
Submitted:
13 March 2024
Posted:
14 March 2024
You are already at the latest version
Abstract
The treatment of acute myeloid leukemia (AML) with adverse genetics remains unsatisfactory, with very low response rates to standard chemotherapy and shorter durations of remission commonly observed in these patients. The complex biology of AML with adverse genetics is continuously evolving. Herein, we discuss recent advances which have investigated the contribution of cell intrinsic mechanisms as well as of the immune system to myeloid leukemogenesis in this specific subset of AML. We focus on the biological rationales for combining targeted therapy and immunotherapy, which are currently being investigated in ongoing trials, and could hopefully ameliorate the poor outcomes for these patients
Keywords:
acute myeloid leukemi
; adverse genetics
; leukemogenesis
; targeted therapy
; immunotherapy
Background
The outcomes for AML patients with adverse genetics remain poor, with a median overall survival (OS) of less than one year. [1,2] Adverse risk or high-risk (HR) genetic AML encompasses several genetically defined entities accounts for approximately 50% of all adult AML cases.[3] HR-AML is more commonly characterized by a poor response to standard chemotherapy, very short period of remission, an increased rate of relapse even after allogeneic stem cell transplantation (allo-HCT).
Novel compounds more recently introduced in the clinic, such as FLT3 or BCL2 inhibitors, have only demonstrated a modest impact on disease course. [4] Currently, allo-HCT represents the sole potentially curative strategy for these patients, though survival rates rarely exceed 30–35%. [5,6,7]
In this review, we present the latest advancements in the understanding of HR-AML biology. We integrate insights from genomic analyses and from studies investigating the contribution of the immune system to myeloid leukemogenesis. Furthermore, we discuss the biological rationales behind the strategy of combining small molecules, which target specific genetic lesion(s), with immunotherapy. These combined treatment approaches are currently being investigated in ongoing clinical trials, holding promise for improving HR-AML patient outcomes.
Genetics of HR AML
HR AML represents an extremely complex subgroup of adult AML, characterized by a variety of well-defined cytogenetic and/or genetic lesions, which contribute to the aggressive course of the disease and its intrinsic resistance to standard chemotherapeutic approaches. In this section, we provide an overview of the current biological knowledge for each specific genetic entity of HR-AML, according to the European LeukemiaNet classification [1] (Table 1).
t(6;9)(p23.3;q34.1)DEK::NUP214. NUP214 is a nucleoporin that binds to the cytoplasmic side of the nuclear pore complex (NPC), that is critical for nucleo-cytoplasmic transport of proteins and mRNA. Defective nuclear export derived from DEK-NUP214 fusion induces the nuclear retention of transcription factors (TFs) that induce sustained HOX gene upregulation.[2] DEK is a chromatin-associated protein critical for the maintenance of chromatin stability.
t(v;11q23.3)KMT2A-rearranged. Acute leukemias carrying KMT2A (MLL) translocations represent 5-10% of acute leukemia in all age, and up to 70% of infantile leukemia.[8] KMT2A fusion supports leukemogenesis by recruiting the superelongation complex (SEC), the histone H3K79 methyltransferase DOT1L and menin (MEN1), to induce the overexpression of AML TFs such as HOXA9, MEIS1 and MEF2C. [9] KMT2A-rearranged leukemias are featured by promiscuous expression of lineage markers and a propensity for lineage switching. [10,11]
t(9;22)(q34.1;q11.2)BCR::ABL1 (BCR-ABL+). This category comprises a subset of de novo AML developed in patients without a history of chronic myeloid leukemia (CML) and lacking recurrent genetic aberrations affecting CEBPA or NPM1 genes, or cytogenetic alterations such as inv(16) or inv(3). Distinguishing BCR-ABL+ AML from a myeloid blast crisis of CML poses challenges. Unique to BCR-ABL+ AML are the loss of IKZF1 and CDKN2A, along with cryptic deletions in IGH and TRG genes, features not observed in myeloid blast crisis of CML. [12] AML blasts in this category often aberrantly express CD19, CD7 and TdT. Although BCR-ABL+ AML generally falls under the adverse-risk category, it should be noted that cases associated with inv(16) or NPM1 mutations may have favorable outcomes. [13,14,15]
t(8;16)(p11.2;p13.3)KAT6A::CREBBP. It is a rare subset, representing 0.2 to 0.4% of all AML cases. CREBBP alterations in de novo AML have been reported to be associated with poor prognosis. [16] KAT6A, also known as MOZ or MYST3, encodes the monocytic leukemia zinc finger protein, a histone acetyltransferase of the MYST family that regulates gene transcription by activating RUNX1 transcription factor complex. CREBBP plays a critical role in transcription regulation. Similar to KAT6A, CREBBP has an intrinsic histone acetyltransferase activity.
EVI1-rearranged.GATA2, MECOM(EVI1) AML is characterized by the reposition of a distal GATA2 enhancer that activates MECOM expression leading to GATA2 haploinsufficiency. About 20% of AML with inv(3)/t(3;3) harbor mutations in RUNX1, while around 25% exhibit mutations in IKZF1. Additionally, a subset of these AML cases presents with activating mutations in the RAS GTPase family member (NRAS or KRAS) or other signaling pathway proteins, such as PTPN11 and NF1, contributing to RAS signaling dysregulation and promoting AML cell proliferation. About 20% of patients have mutations in the polycomb protein ASXL1, and 30-60% has mutations in the spliceosomal machinery components, such as SF3B1 and U2AF1. TP53 mutations are found in approximately 25% of cases.[17] Other mutations, albeit less frequently observed, occur in DNMT3, TET2 and IDH1/2 genes.[18] EVI1r AML often presents with monolobated megakaryocytes, multilineage dysplasia and normal/elevated blood platelet counts. [19]
-5 or del(5q); -7; -17/abn(17p). These abnormalities are commonly observed in AML patients, previously treated with chemotherapy, including alkylating agents, platinum-based agents or antimetabolites. 5q deletion is typically large, involving ∼70 Mb of 5q14-q33 chromosome. This region includes haploinsufficent genes like RPS14 (ribosomal protein S14) and APC (adenomatous polyposis coli), microRNA genes (mir-145 and mir-146A) which are implicated in megakaryocytic dysplasia, as well as genes controlling hematopoietic stem cell expansion, such as EGR1 and CSNK1A1. [20] Monosomy 7, the most common autosomal monosomy in AML, and frequently seen in therapy-related AML.[20], can be also found in congenital diseases predisposing to myeloid neoplasms, such as those bearing germline GATA2 mutations, or affected by neurofibromatosis, and severe congenital neutropenia.[21] The tumor suppressor genes located in chromosome 7 are believed to act in a haploinsufficient manner, and include SAMD9/SAMD9L endosomal proteins, EZH2 histone modifying enzyme and MLL3, that is associated with Ras pathway mutations and TP53 inactivation.[21] 17p deletion or monosomy commonly involves the tumor suppressor gene p53 on band 17p13.1.
Complex karyotype (CK). CK is defined by the presence of ≥ 3 chromosomal abnormalities in the absence of specific recurring translocations or inversions included in the WHO classification, [22] such as t(8;21), inv(16) or t(16;16), t(9;11), t(v;11)(v;q23.3), t(6;9), inv(3) or t(3,3). [23] This subtype accounts for 10-12% of adult AML cases, with the most common chromosomal losses being 5q (80% of cases), 7q and 17p chromosomes. [24] More recently, CK AML has been proposed to be further subclassified into typical CK-defined by the presence of 5q, 7q abnormalities and/or 17p loss- and atypical CK, which lacks these specific chromosomal abnormalities. Typical CK AML, often associated with TP53 mutations (in 80% of cases), have very poor prognosis.[24] In contrast, [25]patients with atypical CK AML, who are generally younger, frequently have mutations in PHF6, FLT3-TKD, MED12 and NPM1, and tend to achieve a longer overall survival compared to those with typical CK AML. [24]
Monosomal karyotype (MK). MK is defined by the presence of ≥2 distinct autosomal monosomies or a single autosomal monosomy accompanied by structural abnormalities (deletions of -X or -Y are not considered monosomies).[26] MK AML occurs more frequently in therapy-related cases compared to de novo AML, and is closely associated with alterations in the TP53 gene, leading to significant chromosomal instability.[27] The most common chromosomal alterations include monosomy 7 (∼35%), monosomy 5 (∼22%) -17 (∼11%). [27]
Mutated RUNX1. RUNX1 mutations typically affect the Rnt Homology Domain (RHD) or the Transactivation Domain (TAD) of the gene (located at 21q22), and encodes the alpha subunit of the Core Binding Factor (CBF). Given the association of RUNX1 mutations with autosomal dominant thrombocytopenia, it is advisable to screen for germline mutations among family members to rule out this hereditary condition. RUNX1-mutated AML is predominantly observed in older male patients. It may be preceded by Fanconi anemia or congenital neutropenia. A prior history of myelodysplastic syndrome or prior exposure to radiation can be present. There is frequent association with MLL-PTD or ASXL1 mutations,[28,29] indicating a complex genetic landscape that influences disease progression and treatment response.
