
Submitted:
15 March 2024
Posted:
18 March 2024
You are already at the latest version
Abstract
Obesity results in hepatic fat accumulation, i.e., steatosis. Besides the fat overload, impaired fatty acid β-oxidation also promotes steatosis. Fatty acid β-oxidation takes place in mitochondria and peroxisomes. Usually very long chain and branched-chain fatty acids are first oxidized in peroxisomes, and the resultant short chain fatty acids are further oxidized in mitochondria. Peroxisome biogenesis is regulated by peroxin 16 (PEX16). In liver-specific PEX16 knockout (Pex16Alb-Cre) mice, hepatocyte peroxisomes were absent, but hepatocytes proliferated, and liver mass was enlarged. These results suggest that normal liver peroxisomes restrain hepatocyte proliferation and liver sizes. After high fat diet (HFD) feeding, body weights were increased in PEX16 floxed (Pex16fl/fl) mice and adipose-specific PEX16 knockout (Pex16AdipoQ-Cre) mice but not in the Pex16Alb-Cre mice, suggesting that the development of obesity is regulated by liver PEX16 but not by adipose PEX16. However, it was previously reported that HFD-induced obesity in the Pex16AdipoQ-Cre mice was more severe than in the wild type mice, which was not observed until 27 weeks of HFD feeding. Thus, liver initiates the development of obesity, which is enhanced by abnormal adipocytes. HFD increased liver mass in the Pex16fl/fl mice but somehow reduced the already enlarged liver mass in the Pex16Alb-Cre mice. Basal levels of serum triglyceride, free fatty acids and cholesterol were decreased whereas serum bile acids were increased in the Pex16Alb-Cre mice, and HFD-induced steatosis was not observed in the Pex16Alb-Cre mice. These results suggest that normal liver peroxisomes contribute to the development of liver steatosis and obesity.
Keywords:
peroxisomes
; bile acid
; fatty acid oxidation
1. Introduction
Obesity is closely linked with metabolic dysfunction-associated steatotic liver disease (MASLD), previously known as non-alcoholic fatty liver disease (NAFLD), which starts from steatosis (fatty liver) [1]. When adipose triglyceride (TG) is degraded into free fatty acids (FFA) and released into blood, livers will reabsorb the FFA for re-synthesis of TG, which binds to apolipoprotein B (apo B) to be released into blood in the form of very low-density lipoprotein (VLDL). However, steatosis is formed if TG is accumulated in liver to produce lipid droplets [2]. The early stages of steatosis are primarily asymptomatic; however, the long-term fatty liver has a progression from steatosis to steatohepatitis, fibrosis, and cirrhosis leading to hepatocellular carcinoma [1]. The impairment of fatty acid β-oxidation (FAO) is one of major reasons for the development of steatosis. Mitochondria are known as major organelles for FAO. Peroxisomes also oxidize fatty acids [3]. Usually, very long chain and branch chain fatty acids are oxidized in peroxisomes and the resultant shorter chain fatty acids will be further oxidized in mitochondria. The first reaction of peroxisomal FAO is catalyzed by the rate-limiting enzyme acyl-CoA oxidase (ACOX), which produces hydrogen peroxide (H2O2) as a byproduct, and the generated H2O2 is locally decomposed by peroxisomal catalase [4].
It is well known that peroxisome proliferator-activated receptor α (PPARα) regulates peroxisomal FAO [5,6], and PPARα agonist WY-14,643 prevents hepatic TG accumulation in the diet-induced obese mice or the ethanol-fed mice [7,8]. Global or organ-specific abrogation of pparα deteriorated abnormal lipid metabolism in mice [9]. PPARα agonist prevented obesity in ob/ob obese mice [10] and pparα absence in ob/ob obese mice make the mice more obese [11], suggesting that PPARα protects against obesity in mice. PPARα also regulates fatty acid oxidation (FAO) and protects against hepatic steatosis in patients. For example, expression of PPARα in human liver is reduced in NAFLD patients and negatively correlates with NAFLD severity [12]. Interestingly, in the mice lacking cytochrome P450 2A5 (CYP2A5) i.e., cyp2a5-/- mice, we observed an elevated basal level of PPARα but more severe ethanol- and HFD-induced steatosis [13,14,15], but when the pparα-/-/cyp2a5-/- mice are created to abrogate the upregulated PPARα, HFD-induced steatosis was more pronounced, and evident liver necroinflammation and fibrosis were observed in the pparα-/-/cyp2a5-/- mice but not in the cyp2a5-/- mice or pparα-/- mice [14,15], suggesting that PPARα is still protective against the HFD-induced steatosis, steatohepatitis, and fibrosis.
PPARα also regulates peroxisome proliferation in liver, and consistently PPARα agonists-induced peroxisome proliferation was not observed in the pparα -/- mice [16]. Peroxisomes can be generated de novo by budding from endoplasmic reticulum (ER). Recently it was reported that newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes [17]. Peroxisomal membrane proteins (PMP) are synthesized in the ER [18]. Peroxisomes require a group of proteins called peroxins (PEX) for their assembly and division. PEX16 is an integral membrane protein and acts as a docking site to recruit PEX3. PEX3 is a docking receptor for PEX19, and PEX19 is an import receptor for newly synthesized peroxisomal membrane proteins [18,19]. Thus, PEX16 plays a pivotal role in the peroxisome biogenesis [20]. The loss of PEX16 results in the complete absence of any peroxisomal structures in patients [21]. In the hepatocyte-specific PEX16 knockout mice (Pex16Alb-Cre), peroxisome marker PMP70 was absent [22], indicating that peroxisomes are also absent. In this study, we applied the Pex16Alb-Cre mice to examine the effects of liver peroxisomes on HFD-induced alterations in hepatic lipid metabolism, steatosis, and development of obesity.
2. Materials and Methods
2.1. Studies in Experimental Rodent Models
Mice and Treatment: Pex16 floxed mice (Pex16fl/fl mice; purchased from Jackson Laboratory, strain number #034155) were crossed with transgenic mice expressing Albumin-Cre recombinase (Alb-Cre mice; purchased from Jackson Laboratory, strain number #003574) and or expressing adiponectin-Cre recombinase (adipoQ-Cre mice; purchased from Jackson Laboratory, strain number #028020) to create the Pex16Alb-Cre mice and adipose-specific PEX16 knockout (Pex16AdipoQ-Cre) mice, respectively. The littermates Pex16fl/fl mice not expressing Alb-Cre or AdipoQ-Cre were used as normal control mice. All the mice were housed in temperature-controlled animal facilities with 12-hour light/dark cycles and were permitted consumption of tap water and Purina standard chow ad libitum. The mice received humane care. All in vivo experiments were approved by the Institution Committee of Animal Use and Care (IACUC) at Marshall University.
Six to eight weeks old male mice were fed HFD for 10 weeks to induce obesity and steatosis. The mice in control groups were fed control diet (CD). The HFD and CD diets were purchased from Bio-Serv company (Frenchtown, NJ, USA). HFD and CD contain the same amount (20.5%) of protein. In HFD, 60% calories are from fat, but in CD only 16% calories are from fat. All mice were permitted consumption of tap water and HFD or CD ad libitum. Mouse body weight and diet consumption were measured weekly. The amount of diet consumption was not significantly different between the HFD and CD.
Glucose tolerance test: After 9 weeks of HFD feeding, the mice were subjected to an overnight fast (15h) followed by glucose injection intraperitoneally at 1 g/kg. Blood was collected from tails before glucose injection for measuring basal levels of blood glucose by Bayer Contour blood glucose meter. After glucose injection, tail blood was collected at 30, 60, 90, and 120 min for glucose assays, respectively.
Liver pathology: After another 6 days of feeding (totally 10 weeks), the mice were sacrificed following an overnight fast (15 h). Blood was collected for isolating serum. The livers and gonadal white adipose tissues were collected and weighted, and liver index and fat index were calculated as liver weight/100g body weight and gonadal adipose tissue weight/100g body weight. One piece of liver and gonadal adipose tissue from each mouse were put in Neutral Formalin Buffer for preparing paraffin sections for Hematoxylin & Eosin (HE) staining and immunohistochemical staining (IHC). Liver sections with HE staining were used for pathological evaluation. The criteria for grade of steatosis are based on the percentage of hepatocytes containing lipid droplets: 0, none; 1, <5%; 2, 5-33%; 3, 34-66%; 4, >67%. For IHC, a Broad Spectrum IHC Select® HRP/DAB kit (from EMD Millipore, Cat#: DAB150) was used. Five 100x fields per liver (one 100x field area = 2.54 mm2) were examined for quantification of positive staining.
Biochemical assays and Western Blotting analysis: The liver tissue aliquots were stored at -80°C. The liver tissues were homogenized in 0.15M potassium chloride (KCL) to make homogenates for TG contents and Western blotting analyses. The isolated serum was used for measuring TG, FFA, β-hydroxybutyrate, bile acid, glucose, and cholesterol. Commercially available assay kits and primary antibodies are listed in Table 1.
Statistical analysis: Results are expressed as mean ± S.D. Statistical evaluation was carried out by using two-way analysis of variance (ANOVA) with subsequent the Student-Newman-Keuls post hoc test. For body weight changes, Repeated Measures ANOVA was carried out. P<0.05 was considered as statistical significance.
