Submitted:
12 March 2024
Posted:
13 March 2024
You are already at the latest version
Abstract
Psoriasis is a lifelong, systemic, immune mediated inflammatory skin condition, affecting 1-3% of the world’s population, with an impact on quality of life similar to diseases like cancer or diabetes. Genetics are the single largest risk factor in psoriasis, with GWAS studies showing that many psoriasis risk genes lie along the IL23/Th17 axis. Potential psoriasis risk genes determined through GWAS can be annotated and characterised using functional genomics, allowing the identification of novel drug targets and the repurposing of existing drugs. The pathogenesis of psoriasis is complex and focusing on the IL23/Th17 axis can provide insight into key cell types, cytokines and intracellular signaling pathways involved. Examination of currently available biological treatments, time to relapse post drug withdrawal and rates of drug primary/secondary drug failure show the need for greater understanding of the underlying genetic mechanisms of psoriasis and how they can impact treatment. This could allow for patient stratification towards the treatment most likely reduce dis-ease burden for the longest period possible.
Keywords:
psoriasis
; pathogenesis
; tailored medicine
; functional genomics
; genetics
1. Introduction
Psoriasis is a systemic, immune mediated, papulosquamous inflammatory skin condition, with a chronic relapsing-remitting course, which may also involve the nails and joints [1]. It affects 1-3% of the world’s population [2,3], with <0.5% being children [4]. Psoriasis occurs evenly between sexes, though early disease and increased severity are associated with being female and having an effected first degree relative [5]. The negative impact on patients’ Health-Related Quality of Life (HRQL) causes disability comparable to major diseases such as cancer and diabetes [6].
Genetics are the single largest risk factor for psoriasis. Family history in psoriasis is positive for 40-50% of patients, and up to 75% in those presenting <30 years [7]. Familial clustering in psoriasis is well established, with twin studies indicating a heritability range of 70-90% [8]. HLA-C*06:02 is the main genetic risk factor for psoriasis. Inheritance of one allele increases the risk of developing psoriasis by 4-5% [9].
2. The Genetics of Psoriasis
GWAS have identified >80 loci associated with psoriasis susceptibility in both European and east Asian populations [10], explaining up to 28% of heritability in psoriasis [11].
2.1. Genes Associated with the IL23 Pathway
The identification of the IL23R, IL12B, IL23A, IRF4, NF-ΚBIZ, SOCS1, STAT3 and TRAF3IP2 loci (Table 1) suggest IL23/Th17 signaling plays a prominent role in disease pathogenesis, with IL12B coding for the p40 subunit found in both IL23 and IL12 and TRAF3IP2 coding for ACT1, an adaptor protein essential in the signal transduction of IL17A [13]. KLF4 upregulates IL17A expression during Th17 differentiation. Significant enrichment of disease risk variants in the active chromatic domains of Th1 and Th17 cells were also found [11]. The gain of function mutation in CARD14 alone can drive IL23/IL17 mediated psoriasiform inflammation [14], this may be due to its role as a key mediator in the pathway through interaction with the ACT1-TRAP6 signaling complex [15], further evidenced in a study by Li et al. where epigenetic regulation of CARD14 through H3K9 demethylation controlled IL23 expression in murine keratinocytes [16]. The loss of function mutation in IL36RN increases IL36 expression, which upregulates IL6, IL23, IL8, and NF-κB signaling [17]. TGF-β and IL-23 can increase HIF-1α expression and promote the interaction between HIF-1α and P300 in CD4+ T cells [18], leading to increased miR-210 expression in CD4+ T cells, which promotes keratinocyte proliferation, increased chemokine secretion and increased production of TGF-β. miR-210 also promotes Th17 and Th1 cell differentiation while inhibiting Th2 differentiation by acting on STAT6 and LYN signaling [19]. The most recent psoriasis GWAS meta analyses found a novel variant at chromosome 22q11.1, an untranslated region of IL17RA, which codes for the most common co-receptor subunit of IL-17A, IL-17C, IL-17E, and IL-17F [20], this unit is targeted by brodalumab, a biologic found to be highly effected in the treatment of psoriasis, providing further evidence for the key nature of the IL-23 pathway [21].
Looking into IL23R more specifically, Tsoi et al. identified a particularly robust susceptibility signal within this gene. The lead psoriasis associated SNP (rs9988642) is in high LD with rs11209026, a missense exonic SNP found within IL23R. The latter SNP is protective for psoriasis, alongside other autoimmune diseases such as inflammatory bowel disease, ankylosing spondylitis, and asthma, and is present in around 7% of the population [22,23].
2.2. Limitations of GWAS
Index SNPs identified through GWAS are not necessarily causal and determining implicated genes in different cell types requires further analysis. Genotyped SNPs are chosen to be part of the array as they are in high linkage disequilibrium (LD) with many SNPs and allow identification of large genomic regions containing unmeasured SNPs who have equal probability of being causal, however they depend on cohort size and ethnicity and therefore the lead SNP can be different for different cohorts. These regions have a high probability of containing the causal SNP, however the association between a tag-SNP and a trait can be indirect, due to the tag-SNP being associated with the causal SNP [24].
A few risk variants are found within coding regions of genes (IL23R, TRAF3IP2, CARD14 and IFIH1) [11], however further characterisation is required to determine the function of intronic and intergenic non-coding variants. While not coding directly for proteins, intronic variants have been found to influence gene expression through enrichment in enhancer regions [25]. Many associated SNPs are found within promotors for candidate genes and implicate that gene in disease development, such as the IL23R, ERAP1 and IL12B loci [11]. However, the vast majority of disease associated variants are not within coding or promoter regions, and even those that may not be implicated in disease, as seen with IL12B, where variants are intronic within RNF145, though the most likely causal gene is IL12B [26]. Intergenic variants present the greatest challenge, here the associated gene is usually postulated based on proximity and biological relevance [27].
2.3. Functional Annotation of SNPs
For intronic and intergenic SNPs, once a set of potential risk SNPS has been compiled through GWAS, bioinformatics can be used to identify SNPs in LD with the lead SNPs found through GWAS, as well as identifying alignment with histone modification or transcription factor binding sites, regulatory features that increase the likelihood of the SNPs having a causal effect. Further functional experimentation for validation of the SNPs identified as most likely to be causal can include techniques such as chromatin immunoprecipitation (ChIP), multiome single cell, CRISPR, Hi-C and eQTL in disease relevant cell types [28].
A key challenge in both bioinformatic and experimental approaches is the requirement of specific cell types, environments and stimulation to bring forth the regulatory mechanisms identified, evidenced in a variety of transcriptome studies [29,30,31].
RegulomeDB [32] and HaploReg [33] are databases of all known SNPs annotated with all known functional elements in a variety of cell lineages, allowing production of a score indicating the likelihood that a given SNP may be causal.
Using databases such as GTex – the most comprehensive eQTL database to date [34], eQTLs can be identified through correlation of the genomes of individuals with the expression levels of genes within specific cell types/ tissues, with the lead disease associated SNP required to correlate with the lead eQTL for strong evidence of correlation with expression. However, Fairfax et al. showed that over half of the eQTLs identified on primary monocytes were present only post-stimulation [35]. Ding J et al. built a dataset mapping eQTLs in psoriasis patient skin tissues and found significant enrichment of psoriasis GWAS SNPs - with FUT2, RPS26 and ERAP2 expression effected [36]. Although GWAS SNPs generally show significant enrichment in eQTLs [35,37,38], only 20–50% of GWAS SNPs overlap with an eQTL, and it must be noted that eQTLs prove only correlation and not causation, therefore further characterisation is required.
Laboratory based approaches can work to characterise the effect of potential causal SNPs on gene expression, alongside the mechanism of action, and relate this back to the disease phenotype.
Capture Hi-C and HiChIP can map active chromatin interactions genome wide with high enough resolution to identify enhancer-promoter interactions, aiding in the identification of causal genes at GWAS loci [39], as noncoding regulatory elements have been shown to interact with genes over long distances through DNA looping [40,41].
Much like eQTL, many studies have shown that chromatin interactions are cell type specific and altered during differentiation and stimulation [42,43,44], and due to the systemic nature of psoriasis, the complex interplay between skin-resident and immune cells may also play a part. ChIP, ChIP-qPCR and/or ChIP-Seq can complement these DNA-DNA interaction studies nicely, through characterisation of DNA-protein interactions at GWAS loci [45] - determining whether a potential causal allele at a risk SNP affects the level of protein binding to DNA
The introduction of CRISPR/cas9 has had a great impact on the functional annotation of putative causal risk SNPs. This method can use fusion proteins to alter the transcriptional activity of the single SNP of interest, either activating or repressing enhancers [46,47], followed by methods such as RT-qPCR and RNA-Seq to identify differential gene expression between modified and control cells, allowing functional validation of putative causal risk SNPs.
