
Submitted:
14 March 2024
Posted:
15 March 2024
You are already at the latest version
Abstract
Evolution remains an incessant process in viruses, allowing them to elude host immune response and induce severe diseases, impacting the diagnostic and vaccine effectiveness. Predicting emerging viral genomes is crucial, particularly in diseases like dengue, where viruses disrupt host cells, leading to fatal outcomes. Deep learning has been applied to predict dengue fever cases; there has been relatively less emphasis on its significance in forecasting emerging Dengue Virus (DENV) serotype. While Recurrent Neural Networks (RNN) were originally designed for modeling temporal sequences, our proposed DL-DVE generative and classification model, trained on complete genome data of DENV, transcends traditional approaches by learning semantic relationships between nucleotides in a continuous vector space instead of representing contextual meaning of nucleotide characters. Leveraging 2000 publicly available DENV complete genome sequences, our Long Short-Term Memory (LSTM) based generative and Feedforward Neural Network (FNN) based classification DL-DVE model showcases proficiency in learning intricate patterns and generating sequences for emerging serotype of DENV. The generative model showed accuracy of 93% and the classification model provided insight into the specific serotype label, corroborated by BLAST search verification. Evaluation metrics such as ROC-AUC value 0.818, accuracy, precision, recall and F1 score all to be around 99.00%, demonstrated the classification model’s reliability. Our model classified the generated sequences as DENV-4, exhibiting 65.99% similarity to DENV-4 and around 63-65% similarity with other serotypes, indicating notable distinction from other serotypes. Moreover, the intra-serotype divergence of sequences with a minimum 90% similarity underscored their uniqueness. We analyzed the conserved motifs in the genome through MEME Suite (version 5.5.5). Our research strives to contribute to the ongoing fight against the Dengue virus by offering predictive insights into its genomic evolution. Looking ahead, proactive predictive modeling before mutations occur holds potential for guiding vaccine design and diagnostic kit development.
Keywords:
Virus forecasting
; DL modeling
; virus classification
; Dengue evolution
; genome sequence
1. Introduction
Understanding the evolutionary dynamics of a virus is crucial for discerning its origin, focusing on key characteristics such as structure, classification and evolution. This knowledge plays a pivotal role in unraveling the fundamental biological mechanisms, thereby advancing vaccine and drug development. Despite the ongoing discoveries related to viruses, the potential existence of unidentified viruses remains a constant concern. The rapid and widespread dissemination of viruses has become robust, presenting formidable challenges in controlling and predicting their expansion. Thus leads to epidemics and pandemics, underscoring the unpredictable risks associated with these agents [1].
Dengue infection is transmitted through the bite of an Aedes mosquito carrying the ~10kb genome-size dengue virus (DENV). Clinically, identifying dengue fever has historically been challenging due to the prevalence of other infections with similar syndromes in tropical environments. The ambiguity in distinguishing dengue from various viral diseases, ranging from yellow fever to tropical influenza has persisted. Additionally, labeling the historical dengue outbreaks as chikungunya particularly in the 1800s is complicated due to the inconsistent and conflicting reporting information. Despite these challenges, the global public health system has remained engaged in addressing the persistent threats posed by the dengue virus [2]. In the landscape of genomic analysis, deep learning (DL) has emerged as a powerful tool, particularly for extracting features and patterns from complex genomic data. Similarly, in the context of infectious diseases like dengue, machine learning applications have gained prominence, leveraging epidemiological and clinical data for predictive modeling. Rachata et al., notably utilized weather data and feature selection algorithms to forecast dengue incidences, employing Artificial Neural Networks (ANNs) and Support Vector Machines (SVMs) to predict number of cases based on weather and gene expression data [3,4]. Despite the success of SVMs using word2vec representation in specific tasks, their efficiency waned when directly applied to nucleic acid sequences. There are multiple methods for viral genome classification, employing both alignment and machine learning approaches. In the alignment based approach, detection of viral sequences is carried out using tools such as USEARCH, SCUEAL [5] and REGA [6], relying on alignment scores for genome classification. However, these alignment based methods have limitations, notably their performance dependence on selection of initial alignments. However, in the machine learning approach, various methods have been proposed for the classification of viral genome sequences. But, these methods face limitations in their ability to detect viral genome contigs and the challenge of extracting useful hidden information. Moreover, those trained exclusively on nucleotide sequence data, further constraints their utility in comprehensive genome analysis.
The deep learning models such as Recurrent Neural Networks (RNNs) have demonstrated their effectiveness in the field of natural language processing. The applications of deep learning in computational biology mainly concentrates in genome analysis and sequencing. However, the RNN are considered black boxes due to the complexity of the model in interpreting the hidden state of the model. Long Short-Term Memory (LSTM) and Convolutional Neural Networks (CNN) have been used to detect viral genome sequences using training of pattern and frequency of branches. Whereas, the Feedforward Neural Network (FNN), a type of CNN model, is adept at evaluating visual patterns while accommodating the inherent heterogeneity within the data. This network is designed to map fixed length inputs to a fixed size output and is trained using a backpropagation algorithm. Consisting of multiple layers, the CNN effectively stores and updates information in filter weights as it learns the intricate relationship between input and output.
This study aims to predict the genomic sequences of emerging serotype to deepen our understanding of dengue virus evolution and also classifies predicted as well as unknown dengue virus genome sequences respective to their serotypes. Utilizing the RNNs trained on complete genome, the model focuses on learning patterns in tokens rather than representing character meanings. LSTM, chosen for genomic prediction tasks, excel in handling sequential data and mitigating the vanishing gradient problem inherent in traditional RNNs.
2. Materials and Methods
2.1. Data Collection and Preprocessing
A dataset comprising 2000 complete genome sequences of dengue virus were assembled from the NCBI [7] and BV-BRC databases [8]. The datasets incorporated 500 sequences for each of the four serotypes of dengue virus. Before further analysis, a preprocessing step was performed to ensure that the data were in suitable format. Subsequently, four distinct datasets representing serotypes separately were built, each containing sequences spanning 10,273 base pairs. All the sequences were concatenated into a single dataset following a labeling process. All nucleotides were selected as features and DNV1, DNV2, DNV3 and DNV4 as serotype labels.