2.2. Studies in Patients with Chronic Liver Diseases
Liver paraffin sections from patients with primary chronic liver diseases including gallstone, cholecystitis, hepatic cyst, hepatic hemangioma, cirrhosis, and hepatocellular carcinoma were collected from Guiqian International General Hospital, Guiyang, China in the past three years. The adjacent or distal tissues of the original liver lesions were blindly diagnosed and scored by a pathologist (L.W.). Thirty-nine cases with typical spectrum of MASLD (steatosis, inflammation, fibrosis) were selected for IHC. Primary antibodies were purchased from international suppliers: ACOX-1(1:300, Atlas Antibodies, Stockholm, Sweden); PEX16, PMP70, and Catalase (1:500, Bioss Antibodies, Beijing, China); Cyclooxygenase 2 (1:500, Abcam, Boston, USA); MDA (1:100, Kanglang Biotechology, Shanghai, China). IHC UltraView Univeral DAB kit was used to detect above markers by polymer method at Ventana ULTRA automatic instrument (Roche Diagnostics, Rotkreuz, Switzerland). Heat-mediated antigen retrieval was performed with epitope retrieval solution Tris-EDTA buffer (pH 9.0), and hematoxylin staining was used as a counterstain.
3. Results
3.1. Resistance to HFD-Induced Body Weight Gain Is Observed in the Pex16Alb-Cre Mice but Not in the Pex16AdipoQ-Cre Mice
The male Pex16fl/fl mice, Pex16AdipoQ-Cre mice (AKO), and Pex16Alb-Cre mice (LKO mice) were fed HFD, and the body weight were measured on a weekly basis. As shown in Figure 1A, the HFD-induced body weight gain gradually increased with a linear relationship in all groups. The slope was about 2.0, 1.6, 0.6 for the Pex16fl/fl mice, Pex16AdipoQ-Cre mice, and Pex16Alb-Cre mice, respectively. We used Repeated Measures ANOVA to compare the 3 groups (Figure 1B). There was no difference in HFD-induced body weight gain between the Pex16fl/fl mice and the Pex16AdipoQ-Cre mice. Liver mass as indicated by liver index and gonadal adipose tissue mass as indicated by fat index were comparable in the Pex16fl/fl mice and the Pex16AdipoQ-Cre mice (Figure 1C). The HE staining in the sections of adipose tissues showed that the sizes of adipocytes in the HFD-fed Pex16fl/fl mice and HFD-fed Pex16AdipoQ-Cre mice, and the inflammation as indicated by “Crown” formation was observed in both the HFD-fed Pex16fl/fl mice and the HFD-fed Pex16AdipoQ-Cre mice (Figure 1D). Similarly, HFD-induced steatosis as indicated by lipid droplet formation was comparable in the Pex16fl/fl mice and the Pex16AdipoQ-Cre mice (Figure 1E). These results suggest that the absence of adipose PEX16 does not have an influence on HFD-induced obesity and the associated steatosis. Body weights were increased in the Pex16Alb-Cre mice to a much lesser extent than in the Pex16fl/fl mice and the Pex16AdipoQ-Cre mice (Figure 1A,B), suggesting that the absence of liver PEX16 leads to a resistance to HFD-induced body weight gain.
HFD feeding induces obesity in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice.
Interestingly, the basal levels of gonadal adipose tissue mass (fat index) were lower in the Pex16Alb-Cre mice than in the Pex16fl/fl mice, and in response to the HFD feeding the gonadal adipose tissues were expanded to a greater extent in the Pex16fl/fl mice than in the Pex16Alb-Cre mice (Figure 2A). The HE staining in the sections of adipose tissues showed that the adipocytes in the HFD-fed Pex16fl/fl mice were about 50-60 µm in diameter, which was larger than those in the HFD-fed Pex16Alb-Cre mice (about 30-40 µm in diameter), and the inflammation as indicated by “Crown” formation was observed in the HFD-fed Pex16fl/fl mice but not in the HFD-fed Pex16Alb-Cre mice (Figure 2B). Like the HFD-induced body weight gain, HFD induced hyperglycemia in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice (Figure 2C). Glucose tolerance test showed an intolerance to the bolus injection of glucose in the Pex16fl/fl mice to a greater extent than in the Pex16Alb-Cre mice (Figure 2D). These results suggest that HFD induces obesity in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice.
3.2. HFD Feeding Induces Steatosis in the Pex16fl/fl Mice but Not in the Pex16Alb-Cre Mice
In contrast to gonadal adipose tissues, basal levels of liver mass (liver index) were lower in the Pex16fl/fl mice than in the Pex16Alb-Cre mice, and HFD feeding lowered the liver index in both the Pex16fl/fl mice and Pex16Alb-Cre mice although it was still higher in the Pex16Alb-Cre mice than in the Pex16fl/fl mice (Figure 3A). The mechanisms by which HFD lowered the liver index are different in the Pex16Alb-Cre mice than in the Pex16fl/fl mice. In the Pex16fl/fl mice, both the body weight and liver weight were increased by the HFD feeding, but the body weights increased (Week 0:18.96 ± 2.21 g vs Week 10: 39.44 ± 2.78 g) to a greater extent than liver weights did (CD: 0.99 ± 0.08 g vs HFD: 1.7 ± 0.26 g), so liver index was lowered. However, in the Pex16Alb-Cre mice, after the HFD feeding, liver weights were not increased but somehow decreased (CD: 2.37 ± 0.013 g vs HFD: 1.75 ± 0.22 g), and body weights were almost not changed (Week 0: 22.48 ± 0.46 g vs Week10: 27.62 ± 3.62 g). Interestingly, liver TG contents were increased by the HFD feeding in the Pex16fl/fl mice to a greater extent than in the Pex16Alb-Cre mice (Figure 3B). The HE staining showed that HFD-induced steatosis as indicated by lipid droplet formation was observed in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice (Figure 3C,D). Very interestingly, the sizes of hepatocyte cell nucleus in the Pex16Alb-Cre mice were smaller than in the Pex16fl/fl mice, and correspondingly, the number of hepatocyte cell nucleus per mm2 was higher in the Pex16Alb-Cre mice than in the Pex16fl/fl mice (Figure 3E).
3.3. The Absence of Liver PEX16 Leads to Hepatocyte Proliferation
To check whether the increase in hepatocyte nuclear number is resulted from hepatocyte proliferation, cell proliferation markers proliferating cell nuclear antigen (PCNA) and Ki67 were detected by IHC. PCNA staining was negative and HFD did not induce PCNA in the Pex16fl/fl mice, but it was positive in the Pex16Alb-Cre mice, fed with CD or HFD (Figure 4A,B). Ki67 staining was negative in the CD-fed Pex16fl/fl mice but HFD slightly induced Ki67 positive staining in the Pex16fl/fl mice. Ki67 was positively stained in the Pex16Alb-Cre mice, but HFD did not further increase the Ki67 staining (Figure 4A,C). These results suggest that absence of PEX16 leads to hepatocyte proliferation, but HFD has no effect on the hepatocyte proliferation.
3.4. The Absence of Liver PEX16 Leads to Alteration of Fatty Acid Metabolism in the Liver
In the Pex16Alb-Cre mice, the absence of PEX16 was confirmed by the Western blotting analysis (Figure 5A). Peroxisome marker PMP70 was undetectable, indicating the absence of peroxisomes in the Pex16Alb-Cre mice. In the peroxisomal matrix, fatty acid β-oxidation enzyme ACOX1 and thiolase oxidize long straight chain fatty acids, and ACOX2 and its downstream enzyme sterol carrier protein x (SCPx) oxidize branched-chain fatty acids [23]. In the Pex16Alb-Cre mice, ACOX2 and SCPx were downregulated. However, ACOX1 and thiolase were upregulated. Aligning with the ACOX1, catalase was also upregulated in the Pex16Alb-Cre mice (Figure 5A). Consistent with the upregulation of ACOX1, thiolase and catalase, basal levels of serum fatty acids were lower in the Pex16Alb-Cre mice than in the Pex16fl/fl mice, and HFD tended to elevate serum fatty acids in the Pex16Alb-Cre mice (Figure 5B). Likewise, basal levels of serum β–hydroxybutyrate, a major component of ketone bodies, were also lower in the Pex16Alb-Cre mice, and HFD tended to elevate serum β–hydroxybutyrate (Figure 5C). FFA is absorbed through fatty acid transporters CD36 or synthesized by fatty acid synthase (FASN) and transferred by liver fatty acid-binding protein (L-FABP) [24]. Basal CD36 expression was not changed but basal expressions of L-FABP and FASN were upregulated in the Pex16Alb-Cre mice (Figure 5D). In the Pex16Alb-Cre mice, HFD feeding induced L-FABP but inhibited liver CD36 and FASN. The 3-Hydroxy-3-methylglutaryl-CoA synthase-2 (HMGCS2) mediates the rate-limiting step in mitochondrial synthesis of ketone bodies [25], but HMGCS2 was downregulated in the Pex16Alb-Cre mice (Figure 5D). ATGL (adipose triglyceride lipase) is responsible for the initial step of TG degradation [26], and MTTP (microsomal triglyceride transfer protein) transfers newly synthesized TG in the ER for assembly of VLDL [27]. Basal levels of ATGL and MTTP were not changed (Figure 5D), although basal serum TG levels were lower in the Pex16Alb-Cre mice than in the Pex16fl/fl mice (Figure 5D).