3. Pathogenesis
The genetics show that the IL23/ Th17 pathway is key to psoriasis pathogenesis, setting it apart, alongside Crohn’s, as the only diseases to be so, with other immune mediated diseases being mainly Treg/Th1 driven. Figure 1 shows a simplified version of the psoriasis axis: When a keratinocyte is injured due to illness, infection or environmental reasons, it released self-DNA/RNA, which forms a complex with the LL37 autoantigen, these complexes activate both mDCs to produce TNF-α, IL-23, IL-12 and pDCs to produce IFN-α via stimulation of TLR9, TLR7 and TLR8, this leads to the activation and migration to the lymph nodes of local myeloid dendritic cells (also known as conventional dendritic cells) (mDCs), which can activate T cells through antigen presentation [48]. mDCs are also activated by INF-γ, TNF-α, IL-1-β, and IL-6 secreted by innate immune cells such as keratinocytes, macrophages and NKT cells (Table 2), and go on to produce TNF-α, IL-12, IFNα and -β, IL-6 and IL-23, these cytokines then cause the differentiation and proliferation of naïve T lymphocytes to varying T cells including Th17 and Th22 lymphocytes, which move into the blood and skin. Th17 lymphocytes release IL-17 alongside γδ T lymphocytes, NK cells, mastocytes, and innate lymphoid cells (ILCs), as well as IL-22, IFNγ, IL-2 and IL-29, whereas Th22 lymphocytes release IL-22 alone. IL22, IL17a and IL17f cause development of the psoriasis phenotype through the proliferation and impaired differentiation of keratinocytes. This process also includes many mechanisms of positive feedback, causing propagation of the disease and increasing inflammation [20,49].
A key cell type seen in Figure 1, DCs provide the link between innate and adaptive immunity, and in psoriasis this manifests as the link between disease initiation and propagation. Studies have shown increased pDC infiltration in psoriasis vs healthy skin [50], pDCs usually have safeguards against recognition of self-nucleic acids, however the large amounts of antimicrobial peptides such as LL-37 produced in psoriasis enables their recognition, leading to the production of vast amounts of IFNα [51]. pDCs are the main producers of IFNα in the skin. Nestle et al. (2005) and Okada et al. (2014) previously determined the importance of IFNα in the development of the psoriasis phenotype [52,53]. This stance is supported by genetic analysis revealing DDX58 and RNF114, both type 1 IFN genes, to confer psoriasis risk. IFNα also stimulates the differentiation of monocytes into inflammatory dendritic cells (iDCs) and CD4+ T cells into Th1 and Th17 cells [54]. iDC levels are reported to be increased in psoriasis and have been shown to present antigens to CD4+ helper and CD8+ cytotoxic cells and produce cytokines such as IL-12, IL-23, TNF-α, IL-1β, IL-6 and TGF-β [51].
There are two types of conventional dendritic cells, also known as myeloid dendritic cells (mDCs). Type 1 mDCs are known as resident dendritic cells, and are APCs that present to T lymphocytes, they are BDCA-1-positive (CD1c+), and numbers are normal in psoriasis [55]. Type 2 mDCs are BDCA-1-negative (CD1c−), with numbers greatly increased in psoriasis lesions, and normalising after treatment with biologics. Also known as inflammatory DCs or TiP-DCs, type 2 mDCs produce TNF-α, inducible nitric oxide synthase (iNOS), IL-6, IL-12, IL-20, and IL-23 [51], and again link the innate and adaptive immune systems through stimulation of naïve T cell to differentiate and presentation of foreign antigens to CD8+ T cells through cross presentation [56]. mDCs can also be directly stimulated by TNFα and LL37-self nuclease complexes [51].
3.1. Main Intracellular Pathways
The IL-23 protein itself is key to the IL-23/Th17 axis, stimulating differentiation of Th22 and Th17 cells, release of inflammatory cytokines and feeding the positive feedback loop propagating inflammation within psoriatic plaques.
IL-23 is a heterodimeric complex of p19 and p40 subunits, p19 is shared with IL-39, whereas the p40 subunit is found in IL-12. The receptor for IL-23 consists of IL-12Rβ1, shared with IL-12, and an IL-23Rα chain. This structural similarity with IL-12 along with IL-12s possible protective role in psoriasis greatly influenced the development of biologics aimed to target the p19 subunit specifically (Table 3) [57,58]. A study by Lee et al. also found that the expression of both p19 and p40 subunits was upregulated in psoriasis, as opposed to the IL-12 specific p35 [59].
In disease status, the JAK/STAT3 pathway is activated by INF-γ, IL-12, IL-22, and IL-23 (Table 2) [49,60]. The binding of IL23 to IL23R attracts a heterodimer of JAK2 and TYK2, which binds to its intracellular domain. The heterodimer then auto-phosphorylates, which activates the receptor and attracts STAT proteins, which bind and are phosphorylated before moving to the nucleus to regulate gene transcription [61]. TYK2 specifically is mainly activated by IL12 and IL23 - the lead receptor dimerises IL-12Rβ1/IL-23R, IL-12Rβ1 associates with Tyk2 and its heterotypic subunits, while IL-23R binds to Jak2. TYK2 deficiency leads to reduced ability to recruit Th17 and Th22 cells [62]. STAT3 is hyperactivated in immune cells and keratinocytes, inhibits cell differentiation, and promotes proliferation and production of antimicrobial proteins (AMPs) in response to IL-23, IL-6, IL-17, IL-21, IL-19 and IL-22 [20]. STAT3 is activated by phosphorylation of a conserved tyrosine residue by JAK kinases [60]. Phosphorylated STAT3 enhances RORγt expression, an intracellular regulator for the proliferation and function of Th17 cells [63], and both bind to promoters of genes such as IL17A, IL17F, IL22, IL26, and IL23R [64]. STAT3 mediates the effects of IL23, so is essential for the amplification and maintenance of Th17 differentiation, it upregulates IL17A and F expression, alongside other genes required for the Th17 pathway, such as RORγT, RORα, BATF, IRF4, AHR, IL-6Rα, and C-MAF, as well as being essential for the function of γδ T cells (Calautti et al., 2018). STAT3 also inhibits the convergence of Tregs downstream of IL6 and IL23 signaling, leading to a loss in suppressive power, as well as mediating IL6 stimulated IL21 secretion by naïve T cells, leading to the induction of IL23R and IL27 expression [60].

Table 2.
A summary of cell types involved in the IL-23 pathway in psoriasis. This table summarises the cell types immediately involved in an IL-23 driven psoriasis pathway, illustrating the relevant stimulants responded to, intracellular pathways activated, and proteins produced.
Table 2.
A summary of cell types involved in the IL-23 pathway in psoriasis. This table summarises the cell types immediately involved in an IL-23 driven psoriasis pathway, illustrating the relevant stimulants responded to, intracellular pathways activated, and proteins produced.
| Cell type | Stimulant | Intracellular signalling | Production | References |
| Keratinocyte | TNFα | NFκB | IL23 | [20,49,65] |
| IL-17 | ACT1/TRAF6 NFκB/MAPK | IL23 | ||
| IL-36 | MyD88/IRAK/ MAPK/ NFκB | IL23 | ||
| IL-23 | JAK/STAT3 | CCL20 | ||
| TGFβ | ||||
| Th17 | IL-36 | MyD88/IRAK/ MAPK/ NFκB | IL-23 | [60,65] |
| IL-23 | JAK/STAT3 | IL-17 | ||
| IL-22 | ||||
| IFNγ | ||||
| IL-2 | ||||
| IL-29 | ||||
| ILC3 | IL-23 | JAK/STAT3 | IL-23 | [57,65]. |
| IL-17 | ||||
| Monocytes | Mycobacterium | NFκB | IL-23 | [65]. |
| IL-23 | JAK/STAT3 | IL-22 | ||
| Macrophage | IFNγ | JAK/STAT1 | IL-23 | [66,67]. |
| Microbial infection | Dependent on microbe | IL-23 | ||
| IL-23 | JAK/STAT3 | Increased IL23R expression | ||
| TNFα | ||||
| IL-36γ | MyD88/IRAK/ MAPK/ NFκB | IL-23 | ||
| IL-23 Macrophage | IL-23 | JAK/STAT3 | IL-17A/F | [68]. |
| IL-22 | ||||
| IFNγ | ||||
| Myeloid dendritic cell | IFNα | JAK/STAT1/2 | IL-23 | [51]. |
| IFNγ | JAK/STAT1 | IL-23 | ||
| TNFα | NFκB | IL-23 | ||
| Langerhans cell | IL-36γ | MyD88/IRAK/ MAPK/ NFκB | IL-23 | [51] |
| Skin resident memory T cells | IL-23 | JAK/STAT3 | Proliferation | [69,70] |
| IL-17 | ||||
| Naïve T cell | IL-23 | JAK/STAT3 | Inhibition of Treg convergence | [71] |
| Th1 | IL-23 | JAK/STAT3 | IFNγ | [65]. |
| IL-26 | ||||
| IL-17 | ||||
| IL-22 | ||||
| IL-29 | ||||
| Th22 | IL-23 | JAK/STAT3 | IL-22 | [72]. |
| Neutrophil | IL-23 | JAK/STAT3 | IL-17 | [57,73]. |
| LL36 | ||||
| Extracellular trap formation | ||||
| Treg | IL-23 | JAK/STAT3 | IFNγ | [74]. |
| TNFα | ||||
| IL-17A | ||||
| γδT cell | IL-23 | JAK/STAT3 | IL-17 | [75,76] [77] |
| IL-22 | ||||
| αβT cell | IL-23 | JAK/STAT3 | IL-17 | [78]. |
| NK22 | IL-23 | JAK/STAT3 | IL-22 | [49,57] |
| NK17 | IL-23 | JAK/STAT3 | Differentiation | |
| IL-17 | ||||
| IFNγ | ||||
| NKT1 | IL-23 | JAK/STAT3 | IFNγ | [79] |
| MAIT17 cells | IL-23 | JAK/STAT3 | IL-17 | [80] |
RORγt is a nuclear receptor required for Th17 cell differentiation from both murine and human CD4+ T cells. Stimulated by IL23 and IL6, it acts on Th17 gene promoters IL17A, IL17F, IL22, IL26, IL23R, Csf-2, CCR6, and CCL20. Success of IL23 targeted biologics, and studies showing that lack of RORγt leads to failure of Th17 cells to differentiate demonstrates its potential as an effective drug target [63,81].
