant for food and drink and the 2-furaldehyde, an oily aldehyde with an aromatic smell and a bitter almond taste. The last molecule is one of the products of the toasting of wooden barrels through the conversion of wood sugars and enters the wine during aging in barrique. Catechin and ascorbic acid oxidize at potentials similar to those of gallic acid, while 2-furaldehyde is not electroactive in the potential range used (since it is reduced to a potential of about - 0.4 V). Figure 8 shows the voltammograms obtained in solutions containing a fixed concentration of GA (red line in the voltammograms) and increasing concentrations of the interferent.
The voltammograms obtained show that the eMIP-modified electrode is highly selective in the presence of electroactive and non-electroactive substances in the measurement's operating conditions.
To test the sensor's applicability in real matrices, soft drinks (green tea) and alcoholic beverages (white wine, rosé wine, red wine, marsala wine) were analyzed. For the quantification of GA, the method of standard additions was used on diluted portions of the samples, using hydroalcoholic solutions as a diluent with a % of ethanol equal to that declared on the product label.
The total polyphenol content of the same drinks was determined by the Folin-Ciocalteau method (expressed as mg/L GAE, Gallic Acid Equivalents).
Table 4 summarizes the results of the analyses and their comparison with the data reported in the literature.
The amount of Gallic Acid found in all the samples examined is similar to the concentrations previously reported in the literature. The values are always lower than the content of total polyphenols. This experimental evidence further demonstrates the good selectivity of the sensor, which can discriminate Gallic Acid from other polyphenols present in the tested beverages.
4. Conclusions
In this work, a modified electrochemical sensor for the analysis of Gallic Acid in non-alcoholic and alcoholic beverages was developed and applied.
The sensor consists of a screen-printed cell for voltammetric analysis in which the graphite-ink working electrode was coated with a molecularly imprinted polymer film obtained by pyrrole electropolymerization in the presence of Gallic Acid as a template (eMIP). To obtain reproducible measurements with stable and low-intensity background currents, it was necessary to overoxidize the polymer film.
From the preliminary characterization of the electrochemical process, it has been observed that Gallic Acid irreversibly oxidizes to potentials of about + 0.2 V and the peak potential is not affected by the presence of eMIP film and alcohol percentages (ethanol) up to 10 % by volume. Higher alcohol percentages lead to a shift of potential to more positive values and a decrease in current. Therefore, it was concluded that the application of the voltammetric technique for the analysis of alcoholic beverages requires quantification either by matrix calibration or by the standard addition method; furthermore, the method is ineffective for quantitative analysis in spirits with an ethanol content of 40% or more or, in those cases, dilution of the sample should be necessary.
The selected voltammetric method is the differential pulse voltammetry (DPV).
Calibration curves were made to assess the method's figures of merit, from which it was verified that the sensitivity of the eMIP-modified sensor is higher than that of the unmodified electrode (bare); moreover, the LOD obtained is amply sufficient to quantify Gallic Acid in non-alcoholic and alcoholic beverages.
It is important to note that sensors’ modification with MIP polymers is generally done not so much to improve sensitivity but rather to increase selectivity. For this purpose, tests were carried out with potential interfering substances and in all cases, the sensor responded positively only to the analyte, while the signal of the interference was found to be absent or negligible.
The application of the sensor for the dosing of Gallic Acid in real matrices was carried out by analyzing non-alcoholic and alcoholic beverages and comparing the results with those reported in the literature and with the content of total polyphenols determined by the Folin-Ciocalteau method. In all cases, a concentration of Gallic Acid in line with what was previously found in the literature for the beverages examined was determined, and the values are always lower than the content of total polyphenols. This demonstrates the good selectivity of the sensor that can discriminate the target molecule from other polyphenols present.
Author Contributions
Conceptualization, GA; methodology, GA and CZ; formal analysis, GA and CZ; investigation, LVD, AC and CC; data curation, GA and CZ; writing—original draft preparation, GA; writing—review and editing, CZ, LVD, AC and CC All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brewer, MS Natural Antioxidants: Sources, Compounds. Mech. Action Potential Appl. R. 2011, 10.
- Roidoung, S.; Dolan, K.D.; Siddiq, M. Gallic acid as a protective antioxidant against anthocyanin degradation and color loss in vitamin-C fortified cranberry juice. Food Chem. 2016, 210, 422–427. [Google Scholar] [CrossRef]
- Wang, L.; Halquist, M.S.; Sweet, D.H. Simultaneous determination of gallic acid and gentisic acid in organic anion transporter expressing cells by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2013, 937, 91–96. [Google Scholar] [CrossRef]
- Koçak, Ç.C.; Karabiberoğlu, Ş.U.; Dursun, Z. Highly sensitive determination of gallic acid on poly (l-Methionine)-carbon nanotube composite electrode. J. Electroanal. Chem., 2019, 853, 113552. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, K.; Ning, Y.; Zhao, G. Effect of the structure of gallic acid and its derivatives on their interaction with plant ferritin. Food Chem. 2016, 213, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Węgiel, J.; Burnat, B.; Skrzypek, S. A graphene oxide modified carbon ceramic electrode for voltammetric determination of gallic acid. Diam. Relat. Mater. 2018, 88, 137–143. [Google Scholar] [CrossRef]
- Ng, LK; Lafontaine, P.; Harnois, J. Gas chromatographic–mass spectrometric analysis of acids and phenols in distilled alcohol beverages. Application of anion-exchange disk extraction combined with in-vial elution and silylation. J. Chromatogr. A 2000, 873, 29–38.
- Song, R.; Cheng, Y.; Tian, Y.; Zhang, Z.J. A validated solid-phase extraction HPLC method for the simultaneous determination of gallic acid, catechin and epicatechin in rhubarb decoction. Chin. J. Nat. Med. 2012, 10(4), 275–278. [Google Scholar] [CrossRef]
- Yue, M.E.; Jiang, T.F. : Shi, Y.P. Determination of gallic acid and salidroside in Rhodiola and its preparation by capillary electrophoresis. J. Anal. Chem. 2006, 61, 365–368. [Google Scholar] [CrossRef]
- Dmitrienko, S.G.; Medvedeva, O.M.; Ivanov, A.A.; Shpigun, O.A.; Zolotov, Y.A. Determination of gallic acid with 4-nitrobenzenediazonium tetrafluoroborate by diffuse reflectance spectrometry on polyurethane foam. Ana. Chim. Acta 2002, 469(2), 295–301. [Google Scholar] [CrossRef]
- Shah, S.N.A.; Li, H.; Lin, J.M. Enhancement of periodate-hydrogen peroxide chemiluminescence by nitrogen doped carbon dots and its application for the determination of pyrogallol and gallic acid. Talanta 2016, 153, 23–30. [Google Scholar] [CrossRef]
- Ermis, N.; Zare, N.; Darabi, R.; Alizadeh, M.; Karimi, F.; et al. Recent advantage in electrochemical monitoring of gallic acid and kojic acid: A new perspective in food science. J. Food Meas. Charact. 2023, 17, 3644–3653. [Google Scholar] [CrossRef]