3.5. The Absence of Liver PEX16 Leads to Alteration of Cholesterol and Bile Acid Metabolism
HMGCS1 is upregulated in the Pex16Alb-Cre mice (Figure 6A). Unlike HMGCS2, which is located in mitochondria for the synthesis of ketone bodies, HMGCS1 is for cholesterol synthesis [29]. The 3-Hydroxy-3-methylglutaryl-CoA reductase (HMGCR) is a rate limiting enzyme of cholesterol synthesis [28]. HMGCR was not altered in the Pex16Alb-Cre mice, but basal levels of serum cholesterol were lower in the Pex16Alb-Cre mice than in the Pex16fl/fl mice (Figure 6B). Cholesterol is used for bile acid synthesis [30]. Correspondingly, serum levels of bile acid were higher in the Pex16Alb-Cre mice than in the Pex16fl/fl mice (Figure 6C). CYP7A1 is a rate limiting enzyme to catalyze bile acid synthesis [30], but CYP7A1 was not changed in the Pex16Alb-Cre mice [30]. As an intracellular bile acid sensor, the nuclear receptor farnesoid X receptor (FXR) is critical for bile acid and lipid homeostasis [31]. FXR was almost undetectable in the Pex16Alb-Cre mice (Figure 6C), implying that the elevation of serum bile acids is associated with the impaired regulation of FXR on bile acid homeostasis.
3.6. Peroxisomes Are Associated with Liver Steatosis in Patients with Typical Spectrum of MASLD
Besides cirrhosis and hepatocellular carcinoma that are classified in MASLD, primary chronic liver diseases like gallstone, cholecystitis, hepatic cyst, hepatic hemangioma also exhibited typical spectrum of MASLD, i.e., steatosis, steatohepatitis and fibrosis. In adjacent tissues and distal tissues of the original lesions, ballooning degeneration, steatosis, necroinflammation, and fibrosis were observed (Figure 7). The patients’ liver sections displaying typical spectrum of MASLD were selected to study the association of peroxisomes with liver steatosis. IHC for peroxisomal membrane proteins (PEX16 and PMP70) and peroxisomal matrix enzymes (catalase and ACOX1) were performed in these liver sections. As shown in Figure 8, PEX16 and PMP70 were stained much stronger in the area full of lipid droplets (steatotic area) than in the area without lipid droplets (non-steatotic area), while catalase was identically stained regardless of lipid droplets. Interestingly, ACOX1 staining was weaker in the steatotic area than non-steatotic area, suggesting that steatosis is associated with the impaired peroxisomal fatty acid β-oxidation.
Here, we also detected inducible cyclooxygenase 2 (COX-2) because COX-2-produced prostaglandins is a class of substrates of ACOX1, and COX-2/ACOX1 formed an alternate pathway for fatty acid β-oxidation [32]. Interestingly, in contrast to ACOX1, COX-2 staining was much stronger in the steatotic area than non-steatotic area. Thus, under the ACOX1 suppression, the induced COX-2 may increase the accumulation of prostaglandins and promote inflammation and lipid peroxidation [33]. Indeed, necroinflammation along with lipid droplets was observed in Figure 7, and lipid peroxidation marker malondialdehyde (MDA) was mainly detected in the steatotic area (Figure 8). These results suggest that the suppressed ACOX1 but not the increased peroxisomal membrane proteins PEX16 and PMP70 leads to steatosis and the subsequent advancement to necroinflammation in the liver.
4. Discussion
There is an interaction between liver and adipose tissues. It is well known that obesity leads to hepatic steatosis. In this study, we investigated the effects of liver peroxisomal function on the development of diet-induced obesity. We found that HFD-induced body weight gain, adipose tissue expansion, and glucose intolerance were observed in the Pex16fl/fl mice rather than in the Pex16Alb-Cre mice that lack structural peroxisomes, and consistently, HFD-induced steatosis was observed in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice. Interestingly, the Pex16Alb-Cre mice displayed lower basal levels of serum TG, cholesterol, free fatty acids, and ketone body, but serum levels of bile acids were higher in the Pex16Alb-Cre mice. What is more, the Pex16Alb-Cre mice exhibited elevated hepatocyte proliferation, which needs more cholesterol and energy (ATP) to construct cell membrane. These results suggest that the absence of liver PEX16 and peroxisomes inhibits the development of obesity through enhancing hepatocyte proliferation and increasing hepatic lipid consumption.
When we observed a difference in HFD-induced body weight gain between the Pex16Alb-Cre mice and the Pex16fl/fl mice, we did not observe a difference between the Pex16AdipoQ-Cre mice and the Pex16fl/fl mice, suggesting that it is liver peroxisomes but not adipose peroxisomes that affect HFD-induced obesity. However, recently, it was reported that HFD-induced obesity was developed in the Pex16AdipoQ-Cre mice to a greater extent than in the WT mice. Actually, HFD-induced body weight gain in the Pex16AdipoQ-Cre mice was not higher than in the WT mice until after 27 weeks of feeding [34]. Previously, we identified in human populations that CYP2A6 (CYP2A5 in mice) was associated with obesity with modest body mass index (BMI) but not with severe obesity with high BMIs, implicating that CYP2A6 is associated with the early stage of obesity; correspondingly, difference in HFD-induced body weight gain between the cyp2a5-/- mice and WT mice was observed as early as 6 weeks [35]. CYP2A5/6 is also mainly expressed in the liver. Thus, it seems that the liver affects early stage of obesity, but adipose tissues exert effect later than the liver. This claim is supported by a recent study showing that hepatic lipid metabolism-associated genes were increased in response to HFD as early as 3 h, and liver TG contents were increased at 6 h whereas adipose TG contents were increased after 12 h [36]. As for peroxisomes, it is possible that absence of liver peroxisomes initiates to speed up the development of obesity, and the expanded adipose tissues further worsen the occurring obesity. Unlike proteins and carbohydrates that are digested into amino acids and glucose and then directly enter the liver via portal vein, food fat is directly absorbed into lymph instead of blood. How the liver responds to HFD earlier than adipose tissues needs further studies.
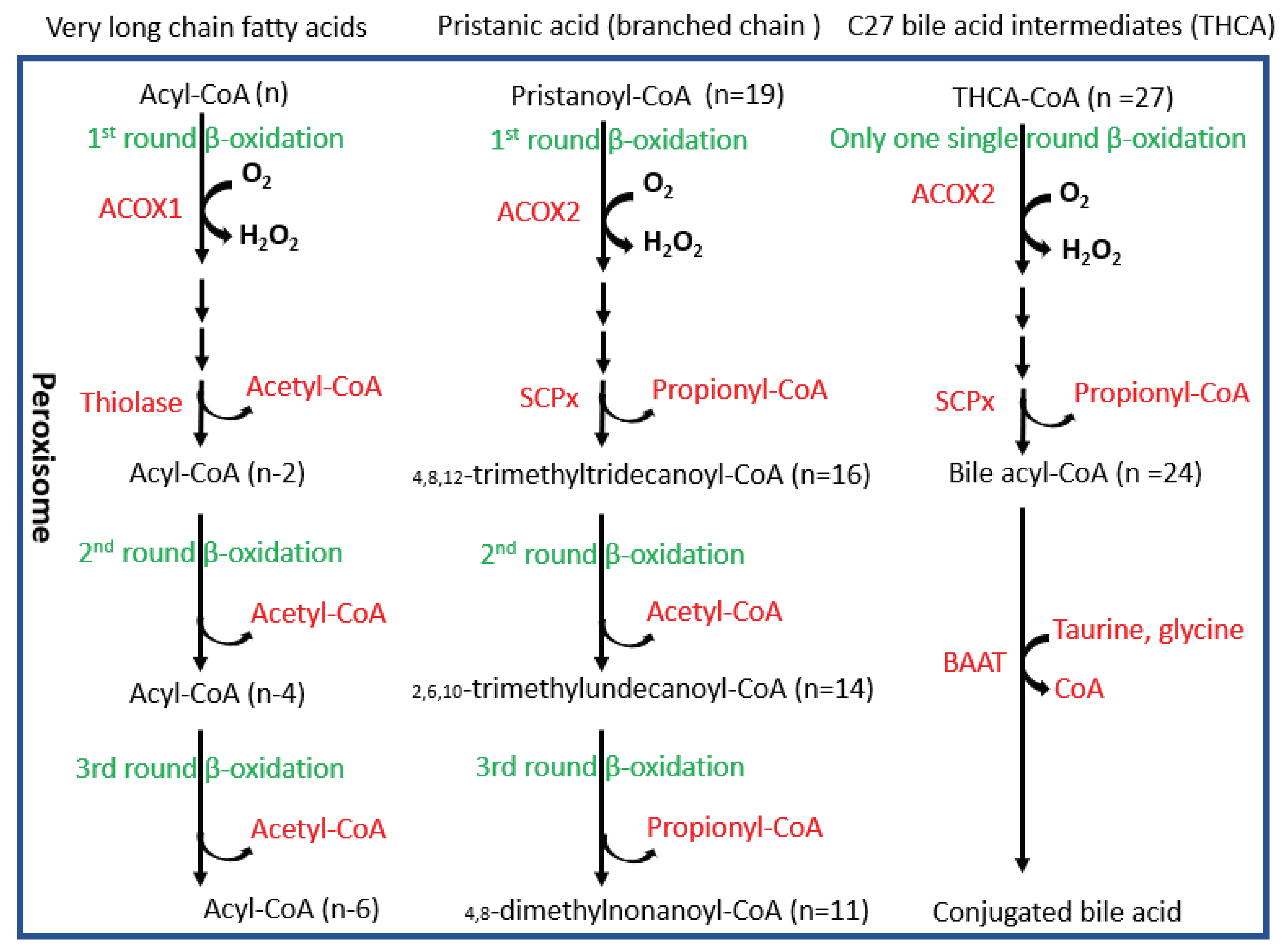
In peroxisomes, at least two fatty acid oxidation pathways are identified (Figure 9). ACOX1 and its downstream thiolase oxidize very long chain fatty acids, and ACOX2 and its downstream SCPx oxidize branched chain fatty acids [23]. Interestingly, ACOX1 is inducible by PPARα agonists but ACOX2 is not inducible [23]. An interesting observation in the Pex16Alb-Cre mice is that ACOX1 and thiolase were upregulated but ACOX2 and SCPx were downregulated. Basal levels of serum fatty acids were lower in the Pex16Alb-Cre mice than in the Pex16fl/fl mice, which is consistent with the upregulated ACOX1 pathway. Among polyunsaturated fatty acids (PUFA), arachidonic acid is special. Very minor free arachidonic acid is detected in cells. Usually, arachidonic acid is released from phospholipid by phospholipase A2 (PLA2) and is used by COX-2 for synthesis of prostaglandins and thromboxane, both of which are substrates of ACOX1 [37]. When expression of COX-2 is elevated but ACOX1 is suppressed, these inflammatory mediators are supposed to be accumulated and subjected to lipid peroxidation, as we observed in human samples (Figure 8).