NF-κB is formed of a group of proteins, including RelA (p65), RelB and c-Rel, together with subunits of NF-κB1 (p105) and NF-κB2 (p100), processed into p50 and p52 (Perkins et al., 1992), it forms dimers, though these are retained in the cytoplasm by IκB proteins. NF-κB signalling is induced by many inflammatory cytokines (Table 2) leading to the phosphorylation of IκBα by IKKβ, degradation of IκB through proteins such as TRAFs and ACT1, and phosphorylation of IKKs for translocation to the nucleus to regulate transcription [82]. Many psoriasis risk genes are involved in this pathway; TNFAIP3, NF-ΚBIZ and TNIP1 are involved in pathway regulation, with NF-ΚBIA inhibiting the pathway, RELA coding for an NF-κB subunit and TRAF3IP2 coding for ACT1 (Table 1). Inhibition of NF-κB signaling has been shown to decrease levels of IL-23 mRNA [83].
Looking specifically at intracellular signaling, genes associated with the signaling pre and post IL-23 production are implicated in psoriasis GWAS. Interestingly, Lysell et al. found that 5 SNPs within the IL23R, IL23A and IL12B genes were only associated with severe psoriasis, alongside a significant difference in NF-ΚB1 when stratifying the cohort based on disease severity. TYK2 also showed higher expression in the severe cohort, with the association disappearing in the milder group. Out of the determined risk genes, only STAT3, TNFAIP3 and TRAF3IP2 associations remained significant in all groups, with no significant difference between disease severities. Most interestingly, interaction between genes associated with the NF-κB pathway and IL-23 signaling was increased in the severe phenotype group, with interaction between risk alleles in IL23R, NF-ΚB1, TNIP1, IL12B, and IL23A only seen in the severe cohort [84]. This study is interesting and provides some support for the link between NF-κB signaling and IL-23 production and downstream signaling shown in Table 2, however it is the only study on this topic and so requires further validation.
4. Treatments
Patient response in psoriasis is commonly measured using the Psoriasis Area and Severity Index (PASI). PASI is calculated through clinician assessment of the percentage body area affected with psoriasis and the severity of each area impacted. The score can range from 0-72, generally a score of 5-10 is considered moderate disease and >10 as severe. A 75% or 90% reduction in PASI is the benchmark in most clinical trials, noted as PASI75 and PASI90 respectively [85].
Comparing currently available psoriasis biologics (Table 3); in TNFα inhibitors etanercept is barely superior against systemic treatment options [86], though infliximab and adalimumab performed better [87,88]. While superior to etanercept, ustekinumab was inferior to all IL17 therapeutics, due to lower specificity and the possible protective effect of IL12 [89]. Risankizumab and guselkumab have proved superior to ustekinumab and TNF inhibitors, with tildrakizumab being the least successful IL23p19 antagonist, possibly due to lower affinity [90,91,92]. There is similarity in efficacy between IL17 and IL23p19 antagonists, with ixekizumab having a faster response, possibly due to IL23p19 inhibitors acting further upstream, but guselkumab having the better long-term result [93,94]. The recently approved bimekizumab works at a faster rate and, based on network meta-analysis, seems to be one of the highest performing biologics to date [95], possibly due to its inhibition of both IL17A and F, whereas IL-23 inhibitors allow for the production of IL17 through other mechanisms. However, it has yet to be compared to risankizumab or guselkumab.
Table 3.
Summary of biologic drugs used in psoriasis treatment. This table summarises the biologic drugs used in psoriasis treatment, alongside their targets and mechanisms of action.
Table 3.
Summary of biologic drugs used in psoriasis treatment. This table summarises the biologic drugs used in psoriasis treatment, alongside their targets and mechanisms of action.
| Drug | Target | Mechanism | References |
|---|---|---|---|
| Ustekinumab | P40 subunit shared by IL12 and IL23 | Disrupts Th1 and Th17 differentiation and IL12 and IL23 signaling | [64,96] |
| GuselkumabTildrakizumabRisankizumab | P19 subunit of IL23 | Disrupt Th17 and IL23 signaling | [57,90,93,97,98] |
| SecukinumabIxekinumab | IL17A | Prevents both IL17A homodimers and IL17a-IL17F heterodimers binding to their receptors. | [21,57,64,99,100] |
| Brodalumab | IL17RA | Due to the commonality of the IL17RA chain in receptor complexes, interrupts signaling of IL-17A, IL-17C and IL-17F homodimers and the IL-17A/F heterodimer | |
| Bimekizumab | IL17A/F | Prevents IL17A and F homodimers and the IL17A-IL17F homodimer binding to their receptors. | [95] |
| EtanerceptAdalimumabInfliximabCertolizumab | TNF-α | Indirect impact on IL17, by regulation of IL23 production from myeloid or CD11c+ dendritic cells. | [57,64,101] |
4.1. The Need for Personalised Stratified Medicine in Psoriasis
As observed commonly with biologics, patients’ initial response tapers off over time (secondary failure) though some do not respond at all (primary failure). The time between first response and withdrawal of the drug due to loss of efficacy differs between biologics, though the risk of treatment failure is positively correlated with the number of biologics the patient has previously tried [102]. A 2022 study by Elberdín et al. [103] found that over 10 years, the median number of biologics patients had been on was 2 (range 1-8), with lack of efficacy being the main reason for switching. It found that ustekinumab had the best drug survival, with efalizumab being withdrawn from the market in 2009 (Figure 3). As IL-23p19 inhibitors show an increased remission period post drug withdrawal compared to ustekinumab, it will be interesting to see whether it would have increased survival in 10 years. The mechanisms leading to treatment failure remain unclear.
One possible reason could be the development of antidrug antibodies (ADAs). Specific to biologic treatments, an immune response can be generated to target the monoclonal antibodies, leading to reduced circulating drug levels, drug efficacy, drug survival and/or adverse effects such as infusion reactions [104]. A possible solution is the administration of immunosuppressants alongside biologic treatment, such as methotrexate/ azathioprine co-prescription with TNF inhibitor treatments, though this does come with the risk of immunosuppression in patients [104]. Interestingly, the development of ADAs can be influenced by genetic factors, with the HLA-DRβ-11, HLA-DQ-03, and HLA-DQ-05 alleles conferring a higher risk of ADA development post anti-TNF treatment [105]. The most consistent genetic association with ADA development is HLA-DQA1∗05 alleles, however the relatively small sample sizes and number of associations, and lack of consistent result replication found in these studies make drawing reliable conclusions difficult [106,107,108].
Another possible mechanism is genetic polymorphisms. With response to biologic drugs typically being heterogenous, one hypothesis is that this response reflects genetic variance between patients or genetically distinct disease subsets with distinct pathogeneses. The effect of genetics on anti-TNF response is well characterized, with TNF-α, TNFRSF1A, TNFRSF1B, TNFAIP3, FCGR2A, FCGR3A, IL-17F, IL-17R, and IL-23R suggested to modulate response [109], however, few studies explore the interaction of IL-17 and IL-23 inhibitors with genetics. Ustekinumab shows a higher efficacy and faster response time in HLA-Cw*06 positive patients than in negative patients [109], and Van den Reek et al. found that the IL12B rs3213094-T allele increased efficacy and TNFAIP3 rs610604-G allele predicting a worse outcome [110], however other studies were unable to replicate this. The SUPREME study found that HLA-Cw*06 status did not influence response to secukinumab [111], however the two Italian studies predicted a higher PASI90 in HLA-Cw*06 positive patients [112,113]. An investigation into the effects of IL-17A polymorphisms on secukinumab and ixekinumab response identified five non-coding SNPs, however none influenced PASI75/90 achievement at 12 or 24 weeks [114]. In conclusion, a link between genetic and treatment response has been found, however, especially in regard to the newer and more effective IL-17 and IL-23 inhibitors, more studies are needed to reliably determine the effects of the polymorphisms identified as modulating treatment response. Discovery of genetic biomarkers for drug response could allow stratification of patients into subgroups to increase response rates, allowing patients an earlier increase in quality of life.
Patients withdraw from therapeutics for a variety of reasons; withdrawal is associated with risk of relapse, though time to relapse varies between person and drug. The median time to relapse was 16-20 weeks for tildrakizumab, an IL-23p19 inhibitor (defined as below PASI 90), or 20-25 weeks for PASI <75 [115], whereas guselkumab had a median relapse time of 15 weeks post withdrawal (PASI <90) [93] and risankizumab a median of 30 weeks (PASI 90) [116]. Ustekinumab’s median time was 15 weeks to PASI <75 post withdrawal, 22-24 weeks for PASI<50 [117,118]. While IL17 inhibitors seem to have a shorter time to relapse and occasional rebound of disease, studies conflict over median time, from 46 days (brodalumab) [119] to 20 weeks (ixekizumab, PASI <50) [120], this difference is likely due to differing relapse criteria. The median time to relapse when withdrawing TNFα inhibitors has been found as 12.1 weeks (etanercept) [121] to 19.5 weeks (infliximab) [122], the shortest post-withdrawal period [123]. The increased time period for IL23 inhibitors may because IL23 is an upstream cytokine of many psoriasis pathways, impacting cytokines such as IL17, and potentially the proliferation and survival of epidermal T cells [115].