2.2. DL-DVE Architecture and Working for Sequence Generation
The genomic sequences of DENV were extracted from FASTA files using the Biopython library. A tokenization approach was used, treating the sequences at the character level, and an n-gram strategy was employed to generate the input sequences for the model from a FASTA file. To ensure uniformity in the sequence length, the data underwent padding. The generative model, implemented as a sequential model in Tensorflow Keras, comprised an embedding layer [9], two LSTM layers for capturing sequential patterns, and a dense layer for output. The model was compiled using sparse categorical cross entropy loss and the Adam optimizer. The sequential model designed for sequence processing with a specific focus on capturing patterns in the sequences related to the classification task.
For training, the model involved utilization of prepared predictors and labels over 15 epochs to efficiently capture underlying patterns in the data. Additionally, a function was used to generate a new sequence based on a given seed text and the trained generative model. The sequence generation process involved predicting the next word with a controlled level of randomness to introduce diversity while preserving the patterns in the entire data [10] enhancing the generative capabilities of the model. A detailed architecture of DL-DVE model for sequence classification and generation is shown in Figure 1.
2.2.1. Comparison of Generative Models2.2.2. ConV1d
We used ConV1d, a CNN based model implemented with an embedding layer to represent words in a continuous vector space. A single Conv1D layer with 128 filters and a kernel size of 5 captured local patterns, GlobalMaxPooling1D reduced the dimensionality, and a dense layer with a softmax activation generated a probability distribution over the entire network.
2.2.3. GRU
The model consisted of an embedding layer for word representation, followed by two GRU layers with 100 units, capturing the sequential patterns. The final dense layer outputs a probability distribution.
2.2.4. Simple RNN
A simple RNN model was implemented using an embedding layer converting nucleotides into 50-dimensional vectors, followed by two simple RNN layers with 100 units each. The first layer returned sequences, capturing the temporal patterns, while the second provided a condensed representation. The final dense layer outputs probabilities making its suitability for our sequence generation task. We further analyzed the generated sequences using BLAST search and Pairwise2 algorithm to assess the most similar type of sequence and similarity scores with other serotypes using Biopython. Furthermore, we used MEME Suite (version 5.5.5) to enrich our understanding of the conserved patterns and motifs in the sequences [11].
2.3. DL-DVE Architecture and Working for Sequence Classification
The Biopython library was used to extract genomic sequences of DENV from FASTA files. Sequence parsing facilitated the extraction of sequences and associated virus types. The unique nucleotide characters within the sequences were identified to assess dataset diversity. To enable the deep learning models utilization, nucleotide characters were mapped to numerical values and vice versa. One- hot encoding transformed the sequences into numerical representations [12]. Dengue virus types underwent conversion to numerical labels using scikit-learn’s LabelEncoder for multi-class classification modeling [13]. The dataset was divided into training and testing sets, with 80% of the data allocated for training and 20% for testing ensuring the model’s ability to generalize to the unseen data.
A FNN model was deployed using Tensorflow Keras library. The model consists of a flattening layer, a dense hidden layer employing Rectified Linear Unit (ReLU) activation, and an output layer utilizing softmax activation for effective multi-class classification. The pivotal role of non-linear activation function is highlighted post-convolution, particularly in comprehending CNN dynamics. Among the most commonly employed activation functions, namely ReLU, Sigmoid, and Tanh, ReLU demonstrates accelerated learning. The outer layer employed Softmax activation, enabling the assessment of class probabilities in prediction scenarios. The FNN integrated multiple filters traversing a one-hot encoded binary vector representing the sequence.
The model underwent compilation utilizing the categorical cross-entropy loss function and the Adam optimizer. During training, the Adam optimizer dynamically adjusted weights and biases, while the sparse categorical cross entropy loss function quantified the dissimilarity between predicted probabilities and true labels. The training spanned 10 epochs with a batch size of 32, incorporating 10% of the training data for validation purposes. Evaluation on the testing set gauged the model’s accuracy in predicting virus type, with predictions made on a subset of testing data compared against actual virus types to assess performance. To comprehensively evaluate the model’s effectiveness, various classification metrics, including accuracy, precision, recall, and F1 score were computed. A user-friendly interface was developed to facilitate the input of new viral sequences. The provided sequence underwent preprocessing and was fed to the trained model to predict the associated virus type. The workflow and architecture of classification and generative model is shown in Figure 1.
3. Results
Complete genome sequence data of DENV from the four existing serotypes was employed in our LSTM model with the aim of generating sequences that exhibit the probability of emerging as a new serotype. Initially, we employed a range of models including ConV1d, GRU, Simple RNN and our proposed LSTM model, aiming to ascertain the most effective approach. Notably, at 10 epoch, our LSTM model demonstrated 37% accuracy, outperforming ConV1d, GRU and Simple RNN. Moreover, when trained at 10 epochs, our LSTM model generated sequences exceeding 10kb in size, which distinguished it from the other models that generated sequences of less than 10kb.
Recognizing the potential for enhancement, we extended the training duration of our LSTM model to 15 epochs. This adjustment yielded a substantial increase in accuracy, reaching 93%. This improvement underscored the efficacy of prolonged training in refining the model’s predictive capabilities as shown in Table 1.
To further analyze the origin of generated sequences, we employed a multi-class classification approach using the FNN model for genomic sequence classification. Our study contributes to the landscape of genomic analysis, employing one-hot encoding, a proven method for next generation sequencing reads and phenotype label abstraction. Our CNN based FFN classification model trained on DENV complete genome data achieved an accuracy of approximately 99.00%. Across the 10 training epochs, the model consistently improved, achieving a final validation accuracy of 98.12% (Figure 2).
The model demonstrated reliable results in achieving high-quality predictions across multiple evaluation metrics with an overall accuracy, precision, recall, and F1 score of 99.00% as shown in Figure 3.
The model’s performance was assessed through the ROC-AUC curve. The loss function appeared to decrease significantly and the area under the ROC-AUC on the validation set showed fluctuations, ultimately stabilizing around 0.818. This robust accuracy, coupled with the low training loss, suggests the model's effectiveness in accurately classifying instances as shown in Figure 4.
We investigated the model’s performance by inserting unknown sequences of the dengue virus genome. The model defined a serotype for that sequence. Which was further cross-checked the predicted serotype with the actual serotype confirmed from its source. The FNN model worked efficiently on the unknown dengue virus sequence data and predicted the actual classes or serotypes of dengue virus Figure 5.