- Qin, F.; Hu, T.; You, L.; Chen, W.; Jia, D.; Hu, N.; Qi, W. . Electrochemical detection of gallic acid in green tea using molecularly imprinted polymers on TiO2@ CNTs nanocomposite modified glassy carbon electrode. Int. J. Electrochem. Sci. 2022, 17(4), 220426. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, Q.; Chen, T.; Wu, W.; Tang, X.; Wang, G.; Feng, J.; Zhang, W. Facile potentiometric sensing of gallic acid in edible plants based on molecularly imprinted polymer. J. Food Sci. 2020, 85(8), 2622–2628. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Nasirizadeh, N.; Entezam, M.; Koosha, M.; Azimzadeh, M. An electrochemical nanosensor based on molecularly imprinted polymer (MIP) for detection of gallic acid in fruit juices. Food Anal. Method. 2016, 9, 2721–2731. [Google Scholar] [CrossRef]
- Suresh, R.R.; Lakshmanakumar, M.; Arockia Jayalatha, J.B.B.; Rajan, K.S.; Sethuraman, S.; Krishnan, U.M.; Rayappan, J.B.B. Fabrication of screen-printed electrodes: Opportunities and challenges. J. Mater. Sci. 2021, 56, 8951–9006. [Google Scholar] [CrossRef]
- Li, M.; Li, Y.-T.; Li, D.-W.; Long, Y.-T. Recent developments and applications of screen-printed electrodes in environmental assays—A review. Anal. Chim. Acta 2012, 734, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Renedo, O.D.; Alonso-Lomillo, M.A.; Martínez, M.A. Recent developments in the field of screen-printed electrodes and their related applications. Talanta 2007, 73, 202–219. [Google Scholar] [CrossRef]
- Haupt, K.; Linares, A.V.; Bompart, M.; Bui, B.T.S. Molecularly Imprinted Polymers. Top. Curr. Chem. 2011, 325, 1–28. [Google Scholar]
- Beltran, A.; Borrull, F.; Marcé-Recasens, R.M.; Cormack, P. Molecularly-imprinted polymers: Useful sorbents for selective extractions. TrAC Trends Anal. Chem. 2010, 29, 1363–1375. [Google Scholar] [CrossRef]
- Alexander, C.; Andersson, H.; Andersson, L.I.; Ansell, R.J.; Kirsch, N.; Nicholls, I.A.; O'Mahony, J.; Whitcombe, M.J. Molecular imprinting science and technology: A survey of the literature for the years up to and including 2003. J. Mol. Recognit. 2006, 19, 106–180. [Google Scholar] [CrossRef]
- Kupai, J.; Razali, M.; Buyuktiryaki, S.; Kecili, R.; Szekely, G. Long-term stability and reusability of molecularly imprinted polymers. Polym. Chem. 2016, 8, 666–673. [Google Scholar] [CrossRef]
- Kadhem, A.J.; Gentile, G.J.; Fidalgo de Cortalezzi, M.M. Molecularly Imprinted Polymers (MIPs) in Sensors for Environmental and Biomedical Applications: A Review. Molecules 2021, 26, 6233. [Google Scholar] [CrossRef]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular imprinting: Perspectives and applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef]
- Leibl, N.; Haupt, K.; Gonzato, C.; Duma, L. Molecularly Imprinted Polymers for Chemical Sensing: A Tutorial Review. Chemosensors 2021, 9, 123. [Google Scholar] [CrossRef]
- Prasad, B.B.; Pandey, I.; Srivastava, A.; Kumar, D.; Tiwari, M.P. Multiwalled carbon nanotubes-based pencil graphite electrode modified with an electrosynthesized molecularly imprinted nanofilm for electrochemical sensing of methionine enantiomers. Sens. Actuators B Chem. 2013, 176, 863–874. [Google Scholar] [CrossRef]
- Cabral-Miranda, G.; Gidlund, M.; Sales, M.G.F. Backside-surface imprinting as a new strategy to generate specific plastic antibody materials. J. Mater. Chem. B 2014, 2(20), 3087–3095. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.T.; Sharma, S.; Dutra, R.A.; Noronha, J.P.; Cass, A.E.; Sales, M.G.F. Protein-responsive polymers for point-of-care detection of cardiac biomarker. Sens. Actuators B Chem. 2014, 196, 123–132. [Google Scholar] [CrossRef]
- Pacheco, J.P.G.; Silva, M.S.V.; Freitas, M.; Nouws, H.P.A.; Delerue-Matos, C. Molecularly imprinted electrochemical sensor for the point-of-care detection of a breast cancer biomarker (CA 15-3). Sens. Actuators B Chem. 2018, 256, 905–912. [Google Scholar] [CrossRef]
- Rebelo, P.; Costa-Rama, E.; Seguro, I.; Pacheco, J.G.; Nouws, H.P.A.; Cordeiro, M.N.D.S.; Delerue-Matos, C. Molecularly imprinted polymer-based electrochemical sensors for environmental analysis. Biosens. Bioelectron. 2021, 172, 112719. [Google Scholar] [CrossRef]
- Lopes, F.; Pacheco, J.; Rebelo, P.; Delerue-Matos, C. Molecularly imprinted electrochemical sensor prepared on a screen printed carbon electrode for naloxone detection. Sens. Actuators B Chem. 2017, 243, 745–752. [Google Scholar] [CrossRef]
- Seguro, I.; Rebelo, P.; Pacheco, J.G.; Delerue-Matos, C. Electropolymerized, Molecularly Imprinted Polymer on a Screen-Printed Electrode—A Simple, Fast, and Disposable Voltammetric Sensor for Trazodone. Sensors 2022, 22, 2819. [Google Scholar] [CrossRef] [PubMed]
- Couto, R.A.; Mounssef Jr, B.; Carvalho, F.; et al. Methylone screening with electropolymerized molecularly imprinted polymer on screen-printed electrodes. Sens. Actuators B Chem. 2020, 316, 128133. [Google Scholar] [CrossRef]
- Rebelo, P.; Pacheco, J.G.; Voroshylova, I.V.; Melo, A.; Cordeiro, M.N.D.; Delerue-Matos, C. A simple electrochemical detection of atorvastatin based on disposable screen-printed carbon electrodes modified by molecularly imprinted polymer: Experiment and simulation. Anal. Chim. Acta 2022, 1194, 339410. [Google Scholar] [CrossRef]
- Gonçalves, L.M. Electropolymerized molecularly imprinted polymers: Perceptions based on recent literature for soon-to-be world-class scientists. Curr. Opin. Electrochem. 2021, 25, 100640. [Google Scholar] [CrossRef]
- Crapnell, R.D.; Hudson, A.; Foster, C.W.; Eersels, K.; Grinsven, B.v.; Cleij, T.J.; Banks, C.E.; Peeters, M. Recent Advances in Electrosynthesized Molecularly Imprinted Polymer Sensing Platforms for Bioanalyte Detection. Sensors 2019, 19, 1204. [Google Scholar] [CrossRef]
- Unger, C.; Lieberzeit, P.A. Molecularly imprinted thin film surfaces in sensing: Chances and challenges. React. Funct. Polym. 2021, 161, 104855. [Google Scholar] [CrossRef]
- Ramanavičius, S.; Morkvėnaitė-Vilkončienė, I.; Samukaitė-Bubnienė, U.; Ratautaitė, V.; Plikusienė, I.; Viter, R.; Ramanavičius, A. Electrochemically Deposited Molecularly Imprinted Polymer-Based Sensors. Sensors 2022, 22, 1282. [Google Scholar] [CrossRef]
- Ramanavicius, S. , Ramanavicius, A. Charge transfer and biocompatibility aspects in conducting polymer-based enzymatic biosensors and biofuel cells. Nanomaterials, 2021, 11(2), 371. [Google Scholar] [CrossRef]
- Sadki, S.; Schottland, P.; Brodie, N.; Sabouraud, G. The mechanisms of pyrrole electropolymerization. Chem. Soc. Rev. 2000, 29, 12. [Google Scholar]
- Burak, D.; Emregul, E.; Emregul, K.C. Copper–zinc alloy nanoparticle based enzyme-free superoxide radical sensing on a screen-printed electrode. Talanta 2015, 134, 206–214. [Google Scholar]
- Pesavento, M.; Merli, D.; Biesuz, R.; Alberti, G.; Marchetti, S.; Milanese, C. A MIP-based low-cost electrochemical sensor for 2-furaldehyde detection in beverages. Anal. Chim. Acta 2021, 1142, 201–210. [Google Scholar] [CrossRef]
- Pesavento, M.; D’Agostino, G.; Alberti, G.; Biesuz, R.; Merli, D. Voltammetric platform for detection of 2,4,6-trinitrotoluene based on a molecularly imprinted polymer. Anal. Bioanal. Chem. 2013, 405, 3559–3570. [Google Scholar] [CrossRef]
- Akhoundian, M.; Alizadeh, T.; Ganjali, M.R.; Rafiei, F. A new carbon paste electrode modified with MWCNTs and nano-structured molecularly imprinted polymer for ultratrace determination of trimipramine: The crucial effect of electrode components mixing on its performance. Biosens. Bioelectron. 2018, 111, 27–33. [Google Scholar] [CrossRef]
- Magar, H.S.; Hassan, R.Y.A.; Mulchandani, A. Electrochemical Impedance Spectroscopy (EIS): Principles, Construction, and Biosensing Applications. Sensors 2021, 21, 6578. [Google Scholar] [CrossRef] [PubMed]
- Craven, J.E.; Kinnamon, D.S.; Prasad, S. Randles Circuit Analysis Toward Investigating Interfacial Effects on Microchannel Electrodes. IEEE Sens. Lett. 2018, 2, 15–18. [Google Scholar] [CrossRef]
- Milanesi, C.; Protti, S.; Chiodi, D.; Profumo, A.; Merli, D. Electrochemical characterization and voltammetric determination of aryl piperazine emerging as designer drugs, J. Electroanal Chem. 2021, 895, 115480. [Google Scholar] [CrossRef]
- Capucciati, A.; Burato, A.; Bersani, C.; Protti, S.; Profumo, A.; Merli, D. Electrochemical Behavior and Voltammetric Determination of Two Synthetic Aroyl Amides Opioids; Chemosensors 2023, 11(3), 198.