A recent study published by Zhang et al. focused on secukinumab, which targets genes thought to confer psoriasis risk both upstream (IL23R, TYK2, JAK2, STAT3) and downstream (TRAF3IP2/ ACT1, TNFAIP3/ A20) of IL-17 production. They found that although genetic variation in the IL-17 pathway impacts psoriasis susceptibility, this same variation does not significantly impact treatment response to secukinumab [124]. However, due to possible conflict of interest, further studies in this subject would be useful.
Together with the highlighted importance of genetics in understanding and determining psoriasis pathogenesis, this review emphasises the need for the use of genetics to stratify patients towards treatment options that are most likely reduce disease burden for the longest period possible, as currently there is no tool or technique in the choice of first biologic, or those that follow, past clinician experience and preference. This review has highlighted the vast differences in patient response to varying biologics, and the varying times to relapse post drug withdrawal, alongside the lack of understanding as to why this is. Greater understanding of the genetics underlying disease pathogenesis and treatment response may elucidate the underlying mechanisms and therefore allow for the generation of more accurate polygenic risk scores than currently exist, patient stratification for treatment, and management of expectations post drug withdrawal for both patient and clinician.
Author Contributions
Conceptualization, E.S. and S.E.; writing—original draft preparation, E.S.; writing—review and editing, S.E, S.S.R, R.B.W.; supervision, S.E., R.B.W, S.S.R. All authors have read and agreed to the published version of the manuscript.
Funding
E.S is funded by The Kennedy Trust for Rheumatology Research, through the Kennedy Trust Inflammation Impact MBPhD Award (KENN 20 21 06).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
This work was supported by Versus Arthritis and by the NIHR Manchester Biomedical Research Centre (NIHR203308). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet. 2021;397(10281):1301-15.
- Myers, W.A.; Gottlieb, A.B.; Mease, P. Psoriasis and psoriatic arthritis: clinical features and disease mechanisms. Clin. Dermatol. 2006, 24, 438–447. [Google Scholar] [CrossRef]
- World Health, O. Global report on psoriasis. Geneva: World Health Organization; 2016 2016.
- Michalek, I.; Loring, B.; John, S. A systematic review of worldwide epidemiology of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2016, 31, 205–212. [Google Scholar] [CrossRef]
- Johnson, M.A.N.; Armstrong, A.W. Clinical and Histologic Diagnostic Guidelines for Psoriasis: A Critical Review. Clin. Rev. Allergy Immunol. 2012, 44, 166–172. [Google Scholar] [CrossRef]
- Rapp SR, Feldman SR, Exum ML, Fleischer AB, Jr., Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol. 1999;41(3 Pt 1):401-7.
- PCDS PCDS-. Psoriasis: an overview and chronic plaque psoriasis [Webpage]. 2022 [cited 2022 22/11/2022]. Available from: https://www.pcds.org.uk/clinical-guidance/psoriasis-an-overview.
- Capon, F.; Semprini, S.; Novelli, G.; Chimenti, S.; Fabrizi, G.; Zambruno, G.; Murgia, S.; Carcassi, C.; Fazio, M.; Mingarelli, R.; et al. Fine Mapping of the PSORS4 Psoriasis Susceptibility Region on Chromosome 1q21. J. Investig. Dermatol. 2001, 116, 728–730. [Google Scholar] [CrossRef]
- Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42(11):985-90.
- Hwang, S.T.; Nijsten, T.; Elder, J.T. Recent Highlights in Psoriasis Research. J. Investig. Dermatol. 2017, 137, 550–556. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Stuart, P.E.; Tian, C.; Gudjonsson, J.E.; Das, S.; Zawistowski, M.; Ellinghaus, E.; Barker, J.N.; Chandran, V.; Dand, N.; et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ray-Jones, H. Defining the key biological and genetic mechanisms involved in psoriasis: University of Manchester; 2018.
- Lambert, S.; Swindell, W.R.; Tsoi, L.C.; Stoll, S.W.; Elder, J.T. Dual Role of Act1 in Keratinocyte Differentiation and Host Defense: TRAF3IP2 Silencing Alters Keratinocyte Differentiation and Inhibits IL-17 Responses. J. Investig. Dermatol. 2017, 137, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Mellett M, Meier B, Mohanan D, Schairer R, Cheng P, Satoh TK, et al. CARD14 Gain-of-Function Mutation Alone Is Sufficient to Drive IL-23/IL-17-Mediated Psoriasiform Skin Inflammation In Vivo. J Invest Dermatol. 2018;138(9):2010-23.
- Wang, M.; Zhang, S.; Zheng, G.; Huang, J.; Songyang, Z.; Zhao, X.; Lin, X. Gain-of-Function Mutation of Card14 Leads to Spontaneous Psoriasis-like Skin Inflammation through Enhanced Keratinocyte Response to IL-17A. Immunity 2018, 49, 66–79. [Google Scholar] [CrossRef]
- Li, H.; Yao, Q.; Mariscal, A.G.; Wu, X.; Hülse, J.; Pedersen, E.; Helin, K.; Waisman, A.; Vinkel, C.; Thomsen, S.F.; et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mössner, R.; Wilsmann-Theis, D.; Oji, V.; Gkogkolou, P.; Löhr, S.; Schulz, P.; Körber, A.; Prinz, J.; Renner, R.; Schäkel, K.; et al. The genetic basis for most patients with pustular skin disease remains elusive. Br. J. Dermatol. 2017, 178, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the Depressed Brain: Role of Histone Acetylation and Methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zeng, J.; Yuan, J.; Deng, X.; Huang, Y.; Chen, L.; Zhang, P.; Feng, H.; Liu, Z.; Wang, Z.; et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J. Clin. Investig. 2018, 128, 2551–2568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: from keratinocyte perspective. Cell Death Dis. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. New Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef]
- Stuart Philip E, Nair Rajan P, Tsoi Lam C, Tejasvi T, Das S, Kang Hyun M, et al. Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. The American Journal of Human Genetics. 2015;97(6):816-36.
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Kullo, I.J. Methods for the selection of tagging SNPs: a comparison of tagging efficiency and performance. Eur. J. Hum. Genet. 2006, 15, 228–236. [Google Scholar] [CrossRef]
- Farh, K.K.-H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef]
- Zuo, X.; Sun, L.; Yin, X.; Gao, J.; Sheng, Y.; Xu, J.; Zhang, J.; He, C.; Qiu, Y.; Wen, G.; et al. Whole-exome SNP array identifies 15 new susceptibility loci for psoriasis. Nat. Commun. 2015, 6, 6793. [Google Scholar] [CrossRef]
- Ray-Jones, H. Defining the key biological and genetic mechanisms involved in psoriasis. Manchester: University of Manchester; 2016.
- Ray-Jones, H.; Eyre, S.; Barton, A.; Warren, R.B. One SNP at a Time: Moving beyond GWAS in Psoriasis. J. Investig. Dermatol. 2016, 136, 567–573. [Google Scholar] [CrossRef]
- Filkor, K.; Hegedűs, Z.; Szász, A.; Tubak, V.; Kemény, L.; Kondorosi. ; Nagy, I. Genome Wide Transcriptome Analysis of Dendritic Cells Identifies Genes with Altered Expression in Psoriasis. PLOS ONE 2013, 8, e73435. [Google Scholar] [CrossRef]
- Jabbari, A.; Suárez-Fariñas, M.; Dewell, S.; Krueger, J.G. Transcriptional Profiling of Psoriasis Using RNA-seq Reveals Previously Unidentified Differentially Expressed Genes. J. Investig. Dermatol. 2012, 132, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Fariñas, M.; Li, K.; Fuentes-Duculan, J.; Hayden, K.; Brodmerkel, C.; Krueger, J.G. Expanding the Psoriasis Disease Profile: Interrogation of the Skin and Serum of Patients with Moderate-to-Severe Psoriasis. J. Investig. Dermatol. 2012, 132, 2552–2564. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2011, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318-30.
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1118. [Google Scholar] [CrossRef] [PubMed]
- Ding J, Gudjonsson JE, Liang L, Stuart PE, Li Y, Chen W, et al. Gene expression in skin and lymphoblastoid cells: Refined statistical method reveals extensive overlap in cis-eQTL signals. Am J Hum Genet. 2010;87(6):779-89.
- Joehanes, R.; Zhang, X.; Huan, T.; Yao, C.; Ying, S.-X.; Nguyen, Q.T.; Demirkale, C.Y.; Feolo, M.L.; Sharopova, N.R.; Sturcke, A.; et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017, 18, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 2018;175(6):1701-15.e16.
- Shi, C.; Ray-Jones, H.; Ding, J.; Duffus, K.; Fu, Y.; Gaddi, V.P.; Gough, O.; Hankinson, J.; Martin, P.; McGovern, A.; et al. Chromatin Looping Links Target Genes with Genetic Risk Loci for Dermatological Traits. J. Investig. Dermatol. 2021, 141, 1975–1984. [Google Scholar] [CrossRef]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014, 24, 1854–1868. [Google Scholar] [CrossRef]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; A Ewels, P.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Burren OS, Rubio García A, Javierre BM, Rainbow DB, Cairns J, Cooper NJ, et al. Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol. 2017;18(1):165.