The downregulation of ACOX2 pathway may lead to accumulation of branched chain fatty acids. Good examples of branched fatty acids are phytanic acid and pristanic acid. Pristanic acid is a 2-methyl-branched chain fatty acid that can be directly β-oxidized by ACOX2. Phytanic acid is a 3-methyl-branched chain fatty acid that needs to be α-oxidized to pristanic acid. Phytanic acid is derived from phytol and phytol is derived from the plant chlorophyll. Humans obtain phytanic acid primarily from dairy products and from fats of ruminant animals. Bacteria in the rumen of these animals can digest plant chlorophyll to release phytol, which is absorbed and further metabolized to phytanic acid by the ruminant animals. Interestingly, phytanic and pristanic acid are PPARα agonists [38]. Phytanic acid accumulation was observed in patients lacking functional peroxisomes [39,40]. Serum fatty acids were decreased in the Pex16Alb-Cre mice, but we couldn’t detect whether phytanic acid accumulated in the Pex16Alb-Cre mice. However, it is reasonable that the upregulation of PPARα-regulated ACOX1 in the Pex16Alb-Cre mice is associated with phytanic acid accumulation resulted from the downregulation of ACOX2.
It is well known that bile acids are synthesized from cholesterol and cholesterol is an important component of bio-membrane. Acetyl CoA produced by peroxisomal fatty acid β-oxidation can be used for cholesterol synthesis [41]. As shown in Figure 9, besides branched fatty acids, C27 bile acid intermediates are also oxidized by ACOX2 and ultimately matured into C24 conjugated bile acids in peroxisomes [42,43]. Interestingly, even though ACOX2 was downregulated, serum bile acids were still elevated in the Pex16Alb-Cre mice. Consistently, serum cholesterol was decreased in the Pex16Alb-Cre mice. Bile acids are metabolic signals to promote liver regeneration [44]. We observed an increased number of hepatocytes and positive staining of cell proliferation markers PCNA and Ki67 in the Pex16Alb-Cre mice, suggesting that hepatocyte proliferation occurs in the Pex16Alb-Cre mice. Thus, cholesterol-derived bile acids may promote liver regeneration and cholesterol itself is used for cell membrane structure material for proliferated hepatocytes. Liver regeneration is critical for survival in drug-induced acute liver failure [45]. It is possible that hepatocyte proliferation is a reason why the Pex16Alb-Cre mice were resistant against the HFD-induced steatosis and obesity. As a principle of evidence, thioacetamide-induced liver injury was not observed in the Pex16Alb-Cre mice while severe liver injury was observed in the Pex16fl/fl mice (Supplemental Figure 1). Furthermore, recently, we showed that the Pex16Alb-Cre mice were resistant to alcohol-induced steatosis [22]. Further studies are needed to address the effects of peroxisomes on fatty acids-cholesterol-bile acid metabolism, hepatocyte proliferation, and resistance against steatosis and obesity.
Financial support statement: This work was supported in part by NIH grant P20 GM103434 to the West Virginia IDeA Network of Biomedical Research Excellence.
Conflicts of Interest
None.
References
- Bhala, N.; Jouness, R.I.; Bugianesi, E. Epidemiology and natural history of patients with NAFLD. Curr Pharm Des 2013, 19, 5169–5176. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.W.; Adams, L.A. Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci 2011, 48, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr 2001, 21, 193–230. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K. Peroxisome proliferators and peroxisome proliferator-activated receptor alpha: biotic and xenobiotic sensing. Am J Pathol. 2004, 164, 2305–2321. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Veiga, F.M.S.; Graus-Nunes, F.; Rachid, T.L.; Barreto, A.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Anti-obesogenic effects of WY14643 (PPAR-alpha agonist): Hepatic mitochondrial enhancement and suppressed lipogenic pathway in diet-induced obese mice. Biochimie. 2017, 140, 106–116. [Google Scholar] [CrossRef]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E. , et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Huang, J.; Jia, Y.; Fu, T. , et al. Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J 2012, 26, 628–638. [Google Scholar] [CrossRef]
- Gao, Q.; Jia, Y.; Yang, G. , et al. PPARα-Deficient ob/ob Obese Mice Become More Obese and Manifest Severe Hepatic Steatosis Due to Decreased Fatty Acid Oxidation. Am J Pathol 2015, 185, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S. , et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ward, S.C.; Cederbaum, A.I. , et al. Alcoholic fatty liver is enhanced in CYP2A5 knockout mice: The role of the PPARα-FGF21 axis. Toxicology 2017, 379, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, X.; Ward, S.C. , et al. CYP2A6 is associated with obesity: studies in human samples and a high fat diet mouse model. Int J Obes (Lond) 2019, 43, 475–486. [Google Scholar] [CrossRef]
- Chen, X.; Acquaah-Mensah, G.K.; Denning, K.L.; Peterson, J.M.; Wang, K.; Denvir, J.; Hong, F.; Cederbaum, A.I.; Lu, Y. High-fat diet induces fibrosis in mice lacking CYP2A5 and PPARα: a new model for steatohepatitis-associated fibrosis. Am J Physiol Gastrointest Liver Physiol. 2020, 319, G626–G635. [Google Scholar] [CrossRef]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature. 2017, 542, 251–254. [Google Scholar] [CrossRef]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef]
- Kim, P.K.; Mullen, R.T. PEX16: a multifaceted regulator of peroxisome biogenesis. Front Physiol. 2013, 4, 241. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Mullen, R.T.; Schumann, U.; Lippincott-Schwartz, J. The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J Cell Biol. 2006, 173, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Honsho, M.; Tamura, S.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; Fujiki, Y. Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am J Hum Genet. 1998, 63, 1622–1630. [Google Scholar] [CrossRef]
- Chen, X.; Denning, K.L.; Mazur, A.; Lawrence, L.M.; Wang, X.; Lu, Y. Under peroxisome proliferation acyl-CoA oxidase coordinates with catalase to enhance ethanol metabolism. Free Radic Biol Med. 2023, 208, 221–228. [Google Scholar] [CrossRef]
- Wanders, R.J.; Denis, S.; Wouters, F.; Wirtz, K.W.; Seedorf, U. Sterol carrier protein X (SCPx) is a peroxisomal branched-chain beta-ketothiolase specifically reacting with 3-oxo-pristanoyl-CoA: a new, unique role for SCPx in branched-chain fatty acid metabolism in peroxisomes. Biochem Biophys Res Commun. 1997, 236, 565–569. [Google Scholar] [CrossRef]
- Xu, Y.; Denning, K.L.; Lu, Y. PPARα agonist WY-14,643 induces adipose atrophy and fails to blunt chronic ethanol-induced hepatic fat accumulation in mice lacking adipose FGFR1. Biochem Pharmacol. 2021, 192, 114678. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARα. Int J Mol Sci. 2016, 17, 2093. [Google Scholar] [CrossRef]
- Schreiber, R.; Xie, H.; Schweiger, M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochim Biophys Acta Mol Cell Biol Lipids. 2019, 1864, 880–899. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Kunze, M.; Stangl, H.; Porter, F.D.; Berger, J. Peroxisomal cholesterol biosynthesis and Smith-Lemli-Opitz syndrome. Biochem Biophys Res Commun. 2006, 345, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xu, J.; Wu, Y.; Liang, B.; Yan, M.; Sun, C.; Wang, D.; Hu, X.; Liu, L.; Hu, W.; Shao, Y.; Xing, D. The potential role and mechanism of circRNA/miRNA axis in cholesterol synthesis. Int J Biol Sci. 2023, 19, 2879–2896. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Houten, S.M. Peroxisomes and bile acid biosynthesis. Biochim Biophys Acta. 2006, 1763, 1427–1440. [Google Scholar] [CrossRef]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Xu, Y.; Denning, K.L.; Lu, Y. PPARα agonist WY-14,643 induces the PLA2/COX-2/ACOX1 pathway to enhance peroxisomal lipid metabolism and ameliorate alcoholic fatty liver in mice. Biochem Biophys Res Commun. 2022, 613, 47–52. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Park, H.; He, A.; Tan, M.; Johnson, J.M.; Dean, J.M.; Pietka, T.A.; Chen, Y.; Zhang, X.; Hsu, F.F.; Razani, B.; Funai, K.; Lodhi, I.J. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J Clin Invest. 2019, 129, 694–711. [Google Scholar] [CrossRef]
- Wang, K.; Chen, X.; Ward, S.C.; Liu, Y.; Ouedraogo, Y.; Xu, C.; Cederbaum, A.I.; Lu, Y. CYP2A6 is associated with obesity: studies in human samples and a high fat diet mouse model. Int J Obes (Lond). 2019, 43, 475–486. [Google Scholar] [CrossRef]
- Ding, L.; Sun, W.; Balaz, M.; He, A.; Klug, M.; Wieland, S.; Caiazzo, R.; Raverdy, V.; Pattou, F.; Lefebvre, P.; Lodhi, I.J.; Staels, B.; Heim, M.; Wolfrum, C. Peroxisomal β-oxidation acts as a sensor for intracellular fatty acids and regulates lipolysis. Nat Metab. 2021, 3, 1648–1661. [Google Scholar] [CrossRef]
- Diczfalusy, U.; Alexson, S.E. Identification of metabolites from peroxisomal beta-oxidation of prostaglandins. Journal of lipid research. 1990, 31, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.W.; van Der Burg, B.; Jansen, G.A.; Wanders, R.J.; Poll-The, B.T.; van Der Saag, P.T. Pristanic acid and phytanic acid: naturally occurring ligands for the nuclear receptor peroxisome proliferator-activated receptor alpha. J Lipid Res. 2000, 41, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, G.P.; Van Veldhoven, P.P.; Casteels, M. Peroxisomal lipid degradation via beta- and alpha-oxidation in mammals. Cell Biochem Biophys. 2000, 32, 73–87. [Google Scholar] [CrossRef]
- Verhoeven, N.M.; Wanders, R.J.; Poll-The, B.T.; Saudubray, J.M.; Jakobs, C. The metabolism of phytanic acid and pristanic acid in man: a review. J Inherit Metab Dis. 1998, 21, 697–728. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Kunze, M.; Stangl, H.; Porter, F.D.; Berger, J. Peroxisomal cholesterol biosynthesis and Smith-Lemli-Opitz syndrome. Biochem Biophys Res Commun. 2006, 345, 205–209. [Google Scholar] [CrossRef]
- Charles, K.N.; Shackelford, J.E.; Faust, P.L.; Fliesler, S.J.; Stangl, H.; Kovacs, W.J. Functional Peroxisomes Are Essential for Efficient Cholesterol Sensing and Synthesis. Front Cell Dev Biol. 2020, 8, 560266. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Houten, S.M. Peroxisomes and bile acid biosynthesis. Biochim Biophys Acta. 2006, 1763, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Wang, X.; Xu, G.; Yan, Q.; Huang, W. Bile acid signaling and liver regeneration. Biochim Biophys Acta. 2015, 1849, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.M.; McGill, M.R.; Apte, U. Mechanisms and biomarkers of liver regeneration after drug-induced liver injury. Adv Pharmacol. 2019, 85, 241–262. [Google Scholar] [CrossRef]
Figure 1.