Mutated EZH2. Enhancer of Zeste Homolog 2 (EZH2) is a key component of the polycomb group (PcG) proteins, which are crucial for gene silencing via histone modifications. [30] EZH2 composed the regulatory hub of PRC2, that functions as a histone H3 lysine 27 methyltransferase. [30] Unlike its role in clonal haematopoiesis (CH), where EZH2 mutations are not typically implicated, these mutations are more commonly associated with the development of overt leukemia. [31] EZH2 mutations could be initiating event or occur later on during leukemogenesis to drive clonal expansions. [31] The prevalence of EZH2 mutations in de novo AML ranges from 1-4% of patients. [32,33,34] The EZH2 gene is located at 7q36.1, a genomic region that is often deleted in AML (-7 or del7q), and associated with an adverse prognosis. In AML, EZH2 frequently undergoes nonsense and frameshift mutations leading to its inactivation. Notably, mutations in the Serine and Arginine Rich Splicing Factor 2 (SRSF2), which is a high-risk genomic entity,[1] could affect EZH2 expression by modifying sequence-specific RNA binding activity of EZH2. This in turn alters the recognition of splicing enhancer motifs, leading to aberrant EZH2 splicing and nonsense mediated decay and decreased the expression of EZH2, thereby influencing H3K27me3 levels. Furthermore, mutations in ASXL1 gene, another polycomb-related protein mutated in HR-AML [33] also decrease H3K27me3 levels by impairing PRC2 recruitment. This mechanisms contributes to the activation of HOXA9-driven leukemogenesis.[35] In myeloid neoplasms, EZH2 mutations tend to be mutually exclusive with SRSF2 and U2AF1 mutations,[36] while it is more frequently co-mutated with ASXL1 and TET2. [36,37]
Mutated ASXL1. Additional sex combs-like 1 (ASXL1) is a critical epigenetic modifier, whose mutations are commonly identified in CH. [38,39,40] In murine models, ASXL1 knockdown leads to a myelodysplastic-like phenotype, primarily due to the loss of interaction with PRC2. [35,41,42,43] In myeloid neoplasms, the majority of ASXL1 mutations consist of frameshift or nonsense mutations at the exon 12. These mutations are mutually exclusive with DNMT3A, FLT3-ITD, and NPM1 mutations, while ASXL1 mutations frequently co-occur with mutations in DNA methylation genes (such as TET2, IDH1-2), spliceosomes (U2AF1, SRSF2), transcription factors (CEBPA, RUNX1, GATA2), signal transducers (NRAS, JAK2, STAG2).[44] In AML, the frequency of ASXL1 mutations is about 5-10%, [33,45] with a higher prevalence in older patients and those with secondary AML. RUNX1 is the most frequent co-mutated gene and cooperates with mutant ASXL1 to support myeloid leukemogenesis in vivo.[46]
Mutated BCOR. The BCL6 corepressor (BCOR) is a tumor suppressor gene, that is dysfunctional in lymphoid and myeloid tumors. [47] BCOR is a critical component of the noncanonical PRC1.1, that is recruited to specific chromatin regions in a context specific manner.[47] Mutations of BCOR are detected in about 5% of adult de novo AML and 4% of AML with myelodysplasia-related changes. [33,48] Frequency of BCOR mutations is even higher in secondary AML. [49] Most commonly, patients with BCOR-mutated AML carries a normal karyotype (NK). In AML with NK, about 45% of BCOR-mutated AML have co-mutations with DNMT3A and/or RUNX1, while are mutually exclusive with NPM1 and FLT3 mutations. [50,51] Patients with BCOR mutations usually have activated RAS signaling, due to high rate of NRAS and KRAS mutations. [47] In vivo, BCOR leads to overt acute leukemia in the presence of co-mutations, such as DNMT3A[51] or RAS mutations. [52]
Spliceosome mutations (SRSF2, SF3B1, U2AF1, ZRSR2). The most commonly mutated genes in this category are splicing factor 3B subunit 1 (SF3B1), serine and arginine rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1) and zinc finger, CCCH type, RNA-binding motif and serine and arginine rich 2 (ZRSR2),[33] which are implicated in the early assembly of the spliceosome machinery. [53] Mutations in splicing factors (SFmut) are predominantly early events in leukemogenesis. [54] Mutations in splicing factors accounts for about 18% of adult AML, [33] are more frequent in older age, and commonly associate with multilineage dysplasia. [55] While mutations of SF3B1, SRSF2 and U2AF1 are gain-of-function, determining a change of amino acid residues,[56] mutations of ZRSR2 are inactivating nonsense or frameshift. [56] Mutations in SF are always heterozygous and mutually exclusive between each other. [56]
However, pattern of co-mutations between STAG2, RUNX1, SRSF2 and ASXL1 (SRSA genes) [57] or between SRSF2 and IDH2 [56] have been described in human AML. In mice, SF3B1, U2AF1 and SRSF2 mutations cause aberrant hematopoiesis and the acquisition of myelodysplastic-like phenotypes. [58,59,60,61] Mechanisms of splicing factors dysregulation in myeloid leukemogenesis have been extensively reviewed. [62] Briefly, several studies have analyzed the impact of mutations of specific splicing gene and implication for leukemogenesis: i) Mutations in SRSF2 and U2AF1 yield alternative exon usage; ii) ZRSR2 mutant induces the retention of minor introns (U12-type); [63] and iii) SF3B1 mutant instigates the usage of alternative branch points to cause an alternative 3′ splice site. [64,65]SF mutations induce mis-splicing of hematopoietic regulators, such as EZH2 in SRSF2-mutated MDS. [58]
Mutated STAG2 (cohesin complex). Mutations in the cohesin subunit SA-2 (STAG2) define AML with myelodysplasia-related gene mutations irrespective of prior MDS [1] and are considered a marker of poor prognosis. STAG2, together with double-strand-break repair rad21 homologue (RAD21), and structural maintenance of chromosomes (SMC1A and SMC3) form the core of the cohesion complex, that surrounds sister chromatids during replication, and support the transition from metaphase to anaphase. [66] The roles of cohesin mutations in leukemogenesis are multiple, as they can induce aneuploidy through mis-segregation of sister chromatids, or remodel 3D chromosome topology and chromatin interactions. [66] In vivo, mutated cohesion subunits induce the acquisition of a pre-leukemic phenotype, with altered erythroid and myeloid lineages differentiation. Mutations in the cohesion genes ranges between 6-13% in AML [67,68] are mutually exclusive, and can be accompanied by NK or CK. Most STAG2 mutations are nonsense or frameshift, leading to protein truncation or loss-of-function. [63] STAG2 mutations are often, if not always, associated with RUNX1, SRSF2 and ASXL1 mutations.[63] Although STAG2 mutations classify within the adverse-risk category, their prognostic significance appears to be linked to the presence of other co-mutations. When multivariate analysis are adjusted for mutation in BCOR, ASXL1 and RUNX1 - which are more commonly found in STAG2-mutated AML compared to other subsets- STAG2 mutations lose their independent prognostic impact. Intriguingly, mutated STAG2 significantly increases the sensitivity of AML cells to poly ADP-ribose polymerase (PARP), such as talazoparib. [69,70] This suggests that the presence of STAG2 mutations could potentially be exploited to tailor more effective therapeutic strategies in this setting.
Mutated TP53. The majority of TP53 mutations are missense, with hotspots in arginine residues, though other mutational events have been reported, including insertions, deletions and frameshift mutations. More frequently, the mutation occurs in the DNA binding domain, with loss of function of p53 tumor suppressor, despite some mutations can lead to gain-of-function through the binding of mutant p53 to other tumor suppressors such as p63 and p73. [71] The frequency of TP53 mutations in de novo AML ranges from 5-10% increasing to approximately 30% in cases of therapy-related AML and AML with complex cytogenetics. TP53 mutations are particularly prevalent in AML cases that exhibit CK, chromotripsis or a monosomal karyotype. [72] Interstingly, TP53 mutations are less commonly found with mutations in DNMT3A, TET2 and IDH1-2. [72] Moreover, the variant allele frequency of TP53 appears to be directly correlated with the level of cytogenetic complexity and inversely correlated with overall survival in AML patients. [73]
Immune Landscapes of AML with Adverse Genetics
HR-AML is distinguished by elevated inflammation (as indicated by a high iScore), greater clonal diversity, and a higher immunogenic potential. [74] AML harboring TP53, RUNX1, ASXL1 and RAS mutations, found in the adverse-risk category, exhibits a higher immune effector dysfunction (IED172) score, and an IFNγ signature, the latter being associated with a positive response to azacytidine (AZA)+pembrolizumab. [75]AML with mutated TP53 is characterized by enrichment for gene programs related to T cell lineage commitment, positive T cell selection and T cell homeostasis, indicating a T-cell rich environment, as well as for an IFNγ dominant tumor microenvironment (TME). [76] TP53-mutated AML is also enriched for tumor inflammation signature (TIS), as well as characterized by the upregulation of immune checkpoints as PD-L1, TIGIT and LAG3 and markers of immune senescence. [77] Interestingly, PD-L1 upregulation is mostly restricted to HSCs in TP53 mutated AML, while T cell immunity is featured by low levels of PD-1 on CD8+ cytotoxic T cells and by an expansion of ICOShi/PD1- Tregs. [78] Further AML with higher number of mutations or HR-AML are more infiltrated by immune cells and have higher expression of PD-L1, FoxP3, GzmB, PTEN and BCL2 genes, as well as of gene networks lined to immune-exhaustion.[76] Importantly, patients with immune-infiltrated AML and adverse ELN characteristics derive significant benefit from allo-HCT.[76] Cytolytic score (geometric mean of GZMA, GZMH, GZMM, PRF1, and GNLY) correlates with TP53 mutations and deletion of chromosome 5, in AML. [79] Analysis of the Hemap AML and BeatAML datasets, have shown that cases with high cytolytic score are characterized by an MDS-like phenotype with complex cytogenetics and history of MDS. [79] Cytolytic score correlated with diagnosis of AML with myelodysplasia-related changes, suggesting a link between an MDS-like/sAML subtype and an increased cytolytic infiltration. The MDS-like subtype has been associated with RUNX1, TP53, U2AF1 and SRSF2 mutations. Leukemic blasts from MDS-like AML more frequently are classified as HSC or progenitor-like cells, such as multipotent progenitors, megakaryocyte-erythroid progenitors, or granulocyte-monocyte progenitors. Further, AML with a higher cytolytic score have a higher infiltration of NK and CD8+ T cells, the latter biased toward a cytotoxic and effector-memory phenotype.[79] These results suggest that leukemia cell state of differentiation may influence the composition of the bone marrow microenvironment as well as the interactions between immune cells. MDS-like AML blasts have higher expression of HLA-II, LGALS9 and TGFB1, while T and NK cells display elevated levels of their cognate receptors LAG3, HAVCR2 and TGFBR3, and secrete more IFNγ, compared to non MDS-like AML.[79] MDS-like AML more frequently express CD274 and ARG1 inhibitory genes and their corresponding receptors. [79]
Rationales to Combine Targeted Therapy to Immunotherapy in AML with Adverse Genetics
While immunotherapy, particularly immune checkpoint blockade (ICB), offers a promising strategy to stimulate the immune system’s natural ability to fight cancer, its effectiveness as a monotherapy in AML is limited, benefiting only a subset of patients. [80] This limitation underscores the necessity for a combination strategy that not only targets the specific genetic abnormalities driving the leukemogenesis but also addresses other aspects of the disease, such as the differentiation state of leukemia cells, their metabolic pathways, and the influence of the bone marrow microenvironment on tumor growth and immune evasion. It is possible to achieve a more robust and durable antitumor response by integrating targeted therapies that directly inhibit the oncogenic drivers or modulate the leukemia cell phenotype and metabolism with immunotherapies that enhance the immune system’s capacity to detect and destroy cancer cells. These combinatorial approaches aim to dismantle the protective barriers erected by the tumor against immune surveillance and to correct the dysfunctional immune response, thereby offering a potent strategy to treat HR-AML. Inizio moduloFine modulo
Exploiting BCL2 Inhibition for Innate and Adaptive Immune Reactivation
Combination of azacytidine (AZA) with venetoclax (VEN) has achieved complete response (CR)/CR with incomplete count recovery (CRi) rates of 70% in patients with adverse-risk cytogenetic AML without TP53 mutations, as well as durable remission (18.4 months) and improved OS (23.4 months). [81] Unfortunately, these results cannot be extended to AML cases with TP53 mutation, where the response rate and overall prognosis remain poor and comparable to the historical results with hypomethylating agents (HMA). Other high-risk genotypes that are sensitive to BCL2 inhibition are those harboring ASXL1 [82]and RUNX1 mutations. [83] Indeed, hematopoietic stem-/progenitor cells from patients with ASXL1-mutated AML have a higher expression of BCL2, [84] and relapsed-refractory ASXL1-mutated AML treated with HMA and VEN had improved CR/CRi rates in a retrospective study.[82] Recent evidences have proven that, beyond direct anticancer effects, BCL2 inhibition is linked too broader immunomodulatory functions: 1) BCL2 inhibition activates dendritic cells to enhance antitumor immunity and sensitize tumors to anti-PD(L)1 immunotherapy (Figure 1); [85] ii) the Bcl2-inhibitor, VEN, has shown to increase the effector activity of antileukemic T cells without inducing T cell apoptosis (Figure 1), through reactive oxygen species release, against AML in vitro and in vivo; [86] iii) VEN can augment the antitumor efficacy of ICB, as it increase the frequency of PD1+ effector-memory T cells in mouse tumor models. [87]
Targeting TP53-Dependent or Independent Mechanisms of Apoptosis with APR-246/Eprenetapopt
APR-246/eprenetapopt is a small molecule that targets TP53 mutated cancers[88,89] which has shown promising results against TP53-mutated MDS and AML. [90,91,92] APR-246 reactivates mutant p53 transcription, by facilitating its binding to DNA sequences, eventually inducing apoptosis.[89] APR-246 can also cause tumor cell death in p53-independent mechanisms, as for instance by impairing the balance between glutathione (GSH) and reactive oxygen species. [93,94] More recently, using AML cell lines and leukemia xenografts, it has been shown that APR-246 depletes intracellular GSH and induces lipid peroxide production, eventually leading to induction of ferroptosis.[95] Ferroptosis is a programmed cell death induced by iron-dependent lipid peroxidation.[96] Importantly, chemotherapy-resistant tumor cells can be instead greatly sensitive to ferroptosis, [97] as it might be the case of HR-AML. Ferroptosis may exert a double-edge function in the tumor microenvironment, by activating or suppressing immunity. Thus, searching for cancer-specific correlations between ferroptosis induction and the microenvironmental dependence on immunostimulatory or immunoinhibitory checkpoints is key to designing rational combinatorial approaches. In this regard, it has been reported that i) ferroptosis may dampen immune tolerance by inducing the death of glutathione peroxidase (GPX4)-deficient Tregs trough CD28-costimulation;[98] GPX4 is the key regulator of ferroptosis, since it interrupts the lipid peroxidation chain reaction;[99] ii) CTLA4 expression is higher in tumors with higher ferroptotic scores (Figure 1); [97,100] iii) ferroptosis can inhibit tumor immune tolerance by recruiting the ATP-P2X7-CD86 axis; [97]iv) immunotherapy-activated CD8+ T cells enhance ferroptosis-specific lipid peroxidation in tumor cells contributing to cancer immunotherapy efficacy;[101] v) early ferroptotic cells undergo immunogenic cell death, associated with the release of damage-associated molecular patterns (DAMPs) and an enhanced maturation of dendritic cell.[102] Even though experimental insights are currently lacking in AML models, the combination of ferroptotic inducing agents as APR-246/eprenetapopt, which is promising in treating TP53-mutated AML [91,92]may benefit from combination with ICB, as anti-CTLA4 or anti-PD(L)1 (Figure 1).