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 2017, 9, 20–32. [Google Scholar] [CrossRef]
- Rubin, A.J.; Barajas, B.C.; Furlan-Magaril, M.; Lopez-Pajares, V.; Mumbach, M.R.; Howard, I.; Kim, D.S.; Boxer, L.D.; Cairns, J.; Spivakov, M.; et al. Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation. Nat. Genet. 2017, 49, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Christova, R. Chapter Four - Detecting DNA–Protein Interactions in Living Cells—ChIP Approach. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. 91: Academic Press; 2013. p. 101-33.
- Simeonov, D.R.; Gowen, B.G.; Boontanrart, M.; Roth, T.L.; Gagnon, J.D.; Mumbach, M.R.; Satpathy, A.T.; Lee, Y.; Bray, N.L.; Chan, A.Y.; et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 2017, 549, 111–115. [Google Scholar] [CrossRef]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33(5):510-7.
- Reali, E.; Ferrari, D. From the Skin to Distant Sites: T Cells in Psoriatic Disease. Int. J. Mol. Sci. 2023, 24, 15707. [Google Scholar] [CrossRef] [PubMed]
- Vičić, M.; Kaštelan, M.; Brajac, I.; Sotošek, V.; Massari, L.P. Current Concepts of Psoriasis Immunopathogenesis. Int. J. Mol. Sci. 2021, 22, 11574. [Google Scholar] [CrossRef]
- Chen J, Tan Z, Liu H, Liu Z, Wu Y, Li J. Expression of plasmacytoid dendritic cells, IRF-7, IFN-alpha mRNA in the lesions of psoriasis vulgaris. J Huazhong Univ Sci Technolog Med Sci. 2006;26(6):747-9.
- Wang, A.; Bai, Y. Dendritic cells: The driver of psoriasis. J. Dermatol. 2020, 47, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202(1):135-43.
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Ueyama, A.; Yamamoto, M.; Tsujii, K.; Furue, Y.; Imura, C.; Shichijo, M.; Yasui, K. Mechanism of pathogenesis of imiquimod-induced skin inflammation in the mouse: A role for interferon-alpha in dendritic cell activation by imiquimod. J. Dermatol. 2014, 41, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the Immunopathogenesis of Psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef]
- Malissen, B.; Tamoutounour, S.; Henri, S. The origins and functions of dendritic cells and macrophages in the skin. Nat. Rev. Immunol. 2014, 14, 417–428. [Google Scholar] [CrossRef]
- Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. The Journal of Immunology. 2018;201(6):1605-13.
- Wojno, E.D.T.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 p19 and p40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Calautti, E.; Avalle, L.; Poli, V. Psoriasis: A STAT3-Centric View. Int. J. Mol. Sci. 2018, 19, 171. [Google Scholar] [CrossRef] [PubMed]
- Kvist-Hansen, A.; Hansen, P.R.; Skov, L. Systemic Treatment of Psoriasis with JAK Inhibitors: A Review. Dermatol. Ther. 2019, 10, 29–42. [Google Scholar] [CrossRef]
- Ishizaki, M.; Muromoto, R.; Akimoto, T.; Sekine, Y.; Kon, S.; Diwan, M.; Maeda, H.; Togi, S.; Shimoda, K.; Oritani, K.; et al. Tyk2 is a therapeutic target for psoriasis-like skin inflammation. Int. Immunol. 2013, 26, 257–267. [Google Scholar] [CrossRef]
- Liu, S.; Liu, D.; Shen, R.; Li, D.; Hu, Q.; Yan, Y.; Sun, J.; Zhang, F.; Wan, H.; Dong, P.; et al. Discovery of a novel RORγ antagonist with skin-restricted exposure for topical treatment of mild to moderate psoriasis. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Balato, A.; Enerbäck, C.; Sabat, R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet 2021, 397, 754–766. [Google Scholar] [CrossRef]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Fearnley, G.W.; Berekmeri, A.; Laws, P.; Macleod, T.; Ponnambalam, S.; Stacey, M.; Graham, A.; Wittmann, M. IL-36γ Is a Strong Inducer of IL-23 in Psoriatic Cells and Activates Angiogenesis. Front. Immunol. 2018, 9, 200. [Google Scholar] [CrossRef]
- Hou, Y.; Zhu, L.; Tian, H.; Sun, H.-X.; Wang, R.; Zhang, L.; Zhao, Y. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell 2018, 9, 1027–1038. [Google Scholar] [CrossRef]
- Cheuk, S.; Schlums, H.; Sérézal, I.G.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef]
- Tokura, Y.; Phadungsaksawasdi, P.; Kurihara, K.; Fujiyama, T.; Honda, T. Pathophysiology of Skin Resident Memory T Cells. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Hu, P.; Wang, M.; Gao, H.; Zheng, A.; Li, J.; Mu, D.; Tong, J. The Role of Helper T Cells in Psoriasis. Front. Immunol. 2021, 12, 788940. [Google Scholar] [CrossRef]
- Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857-63.
- Menter, A.; Krueger, G.G.; Paek, S.Y.; Kivelevitch, D.; Adamopoulos, I.E.; Langley, R.G. Interleukin-17 and Interleukin-23: A Narrative Review of Mechanisms of Action in Psoriasis and Associated Comorbidities. Dermatol. Ther. 2021, 11, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Shi Y, Chen Z, Zhao Z, Yu Y, Fan H, Xu X, et al. IL-21 Induces an Imbalance of Th17/Treg Cells in Moderate-to-Severe Plaque Psoriasis Patients. Front Immunol. 2019;10:1865.
- Cai, Y.; Xue, F.; Quan, C.; Qu, M.; Liu, N.; Zhang, Y.; Fleming, C.; Hu, X.; Zhang, H.-G.; Weichselbaum, R.; et al. A Critical Role of the IL-1β–IL-1R Signaling Pathway in Skin Inflammation and Psoriasis Pathogenesis. J. Investig. Dermatol. 2018, 139, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Hartwig T, Pantelyushin S, Croxford AL, Kulig P, Becher B. Dermal IL-17-producing γδ T cells establish long-lived memory in the skin. Eur J Immunol. 2015;45(11):3022-33.
- Polese, B.; Zhang, H.; Thurairajah, B.; King, I.L. Innate Lymphocytes in Psoriasis. Front. Immunol. 2020, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity. 2011;35(4):596-610.
- Yip, K.H.; Papadopoulos, M.; Pant, H.; Tumes, D.J. The role of invariant T cells in inflammation of the skin and airways. Semin. Immunopathol. 2019, 41, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Pisarska MM, Dunne MR, O’Shea D, Hogan AE. Interleukin-17 producing mucosal associated invariant T cells - emerging players in chronic inflammatory diseases? European Journal of Immunology. 2020;50(8):1098-108.
- Tang, L.; Yang, X.; Liang, Y.; Xie, H.; Dai, Z.; Zheng, G. Transcription Factor Retinoid-Related Orphan Receptor γt: A Promising Target for the Treatment of Psoriasis. Front. Immunol. 2018, 9, 1210. [Google Scholar] [CrossRef] [PubMed]
- Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF. NF-κB: An essential transcription factor in psoriasis. Journal of Dermatological Science. 2013;69(2):89-94.
- Ramnath D, Tunny K, Hohenhaus DM, Pitts CM, Bergot AS, Hogarth PM, et al. TLR3 drives IRF6-dependent IL-23p19 expression and p19/EBI3 heterodimer formation in keratinocytes. Immunol Cell Biol. 2015;93(9):771-9.
- Lysell, J.; Padyukov, L.; Kockum, I.; Nikamo, P.; Ståhle, M. Genetic Association with ERAP1 in Psoriasis Is Confined to Disease Onset after Puberty and Not Dependent on HLA-C*06. J. Investig. Dermatol. 2013, 133, 411–417. [Google Scholar] [CrossRef]
- Mahil SK, Wilson N, Dand N, Reynolds NJ, Griffiths CEM, Emsley R, et al. Psoriasis treat to target: defining outcomes in psoriasis using data from a real-world, population-based cohort study (the British Association of Dermatologists Biologics and Immunomodulators Register, BADBIR). Br J Dermatol. 2020;182(5):1158-66.
- Reich K, Gooderham M, Green L, Bewley A, Zhang Z, Khanskaya I, et al. The efficacy and safety of apremilast, etanercept and placebo in patients with moderate-to-severe plaque psoriasis: 52-week results from a phase IIIb, randomized, placebo-controlled trial (LIBERATE). J Eur Acad Dermatol Venereol. 2017;31(3):507-17.
- Barker, J.; Hoffmann, M.; Wozel, G.; Ortonne, J.-P.; Zheng, H.; van Hoogstraten, H.; Reich, K. Efficacy and safety of infliximab vs. methotrexate in patients with moderate-to-severe plaque psoriasis: results of an open-label, active-controlled, randomized trial (RESTORE1). Br. J. Dermatol. 2011, 165, 1109–1117. [Google Scholar] [CrossRef]
- Saurat JH, Stingl G, Dubertret L, Papp K, Langley RG, Ortonne JP, et al. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs. placebo in patients with psoriasis (CHAMPION). Br J Dermatol. 2008;158(3):558-66.