Using our DL-DVE generative and classification model, the generated sequences were successfully classified as belonging to the DENV-4 serotype. We assessed the similarity between the sequences generated by our LSTM model and DENV-4 serotype. Surprisingly our investigation revealed a similarity of approximately 66%, suggesting that the generated sequences did not align closely with DENV-4 serotype. We checked the similarity of generated sequences with other serotypes and the approximate similarity observed was 63 to 66% suggesting that the generated sequences did not align closely with any known serotype as shown in Figure 6.
The generated sequences met the specified sequence length requirement and emphasize the effectiveness of our model in accurately predicting and classifying DENV serotypes.
The MEME Suite analysis contributed to an enhanced comprehension of conserved patterns shedding light on motifs within all serotypes and the emerging serotype as shown in Figure 7.
4. Discussion
With the advent of next generation sequencing, predictions have become feasible or at least possible using complete genome sequence data [14]. Genomic data have been used to classify COVID-19 variants and other viruses using deep learning approaches [15,16,17]. CNNs and LSTMs have been more frequently used for prediction of dengue cases [3,18,19,20,21,22,23,24]. That underscores the importance of neural networks combined with genomic sequences as a futuristic method capable of revolutionizing virus studies [25,26,27].
Machine learning models go beyond human reasoning and build prediction models from a number of complex combinations. The DL models such as LSTM, GRU and CNN have been used for sequence classification and generative tasks [28]. Complete genome sequencing data was used to detect HCV variants that showed resistance to direct-acting antivirals. And the identified variants were incorporated into machine learning algorithms for assessment of effectiveness of the predictive model [29]. The LSTM model is considered effective in capturing the complex patterns in data and multiple features to make accurate predictions [30]. Being a subtype of RNNs, these models possess an enhanced capability to learn information from distant points in time. While traditional RNNs encounter the vanishing gradient problem impeding their ability to capture changes that occurred in data long ago. LSTMs overcome this challenge through a gating mechanism where the gates open and close based on values learned from each input. This mechanism enables LSTMs to accumulate information over an extended period by dynamically learning to forget certain aspects of information. This aspect of sequence length carried significant implications for the model’s predictive capacity and biological relevance.
In the realm of biological sequence analysis, machine learning and deep learning using CNNs have demonstrated high precision for binary or multi-class classification [31]. Nucleotide sequence based studies have typically employed one-hot encoding vectors to represent each nucleotide and with all unknown nucleotides represented as all zero vectors. Chaos game representation (CGR), particularly Frequency CGR (FCGR) has shown promise in encoding sequences in image format and has been applied to predict drug resistance [32]. The identification of novel genomic regions in viral pathogens using CNNs and LSTMs have emerged as compelling areas of exploration among researchers [1]. In the CNN framework, the initialization of filter weights involves random uniformness, and these weights are subsequently refined through the backpropagation process to minimize the loss or cost function. The iterative learning process allowed the network to adapt and optimize its performance enhancing its ability to discern meaningful patterns and features within the given data.
An approximate sequence similarity of 65% among DENV serotypes has been demonstrated in multiple studies [33,34,35]. The exploration of motifs with statistical significance was integral in our study of complete genome sequence data encompassing DENV 1 to 4 serotypes, juxtaposed with the generated sequences [36].
5. Conclusions
Our study demonstrates the effectiveness of utilizing sequential models for classifying and generating DENV genomic sequences resulting in the generation of sequences resembling a potential emerging serotype. Our predictive model classified the generated sequences as belonging to DENV-4 serotypes, showing a close resemblance to DENV-4 with a 65.99% sequence similarity while diverging significantly from existing serotypes. The intra-serotype divergence was characterized by a sequence similarity of at least 90%, affirming the distinctiveness of generated sequences within the DENV serotype landscape; further exploration includes the analysis of conserved motifs. Considering the complete genome sequence size of DENV genome is approximately 10kb, our trained model is limited to predict DENV sequences of similar size. Nonetheless, in future directions, predictive modeling applied well in advance of mutations holds promise for informing vaccine design and the development of diagnostic kits.