Targeting CD47 Phagocytic Immune Checkpoint in Adverse Risk AML
CD47 plays a crucial role in the evasion of phagocvtosis by AML cells[103]. Its overexpression is associated with a poorer prognosis. [104] Preclinical evidences have found that targeting CD47 with the humanized anti-CD47 antibody magrolimab might represent an effective strategy to treat AML.[105] Magrolimab is a first-in-class investigational monoclonal antibody against CD47 and macrophage checkpoint inhibitor that interferes with the recognition of CD47 by the SIRPα receptor on macrophages, thus blocking the “don’t eat me” signal used by cancer cells to evade phagocytosis (Figure 1). Several clinical trials are currently ongoing to search for AML patients who could benefit more from anti-CD47/SIRPa immunotherapy. Recent findings also suggest that CD47 expression in AML is genotype-dependent, with higher antigenic density observed in cases with CBFB/MYH11 rearrangements or NPM1 mutations. Conversely, AML with adverse risk genetics, such as MLL-rearranged AML, shows less consistent CD47 expression, with some cases nearly negative for CD47 on leukemic blasts. These findings underscore the potential of personalized approaches that might combine CD47-targeting therapies with agents that can increase CD47 expression or enhance “eat me” signals, such as HMA.[106]
Targeting Poly(ADP-ribose) Polymerase in STAG2-Mutated AML
AML with mutated STAG2 appears more sensitive to PARP inhibitors which inhibit the DNA-damage response (DDR), thereby increasing the neoantigen load and mutational burden. PARP inhibitors can generate tumor-derived double-strand DNA in the cytoplasm, that is sensed by cytosolic DNA sensor cyclic GMP-AMP synthase, thus activating the stimulator of interferon (IFN) genes (STING) signaling pathway.[107] STING activation, induces the upregulation of type I IFNs which promote systemic immune response. PARP inhibitors can reprogram the tumor immune microenvironment by sustaining a Th1 immune response and can upregulate PD-L1 expression through GSK3β inactivation [107] (Figure 1). Of note, cohesin (STAG2)-mutated cancers have been reported to display strong activation of IFN and NF-kB expression signatures, along with PD-L1 upregulation, [108] thus providing another rationale for adding anti-PD(L)1 immunotherapy in STAG2-mutated AML. In advanced solid tumors, the anti-PD-L1 avelumab has been recently combined to talazoparib with evidence of better responses in BRCA-altered tumors.[109] Given that cohesin directly regulates the DNA damage checkpoint activation and repair pathways and that tumors deficient in DNA damage response achieve durable benefit from ICB, [107] STAG2-mutated AML might represent a promising subset for immunotherapy with ICB.
Splice-Site Creating Mutations and Sensitivity to Immune Checkpoint Inhibition
Tumors harboring splice-site creating mutations (SCMs) generate more neoepitopes than non-synonymous mutations and possess a higher expression of PD-L1 (compared to tumors without SCMs).[110] This characteristic is of importance considering that an augmented generation of neoantigens can lead to enhanced efficacy of ICB in tumors with low immunogenicity,[111] such as AML. Further reinforcing this evidence, recent bioinformatic analyses have identified that a specific set of splicing mutations correlates with poor prognosis, increased infiltration by myeloid cells with suppressive phenotypes, and elevated expression of immune checkpoints in the leukemic microenvironment. These preliminary observations suggest that AML harboring SCMs could be particularly susceptible to ICB. [112]
Current Treatment Strategies for AML with Adverse Genetics
Based on the recent ELN guidelines, [1] the eligibility for standard intensive chemotherapy depends primarily on the fitness of the patient, based on age and comorbidities.[1] Fit patients, with HR genetics and no targetable lesions are mainly treated with standard regimen based on antracyclines and cytosine arabinoside. These patients , especially with TP53 mutations[113] could not benefit from the addition of the CD33 inhibitor gemtuzumab[114] neither from the use of encapsulated anthracycline-AraC molecules (CPX 351). For patients who respond to induction chemotherapy, allo-HCT remains the only potentially curative treatment because of the immunological effect of the graft versus leukemia [115] and subsequent post-HCT immunomodulatory treatments such as donor lymphocyte infusions or specific drugs could be beneficial in this high risk population. However, even if recent improvements in allo-HCT platforms appear encouraging, [116]outcomes remain unsatisfactory especially in TP53 mutated AML, with a OS of less than 30% at 2 years.[117]
Venetoclax Plus Azacytidine
For patients unfit for intensive chemotherapy, VEN + AZA are now considered the standard front line treatment based on the results of the Viale A trial. [118] Of note, for patients with adverse risk genetic mutations, given the poor prognosis associated with intensive chemotherapy, there has been interest for less intensive targeted therapeutic approaches.
Recently, Pollyea et al. [81]analyzed outcomes of 127 AML patients with HR genetics treated with AZA-VEN in front line treatment compared to 56 patients treated with AZA alone. The combination of AZA-VEN in patients with adverse genetics, allowed achieving complete remission rate in 70% of patients versus 30% of AZA alone, with a median OS of 23 months versus 11.3 months, respectively. Importantly, outcomes of patients treated with AZA-VEN were comparable with similarly treated patients with intermediate-risk cytogenetics. However, for patients with Tp53 mutation, even if CR was achieved in 41% with AZA-VEN versus 17% with AZA alone, no benefit was observed in OS (5.2 months versus 4.9 months).
The use of AZA-VEN is of interest also in the specific context of several adverse genetic mutations. In particular, a retrospective study, conducted by Aldoss at al. [82] reported outcomes on 90 relapsed refractory AML treated with AZA-VEN. The presence of ASXL1 mutation or TET2 was associated with better response. Furthermore, the association of ASXL1 with a better response to AZA-VEN was recently confirmed in the setting of MDS. [119]
However, a more recent study conducted by Cherry et al. [83]which retrospectively compared patients with newly diagnosed AML who received AZA-VEN (n = 143) versus intensive chemotherapy (n = 149) did not confirm the better results for ASX L1 mutations, but showed that RUNX 1 mutations could benefit from the combination of AZA-VEN as first line treatment.
The mutational testing pre-treatment will be more and more important in the treatment planning, but more data are needed to choice the best treatment in HR AML. Novel treatment combinations are needed to improve remission rates, and also recent guidelines [1,120]reflect the need of novel treatment approaches, including combination of target and immunomodulatory agents.
Promising Targeted Approaches for the Treatment of AML with Adverse Genetics
Menin inhibitors are compounds that disrupt the interaction between the scaffolding protein menin and the methyltransferase KMT2A. Among these inhibitors, Revumenib (SNDX-5613) stands out as one of the most prominent, while others like JNJ-75276617 and KO539 show considerable promise in ongoing development efforts. Revumenib is recognized for its potency and selectivity as a small molecule that effectively disrupts the interaction between menin—a crucial scaffold protein—and histone-lysine N-methyltransferase 2A, encoded by the KMT2A gene. Together, these proteins regulate gene expression through epigenetic mechanisms. Certain genetic alterations, such as KMT2A rearrangement and NPM1 mutation, can disrupt proper regulation pf epigenetic programs, leading to an aberrant proliferation of leukemia cells. Menin inhibitors like Revumenib bind to menin, effectively halting this aberrant process and restoring normal blood cell production. More recent milestones include Revumenib’s Orphan Drug Designation from both the FDA and the European Commission for treating AML. Additionally, it has received Fast Track designation from the FDA for treating relapsed/refractory acute leukemias in both adult and pediatric patients who harbor KMT2A rearrangment or NPM1 mutation. These designations underscore the urgent need for innovative treatments in these specific patient populations and emphasize Revumenib’s potential as a promising therapeutic option in the management of AML.
Another interesting targeted approach includes the use of anti-CD123 directed therapies. CD123 is a subunit of the interleukin 3 (IL3) receptor expressed on the surface of blasts in most AML cases, particularly in poor-risk genetic subgroups. CD123 expression is associated with high cell count at diagnosis and poor prognosis. Tagraxofusp (SL-401) is a recombinant protein targeting CD123 and is currently approved as monotherapy for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN). Additionally, Pivekimab Sunirine (PVEK, IMGN632) is an antibody-drug conjugate (ADC) consisting of a high-affinity CD123 antibody, a cleavable linker, and an indolinobenzodiazepine pseudodimer (IGN) payload. Flotetuzumab (MGD006) is a bispecific antibody engineered to bind CD3 and CD123 on AML cells. Both PVEK and flotetuzumab are being investigated as monotherapies and in combination therapies for AML. These agents hold promise in targeting CD123-expressing AML cells and may offer new treatment options for patients with this challenging disease.