- ten Bergen LL, Petrovic A, Krogh Aarebrot A, Appel S. The TNF/IL-23/IL-17 axis—Head-to-head trials comparing different biologics in psoriasis treatment. Scandinavian Journal of Immunology. 2020;92(4):e12946.
- Gordon, K.B.; Strober, B.; Lebwohl, M.; Augustin, M.; Blauvelt, A.; Poulin, Y.; A Papp, K.; Sofen, H.; Puig, L.; Foley, P.; et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 2018, 392, 650–661. [Google Scholar] [CrossRef]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.-P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. New Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Langley RG, Tsai TF, Flavin S, Song M, Randazzo B, Wasfi Y, et al. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double-blind, phase III NAVIGATE trial. Br J Dermatol. 2018;178(1):114-23.
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.-K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator–controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt A, Papp K, Gottlieb A, Jarell A, Reich K, Maari C, et al. A head-to-head comparison of ixekizumab vs. guselkumab in patients with moderate-to-severe plaque psoriasis: 12-week efficacy, safety and speed of response from a randomized, double-blinded trial. Br J Dermatol. 2020;182(6):1348-58.
- Gordon, K.B.; Foley, P.; Krueger, J.G.; Pinter, A.; Reich, K.; Vender, R.; Vanvoorden, V.; Madden, C.; White, K.; Cioffi, C.; et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet 2021, 397, 475–486. [Google Scholar] [CrossRef]
- Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362(2):118-28.
- Sawyer, L.M.; Malottki, K.; Sabry-Grant, C.; Yasmeen, N.; Wright, E.; Sohrt, A.; Borg, E.; Warren, R.B. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: A systematic review and network meta-analysis of PASI response. PLOS ONE 2019, 14, e0220868. [Google Scholar] [CrossRef] [PubMed]
- Armstrong AW, Puig L, Joshi A, Skup M, Williams D, Li J, et al. Comparison of Biologics and Oral Treatments for Plaque Psoriasis: A Meta-analysis. JAMA Dermatol. 2020;156(3):258-69.
- Blauvelt, A.; Reich, K.; Tsai, T.-F.; Tyring, S.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Vender, R.; Hugot, S.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the CLEAR study. J. Am. Acad. Dermatol. 2017, 76, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. New Engl. J. Med. 2016, 375, 345–356. [Google Scholar] [CrossRef] [PubMed]
- NICE) NIfHaCE. Psoriasis 2022 [Available from: https://cks.nice.org.uk/topics/psoriasis/management/trunk-limbs/.
- Menting SP, Coussens E, Pouw MF, van den Reek JM, Temmerman L, Boonen H, et al. Developing a Therapeutic Range of Adalimumab Serum Concentrations in Management of Psoriasis: A Step Toward Personalized Treatment. JAMA Dermatol. 2015;151(6):616-22.
- Elberdín, L.; Fernández-Torres, R.M.; Paradela, S.; Mateos, M.; Blanco, E.; Balboa-Barreiro, V.; Gómez-Besteiro, M.I.; Outeda, M.; Martín-Herranz, I.; Fonseca, E. Biologic Therapy for Moderate to Severe Psoriasis. Real-World Follow-up of Patients Who Initiated Biologic Therapy at Least 10 Years Ago. Dermatol. Ther. 2022, 12, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Tsakok, T.; Rispens, T.; Spuls, P.; Nast, A.; Smith, C.; Reich, K. Immunogenicity of biologic therapies in psoriasis: Myths, facts and a suggested approach. J. Eur. Acad. Dermatol. Venereol. 2020, 35, 329–337. [Google Scholar] [CrossRef]
- Vaisman-Mentesh, A.; Gutierrez-Gonzalez, M.; DeKosky, B.J.; Wine, Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment With Monoclonal Antibodies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Sazonovs, A.; Kennedy, N.A.; Moutsianas, L.; Heap, G.A.; Rice, D.L.; Reppell, M.; Bewshea, C.M.; Chanchlani, N.; Walker, G.J.; Perry, M.H.; et al. HLA-DQA1*05 Carriage Associated With Development of Anti-Drug Antibodies to Infliximab and Adalimumab in Patients With Crohn’s Disease. Gastroenterology 2020, 158, 189–199. [Google Scholar] [CrossRef]
- Hässler, S.; Bachelet, D.; Duhaze, J.; Szely, N.; Gleizes, A.; Abina, S.H.-B.; Aktas, O.; Auer, M.; Avouac, J.; Birchler, M.; et al. Clinicogenomic factors of biotherapy immunogenicity in autoimmune disease: A prospective multicohort study of the ABIRISK consortium. PLOS Med. 2020, 17, e1003348. [Google Scholar] [CrossRef]
- Liu, M.; Degner, J.; Davis, J.W.; Idler, K.B.; Nader, A.; Mostafa, N.M.; Waring, J.F. Identification of HLA-DRB1 association to adalimumab immunogenicity. PLOS ONE 2018, 13, e0195325. [Google Scholar] [CrossRef]
- Berna-Rico, E.; Perez-Bootello, J.; de Aragon, C.A.-J.; Gonzalez-Cantero, A. Genetic Influence on Treatment Response in Psoriasis: New Insights into Personalized Medicine. Int. J. Mol. Sci. 2023, 24, 9850. [Google Scholar] [CrossRef]
- van den Reek J, Coenen MJH, van de L’Isle Arias M, Zweegers J, Rodijk-Olthuis D, Schalkwijk J, et al. Polymorphisms in CD84, IL12B and TNFAIP3 are associated with response to biologics in patients with psoriasis. Br J Dermatol. 2017;176(5):1288-96.
- Costanzo, A.; Bianchi, L.; Flori, M.L.; Malara, G.; Stingeni, L.; Bartezaghi, M.; Carraro, L.; Castellino, G. ; the SUPREME Study Group Secukinumab shows high efficacy irrespective ofHLA-Cw6status in patients with moderate-to-severe plaque-type psoriasis: SUPREME study. Br. J. Dermatol. 2018, 179, 1072–1080. [Google Scholar] [CrossRef]
- Galluzzo, M.; D’adamio, S.; Silvaggio, D.; Lombardo, P.; Bianchi, L.; Talamonti, M. In which patients the best efficacy of secukinumab? Update of a real-life analysis after 136 weeks of treatment with secukinumab in moderate-to-severe plaque psoriasis. Expert Opin. Biol. Ther. 2019, 20, 173–182. [Google Scholar] [CrossRef]
- Morelli M, Galluzzo M, Madonna S, Scarponi C, Scaglione GL, Galluccio T, et al. HLA-Cw6 and other HLA-C alleles, as well as MICB-DT, DDX58, and TYK2 genetic variants associate with optimal response to anti-IL-17A treatment in patients with psoriasis. Expert Opin Biol Ther. 2021;21(2):259-70.
- van Vugt, L.; Reek, J.v.D.; Meulewaeter, E.; Hakobjan, M.; Heddes, N.; Traks, T.; Kingo, K.; Galluzzo, M.; Talamonti, M.; Lambert, J.; et al. Response to IL-17A inhibitors secukinumab and ixekizumab cannot be explained by genetic variation in the protein-coding and untranslated regions of the IL-17A gene: results from a multicentre study of four European psoriasis cohorts. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.; Carrascosa, J.; Fumero, E.; Schoenenberger, A.; Lebwohl, M.; Szepietowski, J.; Reich, K. Time to relapse after tildrakizumab withdrawal in patients with moderate-to-severe psoriasis who were responders at week 28: post hoc analysis through 64 weeks from reSURFACE 1 trial. J. Eur. Acad. Dermatol. Venereol. 2020, 35, 919–927. [Google Scholar] [CrossRef]
- Blauvelt A, Leonardi CL, Gooderham M, Papp KA, Philipp S, Wu JJ, et al. Efficacy and Safety of Continuous Risankizumab Therapy vs Treatment Withdrawal in Patients With Moderate to Severe Plaque Psoriasis: A Phase 3 Randomized Clinical Trial. JAMA Dermatology. 2020;156(6):649-58.
- Leonardi, C.L.; Kimball, A.B.; A Papp, K.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-Y.; Hui, R.C.-Y.; Tsai, T.-F.; Chen, Y.-C.; Liao, N.-F.C.; Chen, P.-H.; Lai, P.-J.; Wang, T.-S.; Huang, Y.-H. Predictors of time to relapse following ustekinumab withdrawal in patients with psoriasis who had responded to therapy: An 8-year multicenter study. J. Am. Acad. Dermatol. 2019, 88, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Regnault, M.M.; Konstantinou, M.; Khemis, A.; Poulin, Y.; Bourcier, M.; Amelot, F.; Livideanu, C.B.; Paul, C. Early relapse of psoriasis after stopping brodalumab: a retrospective cohort study in 77 patients. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, Y.; Torisu-Itakura, H.; Morisaki, Y.; ElMaraghy, H.; Nakajo, K.; Akashi, N.; Saeki, H. ; the Japanese Ixekizumab Study Group Long-term efficacy and safety results from an open-label phase III study (UNCOVER-J) in Japanese plaque psoriasis patients: impact of treatment withdrawal and retreatment of ixekizumab. J. Eur. Acad. Dermatol. Venereol. 2018, 33, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Gottlieb, A.B.; Leonardi, C.L.; Elewski, B.E.; Wang, A.; Jahreis, A.; Zitnik, R. Clinical response in psoriasis patients discontinued from and then reinitiated on etanercept therapy. J. Dermatol. Treat. 2006, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Evans, R.; Li, S.; Dooley, L.T.; Guzzo, C.A.; Baker, D.; Bala, M.; Marano, C.W.; Menter, A. Infliximab induction therapy for patients with severe plaque-type psoriasis: A randomized, double-blind, placebo-controlled trial. J. Am. Acad. Dermatol. 2004, 51, 534–542. [Google Scholar] [CrossRef]
- Stinco, G.; Balato, N.; Buligan, C.; Campanati, A.; Dastoli, S.; Di Meo, N.; Gisondi, P.; Lacarubba, F.; Musumeci, M.L.; Napolitano, M.; et al. A multicenter retrospective case-control study on Suspension of TNF-inhibitors and Outcomes in Psoriatic patients (STOP study). G. Ital. di Dermatol. e Venereol. 2019, 154, 392–399. [Google Scholar] [CrossRef]
- Zhang, C.; Shestopaloff, K.; Hollis, B.; Kwok, C.H.; Hon, C.; Hartmann, N.; Tian, C.; Wozniak, M.; Santos, L.; West, D.; et al. Response to anti-IL17 therapy in inflammatory disease is not strongly impacted by genetic background. Am. J. Hum. Genet. 2023, 110, 1817–1824. [Google Scholar] [CrossRef]
Figure 1.