Author Contributions
Conceptualization, M.Z.Y. and Z.M.; methodology, Z.M. and H.A.S.; software, M.H.Q; validation, R.S., M.Z.Y. and Z.M.; formal analysis, M.H.Q.; investigation, R.S.; data curation, Z.M.; writing—original draft preparation, Z.M.; writing—review and editing, R.S.; visualization, Z.M and H.A.S.; supervision, M.Z.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The source code for this study can be accessed at the GitHub repository: https://github.com/Ziloeuvre/DL-DVE.git.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bartoszewicz, J. M.; Seidel, A.; Renard, B. Y. Interpretable Detection of Novel Human Viruses from Genome Sequencing Data. NAR Genomics Bioinforma. 2021, 3 (1), lqab004. [CrossRef]
- Kuno, G. A Re-Examination of the History of Etiologic Confusion between Dengue and Chikungunya. PLoS Negl. Trop. Dis. 2015, 9 (11), e0004101. [CrossRef]
- Mello-Román, J. D.; Mello-Román, J. C.; Gómez-Guerrero, S.; García-Torres, M. Predictive Models for the Medical Diagnosis of Dengue: A Case Study in Paraguay. Comput. Math. Methods Med. 2019, 2019, 1–7. [CrossRef]
- Pineda-Peña, A.-C.; Faria, N. R.; Imbrechts, S.; Libin, P.; Abecasis, A. B.; Deforche, K.; Gómez-López, A.; Camacho, R. J.; De Oliveira, T.; Vandamme, A.-M. Automated Subtyping of HIV-1 Genetic Sequences for Clinical and Surveillance Purposes: Performance Evaluation of the New REGA Version 3 and Seven Other Tools. Infect. Genet. Evol. 2013, 19, 337–348. [CrossRef]
- Rachata, N.; Charoenkwan, P.; Yooyativong, T.; Chamnongthal, K.; Lursinsap, C.; Higuchi, K. Automatic Prediction System of Dengue Haemorrhagic-Fever Outbreak Risk by Using Entropy and Artificial Neural Network. In 2008 International Symposium on Communications and Information Technologies; IEEE: Vientiane, Laos, 2008; pp 210–214. [CrossRef]
- Kosakovsky Pond, S. L.; Posada, D.; Stawiski, E.; Chappey, C.; Poon, A. F. Y.; Hughes, G.; Fearnhill, E.; Gravenor, M. B.; Leigh Brown, A. J.; Frost, S. D. W. An Evolutionary Model-Based Algorithm for Accurate Phylogenetic Breakpoint Mapping and Subtype Prediction in HIV-1. PLoS Comput. Biol. 2009, 5 (11), e1000581. [CrossRef]
- Altschul, S. F.; Gish, W.; Miller, W.; Myers, E. W.; Lipman, D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215 (3), 403–410. [CrossRef]
- Olson, R. D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J. J.; Dempsey, D. M.; Dickerman, A.; Dietrich, E. M.; Kenyon, R. W.; Kuscuoglu, M.; Lefkowitz, E. J.; Lu, J.; Machi, D.; Macken, C.; Mao, C.; Niewiadomska, A.; Nguyen, M.; Olsen, G. J.; Overbeek, J. C.; Parrello, B.; Parrello, V.; Porter, J. S.; Pusch, G. D.; Shukla, M.; Singh, I.; Stewart, L.; Tan, G.; Thomas, C.; VanOeffelen, M.; Vonstein, V.; Wallace, Z. S.; Warren, A. S.; Wattam, A. R.; Xia, F.; Yoo, H.; Zhang, Y.; Zmasek, C. M.; Scheuermann, R. H.; Stevens, R. L. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A Resource Combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51 (D1), D678–D689. [CrossRef]
- Cui, F.; Zhang, Z.; Zou, Q. Sequence Representation Approaches for Sequence-Based Protein Prediction Tasks That Use Deep Learning. Brief. Funct. Genomics 2021, 20 (1), 61–73. [CrossRef]
- Dasari, C. M.; Bhukya, R. Explainable Deep Neural Networks for Novel Viral Genome Prediction. Appl. Intell. 2022, 52 (3), 3002–3017. [CrossRef]
- Bailey, T. L.; Boden, M.; Buske, F. A.; Frith, M.; Grant, C. E.; Clementi, L.; Ren, J.; Li, W. W.; Noble, W. S. MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37 (Web Server), W202–W208. [CrossRef]
- Choong, A. C. H.; Lee, N. K. Evaluation of Convolutionary Neural Networks Modeling of DNA Sequences Using Ordinal versus One-Hot Encoding Method. In 2017 International Conference on Computer and Drone Applications (IConDA); IEEE: Kuching, 2017; pp 60–65. [CrossRef]
- Langton, J.; Srihasam, K.; Jiang, J. Comparison of Machine Learning Methods for Multi-Label Classification of Nursing Education and Licensure Exam Questions. In Proceedings of the 3rd Clinical Natural Language Processing Workshop; Association for Computational Linguistics: Online, 2020; pp 85–93. [CrossRef]
- Shim, H. Futuristic Methods in Virus Genome Evolution Using the Third-Generation DNA Sequencing and Artificial Neural Networks. In Global Virology III: Virology in the 21st Century; Shapshak, P., Balaji, S., Kangueane, P., Chiappelli, F., Somboonwit, C., Menezes, L. J., Sinnott, J. T., Eds.; Springer International Publishing: Cham, 2019; pp 485–513. [CrossRef]
- Ali, S.; Sahoo, B.; Zelikovsky, A.; Chen, P.-Y.; Patterson, M. Benchmarking Machine Learning Robustness in Covid-19 Genome Sequence Classification. Sci. Rep. 2023, 13 (1), 4154. [CrossRef]
- Basu, S.; Campbell, R. H. Classifying COVID-19 Variants Based on Genetic Sequences Using Deep Learning Models. In System Dependability and Analytics; Wang, L., Pattabiraman, K., Di Martino, C., Athreya, A., Bagchi, S., Eds.; Springer Series in Reliability Engineering; Springer International Publishing: Cham, 2023; pp 347–360. [CrossRef]