HFD induced body weight gain in the Pex16fl/fl mice and Pex16AdipoQ-Cre mice but not in the Pex16Alb-Cre mice. (A) Body weight gain in the HFD-fed mice; (B) Repeated Measures ANOVA analysis; (C) Liver index and fat index in the HFD-fed WT and AKO mice; (D) Lipid droplets in liver sections from the HFD-fed WT and AKO mice; (E) Adipose inflammation (Arrows showing inflammation “Crown”). WT, Pex16fl/fl mice; AKO, adipose-specific PEX16 knockout (Pex16AdipoQ-Cre) mice; LKO, liver-specific PEX16 knockout (Pex16Alb-Cre) mice.
Figure 1.
HFD induced body weight gain in the Pex16fl/fl mice and Pex16AdipoQ-Cre mice but not in the Pex16Alb-Cre mice. (A) Body weight gain in the HFD-fed mice; (B) Repeated Measures ANOVA analysis; (C) Liver index and fat index in the HFD-fed WT and AKO mice; (D) Lipid droplets in liver sections from the HFD-fed WT and AKO mice; (E) Adipose inflammation (Arrows showing inflammation “Crown”). WT, Pex16fl/fl mice; AKO, adipose-specific PEX16 knockout (Pex16AdipoQ-Cre) mice; LKO, liver-specific PEX16 knockout (Pex16Alb-Cre) mice.
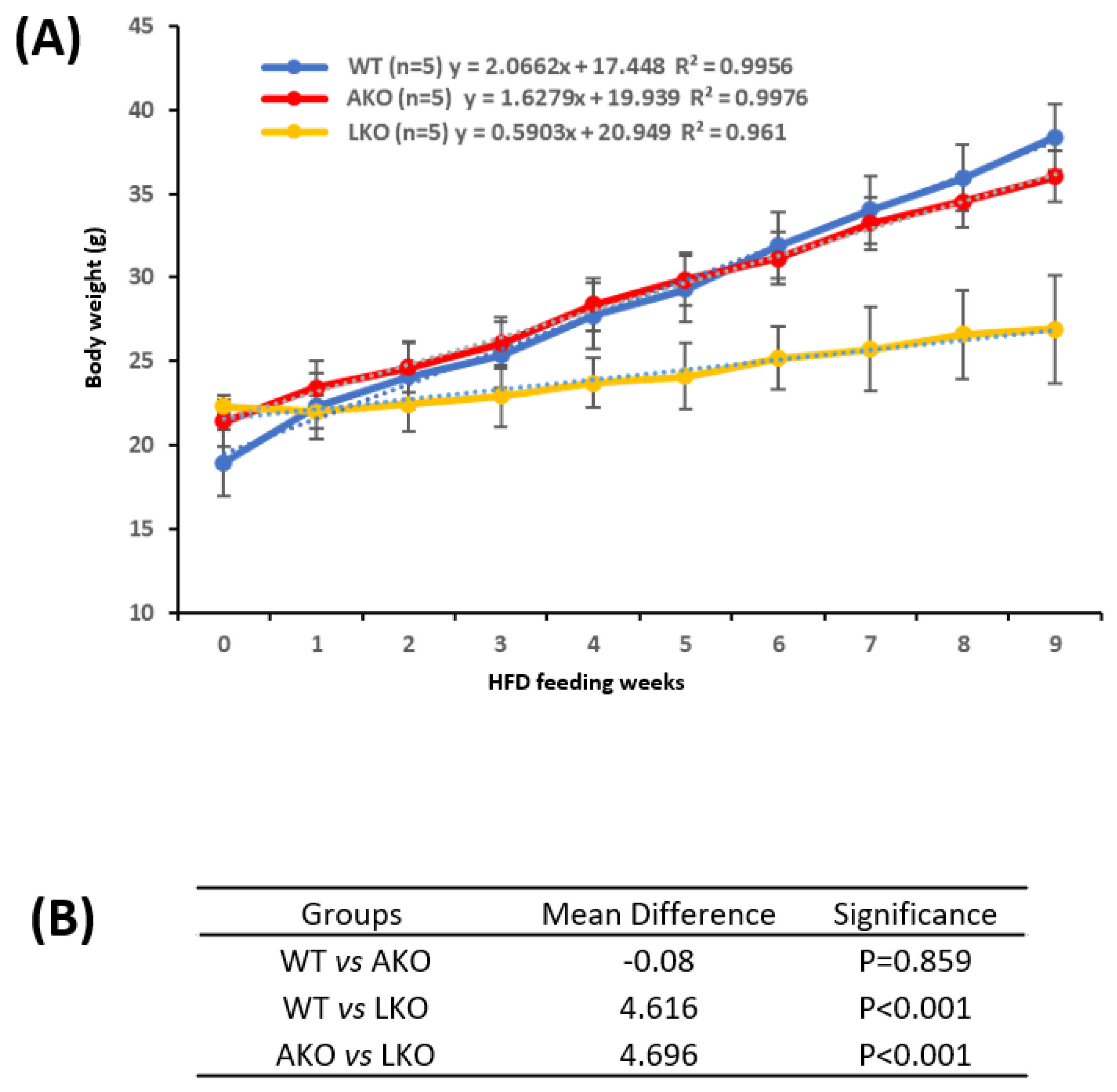
Figure 2.
HFD induced obesity in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice. (A) Gonadal adipose tissue expansion; (B) Adipose inflammation as indicated by “Crown”; (C) Hyperglycemia; (D) Glucose tolerance test.
Figure 2.
HFD induced obesity in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice. (A) Gonadal adipose tissue expansion; (B) Adipose inflammation as indicated by “Crown”; (C) Hyperglycemia; (D) Glucose tolerance test.