A simplified diagram of the main psoriasis pathogenesis axis. Based on majority consensus in literature; the insulted keratinocyte releases self-DNA/RNA, forming a complex with the LL37 autoantigen, which then stimulates pDCs. IFNα released by pDCs alongside cytokines released from a variety of other cells activate mDCs to go on to stimulate the differentiation of naïve T cells into mature ThCs, which go on to propagate the psoriasis phenotype. Created with BioRender.com. [20,49].
Figure 1.
A simplified diagram of the main psoriasis pathogenesis axis. Based on majority consensus in literature; the insulted keratinocyte releases self-DNA/RNA, forming a complex with the LL37 autoantigen, which then stimulates pDCs. IFNα released by pDCs alongside cytokines released from a variety of other cells activate mDCs to go on to stimulate the differentiation of naïve T cells into mature ThCs, which go on to propagate the psoriasis phenotype. Created with BioRender.com. [20,49].
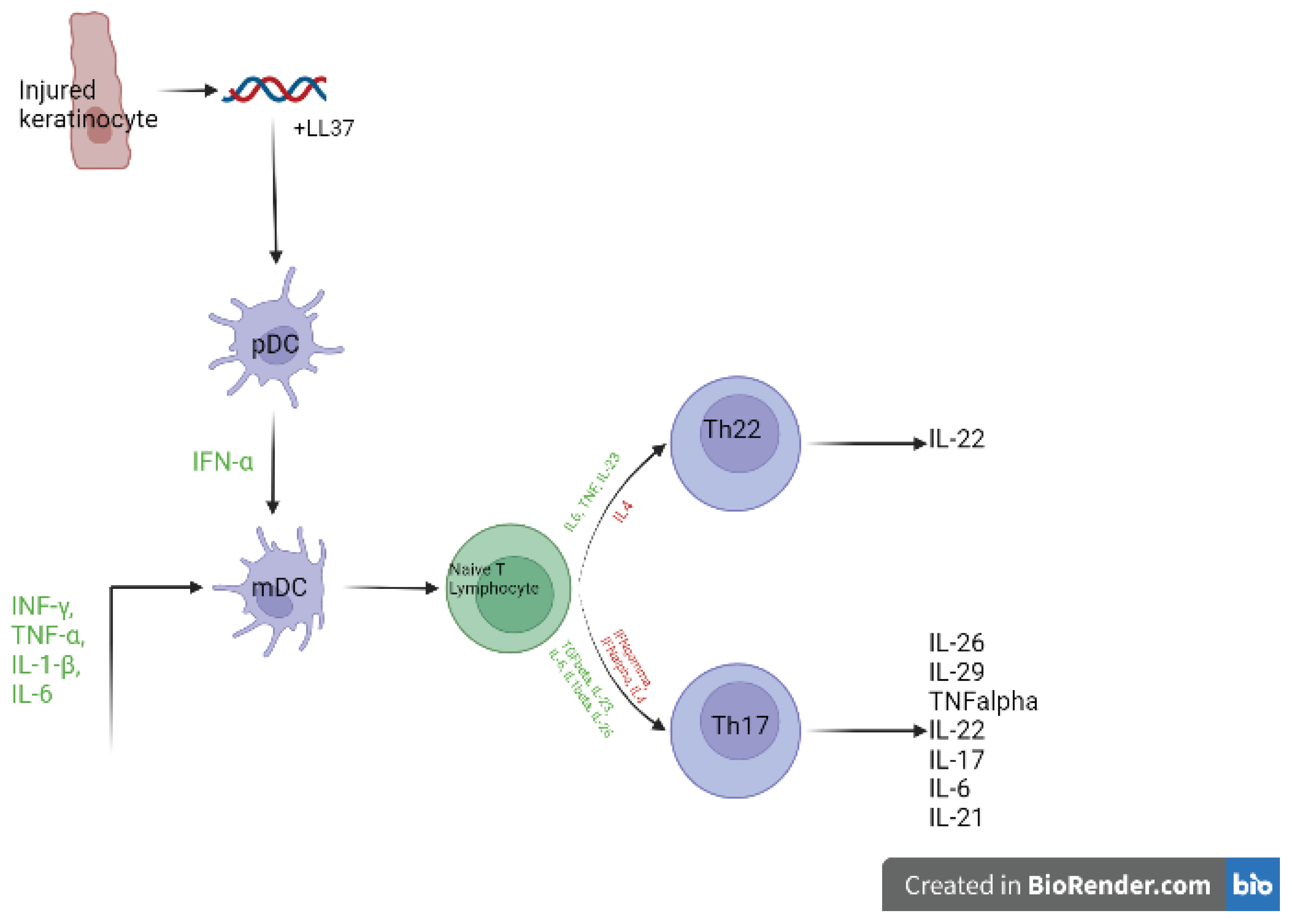
Table 1.
Non-MHC GWAS Loci Associated with Increased Risk of Psoriasis. The risk SNPs identified through various psoriasis GWAS and the potentially associated genes, with genes identified as relevant to the IL-23 pathway highlighted. Adaption of table from Ray-Jones et al. 2018 [12].
Table 1.
Non-MHC GWAS Loci Associated with Increased Risk of Psoriasis. The risk SNPs identified through various psoriasis GWAS and the potentially associated genes, with genes identified as relevant to the IL-23 pathway highlighted. Adaption of table from Ray-Jones et al. 2018 [12].
| Locus | Notable gene(s) in literature | Study Population | Index SNP | Index SNP annotation | P-value |
| 1p36.3 | MTHFR | CHN | rs2274976 | Missense: MTHFR | 2.33 x 10-10 |
| 1p36.23 | SLC45A1, TNFRSF9 | EUR | rs11121129 | Intergenic | 1.7 x 10-8 |
| 1p36 | IL-28RA | EUR | rs7552167 | 4.2kb 5’ of IL-28RA | 8.5 x 10-12 |
| CHN | rs4649203 | 5.5kb 5’ of IL-28RA | 9.74 x 10-11 | ||
| 1p36.11 | RUNX3 | EUR | rs7536201 | 1.5kb 5’ of RUNX3 | 2.3 x 10-12 |
| 1p36.11 | ZNF683 | CHN | rs10794532 | Missense: ZNF683 | 4.18 x 10-8 |
| 1p31.3 | IL-23R | EUR | rs9988642 | 441bp 3’ of IL-23R | 1.1 x 10-26 |
| CHN | chr1: 67,421,184 (build hg18) | Nonsynonymous: IL-23R | 1.94 x 10-11 | ||
| 1p31.3 | C1orf141 | CHN | rs72933970 | Missense: C1orf141 | 1.23 x 10-8 |
| 1p31.1 | FUBP1 | EUR | rs34517439 | Intronic: DNAJB4 | 4.43 × 10−9 |
| 1q21.3 | LCE3B, LCE3D | EUR | rs6677595 | 3.6kb 3’ of LCE3B | 2.1 x 10-33 |
| CHN | rs10888501 | 175bp 3’ of LCE3E | 6.48 x 10-13 | ||
| 1q22 | AIM2 | CHN | rs2276405 | Stop-gained: AIM2 | 3.22 x 10-9 |
| 1q24.3 | FASLG | EUR | rs12118303 | Intergenic | 3.02 × 10−10 |
| 1q31.1 | LRRC7 | EUR | rs10789285 | Intergenic | 1.43 x 10-8 |
| 1q31.3 | DENND1B | EUR | rs2477077 | Intronic: DENND1B | 3.05 x 10-8 (meta) |
| 1q32.1 | IKBKE | EUR | rs41298997 | Intronic: IKBKE | 2.37 × 10−8 |
| 2p16.1 | FLJ16341, REL | EUR | rs62149416 | Intronic: FLJ16341 | 1.8 x 10-17 |
| 2p15 | B3GNT2 | EUR | rs10865331 | Intergenic | 4.7 x 10-10 |
| 2q12.1 | IL1RL1 | CHN | rs1420101 | Intronic: IL1RL1 | 1.71 x 10-10 |
| 2q24.2 | KCNH7, IFIH1 | EUR | rs17716942 | Intronic: KCNH7 | 3.3 x 10-18 |
| CHN | rs13431841 | Intronic: IFIH1 | 2.96 x 10-9 | ||
| 3p24.3 | PLCL2 | EUR | rs4685408 | Intronic: PLCL2 | 8.58 x 10-9 |
| 3q11.2 | TP63 | EUR | rs28512356 | 400bp 3’ of TP63 | 4.31 x 10-8 |
| 3q12.3 | NF-ΚBIZ | EUR | rs7637230 | Intronic: RP11-221J22.1 | 2.07 x 10-9 |
| 3q13 | CASR | CHN | rs1042636 | Missense: CASR | 1.88 x 10-10 |
| 3q26.2-q27 | GPR160 | CHN | rs6444895 | Intronic: GPR160 | 1.44 x 10-12 |
| 4q24 | NF-ΚB1 | CHN | rs1020760 | Intronic: NF-ΚB1 | 2.19 x 10-8 |
| 5p13.1 | PTGER4, CARD6 | EUR | rs114934997 | Intergenic | 1.27 x 10-8 |
| 5q14 | ZFYVE16 | CHN | rs249038 | Missense: ZFYVE16 | 2.14 x 10-8 |
| 5q15 | ERAP1, LNPEP | EUR | rs27432 | Intronic: ERAP1 | 1.9 x 10-20 |
| CHN | rs27043 | Intronic: ERAP1 | 6.50 x 10-12 | ||
| 5q31 | IL13, IL4 | EUR | rs1295685 | 3’-UTR: IL13 | 3.4 x 10-10 |
| 5q33.1 | TNIP1 | EUR | rs2233278 | 5’-UTR: TNIP1 | 2.2 x 10-42 |
| CHN | rs10036748 | Intronic: TNIP1 | 4.26 x 10-9 | ||
| 5q33.3 | IL12B | EUR | rs12188300 | Intergenic | 3.2 x 10-53 |
| CHN | rs10076782 | Intronic: RNF145 | 4.11 x 10-11 | ||