- De Souza, L. C.; Azevedo, K. S.; De Souza, J. G.; Barbosa, R. D. M.; Fernandes, M. A. C. New Proposal of Viral Genome Representation Applied in the Classification of SARS-CoV-2 with Deep Learning. BMC Bioinformatics 2023, 24 (1), 92. [CrossRef]
- Manoharan, S. N.; Kumar, K. M. V. M.; Vadivelan, N. A Novel CNN-TLSTM Approach for Dengue Disease Identification and Prevention Using IoT-Fog Cloud Architecture. Neural Process. Lett. 2023, 55 (2), 1951–1973. [CrossRef]
- Majeed, M. A.; Shafri, H. Z. M.; Zulkafli, Z.; Wayayok, A. A Deep Learning Approach for Dengue Fever Prediction in Malaysia Using LSTM with Spatial Attention. Int. J. Environ. Res. Public. Health 2023, 20 (5), 4130. [CrossRef]
- Nguyen, V.-H.; Tuyet-Hanh, T. T.; Mulhall, J.; Minh, H. V.; Duong, T. Q.; Chien, N. V.; Nhung, N. T. T.; Lan, V. H.; Minh, H. B.; Cuong, D.; Bich, N. N.; Quyen, N. H.; Linh, T. N. Q.; Tho, N. T.; Nghia, N. D.; Anh, L. V. Q.; Phan, D. T. M.; Hung, N. Q. V.; Son, M. T. Deep Learning Models for Forecasting Dengue Fever Based on Climate Data in Vietnam. PLoS Negl. Trop. Dis. 2022, 16 (6), e0010509. [CrossRef]
- Nadda, W.; Boonchieng, W.; Boonchieng, E. Influenza, Dengue and Common Cold Detection Using LSTM with Fully Connected Neural Network and Keywords Selection. BioData Min. 2022, 15 (1), 5. [CrossRef]
- Doni, A.; Sasipraba, T. LSTM-RNN Based Approach for Prediction of Dengue Cases in India. Ingénierie Systèmes Inf. 2020, 25 (3), 327–335. [CrossRef]
- Zhao, X.; Li, K.; Ang, C. K. E.; Cheong, K. H. A Deep Learning Based Hybrid Architecture for Weekly Dengue Incidences Forecasting. Chaos Solitons Fractals 2023, 168, 113170. [CrossRef]
- Gunasekaran, H.; Ramalakshmi, K.; Rex Macedo Arokiaraj, A.; Deepa Kanmani, S.; Venkatesan, C.; Suresh Gnana Dhas, C. Analysis of DNA Sequence Classification Using CNN and Hybrid Models. Comput. Math. Methods Med. 2021, 2021, 1–12. [CrossRef]
- Helaly, M. A.; Rady, S.; Aref, M. M. Convolutional Neural Networks for Biological Sequence Taxonomic Classification: A Comparative Study. In Proceedings of the International Conference on Advanced Intelligent Systems and Informatics 2019; Hassanien, A. E., Shaalan, K., Tolba, M. F., Eds.; Advances in Intelligent Systems and Computing; Springer International Publishing: Cham, 2020; Vol. 1058, pp 523–533. [CrossRef]
- Ao, C.; Jiao, S.; Wang, Y.; Yu, L.; Zou, Q. Biological Sequence Classification: A Review on Data and General Methods. Research 2022, 2022, 0011. [CrossRef]
- Pérez-Enciso; Zingaretti. A Guide for Using Deep Learning for Complex Trait Genomic Prediction. Genes 2019, 10 (7), 553. [CrossRef]
- Tsai, S.-T.; Kuo, E.-J.; Tiwary, P. Learning Molecular Dynamics with Simple Language Model Built upon Long Short-Term Memory Neural Network. Nat. Commun. 2020, 11 (1), 5115. [CrossRef]
- Haga, H.; Sato, H.; Koseki, A.; Saito, T.; Okumoto, K.; Hoshikawa, K.; Katsumi, T.; Mizuno, K.; Nishina, T.; Ueno, Y. A Machine Learning-Based Treatment Prediction Model Using Whole Genome Variants of Hepatitis C Virus. PLOS ONE 2020, 15 (11), e0242028. [CrossRef]
- Lv, Z.; Ao, C.; Zou, Q. Protein Function Prediction: From Traditional Classifier to Deep Learning. PROTEOMICS 2019, 19 (14), 1900119. [CrossRef]
- Ahmad, I.; Iqbal, M. J.; Basheri, M. Biological Data Classification and Analysis Using Convolutional Neural Network. J. Med. Imaging Health Inform. 2020, 10 (10), 2459–2465. [CrossRef]
- Murad, T.; Ali, S.; Khan, I.; Patterson, M. Spike2CGR: An Efficient Method for Spike Sequence Classification Using Chaos Game Representation. Mach. Learn. 2023, 112 (10), 3633–3658. [CrossRef]
- Dieng, I.; Cunha, M. D. P.; Diagne, M. M.; Sembène, P. M.; Zanotto, P. M. D. A.; Faye, O.; Faye, O.; Sall, A. A. Origin and Spread of the Dengue Virus Type 1, Genotype V in Senegal, 2015–2019. Viruses 2021, 13 (1), 57. [CrossRef]
- Sánchez-González, G.; Belak, Z. R.; Lozano, L.; Condé, R. Probability of Consolidation Constrains Novel Serotype Emergence in Dengue Fever Virus. PLOS ONE 2021, 16 (4), e0248765. [CrossRef]
- Katzelnick, L. C.; Fonville, J. M.; Gromowski, G. D.; Arriaga, J. B.; Green, A.; James, S. L.; Lau, L.; Montoya, M.; Wang, C.; VanBlargan, L. A.; Russell, C. A.; Thu, H. M.; Pierson, T. C.; Buchy, P.; Aaskov, J. G.; Muñoz-Jordán, J. L.; Vasilakis, N.; Gibbons, R. V.; Tesh, R. B.; Osterhaus, A. D. M. E.; Fouchier, R. A. M.; Durbin, A.; Simmons, C. P.; Holmes, E. C.; Harris, E.; Whitehead, S. S.; Smith, D. J. Dengue Viruses Cluster Antigenically but Not as Discrete Serotypes. Science 2015, 349 (6254), 1338–1343. [CrossRef]
- Srionrod, N.; Nooroong, P.; Poolsawat, N.; Minsakorn, S.; Watthanadirek, A.; Junsiri, W.; Sangchuai, S.; Chawengkirttikul, R.; Anuracpreeda, P. Molecular Characterization and Genetic Diversity of Babesia Bovis and Babesia Bigemina of Cattle in Thailand. Front. Cell. Infect. Microbiol. 2022, 12, 1065963. [CrossRef]
Figure 1.
Workflow and architecture of DL-DVE for dengue virus genome classification and prediction.
Figure 1.
Workflow and architecture of DL-DVE for dengue virus genome classification and prediction.
Figure 2.
The validation accuracy of FFNN model over 10 epochs.
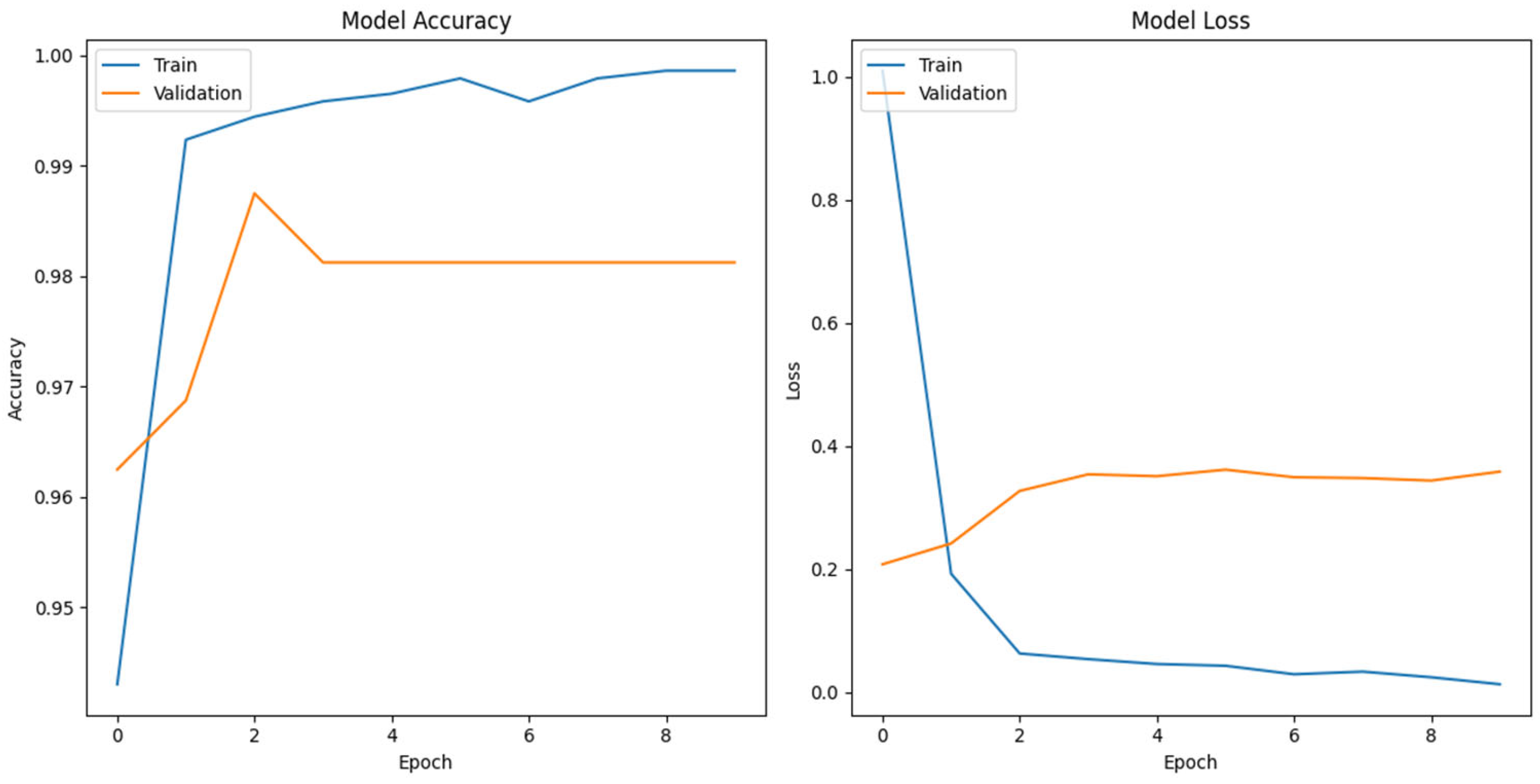
Figure 3.