- Merli, D.; Lio, E.; Protti, S.; Coccia, R.; Profumo, A.; Alberti, G. Molecularly Imprinted Polymer-based voltammetric sensor for amino acids/indazole derivatives synthetic cannabinoids detection. Anal. Chim. Acta 2024, 1288C, 342151. [Google Scholar] [CrossRef]
- Yunhua, W.; Xiaobo, J.; Shengshui, H. Studies on electrochemical oxidation of azithromycin and its interactions with bovine serum albumin. Bioelectrochem. 2004, 64(1), 91–97. [Google Scholar]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. Interfacial Electrochem. 1979, 101(1), 19–28. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Adams, R.N. . Anodic Oxidation Studies of N,N-Dimethylaniline. I. Voltammetric and Spectroscopic Investigations at Platinum Electrodes. J. Am. Chem. Soc. 1962, 84(11), 2058–2061. [Google Scholar] [CrossRef]
- Araújo, S.; Arantes, C.; Faria, V.; Souza, A.O.; Pimentel, M.; Barbosa, L.; Richter, M.; Munoz, A.A.; dos Santos, T.P. . Electrochemistry of 5F-MDMB-PICA synthetic cannabinoid using a borondoped diamond electrode with short anodic-cathodic pretreatment: A simple screening method for application in forensic analysis. Electrochim. Acta 2023, 454, 142356. [Google Scholar] [CrossRef]
- Ding, S.; Lyu, Z.; Li, S.; Ruan, X.; Fei, M.; et al. Molecularly imprinted polypyrrole nanotubes based electrochemical sensor for glyphosate detection. Biosens. Bioelectron. 2021, 191, 113434. [Google Scholar] [CrossRef]
- Maouche, N.; Guergouri, M.; Gam-Derouich, S.; Jouini, M.; Nessark, B.; Chehimi, M.M. Molecularly imprinted polypyrrole films: Some key parameters for electrochemical picomolar detection of dopamine. J. Electroanal. Chem. 2012, 685, 21–27. [Google Scholar] [CrossRef]
- Christensen, P.A.; Hamnett, A. In situ spectroscopic investigations of the growth, electrochemical cycling and overoxidation of polypyrrole in aqueous solution. Electrochim. Acta 1991, 36, 1263–1286. [Google Scholar] [CrossRef]
- Falahi, S.; Falahi, S.; Zarejousheghani, M.; Ehrlich, H.; Joseph, Y.; Rahimi, P. Electrochemical Sensing of Gallic Acid in Beverages Using a 3D Bio-Nanocomposite Based on Carbon Nanotubes/Spongin-Atacamite. Biosensors 2023, 13, 262. [Google Scholar] [CrossRef]
- Clarke, S.; Bosman, G.; du Toit, W.; Aleixandre-Tudo, J.L. White wine phenolics: current methods of analysis. J. Sci. Food Agric. 2023, 103(1), 7–25. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, F.; La Torre, G.L.; Dugo, G.; Pellicanò, T.M. Contenuto di antiossidanti e tecniche colturali e di allevamento della vite. Enologo-Milano 2008, 44(3), 109. [Google Scholar]
- Dugo, G.; Giuffrida, D.; Magnisi, R.; Vilasi, F.; Pellicanò, T.M.; La Torre, G.L. Determinazione HPLC/MS di composti antiossidanti in vini siciliani da dessert. Enologo-Milano 2004, 40(5), 89–94. [Google Scholar]
- Lin, J.K.; Lin, C.L.; Liang, Y.C.; Lin-Shiau, S.Y.; Juan, I.M. Survey of catechins, gallic acid, and methylxanthines in green, oolong, pu-erh, and black teas. J. Agric. Food Chem. 1998, 46(9), 3635–3642. [Google Scholar] [CrossRef]
Figure 1.
Scheme of the possible interaction mechanism between GA and overoxidate polypyrrole.
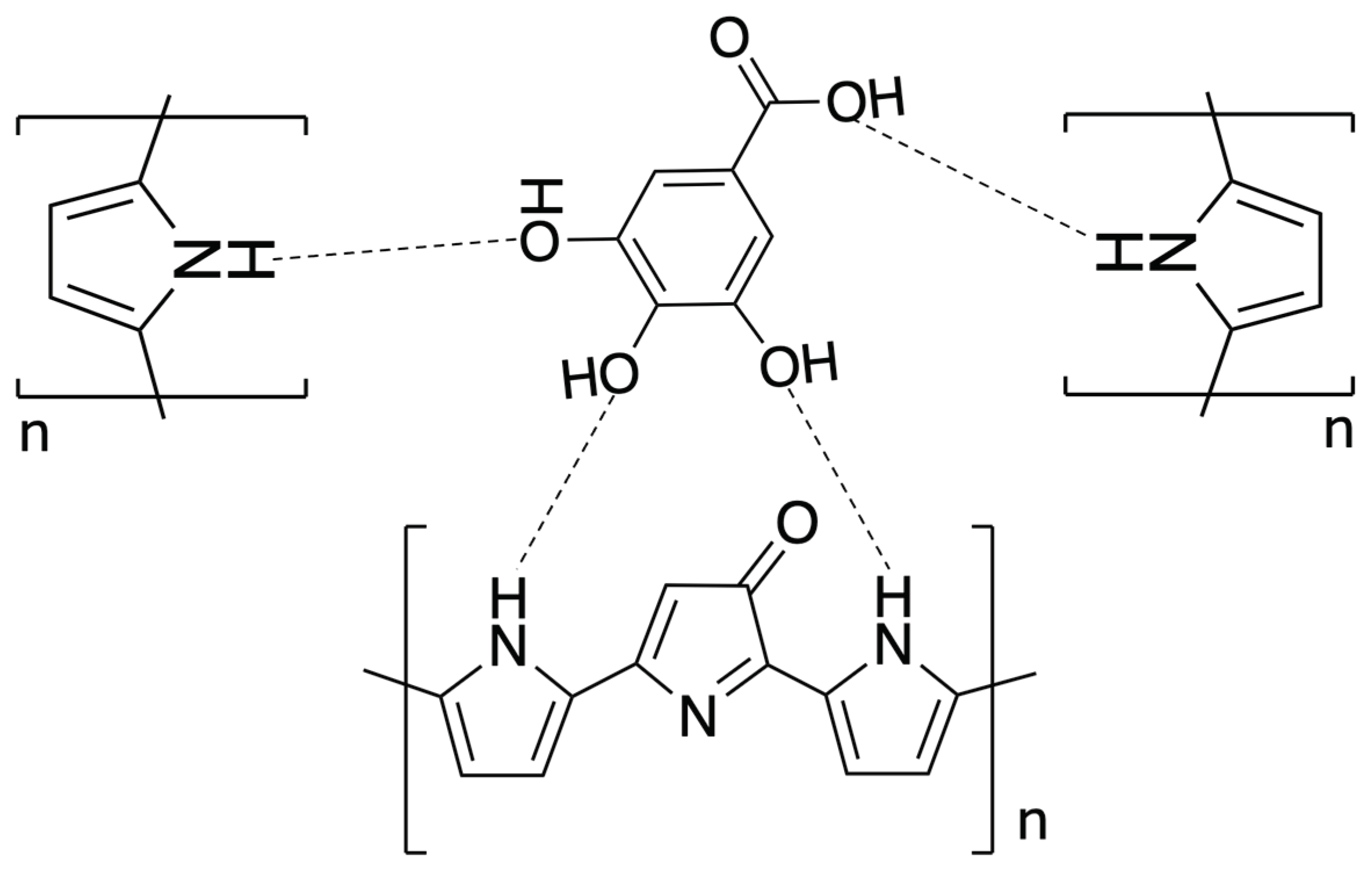
Figure 2.