Novel Investigational Strategies Combining Immunotherapy and Target Therapy in HR Genetic Risk AML
The clinical trials described in this section are summarized in Table 2.
Apr-246 Based Combinations
The first clinical trial which investigated the combination of APR-246 and AZA is a US phase II trial [92,121] (NCT03072043) in which there were enrolled 55 patients with TP53 mutation (40 MDS and 11 AML) with a median age of 66 years. The overall response rate was 71% with a CR rate of 44% and 38% achieved MRD negativity assessed by NGS. The median duration of CR was 7.3 months, with a median follow up of 10.5 months. The median OS was 10.8 months. A French phase II trial [92] (NCT03588078) enrolled 52 patients (34 MDS and 18 AML) with a median age of 74 years. The overall response rate was 52% with a CR rate of 37% with 30% of patients with MRD negativity. The median duration of CR was 11.7months, with a median follow up of 9.7 months. The median OS was 12.1 months. No additional hematological toxicity was reported compared to AZA alone. However neurological effects including ataxia, acute confusion, facial dizziness and paresthesias were reported in 40% of patients. Based on these results a phase III randomized clinical trial was conducted to compare AZA alone + AZA + APR-246 in MDS (NCT03745716). The results have failed to demonstrate the superiority of the combination compared to AZA alone. However, more recently, a phase I trial (NCT04214860) have shown that the addition of APR-246 to VEN and AZA appears encouraging in treating TP53 mutated AML with a well-tolerated toxicity profile and promising efficacy by achieving an overall response of 64% (25/49) and a CR of 38% (15/39). [122] Furthermore, APR-246 has been investigated in the post HCT setting (NCT03931291).[91] 33 Patients (14 AML and 19 MDS) with mTP53 received post HCT maintainance treatment with up to 12 cycles of eprenetapopt 3.7 g once daily intravenously on days 1-4 and AZA 36 mg/m2 once daily intravenously/subcutaneously on days 1-5 in 28-day cycles. The median number of eprenetapopt cycles was 7 (range, 1-12). With a median follow-up of 14.5 months, the median RFS was 12.5 months and the 1-year RFS probability was 59.9%. With a median follow-up of 17.0 months, the OS was 20.6 months and the 1-year OS probability was 78.8% Acute and chronic (all grade) graft-versus-host disease and adverse events were reported in 12% (n = 4) and 33% (n = 11) of patients, respectively.
Innate and Adaptive Immune Checkpoint Inhibition in AML with Adverse Genetics
Magrolimab (anti-CD47) Daver et al. recently published the results of a phase Ib trial (NCT03248479)investigating the safety and efficacy of magrolimab in association with AZA in previously untreated AML ineligible for chemotherapy. [123] 87 patients were enrolled: 82.8% had TP53 mutations. 57 (79.2%) of TP53-mutant patients had adverse-risk cytogenetics. Patients received a median of 4 cycles of treatment. Each cycle consisted in infusion of magrolimab as an initial dose (1 mg/kg, days 1 and 4), followed by 15 mg/kg once on day 8 and 30 mg/kg once weekly or every 2 weeks as maintenance. Azacitidine 75 mg/m2 was administered intravenously/subcutaneously once daily on days 1-7 of each 28-day cycle. The most common treatment-emergent adverse events included constipation, nausea and diarrhea and anemia. 32.2% of patients achieved CR, including 31.9% patients with TP53 mutations. The median OS in TP53-mutant and wild-type patients were 9.8 months and 18.9 months, respectively. Based on these results, new phase III randomized clinical trial are recruiting frontline patients. ENHANCE-2 (NCT04778397) is invetigating the role of Magrolimab plus AZA Versus Physician’s Choice of VEN-AZA or intensive Chemotherapy in Patients With TP53 AML in previously untreated AML; ENHANCE-3 (NCT05079230) the role of Magrolimab Versus Placebo in Combination With Venetoclax and Azacitidine in previously untreated patients with acute myeloid leukemia ineligible for intensive chemotherapy.
Sabatolimab (mb5-453). T-cell immounoglobulin domain and mucin domain-3 (TIM-3) is a T cell immune checkpoint that regulate adaptive and innate immunity and is aberrantly expressed on the surface of leukemic cells and higher levels of expression are associated with poor prognosis [124]Sabatolimab, a novel anti TIM3 monoclonal antibody exerts the antileukemic activity by a direct targeting of TIM-3 on the blast surface, promote antibody dependent phagocytosis and promote the block of TIM-3-GALAECTIN-9 interaction preventing leukemia stem cell renewal [125]Sabatolimab has been investigated in association with HMA in patients with HR-MDS and AML unfit for intensive chemotherapy. Patients with AML were 48. ORR was 40%, of these 30% achieved CR. The median duration of response was 12.6 months with a PFS of 27.9%. Patients with at least one genetic adverse risk mutation the ORR was 53.8% with a median duration of response of 12.6 months. [126]Based on these results the STIMULUS clinical trial program was started in which randomized phase II and phase III clinical trial are investigating multiple combinations sabatolimab based in AML, high risk MDS and chronic myelomonocytic leukemia. STIMULUS-AML1 (NCT04150029) is an ongoing Phase II, single-arm study of sabatolimab + AZA + VEN in adult patients with AML inelegibe for intensive chemotherapy. [127]
Nivolumab. Nivolumab is an antibody that binds to PD-1 and blocks signaling mediated by PD-1/PD-L1 interactions. Also, nivolumab blocks signaling mediated by PD-1/PD-L2 interactions. Nivolumab is used to treat various cancers such as melanoma, Hodgkin’s lymphoma, and nonsmall-cell lung cancer (NSCLC).A phase II trial (NCT02397720) assessed the efficacy and safety of nivolumab in combination with AZA in 70 patients with relapsed refractory AML. ORR was 33% of which 22% achieved CR with a median OS of 6.3 months. Response were higher in patients not pretreated with HMA (ORR: 52%) [128] and ASXL1 mutations were associated with improved ORR and OS. Upregulation Of CTLA-4 expression on T cells was observed in patients which doesn’t achieve remission, suggesting CTLA-4 overespression could be a potential mechanism of resistance of PD1 blockade[128]So a subsequent cohort was added (36 patients) and treated with Ipilimumab (antiCTLA-4) + AZA+ nivolumab with the aim to enhance T cell response. ORR was 46%, of which 36% achieved CR. The median OS was 10.5 months comparing better with AZA-NIVOLUMAB. Two new ongloing clinical trial are further investigating the role of these combinations in post transpant setting for patients with RR AML (NCT3600155) and MDS (NCT02530463). Furthermore, Nivolumab was studied in frontline setting combined with idarubicine and cytarabine. There were enrolled 42 patients with AML , 50% had adverse ELN genetic risk and 18% TP53 mutations. [129] The combination lead to an ORR of 80% including 64% CR and 14% CRi/CRp with a median OS of the whole cohort was 18.5 months and for those who proceed to allo-HCT was 25 months. Finally, a phase II pilot study assessed the role of nivolumab as maintainance therapy in high risk AML showing a modest ability to extend remissions providing no support to use as single agent in post HCT setting. [130]
Pembrolizumab. Pembrolizumab is a monoclonal antibody targeting the anti–programmed death-1 (anti-PD1) protein found on T cells. The combination of pembrolizumab + AZA was studied in a multicentric phase II study [131]in 37 patients with newly diagnosed and relapsed refractory AML aged >65y 29 of 37 patients were evaluable for response with ORR of 55% (CR/CRI: 14%, PR: 4%, hematological improvement: 14%, stable 24%) with median OS of 10.8 months. 17 of 22 patients with newly diagnosed AML were evaluable for response with 0RR of 94% (CR/Cri: 47%) with a median OS of 13 months. [131]The combination was well tolerated without major toxicities, with better efficacy in first line setting. A smaller study investigated the role of [132]decitabine + pembrolizumab in 10 patients with relapsed AML. ORR was observed in 6 patients with a median OS of 10 months. Zeidner et al. [133]conducted a phase II study in 37 relapsed refractory AML treated with high dose cytarabine + pembrolizumab. The ORR was 46% (Cr/cri: 38%) with a median OS of 11.1 months. The greatest benefit was observed in patients treated as first salvage regimen. Patients with ASXL1 mutations achieved the better ORR (50%) and two of five patients enrolled with TP53 mutations achieved CRc. A retrospective analysis[134] investigated the potential benefit of the use of pembrolizumab prior to allo-HCT. The results did not show benefit in terms of OS and RFS and no increase in grade III-IV acute graft-versus-host disease was seen in those who received ICI prior to allo HCT compared with historical controls. To date there are many trials that will better elucidate the role pembrolizumab based combinations in the setting of newly diagnosed and relapsed AML combined with HMA and venetoclax (NCT03969446; NCT04284787) and for eradicate MRD pretransplant combined with chemotherapy (NCT04214249). Pembrolizumab and azacytidine (AZA) were also studied in high risk MDS showing no benefit in patients with high risk MDS after a failure of hypometilating (HMA) agents. 17 patients not pretreated with HMA ORR was 76% (cr:18%) whereas in the cohort of patients pretreated with HMA the ORR was only 25% (CR:5%)[135]
Poly(ADP-ribose) Polymerase (PARP) Inhibitors Based Combinations
Talazoparib has been studied in early Phase I-II clinical trials for AML as a monotherapy, revealing limited efficacy (NCT01399840). [136] Better results will be expected in cohesin mutant AML (NCT03974217) characterized by mutations in genes such as STAG2, SMC1A, RAD21, PDS5B, SMC3 as previously described. Preclinical research indicates that combining talazoparib with decitabine, a DNA demethylating agent, enhances PARP1 recruitment and inhibits DNA repair, leading to synergistic cytotoxicity in AML cells. [137]A phase I clinical trial reported the results of decitabine combine with talozoparib in relapsed/refractory AML.[138] Responses included complete remission with incomplete count recovery was observed in two patients (8%) of 24 and hematologic improvement in three. The combination resulted well tolerated. Furthermore, talazoparib is being investigated in combination with Gemtuzumab ozogamicin (GO), an anti-CD33 antibody conjugated to calicheamicin, recently FDA approved for treating CD33-positive AML. (NCT04207190). [139] Despite the lack of robust data supporting the use of PARP inhibitors in AML, there is potential for successful treatment, particularly in cohesin mutant AML and through combination therapies involving agents like decitabine. As previously discussed, STAG2-mutated AML can be more sensitive to immune checkpoint inhibition, in particular to anti-PD(L)1 immunotherapy. The efficacy of combinatorial approaches including PARPi and ICB remains to be assessed in this specific setting.