Classification metrics illustrating accuracy, precision, recall and F1 score.
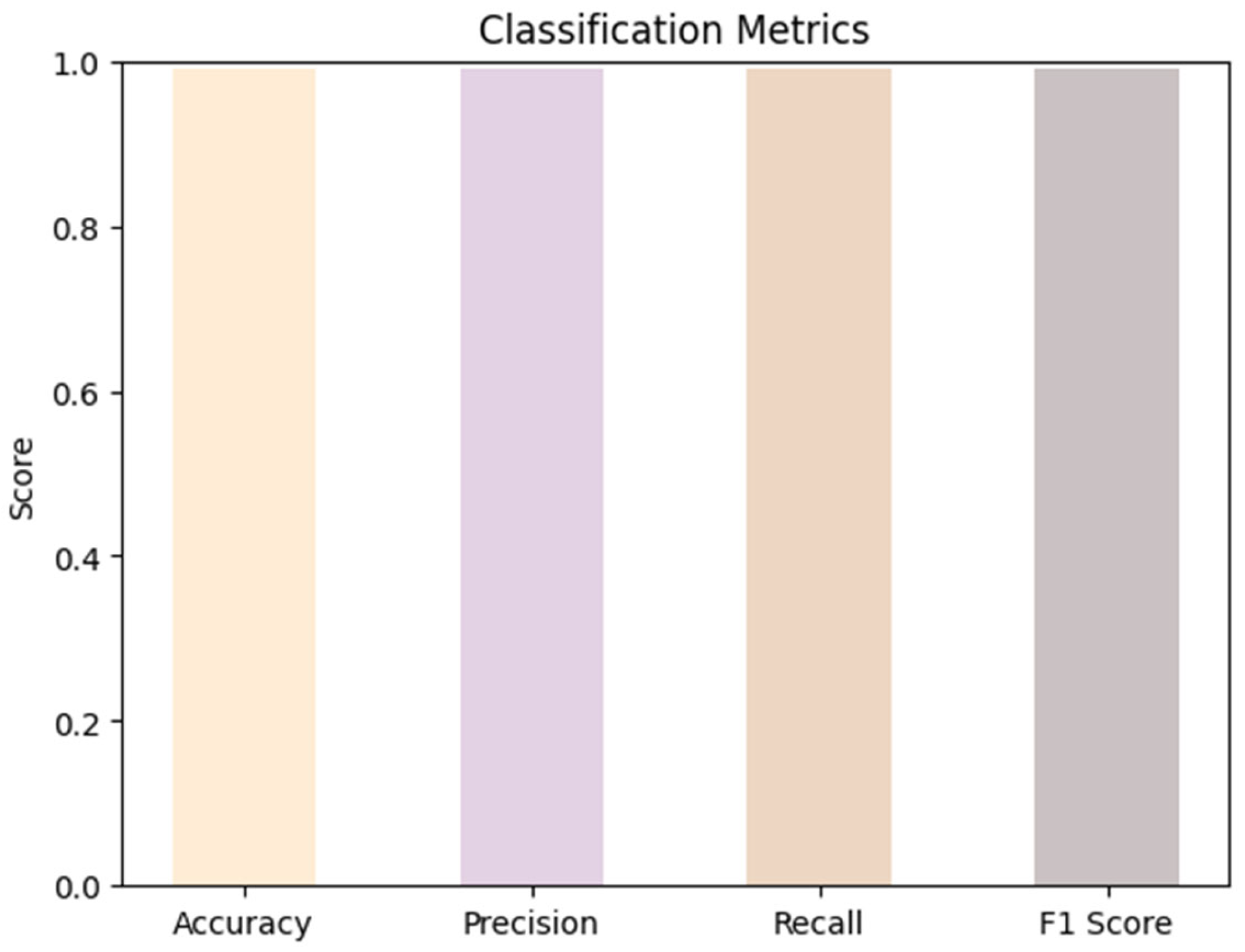
Figure 4.
ROC-AUC for FFNN model.