| 5q33.3 | PTTG1 | CHN | rs2431697 | Intergenic | 1.11 x 10-8 |
| 6p25.3 | EXOC2, IRF4 | EUR | rs9504361 | Intronic: EXOC2 | 2.1 x 10-11 |
| 6p22.3 | CDKAL1 | EUR | rs4712528 | Intronic: CDKAL1 | 8.4 x 10-11 |
| 6q23.3 | TRAF3IP2 | EUR | rs33980500 | Missense: TRAF3IP2 | 4.2 x 10-45 |
| TNFAIP3 | EUR | rs582757 | Intronic: TNFAIP3 | 2.2 x 10-25 | |
| 6q25.3 | TAGAP | EUR | rs2451258 | Intergenic | 3.4 x 10-8 |
| 7p14.3 | CCDC129 | CHN | rs4141001 | Missense: CCDC129 | 1.84 x 10-11 |
| 7p14.1 | ELMO1 | EUR | rs2700987 | Intronic: ELMO1 | 4.3 x 10-9 |
| 8p23.2 | CSMD1 | CHN | rs10088247 | Intronic: CSMD1 | 4.54 x 10-9 |
| 9p21.1 | DDX58 | EUR | rs11795343 | Intronic: DDX58 | 8.4 x 10-11 |
| 9q31.2 | KLF4 | EUR | rs10979182 | Intergenic | 2.3 x 10-8 |
| 10q21.2 | ZNF365 | EUR | rs2944542 | Intronic: ZNF365 | 1.76 × 10−8 |
| 10q22.2 | CAMK2G, FUT11 | EUR | rs2675662 | Intronic: CAMK2G | 7.35 x 10-9 |
| 10q22.3 | ZMIZ1 | EUR | rs1250544 | Intronic: ZMIZ1 | 3.53 x 10-8 |
| 10q23.31 | PTEN, KLLN, SNORD74 | EUR | rs76959677 | Intergenic | 2.75 × 10−8 |
| 10q24.31 | CHUK | EUR | rs61871342 | Intronic: BLOC1S2 | 1.56 × 10−9 |
| 11p15.4 | ZNF143 | CHN | rs10743108 | Missense: ZNF143 | 1.70 x 10-8 |
| 11q13 | RPS6KA4, PRDX5 | EUR | rs694739 | 256bp 5’ of AP003774.1 | 3.71 x 10-9 |
| 11q13.1 | CFL1, FIBP, FOSL1 | EUR | rs118086960 | Intronic: CFL1 | 6.89 × 10−9 |
| 11q13.1 | AP5B1 | CHN | rs610037 | Synonymous: AP5B1 | 4.29 x 10-11 |
| 11q22.3 | ZC3H12C | EUR | rs4561177 | 1.7kb 5’ of ZC3H12C | 7.7 x 10-13 |
| 11q24.3 | ETS1 | EUR | rs3802826 | Intronic: ETS1 | 9.5 x 10-10 |
| 12p13.3 | CD27, LAG3 | CHN | rs758739 | Intronic: NCAPD2 | 4.08 x 10-8 |
| 12p13.2 | KLRK1, KLRC4 | EUR | rs11053802 | Intronic: KLRC1 | 4.17 × 10−9 |
| 12q13.3 | IL-23A, STAT2 | EUR | rs2066819 | Intronic: STAT2 | 5.4 x 10-17 |
| 12q24.12 | BRAP, MAPKAPK5 | EUR | rs11065979 | Intergenic | 1.67 × 10−8 |
| 12q24.31 | IL31 | EUR | rs11059675 | Intronic: LRRC43 | 1.50 × 10−8 |
| 13q12.11 | GJB2 | CHN | rs72474224 | Missense: GJB2 | 7.46 x 10-11 |
| 13q14.11 | COG6 | EUR | rs34394770 | Intronic: COG6 | 2.65 x 10-8 |
| 13q14.11 | LOC144817 | EUR | rs9533962 | Within LOC144817 | 1.93 x 10-8 |
| 13q32.3 | UBAC2, RN7SKP9 | EUR | rs9513593 | Intronic: UBAC2 | 3.60 × 10−8 |
| 14q13.2 | NF-ΚBIA | EUR | rs8016947 | Intronic: RP11-56B11.3 | 2.5 x 10-17 |
| 13q14.11 | LOC144817 | CHN | rs12884468 | Intergenic | 1.05 x 10-8 |
| 14q23.2 | SYNE2 | CHN | rs2781377 | Stop-gained: SYNE2 | 4.21 x 10-11 |
| 14q32.2 | RP11-61O1.1 | EUR | rs142903734 | Intronic: RP11-61O1.1 | 7.15 × 10−9 |
| 15q13.3 | KLF13 | EUR | rs28624578 | Intronic: KLF13 | 9.22 × 10−10 |
| 16p13.13 | PRM3, SOCS1 | EUR | rs367569 | 1.6kb 3’ of PRM3 | 4.9 x 10-8 |
| 16p11.2 | FBXL19, PRSS53 | EUR | rs12445568 | Intronic: STX1B | 1.2 x 10-16 |
| 17q11.2 | NOS2 | EUR | rs28998802 | Intronic: NOS2 | 3.3 x 10-16 |
| 17q12 | IKZF3 | CHN | rs10852936 | Intronic: ZPBP2 | 1.96 x 10-8 |
| 17q21.2 | PTRF, STAT3, STAT5A/B | EUR | rs963986 | Intronic: PTRF | 5.3 x 10-9 |
| 17q25.1 | TRIM47, TRIM65 | EUR | rs55823223 | Intronic: TRIM65 | 1.06 × 10−8 |
| 17q25.3 | CARD14 | EUR | rs11652075 | Missense: CARD14 | 3.4 x 10-8 |
| 17q21.2 | PTRF, STAT3, STAT5A/B | CHN | rs11652075 | Missense: CARD14 | 3.46 x 10-9 |
| 17q25.3 | TMC6 | CHN | rs12449858 | Missense: TMC6 | 2.28 x 10-8 |
| 18p11.21 | PTPN2 | EUR | rs559406 | Intronic: PTPN2 | 1.19 × 10−10 |
| 18q21.2 | POL1, STARD6, MBD2 | EUR | rs545979 | Intronic: POL1 | 3.5 x 10-10 |
| 18q22.1 | SERPINB8 | CHN | rs514315 | 3′ of SERPINB8 | 5.92 x 10-9 |
| 19p13.2 | TYK2 | EUR | rs34536443 | Missense: TYK2 | 9.1 x 10-31 |
| 19p13.2 | ILF3, CARM1 | EUR | rs892085 | Intronic: QTRT1 | 3 x 10-17 |
| 19q13.33 | FUT2 | EUR | rs492602 | Synonymous: FUT2 | 6.57 × 10−13 |
| 19q13.41 | ZNF816A | CHN | rs12459008 | Missense: ZNF816 | 2.25 x 10-9 |
| 20q13.13 | RNF114 | EUR | rs1056198 | Intronic: RNF114 | 1.5 x 10-14 |
| 21q22 | RUNX1 | EUR | rs8128234 | Intronic: RUNX1 | 3.74 x 10-8 |
| 21q22.11 | IFNGR2 | CHN | rs9808753 | Missense: IFNGR2 | 2.75 x 10-8 |
| 21q22.11 | SON | CHN | rs3174808 | Missense: SON | 1.15 x 10-8 |
| 22q11.21 | UBE2L3, YDJC | EUR | rs4821124 | 1kb 3’ of UBE2L3 | 3.8 x 10-8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