Nyquist plot and Randles equivalent circuit of the bare electrode, eMIP-modified electrode after template removal, eMIP-modified electrode before the template removal and eNIP-modified electrode. Measurements in 0.1 M KCl/0.05 M K3Fe(CN)6 electrochemical probe solution.
Figure 2.
Nyquist plot and Randles equivalent circuit of the bare electrode, eMIP-modified electrode after template removal, eMIP-modified electrode before the template removal and eNIP-modified electrode. Measurements in 0.1 M KCl/0.05 M K3Fe(CN)6 electrochemical probe solution.
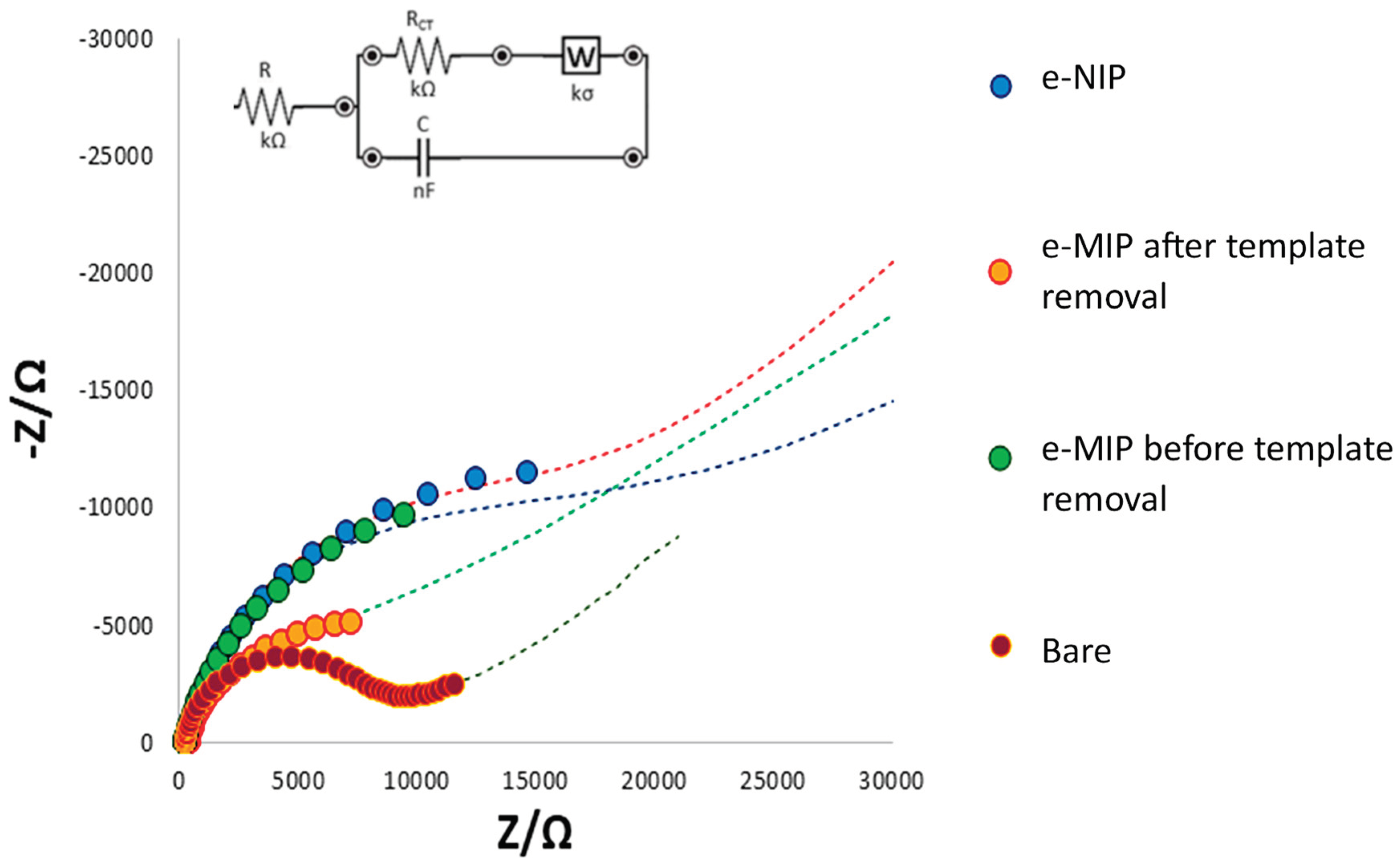
Figure 3.
The electrochemical oxidation reaction of Gallic Acid. (Reproduced with permission from [57], open access Creative Common CC licensed 4.0, MDPI).
Figure 3.
The electrochemical oxidation reaction of Gallic Acid. (Reproduced with permission from [57], open access Creative Common CC licensed 4.0, MDPI).
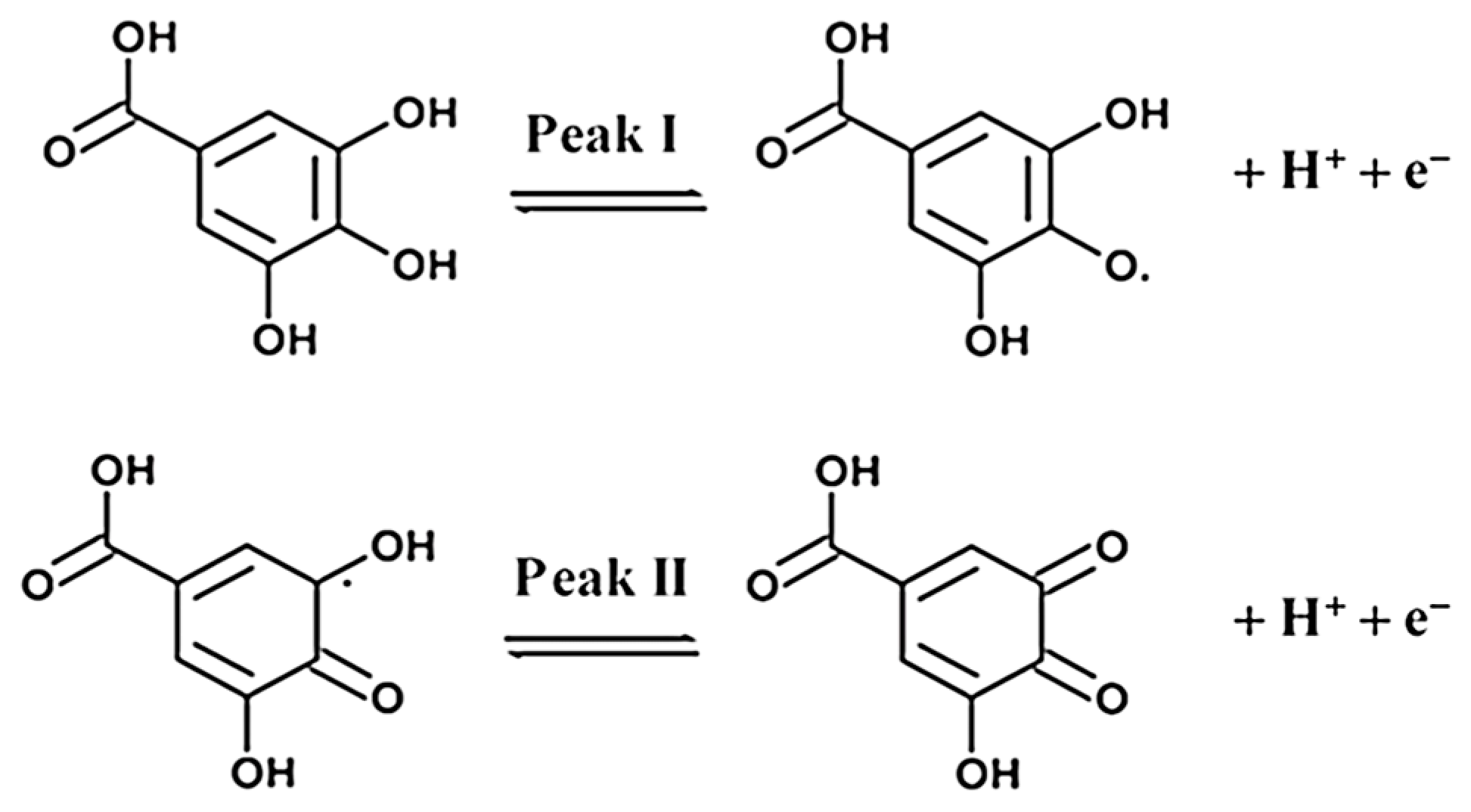
Figure 4.