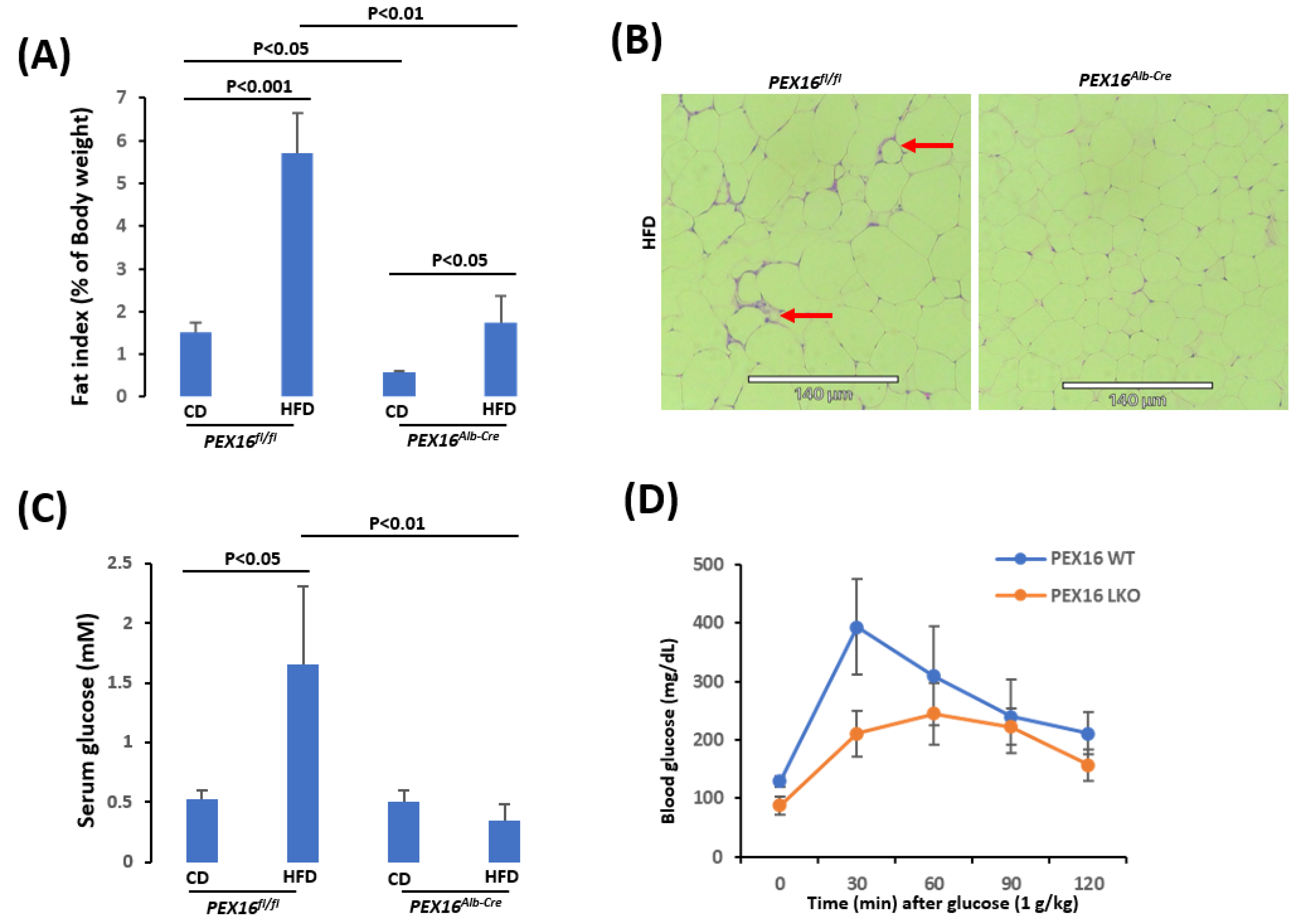
Figure 3.
HFD induced steatosis in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice. (A) Liver index; (B) Liver TG contents; (C) HE staining showing lipid droplets (Arrows) in liver sections; (D) Steatosis quantification; (E) Hepatocyte nuclear number.
Figure 3.
HFD induced steatosis in the Pex16fl/fl mice but not in the Pex16Alb-Cre mice. (A) Liver index; (B) Liver TG contents; (C) HE staining showing lipid droplets (Arrows) in liver sections; (D) Steatosis quantification; (E) Hepatocyte nuclear number.
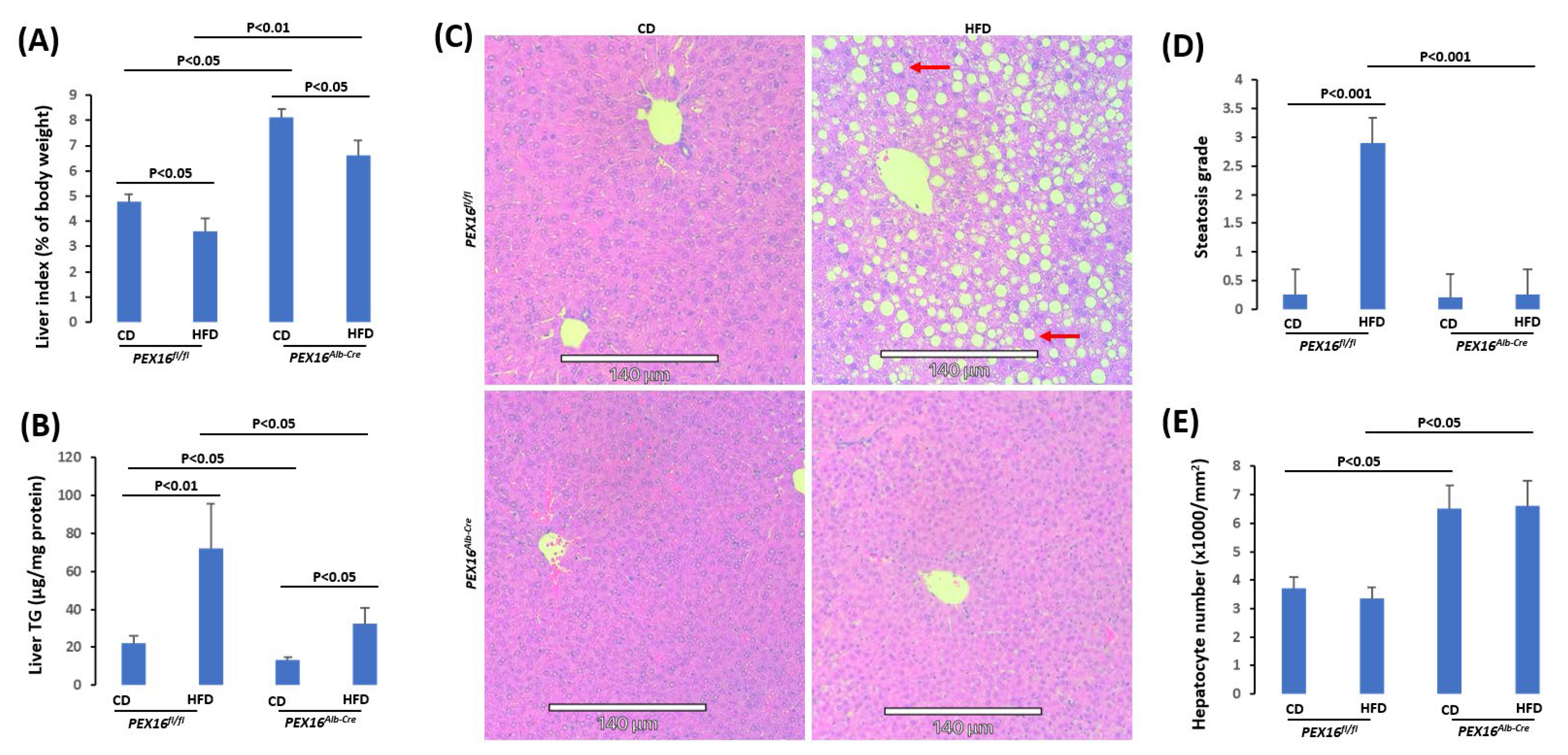
Figure 4.
The absence of liver PEX16 leads to hepatocyte proliferation. (A) PCNA positive staining was observed in the Pex16Alb-Cre mice but not in the Pex16fl/fl mice; (B) PCNA staining quantification; (C) Ki67 staining was observed in the Pex16Alb-Cre mice but not in the Pex16fl/fl mice; (D) Steatosis quantification; (E) Ki67 staining quantification. Arrows show representative positive staining.
Figure 4.
The absence of liver PEX16 leads to hepatocyte proliferation. (A) PCNA positive staining was observed in the Pex16Alb-Cre mice but not in the Pex16fl/fl mice; (B) PCNA staining quantification; (C) Ki67 staining was observed in the Pex16Alb-Cre mice but not in the Pex16fl/fl mice; (D) Steatosis quantification; (E) Ki67 staining quantification. Arrows show representative positive staining.
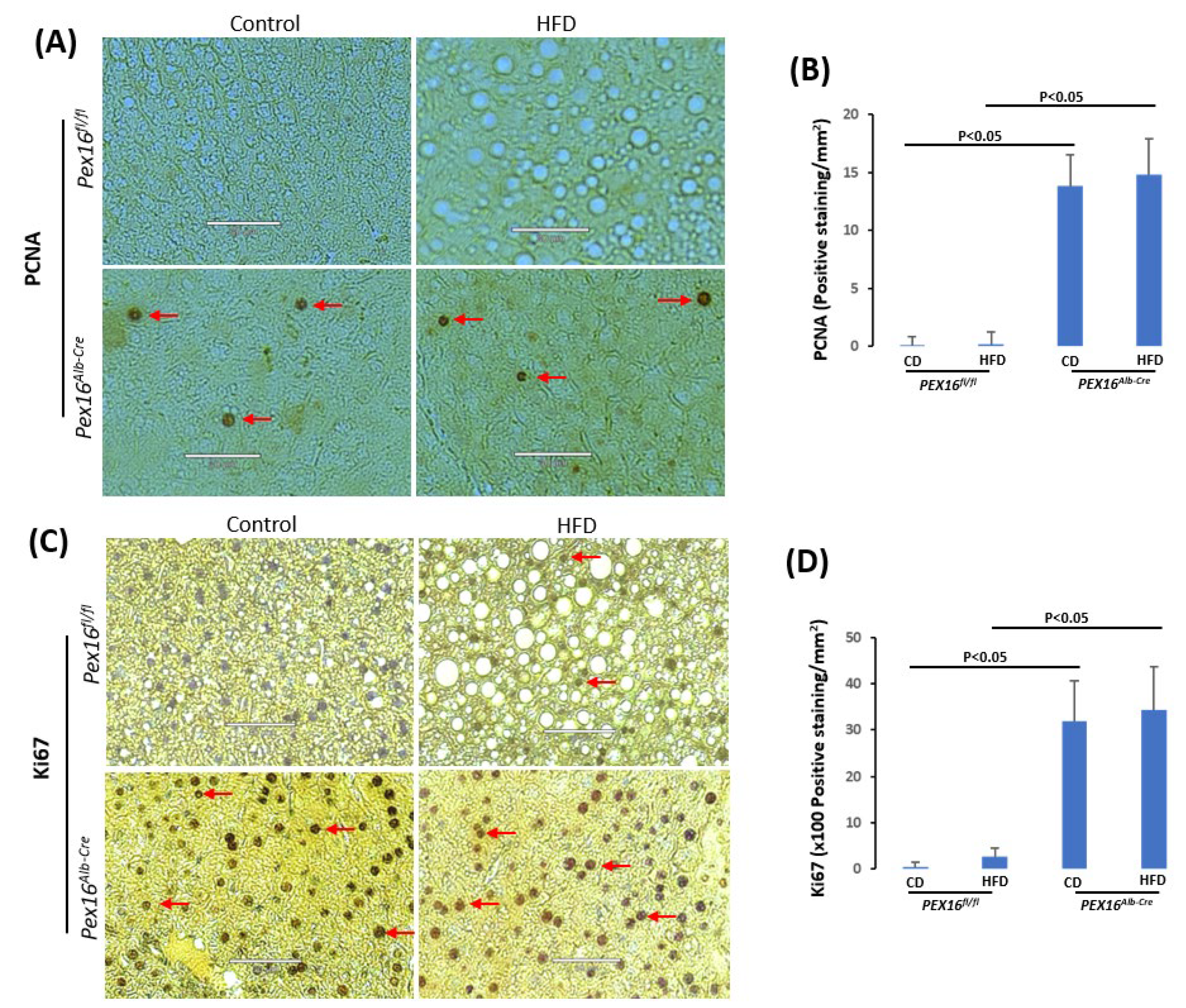
Figure 5.