Regimens Including Menin Inhibitors for KMT2A Mutated AML
The Phase I/II AUGMENT-101 trial (NCT04065399) is currently assessing the efficacy of revumenib monotherapy in adult and pediatric patients with relapsed or refractory acute leukemia characterized by a KMT2A rearrangement or NPM1 mutation. Recently updated findings from this trial were presented at the ASH meeting 2023 [140] where 94 patients were enrolled, with a median age of 37 years. These patients had undergone extensive prior treatments, with a median of 2 prior lines of therapy. With a median follow-up of 6.1 months in the efficacy population, the overall response rate was found to be 63%, with 23% of patients achieving complete remission or complete remission with partial hematologic recovery. Moreover, recognizing the heightened susceptibility of KMT2A rearranged (KMT2Ar) leukemias to apoptosis induction through BCL2 inhibition, recent observations have shown synergistic activity in models of KMT2Ar or NPM1-mutated (NPM1mt) leukemia with dual Bcl-2 and menin inhibition. [141] As a result, the phase I/II SAVE trial (NCT05360160) is investigating the combination of revumenib with venetoclax and the hypomethylating agent ASTX727, showing promising results. Further expanding on this approach, another study (NCT06177067) is evaluating the combination of revumenib with venetoclax and azacytidine in frontline AML patients to assess both safety and efficacy profiles of this triplet regimen. These collective findings underscore the potential significance of menin inhibitors as crucial therapeutic targets for patients with KMT2A mutated acute leukemia, with ongoing evaluation of combinatorial strategies offering promising avenues for further exploration and potential clinical benefit.
Combinatorial Strategies Targeting the Interleukin 3 Receptor CD123
CD123 is a subunit of the interleukin 3 (IL-3) receptor expressed on the surface of blasts in most AML and in particular in poor risk genetic subgroups and high cell count at diagnosis (Figure 1). [142]Tagraxofusp (sl-401) is a recombinant protein drug targeting CD123 and is currently approved as monotherapy for treatment of blastic plasmocitoid dentritic cell neoplasm (BPDCN).
In a Phase Ib trial (NCT03113643), the combination of TAG + AZA and VEN showed promising results in AML, MDS, and BPDCN, with 89% of patients achieving complete responses. This activity was observed across all genetic subgroups, including TP53-mutated AML/MDS and secondary AML. An expansion cohort in newly diagnosed AML, reported by Lane et al. [143] treated 26 adverse-risk patients according to ELN 2022 criteria, with 50% having TP53 mutations. Of these, 39% achieved complete remission (CR), with an additional 19% achieving incomplete CR, and a median overall survival (OS) of 14 months in the overall population, reduced to 9.5 months in the TP53-mutated subgroup. Ongoing trials, such as NCT05442216, are investigating the role of TAG in combination with AZA ± VEN specifically in secondary AML. Moreover, TAG has been studied as a single agent for consolidation therapy in AML patients at high risk of relapse and with measurable residual disease (MRD+) (NCT02270463).
Pivekimab Sunirine (PVEK, IMGN632) is an antibody-drug conjugate (ADC) consisting of a high-affinity CD123 antibody, a cleavable linker, and an indolinobenzodiazepine pseudodimer (IGN) payload. The IGN payload induces DNA alkylation and single-strand breaks without crosslinking, demonstrating high potency against tumor cells while exhibiting reduced toxicity to normal marrow progenitors compared to other DNA-targeting payloads. Preliminary clinical data for relapsed/refractory AML (R/R AML) [144]support the ongoing investigation of the PVEK+AZA+VEN triplet combination therapy (NCT04086264).
Flotetuzumab, a bispecific antibody (MGD006) engineered to bind both CD3 and CD123 on AML cells, is currently undergoing investigation in a Phase I/II trial (NCT02152956) for R/R AML.[145]Among the 88 patients enrolled in the trial, the ORR was reported as 13.6%, with 11.7% achieving CR. Across all dosing cohorts, a reduction in BM blasts has been observed, indicating potential efficacy of the treatment.
These findings suggest that anti CD123 directed therapies (Figure 1) hold promise as a therapeutic option for patients with R/R AML and high-risk genetic profiles, demonstrating activity in reducing leukemic cell burden and achieving complete remission in a subset of patients. Further investigation through ongoing clinical trials will provide additional insights into its safety and efficacy profile, potentially leading to improved outcomes for AML patients.
Conclusions and Perspectives
HR genetic AML represents a complex and heterogeneous disease driven by genetic mutations in stem cells and sustained by various molecular pathways within the microenvironment. Despite ongoing research, the current standard treatments often fail to provide satisfactory outcomes. In this complex landscape, combinatorial strategies involving targeted therapies and immunotherapy hold promise for improving patient’s outcomes. However, few combinations have demonstrated deep remissions thus far, and no drugs have been approved specifically for this high risk AML setting. Several compounds are currently being investigated, with the most promising those targeting KMT2A rearranged AML (menin inhibitor) and TP53 mutated AML (magrolimab and APR-246/eprenetapopt). Moving forward, concerted efforts to design tailored clinical trials for AML with adverse genetics are urgently needed.
Author Contributions
Conceptualization, NS and AM; methodology, NS and AM; original draft preparation, NS and AM, review and editing NS, PS, MDI and AM; visualization, AM; supervision, MDI and PS. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not applicable.
References
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Mendes, A.; Fahrenkrog, B. NUP214 in Leukemia: It’s More than Transport. Cells 2019, 8, 76. [Google Scholar] [CrossRef]
- Aparicio-Pérez, C.; de la Torre, E.P.; Sanchez-Garcia, J.; Martín-Calvo, C.; Martínez-Losada, C.; Casaño-Sanchez, J.; Serrano-López, J.; Serrano, J. Evolving Risk Classifications in AML in a Real-Life Scenario: After Changes upon Changes, Is It More and More Adverse? Cancers 2023, 15, 1425. [Google Scholar] [CrossRef]
- Döhner, H.; Pratz, K.W.; DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Recher, C.; Schuh, A.C.; Babu, S.; Dail, M.; et al. ELN Risk Stratification Is Not Predictive of Outcomes for Treatment-Naïve Patients with Acute Myeloid Leukemia Treated with Venetoclax and Azacitidine. Blood 2022, 140, 1441–1444. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Labopin, M.; Socie, G.; Volin, L.; Passweg, J.; Chevallier, P.; Beelen, D.; Milpied, N.; Blaise, D.; Cornelissen, J.J.; et al. Relapse and survival after transplantation for complex karyotype acute myeloid leukemia: A report from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation and the University of Texas MD Anderson Cancer Center. Cancer 2018, 124, 2134–2141. [Google Scholar] [CrossRef]
- Pasquini, M.C.; Zhang, M.-J.; Medeiros, B.C.; Armand, P.; Hu, Z.-H.; Nishihori, T.; Aljurf, M.D.; Akpek, G.; Cahn, J.-Y.; Cairo, M.S.; et al. Hematopoietic Cell Transplantation Outcomes in Monosomal Karyotype Myeloid Malignancies. Biol. Blood Marrow Transplant. 2016, 22, 248–257. [Google Scholar] [CrossRef]
- Middeke, J.M.; Herold, S.; Rücker-Braun, E.; Berdel, W.E.; Stelljes, M.; Kaufmann, M.; Schäfer-Eckart, K.; Baldus, C.D.; Stuhlmann, R.; Ho, A.D.; et al. TP53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2016, 172, 914–922. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Harada, T.; Heshmati, Y.; Kalfon, J.; Perez, M.W.; Ferrucio, J.X.; Ewers, J.; Engler, B.H.; Kossenkov, A.; Ellegast, J.M.; Yi, J.S.; et al. A distinct core regulatory module enforces oncogene expression in KMT2A-rearranged leukemia. Genes Dev. 2022, 36, 368–389. [Google Scholar] [CrossRef]
- Stasik, C.; Ganguly, S.; Cunningham, M.T.; Hagemeister, S.; Persons, D.L. Infant acute lymphoblastic leukemia with t(11;16)(q23;p13.3) and lineage switch into acute monoblastic leukemia. Cancer Genet. Cytogenet. 2006, 168, 146–149. [Google Scholar] [CrossRef]
- Neuendorff, N.R.; Hemmati, P.; Arnold, R.; Ihlow, J.; Dörken, B.; Müller-Tidow, C.; Westermann, J. BCR-ABL + acute myeloid leukemia: are we always dealing with a high-risk disease? Blood Adv. 2018, 2, 1409–1411. [Google Scholar] [CrossRef]
- Soupir, C.P.; Vergilio, J.-A.; Cin, P.D.; Muzikansky, A.; Kantarjian, H.; Jones, D.; Hasserjian, R.P. Philadelphia Chromosome–Positive Acute Myeloid leukemia: A rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am. J. Clin. Pathol. 2007, 127, 642–650. [Google Scholar] [CrossRef]
- Konoplev, S.; Yin, C.C.; Kornblau, S.M.; Kantarjian, H.M.; Konopleva, M.; Andreeff, M.; Lu, G.; Zuo, Z.; Luthra, R.; Medeiros, L.J.; et al. Molecular characterization ofde novoPhiladelphia chromosome-positive acute myeloid leukemia. Leuk. Lymphoma 2012, 54, 138–144. [Google Scholar] [CrossRef]