Comparison among three calibration curves performing DPV measurements in different ionic media.
Figure 4.
Comparison among three calibration curves performing DPV measurements in different ionic media.
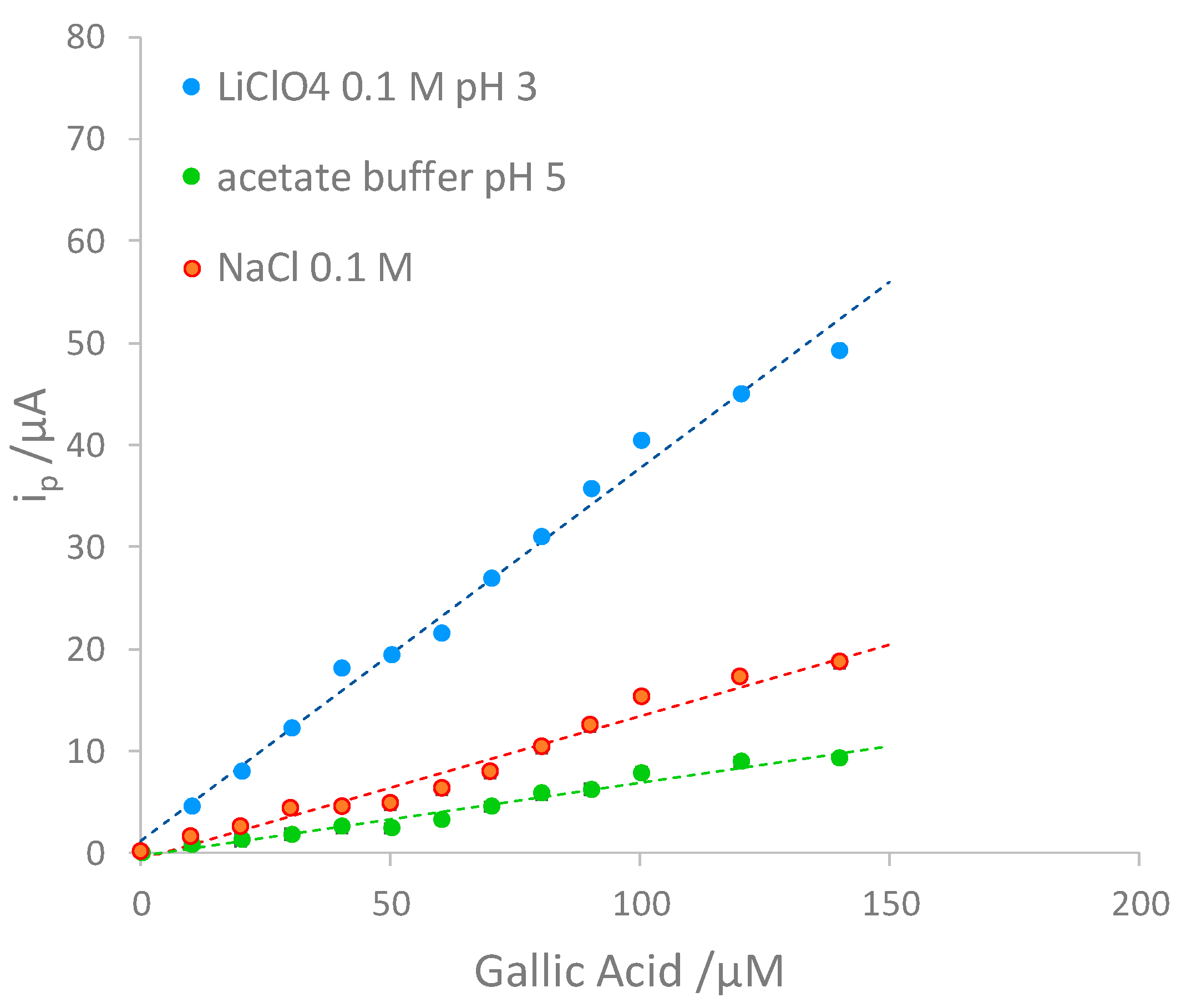
Figure 5.
Histograms reporting peak potential (Ep, V) and peak current (ip, μa) for 0.2 mM GA solution in LiClO4 0.1 M at pH 3 and containing ethanol at different percentages from 0 to 40 %. DPV measurements: potential scan from -1 V to +1 V, potential step 0.015 V, pulse time 0.02 s and scan speed 0.05 v/s.
Figure 5.
Histograms reporting peak potential (Ep, V) and peak current (ip, μa) for 0.2 mM GA solution in LiClO4 0.1 M at pH 3 and containing ethanol at different percentages from 0 to 40 %. DPV measurements: potential scan from -1 V to +1 V, potential step 0.015 V, pulse time 0.02 s and scan speed 0.05 v/s.
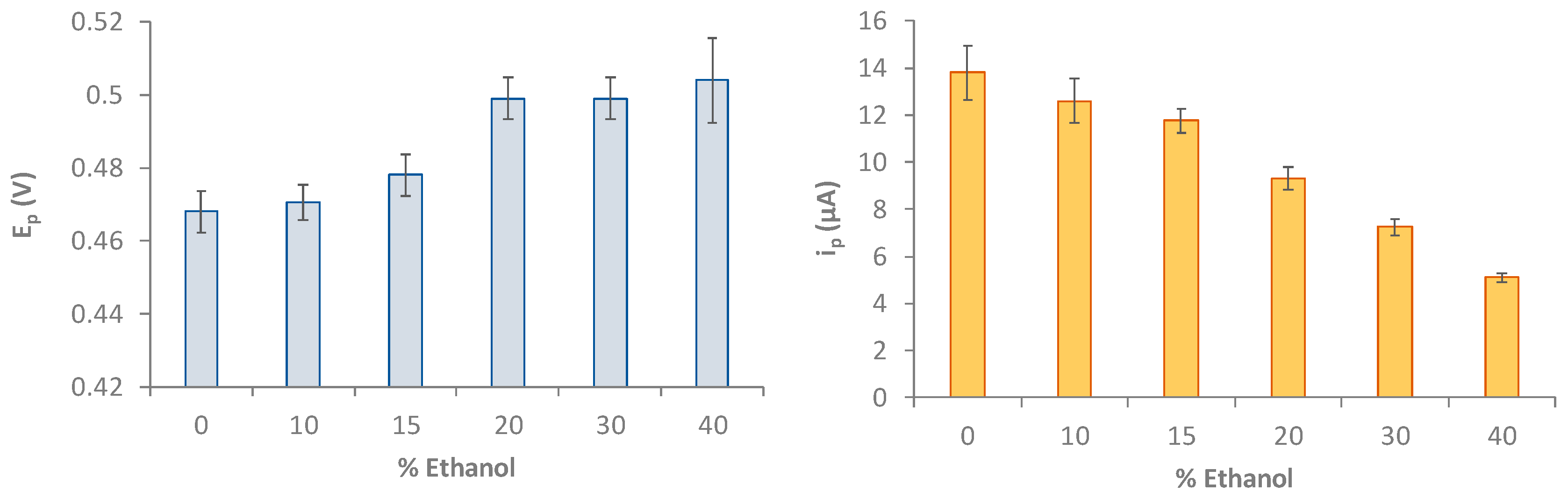
Figure 6.