Absence of liver PEX16 lowered serum free fatty acids, ketone bodies and TG. (A) Expression of liver peroxisomal fatty acid β-oxidation enzymes; (B) Serum fatty acids; (C) Serum β-hydroxybutyrate; (D) Expression of fat metabolism enzymes; (E) Serum TG.
Figure 5.
Absence of liver PEX16 lowered serum free fatty acids, ketone bodies and TG. (A) Expression of liver peroxisomal fatty acid β-oxidation enzymes; (B) Serum fatty acids; (C) Serum β-hydroxybutyrate; (D) Expression of fat metabolism enzymes; (E) Serum TG.
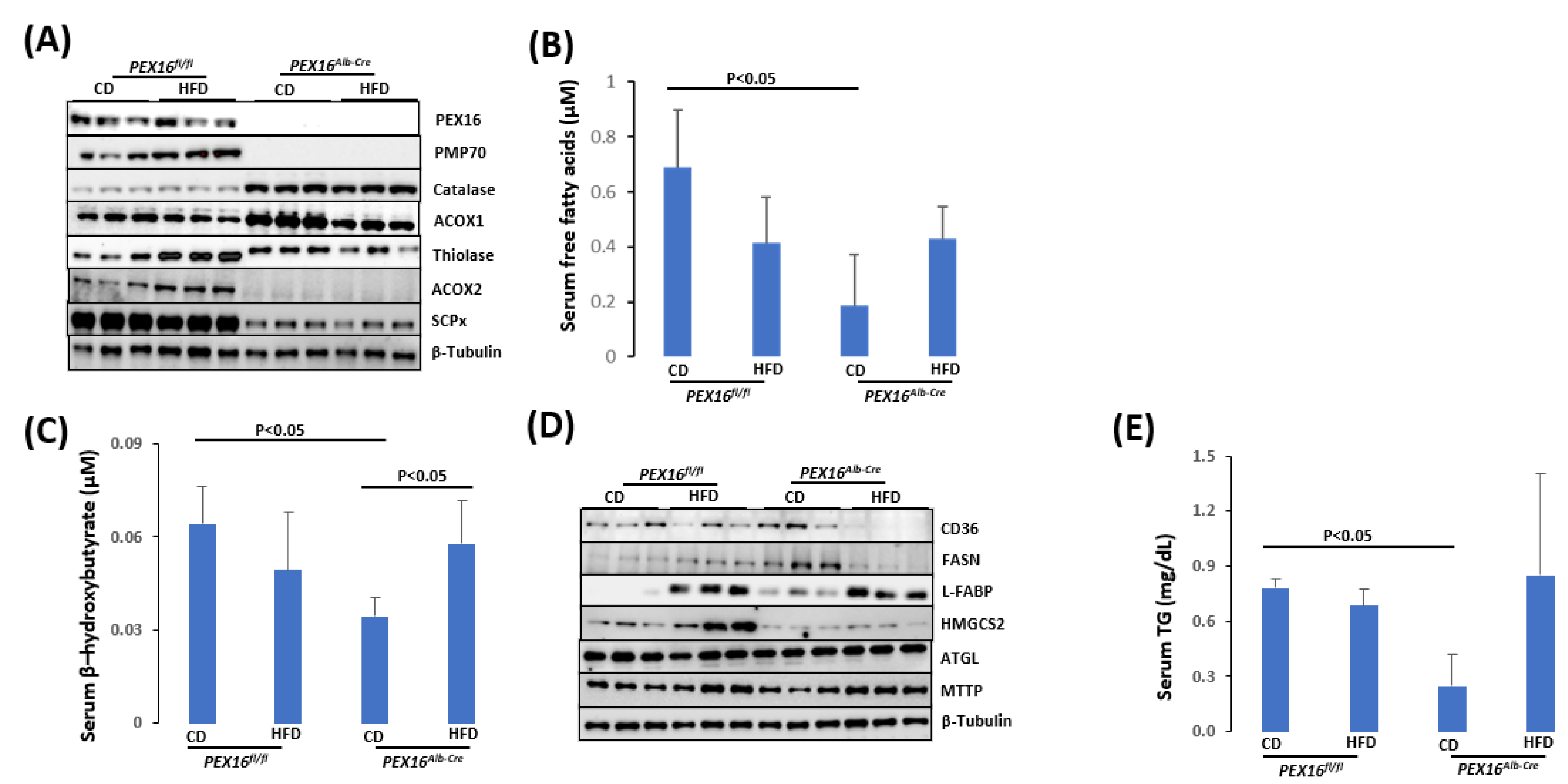
Figure 6.
The absence of liver PEX16 altered cholesterol and bile acid metabolism. (A) Expression of liver cholesterol and bile acid synthetic enzymes; (B) Serum cholesterol; (C) Serum bile acids.
Figure 6.
The absence of liver PEX16 altered cholesterol and bile acid metabolism. (A) Expression of liver cholesterol and bile acid synthetic enzymes; (B) Serum cholesterol; (C) Serum bile acids.
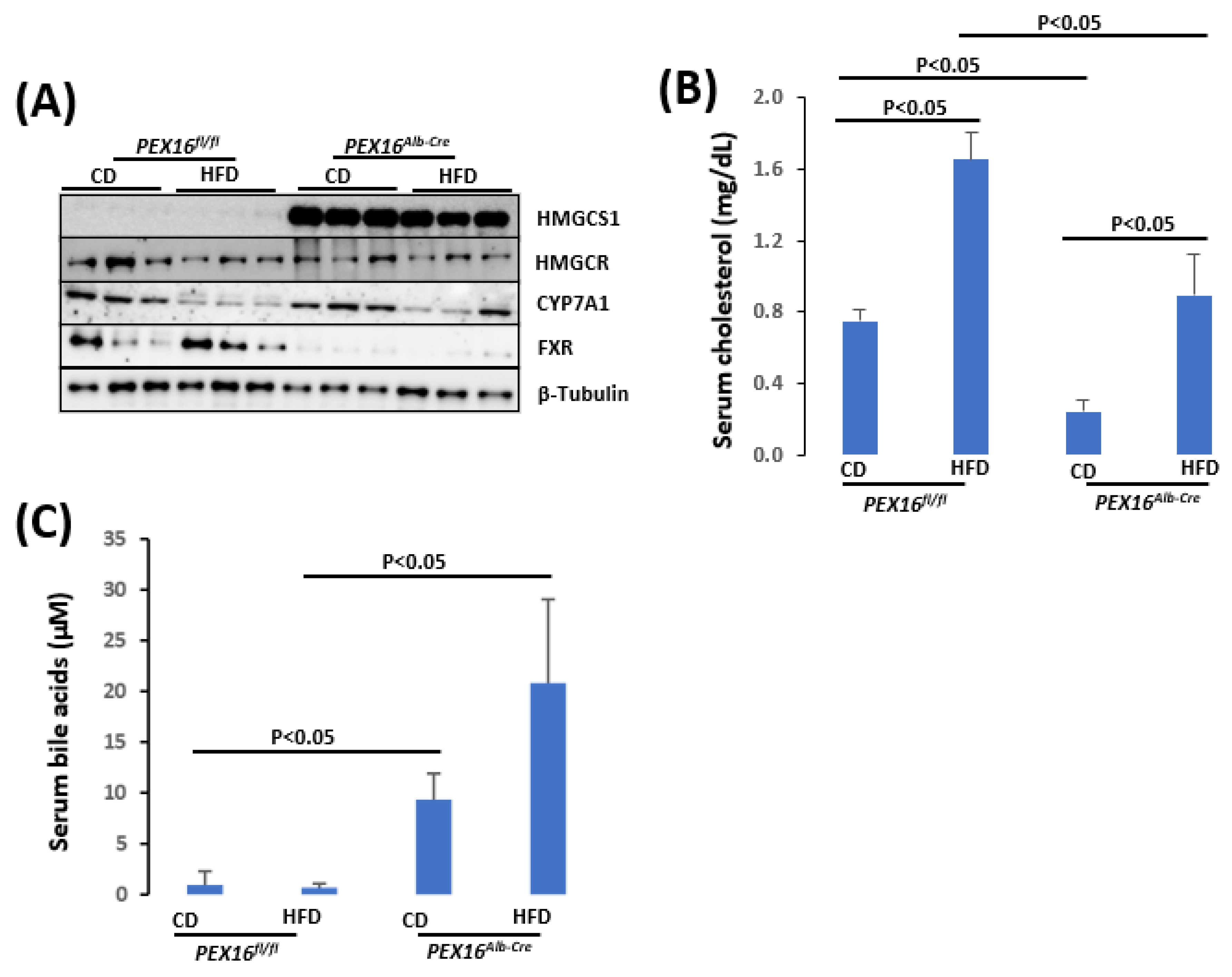
Figure 7.