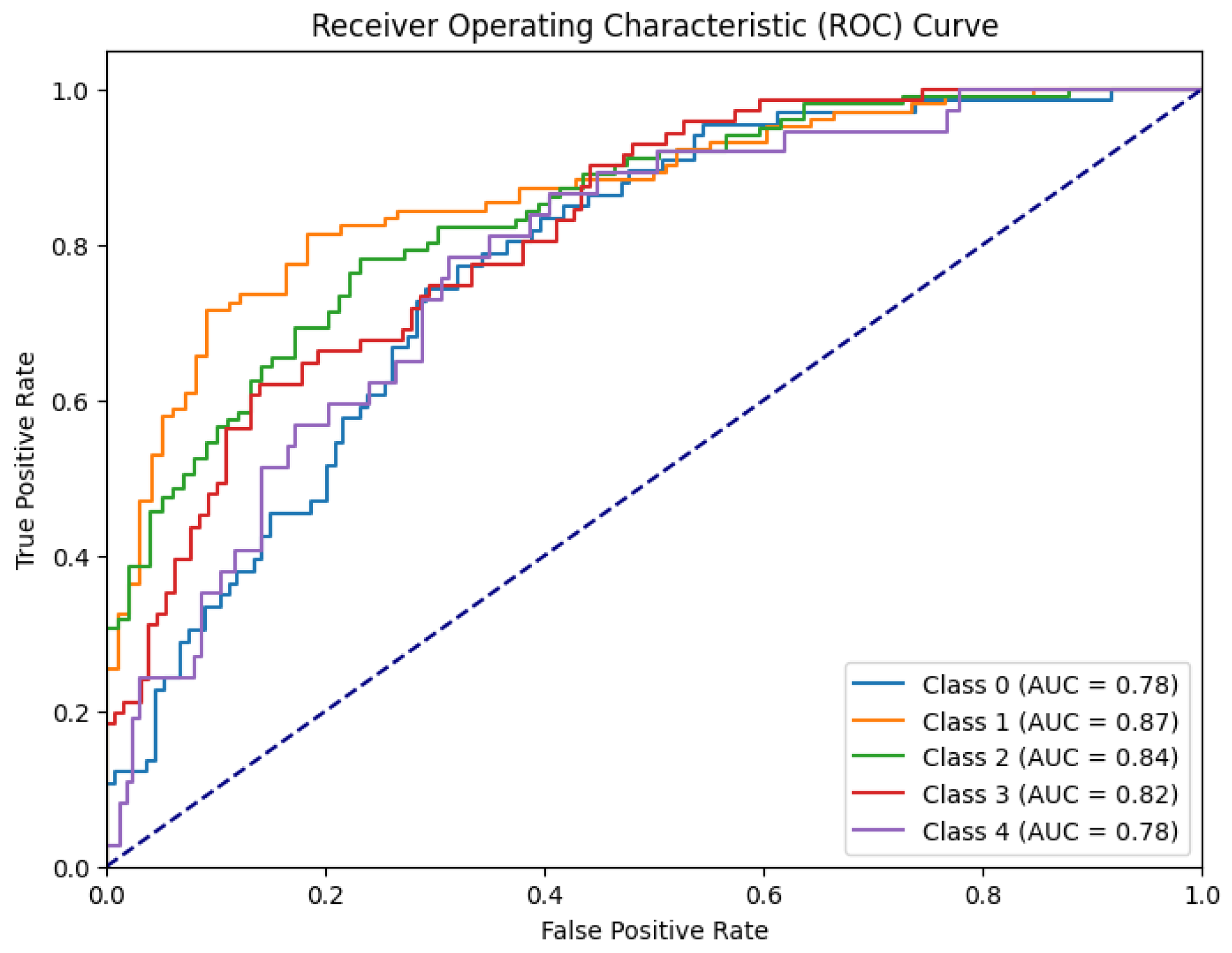
Figure 5.
FFNN model accuracy through predicted vs actual virus types.
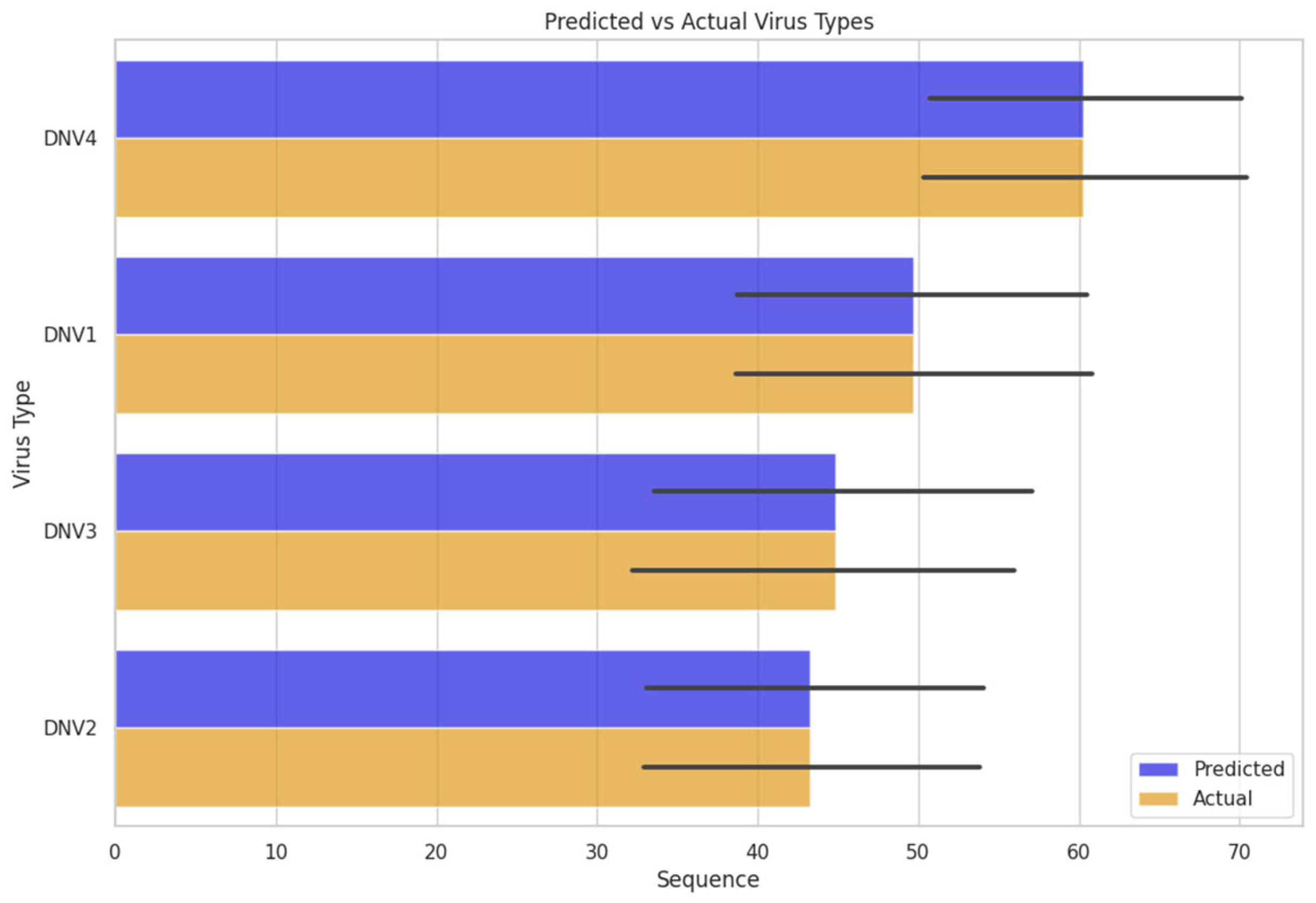
Figure 6.