DPV voltammograms in LiClO4 0.1 M at pH 3 with increasing concentrations of GA. DPV measurements: potential scan from -1 V to 1 V, potential step 0.015 V, pulse width 0.2 V, pulse duration 0.02 s and scan speed 0.05 v/s.
Figure 6.
DPV voltammograms in LiClO4 0.1 M at pH 3 with increasing concentrations of GA. DPV measurements: potential scan from -1 V to 1 V, potential step 0.015 V, pulse width 0.2 V, pulse duration 0.02 s and scan speed 0.05 v/s.
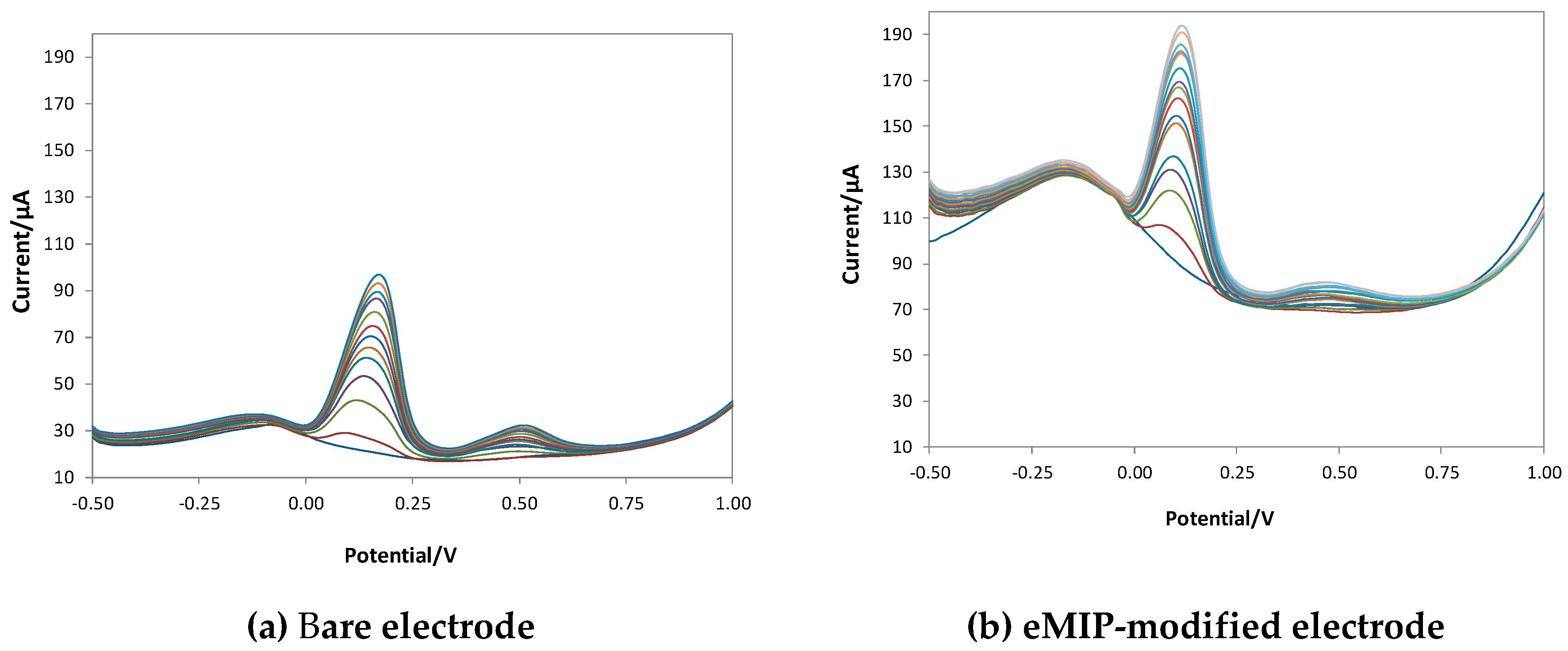
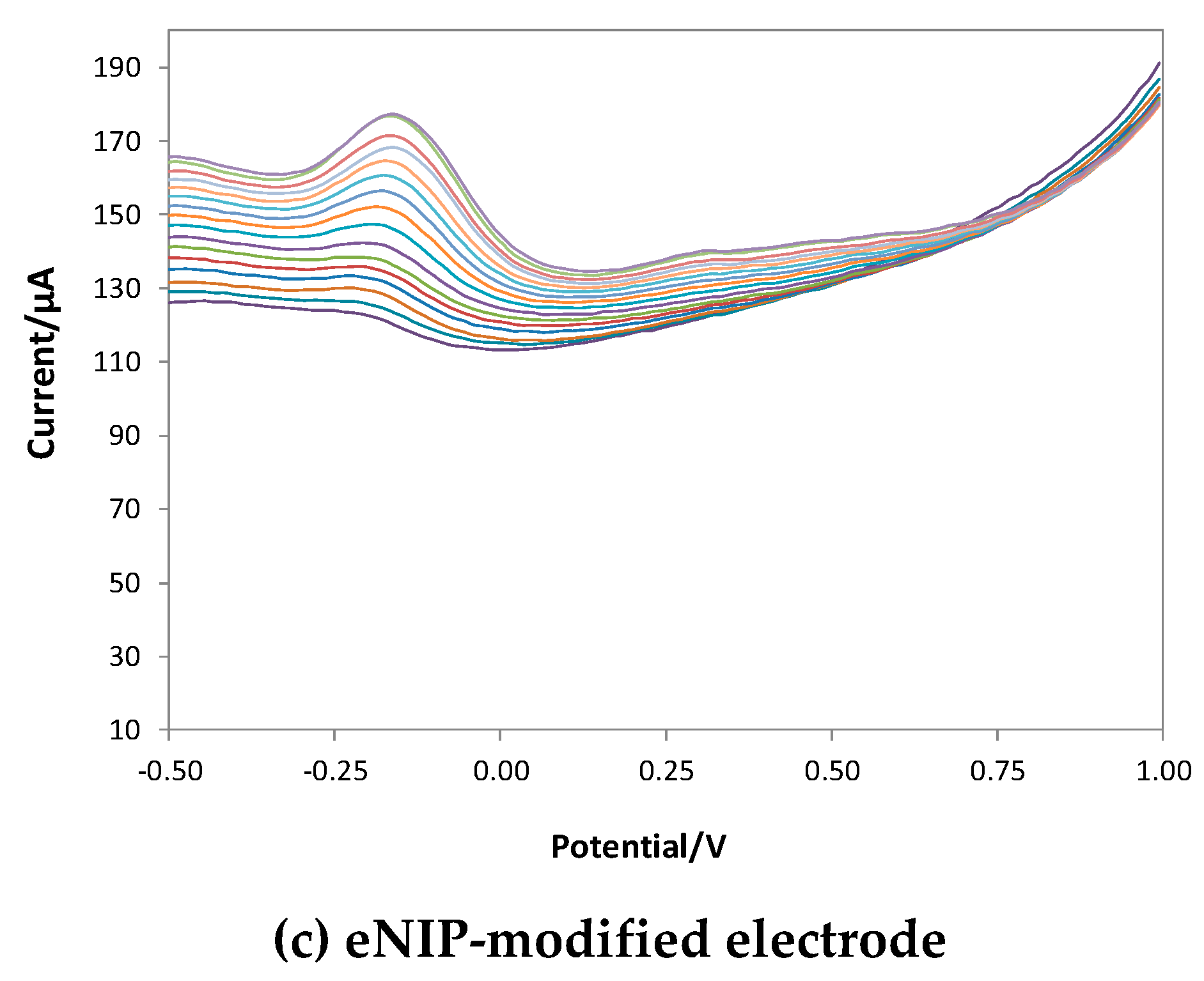
Figure 7.
Calibration curves for bare, eMIP- and eNIP-modified electrodes. The error bars correspond to the standard deviation of the measurements performed with three electrodes of each kind.
Figure 7.
Calibration curves for bare, eMIP- and eNIP-modified electrodes. The error bars correspond to the standard deviation of the measurements performed with three electrodes of each kind.