HE staining in liver sections from patients with chronic liver diseases. Yellow arrows, green arrows and red arrows indicating lipid droplets, inflammation foci, and fibrosis, respectively. Black arrow shows ballooning degeneration.
Figure 7.
HE staining in liver sections from patients with chronic liver diseases. Yellow arrows, green arrows and red arrows indicating lipid droplets, inflammation foci, and fibrosis, respectively. Black arrow shows ballooning degeneration.
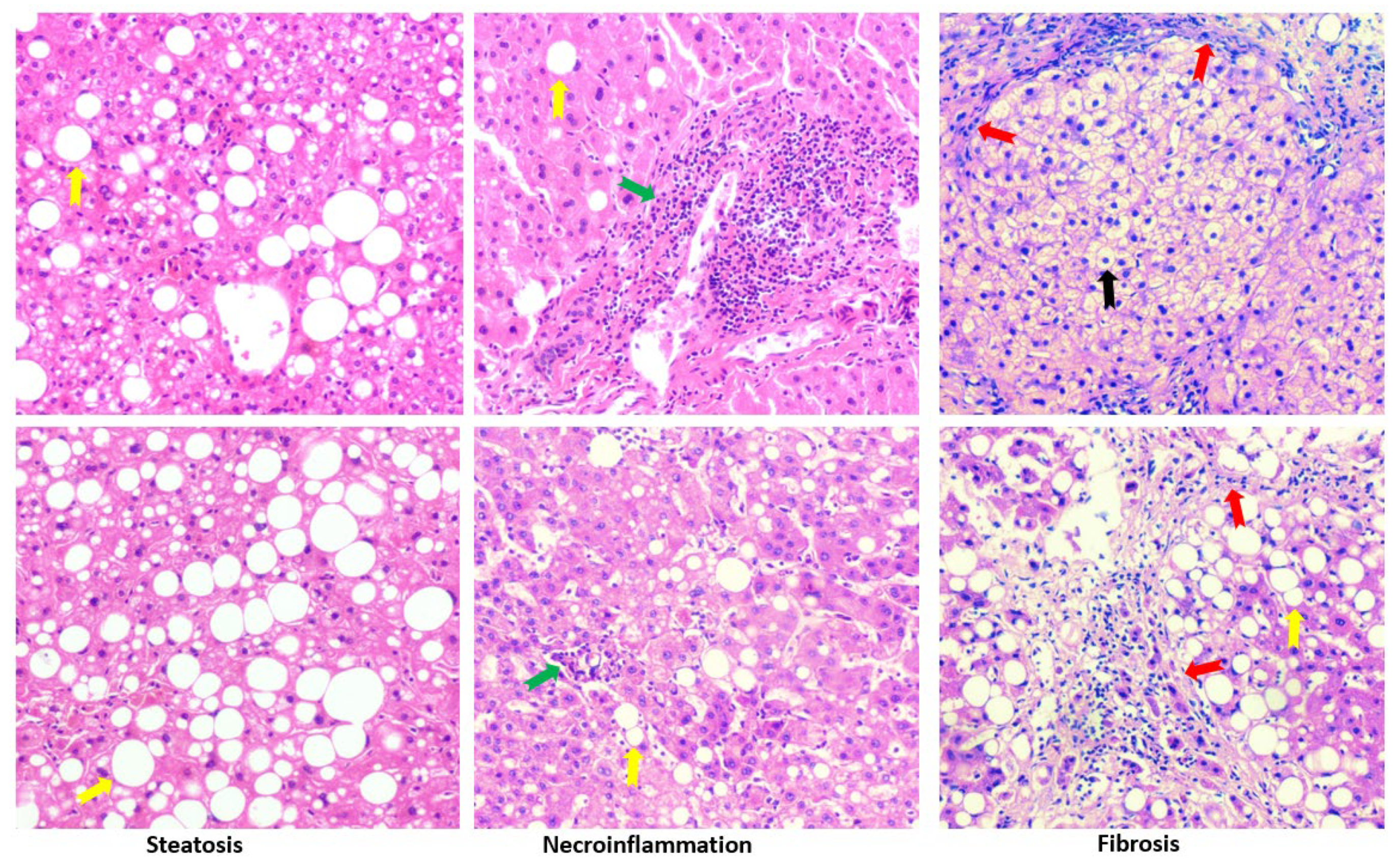
Figure 8.
IHC staining in liver sections from patients with chronic liver diseases.
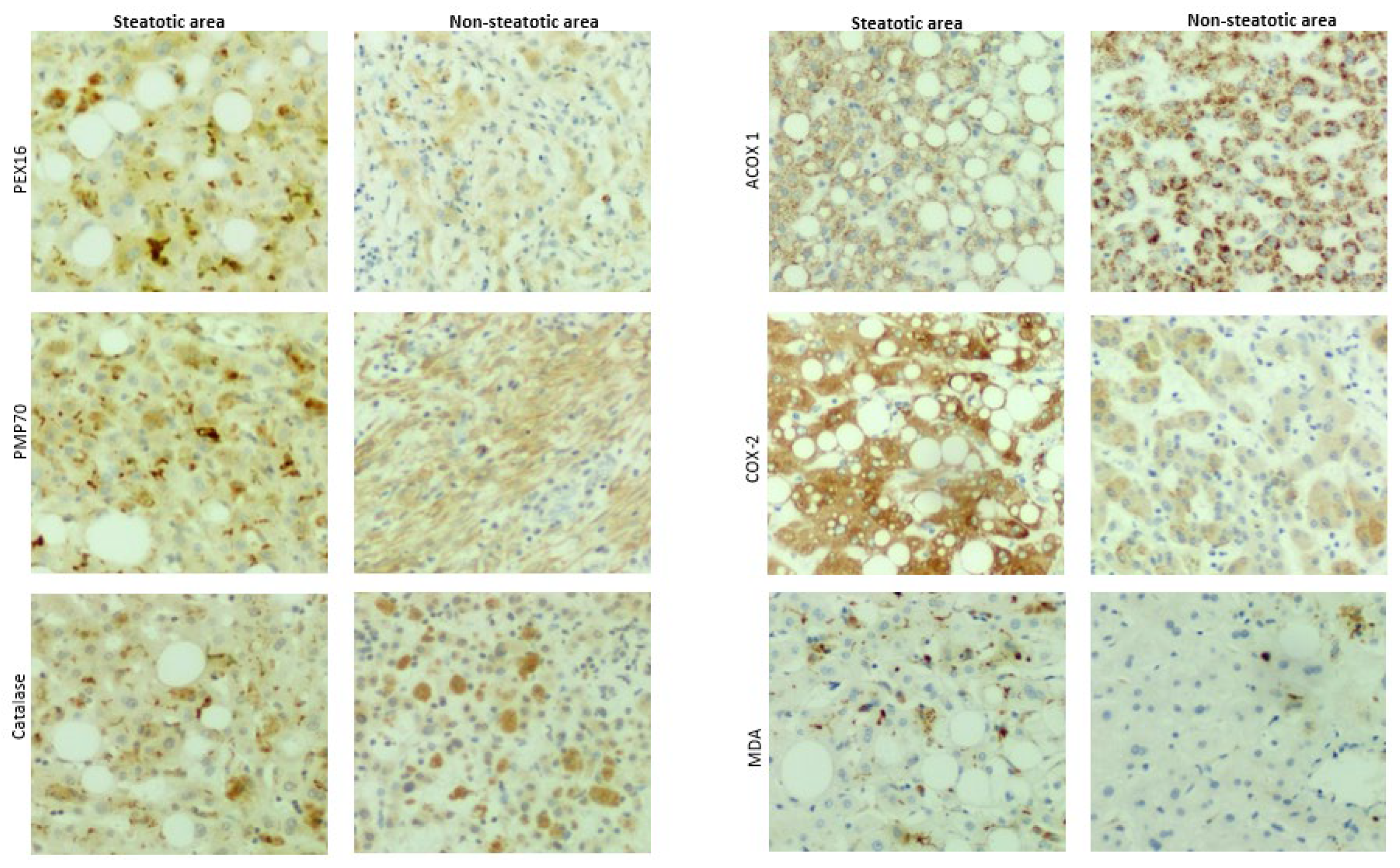
Figure 9.
Pathways for peroxisomal β-oxidation. BAAT, bile acyl-CoA: amino acid acyltransferase; THCA, 3α,7α,12α- trihydroxycholestanic acid.
Figure 9.
Pathways for peroxisomal β-oxidation. BAAT, bile acyl-CoA: amino acid acyltransferase; THCA, 3α,7α,12α- trihydroxycholestanic acid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