- Nacheva, E.P.; Grace, C.D.; Brazma, D.; Gancheva, K.; Howard-Reeves, J.; Rai, L.; Gale, R.E.; Linch, D.C.; Hills, R.K.; Russell, N.; et al. Does BCR/ABL1 positive Acute Myeloid Leukaemia Exist? Br. J. Haematol. 2013, 161, 541–550. [Google Scholar] [CrossRef]
- Lamble, A.J.; Hagiwara, K.; Gerbing, R.B.; Smith, J.L.; Kolekar, P.; E Ries, R.; A Kolb, E.; Alonzo, T.; Ma, X.; Meshinchi, S. CREBBP Alterations are Associated with A Poor Prognosis in de novo AML. Blood 2023, 141, 2156–2159. [Google Scholar] [CrossRef]
- Lavallée, V.-P.; Gendron, P.; Lemieux, S.; D’angelo, G.; Hébert, J.; Sauvageau, G. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood 2015, 125, 140–143. [Google Scholar] [CrossRef]
- Birdwell, C.; Fiskus, W.; Kadia, T.M.; DiNardo, C.D.; Mill, C.P.; Bhalla, K.N. EVI1 dysregulation: impact on biology and therapy of myeloid malignancies. Blood Cancer J. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Lugthart, S.; Gröschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.; van Zelderen-Bhola, S.L.; Ossenkoppele, G.J.; Vellenga, E.; Ruiter, E.v.D.B.-D.; Schanz, U.; et al. Clinical, Molecular, and Prognostic Significance of WHO Type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and Various Other 3q Abnormalities in Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef]
- Inaba, T.; Honda, H.; Matsui, H. The enigma of monosomy 7. Blood 2018, 131, 2891–2898. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Mrózek, K.; Eisfeld, A.-K.; Kohlschmidt, J.; Carroll, A.J.; Walker, C.J.; Nicolet, D.; Blachly, J.S.; Bill, M.; Papaioannou, D.; Wang, E.S.; et al. Complex karyotype in de novo acute myeloid leukemia: typical and atypical subtypes differ molecularly and clinically. Leukemia 2019, 33, 1620–1634. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.T.; Park, H.J.; Kim, B.K.; An, H.Y.; Choi, J.Y.; Kang, H.J. Post-Transplantation Cyclophosphamide-Based Haploidentical versus Matched Unrelated Donor Peripheral Blood Hematopoietic Stem Cell Transplantation Using Myeloablative Targeted Busulfan-Based Conditioning for Pediatric Acute Leukemia. Biol. Blood Marrow Transplant. 2022, 28, 195–e1. [Google Scholar] [CrossRef] [PubMed]
- Breems, D.A.; Van Putten, W.L.; De Greef, G.E.; Van Zelderen-Bhola, S.L.; Gerssen-Schoorl, K.B.; Mellink, C.H.; Nieuwint, A.; Jotterand, M.; Hagemeijer, A.; Beverloo, H.B.; et al. Monosomal Karyotype in Acute Myeloid Leukemia: A Better Indicator of Poor Prognosis Than a Complex Karyotype. J. Clin. Oncol. 2008, 26, 4791–4797. [Google Scholar] [CrossRef] [PubMed]
- Anelli, L.; Pasciolla, C.; Zagaria, A.; Specchia, G.; Albano, F. Monosomal karyotype in myeloid neoplasias: a literature review. OncoTargets Ther. 2017, 10, 2163–2171. [Google Scholar] [CrossRef]
- Tang, J.-L.; Hou, H.-A.; Chen, C.-Y.; Liu, C.-Y.; Chou, W.-C.; Tseng, M.-H.; Huang, C.-F.; Lee, F.-Y.; Liu, M.-C.; Yao, M.; et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood 2009, 114, 5352–5361. [Google Scholar] [CrossRef]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 Mutations Are Associated With Poor Outcome in Younger and Older Patients With Cytogenetically Normal Acute Myeloid Leukemia and With Distinct Gene and MicroRNA Expression Signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef]
- Hanaki, S.; Shimada, M. Targeting EZH2 as cancer therapy. J. Biochem. 2021, 170, 1–4. [Google Scholar] [CrossRef]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in Myeloid Malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef]
- Wang, X.; Dai, H.; Wang, Q.; Wang, Q.; Xu, Y.; Wang, Y.; Sun, A.; Ruan, J.; Chen, S.; Wu, D. EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -7/del(7q) in De Novo Acute Myeloid Leukemia. PLOS ONE 2013, 8, e61341. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 mutations and impact on clinical outcome: an analysis in 1,604 patients with newly diagnosed acute myeloid leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; A Miller, C.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; LaFave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 leads to myelodysplastic syndrome–like disease in mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef]
- Inoue, D.; Kitaura, J.; Togami, K.; Nishimura, K.; Enomoto, Y.; Uchida, T.; Kagiyama, Y.; Kawabata, K.C.; Nakahara, F.; Izawa, K.; et al. Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J. Clin. Investig. 2013, 123, 4627–4640. [Google Scholar] [CrossRef]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 2019, 76, 2511–2523. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.-A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef]
- Sportoletti, P.; Sorcini, D.; Falini, B. BCORgene alterations in hematologic diseases. Blood 2021, 138, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Class, C.A.; Sasaki, K.; Ravandi, F.; Cortes, J.E.; Daver, N.; Takahashi, K.; Short, N.J.; DiNardo, C.D.; et al. Outcomes of acute myeloid leukemia with myelodysplasia related changes depend on diagnostic criteria and therapy. Am. J. Hematol. 2020, 95, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Tiacci, E.; Holmes, A.B.; Kohlmann, A.; Martelli, M.P.; Kern, W.; Spanhol-Rosseto, A.; Klein, H.-U.; Dugas, M.; Schindela, S.; et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 2011, 118, 6153–6163. [Google Scholar] [CrossRef]
- Sportoletti, P.; Sorcini, D.; Guzman, A.G.; Reyes, J.M.; Stella, A.; Marra, A.; Sartori, S.; Brunetti, L.; Rossi, R.; Del Papa, B.; et al. Bcor deficiency perturbs erythro-megakaryopoiesis and cooperates with Dnmt3a loss in acute erythroid leukemia onset in mice. Leukemia 2020, 35, 1949–1963. [Google Scholar] [CrossRef]
- Kelly, M.J.; So, J.; Rogers, A.J.; Gregory, G.; Li, J.; Zethoven, M.; Gearhart, M.D.; Bardwell, V.J.; Johnstone, R.W.; Vervoort, S.J.; et al. Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, C.; Lussana, F.; Salmoiraghi, S.; Cavagna, R.; Buklijas, K.; Elidi, L.; Zanghi’, P.; Michelato, A.; Delaini, F.; Oldani, E.; et al. Clinical significance of chromatin-spliceosome acute myeloid leukemia: a report from the Northern Italy Leukemia Group (NILG) randomized trial 02/06. Haematologica 2021, 106. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin–RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef]
- Craddock, C.F. Full-intensity and reduced-intensity allogeneic stem cell transplantation in AML. Bone Marrow Transplant. 2008, 41, 415–423. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1 K700E Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef]
- Saez, B.; Walter, M.J.; Graubert, T.A. Splicing factor gene mutations in hematologic malignancies. Blood 2017, 129, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Ochi, Y.; Ogawa, S. Chromatin-Spliceosome Mutations in Acute Myeloid Leukemia. Cancers 2021, 13, 1232. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. Cancer 2020, 20, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Hou, H.-A.; Tang, J.-L.; Kuo, Y.-Y.; Chiu, Y.-C.; Liu, C.-Y.; Tseng, M.-H.; Lin, T.-Y.; Liu, M.-C.; Liu, C.-W.; et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, J.-N.; Stasik, S.; Röllig, C.; Sauer, T.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; Steffen, B.; et al. Alterations of cohesin complex genes in acute myeloid leukemia: differential co-mutations, clinical presentation and impact on outcome. Blood Cancer J. 2023, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Black, H.E.; Jhujh, S.; Stewart, G.S.; I Savage, K.; I Mills, K. STAG2 Loss Gives Rise to Therapeutically Targetable DNA Damage Repair Defects and Altered Replication Fork Dynamics in Acute Myeloid Leukaemia. Blood 2019, 134, 1255–1255. [Google Scholar] [CrossRef]
- J, G. A pilot proof-of-concept study of talazoparib for cohesin-mutated AML and MDS with excess blasts. https://clinicaltrials.gov/ct2/show/NCT03974217. 2022; NCT0397421.
- Perri, F.; Pisconti, S.; Scarpati, G.D.V. P53 mutations and cancer: a tight linkage. Ann. Transl. Med. 2016, 4, 522–522. [Google Scholar] [CrossRef]
- George, B.; Kantarjian, H.; Baran, N.; Krocker, J.D.; Rios, A. TP53 in Acute Myeloid Leukemia: Molecular Aspects and Patterns of Mutation. Int. J. Mol. Sci. 2021, 22, 10782. [Google Scholar] [CrossRef]
- A Sallman, D.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef]
- Lasry, A.; Nadorp, B.; Fornerod, M.; Nicolet, D.; Wu, H.; Walker, C.J.; Sun, Z.; Witkowski, M.T.; Tikhonova, A.N.; Guillamot-Ruano, M.; et al. An inflammatory state remodels the immune microenvironment and improves risk stratification in acute myeloid leukemia. Nat. Cancer 2022, 4, 1–16. [Google Scholar] [CrossRef]
- Rutella, S.; Vadakekolathu, J.; Mazziotta, F.; Reeder, S.; Yau, T.-O.; Mukhopadhyay, R.; Dickins, B.; Altmann, H.; Kramer, M.; Knaus, H.A.; et al. Immune dysfunction signatures predict outcomes and define checkpoint blockade–unresponsive microenvironments in acute myeloid leukemia. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Dufva O, Pölönen P, Brück O, Keränen MAI, Klievink J, Mehtonen J et al. Immunogenomic Landscape of Hematological Malignancies. Cancer Cell 2020; 38: 380-399.e13.