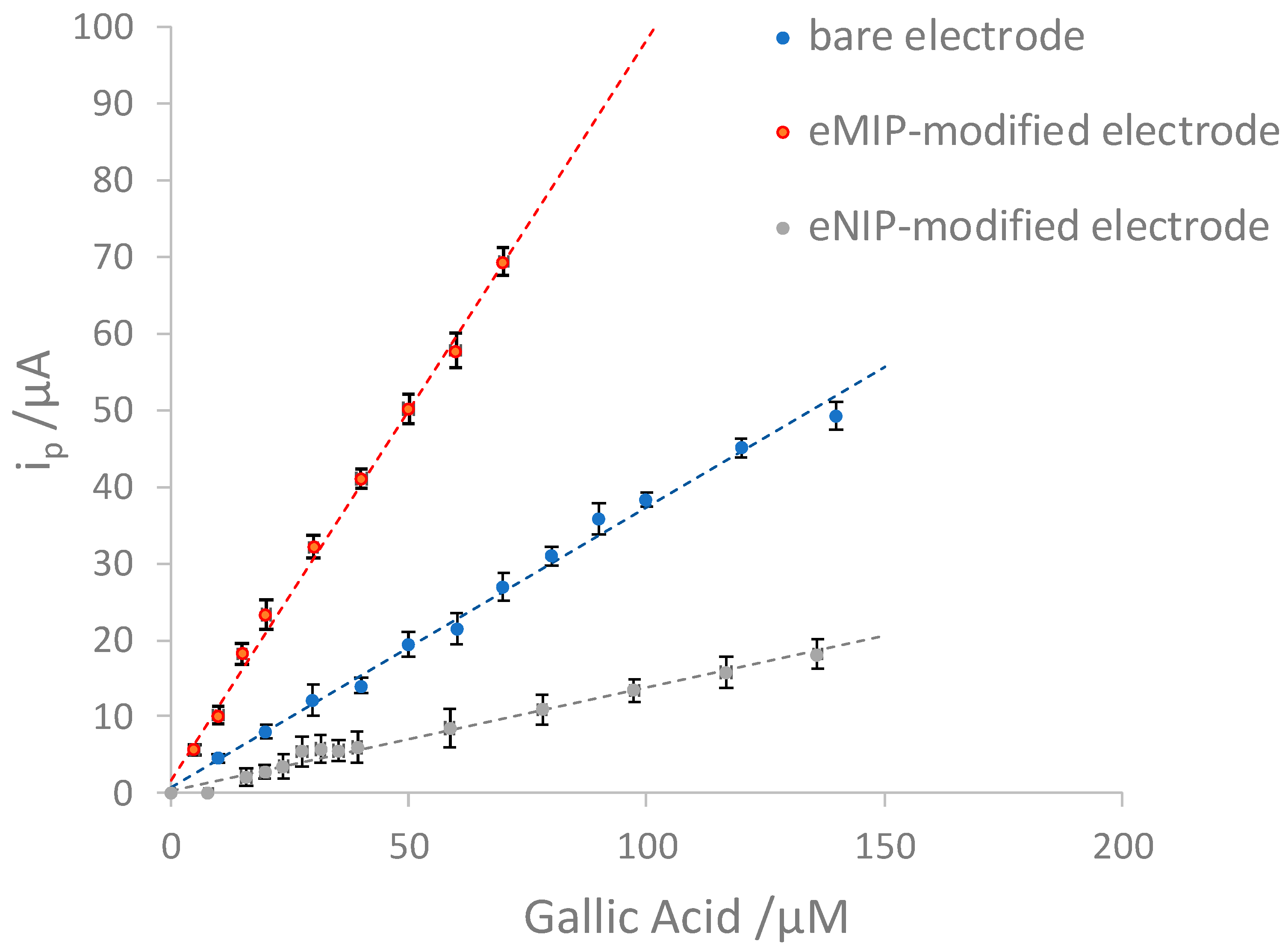
Figure 8.
DPV voltammograms recorded in LiClO4 0.1 M at pH 3, with a fixed GA concentration (red line) and increasing quantities of interferent. DPV measurements: potential scan from -1 V to +1 V, potential step 0.015 V, pulse width 0.2 V, pulse duration 0.02 s and scan speed 0.05 v/s.
Figure 8.
DPV voltammograms recorded in LiClO4 0.1 M at pH 3, with a fixed GA concentration (red line) and increasing quantities of interferent. DPV measurements: potential scan from -1 V to +1 V, potential step 0.015 V, pulse width 0.2 V, pulse duration 0.02 s and scan speed 0.05 v/s.
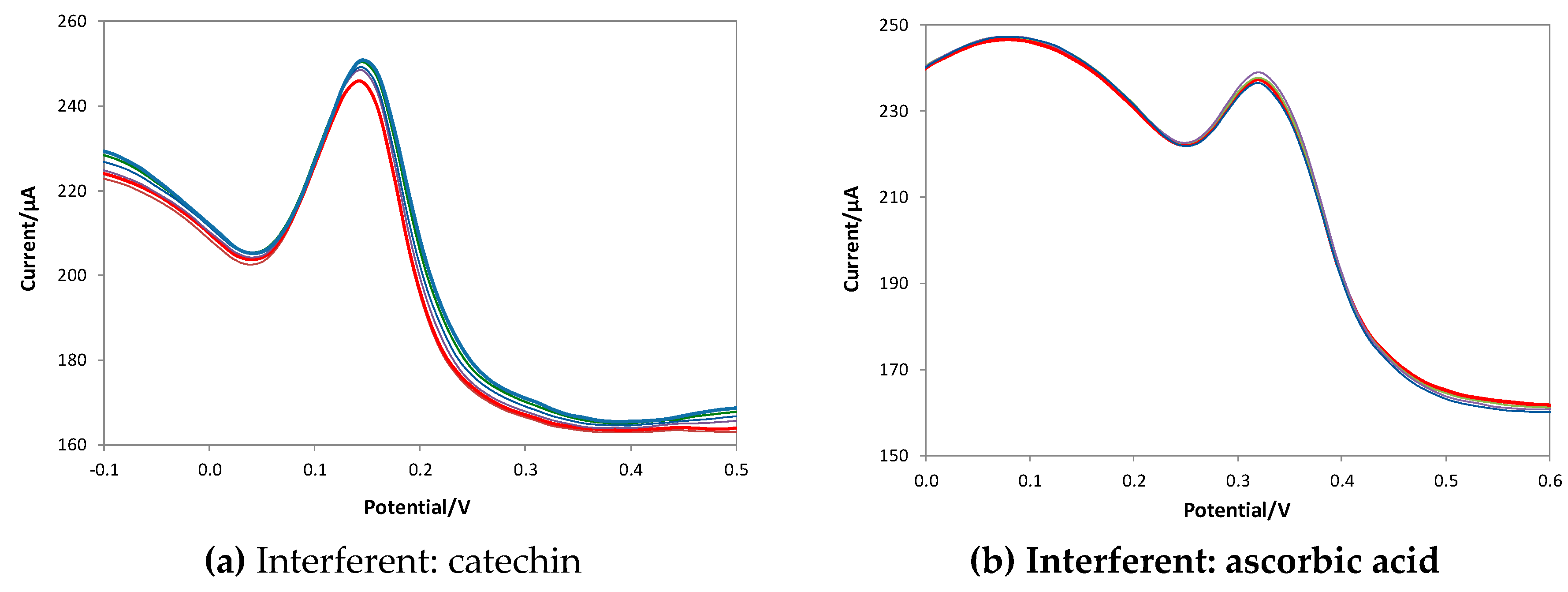
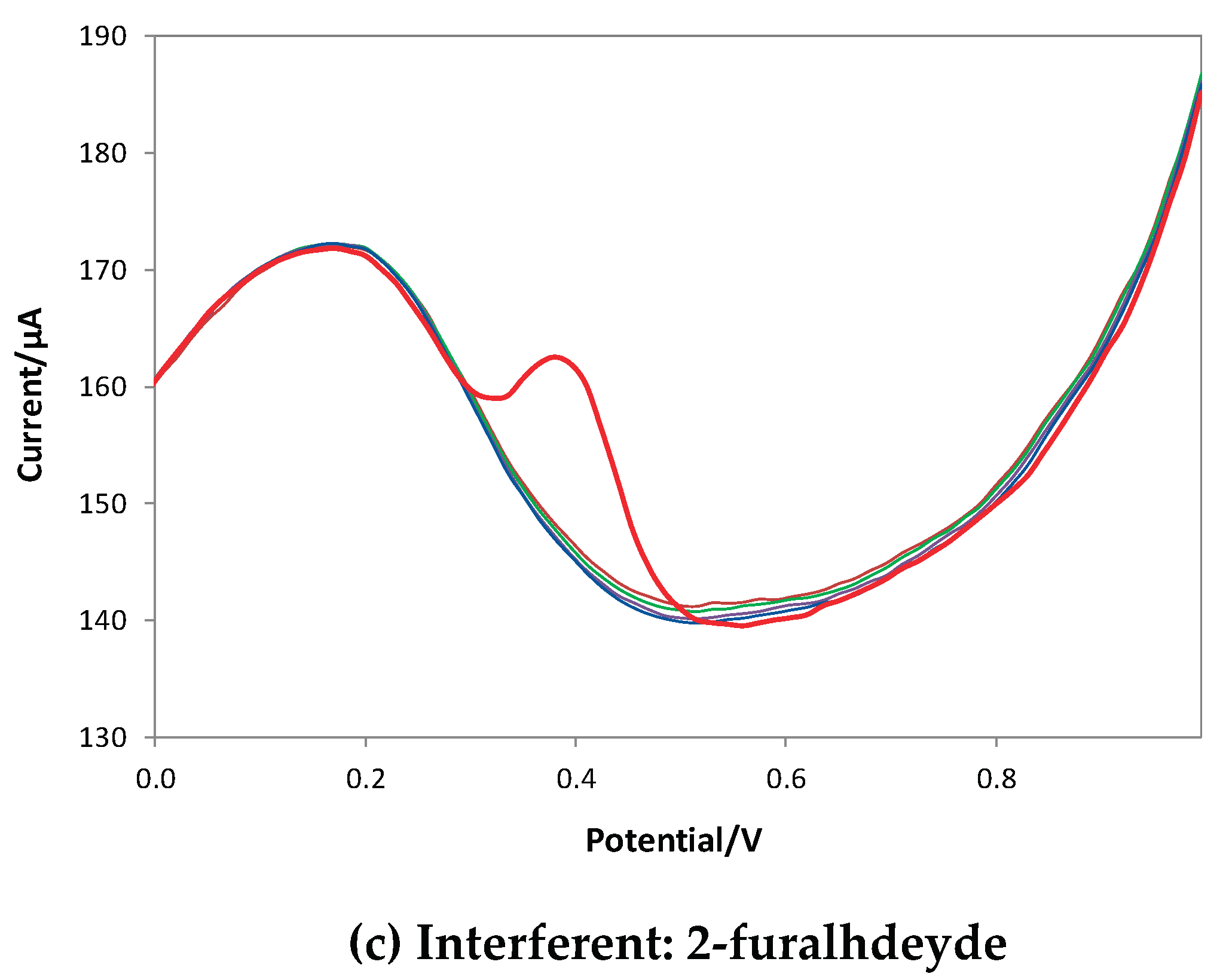

Table 1.
Active area values calculated by Randles–Sevick's equation (Eq. 1). Electrochemical probe solution: 5 mM K3Fe(CN)6/0.1 M KCl, pH 7.5. Potential scan from −1 to +1 V; scan rate from 0.025 to 0.5 V/s. Double-layer capacitance measured by CV in 0.1 M NaCl solution. Potential scan from −0.05 to +0.05 V; scan rate from 0.025 to 0.5 V/s.
Table 1.
Active area values calculated by Randles–Sevick's equation (Eq. 1). Electrochemical probe solution: 5 mM K3Fe(CN)6/0.1 M KCl, pH 7.5. Potential scan from −1 to +1 V; scan rate from 0.025 to 0.5 V/s. Double-layer capacitance measured by CV in 0.1 M NaCl solution. Potential scan from −0.05 to +0.05 V; scan rate from 0.025 to 0.5 V/s.
| Active area (mm2)† | Capacitance (μF) | |
|---|---|---|
| bare electrode | 3.8(2) | 0.7(5) |
| eMIP-modified electrode | 2.4(2) | 4.11(3) |
| eNIP-modified electrode | 1.3(1) | 1.49(3) |
†mean values obtained by plotting both the cathodic and the anodic peaks vs. (scan rate)0.5; the number in parenthesis is the standard deviation on the last digit.
Table 2.
Characterization of the oxidation process at the electrode surface. The parameters are obtained from CV scans (from -1 V to +1 V) at different scan rates (from 0.01 V/s to 2 V/s).
Table 2.
Characterization of the oxidation process at the electrode surface. The parameters are obtained from CV scans (from -1 V to +1 V) at different scan rates (from 0.01 V/s to 2 V/s).
| α (V) | E0 (V) | D (cm2/s) | k0 | |
|---|---|---|---|---|
|
bare electrode (LiClO4 0.1M, Gallic Acid 2.5 mM) |
0.49(3) | 0.265(5) | 3.6(2)∙ 10-4 | 0.023(1) |
|
eMIP-modified electrode (LiClO4 0.1M, Gallic Acid 2.5 mM) |
0.49(2) | 0.243(6) | 3.1(1)∙ 10-4 | 0.09(7) |
|