Sequence similarity of intra and inter DENV serotypes and comparison with generated sequences.
Figure 6.
Sequence similarity of intra and inter DENV serotypes and comparison with generated sequences.
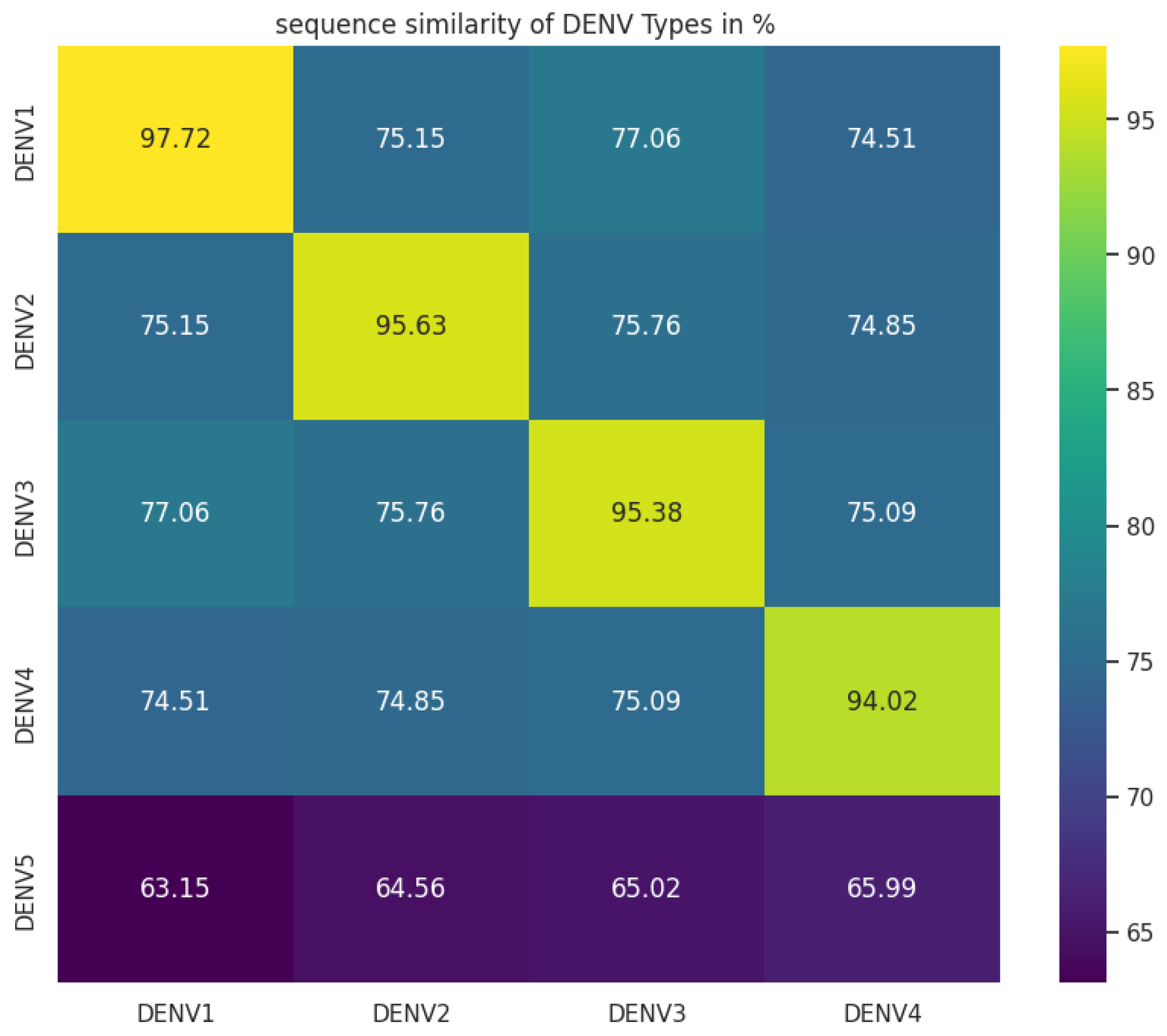
Figure 7.
Sequence Logo Analysis: A visual representation of conserved motifs in the nucleotide sequences, where the y-axis depicts information content in bits (0, 1, 2), and the x-axis represents nucleotides. E-values highlight the statistical significance of motifs, with the first motif at 1.0e-028, the second at 3.8e-026, the third at 1.8e-019, and the fourth at 3.7e-018. This sequence logo provides insights into the nucleotide composition and conservation within the analyzed motifs.
Figure 7.
Sequence Logo Analysis: A visual representation of conserved motifs in the nucleotide sequences, where the y-axis depicts information content in bits (0, 1, 2), and the x-axis represents nucleotides. E-values highlight the statistical significance of motifs, with the first motif at 1.0e-028, the second at 3.8e-026, the third at 1.8e-019, and the fourth at 3.7e-018. This sequence logo provides insights into the nucleotide composition and conservation within the analyzed motifs.

Table 1.
Performance comparison of different models for emerging sequence generation.
| Sr. no. | Model | Accuracy(%) | Epoch | Sequence generated (length) |
|---|---|---|---|---|
| 1 | ConV1d | 29 | 10 | <10kb |
| 2 | GRU | 35 | 10 | <10kb |
| 3 | Simple RNN | 30 | 10 | <10kb |
| 4 | Our proposed LSTM model | 37 | 10 | >10kb |
| 93 | 15 | >10kb |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