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.d.l.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Pratz, K.W.; Wei, A.H.; Pullarkat, V.; Jonas, B.A.; Recher, C.; Babu, S.; Schuh, A.C.; Dail, M.; Sun, Y.; et al. Outcomes in Patients with Poor-Risk Cytogenetics with or without TP53 Mutations Treated with Venetoclax and Azacitidine. Clin. Cancer Res. 2022, 28, 5272–5279. [Google Scholar] [CrossRef]
- Aldoss, I.; Yang, D.; Pillai, R.; Sanchez, J.F.; Mei, M.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Salhotra, A.; et al. Association of leukemia genetics with response to venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Am. J. Hematol. 2019, 94, E253–E255. [Google Scholar] [CrossRef]
- Cherry, E.M.; Abbott, D.; Amaya, M.; McMahon, C.; Schwartz, M.; Rosser, J.; Sato, A.; Schowinsky, J.T.; Inguva, A.; Minhajuddin, M.; et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021, 5, 5565–5573. [Google Scholar] [CrossRef]
- Rahmani, N.E.; Ramachandra, N.; Sahu, S.; Gitego, N.; Lopez, A.; Pradhan, K.; Bhagat, T.D.; Gordon-Mitchell, S.; Pena, B.R.; Kazemi, M.; et al. ASXL1 mutations are associated with distinct epigenomic alterations that lead to sensitivity to venetoclax and azacytidine. Blood Cancer J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, P.; Mao, M.; Zhang, S.; Bigenwald, C.; Dutertre, C.-A.; Lehmann, C.H.; Pan, H.; Paulhan, N.; Amon, L.; et al. BCL2 Inhibition Reveals a Dendritic Cell–Specific Immune Checkpoint That Controls Tumor Immunosurveillance. Cancer Discov. 2023, 13, 2448–2469. [Google Scholar] [CrossRef]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.V.; Jitkova, Y.; et al. Venetoclax enhances T cell-mediated anti-leukemic activity by increasing ROS production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Haribhai, D.; Mathew, R.; Duggan, R.; Ellis, P.A.; Wang, R.; Lasater, E.A.; Shi, Y.; Dave, N.; Riehm, J.J.; et al. Venetoclax Increases Intratumoral Effector T Cells and Antitumor Efficacy in Combination with Immune Checkpoint Blockade. Cancer Discov. 2021, 11, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2021, 107, 403–416. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free. Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Dang, Q.; Sun, Z.; Wang, Y.; Wang, L.; Liu, Z.; Han, X. Ferroptosis: a double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2018, 133, 144–152. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Zhang, W.; Meng, L.; Wang, J.; Lv, Z.; Xia, H.; Wu, M.; Zhang, Y.; Wang, J. Ferroptosis Mediation Patterns Reveal Novel Tool to Implicate Immunotherapy and Multi-Omics Characteristics in Bladder Cancer. Front. Cell Dev. Biol. 2022, 10, 791630. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; A Mishchenko, T.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLOS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Marra, A.; Akarca, A.U.; Martino, G.; Ramsay, A.; Ascani, S.; Perriello, V.M.; O’nions, J.; Wilson, A.J.; Gupta, R.; Childerhouse, A.; et al. CD47 expression in acute myeloid leukemia varies according to genotype. Haematologica 2023, 108, 3491–3495. [Google Scholar] [CrossRef]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Oreskovic, E.; Wheeler, E.C.; Mengwasser, K.E.; Fujimura, E.; Martin, T.D.; Tothova, Z.; Elledge, S.J. Genetic analysis of cancer drivers reveals cohesin and CTCF as suppressors of PD-L1. Proc. Natl. Acad. Sci. 2022, 119. [Google Scholar] [CrossRef]
- Schram, A.M.; Colombo, N.; Arrowsmith, E.; Narayan, V.; Yonemori, K.; Scambia, G.; Zelnak, A.; Bauer, T.M.; Jin, N.; Ulahannan, S.V.; et al. Avelumab Plus Talazoparib in Patients With BRCA1/2- or ATM-Altered Advanced Solid Tumors. JAMA Oncol. 2022, 9, 29–39. [Google Scholar] [CrossRef]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-González, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281. [Google Scholar] [CrossRef]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Zhong, F.-M.; Yao, F.-Y.; Liu, J.; Li, M.-Y.; Jiang, J.-Y.; Cheng, Y.; Xu, S.; Li, S.-Q.; Zhang, N.; Huang, B.; et al. Splicing factor-mediated regulation patterns reveals biological characteristics and aid in predicting prognosis in acute myeloid leukemia. J. Transl. Med. 2023, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chiche, E.; Rahmé, R.; Bertoli, S.; Dumas, P.-Y.; Micol, J.-B.; Hicheri, Y.; Pasquier, F.; Peterlin, P.; Chevallier, P.; Thomas, X.; et al. Real-life experience with CPX-351 and impact on the outcome of high-risk AML patients: a multicentric French cohort. Blood Adv. 2021, 5, 176–184. [Google Scholar] [CrossRef]
- Hui, G.; Ladha, A.; Cheung, E.; Berube, C.; Coutre, S.; Gotlib, J.; Liedtke, M.; Zhang, T.Y.; Muffly, L.S.; Mannis, G.N. Routine Use of Gemtuzumab Ozogamicin in 7+3-Based Inductions for All "Non-Adverse" Risk AML. Blood 2020, 136, 36–37. [Google Scholar] [CrossRef]
- Loke, J.; Buka, R.; Craddock, C. Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia: Who, When, and How? Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Badar, T.; Atallah, E.L.; Shallis, R.M.; Saliba, A.N.; Stahl, M.F.; Bewersdorf, J.P.; Grenet, J.; Patel, A.A.; Abaza, Y.; Murthy, G.S.G.; et al. Predictors of Long-Term Outcome in TP53-Mutated Acute Myeloid Leukemia Patients Receiving Allogeneic Stem Cell Transplant after First- or Second-Line Therapy: Results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Blood 2022, 140, 1435–1437. [Google Scholar] [CrossRef]
- Britt, A.; Mohyuddin, G.R.; McClune, B.; Singh, A.; Lin, T.; Ganguly, S.; Abhyankar, S.; Shune, L.; McGuirk, J.; Skikne, B.; et al. Acute myeloid leukemia or myelodysplastic syndrome with chromosome 17 abnormalities and long-term outcomes with or without hematopoietic stem cell transplantation. Leuk. Res. 2020, 95, 106402. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; McCullough, K.; Johnson, I.; Al-Kali, A.; Begna, K.H.; Patnaik, M.M.; Litzow, M.R.; Hogan, W.; Shah, M.; Alkhateeb, H.; et al. Real-world experience with venetoclax and hypomethylating agents in myelodysplastic syndromes with excess blasts. Am. J. Hematol. 2022, 97, E214–E216. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Altman, J.K.; Assi, R.; Bixby, D.; Fathi, A.T.; Foran, J.M.; Gojo, I.; Hall, A.C.; Jonas, B.A.; Kishtagari, A.; et al. Acute Myeloid Leukemia, Version 3.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Komrokji, R.S.; DeZern, A.E.; Sebert, M.; Garcia-Manero, G.; Rahmé, R.; Steensma, D.P.; Che, J.L.; Roboz, G.J.; Madelaine, I.; et al. Long Term Follow-up and Combined Phase 2 Results of Eprenetapopt (APR-246) and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2021, 138, 246–246. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Sweet, K.; Miller, C.; Wennborg, A.; et al. Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: a phase 1, dose-finding and expansion study. Lancet Haematol. 2023, 10, e272–e283. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.A.; Asch, A.S.; Mannis, G.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and Efficacy of the Anticluster of Differentiation 47 Antibody Magrolimab Combined With Azacitidine in Patients With Previously Untreated AML: Phase Ib Results. J. Clin. Oncol. 2023, 41, 4893–4904. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. 2014, 7, 6880–6888.
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients (Pts) with Very High/High-Risk Myelodysplastic Syndrome (vHR/HR-MDS) and Acute Myeloid Leukemia (AML): Final Analysis from a Phase Ib Study. Blood 2021, 138, 244–244. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Westermann, J.; Kovacsovics, T.; Assouline, S.; Schuh, A.C.; Kim, H.-J.; Macias, G.R.; Sanford, D.; Luskin, M.R.; Stein, E.M.; et al. P582: FIRST RESULTS OF A PHASE II STUDY (STIMULUS-AML1) INVESTIGATING SABATOLIMAB + AZACITIDINE + VENETOCLAX IN PATIENTS WITH NEWLY DIAGNOSED ACUTE MYELOID LEUKEMIA. HemaSphere 2022, 6, 481–482. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; A Thompson, P.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a single-arm, phase 2 study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Reville, P.K.; Kantarjian, H.M.; Ravandi, F.; Jabbour, E.; DiNardo, C.D.; Daver, N.; Pemmaraju, N.; Ohanian, M.; Alvarado, Y.; Xiao, L.; et al. Nivolumab maintenance in high-risk acute myeloid leukemia patients: a single-arm, open-label, phase II study. Blood Cancer J. 2021, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.; Morrow, J.; DeZern, A.E.; Foster, M.C.; Levis, M.J.; Coombs, C.C.; et al. Multi-Center Phase 2 Study of Pembroluzimab (Pembro) and Azacitidine (AZA) in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Newly Diagnosed (≥65 Years) AML Patients. Blood 2019, 134, 832–832. [Google Scholar] [CrossRef]
- Goswami, M.; Gui, G.; Dillon, L.W.; E Lindblad, K.; Thompson, J.; Valdez, J.; Kim, D.-Y.; Ghannam, J.Y.; A Oetjen, K.; Destefano, C.B.; et al. Pembrolizumab and decitabine for refractory or relapsed acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e003392. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Vincent, B.G.; Ivanova, A.; Moore, D.; McKinnon, K.P.; Wilkinson, A.D.; Mukhopadhyay, R.; Mazziotta, F.; Knaus, H.A.; Foster, M.C.; et al. Phase II Trial of Pembrolizumab after High-Dose Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Tschernia, N.P.; Kumar, V.; Moore, D.T.; Vincent, B.G.; Coombs, C.C.; Van Deventer, H.; Foster, M.C.; DeZern, A.E.; Luznik, L.; Riches, M.L.; et al. Safety and Efficacy of Pembrolizumab Prior to Allogeneic Stem Cell Transplantation for Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. 2021, 27, 1021–e1. [Google Scholar] [CrossRef]
- Chien, K.S.; Kim, K.; Nogueras-Gonzalez, G.M.; Borthakur, G.; Naqvi, K.; Daver, N.G.; Montalban-Bravo, G.; Cortes, J.E.; DiNardo, C.D.; Jabbour, E.; et al. Phase II study of azacitidine with pembrolizumab in patients with intermediate-1 or higher-risk myelodysplastic syndrome. Br. J. Haematol. 2021, 195, 378–387. [Google Scholar] [CrossRef]
- Gopal, A.K.; Popat, R.; Mattison, R.J.; Menne, T.; Bloor, A.; Gaymes, T.; Khwaja, A.; Juckett, M.; Chen, Y.; Cotter, M.J.; et al. A Phase I trial of talazoparib in patients with advanced hematologic malignancies. Int J Hematol Oncol 2021; 10.
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents – A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O'Connell, C.L.; Youngblood, B.A.; et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 1313–1322. [Google Scholar] [CrossRef]
- Portwood, S.M.; Cantella, M.C.; Cronin, T.L.; Wang, E.S. Addition of the PARP Inhibitor, Talazoparib, to Gemtuzumab Ozogamicin Significantly Enhances Anti-Leukemic Activity in Human CD33+ Acute Myeloid Leukemia. Blood 2019, 134, 1371–1371. [Google Scholar] [CrossRef]
- Ibrahim Aldoss,, Ghayas C. Issa, Michael Thirman, John DiPersio, Martha Arellano, James S. Blachly, Gabriel N. Mannis, Alexander Perl, David S. Dickens*, Christine M. McMahon, Elie Traer, C. Michel Zwaan, Carolyn Grove, Richard Stone, Paul J. Shami, Ioann NM and EMS. LBA-5 Revumenib Monotherapy in Patients with Relapsed/Refractory KMT2Ar Acute Leukemia: Topline Efficacy and Safety Results from the Pivotal Augment-101 Phase 2 Study. https://ash.confex.com/ash/2023/webprogram/Paper192042.html.
- Carter, B.Z.; Tao, W.; Mak, P.Y.; Ostermann, L.B.; Mak, D.H.; McGeehan, G.M.; Ordentlich, P.; Andreeff, M. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood 2021, 138, 1637–1641. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Mughal, T.I.; Brooks, C.; Lindsay, R.; Pemmaraju, N. Targeting CD123 in hematologic malignancies: identifying suitable patients for targeted therapy. Leuk. Lymphoma 2021, 62, 2568–2586. [Google Scholar] [CrossRef]
- Lane, A.A.; Garcia, J.S.; Raulston, E.G.; Garzon, J.L.; Galinsky, I.; Baxter, E.W.; Leonard, R.; DeAngelo, D.J.; Luskin, M.R.; Reilly, C.R.; et al. Tagraxofusp in Combination with Azacitidine and Venetoclax in Newly Diagnosed CD123+ Acute Myeloid Leukemia, Expansion Cohort of a Phase 1b Multicenter Trial. Blood 2023, 142, 4277–4277. [Google Scholar] [CrossRef]
- Daver, N.G.; Montesinos, P.; Aribi, A.M.; Martinelli, G.; Wang, E.S.; Altman, J.K.; Roboz, G.J.; Burke, P.W.; Walter, R.B.; Begna, K.; et al. A phase 1b/2 study of pivekimab sunirine (PVEK, IMGN632) in combination with venetoclax/azacitidine or magrolimab for patients with CD123-positive acute myeloid leukemia (AML). J. Clin. Oncol. 2023, 41, TPS7073–TPS7073. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
Figure 1.