bare electrode (LiClO4 0.1M, EtOH 20% Gallic Acid 2.5 mM) |
0.50(4) | 0.508(2) | 2.6(5)∙ 10-4 | 8.6(3)∙ 10-6 |
|
eMIP-modified electrode (LiClO4 0.1M, EtOH 20% Gallic Acid 2.5 mM) |
0.44(3) | 0.501(5) | 3.1(5)∙ 10-4 | 6.8(2)∙ 10-6 |
†mean values obtained by plotting both the cathodic and the anodic peaks vs. (scan rate)0.5; the number in parenthesis is the standard deviation on the last digit.
Table 3.
Sensitivity, Detection limit (LOD), Quantification limit (LOQ) and Linear range for bare eMIP- and eNIP-modified electrodes. LOD and LOQ have been calculated from the parameters of the lines interpolating the mean values of the measurements of three electrodes of each type (the dotted lines in Figure 6). The number in parenthesis is the standard deviation on the last digit.
Table 3.
Sensitivity, Detection limit (LOD), Quantification limit (LOQ) and Linear range for bare eMIP- and eNIP-modified electrodes. LOD and LOQ have been calculated from the parameters of the lines interpolating the mean values of the measurements of three electrodes of each type (the dotted lines in Figure 6). The number in parenthesis is the standard deviation on the last digit.
| Electrode | Sensitivity (µA μM-1) |
R2 | LOD (µM) |
LOQ(µM) | Linear range (µM) |
|---|---|---|---|---|---|
| Bare | 0.37(1) | 0.994 | 12 | 35 | 12-140 |
| eMIP | 0.97(2) | 0.996 | 5 | 16 | 5-70 |
| eNIP | 0.134(5) | 0.985 | 18 | 55 | 18-140 |
Table 4.
Gallic Acid concentration in the real samples analyzed and comparison with the total polyphenols content (Folin-Ciocalteau methods) and the mean values of Gallic Acid concentration reported in the literature. Numbers in parentheses are the standard deviations on the last digit.
Table 4.
Gallic Acid concentration in the real samples analyzed and comparison with the total polyphenols content (Folin-Ciocalteau methods) and the mean values of Gallic Acid concentration reported in the literature. Numbers in parentheses are the standard deviations on the last digit.
| Beverage | [Gallic Acid] mg L-1 |
Total polyphenols GAE, mg L-1 |
[Gallic Acid] mean value, mg L-1 |
Ref. |
|---|---|---|---|---|
| White wine | 14(4) | 181.3 | 10 | [58] |
| Red wine | 145(15) | 1285.1 | 7.76-172.01 | [59] |
| Rosè wine | 120(13) | 1081.1 | 7.76-172.01 | [59] |
| Marsala | 17(2) | 281 | 13 | [60] |
| Green tea | 138(26) | 1535 | 104 | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