Biological rationales for combining targeted therapies and immunotherapies in AML with.adverse genetics. Displayed are multiple signaling pathways that are often activated in high-risk AML and can be targeted by a combination of small molecule drugs and immunotherapies. The BCL2 inhibitor venetoclax effectively induces mitochondrial apoptosis in leukemic cells and activates conventional dendritic cells to enhance anti-tumor T cell immunity. APR246/eprenetapopt reestablishes wild-type TP53 tumor suppressor function in TP53mut AML, inducing apoptosis and upregulating co-inhibitory molecules such as CTLA-4 within the leukemic microenvironment, thereby increasing sensitivity to immune checkpoint blockade (ICB) therapy. In cohesin-mutated AML, particularly in cells with STAG2 mutations, DNA repair and replication pathways are identified as genetic vulnerabilities. Consequently, STAG2mut AML cells exhibit increased sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition. Cohesin-deficient leukemic cells also demonstrate elevated expression of the PD-L1 immune checkpoint molecule, which can be targeted by ICB therapy, as with anti-PD(L)1 inhibitors. Another strategy against HR-AML is represented by targeting CD123 molecule, which is found over-expressed in AML. CD123 expression can be further enhanced by hypomethylating agent, such as azacytidine (not shown).
Figure 1.
Biological rationales for combining targeted therapies and immunotherapies in AML with.adverse genetics. Displayed are multiple signaling pathways that are often activated in high-risk AML and can be targeted by a combination of small molecule drugs and immunotherapies. The BCL2 inhibitor venetoclax effectively induces mitochondrial apoptosis in leukemic cells and activates conventional dendritic cells to enhance anti-tumor T cell immunity. APR246/eprenetapopt reestablishes wild-type TP53 tumor suppressor function in TP53mut AML, inducing apoptosis and upregulating co-inhibitory molecules such as CTLA-4 within the leukemic microenvironment, thereby increasing sensitivity to immune checkpoint blockade (ICB) therapy. In cohesin-mutated AML, particularly in cells with STAG2 mutations, DNA repair and replication pathways are identified as genetic vulnerabilities. Consequently, STAG2mut AML cells exhibit increased sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition. Cohesin-deficient leukemic cells also demonstrate elevated expression of the PD-L1 immune checkpoint molecule, which can be targeted by ICB therapy, as with anti-PD(L)1 inhibitors. Another strategy against HR-AML is represented by targeting CD123 molecule, which is found over-expressed in AML. CD123 expression can be further enhanced by hypomethylating agent, such as azacytidine (not shown).
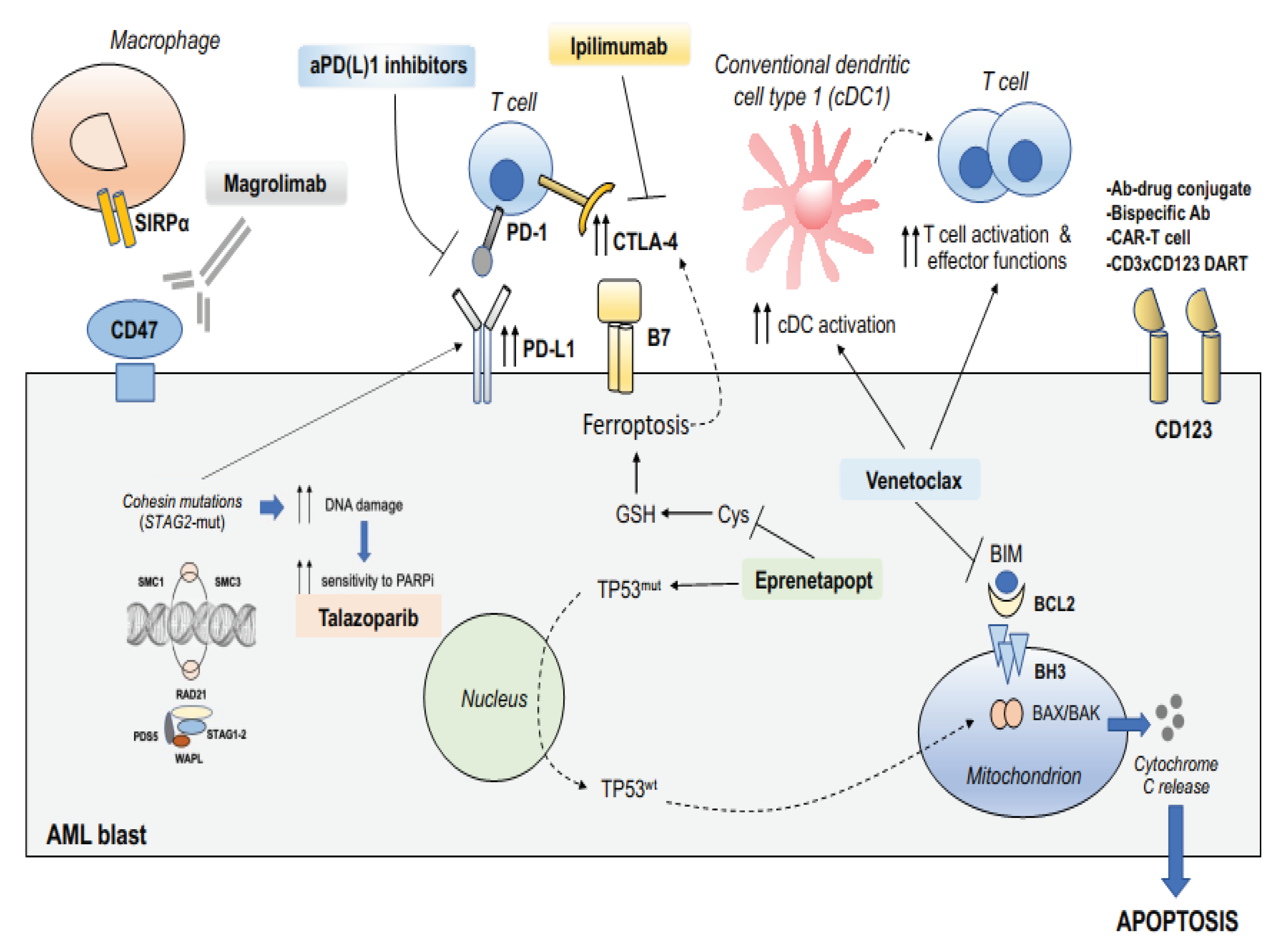

Table 1.
High risk genetic features at diagnosis in AML according to ELN 2022.
| High risk genetic features |
| t(6;9)(p23.3;q34.1)/DEK::NUP214 t(v;11q23.3)KMT2A-rearranged t(9;22)(q34.1;q11.2)/BCR::ABL1 (BCR-ABL+) t(8;16)(p11.2;p13.3)/KAT6A::CREBBP inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2) GATA2, MECOM(EVI1) t(3q26.2;v)/MECOM(EVI1)-rearranged -5 or del(5q); -7; -17/abn(17p) Complex karyotype (CK) Monosomal karyotype (MK) Mutated RUNX1 Mutated EZH2 Mutated ASXL1 Mutated BCOR Spliceosome mutations (SRSF2, SF3B1, U2AF1, ZRSR2) Mutated STAG2 Mutated TP53 |
Table 2.
Clinical trials combining immunotherapy and target therapy in HR genetic AML.
| Drugs | Mutation | Clinical trials | population | Line of treatment | Outcomes |
| APR-246 + AZA APR-246 + AZA APR 246+ VEN + AZA |
TP53 TP53 TP53 |
NCT03072043 (PHASE IB-II) NCT03588078 (PHASE II) NCT04214860 (PHASE I) |
40 MDS 11 AML 34 MDS 18 AML 49 AML |
1 1 1 |
ORR 71%, CR 44% Median OS 10.8 mo ORR 52% CR 37% Median OS 12.1 mo ORR 64% CR 38% |
| MAGROLIMAB+ AZA MAGROLIMAB+AZA Vs VEN-AZA or chemo MAGROLIMAB+AZA-VEN vs placebo + AZA+VEN |
TP53 TP53 TP53 |
NCT03248479 (PHASE I) NCT04778397 (PHASE III) NCT05079230 (PHASE III) |
87 AML (82.8% TP53) Ongoing Ongoing |
1 1 1 |
ORR 47.2% CR 31.9% Median OS 9.8 mo Ongoing Ongoing |
| SABATOLIMAB+ HMA SABATOLIMAB+AZA+VEN |
All HR AML All |
NCT03066648 (PHASE Ib) NCT04150029 (PHASE II) |
53 MDS 48 AML Ongoing |
1 | ORR AML 40% CR30% adverse risk AML ORR 53% median duration of response 12 mo Ongoing |
| NIVOLUMAB + AZA NIVOLUMAB + AZA+IPILIMUMAB NIVOLUMAB + CHEMO |
All All All (50% HR) |
NCT02397720 (PHASE II) NCT02397720 (PHASE II) NCT02464657 (PHASE II) |
70 AML 31 AML 42 AML |
>1 >1 1 |
ORR 33%, CR22% Median OS 6.2 mo (ASLX1 better response) ORR 46%, CR36% Median OS 10.5 mo ORR 80%, CR64% Median OS 18.5 mo |
| PEMBRO+AZA PEMBRO + ARA C PEMBRO + DEC+/-VEN PEMBRO + AZA+ VEN PEMBRO + CHEMO |
All All All All All |
NCT02845297 (PHASE II) NCT02768792 (PHASE II) NCT03969446 (PHASE II) NCT04284787 (PHASE II) NCT04214249 (PHASE II) |
37 AML (17 newly diagnosed) 37 AML Ongoing Ongoing Ongoing |
≥1 >1 ≥1 ≥1 1 |
ORR 55%, CR14% Median OS 10.8 mo newy diagnosed ORR94% CR47% median OS 13 mo ORR46% CR38% median OS 11 mo (ASLX1 better response) Ongoing Ongoing Ongoing |
| TALAZOPARIB + DEC TALAZOPARIB BASED TALAZOPARIB + GENTUZUMAB |
All Cohesin mutated Cd33+ |
NCT02878785 (PHASE I) NCT03974217 (PHASE I) NCT04207190 (PHASE I) |
24 AML Ongoing Ongoing |
>1 ≥1 >1 |
CR 8% Ongoing Ongoing |
| REVUNEMIB + VEN+ ASX727 REVUNEMIB + VEN+ AZA |
All All |
NCT05360160 (PHASE II) NCT06177067 (PHASE II) |
Ongoing Ongoing |
1 >1 |
Ongoing Ongoing |
| TAGRAXOFUSP+AZA+VEN PIVEKIMAB +AZA + VEN |
HR AML Cd123+ |
NCT03113643 (PHASE IB) NCT04086264 (PHASE IB-II) |
Ongoing (preliminary results 26 AML HR) Ongoing |
1 ≥1 |
Ongoing preliminary results CR 39% median OS 14 mo; median OS TP53 9.5 mo Ongoing |
Abbreviations: APR-246: eprenetapopt; AZA: azacytidine; VEN: venetoclax, MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; HR: high risk, ORR: overall response rate, CR: complete remission; OS: overall survival, PEMBRO: pembrolizumab; DEC: decytabine.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



